K 0400-52-20 : 1998 (ISO 8288 : 1986)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が制定した日
本工業規格である。
JIS K 0400-52-20には,次に示す附属書がある。
附属書A(規定) 全金属定量のための試料の前処理
附属表B(規定) 完全抽出の確認方法(方法C)
K 0400-52-20 : 1998 (ISO 8288 : 1986)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 適用分野 ························································································································ 1
2A. 引用規格 ······················································································································ 2
第1章:方法A−フレーム原子吸光法による直接定量 ·································································· 3
3. 原理 ······························································································································ 3
4. 試薬 ······························································································································ 3
5. 装置 ······························································································································ 4
6. サンプリング方法及び試料 ································································································ 4
7. 手順 ······························································································································ 4
7.1 測定試料 ······················································································································ 4
7.2 空試験 ························································································································· 4
7.3 検量線用溶液の調製 ······································································································· 4
7.4 校正及び定量 ················································································································ 4
7.5 照合試験 ······················································································································ 5
8. 試験結果の表現 ··············································································································· 5
9. 試験報告 ························································································································ 5
第2章:方法B−錯価(APDC)及び抽出(MIBK)後のフレーム原子吸光法による定量 ···························· 6
10. 原理 ···························································································································· 6
11. 試薬 ···························································································································· 6
12. 装置 ···························································································································· 6
13. サンプリング方法及び試料······························································································· 6
14. 手順 ···························································································································· 6
14.1 測定試料 ····················································································································· 6
14.2 錯化及び抽出 ··············································································································· 6
14.3 空試験 ························································································································ 7
14.4 検量線用溶液の調製 ······································································································ 7
14.5 校正及び定量 ··············································································································· 7
15. 試験結果の表現 ············································································································· 8
15.1 計算 ··························································································································· 8
15.2 精度 ··························································································································· 8
15.3 妨害物質 ····················································································································· 8
第3章:方法C−錯価(HMA-HMDC)及び抽出(DIPK-キシレン)後のフレーム原子吸光法による定量 ······· 8
16. 試験報告 ······················································································································ 8
17. 原理 ···························································································································· 8
K 0400-52-20 : 1998 (ISO 8288 : 1986) 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
18. 試薬 ···························································································································· 9
19. 装置 ···························································································································· 9
20. サンプリング方法及び試料······························································································· 9
21. 手順 ···························································································································· 9
21.1 測定試料 ····················································································································· 9
21.2 錯化及び抽出 ··············································································································· 9
21.3 空試験 ······················································································································· 10
21.4 検量線用溶液の調製 ····································································································· 10
21.5 校正及び定量 ·············································································································· 10
22. 試験結果の表現 ············································································································ 10
22.1 計算 ·························································································································· 10
22.2 繰返し性及び再現性 ····································································································· 10
22.3 妨害物質 ···················································································································· 11
23. 試験報告 ····················································································································· 11
附属書A(規定) 全金属定量のための試料の前処理 ································································· 12
附属書B(規定) 完全抽出の確認方法(方法C) ···································································· 13
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 0400-52-20 : 1998
(ISO 8288 : 1986)
水質−コバルト,
ニッケル,銅,亜鉛,
カドミウム及び
鉛の定量−原子吸光法
Water quality−Determination of cobalt, nickel, copper, zinc, cadmium
and lead−Flame atomic absorption spectrometric methods
序文 この規格は,1986年に発行されたISO 8288, Water quality−Determination of cobalt nickel, copper, zinc,
cadmium and lead−Flame atomic absorption spectrometric methodsを翻訳し,技術的内容及び規格票の様式を
変更することなく作成した日本工業規格である。
なお,この規格で点線の下線を施してある箇所は,原国際規格にない事項である。
1. 適用範囲 この規格は,コバルト,ニッケル,銅,亜鉛,カドミウム及び鉛のフレーム原子吸光法に
関する3種類の方法について規定する。
第1章:方法A,フレーム原子吸光法による直接定量
第2章:方法B,錯化 (APDC) 及び抽出 (MIBK) 後のフレーム原子吸光法による定量
第3章:方法C,錯化 (HMA-HMDC) 及び抽出(DIPK−キシレン)後のフレーム原子吸光法による定
量
2. 適用分野
2.1
方法Aは,対象元素の濃度が比較的高く,妨害がない場合に特に適している。
試料が複雑な組成又は性質が未知のとき,若しくは溶存成分濃度が高い場合(かん水又は汽水)は,方
法Aは不適当で,方法B又は方法Cを用いるとよい。
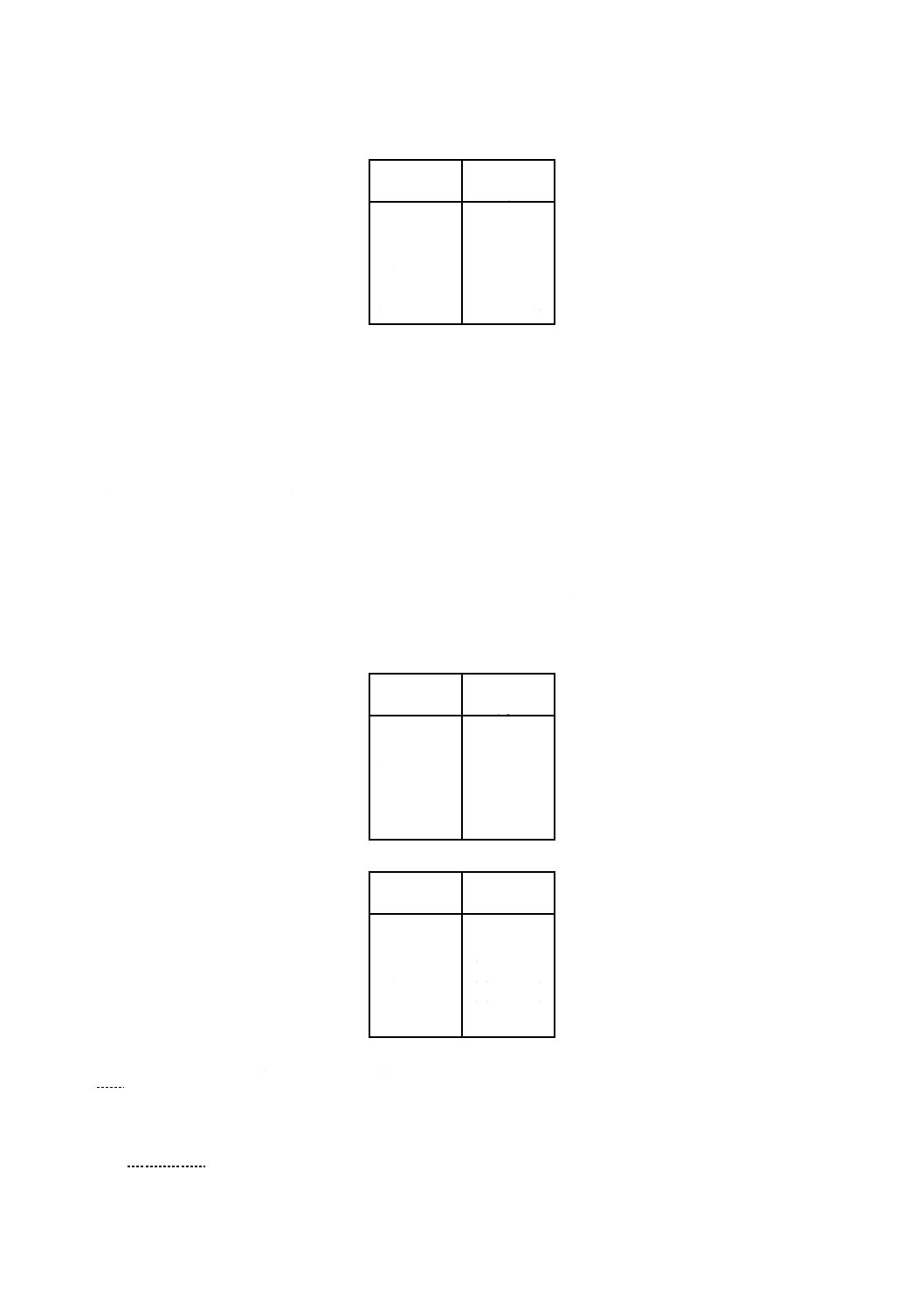
方法Aの定量範囲は,原子吸光分析装置の性能によって異なるが,おおむね表1に示す範囲である。
濃度が表の上限を超えるときは,分析の前に試料を薄めるとよい。
2
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1
対象元素
定量範囲
mg/l
コバルト
0.1 〜 10
ニッケル
0.1 〜 10
銅
0.05〜
6
亜鉛
0.05〜
2
カドミウム
0.02〜 10
鉛
0.2 〜 10
2.2
方法B及び方法Cは,試料(又は希釈試料)中の対象元素濃度が0.5μg/lを超えるときに適用する。
2.2.1
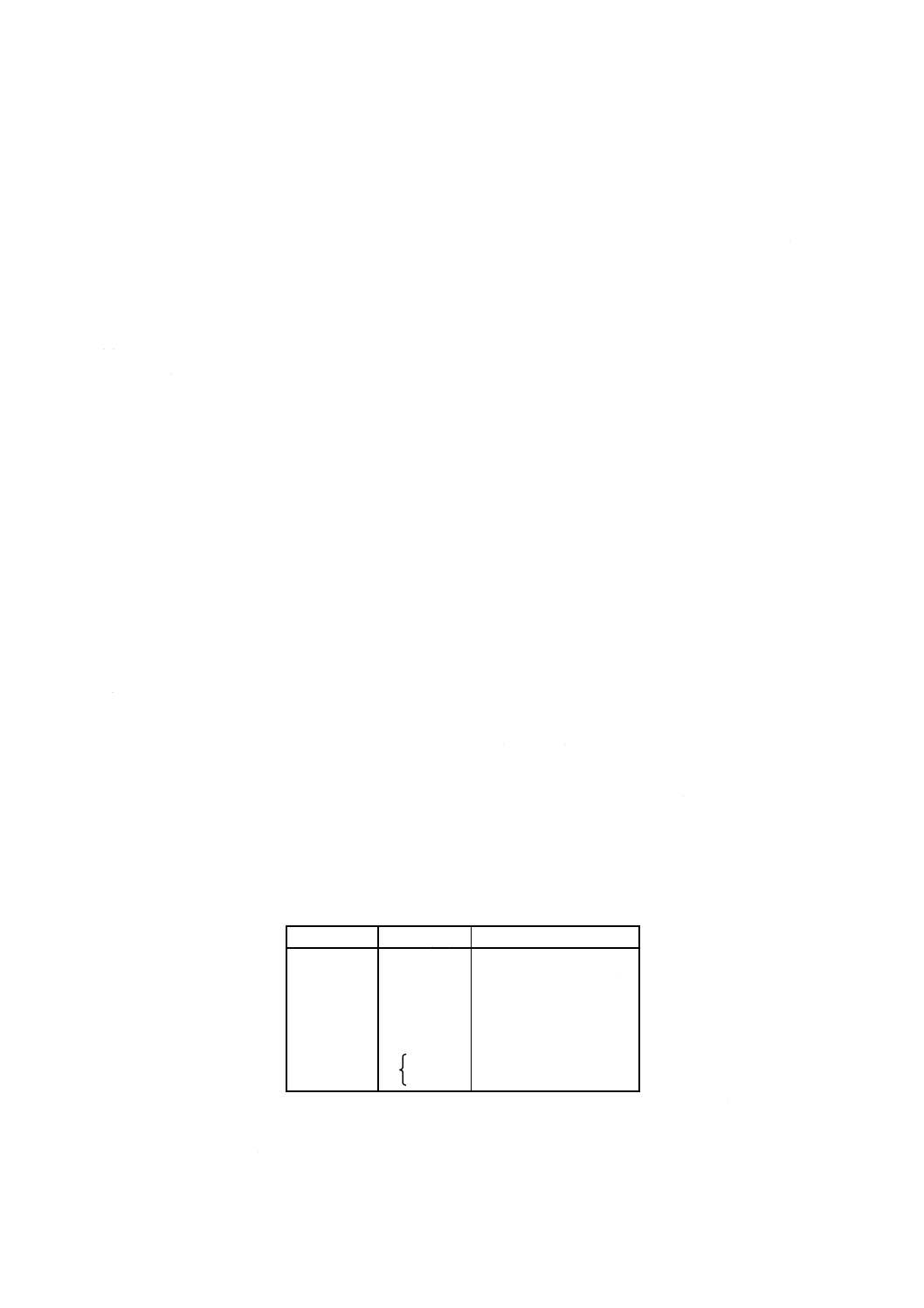
方法B 方法Bの定量範囲は,使用する原子吸光分析装置の性能によって異なるが,おおむね表2
に示す範囲である。
2.2.2
方法C 測定試料と抽出液の液量比が,22.2に示すように20 : 1の場合は,方法Cの定量範囲は表
3に示すとおりである。
これよりも低い濃度は,測定試料と抽出液の液量比を高くすることによって定量できる。この混合有機
溶媒は水にごくわずかしか溶けないので,液量比50 : 1が可能である。
方法Cでは,水相と有機相の分離がより速い。金属キレート,特にカドミウムキレートは,この混合有
機溶媒中ではより安定である。
備考1. 全金属を定量する場合は,分析前に試料の前処理(附属書Aの処理例を参照)が必要である。
2. 方法B及び方法Cは,試料(又は希釈試料)のCOD (ISO 6060) が500mg/lを超えるときは
不適当である。
表2
対象元素
定量範囲
μg/l
コバルト
1
〜200
ニッケル
1
〜200
銅
1
〜200
亜鉛
0.5
〜 50
カドミウム
0.5
〜 50
鉛
5
〜200
表3
対象元素
定量範囲
μg/l
コバルト
0.5
〜100
ニッケル
0.5
〜100
銅
0.5
〜100
亜鉛
0.2
〜 50
カドミウム
0.2
〜 50
鉛
2
〜200
2A. 引用規格 次に掲げる規格は規定を含んでおり,この規格に引用することによって,この規格の規定
を構成する。この規格制定の時点では,次に示す版が有効であった。すべて規格は改正されることがあり,
この規格に基づいて契約を結ぶ関係者は次の規格の最新版の適用の可能性を調査されたい。
JIS K 8005 容量分析用標準物質

3
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8103 ジエチルエーテル(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8264 ぎ酸(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8283 くえん酸一水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8701 鉛(試薬)
JIS K 8732 二硫化炭素(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8889 メタクレゾールパープル(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8903 4−メチル−2−ペンタノン(試薬)
JIS K 9062 ニッケル(試薬)
ISO 5725 : 1986, Precision of test methods−Determination of repeatability and reproducibility for a
standard test method by inter-laboratory tests1)
ISO 6060 : 1986, Water quality−Determination of the chemical oxygen demand
第1章:方法A−フレーム原子吸光法による直接定量
3. 原理 試料(又は希釈試料)の酸性ろ液の測定試料を原子吸光装置のフレーム中へ噴霧。
連続バックグラウンド補正システムを備えた分光器を用いるか,又はこのようなシステムがない場合は,
非固有吸収に対する補正を行った後,各元素の固有吸収の測定による各元素濃度の直接定量。
4. 試薬 試薬は,その使用が定量の正確さに影響を与えないように,すべて分析用と認められたもので
なければならない。用いる水は空試験を行ったとき,検出できる濃度の対象金属を含まないイオン交換水
又は蒸留水でなければならない。
4.1
硝酸ρ=1.4g/ml JIS K 8541に規定する硝酸。
4.2
硝酸c (HNO3) 1.5mol/l 硝酸 (4.1) 100mlを水600mlに加え,1000mlに薄める。
4.3
硝酸c (HNO3) 0.03mol/l 硝酸 (4.1) lmlを水400mlに加え,水で500mlに薄める。
4.4
金属 1lにつき金属1.000gを含む標準液2)
各元素について,[JIS K 9062に規定するニッケル(99.5%以上),JIS K 8005に規定する容量分析用標
準物質の亜鉛及び銅,JIS K 8701に規定する鉛(特級)]純金属1.000gをはかりとり,硝酸 (4.1) に溶か
し,加熱して完全に溶かす。冷後,各溶液を全量フラスコ1000mlに移し,水で標線まで薄め,混合する。
標準液の調製には,正確な組成の分かった金属塩を用いてもよい。
各標準液は,ポリエチレン又はほうけい酸ガラス容器に貯蔵する。
これらの標準液1mlは,それぞれの金属1.00mgを含む。
1) 現在はISO 5725-1〜4,-6 : 1994が発行されている。
2) 標準液は市販されている。
4
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 装置 通常の実験室設備,及び
原子吸光分析装置 対象金属用の中空陰極ランプ又は無電極放電ランプ,非固有吸収補正装置及びアセチ
レン−空気フレーム用のネブライザーバーナーを備えたもの。
機器条件の設定は,すべて製造業者の取扱説明書に従う。
備考 ガラス器具の清浄に関する注意 すべてのガラス器具は,硝酸 (4.2) に浸した後,水で洗わな
ければならない。
6. サンプリング方法及び試料
6.1
サンプリングには,硝酸 (4.2) で洗い,水ですすいだポリエチレン又はほうけい酸ガラス容器を用
いなければならない。
6.2
全金属を定量する場合は,試料採取直後に硝酸 (4.1) を加えてpH1〜2(通常,試料1lにつき酸2ml
で十分である。)にする。加えた酸の量を記録し,空試験 (7.2) にも同量の酸を用いる。
溶存金属だけを定量する場合は,試料採取直後できるだけ早く,孔径0.45μmのメンブレンフィルター
でろ過し,直ちにろ液に硝酸 (4.1) を加えてpH1〜2にする。
フィルターは,使用前に硝酸 (4.2) でよく洗い,水ですすいでおかなければならない。
7. 手順
7.1
測定試料 酸性にした試料 (6.2) から金属0.2〜1mgを含む測定試料(各金属の上限については,表
1参照)を全量フラスコ100mlにとる。水を標線まで加える。
7.2
空試験 定量と並行して,サンプリング及び定量の場合と同量の試薬を用いて同じ操作を行う。た
だし,測定試料の代わりに水を用いる。
7.3
検量線用溶液の調製 一連の定量に先立って,各金属の定量範囲にわたって少なくとも四つの検量
線用溶液を,各標準液 (4.4) から調製する。
これらの検量線用溶液は,標準液 (4.4) を硝酸 (4.3) で薄めて調製する。
7.4
校正及び定量
− 各対象金属に対して次のように操作する。吸光度の測定前に,対象金属の検量線用溶液 (7.3) を噴霧
し,表4の情報を用い,製造業者の指示に従って分光計を調整する。噴霧及びフレームの条件(噴霧
量,フレームの種類,フレーム中の光束の位置)を最適にする。水を用いてゼロ吸収の応答の調節を
行う。
表4
対象元素
波長 (nm)
フレーム
コバルト
240.7
アセチレン−空気
ニッケル
232.0
酸化性アセチレン−空気
銅
324.7
酸化性アセチレン−空気
亜鉛
213.8
アセチレン−空気
カドミウム
228.8
アセチレン−空気
鉛
283.3
アセチレン−空気
217.0
− 各対象金属について,検量線用溶液 (7.3) 及びゼロメンバーとして空試験液 (7.2) を噴霧する。検量
線用溶液の金属含有量,mg/lを横軸に,対応する吸光度を縦軸にプロットする。検量線は,例えば,
5試料ごとに検量線用溶液の吸光度を測定して,確認することが望ましい。
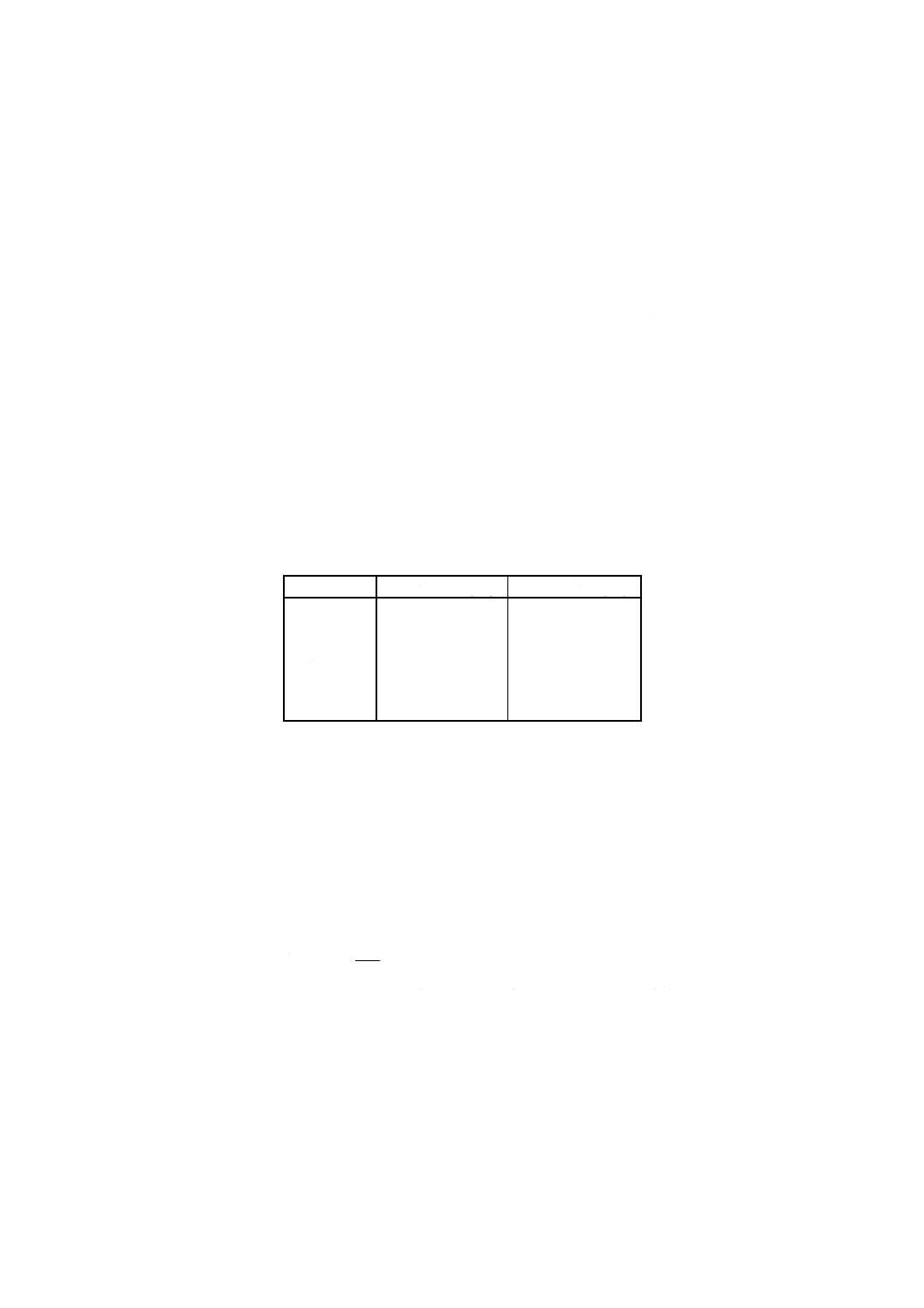
5
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 測定試料 (7.1) をバーナーのフレームに噴霧する。
− 対象金属の吸光度を測定し,各測定後,硝酸 (4.3) を噴霧してネブライザー系を洗浄する。
備考 非固有吸収の補正に関する注意 使用する分光計が,対象金属の固有吸収Aの信号を自動的に
示すバックグラウンド補正システムを備えていない場合は,非固有吸収A0を測定する必要があ
る。これは次のように行う。
− 対象金属の分析線の近くにあるスペクトル線,二つのスペクトル線の波長差はlnmを超えな
い線を選ぶ。
− 中空陰極ランプの内蔵ガス(アルゴン又はネオン)のスペクトル線,又はジルコニウム,重
水素の中空陰極ランプから放射されるスペクトル線を用いる(表5参照)。
− 測定試料を噴霧して分析線に対応する吸光度A0を測定する。
− 固有吸収を次の式から計算する。
A=A1−A0
ここに, A1: 分析波長における全吸光度
− フレーム条件及びランプに供給されるエネルギーは,吸光度A1,A0の測定の間変化してはな
らない。
表5
対象元素
A1測定波長 (nm)
A0測定波長 (nm)
コバルト
240.72
241 (D)
ニッケル
232.00
232 (D)
銅
324.75
325 (Zr)
亜鉛
213.86
214 (D)
カドミウム
228.80
229 (D)
鉛
283.30
283.7 (Zr)
7.5
照合試験
− マトリックス効果の有無を調べるために照合試験を行う。これには,標準添加法を用いる。
− マトリックス効果があることが分かった場合は,この方法は不適当であり,方法B,方法Cを用いて
再試験するか,又は標準添加法による結果を採用する。
8. 試験結果の表現 検量線を用い,各金属について測定試料 (7.4) 及び空試験液 (7.2) の吸光度に相当
する濃度を求める。
対象金属について,試料の濃度,mg/l,は,次の式で与えられる。
V
b
t
100
)
(
×
−ρ
ρ
ここに,
ρt: 測定試料の吸光度に相当する金属の濃度,mg/l
ρb: 空試験の吸光度に相当する金属の濃度,mg/l
9. 試験報告 報告書には,次の事項を含めなければならない。
− この規格の引用
− 用いた試験方法の引用
− 試料の完全な確認
− 試験結果
6
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− この規格に規定されていないこと及び応用操作の詳細,並びに結果に影響する可能性があるすべての
要因の詳細。
第2章:方法B−錯化 (APDC) 及び抽出 (MIBK) 後のフレーム原子吸光法による定量
10. 原理 対象金属と1−ピロリジンジチオカルバミド酸アンモニウム (APDC) の錯体の生成,及び
pH2.5で4−メチル−2−ペンタノン[メチルイソブチルケトン (MIBK)]による抽出。
有機相中の金属のフレーム原子吸光法による定量。
11. 試薬 4. 参照。
11.1 硝酸ρ=1.4g/ml 4.1と同じ。
11.2 水酸化ナトリウム溶液c (NaOH) =2.5mol/l JIS K 8576に規定する水酸化ナトリウム100gを注意し
ながら水に溶かし,1lに薄める。
11.3 塩酸c (HCl) =0.3mol/l JIS K 8180に規定する塩酸 (Q=1.19g/ml) 25mlを注意しながら水と混ぜ,
1lに薄める。
11.4 メチルイソブチルケトン (MIBK) 1) JIS K 8903に規定する4−メチル−2−ペンタノン
11.5 1−ピロリジンジチオカルバミド酸アンモニウム (APDC) 2) 20g/l溶液 APDC2gを水に溶かし,水で
100mlとし,混合する。沈殿があるときはろ過する。溶液が着色しているときは,無色になるまでMIBK
(11.4) で繰り返し抽出して精製する。
この溶液は試料のバッチごとに新たに調製する。
11.6 ブロモフェノールブルー指示薬溶液 エタノール溶液 [50% (v/v)] 1lに,JIS K 8844に規定するブロ
モフェノールブルー1gを含む。
11.7 金属標準液 金属1.000mg/lを含む。4.4参照。
12. 装置 5.参照
13. サンプリング方法及び試料 6.参照
14. 手順
14.1 測定試料 酸性試料(5.参照)から対象金属5〜20μgを含む測定試料(各金属の上限は表2参照)
を全量フラスコ100mlにとる。水を標線まで加える。
14.2 錯化及び抽出
− 測定試料 (14.1) 及び各検量線用溶液 (14.4) 100mlを四ふっ化エチレン樹脂 (PTFE) 製の口つきの分
液漏斗250mlにとる。
− 各分液漏斗にブロモフェノール指示薬 (11.6) 2,3滴を加え,水酸化ナトリウム (11.2) を青が残るま
で加える。
− かき混ぜながら,青がちょうど消えるまで塩酸 (11.3) を滴加し,さらに塩酸 (11.3) 2mlを過剰に加え
1) 4−メチル−2−ペンタノン
2)ピロリジンカルボジチオ酸アンモニウム
7
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。pHは2.3〜2.5にならなければならない(備考1.参照)。
− APDC (11.5) 5mlを加え,次に,MIBK (11.4) 10.0mlを加え,2分間激しく振り混ぜる。pHは約2.8に
ならなければならない。
− 混合溶液は,栓をした分液漏斗中で少なくとも1時間光及び熱を遮断して放置する。放置時間はすべ
ての溶液について全く同じでなければならない。わずかの水相も混入しないように注意しながら有機
相を集める(必要があれば,遠心分離する。)(備考2.参照)。
備考1. 指示薬の代わりにpH計を用いてもよい。
2. 約5℃の暗所におけば,放置時間を延長しても差し支えない。この場合は有機相を遠心分離
する必要はない。
14.3 空試験 空試験は,定量と並行してサンプリング,錯化,抽出の過程を,同量の試薬を用いて同じ
操作 (14.2) を行う。ただし,測定試料の代わりに,水を用いる。
14.4 検量線用溶液の調製
− 使用直前に対象元素の標準液 (4.4) を水3) で薄めて10mg/lの溶液を調製する。
− 全量フラスコ500mlに次の溶液を加える。
− 各10mg/lの亜鉛,カドミウムを含む溶液5mlずつ
− 各10mg/lの銅,コバルト,ニッケル,鉛を含む溶液20mlずつ
− 硝酸 (11.1) 0.5ml。
− 水を標線まで加える。これを溶液Sとする。溶液Sを水で薄め,次の濃度範囲にわたって少なくとも
4種類の検量線用溶液を調製する。
Zn, Cd=0〜50μg/l
Co, Cu, Ni, Pb=0〜200μg/l
− 各検量線用溶液に,試料の保存のために加えた(6.2参照)と同量の硝酸 (11.1) を加えて酸性にする。
添加量は試料と検量線用溶液の硝酸の濃度が同じになるようにしなければならない。
14.5 校正及び定量
− 各対象金属に対して次のように操作する。吸光度の測定に先立って,対象金属の検量線用溶液の抽出
有機相 (14.2) を噴霧し,表4の情報を参考にし,製造業者の指示に従って分光計を調整する。さきの
(7.4) と同様に噴霧及びフレームの条件を最適にする。MIBK (11.4) を用いてゼロ吸収に対する機器の
応答を調節する。
− 各対象金属に対して,検量線用溶液の抽出有機相を噴霧する。検量線用溶液の金属含有量μg/lを横軸
に,対応する吸光度を縦軸にプロットして検量線を作成する。検量線は,例えば,5試料ごとに検量
線用溶液の吸光度を測定して,確認することが望ましい。
− 測定試料の抽出有機相を噴霧する。
− 対象金属の吸光度を測定し,各測定後にMIBKを噴霧してネブライザー系を洗う。非固有吸収の補正
については7.4の備考参照。
備考 有機溶液を光,熱にさらさないことが重要である。コバルト,銅,亜鉛,特にカドミウムの錯
体はMIBK中で不安定である。カドミウムは直ちに測定しなければならない。その他の金属は
数時間は貯蔵できる。
3) 海水その他高濃度の塩化ナトリウムを含む水中の金属を定量するときは,対象試料と同量の塩化ナトリ
ウムを含む水を用いて検量線用液及び空試験液を調製する。
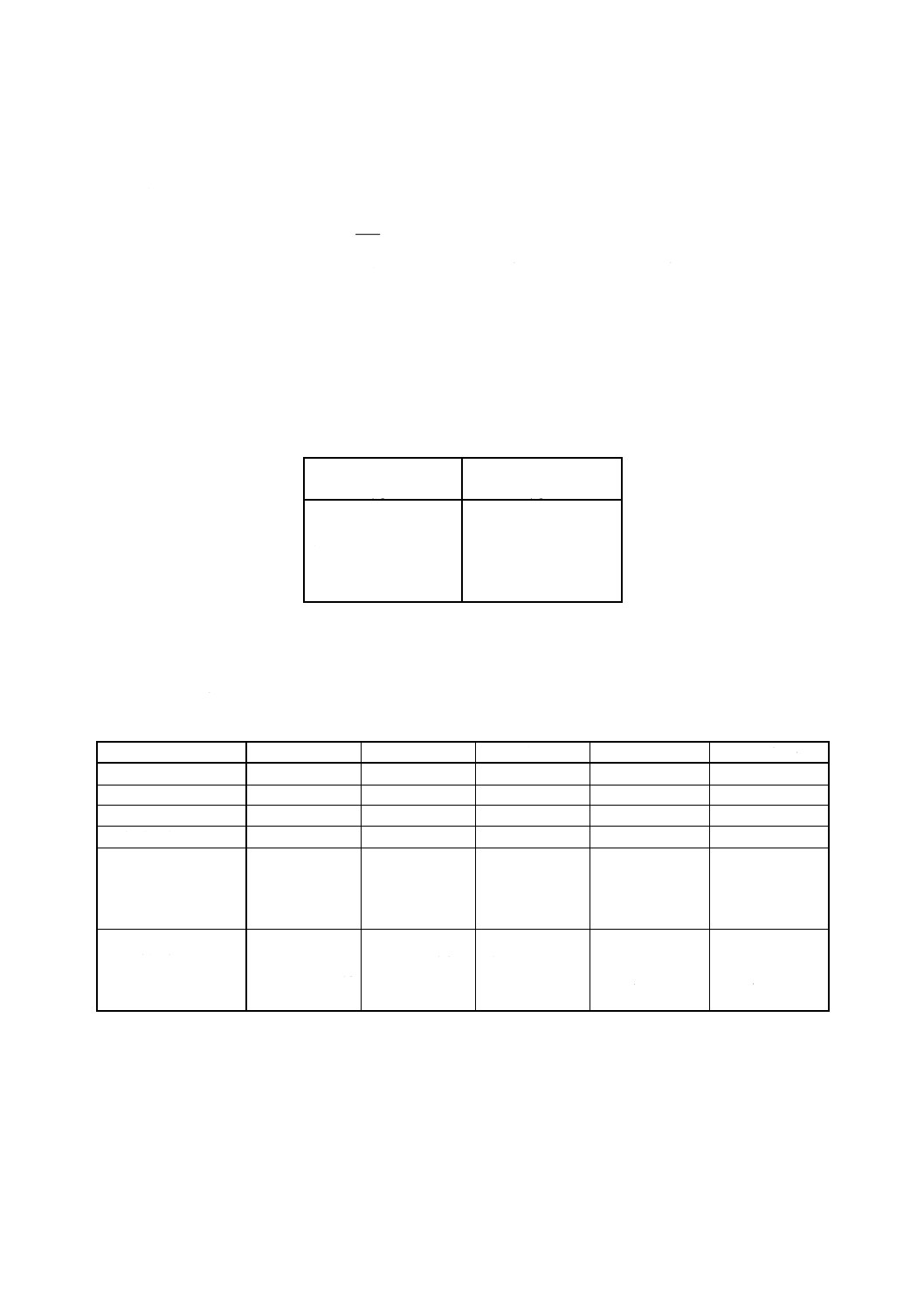
8
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15. 試験結果の表現
15.1 計算 検量線を用いて,各金属について測定試料及び空試験液の吸光度に相当する濃度を求める。
各対象金属について,試料の濃度,μg/l,は次の式で示される。
V
b
t
100
)
(
×
−ρ
ρ
ここに,
ρt: 測定試料の吸光度に相当する金属の濃度,μg/l
ρb: 空試験の吸光度に相当する金属の濃度,μg/l
V: 分析用に採取した酸性試料の体積,ml
15.2 精度 二つの抽出法(方法B及び方法C)の繰返し性,再現性を比較するために,1981年に国際室
間試験が組織された。
− 二つの分析試料の組成を表6に示す。
表6
試料L(低レベル)
μg/l
試料H(高レベル)
μg/l
鉛
20
100
カドミウム
4
20
銅
6
40
コバルト
5
120
ニッケル
10
100
− ISO 5725による統計解析を表7に示す。
15.3 妨害物質 5mg/l未満のほか物質は通常,妨害しない。
16. 試験報告 9. 参照。
表7
金属
鉛
カドミウム
銅
コバルト
ニッケル
水準
L
H
L
H
L
H
L
H
L
H
試験室数
14
14
14
14
14
14
14
14
14
14
棄却後の試験室数
14
12
11
13
12
14
13
14
14
14
平均(μg/l)
19.7
96
4
30.2
5.7
40.6
5.1
121.3
10.7
103.2
繰返し性
標準偏差,σr
1.5
2.6
0.1
0.7
0.2
1.7
0.4
1.9
0.4
2.3
繰返し性の変動係数
7.6%
2.7%
2.5%
2.3%
3.5%
4.2%
7.8%
1.6%
3.7%
2.2%
繰返し性r(=2.83σr)
4.24
7.36
0.28
1.98
0.57
4.8
1.13
5.38
1.13
6.51
再現性
標準偏差,σR
3.2
5.3
0.3
1.3
0.7
5.9
1.4
7.6
1.4
16.2
再現性の変動係数
16.2%
5.5%
7.5%
4.3% 12.3%
14.5% 27.5%
6.9% 13.1%
15.7%
再現性,R(=2.83σR)
9.05
15
0.85
3.68
1.98
16.7
3.96
21.51
3.96
48.85
第3章:方法C−錯化 (HMA-HMDC) 及び抽出(DIPK-キシレン)後のフレーム原子吸光法による定量
17. 原理 金属とヘキサメチレンアンモニウム−ヘキサメチレンジチオカルバミド酸 (HMA-HMDC) の
錯体の生成及びpH2〜4の緩衝液中でジイソプロピルケトン−キシレンによる抽出。
有機相中の金属のフレーム原子吸光法による定量。
9
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
18. 試薬 4.参照。
18.1 硝酸ρ=1.4g/ml JIS K 8541に規定するもの。
18.2 ヘキサメチレンジチオカルバミド酸−ヘキサメチレンアンモニウム (HMA-HMDC) 1)
− キシレン300mlに蒸留したヘキサメチレンイミン (bp136〜139℃) 224mlを加えた溶液を氷浴中で冷却
し,これに蒸留した二硫化炭素 (bp46.2℃) (JIS K 8732に規定する二硫化炭素を用いる。)60mlを絶
えず冷却し,かき混ぜながら30分間以内に加える。
− 1時間冷却し,かき混ぜる。溶液をろ過し,凝集した白い沈殿を集め,JIS K 8103に規定するジエチ
ルエーテルで3回洗浄し,ろ紙に挟んで乾かす。
18.3 HMA-HMDC抽出液6.8g/l 乾かした全量フラスコ250ml中でHMA-HMDC (18.2) 1.7gをJIS K 8271
に規定するキシレン75mlに静かに加熱して溶かす。ジイソプロピルケトン2) (DIPK) (bp 124.5℃) を標線
まで加える。
この溶液は光を避け,5℃で貯蔵すれば1週間は安定である。
18.4 HMA-HMDCメタノール溶液55g/l 乾いた全量フラスコ100ml中で,HMA-HMDC (18.2) 5.5gをJIS
K 8891に規定するメタノールに静かに加熱して溶かす。室温に冷却し,標線までメタノールを加える。
18.5 ぎ酸塩緩衝液 JIS K 8264に規定するぎ酸 [98〜100% (m/m)] 368g及びJIS K 8283に規定するくえ
ん酸一水和物14gを水350mlに溶かす。絶えずかき混ぜ,冷却しながらJIS K 8576に規定する水酸化ナト
リウム243gを徐々に加える。m−クレゾールスルホンフタレイン(JIS K 8889に規定するメタクレゾール
パープル)50mgを加える。この溶液をよくかき混ぜながら,抽出液 (18.3) で連続2回抽出して精製する。
18.6 金属標準液 金属1.000g/l各対象金属1.000gを一緒に硝酸 (18.1) と加熱して完全に溶かす。冷却し,
水で1000mlにする。この溶液の酸濃度は約0.1〜0.5mol/lでなければならない。
標準液の調製には正確な組成の分かった金属塩を用いてもよい。
18.7 金属有機標準液 金属50mg/l乾いた全量フラスコ100mlに標準液 (18.6) 5mlをとる。JIS K 8264に
規定するぎ酸 [98〜100% (m/m)] 50ml及びくえん酸一水和物0.2〜0.5gを加える。ジイソプロピルケトンを
標線まで加える。
19. 装置 5.に規定する装置及び
マイクロピペット マイクロピペットのプラスチックチップを硝酸 (4.2) に数時間浸して清浄にする。
40℃を超える温度は避ける。使用前に水ですすぐ。
20. サンプリング方法及び試料 6.参照。
21. 手順
21.1 測定試料 酸性にした試料(6.参照)からの測定試料は,通常約400mlである。
濃縮倍数を上下させたい場合は,水相と有機相の液量比50 : 1までの範囲で異なる液量を用いてもよい。
21.2 錯化及び抽出
− 測定試料 (21.1) を全量フラスコ500mlにとる。ぎ酸塩緩衝液 (18.5) 20mlを加える。指示薬の色は純
黄色でなければならない。赤が現れたときは,ぎ酸緩衝液20mlを追加する。
1) この試薬には市販品がある。
2) 2,4−ジメチル−3−ヘプタン
10
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− HMA-HMDCメタノール溶液 (18.4) 2.0mlを加えて振り混ぜた後3〜5分間放置する。
− 抽出液 (18.3) 20.0mlを加え,少なくとも3分間フラスコを激しく振り混ぜる。
− 良好な層分離を得るために,混合液を10〜15分間放置する。次に,フラスコの首の部分に有機層がく
るまで慎重に水を加える。
− 定量 (21.5) のため有機層の噴霧は,フラスコの首の部分から直接行ってもよい。
− 有機層を長時間保存するときは,わずかの水相も入らないように注意しながらピペットでこれを取り
出し,冷暗所に貯蔵する。
21.3 空試験 定量操作と並行して,サンプリング,錯化,抽出の場合と同量の試薬を用いて空試験を行
う。ただし,測定試料の代わりに硝酸 (4.3) を用いる。
21.4 検量線用溶液の調製
21.4.1 水溶液 一連の定量前に,対象濃度範囲にわたって,少なくとも四つの校正用水溶液を調製する。
これらの検量線用溶液は,標準液 (18.6) を硝酸 (4.3) で薄めて調製する。
備考 これらの水溶液セットは,抽出の完全性を確認するのに用いる。抽出操作に不慣れな試験室で
は附属書Bに示す操作に従って,抽出の完全性を確かめるとよい。
21.4.2 有機溶液 使用直前に,全定量範囲にわたって,少なくとも四つの有機検量線用溶液を調製する。
これらの検量線用溶液は,有機標準液 (18.7) を抽出液 (18.3) で薄めて調製する。この場合,乾いた全量
フラスコ25ml及びマイクロピペット (19. ) を用いる。
21.5 校正及び定量
− 各金属に対して,次のように操作する。吸光度の測定に先立って,有機検量線用溶液 (21.4.2) の一つ
を噴霧し,表4を用い,製造業者の指示に従って分光計を調製する。さきと同様に (7.4) ,噴霧及び
フレームの条件を最適にする。抽出液 (18.3) を用いてゼロ吸収に対する機器の応答を調節する。
− 各対象金属について,有機検量線用溶液 (21.4.2) を噴霧する。有機検量線用溶液の金属含有量,mg/l
を横軸に,対応する吸光度を縦軸にプロットして検量線を作成する。
− 測定試料 (21.1) の有機抽出液を噴霧する。
− 対象金属の吸光度を測定する。各測定後にメタノールを噴霧してネブライザー系を洗い,目詰まりを
防止する。
必要があれば,非固有吸収を補正する(7.4の備考参照)。
22. 試験結果の表現
22.1 計算 検量線を用いて,各金属について測定試料及び空試験の吸光度に相当する濃度を求める。各
対象金属について,試料の金属濃度,μg/l,は,次の式で示される。
V
b
t
20
)
(
×
−ρ
ρ
ここに,
ρt: 測定試料の吸光度に相当する金属の濃度,μg/l
ρb: 空試験の吸光度に相当する金属の濃度,μg/l
V: 分析用に採取した酸性試料の体積,ml
22.2 繰返し性及び再現性 二つの抽出法(方法B及び方法C)の繰返し性,再現性を比較するために1981
年に国際室間試験が組織された。
二つの分析試料の組成は,表6に示した。
ISO 5725による統計解析を,表8に示す。
11
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表8
金属
鉛
カドミウム
銅
コバルト
ニッケル
水準
L
H
L
H
L
H
L
H
L
H
試験室の数
16
16
16
16
16
16
16
16
16
16
棄却後の試験室数
14
14
13
13
15
16
14
14
11
14
平均(μg/l)
20.3
97.7
4
29.8
6.4
41
5.8
115.8
10.9
100.2
繰返し性
標準偏差,σr
1.06
3.8
0.3
0.8
0.5
3.3
0.6
3.8
1.8
4.7
繰返し性の変動係数
5.2%
3.9% 7.5%
2.7% 7.8%
8.0% 0.3%
3.1% 16.5%
4.7%
繰返し性,r (:2.83σr)
3.0
10.7
0.85
2.26
1.41
9.34
1.7
10.2
5.1
13.3
再現性
標準偏差,σR
2.8
3.4
0.4
2.7
1.1
4.8
1.2
8.2
4
13.7
再現性の変動係数
13.8%
3.5% 10%
9.1% 17.2%
11.7% 20.7%
7.1% 36.7%
13.7%
再現性,R (=2.83σR)
7.9
9.62
1.13
7.64
3.11
13.6
3.4
23.21
11.3
38.8
22.3 妨害物質
− 鉄を含めて重金属は,20mg/lまで許容できる。重金属の全濃度が20mg/lを超える場合は,測定試料と
抽出液の体積比を20 : 1未満にしなければならない。
− ニトリロ三酢酸の許容濃度は,250mg/lである。エチレンジアミン四酢酸 (EDTA) はニッケルの抽出
を妨害する。EDTA・2Na二水和物 (C10H14N2O8Na2・2H2O) 25mg/lは他の5金属の抽出には影響しな
い。
− 天然水試料中に含まれるフミン酸は酸性にすると沈殿する。この沈殿はろ過によって除去するとよい。
沈殿には重金属は取り込まれない。ろ液中の金属は,ヘキサメチレンジチオカルバミド酸のジイソプ
ロピルケトン−キシレン抽出によって完全に回収できる。
23. 試験報告 9.参照。
12
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A(規定) 全金属定量のための試料の前処理
1. 適用範囲 この附属書は,全金属定量のための試料の前処理について規定する。
A.0 大多数の試料に対して,塩酸と硝酸による無機質化で十分である。しかし,ときに例えば,著しく汚
染された廃水などでは,より激しい処理が必要になる。
無機質化の例として,次の操作がある。
A.1 測定試料100mlにつき塩酸 (ρ=1.19g/ml) 5mlを加える。
− 水蒸気浴で15〜20mlになるまで,沸騰させることなく加熱する。
− 冷後,不溶解物をろ別してネブライザーの目詰まりを防止する。ろ液を全量フラスコ100mlに集める。
− ろ紙を水で数回洗う。
A.2 測定試料100mlにつき硝酸4ml (15mol/lを加え,液量50mlになるまで加熱する。
− 処理した試料を沸騰フラスコに入れ,塩酸 (ρ=1.19g/ml) 12mlを加える。沸騰フラスコと冷却管を連
結し,2.5時間溶液を還流させる。
− 冷後,ネブライザーの目詰まりを防止するために不溶解物をろ別する。ろ液を全量フラスコ100mlに
集める。
− 冷却管とろ紙を水で数回洗い,これを全量フラスコ100mlに合わせ,水を標線まで加える。
13
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B(規定) 完全抽出の確認方法(方法C)
1. 適用範囲 この附属書は,方法Cによる場合の各金属の有機相への完全抽出の確認について規定する。
− 試料を分析する前に,試験と並行して試料の予想濃度の範囲で少なくとも二つの検量線用溶液(本体
21.4.1)を調製する。
− 本体21.2に従ってこれらの検量線用溶液を錯化,抽出する。
− 有機抽出液をフレーム中に噴霧し,本体21.5によって有機抽出液の吸光度を測定する。
− これと並行して,対応する検量線用溶液の濃度のそれぞれ20倍,50倍の濃度(濃縮係数によって異
なる)の少なくとも二つの有機検量線用溶液(本体21.4.2)を調製する。これらの有機溶液を噴霧し,
本体21.5によって吸光度を測定する。
− 検量線用溶液の有機抽出液が有機検量線用溶液と同じ吸光度値を示すならば,抽出は完全である。
14
K 0400-52-20 : 1998 (ISO 8288 : 1986)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
平成8年度JIS K 0102改正原案作成委員会 構成表
氏名
所属
(委員長)
○ 並 木 博
工学院大学工学部
○ 佐 藤 寿 邦
横浜国立大学工学部
○ 西 出 徹 雄1) 工業技術院標準部消費生活規格課
乾 敏 一2) 通商産業省環境立地局産業施設課
○ 畑 野 浩3) 環境庁水質保全局水質規制課
中 村 進
工業技術院物質工学工業技術研究所計測化学部
中 村 和 憲
工業技術院生命工学工業技術研究所
○ 田 尾 博 明
工業技術院資源環境技術総合研究所水圏環境保全部
田 中 宏 明
建設省土木研究所下水道部
柴 田 康 行
国立環境研究所化学環境部
○ 土 屋 悦 輝
東京都立衛生研究所環境保健部
渡 辺 真利代
東京都立衛生研究所環境保健部
○ 日 野 隆 信
千葉県衛生研究所
小 倉 光 夫
神奈川県環境科学センター水質環境部
西 尾 高 好
財団法人日本環境衛生センター東日本支局環境科学部
○ 坂 本 勉
財団法人日本規格協会技術部
山 村 修 蔵
財団法人日本規格協会技術部
浅 田 正 三
財団法人日本品質保証機構環境計画センター
○ 梅 崎 芳 美
社団法人産業環境管理協会
横 倉 清 治
社団法人日本環境測定分析協会(三菱マテリアル株式会
社)
神 代 啓
社団法人日本化学工業協会
池 田 久 幸
社団法人日本分析機器工業会(横河アナリティカルシス
テムズ株式会社)
長 澤 忠 彦
社団法人日本鉄鋼連盟(住友金属工業株式会社)
山 田 昭 捷
社団法人日本下水道協会(東京都下水道局)
土 屋 徳 之
石油連盟(興亜石油株式会社)
松 谷 成 晃
日本石鹸洗剤工業会(ライオン株式会社)
波多江 正 和
日本製紙連合会技術環境部
佐 山 恭 正
日本鉱業協会(三菱マテリアル株式会社)
狩 野 久 直
日本練水株式会社研究所
久 島 俊 和
オルガノ株式会社総合研究所
○ 川 瀬 晃
セイコー電子工業株式会社科学機器事業部
○ 米 倉 茂 男
元 東京都立工業技術センター
岩 崎 岩 次
社団法人日本工業用水協会
(事務局)
秋 本 孝
社団法人日本工業用水協会
飛 渡 祥 弘
社団法人日本工業用水協会
本 郷 秀 昭
社団法人日本工業用水協会
備考 1):発足当初は岡林哲夫(工業技術院繊維化学規格課)
2):発足当初は相澤徹(通商産業省環境立地局産業施設課)
3):発足当初は飯島孝(環境庁水質保全局水質規制課)
○は幹事兼任
文責 並木 博