K 0150:2020 (ISO 16962:2017)
(1)
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 原理······························································································································· 2
5 装置······························································································································· 2
5.1 グロー放電発光分光分析装置 ··························································································· 2
6 グロー放電発光分光分析装置の調整 ····················································································· 4
6.1 概要 ···························································································································· 4
6.2 直流放電源の制御パラメータの設定··················································································· 4
6.3 高周波放電源の制御パラメータの設定················································································ 7
6.4 必要とする装置性能 ······································································································· 9
7 試料の準備とその調製 ······································································································ 10
8 検量線の作成 ·················································································································· 11
8.1 概要 ··························································································································· 11
8.2 検量線作成用試料 ········································································································· 11
8.3 検量線の妥当性確認用試料及び検量線作成用のめっき標準物質 ·············································· 12
8.4 検量線作成用試料のスパッタリング速度の測定 ··································································· 13
8.5 検量線作成用試料の発光強度の測定·················································································· 14
8.6 検量線定数の計算 ········································································································· 14
8.7 標準物質を用いた検量線の妥当性確認··············································································· 14
8.8 検量線の検証及びドリフト補正 ······················································································· 15
9 試験試料の分析 ··············································································································· 16
9.1 放電制御パラメータの調整 ····························································································· 16
9.2 測定時間及びデータ取得速度(サンプリングレート)の設定 ················································· 16
9.3 試験試料の定量深さ方向プロファイルの定量化 ··································································· 16
10 結果の表記 ··················································································································· 16
10.1 定量深さ方向プロファイル ···························································································· 16
10.2 単位面積当たりのめっき付着量の定量 ············································································· 16
10.3 各元素の平均質量分率の決定 ························································································· 17
11 精度 ···························································································································· 18
12 試験報告書 ··················································································································· 18
附属書A(規定)検量線定数の算出及び深さ方向プロファイルの定量化 ········································· 19
附属書B(参考)分析線として使用できるスペクトル線 ······························································ 29
附属書C(参考)単位面積当たりのめっき質量(めっき付着量)の定量 ········································· 30
K 0150:2020 (ISO 16962:2017)
(2)
ページ
附属書D(参考)共同実験に関する追加資料 ············································································ 35
参考文献 ···························································································································· 37
K 0150:2020 (ISO 16962:2017)
(3)
まえがき
この規格は,産業標準化法第16条において準用する同法第12条第1項の規定に基づき,一般社団法人
表面化学分析技術国際標準化委員会(JSCA)及び一般財団法人日本規格協会(JSA)から,産業標準原案
を添えて日本産業規格を改正すべきとの申出があり,日本産業標準調査会の審議を経て,経済産業大臣が
改正した日本産業規格である。これによって,JIS K 0150:2009は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本産業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本産業規格 JIS
K 0150:2020
(ISO 16962:2017)
表面化学分析−亜鉛及び/又はアルミニウム基金属
めっきのグロー放電発光分光分析方法
Surface chemical analysis-Analysis of zinc- and/or aluminium-based
metallic coatings by glow-discharge optical-emission spectrometry
序文
この規格は,2017年に第2版として発行されたISO 16962を基に,技術的内容及び構成を変更すること
なく作成した日本産業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
1
適用範囲
この規格は,亜鉛及び/又はアルミニウム基材料からなる金属めっきの厚さ,単位面積当たりの付着量,
及び化学組成の定量のためのグロー放電発光分光分析方法について規定する。合金元素として対象とする
ものは,ニッケル,鉄,けい素,マグネシウム,鉛及びアンチモンである。
この分析方法は,次の質量分率の含有量をもつ元素に適用できる。
− 亜鉛
:質量分率 0.01 %〜100 %
− アルミニウム :質量分率 0.01 %〜100 %
− ニッケル
:質量分率 0.01 %〜20 %
− 鉄
:質量分率 0.01 %〜20 %
− けい素
:質量分率 0.01 %〜15 %
− マグネシウム :質量分率 0.01 %〜20 %
− 鉛
:質量分率 0.005 %〜2 %
− アンチモン
:質量分率 0.005 %〜2 %
注記1 鉛及びアンチモンは,環境汚染及び健康被害の原因となるため最近の素材では添加されない
が,この規格は,これらの元素を含有する古い素材にも適用できる。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 16962:2017,Surface chemical analysis−Analysis of zinc- and/or aluminium-based metallic
coatings by glow-discharge optical-emission spectrometry(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
2
K 0150:2020 (ISO 16962:2017)
JIS G 0417 鉄及び鋼−化学成分定量用試料の採取及び調製
注記 対応国際規格:ISO 14284,Steel and iron−Sampling and preparation of samples for the
determination of chemical composition
ISO 17925,Zinc and/or aluminium based coatings on steel−Determination of coating mass per unit area and
chemical composition−Gravimetry, inductively coupled plasma atomic emission spectrometry and flame
atomic absorption spectrometry
3
用語及び定義
この規格には,定義する用語はない。
4
原理
この分析方法は,次の過程から構成される。
a) 分析対象となる試料の準備。通常は,平板又は円盤試料を分析装置又は分析目的に合わせて調製する
(幅又は直径が5 mm以上,一般的には20 mm〜100 mmの円形又は長方形の試料が適している。)。
b) 直流又は高周波のグロー放電による表面めっき層の陰極スパッタリング
c) グロー放電によって生成するプラズマ中の分析対象原子の励起
d) スパッタリングによる,分析対象原子及びイオンの特性発光線の時間経過に伴う強度変化の測定(深
さ方向の定性的な元素分布)
e) 発光強度と時間との測定結果から,検量線を用いた,質量分率と深さ方向分布との関係への変換(定
量的な深さ方向元素分布)。定量のための検量線は,化学組成が既知の標準物質の測定と,スパッタリ
ング速度の測定とをすることによって作成する。
5
装置
5.1
グロー放電発光分光分析装置
5.1.1
概要
必要な分析装置は,グリムタイプ[1]又は類似した構造をもつグロー放電発光源(直流又は高周波電力が
印加される。),及び分析元素の適切なスペクトル線(推奨する線については附属書B参照)を分光検出で
きるJIS K 0144に規定する多元素同時検出型分光器を含む。また,深さ方向元素分析を行う場合,特殊な
スペクトル線を追加できるように,この分光器と別に走査型分光器(モノクロメータ)を用意して測定す
ることも一般的である。さらに,電荷結合型素子(CCD)又は電荷注入型素子(CID)のようなアレイ型
半導体検出器も,広い波長範囲にある複数の分析線を同時測定するために用いることができる。
グロー放電発光源の中空陽極の内径は,2 mm〜8 mmの範囲とすることが望ましい。試料が薄い場合,
冷却液が循環する金属ブロックのような冷却器具で試料を冷やすことが望ましいが,この分析を行うに当
たっては必須ではない。
定量の原理は,めっき層の連続的スパッタリングに基づいているため,分光器は,発光強度の時間分解
測定のためのデジタル読出し部を備えていなければならない。スペクトル線当たり,少なくとも1秒間当
たり300点のデータ取込みが可能な装置が望ましいが,大多数の分析対象試料では1秒間当たり50点を超
える取込み速度であれば十分である。実際には,スペクトル線当たり10点/秒〜100点/秒のサンプリン
グ速度が適している。
3
K 0150:2020 (ISO 16962:2017)
5.1.2
スペクトル線の選択
定量する元素それぞれについて使用できるスペクトル線が多数あるため,使用する分光器の波長範囲,
分析対象元素の質量分率,スペクトル線の感度,及び試料中に共存するその他の元素による分光干渉を含
む因子を考慮して適切な分析線を選択しなければならない。定量を行う元素が試料中の主成分である場合,
自己吸収に対して極めて敏感なスペクトル線(基底電子準位に遷移するスペクトル線,いわゆる共鳴線)
があることに特に留意しなければならない。自己吸収が起きると,分析対象元素の高質量分率側で検量線
の直線相関を失う原因となるため,強い自己吸収があるスペクトル線は,主成分元素の定量に使用するの
を避けることが望ましい。スペクトル線の選択に関して,附属書Bに分析線として使用できるスペクトル
線を示す。この表に記載されていない他のスペクトル線についても,それが良好な特性をもつ場合には,
分析線として使用してもよい。
5.1.3
グロー放電励起源の選択
5.1.3.1
陽極の内径
市販されているほとんどのグロー放電発光分光分析装置において,陽極の内径を選択できるような設計
が用意されており,2 mm,4 mm及び8 mmが一般的である。旧型装置には,陽極の内径が固定のものが
あるが(通常は8 mm),最近の装置では4 mmの陽極が最も普及している。陽極の内径が大きくなるほど
大きな試料を必要とし,分析中の高電力を必要とするので,試料温度が上昇する。一方,大口径の陽極に
なるほど大容量プラズマが生成し,より強く発光して検出限界がより低くなる(すなわち,分析感度がよ
り高くなる。)。さらに,陽極の大口径化は,表面の元素分布に揺らぎがある試料において,平均的な元素
濃度を求めることに寄与するが,適用する分析対象によって,この特性が有利な場合とならない場合があ
る。表面分析に適用した場合,試料の過熱が問題となる場合がある。例えば,表層の熱伝導度が小さいこ
と及び/又は試料が非常に薄いことによって,試料の過熱が問題になることがある。このような場合には,
ある程度分析感度の低下があるとしても,陽極の内径がより小さいもの(例えば,2 mm又は2.5 mm)が
望ましい。
5.1.3.2
放電用電源
放電用電源は,直流(DC)型又は高周波(RF)型のいずれかとすることができる。RF型は,導電性及
び絶縁性のいずれの試料に対してもスパッタリングすることができるため,高分子皮膜,絶縁性の酸化物
層などに使用できる。一方,DC型は,電気的パラメータ(電圧,電流,電力など)の測定及び制御が技
術的により簡単である。DC放電とRF放電との切替えができるグロー放電発光分光分析装置が市販されて
いるが,RF放電の専用装置が標準となってきている。
5.1.3.3
装置の制御モード
電源の電気的パラメータ(電流,電圧及び電力)及びプラズマガスの導入圧力に関して,DC型及びRF
型の放電の制御は,幾つかの異なる方法で行うことができる。幾つか異なった方法で行われている理由と
しては,次のようなものが挙げられる。
− “歴史的”理由によって,旧型の装置は,よりシンプルだが機能的な電源をもつ一方,技術の進化に
伴い,新型の機種では,より正確で簡単な放電制御ができる。
− 分析機器の製造業者(以下,機器製造業者という。)ごとに,放電制御に関して異なった方法を取って
きた経緯がある。
− 個々の分析事例に関わる問題点から,特定の放電制御モードが適している場合がある。
この規格では,数種の利用可能な放電制御モードに基づき,放電源の電気的パラメータの最適化を行う
方法を,箇条6に規定する。幾つかの異なった分析装置の全般にわたり,包括的に適用できる方法を示す。
4
K 0150:2020 (ISO 16962:2017)
大部分の分析対象において,分析特性に関してはこれらの制御モードによる大きな差異はないが,実際の
運用及び手法の難易については差異が生じる。例えば,導入ガス圧力を能動的に制御できる測定装置では,
個々の分析操作ごとに自動的に同じ測定パラメータを用いて測定ができる。この制御機能がない場合には,
測定パラメータを最適化するために手動で圧力調整を行う必要がある。
注記 様々な放電制御モードが使われている背景として,発光収率[2][3][4][7]と呼ばれるパラメータ
が校正及び定量の基礎となっている。発光収率は,電流,電圧及び程度は低いがガス圧力によ
って変化する[4][7]。測定対象物質によってその電気的特性が変化することから,実操作におい
て,全ての分析対象試料に対して,これら三つのパラメータを一定に制御することは不可能で
ある。分析装置の中には,測定中にプラズマガス圧力を自動制御する機構を付加して,放電源
の電気的パラメータ(プラズマ抵抗)を一定に維持するものがある。その他の方法として,経
験的に得られた関係式を用いてプラズマ抵抗の変動を補正する方法[4][7]があり,この補正法を
採用している市販のグロー放電発光分光分析装置ではソフトウエアに組み込まれている。
6
グロー放電発光分光分析装置の調整
6.1
概要
分析装置の使用準備は,機器製造業者の取扱説明書又は個々に文書化した手順に従って行う。
測定の準備段階で,分光システムの調整に関する最も重要な作業は,機器製造業者から提供された手順
に従って,分光器の入口スリットが正しく調整されているかを確認することである。この調整によって,
最適な信号対バックグラウンド比及び良好な再現性を得るためのスペクトル線のピーク位置で発光強度を
測定できる(JIS K 0144参照)。
放電制御パラメータは,次の要件を満たすように選択しなければならない。
a) めっき皮膜を過度の熱を与えないような適切なスパッタリングを行う。
b) できるだけ良好な深さ分解能を得るためのクレータ形状を得る。
c) 最適な精確さ(accuracy)を得るための検量線作成及び分析における放電条件を得る。
放電制御パラメータの選択方法の詳細を6.2及び6.3に示す。
検出器に印加する高電圧(例えば,光電子増倍管の印加電圧)の設定は,放電制御パラメータに依存す
るが,設定手順は,全ての放電制御モードで同じであるので,6.2.1.2に共通の手順として記載する。
同様に,発光信号の安定性及びスパッタリング痕形状に関して,放電源を調整しその制御パラメータを
最適化するための作業も,放電励起源の全ての制御モードで類似した手順をとる。
注記 プラズマガス圧力の測定に関しては,DC放電とRF放電とでは違いはない。ただし,グリム型
放電管では,その構造部位によってガス圧力勾配が生じ,このためガス圧力の判読値は,測定
系中での圧力計の挿入位置によって変わる。一部の分析装置では,プラズマ本体の実際圧力が
測定できるように圧力計を設置しているものがあるが,分析装置の中には真空ポンプに近い側
に圧力計を置いているため,実際圧力より低い値を示すものもある。このような測定装置に関
しては,プラズマガス圧力の判読値は,その測定系の圧力測定位置によって変わるためプラズ
マ本体の実際のガス圧力を測定するためではなく,他の放電制御パラメータを調整するための
指標としてだけ使用することができる。
6.2
直流放電源の制御パラメータの設定
6.2.1
固定印加電流−定電圧モード又は固定印加電圧−定電流モード
6.2.1.1
概要
5
K 0150:2020 (ISO 16962:2017)
この制御モードで調整が必要な制御パラメータは,印加電流及びプラズマガス圧力である。まず,グロ
ー放電を維持するための供給電源を定電流で動作するように設定する。最初に印加電流値を固定し,その
後,ガス圧力を調節して印加電圧を決める(電流を,電源の操作パネル上で設定し,電圧は,プラズマガ
ス圧力,又は流量を変化させることで調整する。)。この場合,印加電流及び電圧は,機器製造業者が推奨
する代表的な値に設定する。代わりに,供給電源を定電圧で動作するように設定してもよい。この場合,
最初に印加電圧値を固定し,その後,ガス圧力を調節して印加電流を決める(電圧を,電源の操作パネル
上で設定し,電流を,プラズマガス圧力,又は流量を変化させることで調整する。)。機器製造業者からの
推奨値が得られない場合,電圧は700 Vとし,電流は次の範囲に設定する。
− 陽極の内径が2 mm又は2.5 mmの場合 :5 mA〜10 mA
− 陽極の内径が4 mmの場合
:15 mA〜30 mA
− 陽極の内径が7 mm又は8 mmの場合
:40 mA〜100 mA
最適な電流値が事前に分からない場合,推奨範囲のほぼ中央の電流値で始めるのがよい。
これら二つの制御方法のいずれを採用しても差異はない。ただし,極薄膜への適用の場合には,分析結
果に多少影響を与える放電の開始時の立ち上がり特性に,非常に小さな差異が生じてもよい。
注記 (対応国際規格のNOTEは許容事項のため,本文に移動した。)
6.2.1.2
検出器の印加高電圧の設定
多様な形態及び組成の表面皮膜(表面層)をもつ試料を選択し,その分析対象元素の発光信号を確実に
検出するために検出器電源を制御する。光電子増倍管(PMT)に印加する高電圧は,最も含有量が少ない
元素に対して十分な感度が得られ,かつ,最も含有量が多い元素に対しては検出器に飽和が起こらないよ
うに設定する。また,アレイ型半導体検出器(CCD又はCID)の場合には,その積分時間をPMT検出器
の印加電圧と同様に設定する。
6.2.1.3
放電励起源の制御パラメータの設定
放電励起源の制御パラメータの設定は,次の手順によって行う。
a) 放電励起源の最適化を行うために,個々の試料に対して,表面めっき層を完全に超えて下地素材に達
するまで十分な時間をかけてスパッタリングをしながら,全体を通した深さ方向分析を行う。
b) スパッタリング時間に対する発光強度の変化(“深さ方向定性プロファイル”ともいう。)をみて,選
択した放電励起源の設定で常に安定した発光信号が表面層から下地素材まで得られていることを確認
する。
c) 安定した発光信号が得られない場合には,放電パラメータのいずれか一つの値を調整して,a) 及びb)
を再度行う。
d) さらに,安定した信号が得られない場合は,その他の制御パラメータまで含めて調整を行い,測定を
繰り返す。
e) 必要と認められる場合には,制御パラメータの組合せを変えて様々な可能性を考慮し,a) 及びb) の測
定を安定な発光信号が得られるまで繰り返す。
注記 試料の熱的な不安定性によって発光信号が不安定になる場合がある。この場合には,試料の
冷却が有効である。
6.2.1.4
スパッタリング痕形状の最適化
適切な表面粗さ計が使用できる場合には,スパッタリングによって生じたクレータ形状を,次の手順に
よって確認する。
a) 試験試料の中から代表的なものを選び,その試料を深さが約10 µm〜20 µmの範囲でスパッタリング
6
K 0150:2020 (ISO 16962:2017)
を行う。ただし,このときスパッタリング痕は,まだ表面めっき層の内部にとどまっている。そのた
め,めっき層厚が10 µm〜20 µmの試料の場合に限り実施可能である。
b) 条件に合う試料がない場合には,鉄鋼又は黄銅を試験試料として用いる。
c) 表面粗さ計を用いてそのクレータ形状を計測する。
d) a)〜c) の操作を,放電励起源の各制御パラメータを一つずつ変えながら,数回繰り返して行う。
e) スパッタリング痕の底面が平たん(坦)な形状となる放電条件を,最適値として採用する。
注記 試料によっては,最適クレータ形状と信号安定性とに係る二つの条件は必ずしも一致せず,
優先順位を付けて調整が必要な場合がある。
f)
a)〜e) の操作によって決定した測定条件が,6.2.1.3に規定した発光信号が安定して得られる放電条件
と大きな差異がなければ,検量線作成用及び試験試料の分析に使用する。
6.2.2
固定印加電流−定ガス圧力制御モード
この制御モードでは,まず印加電流値を定め,その後,最適な電圧値が得られるプラズマガス圧力を調
整して,次の手順によって測定を行う。
a) グロー放電を維持するための供給電源を,定電流で動作するように設定する。この場合,電流値は,
機器製造業者が典型例として推奨するものを採用する。機器製造業者からの推奨値が得られていない
場合には,電流は,次の範囲に設定する。最適な電流値が決まっていない場合には,これらの電流値
の中間値を用いて測定するのが望ましい。
− 陽極の内径が2 mm又は2.5 mmの場合:5 mA〜10 mA
− 陽極の内径が4 mmの場合:15 mA〜30 mA
− 陽極の内径が7 mm又は8 mmの場合:40 mA〜100 mA
b) 代表的なめっき試料にスパッタリングを行い,表面めっき層で,放電電圧が約700 Vに達するまでプ
ラズマガス圧力を調整する。
c) 検出器に印加する高電圧は,6.2.1.2に従って設定する。
d) 放電パラメータは,6.2.1.3と同様な手順で順次調整を行う。ただし,最初に放電電流値を調整し,必
要に応じてガス圧力を変える。
e) スパッタリング痕形状を,6.2.1.4と同様な手順で最適化する。この場合に調整する放電パラメータは,
プラズマガス圧力である。この最適化によって得られた放電条件は,検量線作成用及び試験試料の分
析において使用する。
f)
組成及び履歴の異なる試料を新たに測定する前に,それ以前の測定試料群で得られている放電電圧と
5 %以上変わっていないかを調べるために,試験的な放電操作を行って得られる電圧値を確認するこ
とが望ましい。大きな変動がある場合には,放電電圧を目標値近傍に設定するためにガス圧力を再調
整する。
注記 (対応国際規格のNOTEは要求事項のため,本文に移動した。)
6.2.3
固定印加電圧−定ガス圧力制御モード
この制御モードでは,まず印加電圧値を定め,その後,最適な電流値が得られるプラズマガス圧力に調
整して,次の手順によって測定を行う。
a) グロー放電を維持するための供給電源を定電圧で動作するように設定する。この場合,電圧値は,機
器製造業者が典型例として推奨するものを採用する。機器製造業者からの推奨値が得られない場合に
は,電圧値を700 Vに設定する。
b) 代表的なめっき皮膜試験試料にスパッタリングを行い,表面めっき層の部位で,放電電流が次の範囲
7
K 0150:2020 (ISO 16962:2017)
になるようにガス圧力を調整する。最適な電流値が決まっていない場合には,これらの電流範囲の中
間値を用いて測定をすることが望ましい。
− 陽極の内径が2 mm又は2.5 mmの場合:5 mA〜10 mA
− 陽極の内径が4 mmの場合:15 mA〜30 mA
− 陽極の内径が7 mm又は8 mmの場合:40 mA〜100 mA
c) 6.2.1.2と同様な手順で,検出器に印加する高電圧を設定する。
d) 6.2.1.3と同様な手順で,放電パラメータを順次調整する。ただし,最初に放電電圧値を調整し,必要
に応じてガス圧力を変える。
e) 6.2.1.4と同様な手順で,スパッタリング痕形状を最適化する。この場合に調整する放電パラメータは,
プラズマガス圧力である。この最適化によって得られた放電条件は,検量線作成用及び試験試料の分
析において使用する。
f)
組成及び履歴の異なる試料を新たに測定する前に,それ以前の測定試料群で得られている放電電流と
5 %以上変わっていないかを調べるために,試験的な放電操作を行い得られる電流値を確認すること
が望ましい。大きな変動がある場合には,放電電流を目標値近傍に設定するためにガス圧力を再調整
する。
注記 (対応国際規格のNOTEは要求事項のため,本文に移動した。)
6.3
高周波放電源の制御パラメータの設定
6.3.1
概要
一般に使用されている高周波放電制御パラメータの組合せは,定印加電力−定プラズマガス圧力,定印
加電圧−定プラズマガス圧力,又は定実効電力−定印加電圧の各制御モードである。その他の制御モード
も含めて,6.1に規定した放電励起源に求められる3項目を満足するものであれば,いずれのモードを使用
してもよい。6.3.2〜6.3.4に異なる制御モードのパラメータを設定する方法を記載する。
注記 高周波放電源が直流放電源と異なる点は,測定装置にもよるが,高周波放電源では電源から供
給される(進行波)高周波電力を測定できるが,プラズマの維持に関わる実効電力は,測定で
きないことである。印加高周波電力は,通常10 W〜100 Wの範囲であるが,接続部,ケーブル
などでの電力損失は,装置の電極構造及び試料接点によって大きく変化する。電力損失は一般
的に印加電力の10 %〜50 %となる。さらに,プラズマ内で発生した電圧及び電流の測定は,高
周波システムの技術的な難しさによって多少制限を受け,市販の測定装置では,印加高周波電
力の測定だけしかできないものもある。
6.3.2
固定印加電力−定ガス圧力制御モード
この制御モードでは,まず印加電力値を定め,その後,プラズマガス圧力が一定となるように調整して
測定を行う。
a) 印加電力を機器製造業者が推奨する値に設定し,最適測定条件が得られるようにガス圧力を調節する。
機器製造業者からの推奨値が得られていない場合には,印加電力及びガス圧力は,使用する装置で可
変可能なそれぞれの範囲で中間付近の数値を選択する。
b) 鉄又は鉄鋼試料を用いてスパッタリング速度(すなわち,単位時間当たりのクレータ深さ)を測定す
る。
c) 印加電力は,スパッタリング速度が約2 µm/min〜3 µm/minとなるように調整する。
d) 6.2.1.2と同様な手順で,検出器に印加する高電圧を設定する。
e) 6.2.1.3と同様な手順で,放電パラメータを順次調整する。ただし,最初に印加電力値を調整し,必要
8
K 0150:2020 (ISO 16962:2017)
に応じてガス圧力を変える。
f)
6.2.1.4と同様な手順で,スパッタリング痕形状を最適化する。この場合に調整する放電パラメータは,
ガス圧力である。
g) 必要に応じて,鉄又は鉄鋼試料を用いてスパッタリング速度を再測定し,その値が約2 µm/min〜3
µm/minとなるように印加電力を調整する。
h) 印加電力及びガス圧力の調整を繰り返して,スパッタリング速度とクレータ形状とがほぼ一定となる
ような放電条件を見いだす。この場合に,印加電力及びガス圧力の可変範囲は使用する装置によって
変わることに留意する。
i)
a)〜h) の操作によって得られた放電条件を,検量線作成用及び試験試料の分析において使用する。
6.3.3
固定印加電力−定直流バイアス電圧制御モード
この制御モードでは,まず印加電力値を定め,その後,ガス圧力を直流バイアス電圧が一定となるよう
に調整して測定を行う。
a) 最初に,高周波の印加電力値を設定し,プラズマガス圧力を調整することで機器製造業者が推奨する
直流バイアス電圧値となるようにする。機器製造業者からの推奨値が得られていない場合には,金属
試料の深さ方向分析で通常使用される設定範囲の中間値を目安に印加電力値及びバイアス電圧値を設
定する。プラズマガス圧力について追従可変機構が装備されている分析装置では,この操作は,自動
的に行われる。
b) 鉄又は鉄鋼試料を用いてスパッタリング速度(すなわち,単位時間当たりのクレータ深さ)を測定し,
その値が約2 µm/min〜3 µm/minとなるように印加電力を調整する。
c) 6.2.1.2と同様な手順で,検出器に印加する高電圧を設定する。
d) 6.2.1.3と同様な手順で,放電パラメータを順次調整する。ただし,最初に入力する電力値を調整し,
必要に応じてバイアス電圧を変える。
e) 6.2.1.4と同様な手順で,スパッタリング痕形状を最適化する。この場合に調整する放電パラメータは,
直流バイアス電圧である。
f)
必要に応じて,鉄又は鉄鋼試料を用いてスパッタリング速度を再測定し,その値が約2 µm/min〜3
µm/minとなるように印加電力を調整する。
g) 印加電力及びバイアス電圧の調整を繰り返して,スパッタリング速度とクレータ形状とがほぼ一定と
なるような放電条件を見いだす。この場合に,実際の印加電力及びバイアス電圧の可変範囲は使用す
る装置によって変わることに留意する。
h) a)〜g) の操作によって得られた放電条件を,検量線作成用及び試験試料の分析において使用する。
6.3.4
固定実効電力−定高周波実効電圧制御モード
実効電力及び高周波実効電圧の二つの制御モードがある。実効電力は,印加電力から反射電力と試料を
プラズマが生成していない状態(真空条件)で測定した暗電力とを差し引いた値として定義する。高周波
実効電圧は,直流バイアス電圧がない状態で対電極部で測定される高周波電力波形のRMS
(root-mean-square)値として定義する[8][9]。この制御モードでは,まず実効電力値を定め,その後,高周
波実効電圧が一定となるようにガス圧力を調整して測定を行う。
a) 最初に,実効電力を機器製造業者が推奨する電力値に設定する。機器製造業者からの推奨値が得られ
ていない場合には,高周波電圧は,700 Vに設定する。
b) 実効電力は,陽極の内径が4 mmの場合は10 W〜15 Wに,2 mmの陽極の場合は5 W〜10 Wに調整
する。実効電力に関して,最適値が分かっていない場合には,これらの電力範囲の中間付近の数値を
9
K 0150:2020 (ISO 16962:2017)
選択する。
c) 6.2.1.2と同様な手順で,検出器に印加する高電圧を設定する。
d) 6.2.1.3と同様な手順で,放電パラメータを順次調整する。ただし,最初に実効電力値を調整し,必要
に応じて実効電圧値を調整する。
e) 6.2.1.4と同様な手順で,スパッタリング痕形状を最適化する。この場合に調整する放電パラメータは,
高周波実効電圧である。
f)
高周波実効電圧を調整することによって,クレータの底面形状ができるだけ平たんとなるような条件
を見いだす。
g) a)〜f) の操作によって得られた放電条件を,検量線作成用及び試験試料の分析において使用する。
6.4
必要とする装置性能
6.4.1
概要
分析装置は,6.4.2及び6.4.3 1)に記載する性能仕様を満足することが望ましい。
注記 分析条件の設定には,通常,多くの装置パラメータの調整を繰り返す必要がある。
注1) 対応国際規格では“in 6.3.2 and 6.3.3 below”と記載していたが,明らかな誤記のため,この規格
では修正して記載した。
6.4.2
併行精度
分析装置が要求される併行精度を満たしていることを確かめるために,次の試験を行わなければならな
い。
質量分率1 %以上の分析対象元素を含む均質なバルク試料を用いて,10回の発光強度測定を行う。その
グロー放電条件は,実際の分析の条件と同じにする。測定は,少なくとも60秒間の放電(以下,予備放電
という。)の後に,5秒間〜20秒間のデータ取り込みを行う。各測定においては,試料の新しい測定部位を
用いる。10回の測定の相対標準偏差を計算する。相対標準偏差は,分析目的に沿った要求水準及び/又は
仕様を満足しなければならない。
注記 この試験で通常得られる相対標準偏差は,2 %以下である。
6.4.3
検出限界
6.4.3.1
概要
検出限界は,分析装置及び試料のマトリックス構成に依存するため,ある分析対象元素の検出限界は,
全ての装置に一律に決まらず,また,深さ方向に一定でない。この規格の目的からみて,各分析物の検出
限界は,分析対象の表面層で予想される最低質量分率の5分の1以下であれば,許容範囲である。
6.4.3.2
信号対ノイズ比(SNR)法
特定の分析対象元素の検出限界を求めるための一つ目の方法として,信号対ノイズ比(SNR)法による
手順を次に示す。
a) ブランク信号を求めるために適切なバルク試料を選定する。このバルク試料は,マトリックス成分の
化学組成が,深さ方向分析の対象とするめっきのマトリックス成分の化学組成と類似していることが
望ましい。さらに,ブランク試料中の分析対象元素の含有量は,1 µg/g未満とする。
b) ブランク試料を10回繰り返して放電する。各放電において,分析線の波長位置で10秒間発光強度を
取得し,バックグラウンド強度とする。用いるグロー放電の放電条件は,実試料の分析で用いる条件
と同じにすることが望ましい。安定な信号が得られるまで,予備放電を実施する。測定1回ごとに放
電位置を変えて,ブランク試料表面のスパッタリングしていない部位を用いる。
c) 検出限界は,質量分率を用いて,式(1)によって算出する。
10
K 0150:2020 (ISO 16962:2017)
3
DL
S
σ
×
=
··············································································· (1)
ここに,
DL: 検出限界
σ: バックグラウンド強度の標準偏差
S: 使用した分析装置で得られた分析の感度。標準物質群か
ら得られた検量線によって求められ,任意単位で測定し
た発光強度と質量分率との比で表される。
算出した検出限界が許容できない場合,この試験を繰り返さなければならない。その結果も許容できな
い場合は,実試料の分析前にその原因について調査し是正する。
6.4.3.3
信号対バックグラウンド強度比−バックグラウンド強度の相対標準偏差(SBR-RSDB)法
特定の分析対象元素の検出限界を求めるための二つ目の方法として,信号対バックグラウンド強度比−
バックグラウンド強度の相対標準偏差(SBR-RSDB)法による手順を,次に示す。
a) 分析するめっきと類似したマトリックス構成をもち,分析元素の含有量が質量分率0.1 %超で,その
定量値が既知のバルク試料を選択する。自己吸収が起こりやすい分析線(5.1.2参照)を用いる場合に
は,分析元素の質量分率が1 %以下のものを選択することが望ましい。
b) 選択したバルク試料で,3回の繰返し放電を行う。各放電において,分析線の波長位置で発光強度を
10秒間積分した発光強度を求める。この場合のグロー放電の条件は,実試料の分析と類似した放電条
件を採用する。個々の発光強度を測定して定量化を行う前に,安定な信号を得るために,ブランク試
料の予備放電を十分な時間をかけて実施する。測定1回ごとに放電位置を変えて,ブランク試料表面
のスパッタリングしていない部位を用いる。3回の繰返し測定を行い,その発光強度の積分値の平均
をとる。
c) 分析線のピーク位置から0.2 nm以内の波長範囲で,発光線のピークがない波長位置を選択する。10
回の繰返し放電を行い,バックグラウンド強度とする。グロー放電の条件及び予備放電は,手順b) で
用いたものと同じとする。この場合も,測定1回ごとに放電位置を変えて,ブランク試料表面のスパ
ッタリングしていない部位を用いる。10回の繰返し測定の発光強度の平均及びその相対標準偏差を計
算する。
d) 検出限界は,式(2)によって算出する。
(
)
(
)
A
rel,B
B
B
3
100
DL
w
II
I
σ
×
×
=
−
······························································ (2)
ここに,
DL: 検出限界
wA: 試料中の分析対象元素の質量分率
IB: 手順c) で求めた平均バックグラウンド強度
σrel,B: 手順c) で求めたバックグラウンド強度の相対標準偏差
(%)
I: 手順b) で求めた平均ピーク強度
算出した検出限界が許容できない場合には,この試験を繰り返さなければならない。その結果も許容で
きない場合には,実試料の分析前にその原因について調査し,是正する。
7
試料の準備とその調製
JIS G 0417及び/又は関連する規格によって,試料を準備する。このような規格を利用できない場合は,
11
K 0150:2020 (ISO 16962:2017)
めっき材の製造業者が推奨する方法又は他の適切な手順に従う。めっき鋼板の端部からの試料採取は,避
けることが望ましい。試料の寸法は,分析装置のグロー放電励起源に装着できる大きさとする。試料とし
て,20 mm〜100 mmの円形又はく(矩)形の形状(直径,幅及び/又は長さ)をもつものを用いる。これ
は,放電源に試料を固定し,真空を保持するためのOリングの直径が,通常,8 mm〜20 mmであること
による制約であり,個々の試料に対して少なくても2回の繰返し測定ができる程度の表面面積があること
が望ましい。
油を取り除くために,適切な溶媒(高純度アセトン,エタノール又はn-ヘプタン)で試料表面を洗浄す
る。不活性ガス(アルゴン又は窒素)又は油を含まない清浄な圧縮空気を,ガス管が表面に接しないよう
に,試料表面に吹き付け乾燥させる。油分の除去を効率的に行うため,試料表面を溶媒をしみ込ませた柔
らかい毛羽のない布又は紙で軽く拭き取ってもよい。拭き取った後,適切な溶媒(高純度アセトン,エタ
ノール又はn-ヘプタン)で表面を洗浄し,乾燥させる。
8
検量線の作成
8.1
概要
検量線は,各分析元素及び個々の分析線ごとに,A.3又はA.4に規定する方法に従って,応答信号と分
析元素の含有量の相関関係の決定とから成る。そのためには,検量線作成用試料の化学組成及びスパッタ
リング速度(質量減少率)の二つが必要である。
8.2
検量線作成用試料
8.2.1
概要
検量線作成に用いる試料群は,可能な限りCRM(認証標準物質)を使用することが望ましい。発光収率
を用いた定量方法を採用するため,その試料群の化学組成は,分析対象のめっき材と類似したものでなく
てもよいが,スパッタリング速度に関しては,精確な値が再現性よく得られていなければならない。高純
度金属は,それが発光線をもたない波長領域において,バックグラウンド強度を決めるために極めて有効
である。ただし,融点が800 K以下の一部の純金属,例えば,Zn,Sn及びPbは,再現性のあるスパッタ
リング速度及び発光強度を得ることが困難であるため,高質量分率域で検量線を作成するためには検量線
作成用試料として推奨できない場合がある。検量線作成用試料群の選択は,次による。
a) 分析元素ごとに,その含有量がゼロから最大の質量分率の範囲で,少なくても3点の検量点が得られ
るように試料群を準備する。これは,検量線作成のための最低限の条件であり,実際には,少なくて
も5点の検量点が得られるように,可能な限り多くの試料を準備することが望ましい。
b) 試料は,検量線作成のためにスパッタリングを行っている間で,分析対象元素の含有量がそのバルク
化学組成を代表する程度の均質性をもたなければならない。
これらの必要事項に基づき,8.2.2〜8.2.9に示す検量線作成用の標準物質を使用することが望ましい。分
析対象元素を含有していれば,これ以外の合金を検量線作成に使用してもよい。
8.2.2
黄銅標準物質
亜鉛の質量分率が25 %〜50 %,アルミニウムの質量分率が1 %〜4 %,鉛の質量分率が1 %〜4 %の黄銅
標準物質を,二つ以上選定する。
8.2.3
亜鉛アルミニウム合金標準物質
亜鉛の質量分率が40 %〜90 %の亜鉛アルミニウム合金標準物質を,二つ以上選定する。
8.2.4
鉄又は低合金鋼標準物質
鉄含有量が98 %を超える鉄又は低合金鋼標準物質を,二つ以上選定する。鉄の質量分率は,100 %から
12
K 0150:2020 (ISO 16962:2017)
鉄以外の全ての成分の質量分率を差し引いた値としてもよい。
8.2.5
ステンレス鋼標準物質
ニッケルの質量分率が10 %〜40 %のステンレス鋼標準物質を,二つ以上選定する。
8.2.6
ニッケル合金標準物質
ニッケルの質量分率が70 %を超えるニッケル合金標準物質を,一つ以上選定する(検量線の点は,スパ
ッタリング速度と質量分率との積で表されるので,標準物質のスパッタリング速度も関係する。亜鉛ニッ
ケル合金を選定するときには,そのスパッタリング速度はニッケル合金よりも大きくなることが予想され
る。このような場合,検量線作成用に準備する合金標準物質のニッケル質量分率は,箇条1に記載の20 %
よりも高いものが必要である。)。
8.2.7
アルミニウムけい素合金標準物質
けい素の質量分率が5 %〜10 %のアルミニウムけい素合金標準物質を,一つ以上選定する。
8.2.8
アルミニウムマグネシウム合金標準物質
マグネシウムの質量分率が3 %〜10 %のアルミニウムマグネシウム合金標準物質を,一つ以上選定する。
8.2.9
高純度銅及び高純度亜鉛標準物質
分析元素の質量分率が0.001 %未満の高純度銅及び/又は高純度亜鉛を,標準物質として一つ選定する。
これらの標準物質は,分析対象元素が銅及び亜鉛以外の場合に,検量線のゼロ点を決めるために使うこと
ができる。純鉄よりも純銅又は純亜鉛を標準物質として用いる理由は,これらの元素では発光線の本数が
少なく,分析対象元素の発光スペクトルの波長領域の全般にわたって,そのバックグラウンド強度への寄
与が非常に小さいことによる。
8.3
検量線の妥当性確認用試料及び検量線作成用のめっき標準物質
8.3.1
概要
分析結果の精確さを検証するために,検量線確認(8.7参照)用の標準物質を用意することが望ましい。
8.3.2〜8.3.5の標準物質を使用するのが望ましいが,他の適切な標準物質を使用してもよい。これらの標準
物質は,検量線作成用試料(8.2参照)として使用してもよい。
8.3.2
ニッケル亜鉛電気めっき標準物質
ニッケルの質量分率が20 %未満の電気めっき標準物質を用意する。単位面積当たりのめっき付着量及び
化学組成は,ISO 17925などで規定する方法によって値付けを行わなければならない。
8.3.3
亜鉛電気めっき標準物質
亜鉛の質量分率が30 %を超え,鉄の質量分率が5 %を超える電気めっき標準物質を用意する。単位面積
当たりのめっき付着量及び化学組成は,ISO 17925などで規定する方法によって値付けを行わなければな
らない。
8.3.4
亜鉛アルミニウム溶融めっき標準物質
亜鉛の質量分率が10 %を超え,アルミニウムの質量分率が5 %を超える標準物質を用意する。単位面積
当たりのめっき付着量及び化学組成は,ISO 17925などで規定する方法によって値付けを行わなければな
らない。
8.3.5
亜鉛鉄溶融めっき(焼鈍処理)標準物質
鉄の質量分率が10 %程度の亜鉛合金溶融めっき,及びそれに焼鈍処理を施した溶融めっき標準物質を用
意する。その単位面積当たりのめっき付着量及び化学組成は,ISO 17925などで規定する方法によって値
付けを行わなければならない。
注記1 標準物質(RM)とは,定量対象となる一つ以上の物性値が十分に均質かつ安定であり,装
13
K 0150:2020 (ISO 16962:2017)
置校正,測定方法の評価,又はその物性値の値付けのために作製された物質である。
注記2 認証標準物質(CRM)とは,定量対象となる一つ以上の物性値が認証値として値付けされて
いるものである。この場合の“認証”とは,その定量手続きが計量学的に定義されている明
確な単位系に従ったトレ−サビリティによって行われ,文書によってその認証された物性値
及びその不確かさを記載することである。商標としてのSRM○
R2)は,アメリカ国立標準技術
研究所(National Institute of Standards and Technology,NIST)から頒布されているCRMの一
例である。
注2) SRM(Standard Reference Material)は,NISTによって頒布されている標準物質の商標である。
この情報は,この規格の利用者の便宜を図って記載するものである。
8.4
検量線作成用試料のスパッタリング速度の測定
“スパッタリング速度”とは,グロ−放電でスパッタリングされる質量の減少率をいう。また,“相対ス
パッタリング率”とは,同一スパッタリング条件下における試料のスパッタリング速度を標準物質のスパ
ッタリング速度で除した値をいう。試料及び標準物質のスパッタリング面積が同じであれば,相対スパッ
タリング率は,単位面積当たりの相対スパッタリング率に等しい。スパッタリング速度は,次の手順で求
める。
a) 機器製造業者の推奨する方法又は他の適切な手順に従って,試料表面を仕上げる。
b) 放電制御パラメータを,6.2又は6.3に規定した手順によって決定した値に設定する。
c) クレータ深さが20 μm〜40 μmの範囲となる時間を推定し,試料をスパッタリングし,個々の測定に
ついてそれぞれ全スパッタリング時間を記録する。
d) 試料表面に複数回測定を行うだけの余裕があれば,c) を数回繰り返し,各クレータについて全スパッ
タリング時間を記録する。
e) 光学型又は触針型の表面粗さ計を用いて,中心を通り,異なった方向に4回以上深さを測定し,各ク
レータの平均深さを計測する。
f)
絶対スパッタリング速度を求める場合は,次による。
1) 一つ以上のクレータの面積を測定する。
2) この面積に平均スパッタ深さを乗じて,各クレータのスパッタリング体積を計算する。
3) このスパッタリング体積に試料の密度を乗じて,スパッタリング質量を計算する。
4) このスパッタリング質量を全スパッタリング時間で除して,各クレータのスパッタリング速度を計
算する。
5) 各クレータの測定結果から,スパッタリング速度の平均値及び標準偏差を算出する。
g) 相対スパッタリング率を求める場合は,次による。
1) スパッタリング深さに試料の密度を乗じて,各クレータの単位面積当たりのスパッタリング質量を
計算する。
2) この単位面積当たりのスパッタリング質量を全スパッタリング時間で除して,各クレータの単位面
積当たりのスパッタリング率を計算する。
3) 基準とする試料(鉄又は低合金鋼が望ましい。)を選び,1) 及び2) の方法で,この基準試料の単位
面積当たりのスパッタリング率を測定して平均値を求める。
4) 試料の単位面積当たりのスパッタリング率を,基準とする試料の単位面積当たりの平均スパッタリ
ング率で除して,各クレータの相対スパッタリング率を計算する。
5) 各クレータの測定結果から,相対スパッタリング率の平均値及び標準偏差を求める。
14
K 0150:2020 (ISO 16962:2017)
使用する表面粗さ計の深さ測定値の校正精度は,5 %よりも良好なものとすることが望ましい。
注記1 クレータの平均深さの真値は,クレータの中心を通る一次元走査から得られた平均深さと厳
密には一致せず,スパッタリングされた領域について実測された全ての値の平均である。近
年,スパッタリング領域の二次元・三次元の全体像を計測できる,新型の表面形状計測シス
テムが開発されている[9][10]。
注記2 スパッタリングされた物質の質量は,スパッタリング前後で試料をひょう(秤)量して得ら
れる。しかし,これには非常に高精度の天びん(秤)を必要とし,かつ,測定の不確かさは
クレータ深さ測定方法よりも一般に大きな値となる。
注記3 標準物質の中には,そのスパッタリング率の値を機器製造業者から得られるものがある。
利用できる測定設備があれば,個々の検量線作成用試料の密度を測定する。均質な試料では,試料の質
量をその体積で除して求めることができる。この場合,試料体積は,試料を水中に浸せき(漬)しアルキ
メデスの原理によって求める。そのほか,試料体積は,その寸法からも算出することができる。また,式
(A.34)に従って,試料の化学組成から見積もられた密度を用いることもできる。実測値又は計算値として
得られる密度の精確さは,5 %より良好であることが望ましい。
8.5
検量線作成用試料の発光強度の測定
標準物質の発光強度の測定は,次による。
a) 機器製造業者が推奨する方法によって,標準物質表面を仕上げる。特にその手順がない場合には,バ
ルク標準物質の場合は,通常220番の乾式研磨紙による仕上げで十分であるが,湿式研磨による方法
としてもよい。湿式研磨の場合には,その表面をエタノールで十分に洗浄し,アルゴン又は窒素のよ
うな不活性ガス,又は清浄で油分を含まない圧縮空気を吹き付けて完全に乾燥させる。試料の表面が
ガスの吹出口に触れないように注意する。
b) 6.2又は6.3に規定した放電条件を参照して,放電励起源の制御パラメータを調整する。予備放電時間
を50秒〜200秒に,信号積分時間を5秒〜30秒に設定する。
c) 分析対象元素の発光強度を測定する。強度の単位は任意でよいが,通常1秒当たりのカウント数(cps)
又は電圧(V)が使われる。各試料を2回以上測定し,平均値を求める。
8.6
検量線定数の計算
A.3又はA.4のいずれかによって,検量線を作成する。市販されている全ての分析装置には,A.3及び
A.4の検量線定数の算出方法のうち,少なくとも一つがソフトウエアとして組み込まれている。
8.7
標準物質を用いた検量線の妥当性確認
8.7.1
概要
検量線を作成した後に,その精確さを確かめるために,直ちに検量線の妥当性確認を行う。新しい試料
を測定するごとに妥当性確認を行う必要はない。日常の分析では,測定機器の感度等の時間変動(ドリフ
ト)を補正するため,8.8で規定する検量線の検証を行わなければならない。妥当性確認を行うためには二
つの方法がある。第一の方法は,バルク標準物質を使用する方法(8.7.2)であり,第二の方法は,めっき
標準物質を使用する方法(8.7.3)である。めっき標準物質を作製するのが難しい場合があるため,第二の
方法(8.7.3)は任意とする。
注記 妥当性確認とは,客観的証拠を提示することによって,特定の意図された用途又は適用に関す
る要求事項が満たされていることを確認することである(JIS Q 9000:2015の3.8.13参照)。方
法の妥当性確認は,JIS Q 17025:2018の7.2.2に定義されている。検量線の妥当性確認は,その
考え方に沿っている(8.8の注記参照)。
15
K 0150:2020 (ISO 16962:2017)
8.7.2
バルク標準物質による精確さの評価
a) 検量線の妥当性確認を行うため,幾つかのバルク標準物質を8.3 3)の規定によって選択する。
注3) 対応国際規格では“8.1”としていたが,該当の記載がなく誤記のため,この規格では修正し
て記載した。
b) 検量線作成時と同じグロー放電条件,予備放電時間及び積分時間でこれらの標準物質の発光強度を測
定する。各試料について,放電ごとに試料表面の測定部位を変えて,3回以上の放電を行わなければ
ならない。
c) 検量線を使って,各妥当性確認試料中の分析対象元素の平均質量分率を計算する。
d) 求めた分析対象元素の平均質量分率と既知の値(例えば,認証値)とが,統計的な許容差内にあるこ
とを確認する。許容差内になければ,その原因を究明し,場合によっては,検量線を再度作成する。
8.7.3
めっき標準物質による精確さの評価
a) 機器製造業者の説明書によって,深さ方向分析の準備をする。
b) 検量線作成時と同じグロー放電の制御パラメータを使用する。
c) 表面めっき層が完全にスパッタリングされ,基板に達するまで,放電を行う。
d) 検量線を使って,分析対象元素の質量分率と分析深さとの定量的な関係を求める。通常の分析装置は,
各測定が完了すると,この定量関係を自動的に計算するソフトウエアを装備している。
e) 定量した全ての分析対象元素を合算して,めっきの質量をグラム毎平方メートル(g/m2)の単位で計
算する。市販の分析装置は,この計算を行う機能をもつソフトウエアを装備している。計算値とめっ
き標準物質の付与値との差は,±5 %を超えてはならない。
f)
めっき厚さをマイクロメートル(μm)単位で計算する。標準物質の厚さの付与値と計算値との差は,
±5 %を超えてはならない。また,市販のめっき鋼鈑においては,化学分析によって求めた厚さとグ
ロー放電による計算値との差は,±10 %でなければならない。
g) 表面粗さ計を使用する場合は,めっき厚みの検証が即座にできる。めっき標準物質の付与値,グロー
放電による計算値,及び表面粗さ計で得られた値が,統計的な許容差内にあることを確認する。
h) 許容差内になければ,その原因を究明し,場合によっては,検量線を再度作成する。
8.8
検量線の検証及びドリフト補正
分析機器からの発光強度は,時間とともに変動する。標準物質を使って検量線を求め,その妥当性確認
が完了している場合でも,試験試料を定量する前には,作業日ごと又は分析担当者の交替ごとに,検量線
が管理範囲内にあることを検証する必要がある。機器製造業者の説明書に検証の手順が示されていない場
合は,次の手順による。
a) 検証のために,数個の均質な試料を選定する。この試料の元素組成は,試験試料中に含まれる分析対
象元素の質量分率範囲を含むことが望ましい。
b) 検量線作成時と同じ,グロー放電条件,予備放電時間及び積分時間で,検証用試料の発光強度を測定
する。各試料を,放電ごとに新しい表面を使って,2回以上の放電を行わなければならない。
c) 検量線を使って,試料ごとに分析対象元素の質量分率の平均値を計算する。
d) 求めた分析対象元素の質量分率の平均値と既知の値とが,統計的な許容差内にあることを確認する。
許容差内になければ,機器製造業者の指示に従って検量線の各パラメータを調整するか,又は発光強
度を補正することによってドリフト補正を行う。
ドリフト補正の後に,検量線の精確さを確認するために,検証用試料をもう一度分析することが望まし
い。
16
K 0150:2020 (ISO 16962:2017)
注記 検証とは,客観的証拠を提示することによって,規定要求事項が満たされていることを確認す
ることである(JIS Q 9000:2015の3.8.12及びこの規格の8.7.1の注記参照)。
9
試験試料の分析
9.1
放電制御パラメータの調整
放電励起源の制御パラメータを,検量線作成時の放電条件と可能な限り同じになるように,調整する。
9.2
測定時間及びデータ取得速度(サンプリングレート)の設定
測定する試料に合わせて,適切な全測定時間及び発光強度のデータ取得速度(以下,サンプリングレー
トという。)を選ぶように,注意しなければならない。市販されているグロー放電発光分光分析装置のソフ
トウエアでは,サンプリングレートを自由に変えられる設計がなされている。ソフトウエアの多くは,全
測定時間を分割して,区間ごとにサンプリングレートを調整できる。サンプリングレートは,分析試料の
めっきの公称厚さを通過してスパッタリングが完了するのに要する時間から決める。非常に大きいスパッ
タリング速度を選択した場合は,発光強度の積分時間は短くなり,その信号は比較的大きなノイズ成分を
含むことに注意することが望ましい。一般的に推奨できるサンプリングレートは,構成元素の深さ方向組
成変化(以下,深さ方向プロファイルという。)を記録する場合に,単一層中又は何らかの遷移領域から
10点以上の測定点となるように選択することが望ましい。全測定時間を二つ以上の時間領域に分割し,試
料の最表面部位では深さ方向プロファイルに確実に追随できるように大きいサンプリングレートを設定し,
その後はサンプリングレートを小さくして,発光信号の信号対ノイズ比を改善するのが望ましい。
例として,厚さ5 nmの非常に薄い表面層をもつ試料を,スパッタリング速度50 nm/sで分析する場合,
表面層の厚さの2倍程度(10 nm)までは,大きいサンプリングレートを設定することを推奨する。少な
くても最初の0.2秒間は,大きいサンプリングレートを用い,この経過時間内に少なくても20点測定する。
このために,サンプリングレートを毎秒100点以上とする。スパッタリング深さが大きくなると深さ方向
分解能が低下するので,サンプリングレートを減じても,深さ方向の情報に影響を与えることは少ない。
9.3
試験試料の定量深さ方向プロファイルの定量化
箇条8の手順と,附属書Aの計算方法とによって,定量深さ方向プロファイルを作成する。
10
結果の表記
10.1
定量深さ方向プロファイル
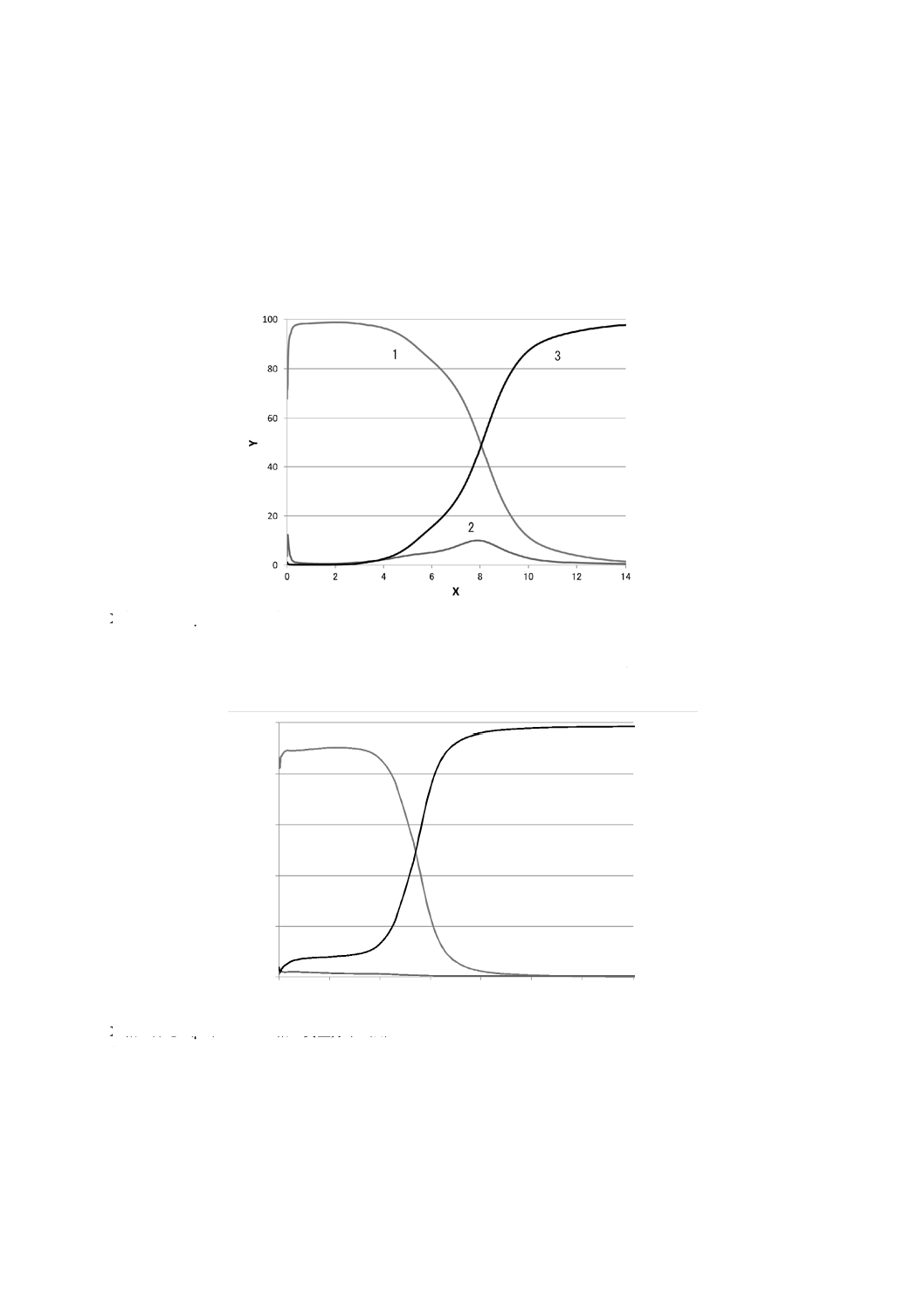
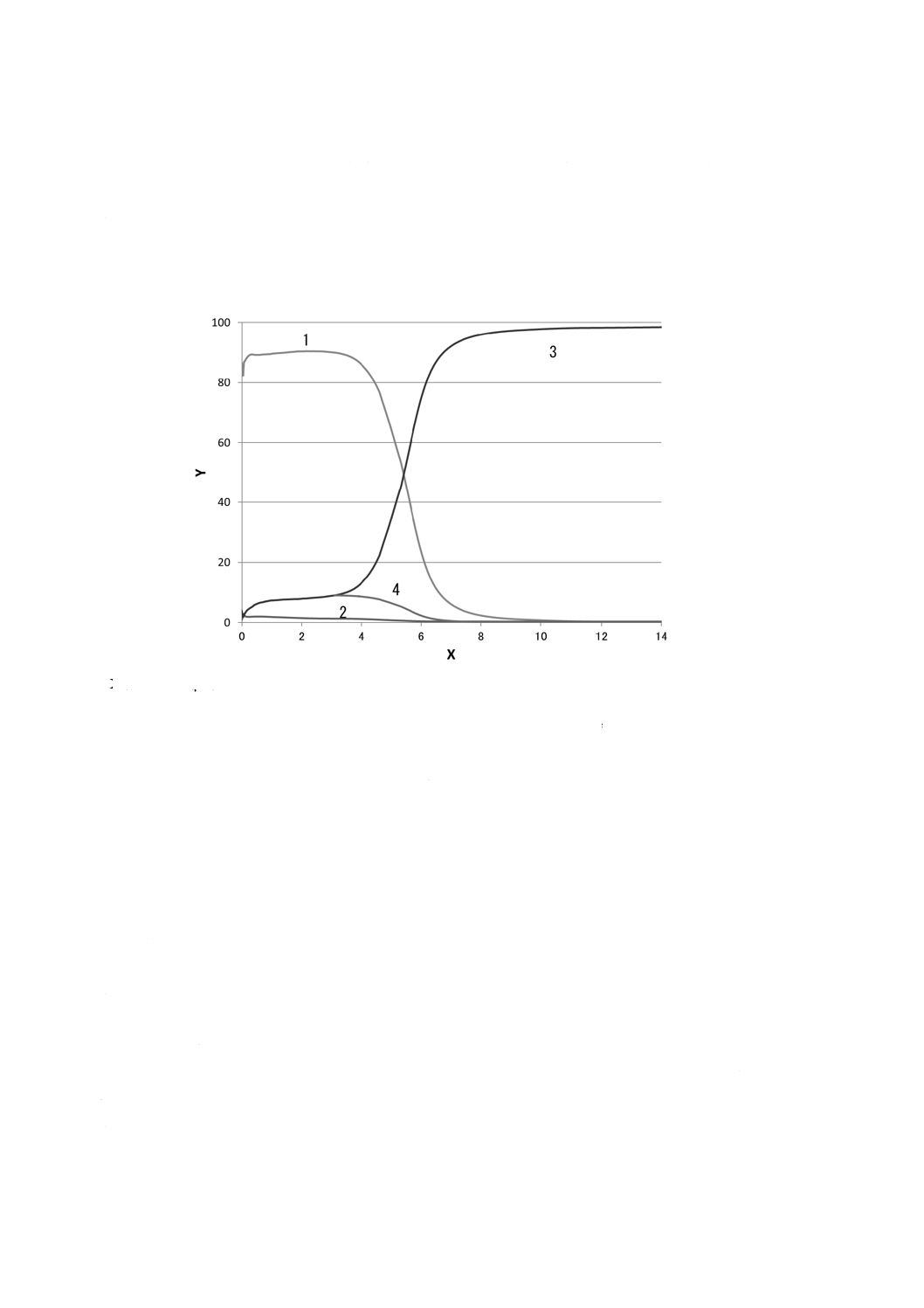
定量深さ方向プロファイルの例を,図1及び図2に示す。
10.2
単位面積当たりのめっき付着量の定量
10.2.1
一般的方法
めっき層に存在する元素だけを対象とする。めっき層を構成する全ての元素が,基板にほとんど含まれ
ない場合(例えば,鋼板上の溶融亜鉛めっき層),表面層の全質量は,各分析元素の深さ方向プロファイル
を積分して求める。この場合,定量深さ方向プロファイルは,分析元素ごとの質量分率及びめっき層厚を
表示する。市販の分析装置は,この計算を行うソフトウエアが附属している。
深さ方向に積分して全質量を求める方法は,次による。
a) めっき厚さは,めっき中の主元素のいずれかを選択して,その質量分率が表面層の値(深さ方向プロ
ファイルの平たんな部位の値)の50 %に減少した距離(深さ)として定義する。
b) めっき層から基板へ移行する界面の幅は,選択した元素の質量分率が,めっき層の値(深さ方向プロ
ファイルの平たんな部位の値)の84 %から16 %になるまでの2点間の距離(深さ)と定義する。こ
17
K 0150:2020 (ISO 16962:2017)
の平たんな部位の質量分率は,完全に平たんでない場合又は非常に薄い場合,ソフトウエアによらず
分析者の判断によって決定する。
c) 積分深さは,めっき厚さと界面の幅との和とする。
10.2.2
10.2.1が当てはまらない試料の場合の方法
めっき層の主要元素のいずれかが基板と同じ場合(例えば,鋼板上の鉄を含んだめっき層,鉄酸化物層
など)は,試料に応じて計算する方法を附属書Cに参考として示す。
X軸 深さ(μm) Y軸 質量分率(%)
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
図1−鋼板上の溶融亜鉛めっき(Zn-Al)皮膜の定量深さ方向プロファイルの例
0
20
40
60
80
100
0
2
4
6
8
10
12
14
Y
X
1
3
2
X軸 深さ(μm) Y軸 質量分率(%)
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
図2−鋼板上の合金化溶融亜鉛めっき(Zn-Fe)皮膜の定量深さ方向プロファイルの例
10.3
各元素の平均質量分率の決定
めっき層内の各元素の平均質量分率は,その元素の単位面積当たりの質量を,全ての元素の単位面積当
18
K 0150:2020 (ISO 16962:2017)
たりの質量の合計で除した値とする。
11
精度
このグロー放電発光分光分析方法を使った共同実験が,4試験所で,7元素について計画され,各試験所
が各元素を二つ以上定量した。使用した試料及び得られた平均値を,表D.1に示す。得られた結果は,JIS
Z 8402-2に従い,統計的に処理した。JIS Z 8402-1で規定する併行条件,すなわち,一人のオペレータが
同じ機器,同じ操作条件,同じ検量線を使って,最小の期間で2回以上の定量を行った。併行許容差は,
JIS Z 8402-6によって計算した。得られた結果を,表1及び表2に示す。また,それらをグラフ化したも
のを附属書Dに示す。
注記 溶融めっき鋼鈑について得られた精度は,主に工業製品がもつめっき層の不均質性によるもの
であり,測定能力を表すものではない。
表1−単位面積当たりのめっき付着量の併行標準偏差及び併行許容差の結果
単位 g/m2
めっきの種類
併行標準偏差
sr
併行許容差
r (=2.8×sr)
電気めっき
0.75
2.1
溶融めっき
4.5
12.6 a)
注a) 対応国際規格では“16.0”としているが,rの計算式の結果と異なる
ため,この規格では修正して記載した。
表2−めっき中元素質量分率の併行標準偏差及び併行許容差の結果
単位 g/m2
元素質量分率
%
併行標準偏差
sr
併行許容差
r (=2.8×sr)
<0.1
0.006
0.017
1
0.03
0.084
10
0.2
0.56
99
1
2.8
12
試験報告書
試験報告書には,次の事項を記載することが望ましい。
a) 試料を特定するための全情報
b) 試験所機関名及び分析した年月日
c) 分析方法(この規格又は別の規格を適用した場合の番号)
d) 分析結果及び分析結果を表す図表など
e) 分析の際に何らかの特記するべき事象が起こった場合には,その内容
f)
この規格に規定のない操作,又は結果に影響を及ぼした可能性がある操作
19
K 0150:2020 (ISO 16962:2017)
附属書A
(規定)
検量線定数の算出及び深さ方向プロファイルの定量化
A.1 概要
市販されているグロー放電発光分光分析装置において,検量線作成及び深さ方向プロファイルの定量化
は,発光収率の概念に基づいている。発光収率は,分析対象元素のある発光線を測定した場合に,その元
素の単位スパッタリング量当たりの発光強度と定義される[2][3]。発光収率は,分析される元素に依存して
変化する量であり,測定装置ごとに変わるため,スペクトル線ごとに測定装置を特定して独立に求めなけ
ればならない。ある分析対象元素のスペクトル線の積分発光強度は,その元素のスパッタリング量に比例
し,発光収率は試料組成(マトリックス構成)には依存しないことを前提としている。この前提について
は,既に幾つかの研究がなされており[4][5][6],少なくとも第一近似としては良好な結果が得られるものと
して,その有効性が幅広く認められている。また,マトリックス構成に依存しない発光収率の考え方に基
づいて,検量線作成及び深さ方向プロファイルの定量化を行うための幾つかの異なった数学モデルが提案
されている。このため,市販の分析装置に組み込まれているソフトウエアの中には,この規格の計算方法
とは必ずしも一致しないものもある。また,装置の中には,実験的なモデルに基づき,放電励起源の制御
パラメ−タ補正の計算方法を行うものもある。最近,水素の影響を考慮した発光収率の補正方法が考案さ
れ,市販装置のソフトウエアに組み込まれている。次に,これらの定量化計算方法に使用する諸変数の詳
細を記載する。これらの変数は,この規格を構成するものとして,全て有効で,かつ,実際に使用できる。
実際の分光器システムにおける検量線の妥当性確認は,8.7で規定した一連の妥当性確認手順によって行わ
れ,この妥当性確認で確認されることが評価変数の妥当性を証明する。今後,定量化のための計算方法が
新たに提案される場合においても,妥当性確認で満足できる結果が得られれば,その方法を用いてもよい。
A.2 記号の定義
A.2.1 この附属書で全般的に用いている記号
Aij
領域jにおける元素iの原子分率
AD
試料Dのスパッタリング面積
aλ
波長λの発光強度に与える放電電流の影響を補正するための定数
bλ
波長λの発光強度に与える水素の影響を補正するための定数
Iiλ
元素iの波長λの発光強度
I'iλ
放電制御パラメータの変動に対して補正した元素iの波長λの発光強度
I''iλ
水素の影響を補正した元素iの波長λの発光強度
IH
水素の発光強度
IHref
標準物質[例えば,水素化チタン(TiH2)薄膜又はポリマー皮膜]における水素の
発光強度
IHD
検量線作成用試料Dにおいて測定された水素の発光強度
IBλ
波長λにおいて測定されたバックグラウンド強度の平均値
miDj
試料Dの領域jにおける元素iの単位面積当たりのスパッタリング質量
mjtot
領域jの単位面積当たりの全スパッタリング質量
20
K 0150:2020 (ISO 16962:2017)
rUiλ
最小二乗法を使い推定された検量線の回帰曲線における係数
sj
領域jにおける放電電流値
s0
機器製造業者によって決められた参照電流値
scal
定量方法の校正のために使用される電流値
Uj
領域jにおける放電電圧
U0
スパッタリング開始の電圧いき(閾)値
UD
検量線作成用試料Dにおいて測定された放電電圧
Uav
検量線作成に用いられた試料群において測定された放電電圧の平均値
Ucal
定量方法の校正のために使用される放電電圧
wiD
試料Dにおける元素iの質量分率
wiDj
試料Dの領域jにおける元素iの質量分率
Wi
元素iの原子量
zj
領域jの厚さ
∆tj
スパッタリング時間の増分
ρi
各構成元素iの密度(純物質)
ρj
領域jの密度
A.2.2 A.3及びA.7で相対スパッタリング速度に関連して使用される記号
Bλ
スペクトル線波長λにおけるバックグラウンド強度の補正項(質量分率 %換算)
Bλrel
スペクトル線波長λにおける相対バックグラウンド強度の補正項(質量分率 %換算)
eiλ
元素iの波長λのスペクトル線の検量線において線形相関からのずれを表す係数
kiλ
元素iの波長λのスペクトル線の発光収率の逆数を相対スパッタリング速度で除し
た値として得られる感度係数
qD
試料Dの単位面積当たりの質量減少速度で表したスパッタリング速度
qref
標準物質(例えば,純鉄試料)のスパッタリング速度
qDj
試料Dの領域jにおける単位面積当たりの質量減少速度(スパッタリング速度)
Riλ
元素iの波長λのスペクトル線の逆発光収率
iλ
元素iの波長λのスペクトル線の発光収率
A.2.3 A.4及びA.8で絶対スパッタリング速度に関連して使用される記号
B'λ
スペクトル線波長λにおけるバックグラウンド強度の補正項(質量分率と試料のス
パッタリング速度との積の次元をもつ。)
B'λrel
スペクトル線波長λにおける相対バックグラウンド強度の補正項(質量分率の次元
をもつ。)
e'iλ
元素iの波長λのスペクトル線の検量線において線形相関からのずれを表す係数
k'iλ
元素iの波長λのスペクトル線の発光収率の逆数をスパッタリング速度で除した値
として得られる感度係数
q'D
試料Dのスパッタリング速度
q'Dj
試料Dの領域jにおけるスパッタリング速度
R'iλ
元素iの波長λのスペクトル線の逆発光収率
21
K 0150:2020 (ISO 16962:2017)
'iλ
元素iの波長λのスペクトル線の発光収率
rHiλ
元素iの波長λのスペクトル線の検量線に水素の影響を加えるための回帰係数
A.3 相対スパッタリング速度を用いた検量線定数の算出方法
検量線の作成は,式(A.1)又は式(A.2)によって行う。
(
)
D
D
ref
i
i
i
w
qq
R
I
B
λ
λ
λ
×
=
×
−
······················································ (A.1)
(
)
D
ref
D
rel
i
i
i
w
R
I
q
q
B
λ
λ
λ
=
×
×
−
···················································· (A.2)
ここに,
wiD: 試料Dにおける元素iの質量分率
qD/qref: 標準物質に対する試料Dの相対スパッタリング率
qD: 試料Dの単位面積当たりの質量減少速度で表したスパ
ッタリング速度
qref: 標準物質(例えば,純鉄試料)スパッタリング速度
Riλ: 元素iの波長λのスペクトル線の逆発光収率(注記1を
参照)
Iiλ: 元素iの波長λのスペクトル線の発光強度
Bλ: スペクトル線波長λにおけるバックグラウンド強度の補
正項
Bλrel: スペクトル線波長λにおける相対バックグラウンド強度
の補正項。式(A.2)で用いられ,分析元素の質量分率の関
数となる。“バックグラウンド等価濃度(BEC)”ともい
う(注記2を参照)。
qref/qD: スパッタリング速度の補正因子
注記1 逆発光収率Riλと発光収率
iλとの関係は,式(A.3)で表される。
(
)
ref
1
i
i
R
q
λ
λ
=
×
··································································· (A.3)
ここで発光収率は,式(A.4)で定義される。
(
)(
)
B
D
D
i
i
i
I
I
w
q
λ
λ
λ
≡
−
×
························································ (A.4)
ここに,
IBλ: 波長λにおいて測定されたバックグラウンド強度の平均
値
注記2 二つのバックグラウンド強度の補正項は,式(A.5)の関係がある。
(
)
rel
rel
D
B
qq
B
λ
λ
=
×
································································· (A.5)
実際の操作においては,式(A.1)及び式(A.2)は,非線形の検量線,例えば,2次及び高次の項を含む関係
式によって表現できる。このような直線相関がない検量線を用いるときの解析例として,検量線が二次曲
線に近似できる場合には,式(A.1)及び式(A.2)は,それぞれ式(A.6)及び式(A.7)のように表される。
(
)
2
D
D
ref
i
i
i
i
i
w
qq
R
I
e
I
B
λ
λ
λ
λ
λ
×
=
×
+
×
−
··········································· (A.6)
(
)
(
)
2
D
ref
D
ref
D
rel
i
i
i
i
i
w
R
I
q
q
e
I
q
q
B
λ
λ
λ
λ
λ
=
×
×
+
×
×
−
··························· (A.7)
22
K 0150:2020 (ISO 16962:2017)
ここに,
eiλ: 元素iの波長λのスペクトル線の検量線において線形相
関からのずれを表す係数
これらの検量線定数を求めるためには,実測された検量線データを用いて最小二乗法による回帰計算を
行わなければならない。
標準物質のスパッタリング速度qrefは,通常,低合金鋼のように検量線の基準として幅広く使用されて
いる試料について測定された,単位面積当たりのスパッタリング速度を用いる。これは,幾つかの鉄基の
検量線試料では,相対スパッタリング率はほぼ等しく(スパッタリング率の補正因子を1とみなせる。),
プラズマ条件によってあまり変動しないためである。
式(A.1)及び式(A.2)のバックグラウンド強度の補正項は,完全に定数ではなく,試料のマトリックス構成
に依存して変動する。実際の定量操作では,個々の分析線においてバックグラウンド強度の補正項が最小
となるような測定条件を選択することが常に推奨される。
市販の分析装置は,共存元素のスペクトル線の重なり(分光干渉)に起因する発光強度を,バックグラ
ウンド加算分として差し引く機能をもっている。この機能が利用できる場合には,その補正を利用するこ
とが望ましい。
A.4 絶対スパッタリング速度を用いた検量線定数の算出方法
検量線の作成は,式(A.8)又は式(A.9)によって行う。
D
D
i
i
i
w
q
R
I
B
λ
λ
λ
′
′
′
×
=
×
−
······························································ (A.8)
D
D
rel
i
i
i
w
R
Iq
B
λ
λ
λ
′
′
′
=
×
−
····························································· (A.9)
ここに,
wiD: 試料Dにおける元素iの質量分率
q'D: 試料Dのスパッタリング速度
R'iλ: 元素iの波長λのスペクトル線の逆発光収率(注記1を
参照)
Iiλ: 元素iの波長λの発光強度
B'λ: スペクトル線波長λにおけるバックグラウンド強度の補
正項。分析元素の質量分率にスパッタリング速度を乗じ
た値で表され,通常は定数又は実測に合わせた複雑な関
数形をとる。機器製造業者から提供される。
B'λrel: 式(A.9)で使用するスペクトル線波長λにおける相対バ
ックグラウンド強度の補正項。分析元素の質量分率の関
数となり,通常は定数又は実測に合わせた複雑な関数形
をとる。“バックグラウンド等価濃度(BEC)”ともいう
(注記2を参照)。機器製造業者から提供される。
注記1 逆発光収率R'iλと発光収率'iλとの関係は,次の式(A.10)で表される。
1
i
i
Rλ
λ
′
′
=
···········································································(A.10)
ここで,発光収率は,次の式(A.11)で定義される。
(
)(
)
B
D
D
i
i
i
I
I
w
q
λ
λ
λ
′
′
=
−
×
······················································· (A.11)
ここに,
IBλ: 波長λにおいて測定されたバックグラウンド強度の平均
値
注記2 二つのバックグラウンド強度の補正項は,式(A.12)の関係がある。
rel
D
B
Bq
λ
λ
′
′
′
=
········································································(A.12)
23
K 0150:2020 (ISO 16962:2017)
実際の操作においては,式(A.8)及び式(A.9)は,非線形の検量線,例えば,2次及び高次の項を含む関係
式によって表現できる。このような直線相関がない検量線を用いるときの解析例として,検量線が二次曲
線に近似できる場合には,式(A.8)及び式(A.9)は,それぞれ式(A.13)及び式(A.14)のように表される。
2
D
D
i
i
i
i
i
i
w
q
R
I
e
I
B
λ
λ
λ
λ
λ
′
′
′
′
×
=
×
+
×
−
·················································(A.13)
2
D
D
D
rel
i
i
i
i
i
i
w
R
Iq
e
Iq
B
λ
λ
λ
λ
λ
′
′
′
′
′
=
×
+
×
−
···········································(A.14)
ここに,
e'iλ: 元素iの波長λのスペクトル線の検量線において線形相
関からのずれを表す係数
これらの検量線定数を求めるためには,実測された検量線データを用いて最小二乗法による回帰計算を
行わなければならない。
式(A.8)及び式(A.9)のバックグラウンド強度の補正項は,完全に定数ではなく,試料のマトリックス構成
に依存して変動する。実際の定量操作では,個々の分析線においてバックグラウンド強度の補正項が最小
となるような測定条件を選択することが常に推奨される。
市販の分析装置は,共存元素のスペクトル線の重なり(分光干渉)に起因する発光強度を,バックグラ
ウンド加算分として差し引く機能をもっている。この機能が利用できる場合には,その補正を利用するこ
とが望ましい。
A.5 放電制御パラメータの変動による発光強度及びスパッタリング質量の補正
A.5.1 概要
大多数の市販の分析装置では,検量線作成時に用いられた放電制御パラメータが変動した際に,発光線
の実測強度及びスパッタリング質量を補正する機能をもっている。酸化物薄膜が100 nm未満の場合,放
電パラメータが安定しないときに,この極薄膜のスパッタリングが,短い時間間隔で少なくとも部分的に
発生することから,このような補正を行うことが望ましい。この補正計算は,次による。
A.5.2 放電制御パラメータの変動に対して求められた補正係数に基づいた発光強度の補正
各元素iのスペクトル線λについて,分析対象領域jにおいて,補正された発光強度I'iλは,式(A.15)に
よって求める。
(
)
()
0
a
i
j
j
I
ss
fU
λ
λ′=
×
····························································(A.15)
ここに,
aλ: 波長λの発光強度に与える放電電流の影響を補正するた
めの定数
f(Uj): 領域jにおける放電電圧Uの関数として多項式(通常,
1次式〜3次式)展開した,発光線λの補正係数
sj: 領域jにおける放電電流値
s0: 機器製造業者によって決められた参照電流値
べき乗数aλ及び強度多項式の係数の具体的値は,通常,機器製造業者によって提供される。
実測強度Iiλの代わりに,補正された発光強度I'iλを,式(A.8),式(A.9),式(A.13)及び式(A.14)に代入して
計算する。
注記 べき乗数aλ及び強度多項式の係数の値は,スペクトル線ごとの測定装置に依存しない定数であ
る。
24
K 0150:2020 (ISO 16962:2017)
A.5.3 放電電圧に起因する発光収率変動の補正
プラズマガス圧力を一定とした場合には,放電電圧及び電流は,プラズマ抵抗値に相互に依存して変化
する。したがって,この場合には,電圧又は電流のいずれか一つのパラメータに対して発光収率の補正を
行うことができる。検量線式に含まれる発光収率を放電電圧に対して補正する方法の一つは,式(A.16)で
示す,逆発光収率に電圧依存項を組み入れるものである。
(
)
D
av
1
U
i
i
R
r
U
U
λ
λ
′′=+
×
−
····························································(A.16)
ここに,
rUiλ: 最小二乗法を使い推定された検量線の回帰曲線におけ
る係数
UD: 検量線作成用試料Dにおいて測定された放電電圧
Uav: 検量線作成に用いられた試料群において測定された放
電電圧の平均値
この式形は乗法補正というものであり,放電電圧を新たなパラメータとして,そのほかの変数との依存
関係をその積としての検量線式に組み入れたものである。
この電圧補正項は,検量線の式に組み込まれるため,次のa)〜c) の手順を行うことで,発光収率の電圧
補正に関して新たな補正計算を行う必要はない。
a) 発光励起源は,プラズマガス圧力を一定として放電を行う。
b) 直流バイアス電圧又は印加電圧を検量線作成時及び実際試料測定時に測定する。
c) 試料に依存する放電電圧の変化が把握できるように,検量線作成のために十分な個数の試料を準備し
て測定する。
補正係数rUiλは,検量線作成時に個別に評価するものであり,測定装置の間で共通の値として使用する
ことはできない。
注記 (対応国際規格のNOTEは許容事項のため,A.5.3本文に移動した。)
A.5.4 放電制御パラメータに起因するスパッタリング速度変動の補正
A.5.2の手順に従って発光強度の変動補正が行われた場合には,放電制御パラメータの変化によるスパッ
タリング速度の変動についても補正を行う必要がある。領域jにおける標準物質のスパッタリング速度の
補正値q'refは,(A.17)によって求める。
0
ref
cal
cal
0
j
j
s
U
U
q
s
U
U
−
′=
−
·······························································(A.17)
ここに,
sj: 領域jにおける放電電流値
scal: 定量方法の校正のために使用される電流値
Uj: 領域jにおける放電電圧
Ucal: 定量方法の校正のために使用される放電電圧
U0: スパッタリング開始の電圧いき値
標準物質のスパッタリング速度の補正値q'refは,スパッタリング速度qrefの代わりに,式(A.20)又は式
(A.22)に代入して用いる。
A.6 水素の影響による発光収率の補正
A.6.1 概要
グロー放電プラズマが水素をある程度含有する場合には,水素は,分析対象元素のスペクトル線によっ
てはその発光収率に大きな影響を与えることがあり,その測定強度に影響する。酸化物薄膜が100 nm未
25
K 0150:2020 (ISO 16962:2017)
満の試料では,水素原子がその酸化物に含まれる場合があるため,この現象を補正することが望ましい。
さらに,放電の開始時に,水素原子を含む化合物(水及び炭化水素化合物)がグロー放電管の内壁から放
出される。この補正計算は,次による。
A.6.2 水素スペクトル線の発光強度及び補正係数を用いた分析線の発光強度の補正
分析元素iのスペクトル線λごとに,領域jにおける発光強度I''iλを,式(A.18)によって求める。
H
Href
exp
i
i
I
I
I
bI
λ
λ
λ
′′
′
=
······························································(A.18)
ここに,
bλ: 波長λの発光強度に与える水素の影響を補正するための
定数
IH: 水素の発光強度
IHref: 標準物質[例えば,水素化チタン(TiH2)薄膜又はポリ
マー皮膜]における水素の発光強度
実測強度Iiλ又はI'iλの代わりに,この補正した強度I''iλを,式(A.6)及び式(A.7)に代入して質量分率及び
スパッタリング質量を計算する。
定数bλは,個々のスペクトル線に対して固有の値をもち測定装置には依存しないが,標準物質には左右
される。したがって,定数bλを複数の測定装置で使用する場合には,同じ標準物質でその発光強度IHrefを
評価する必要がある。
注記 (対応国際規格のNOTEは要求事項のため,本文に移動した。)
A.6.3 水素の影響による検量線式内の発光収率の補正
発光収率を水素スペクトル線の強度を因子とした発光収率の乗法補正式[式(A.19)]を,検量線式に組
み入れる。
(
)
H
HD
1
i
i
R
r
I
λ
λ
′′=
×
+
··································································(A.19)
ここに,
rHiλ: 元素iの波長λのスペクトル線の検量線に水素の影響を
加えるための回帰係数
IHD: 検量線作成用試料Dにおいて測定された水素の発光強
度
この補正項は,検量線式に組み込まれるため,次のa) 及びb) の要求事項を満たせば発光収率の水素補
正に関する新たな補正計算を行う必要はない。
a) 検量線作成のための試料群の中に,十分な水素含有量をもつ試料が少なくとも1個ある。
b) 回帰係数rHiλは,水素含有量が多い試料中に,ある程度以上含まれる分析元素だけで決定できる。係
数rHiλは,検量線作成時に個別に評価できるものであり,測定装置の間で共通の値として使用するこ
とはできない。
注記 [対応国際規格のNOTEは許容事項のため,A.6.3 b) 本文に移動した。]
A.7 相対スパッタリング率を用いた質量分率及びスパッタリング質量の算出
A.7.1 概要
深さ方向分布の各測定点におけるスパッタリング質量及び構成元素の質量分率は,使用する検量線によ
って様々なアルゴリズムを用いて算出できる。ただし,いずれの方法によっても得られる結果は等しい。
A.7.2 相対元素スパッタリング率を用いる方法
式(A.1)に示す検量線を使用する場合には,次の計算方法によって,発光強度からスパッタリング質量及
26
K 0150:2020 (ISO 16962:2017)
び構成元素の質量分率を算出する。
試料Dの深さ方向プロファイルの各領域jについて,測定元素iごとに検量線を用いて [wiD×(qD/qref)]j
を算出する。この量を,“相対元素スパッタリング率”と定義する。
測定した元素の質量分率の総和が分析試料の全元素構成の98 %を超える場合には,試料Dの深さ方向
プロファイルの領域jにおける相対スパッタリング率 (qD/qref)jは,式(A.20)によって求める。
(
)
(
)
D
ref
D
D
ref
/100
i
j
i
j
qq
w
qq
=
×
∑
········································(A.20)
試料Dの領域jにおける元素iの質量分率wiDjは,式(A.21)で算出される。
(
)
(
)
D
D
D
ref
D
ref
ij
i
j
j
w
w
qq
qq
=
×
·············································(A.21)
wiDjの単位は,百分率(%)である。
スパッタリング時間の増分∆tjにおける,領域jの単位面積当たりの全スパッタリング質量mjtotは,式
(A.22)によって求める。
(
)
tot
ref
D
ref
j
j
j
m
q
qq
t
=
×
×∆ ·······················································(A.22)
A.7.3 構成元素の質量分率を用いる方法
式(A.2)に示す検量線を使用する場合には,次の計算方法によって,発光強度からスパッタリング質量及
び構成元素の質量分率を算出する。
測定した元素の質量分率の総和が分析試料の全元素構成の98 %を超える場合には,試料Dの領域jにお
ける元素iの質量分率wiDj(%)は,式(A.23)によって求める。
(
)
(
)
rel
D
rel
100
i
i
j
ij
i
i
j
i
k
I
B
w
k
I
B
λ
λ
λ
λ
λ
λ
×
−
=
×
×
−
∑
················································(A.23)
この式において,kiλは,Riλ×(qref/qD) に等しい。
注記 式(A.23)は,測定した元素の質量分率の総和を100 %として正規化したものである。
非線形の検量線を用いる場合には,式(A.23)における線形関数を対応する非線形の関数に置き換える。
試料Dの深さ方向分布の各領域jにおいて,単位面積当たりのスパッタリング速度qDjは,式(A.24)で算
出する。
(
)
D
ref
rel/100
j
i
i
i
q
q
k
I
B
λ
λ
λ
=
×
×
−
∑
·············································(A.24)
スパッタリング時間の増分∆tjにおける,試料Dの各領域jにおける元素iの単位面積当たりのスパッタ
リング質量miDjは,式(A.25)で算出する。
D
D
D
100
ij
j
ij
j
m
q
w
t
=
×
×∆
··························································(A.25)
領域jの単位面積当たりの全スパッタリング質量mjtotは,式(A.26)によって求める。
tot
D
j
ij
i
m
m
=∑
······································································(A.26)
A.8 絶対スパッタリング速度を用いた質量分率及びスパッタリング質量の算出
A.8.1 概要
深さ方向分布の各測定点におけるスパッタリング質量及び構成元素の質量分率は,使用する検量線によ
27
K 0150:2020 (ISO 16962:2017)
って様々なアルゴリズムを用いて算出できる。ただし,いずれの方法によっても得られる結果は等しい。
A.8.2 元素スパッタリング率を用いる方法
式(A.8)に示す検量線を使用する場合には,次の計算方法によって,発光強度からスパッタリング質量及
び構成元素の質量分率を算出する。
試料Dの深さ方向分布の各領域jについて,測定元素iごとに検量線を用いて (wiD×q'D)jを算出する。
この量を元素スパッタリング率と定義する。
測定した元素の質量分率の総和が分析試料の全元素構成の98 %を超える場合には,試料Dの深さ方向
分布の領域jにおけるスパッタリング速度q'Djは,式(A.27)で求める。
(
)
D
D
D
/100
j
i
j
i
q
w
q
′
′
=
×
∑
·························································(A.27)
試料Dの領域jにおける元素iの質量分率wiDjは,式(A.28)で求める。
(
)
D
D
D
D
ij
i
j
j
w
w
q
q
′
′
=
×
······························································(A.28)
ここでwiDjの単位は,百分率(%)である。
スパッタリング時間の増分∆tjにおける,領域jの単位面積当たりの全スパッタリング質量mjtotは,式
(A.29)によって算出する。
tot
D
D
j
j
j
m
q
tA
′
=
×∆
·································································(A.29)
ここに,
AD: 試料Dのスパッタリング面積
A.8.3 構成元素の質量分率を用いる方法
式(A.9)に示す検量線を使用する場合には,次の計算方法によって,発光強度からスパッタリング質量及
び構成元素の質量分率を算出する。
測定した元素の質量分率の総和が分析試料の全元素構成の98 %を超える場合には,試料Dの領域jにお
ける元素iの質量分率wiDj(%)は,式(A.30)で求める。
(
)
(
)
D
100
i
i
j
ij
i
i
j
i
k
I
B
w
k
I
B
λ
λ
λ
λ
λ
λ
′
′
×
−
=
×
′
′
×
−
∑
··················································(A.30)
ここに,k'iλは,R'iλ/q'Dに等しい。
注記 式(A.30)は,測定した元素の質量分率の総和を100 %として正規化したものである。
非線形の検量線を用いる場合は,式(A.30)における線形関数を対応する非線形関数に置き換える。
深さ方向分布の各領域jにおいて,スパッタリング速度q'Djは,式(A.31)で求める。
(
)
D
/100
j
i
i
i
q
k
I
B
λ
λ
λ
′
′
′
=
×
−
∑
·····················································(A.31)
深さ方向分布の各領域j,スパッタリング時間の増分∆tjにおける,元素iのスパッタリング質量miDjは,
式(A.32)によって算出する。
D
D
D
100
ij
j
ij
j
m
q
w
t
′
=
×
×∆
··························································(A.32)
領域jの単位面積当たりの全スパッタリング質量mjは,式(A.33)で求める。
D
D
j
ij
i
m
m
A
=∑
···································································(A.33)
28
K 0150:2020 (ISO 16962:2017)
A.9 スパッタリング深さの算出方法
A.9.1 概要
A.7又はA.8に示した計算方法によって,試料のスパッタリング質量及び構成元素の質量分率を決める。
さらに,スパッタリング深さを求めるためには,スパッタされた試料の密度が必要である。ここで扱う試
料については,試料の密度は,元素組成及び純物質の密度から算出できる。
スパッタリング深さを算出するために二つの方法があり,試料の密度を得るためにいずれかの方法が利
用できる。
A.9.2 原子容積を用いる方法
試料Dの深さ方向プロファイルの各領域jにおいて,その密度ρjは,式(A.34)で求める。
D
100
ij
j
i
i
w
ρ
ρ
=
∑
··································································(A.34)
ここに,
ρi:
各構成元素iの密度(純物質)
各領域jの厚さzjは,式(A.35)で求める。
tot
D
j
j
j
m
z
A
ρ
=
×
·········································································(A.35)
分析された領域全体の深さは,式(A.35)によって領域jごとに得た厚さzjの総和から得ることができる。
mjtotを∆tjで除すことによって,領域jにおける単位面積当たりのスパッタリング速度を求めることができ
る。
A.9.3 平均密度を用いる方法
試料Dの深さ方向分布の各領域jにおける,元素iごとの原子分率Aijは,式(A.36)で求める。
(
)
(
)
D
D
ij
i
ij
ij
i
i
w
W
A
w
W
=∑
·································································(A.36)
ここに,
Wi: 元素iの原子量
各領域jにおいて,その推定される密度ρjは,式(A.37)で求める。
j
ij
i
iA
ρ
ρ
=
×
∑
·····································································(A.37)
各領域jの厚さzjは,式(A.35)で算出する。また,分析された領域の全深さは各領域ごとに得た厚さzj
の総和から求める。
29
K 0150:2020 (ISO 16962:2017)
附属書B
(参考)
分析線として使用できるスペクトル線
B.1
推奨スペクトル線の波長表
分析線として使用できる発光線を,表B.1に示す。
表B.1−元素ごとに使用できるスペクトル線
元素
波長
(nm)
使用可能な質量分率の範囲
(%)
備考
Zn
330.26
0.001〜100
−
Zn
334.50
0.001〜100
−
Zn
481.053
0.001〜100
−
Al
172.50
0.1〜100
−
Al
396.15
0.001〜100 a)
自己吸収
Ni
231.603
0.01〜100
−
Ni
341.78
0.001〜100 a)
弱い自己吸収
Ni
349.30
0.005〜100 a)
弱い自己吸収
Si
212.41
−
−
Si
251.61
−
−
Si
288.16
0.001〜20
−
Fe
249.318
0.01〜100
−
Fe
259.94
0.01〜100
−
Fe
271.44
0.1〜100
−
Fe
371.94
0.005〜100 a)
弱い自己吸収
Fe
379.50
0.01〜100
−
Cu
296.12
0.01〜100
−
Cu
327.40
0.001〜5 a)
強い自己吸収
Mg
383.83
0.001〜100
−
注a) 非線形の検量線を使用する。
30
K 0150:2020 (ISO 16962:2017)
附属書C
(参考)
単位面積当たりのめっき質量(めっき付着量)の定量
C.1 概要
ある元素の単位面積当たりのめっき付着量は,めっき層の深さ(又はスパッタリング時間)範囲で,そ
の元素の分布領域を深さ方向に積分して算出する。市販のグロー放電発光分光分析装置は,この計算ソフ
トウエアを備えている。この積分演算は,縦軸の単位[グラム毎平方メートル毎秒(g/m2/s)],及び横軸
の単位[秒(s)]で示した定量深さ方向プロファイルの範囲で行う。分析装置には,この積分を,画面に
表示された分布図上で,カーソル操作によって行うソフトウエアを備えているものもある(図C.1参照)。
必要な場合は,オプションでソフトウエアを追加することもできる。ある元素のめっき質量は,めっき層
の範囲内で,スパッタリング時間(又は深さ)で分割された領域に関してめっき質量の寄与分を積算して
求める。めっき層の総質量(付着量)は,個々の元素の積分めっき質量を合計して求める。分析対象元素
がめっき層と基板との両方に相当量含まれている場合,例えば,鋼基板上の亜鉛−鉄(Zn-Fe)合金めっき
又はアルミニウム基板上のアルミニウム−シリコン(Al-Si)合金めっきでは,めっき層と基板との境界域
では分析対象元素の深さ方向分布がめっき層によるものか,基板の金属によるものかを判断することは困
難である。このような場合は,めっき層中に含まれる他の分析主元素の深さ方向分布を参照して,間接的
にめっき層と基板金属との区別をする。C.2及びC.3に示す二つの方法がある。
C.2 方法1
最初に,めっき層中よりも基板中の質量分率が高い元素に注目する。このような場合に当てはまる代表
的なめっき組成として,図C.1(図2も参照)に示す亜鉛−鉄(Zn-Fe)合金化溶融亜鉛めっき鋼板がある。
この例では,鉄が注目される分析対象元素である。下地鋼板に由来する鉄原子が測定され始める測定点と
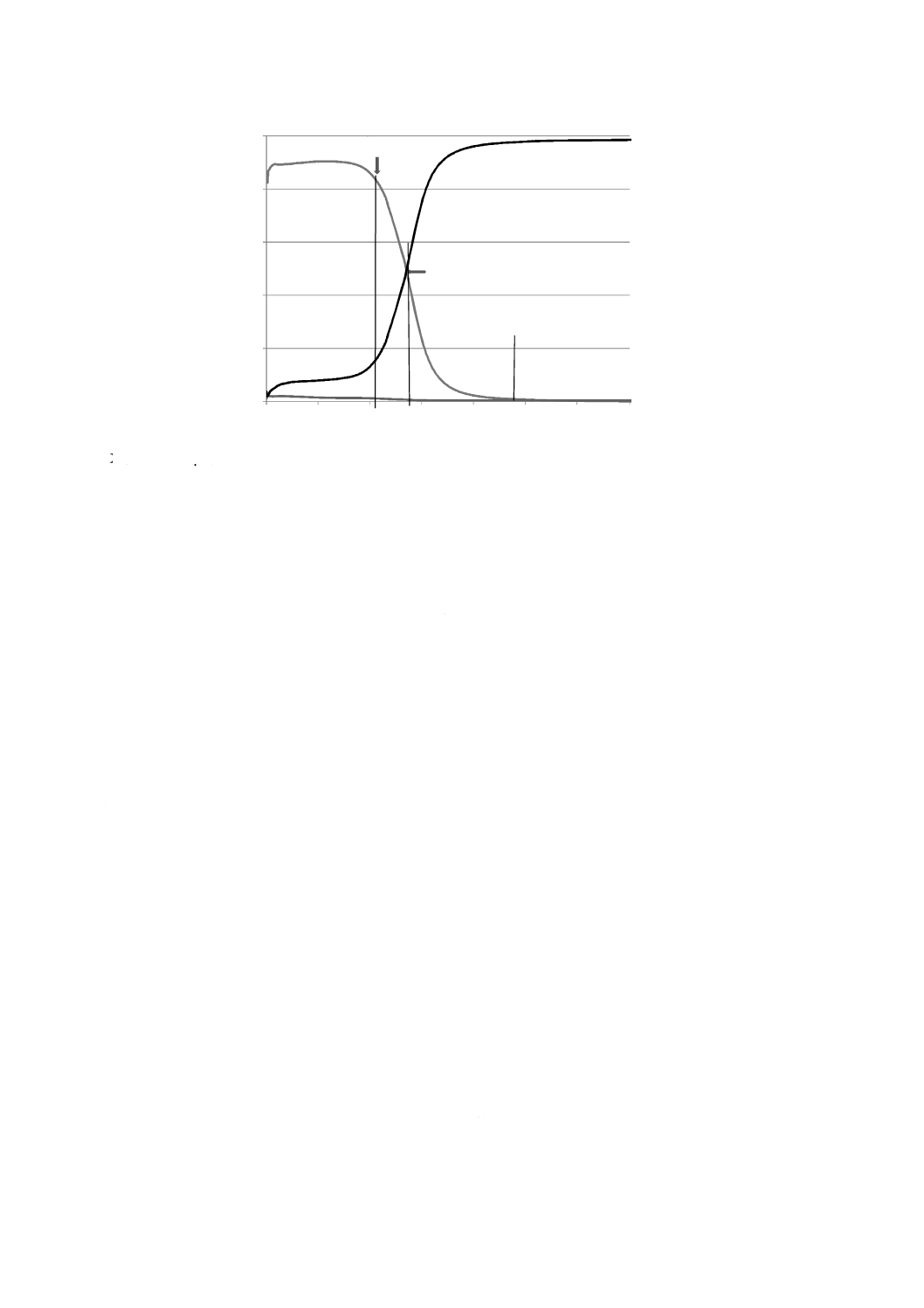
して,その時間tt又はそれに対応する深さddは,めっき層の主成分元素(亜鉛)の縦軸の値が,めっき層
中における最大値又は平たん部の値の95 %となる時間と定義する。このtt又はddは,図C.1に示す三つの
例のスパッタリング時間(又は深さ)分布上に図示したように決定することができる。時間ttを過ぎると,
めっき層中の鉄は,亜鉛の分布と比例して減少し,ゼロに近づくと仮定する。したがって,この遷移領域
の鉄のめっき質量は,時間ttから亜鉛がその定量限界以下になる時間までの亜鉛のめっき質量に,時間tt
(深さdd)における亜鉛に対する鉄の比率(定量深さ方向分布の縦軸値の比)を乗じた値に等しい。亜鉛
のめっき質量算出を行う積分終了点(深さD)は,亜鉛の質量分率がめっき層と比較して50 %となる深さ
に,95 %深さ(dd)と50 %深さとの間隔値を3倍したものを加えた位置として定義する。深さDよりも基
板側で測定される亜鉛は,無視してよいものとする。また,めっき層中の単位面積当たりの鉄の全質量は,
深さDまでの遷移領域における鉄の質量分率の時間積分値(質量)と時間ttより前の鉄の質量との和とな
る(図C.2参照)。
注記 時間tt(深さdd)を決めるための方法として,めっき層中には含まれないが基板中には存在す
る元素の深さ方向プロファイルを用いることができる。この場合は,時間ttは,その元素が最
初に検出される時間としてもよい。ニオブ,モリブデン,マンガン,銅,コバルトは,適用可
能な元素の例である。
めっき層中の質量分率が基板材料の中の質量分率よりも大きい元素については,その元素のめっき層中
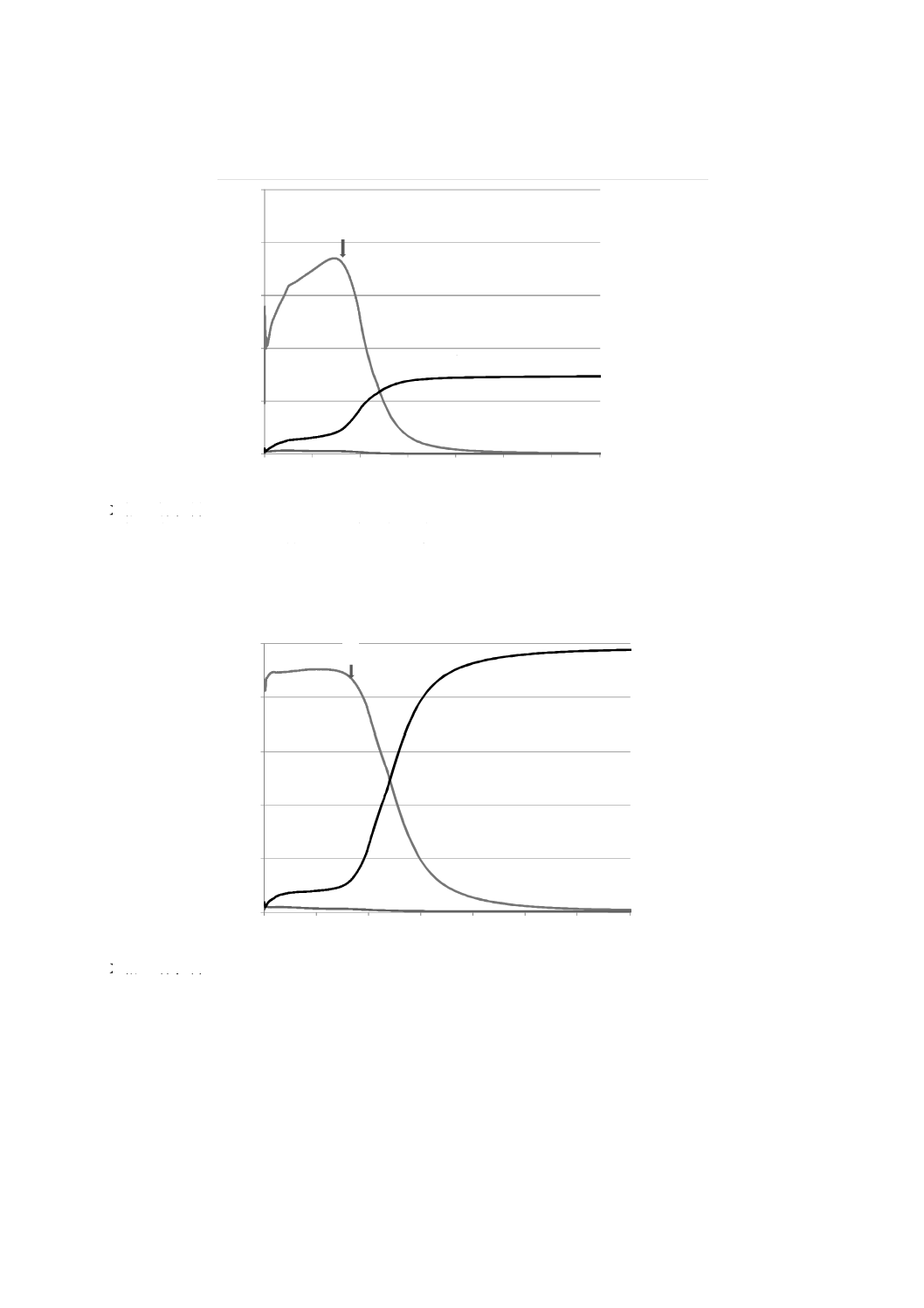
31
K 0150:2020 (ISO 16962:2017)
の単位面積当たりの全質量は,時間ゼロから積分終了点(深さD)となる時間までの積分値として求める。
0.0
0.2
0.4
0.6
0.8
1.0
0
20
40
60
80
100
120
140
Y
X
tt
1
2
3
X軸 時間(s)
Y軸 単位面積当たりのスパッタリング率(g/m2/s)
tt 亜鉛のスパッタリング率最大値の95 %となる時間
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
a) 元素スパッタリング率による定量深さ方向プロファイルの例
0
20
40
60
80
100
0
20
40
60
80
100
120
140
Y
X
tt
3
1
2
X軸 時間(s)
Y軸 元素の質量分率(%)
tt 亜鉛の平たん部の値の95 %となる時間
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
b) 元素質量分率による定量深さ方向プロファイルの例
図C.1−亜鉛−鉄(Zn-Fe)合金化溶融亜鉛めっきの亜鉛平たん部の値の95 %点を求めるための
定量深さ方向プロファイル
32
K 0150:2020 (ISO 16962:2017)
0
20
40
60
80
100
0
2
4
6
8
10
12
14
Y
X
dd
Zn 50%
D
1
2
3
X軸 深さ(μm)
Y軸 元素の質量分率(%)
dd 亜鉛の平たん部の値の95 %となる深さ
D 亜鉛の積分の最終点
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
c) めっき皮膜に含まれる成分元素の積分終了点の決定方法
図C.1−亜鉛−鉄(Zn-Fe)合金化溶融亜鉛めっきの亜鉛平たん部の値の95 %点を求めるための
定量深さ方向プロファイル(続き)
C.2.1 鉄の検量線の調整
この規格で記載する定量方法による結果を,統計的な許容範囲内でISO 17925で記載されているような
基準化学分析法の結果と良好な一致を得るためには,通常に鋼試料を用いて得られた鉄の検量線を,亜鉛
−鉄(Zn-Fe)めっき標準物質を用いて調整する必要がある。グロー放電発光分光分析の定量結果が化学分
析値と一致しない理由は,様々考えられる。最も起こり得る要因を記載する。
a) 鉄の発光収率が,鋼試料と亜鉛−鉄(Zn-Fe)めっき試料との間で差異がある場合がある。これは,基
準として用いた鋼試料では,鉄の質量分率がかなり高い条件で発光収率が見積もられることによる
(8.4参照)。
b) 深さddよりも深い鋼基板との界面領域では,界面層には鉄の含有量が高い亜鉛−鉄(Zn-Fe)金属間
化合物相ができるため,鉄の質量分率は大きくなる傾向となる。これは,深さddからDの領域で鉄の
めっき質量を低く評価することになる。このようなグロー放電発光分光分析の定量値と化学分析値と
の不一致を補正するために,次のc)〜g) に示すような手順を取ることが望ましい。
c) バルクの検量線作成用試料を用い,箇条8に規定した方法・手順に従い,適切な方法で検量線の作成
を行う。
d) 亜鉛−鉄(Zn-Fe)めっき標準物質を数個選び,これらを試験試料として分析を行い,方法1に従って
めっき層中の鉄の質量分率を求める。
e) 試験試料として測定した亜鉛−鉄(Zn-Fe)めっき標準物質全てに対して,化学分析で決められている
認証値との比,Fe(wet chemical) / Fe(GD-OES) を計算して,その平均値を求める。
f)
GD-OESの鉄検量線の傾きの値を,e) で求めたFe(wet chemical) / Fe(GD-OES) の平均値で乗じて補正
33
K 0150:2020 (ISO 16962:2017)
する。一連の分析操作においてどの程度まで補正ができるかについては,個々の分析装置に組み込ま
れているソフトウエアの仕様に依存する。したがって,この規格では,これ以上詳細には言及できな
い。
g) 補正した鉄の検量線を使用してもう一度d) の分析操作を行い,化学分析値と良好な一致があることを
確認する。満足できる結果が得られない場合には,e)〜f) の手順を良好な一致が得られるまで繰り返
す。
X軸 深さ(μm)
Y軸 元素の質量分率(%)
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe 4 めっき中のFe
図C.2−方法1におけるめっき中のFeの寄与を示すための合金化溶融亜鉛めっきの定量深さ方向
プロファイル
C.3 方法2
方法2の手順は,質量分率を縦軸に,深さを横軸にとった組成深さ方向分布について適用できるもので
ある。方法1の場合と同様に亜鉛−鉄(Zn-Fe)合金化溶融亜鉛めっき鋼板を例として,計算手順を次に示
す。定量深さ方向プロファイルについて,計算に必要となる評価パラメータを図C.3に示す。
a) 計算の最初の段階として,亜鉛の質量分率がめっき層中で平たんとなる部位の値の84 %及び16 %に
なる深さを求めて,各々の深さをZn 84 %及びZn 16 %とする。
b) 境界領域におけるZn 84 %とZn 16 %との間の間隔を境界深さWとする。
c) 亜鉛と鉄との質量分率が等しくなる深さを,Sとする。
d) 深さSに境界深さWを加えた深さを,Lとする。
e) 鉄について,0から深さSまでのめっき中の単位面積当たりの質量を全鉄量Fe0-Sとして算出する。
f)
亜鉛について,深さSから深さLまでのめっき中の単位面積当たりの質量ZnS-Lを算出する。
g) ZnとFeとの深さ方向プロファイルが対称形となることに注目して,深さSまでのめっき層中で測定
される鉄の単位面積当たりの質量について下地基板側からの寄与分を,基板側で測定される亜鉛の単
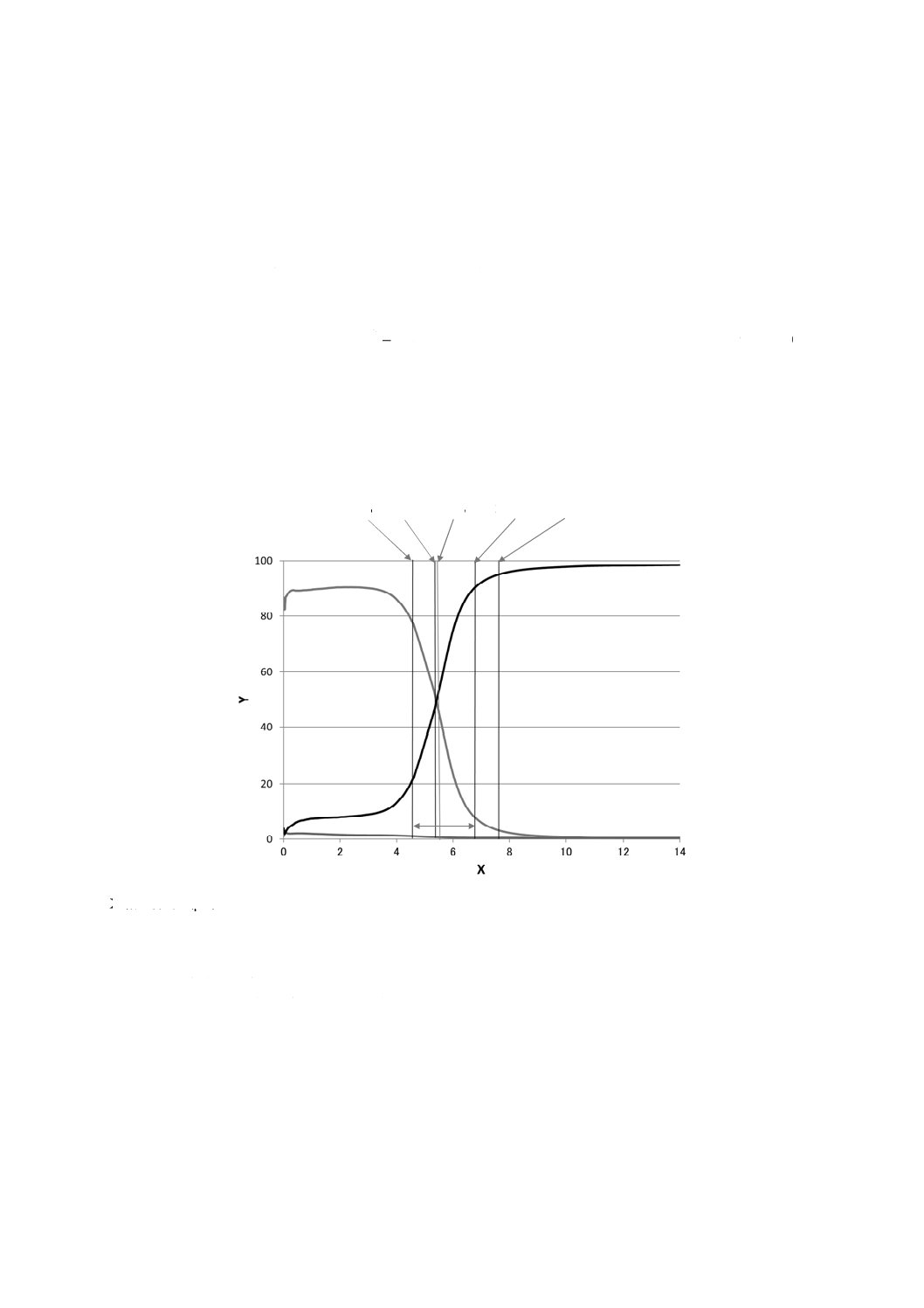
34
K 0150:2020 (ISO 16962:2017)
位面積当たりの質量と比例関係にあるものとして算出する。すなわち,数式ではFe0-S (substrate)=α1
×ZnS-Lである。
h) 同様に,深さSからLの領域で測定される鉄の質量のうち,めっき層からの寄与分を,基板側で測定
される亜鉛の質量と比例関係にあるものとして算出する。すなわち,数式ではFeS-L (coating)=α2×ZnS-L
である。
i)
この数量関係を組み合わせて,めっき層中の鉄の単位面積当たりの質量Fe0-L (coating) は,式(C.1)から
求める。
(
)
(
)
0
0
Fe
coating
Fe
total
αZn
L
S
SL
−
−
−
=
−×
········································ (C.1)
この式で,係数αは,個々の分析において実験によって決める必要がある。鉄の質量分率が10 %程度の
Zn-Feめっきでは,α値を0.85とすると分析結果が化学分析値と良好な一致を得ることが報告されている。
しかしながら,最適なα値は,幾つかの因子,例えば,測定装置,グロー放電励起源の制御パラメータ及
びめっき層の種類によって変動する。
Zn 84 % S
Zn 50 % Zn 16 % L
1
3
2
W
X軸 深さ(μm)
Y軸 元素の質量分率(%)
W 境界領域の深さ
S 亜鉛と鉄との質量分率が等しくなる深さ
L SにWを加えた深さ
1 Zn 2 Al×10(Alの値の10倍表示) 3 Fe
図C.3−方法2における合金化溶融亜鉛めっきの定量深さ方向プロファイル
35
K 0150:2020 (ISO 16962:2017)
附属書D
(参考)
共同実験に関する追加資料
D.1 共同実験の結果
表1及び表2のデータは,亜鉛アルミニウムめっき試料を使って2001年及び2002年に3か国の4試験
所が参加して行った国際共同実験の結果から導かれたものである。
これらの結果の詳細は,ISO/TC 201/SC 8の資料番号N 38の改訂版及びN 55に記載されている。
共同実験に用いた試料について,得られた単位面積当たりのめっき付着量及び元素質量分率の平均値を
表D.1に示す。
精度に関するデータは,図D.1及び図D.2に示す。この中には,バルク試料での結果及びECISS 4)/TC 20
で行った共同実験の結果も合わせて示す。
注4) ECISS:欧州鉄鋼標準化委員会(共同実験実施当時)
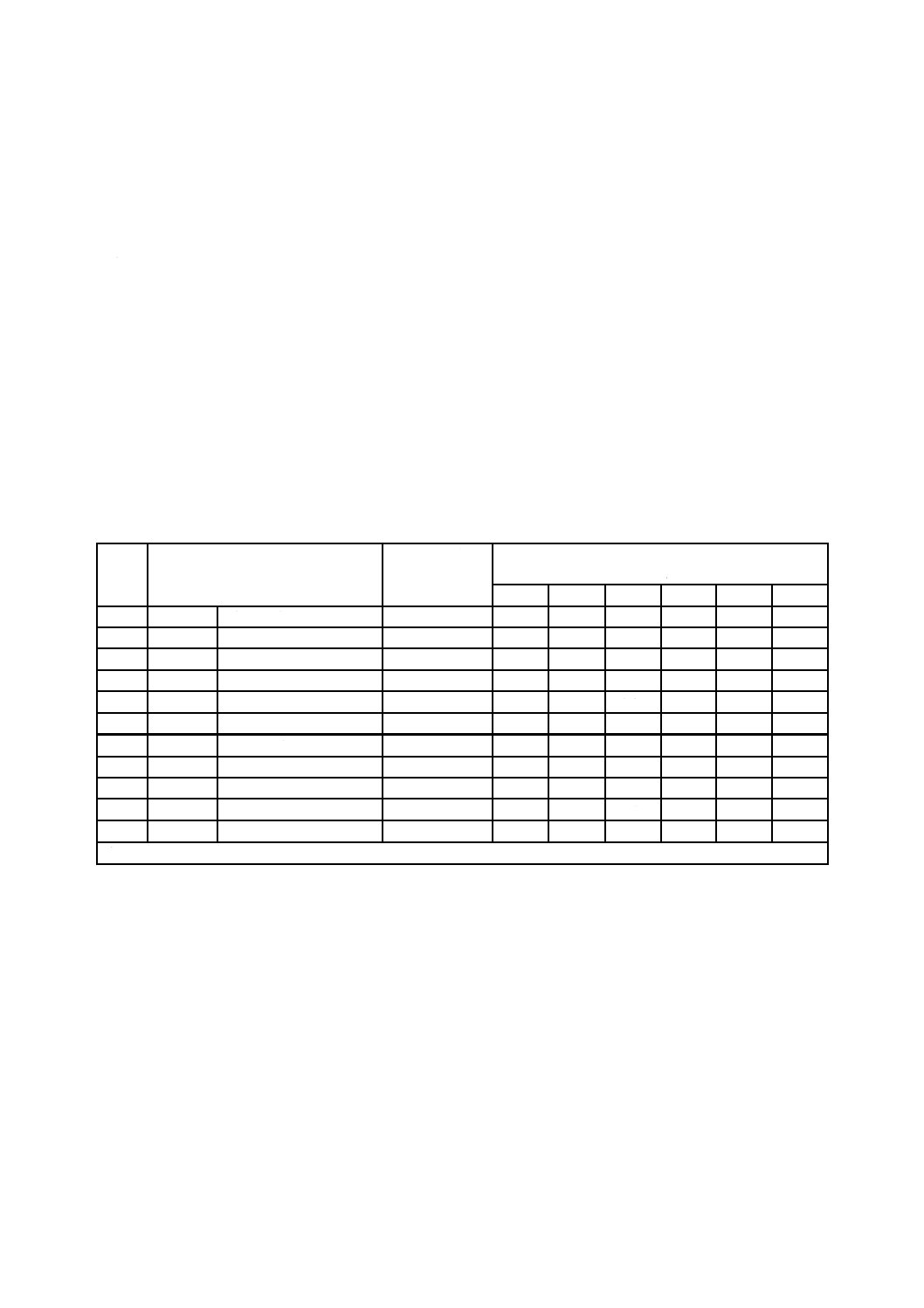
表D.1−共同実験に用いた試料名及び実験結果
試料
番号
めっきの種類
めっき付着量
g/m2
元素質量分率
%
Zn
Fe
Al
Ni
Si
Pb
3
Zn-Fe
合金化溶融亜鉛めっき
57
89.1
10.23
0.210
−
−
−
101
Zn-Fe
合金化溶融亜鉛めっき
49.0
88.3
11.3
0.37
−
−
−
102
Zn-Fe
合金化溶融亜鉛めっき
50.7
89.5
10.05
0.38
−
−
−
103
Zn-Fe
合金化溶融亜鉛めっき
49.7
90.6
9.0
0.39
−
−
−
104
Zn-Fe
合金化溶融亜鉛めっき
53.3
86.6
13.03
0.37
−
−
−
4
Al-Zn
溶融めっき
91.4
42.6
−
54.9
−
1.29
−
12
Zn-Ni
電気めっき
17.81
86.2
−
−
12.5
−
−
201
Zn
溶融めっき
113
99.5
−
0.35
−
−
0.11
202
Zn-Ni
電気めっき
44
86.7
−
−
13.2
−
−
203
Al-Zn
溶融めっき
110
94.9
−
5.1
−
−
−
204
Al-Zn
溶融めっき
81
45.4
−
53.2
−
1.9
−
注記 合金化溶融亜鉛めっきとは,溶融亜鉛めっき処理後に焼鈍処理を施しためっきの種類である。
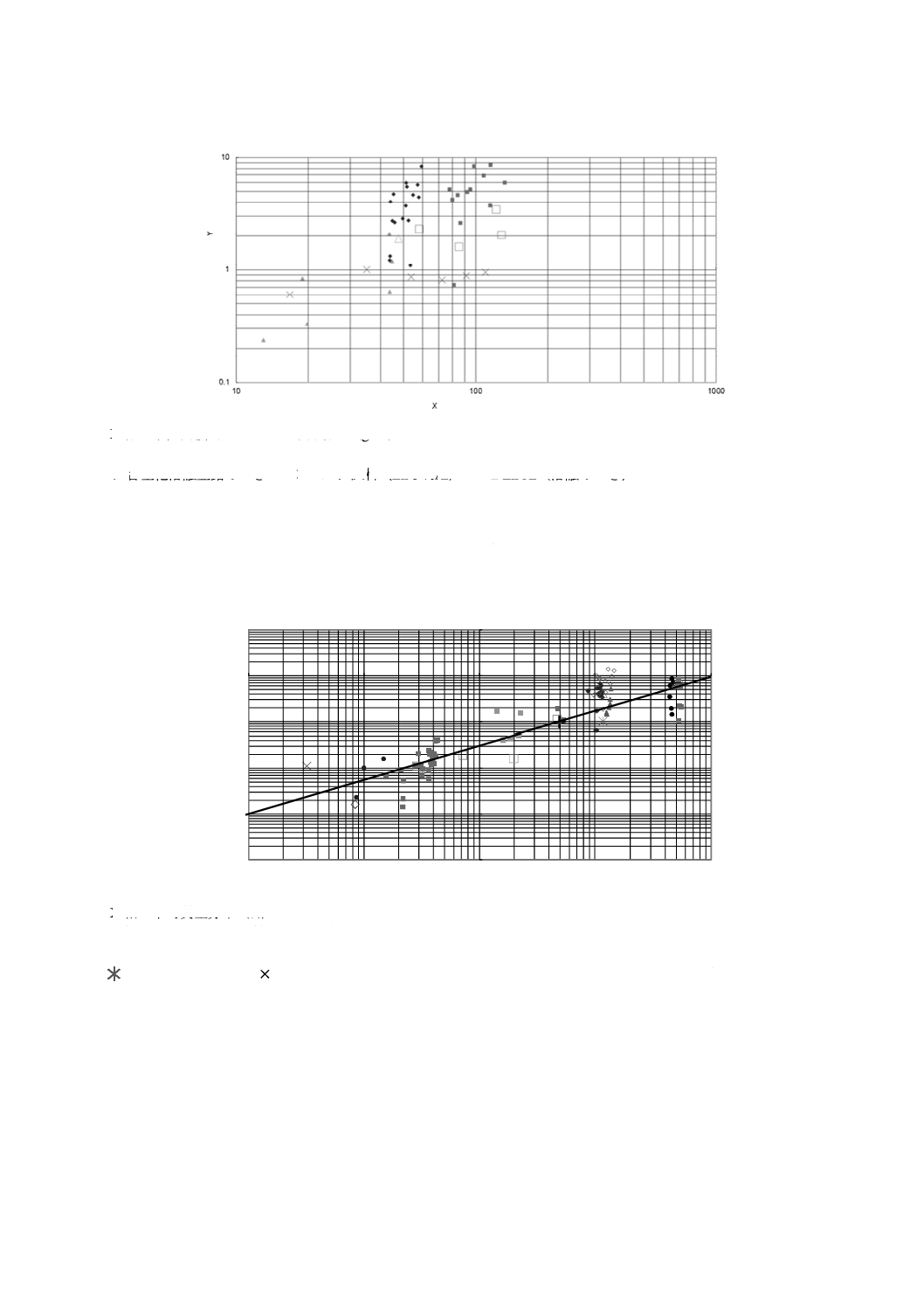
36
K 0150:2020 (ISO 16962:2017)
X軸 単位面積当たりのめっき付着量(g/m2)
Y軸 併行標準偏差(g/m2)
◆ 合金化溶融亜鉛めっき ×バルク試料(Zn-5 %Al) ■ Zn-Al(溶融めっき)
□ Zn-Al(溶融めっき,ECISSデータ) ▲ Zn-Ni(電気めっき)
△ Zn-Ni(電気めっき,ECISSデータ)
図D.1−単位面積当たりのめっき付着量と併行標準偏差との関係
0.0001
0.001
0.01
0.1
1
10
0.01
0.1
1
10
100
Y
X
X軸 平均質量分率(%)
Y軸 併行標準偏差 質量分率(%)
◆Fe
■Al
▲Ni
■Si
●Zn
Ni(バルク試料)
Si(バルク試料)
●Pb
+Al(バルク試料)
◇Fe(不均質試料)
□Al(ECISSデータ)
△Ni(ECISSデータ)
□Si(ECISSデータ)
◇Pb(ECISSデータ)
図D.2−元素の質量分率と併行標準偏差との関係
37
K 0150:2020 (ISO 16962:2017)
参考文献
[1] Grimm, W. Spectrochim. Acta, 23B, 443 (1968)
[2] Takadoum, J., Pirrin, J.C., Pons Corbeau, J., Berneron, R. and Charbonnier, J.C. Surf. Interf. Anal., 6, 174
(1984)
[3] Takimoto, K., Nishizaka, K., Suzuki, K. and Ohtsubo, T. Nippon Steel Technical Report 33, 28 (1987)
[4] Bengtson, A., Eklund, A. and Saric, A. J. Anal. At. Spectrom., 5, 563 (1991)
[5] Naoumidis, A., Guntur, D., Mazurkiewicz, M., Nickel, H. and Fischer, W. Proceedings of 3rd User-Meeting
“Analytische Glimentladungs-Spektroskopie”, p. 138, Jülich (1990)
[6] Payling, R. Spectroscopy, 13, 36 (1998)
[7] Bengtson, A., and Nelis, T., Anal. Bioanal. Chem. 385, 568 (2006)
[8] Wilken, L, Hoffmann, V., Wetzig, K., Spectrochim. Acta B, 62, 1085 (2007)
[9] Marshall, K.A., Casper, T.J., Brushwyler, K.R., Mitchell, J.C., J. Anal. At. Spectrom., 18, 637 (2003)
[10] Wilken, L, Hoffmann, V., Wetzig, K., J. Anal. At. Spectrom., 18, 1141 (2003)
[11] Martin, A., Martinez, A., Pereiro, R., Bordel, N. and Sanz-Medel, A., Spectrochim. Acta B, 62, 1263 (2007)
[12] JIS Z 8402-1 測定方法及び測定結果の精確さ(真度及び精度)−第1部:一般的な原理及び定義
注記 原国際規格では,ISO 5725-1,Accuracy (trueness and precision) of measurement methods and
results−Part 1: General principles and definitionsを記載している。
[13] JIS Z 8402-2 測定方法及び測定結果の精確さ(真度及び精度)−第2部:標準測定方法の併行精度
及び再現精度を求めるための基本的方法
注記 原国際規格では,ISO 5725-2,Accuracy (trueness and precision) of measurement methods and
results−Part 2: Basic method for the determination of repeatability and reproducibility of a standard
measurement methodを記載している。
[14] JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用
的な使い方
注記 原国際規格では,ISO 5725-6,Accuracy (trueness and precision) of measurement methods and
results−Part 6: Use in practice of accuracy valuesを記載している。
[15] JIS Q 9000:2015 品質マネジメントシステム−基本及び用語
注記 原国際規格では,ISO 9000:2015,Quality management systems−Fundamentals and vocabulary
を記載している。
[16] ISO 11505,Surface chemical analysis−General procedures for quantitative compositional depth profiling by
glow discharge optical emission spectrometry
[17] JIS K 0144 表面化学分析−グロー放電発光分光分析方法通則
注記 原国際規格では,ISO 14707,Surface chemical analysis−Glow discharge optical emission
spectrometry (GD-OES)−Introduction to useを記載している。
[18] JIS Q 17025:2018 試験所及び校正機関の能力に関する一般要求事項
注記 原国際規格では,ISO/IEC 17025:2005,General requirements for the competence of testing and
calibration laboratoriesを記載している。
[19] JIS K 0147-1 表面化学分析−用語−第1部:一般用語及び分光法に関する用語
38
K 0150:2020 (ISO 16962:2017)
注記 原国際規格では,ISO 18115-1,Surface chemical analysis−Vocabulary−Part 1: General terms and
terms used in spectroscopyを記載している。
[20] ISO/TS 25138,Surface chemical analysis−Analysis of metal oxide films by glow-discharge optical-emission
spectrometry