10
K 0138:2018
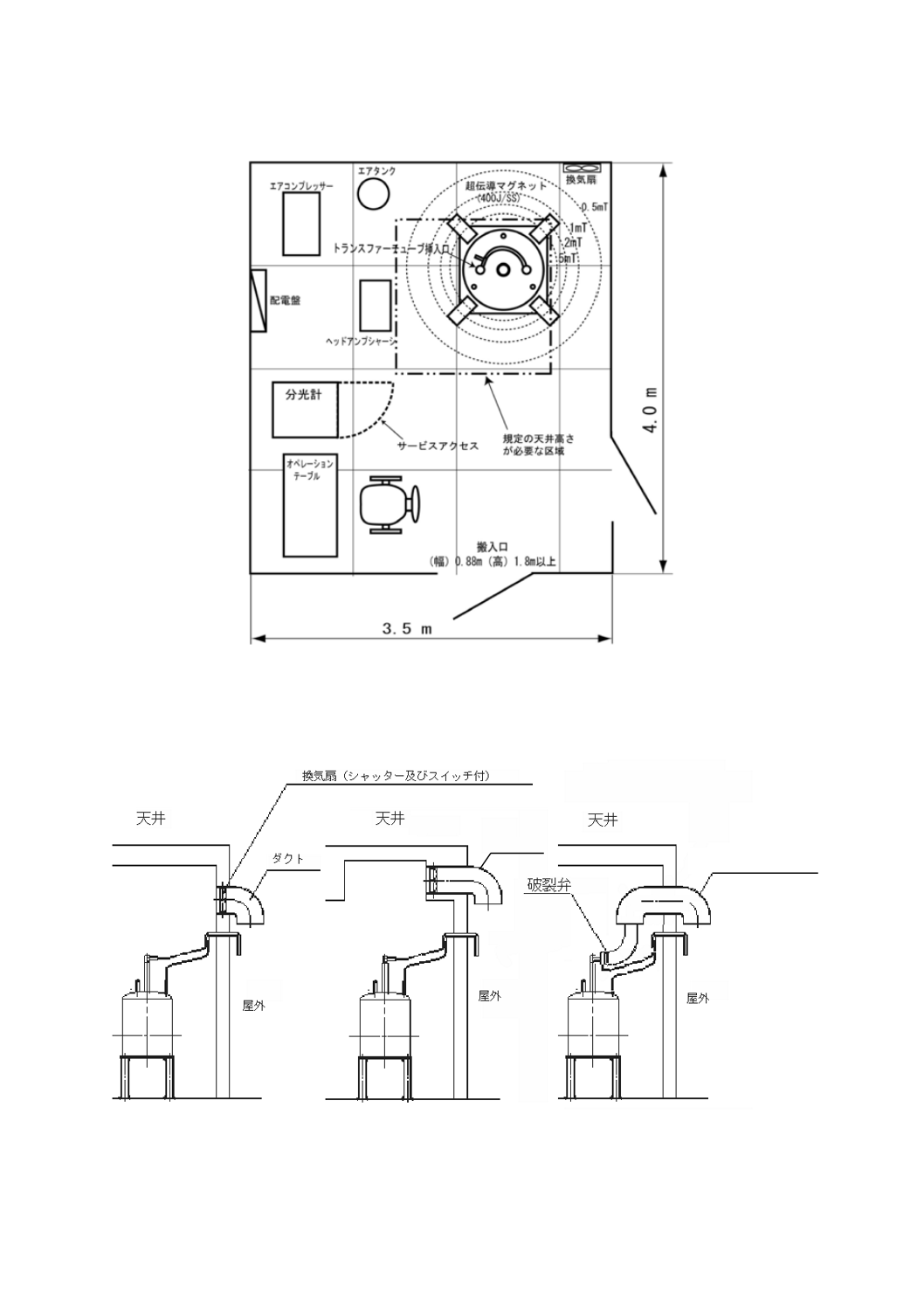
図1−400 MHzのNMR装置における標準的な設置条件の例
図2−NMR装置の標準的な超伝導磁石の排気口とダクトとの連結の例
クエンチダクト
ダクト
K 0138:2018
(1)
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 1
4 qNMRの概要 ·················································································································· 6
4.1 qNMRの原理 ················································································································ 6
4.2 1H qNMRの原理 ············································································································ 7
5 装置······························································································································· 9
5.1 一般事項 ······················································································································ 9
5.2 設置条件及び安全 ·········································································································· 9
5.3 装置構成の概要 ············································································································ 11
6 1H qNMRの準備 ············································································································· 12
6.1 一般的事項 ·················································································································· 12
6.2 試料及びqNMR用基準物質のひょう量 ············································································· 12
6.3 基準物質 ····················································································································· 12
6.4 重水素化溶媒の選定 ······································································································ 13
6.5 NMR試料管 ················································································································ 13
6.6 NMR装置の最適化········································································································ 13
6.7 試料溶液の調製 ············································································································ 14
6.8 1H qNMR測定条件の設定 ······························································································· 14
7 測定······························································································································ 16
7.1 1H qNMR測定条件 ········································································································ 16
7.2 測定操作 ····················································································································· 16
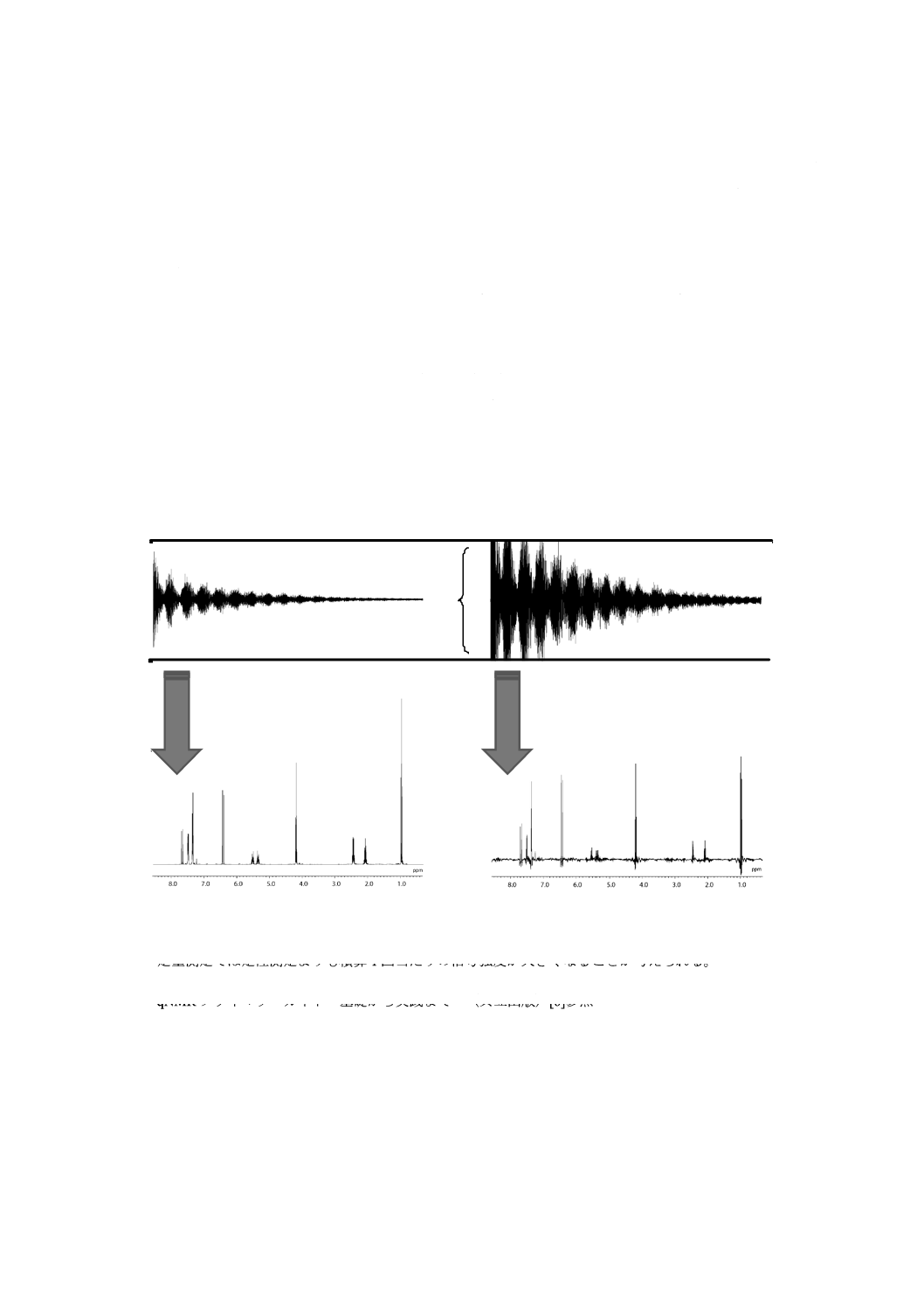
7.3 FIDデータの処理及び保存······························································································ 16
8 解析······························································································································ 16
8.1 一般 ··························································································································· 16
8.2 計算式 ························································································································ 17
8.3 分析値の表し方 ············································································································ 17
8.4 データの整理及び保存 ··································································································· 17
9 データの質の管理 ············································································································ 17
9.1 一般事項 ····················································································································· 17
9.2 NMR装置の適格性確認·································································································· 18
9.3 システム適合性試験 ······································································································ 18
9.4 トレーサビリティの確保 ································································································ 18
9.5 分析値の信頼性の確保 ··································································································· 18
10 個別規格で1H qNMRとして取り入れる際に記載すべき事項 ·················································· 19
K 0138:2018 目次
(2)
ページ
附属書A(参考)試料溶液の調製 ··························································································· 20
附属書B(参考)1H qNMRスペクトルの測定 ··········································································· 28
附属書C(参考)1H qNMRスペクトル解析 ·············································································· 33
附属書D(参考)試料のひょう量 ··························································································· 38
附属書E(参考)不確かさの求め方 ························································································ 42
K 0138:2018
(3)
まえがき
この規格は,工業標準化法に基づき,日本工業標準調査会の審議を経て,経済産業大臣が制定した日本
工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
K 0138:2018
定量核磁気共鳴分光法通則(qNMR通則)
General rules for quantitative nuclear magnetic resonance spectroscopy
1
適用範囲
この規格は,核種プロトン(1H)を用いた定量核磁気共鳴分光法(以下,qNMRという。)によって,
化学物質の純度又は含有率を内標準法に基づいて定量分析する場合の通則について規定する。
なお,溶媒に溶解する非交換性1Hをもつ物質に適用できる。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS B 7609 分銅
JIS K 0050 化学分析方法通則
JIS K 0211 分析化学用語(基礎部門)
JIS K 0215 分析化学用語(分析機器部門)
ISO 17034,General requirements for the competence of reference material producers
3
用語及び定義
この規格で用いる主な用語,定義,記号及び略号は,JIS K 0050,JIS K 0211及びJIS K 0215によるほ
か,次による。
3.1
核磁気共鳴,NMR(nuclear magnetic resonance)
原子核が磁場中においてラジオ波を吸収する共鳴現象。
3.2
定量核磁気共鳴分光法,qNMR(quantitative NMR)
NMRを用いた定量分析法。
3.3
1H核定量核磁気共鳴分光法,1H qNMR(1H quantitative NMR)
プロトン(1H)を観測核としたqNMR分光法。
3.4
シグナル(signal)
NMRの共鳴信号。信号ともいう。
3.5
ラジオ波(radio frequency,RF)
2
K 0138:2018
300 Hz〜3 THzの周波数をもつ電磁波。
NMRで使用されるのは数十MHz〜1 GHz程度の範囲。
3.6
核スピン(nuclear spin)
荷電粒子である原子核の自転運動する性質。
3.7
静磁場(static magnetic field)
時間的に変動しない磁場。
3.8
外部磁場(external magnetic field)
NMR装置にかかる外部からの磁場。
3.9
磁気回転比(gyromagnetic ratio)
原子核が磁場中で歳差運動する角周波数を決定する,原子核種ごとに固有の定数。
核スピンの回転の速さと外部磁場の関係を示した定数。
3.10
緩和(relaxation)
NMRにおいて,原子核がラジオ波を吸収して励起された状態から熱平衡状態に戻る過程。
3.11
緩和時間(relaxation time)
緩和に要する時間,又は緩和に要する時間を表す時定数。
3.12
化学シフト(chemical shift)
基準物質の共鳴周波数(化学シフト基準周波数δ 0 ppmとして)からのずれ(相対的位置)。
NMRスペクトルにおける,各シグナルの周波数と化学シフト基準周波数との周波数差を化学シフト基
準周波数で除したもので定義し,単位はppmである。
3.13
スピン−スピン結合(spin-spin coupling)
共有結合した核スピン同士が電子を介して相互作用し,シグナルの分裂が起こる現象。J coupling,J結
合又はスピン結合ともいう。
3.14
スピン−スピン結合定数(spin-spin coupling constant)
スピン結合によって分裂したシグナルの間隔。J結合定数,J値又はスピン結合定数ともいう。
3.15
NMRスペクトル(NMR spectrum)
共鳴周波数のエネルギー吸収は,10−5秒程度の短時間のパルス波で行い,その後の遷移状態から熱平衡
状態への10ミリ秒〜10秒オーダーの磁気緩和を時間−強度分布として検出する。この分布に対してフー
リエ変換を行い,周波数(化学シフト)−強度分布としたもの。
3.16
シグナル面積(signal area)
3
K 0138:2018
設定したベースラインとシグナルとを囲む範囲を積分して得られる値。信号面積ともいう。
3.17
核オーバーハウザー効果(nuclear Overhauser effect)
ある核スピンが緩和する過程で,磁気的に相互作用のある別のスピンのエネルギー準位に対して占有数
を変化させる効果。
3.18
フリップ角(flip angle)
静磁場中の熱平衡状態における核磁化ベクトルが,ラジオ波パルスの印加によって静磁場の方向から倒
れた角度。励起パルス角(exciting pulse angle)ともいう。
3.19
平衡磁化(equilibrium magnetization)
物体を静磁場に置いてから励起パルスを印加するまでの間に存在する磁化の状態。
3.20
遅延時間(pulse delay)
パルス列の最初のパルスを打ってから,次の積算で同じパルスを打つまでの時間。
3.21
1H qNMRスペクトル(1H qNMR spectrum)
プロトン(1H)を観測核としてqNMRによって得られたNMRスペクトル。
3.22
NMR装置(nuclear magnetic resonance spectrometer)
原子核が磁場中においてラジオ波を吸収する共鳴現象を利用した分光分析装置。
3.23
超伝導磁石(superconducting magnet)
超伝導現象によって,超伝導材料コイルに流れる永久電流によって作られる磁場を用いた磁石。
3.24
NMRプローブ(NMR probe)
ラジオ波(パルス)を照射したり,放出されるラジオ波(NMR信号)を検出したりするもの(NMR装
置における検出器)。
3.25
高周波発生部(high-frequency pulse)
高周波パルスを生成するために,発振器(oscillator),パルス発生器(pulse generator)などで構成される
機器。
3.26
パルスフーリエ変換NMR(FT-NMR)装置(Fourier transform NMR,FT-NMR)
ラジオ波パルスを用いて核磁気共鳴吸収現象を起こし,緩和過程に放出されるラジオ波を得て,その波
をフーリエ変換(Fourier transformation)して横軸が周波数のスペクトルに変換するNMR装置。パルス−
フーリエ変換型NMR装置ともいう。
3.27
スピニングサイドバンド(spinning sideband)
試料回転時に,回転速度(単位:Hz)と等距離に現れるシグナル。
4
K 0138:2018
3.28
ラーモア周波数(Larmor frequency)
磁場中に置かれた原子核などが起こすラーモア歳差運動の角周波数。
3.29
シム(shim)
超電導電磁石が作る磁場においてNMRの観測領域の磁場の不均一性を補正して均一にするためのコイ
ル群(シムコイル)。
3.30
シム調整(shimming)
各シムコイルは様々な勾配磁場を発生するように設計されており,それらのシムコイル群に流す電流を
調整して磁場を均一にする調整作業。
3.31
デジタル分解能(digital resolution)
NMRスペクトル上で隣り合うデータ点間の周波数差。
3.32
スペクトル幅(spectral width)
観測範囲。フーリエ変換後の表示範囲。
3.33
デカップリング(decoupling)
NMRスペクトル上でスピン結合によって起こる信号の分裂を解消する手法。
3.34
自由誘導減衰,FID(free induction decay)
磁場中におかれた磁化にラーモア周波数に近い周波数のパルスで励起されることによって誘導される減
衰振動。
3.35
積算回数(number of scan)
同一の条件で測定を繰り返したときのFID信号を積算する回数。
3.36
ダミースキャン(dummy scan)
FID信号の取得を開始する前に,信号を取得せずに同一条件で行う測定。
3.37
サテライトサイドバンド(satellite sideband)
天然存在比の低い同位体とカップリングしたシグナル。
3.38
システム適合性試験(system suitability)
試験を行う施設の分析システムを使って,当該試験法が目的にかなう試験結果を与えることをあらかじ
め検証し,分析システムの稼働状態を日常的に確認する試験。
3.39
検出の確認(detection)
限度値レベルでのレスポンスの数値的信頼性の確認。
5
K 0138:2018
3.40
システムの性能(performance)
試験対象の分析種を特異的に分析し得ることの確認。
3.41
システムの再現性(repeatability)
繰返し測定におけるばらつきの程度の度合。
3.42
ゼロフィリング(zero filling)
FID信号の末尾に強度ゼロのデータを補完し,見掛け上のデジタル分解能を向上させる方法。
3.43
ベースライン補正(baseline correction)
NMRスペクトルのベースラインのひずみの補正。
3.44
クエンチ(quench)
超伝導磁石が何らかの理由で超伝導状態を維持できなくなったとき,メインコイルを流れる大電流が熱
に変換され,周囲の液体ヘリウムを一気に蒸発させる現象。
3.45
パルスシーケンス(pulse sequence)
一つ以上のパルスを一定の順序に並べFIDを取り込む一連の時間系列。
3.46
窓関数(window function)
FIDをフーリエ変換するとき,スペクトルのSN比(信号雑音比),線幅などを改善させるためにFID信
号に掛け合わせる関数。ウィンドウ関数ともいう。
3.47
適格性確認(qualification)
機器が所定の機能をもち,日常の試験に必要とされる性能を維持していることの確認。
3.48
化学シフト基準物質(primary standard: reference standard for chemical shift)
化学シフトの内部基準となるシグナルを与える化学物質。
3.49
qNMR用基準物質(calibration standard: reference standard for qNMR)
qNMR分光法に用いられる標準物質であり,物質量の基準となる化学物質。
3.50
重水素化溶媒(deuterated solvent)
水及び有機溶媒の水素原子の一部又は全部を重水素に置き換えた溶媒。重溶媒ともいう。
3.51
天びん(balance)
質量を測定する計量器。微量又は極微量のひょう量には電磁式電子天びん又は電磁式電子はかりが通常
用いられる。
6
K 0138:2018
3.52
最小計量値(minimum weight)
天びんの精確さを確保するためのひょう量の下限を示す算出値。
4
qNMRの概要
4.1
qNMRの原理
NMRは,静磁場に置かれた物質の構成原子核が,その核特有の周波数のラジオ波に共鳴して,低エネ
ルギーの核スピン状態から高エネルギーの核スピン状態に遷移することに伴ってラジオ波を吸収する現象
を利用した分析法である。測定対象とする核は,主に1H,13C,15N,19F,29Si,31Pなどである。
全ての核は電荷をもっているが,ある種の核ではこの電荷は核の軸上で回転(スピン)し,核の電荷が
回転(スピン)することによって,この軸に沿って磁気が生じる。回転する電荷の核運動量,すなわち,
原子核の核スピン(I)は,1/2,1,3/2,…,n/2(ただし,nは整数)などの値(1H及び13Cでは,I=1/2)
をとる。I=0の核は,スピンがないことを意味する。量子力学的には,スピンIの核では,式2I+1によ
って均一な外部磁場の中で核がとり得る方向の数が決まる。言い換えれば,核を磁場の中に置くと,核モ
ーメント(陽子及び中性子のスピンから生じる。)は,2I+1(1H,13Cなどは,I=1/2)個のエネルギー準
位に配向する。磁気回転比γの核を外部磁場H0の中に置いた場合,これらのエネルギー準位間の遷移と照
射する周波数vのラジオ波の関係は,次の式で表わされる。
π
2
0
H
vγ
=
ここに,
v: 電磁波の周波数
γ: 磁気回転比
H0: 外部磁場
共鳴(エネルギー準位間の遷移)を起こす周波数vのラジオ波の照射によって,その周波数のラジオ波
の吸収[NMRシグナル(シグナル,信号)]が観測される。どのような環境の核に対しても吸収の係数(遷
移の確率)は一定であるので,得られたシグナル面積は基本的に共鳴核の数に比例する。このような遷移
によって高エネルギー準位に偏った核スピンは,一定時間後に再び熱平衡分布に戻る[緩和する(これに
要する時間を緩和時間という。)]。
分子を磁場の中に置くと,分子内の電子が核を外部磁場から遮蔽する。分子内での核の環境が異なると
その遮蔽の度合も異なるので,異なる環境の核の共鳴周波数も異なることになり,別々のシグナルとして
観測される。シグナルの位置は,化学シフトδとして表現される。共鳴周波数は,磁場に比例して変化す
るので,磁場によらない量として,化学シフトxは,次の式で定義する。
R
R
R
S
δ
+
v
v
v
x
−
=
ここに,
x: 化学シフト
vS: 試料核の共鳴周波数
vR: 基準核の共鳴周波数
δR: 基準核の化学シフト(0ではない場合)
化学シフトは,通例,基準物質(基準核)のシグナル位置を0 ppmとしたppm単位で表すが,基準物質
のシグナル位置が0とできない場合は,その基準物質のあらかじめ定められている化学シフトδRを用いて
補正する。
7
K 0138:2018
分子内の各核における磁場は,周囲の電子の寄与(核遮蔽)だけではなく,分子中の他の核磁石(核ス
ピンをもっている核は,それ自身が一つの磁石である。)の影響下にもあるので,核磁石間の化学結合によ
るカップリングによってシグナルは分裂する。この分裂の間隔をスピン−スピン結合定数(J値)という。
J値は,ヘルツ(Hz)で表す。J値は外部磁場の大きさに依存せず,分裂のパターンは相互作用する核の
数が増すにつれて複雑になる。
NMRスペクトルからは,基本的には化学シフト,スピン−スピン結合定数,シグナル面積[1H核では
数に比例するが,13C核などでは核オーバーハウザー効果(NOE),緩和などの影響を受ける。]などの情
報が得られ,これらを利用して化学物質の構造解析,確認又は定量を行うことができる。
1H NMRでは,定量性を確保した条件で測定したとき,スペクトル上に観察される化合物中の1H核の数
の比がシグナル面積比に対応する特性をもち,国際単位系(SI)へのトレーサビリティが確保されたqNMR
用基準物質を用いることで,純度又は含有率について物質量(モル)に基づいた信頼性の高い値を求める
ことができる。この測定法を,1H核定量核磁気共鳴分光法(1H qNMR)と呼ぶ。
4.2
1H qNMRの原理
物質を溶媒に溶解し,1H NMRを測定して得られるスペクトルは,次の特徴があり,この特徴を利用し
て,化学構造解析の分析法として広く活用されている。
− 測定した分析種の化学構造によって異なる化学シフトに共鳴シグナルを与える。
− 化学結合を通して隣接する炭素に結合する1Hなどの数に応じてシグナルが分裂する。
− シグナル面積が共鳴する1Hの数に比例する。
1H NMRスペクトルでは,同一分子内であっても異なる環境にある水素核は,共鳴周波数に応じて異な
る化学シフトをもつ分離したシグナルとして観測される。各シグナル面積Siは,共鳴する1H核の数Ni(プ
ロトン数),溶液体積V,試料の質量m,モル質量M,分析種の純度又は含有率P,励起パルス角β(フリ
ップ角),シグナルを与える核の縦緩和時間T1i,平衡磁化M0及び測定パラメータとして設定される遅延時
間Trから,式(1)で表わされる。
0
/
/
i
i
)
(cos
1
1
sin
1i
r
1i
r
M
e
e
P
VM
m
KN
S
T
T
T
T
β
β
−
−
−
−
=
············································ (1)
ここに,
Si: 各シグナル面積
K: 定数
Ni: 共鳴する1H核の数(プロトン数)
V: 溶液体積
m: 試料の質量
M: モル質量
P: 分析種の純度又は含有率
β: 励起パルス角(フリップ角)
T1i: シグナルを与える核の縦緩和時間
Tr: 遅延時間
M0: 平衡磁化
ここで,添字のiは異なるシグナルを示し,縦緩和時間T1は1Hの環境によって異なる。NMRは一般に
測定感度がよくないことからスペクトルを取得する際には積算してSN比を向上させている。このとき,
測定対象とする分析種の中で最も長い縦緩和時間T1より十分長い遅延時間Trで積算すると,分析種の全て
のシグナルに対して
1
1
1
r/≈
−
−
T
T
e
の条件を満たすことが可能であり,シグナル面積は共鳴核の個数に比例す
る強度を示し,式(1)は,式(2)で表される。
8
K 0138:2018
i
kN
S=
··················································································· (2)
ここに,
S: シグナル面積
Ni: 共鳴核の個数
k: 対象とする試料溶液の測定に関わる定数
構造解析に利用する場合には遅延時間を十分長くとらず,SN比を向上するために積算回数を多くする
条件,すなわち,検出感度優先の測定が行われているため,分子内のシグナル面積とプロトン数の比,す
なわち,分子内シグナル面積比は精確に求められていない。しかし,定量性を確保できる条件下で分子内
の異なる化学シフトを示すシグナルi及びシグナルjの面積を比較すると,式(3)の比例関係が成り立つ。
j
i
j
i
N
N
S
S=
·················································································· (3)
ここに,
Si: 分子内の共鳴シグナルiの面積
Ni: 分子内の共鳴シグナルiに対応するプロトン数
Sj: 分子内の共鳴シグナルjの面積
Nj: 分子内の共鳴シグナルjに対応するプロトン数
このような定量性の関係は,同じ測定系内の異なる2分子間に由来するシグナルにも適用することがで
きる。この場合,試料溶液を測定する際の励起パルス角及び溶液の体積は化合物によらず一定と考えられ
るので,得られる面積Sは,式(2)及び式(3)から,分析種の純度又は含有率P,モル質量M,質量mなど測
定する化合物だけに依存する値に比例した式(4)を得る。
s
a
s
a
s
a
c
c
N
N
k
k
S
S
×
×
=
s
a
s
s
a
a
c
N
N
S
S
c
×
×
=
s
a
s
s
a
a
s
s
a
a
P
m
m
M
M
N
N
S
S
P
×
×
×
×
=
·························································· (4)
ここに,
Pa: 分析種の純度又は含有率
Ps: qNMR用基準物質の純度
Sa: 分析種のシグナル面積
Ss: qNMR用基準物質のシグナル面積
k: 対象とする試料溶液の測定に関わる定数
Na: 分析種のプロトン数
Ns: qNMR用基準物質のプロトン数
ca: 分析種のモル濃度
cs: qNMR用基準物質のモル濃度
Ma: 分析種のモル質量
Ms: qNMR用基準物質のモル質量
ma: 試料の質量
ms: qNMR用基準物質の質量
注記 分析種とは,分析試料又は試料溶液中の被検査成分,分析対象成分をいう。また,試料とは,
試験・分析・検査に供される物質をいう。すなわち,分析種と分析対象としない成分の混合物
を表す。
それぞれの分子が溶液中で反応などの相互作用を起こさないこと,異なる化学シフトに分離したシグナ
9
K 0138:2018
ルをもつことなど必要な条件はあるものの,この式(4)は定量測定条件下で1H NMR測定を行うことで,純
度既知のqNMR用基準物質があれば,分析種の純度又は含有率を測定できることを示している。特にqNMR
用基準物質がSIへのトレーサビリティを確保している場合には,これを使用することで,分析種の純度又
は含有率をSIにトレーサブルな値として間接的に算出することができる。
1H qNMRにおいて精確な分析結果を得るための大きな要点は,試料及びqNMR用基準物質それぞれの
質量を精密にはかりとること,測定溶媒に完全に溶解させること,定量に適した測定条件で1H qNMRス
ペクトルを取得すること,及び不純物のシグナルを含まないシグナルを定量用シグナルとして選択するこ
とである。
5
装置
5.1
一般事項
NMR装置は,パルスフーリエ変換(パルスFT)法を用いているものとする。
5.2
設置条件及び安全
設置室の環境は,次による。
a) 設置室の広さ 設置室には,装置の設置・運転に必要なスペースに加えて漏れ磁場を管理できる領域
が必要となる。また,部屋の外に管理区域が取れない場合には,超伝導磁石の漏れ磁場の基準値が室
内に収まるだけの広さが必要となる。ただし,漏れ磁場の大きさは磁場の大きさ及び磁石の種類によ
って異なる。
b) 天井の高さ 納入設置,液体ヘリウム充塡などの作業のために,部屋には規定の高さが必要となる。
ただし,必要な天井高は磁場の大きさ及び磁石の種類によって異なる。
c) 床面 床面の振動は少ない方がよい。また,床面の強度も規定値が必要となる。
d) 空調設備及び排気設備 NMR装置は空調された部屋に設置する。また,マグネットがクエンチした
場合に備えた安全対策(酸素濃度計と換気扇を備えた部屋)を行う1)。さらに,居室の入口及び超伝
導磁石には,強磁場発生に関する危険表示を示し,関係者以外を入室させない。超伝導磁石の周囲0.5
mTの範囲が明確になるようにする2)。
一般的な400 MHzのNMR装置の設置条件の例として,床振動0.2 cm/s2(Gal)以下,床耐荷重600
kg以上,温度範囲17 ℃〜27 ℃(±1 ℃/h)及び相対湿度70 %以下がある(図1参照)。また,超電
導磁石がアクシデントによって磁場を失った場合は,ヘリウムガス及び/又は窒素ガスが大量に放出
されるため,ダクトを超伝導磁石の排気口に連結したクエンチダクトが必要になる場合もある(図2
参照)。
注1) NMR装置は,通常,超伝導磁石を使用するため,強磁場に関する危険表示を示し,関係者以
外を入室させない。また,超伝導磁石の設置時又はクエンチ時には数m2のヘリウムガスが室
内に放出される。酸素欠乏を防止するため,十分な換気量をもつ換気扇を設置する。換気容
量及び換気扇の性能に関しては,NMR装置の設置要領書に従う。
2) 平成29年(2017年)現在,強磁場の人体への影響は不明な点が多いが,疫学調査,動物実
験などから障害発生の可能性も議論されている。確実に人体の影響が指摘されているのは,
0.5 mTでペースメーカー誤動作が起きた事例であり,0.5 mTの境界を明示することを推奨す
る。
10
K 0138:2018
図1−400 MHzのNMR装置における標準的な設置条件の例
図2−NMR装置の標準的な超伝導磁石の排気口とダクトとの連結の例
クエンチダクト
ダクト
11
K 0138:2018
5.3
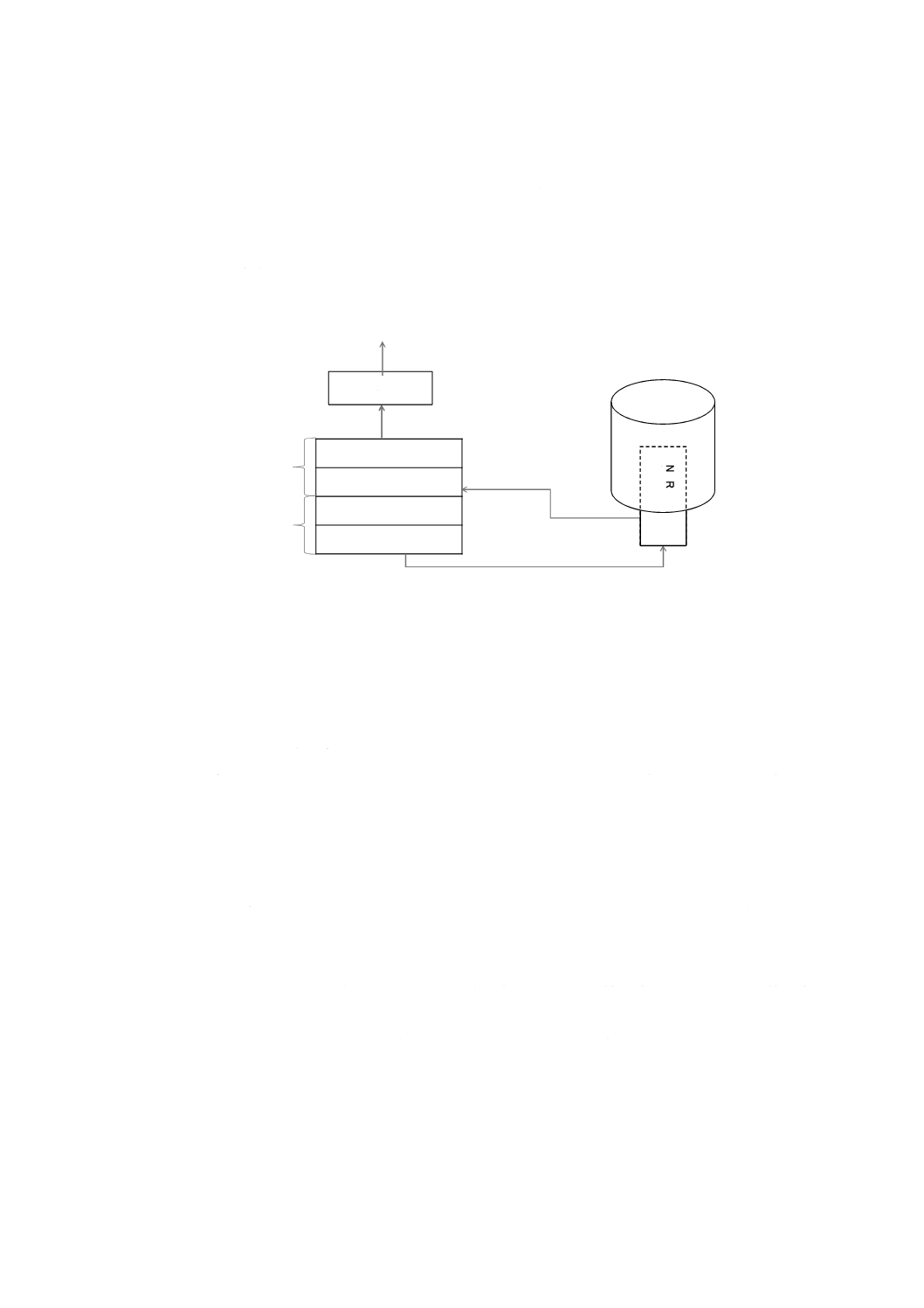
装置構成の概要
NMR装置は,冷却式超伝導磁石,NMRプローブ,高周波発生部,受信部,データ処理部などで構成さ
れ,次の装置を用いる。主な分光計として,パルスフーリエ変換NMR(FT-NMR)装置がある。FT-NMR
装置は,強力なラジオ周波数パルスを用いて観測核を全周波数領域にわたって同時に励起する。パルス照
射後のFID(free induction decay,自由誘導減衰)を観測し,強度の時間関数であるFIDをフーリエ変換に
よって周波数関数に変換してスペクトルを得る(図3参照)。
デジタル検波器
ラジオ波受信装置
ラジオ波発信装置
パルスプログラマ
冷却式
超伝導磁石
N
M
R
プ
ロ
ー
ブ
データ処理部
(コンピュータ)
フーリエ変換
ラジオ波
FID
(自由誘導減衰)
NMRスペクトル
高周波発生部
受信部
図3−パルスフーリエ変換NMR装置(FT-NMR装置)の例
a) 冷却式超伝導磁石 ヘリウム冷却式超伝導磁石。
b) NMRプローブ 試料にラジオ波(パルス)を照射し,試料から放出されるラジオ波(NMRシグナル)
を検出する装置(NMR装置における検出器)。
c) 高周波発生部 パルスプログラム(pulse program)に従って発生したラジオ波のパルス列を,周波数
シンセサイザ(frequency synthesizer)を使って作成したラジオ周波数と混合して,核磁気共鳴周波数
に相当する周波数パルス列を発生させる装置。発生させたパルス列を検出器に導入して,磁場中の試
料に照射する。
d) 受信部 プローブで受信した微弱なFID信号を増幅し,信号をデジタル化後,データ処理部に転送す
る装置。
e) データ処理部 デジタル化したFID信号をフーリエ変換によって,NMRスペクトルに変換し,表示,
解析,記録媒体に保存するための処理装置。
f)
附属装置 主な附属装置を,次に示す。
1) オートサンプルチェンジャ(auto sample changer) 自動NMR試料管交換装置。NMR試料管を自動
で交換する装置。
2) オートチューニング装置 試料を交換するごとに,試料とプローブの電気回路とのチューニングを
自動で行う装置。
3) 温度制御装置 測定試料の温度を制御する装置。
12
K 0138:2018
6
1H qNMRの準備
6.1
一般的事項
1H qNMRには,内標準法及び外標準法がある。内標準法では,試料と少量のqNMR用基準物質とを溶
媒に溶かした試料溶液をNMR試料管に入れて測定する。このため,完全に同一環境下で試料とqNMR用
基準物質のシグナルが得られる。一方,外標準法では,試料を溶媒に溶かした試料溶液の試料管又はqNMR
用基準物質を溶媒に溶かした基準溶液の試料管を別々に調製し,各々測定する。すなわち,試料溶液と基
準溶液の測定データは完全に同一環境下で測定されたものではないことから,測定環境の差異による誤差
要因を排除することが容易ではない。したがって,1H qNMRでは,高精度の定量分析結果が得られる内標
準法を通常用いる。ただし,内標準法による測定が困難であり,外標準法を用いなければならない場合に
は,別に濃度既知の物質を用いて外標準法で測定を行い,目標の精確さに達していることが確認できたと
き,その設定した測定法を用いることができる。
なお,良いスペクトルを得るためには,溶液中の固体の異物,ごみなどの浮遊物が混入することを防ぐ
必要がある。固形分の存在は,シグナルの線幅が広がり分解能の低下を引き起こすことがある。また,溶
液濃度の均一性も重要であり,特に高磁場装置では,NMR試料管内の試料溶液濃度が不均一であること
によって,顕著な分解能の低下が認められることがある。実際に溶液濃度が高すぎる場合,溶液の粘性が
高まり分解能の低下が生じる。NMR試料管は種々あるが,試料管の径は一般に細いものを用いた方が,
局所磁場の均一性がよく,分解能に優れた良質のスペクトルが得られる。分解能の高いスペクトルを得る
ためには,高磁場の装置になるほど,ひずみがなく,真円度が高いNMR試料管を用いる必要がある。NMR
試料管の選択が不適切な場合,良好なスペクトルが得られないことがある。
6.2
試料及びqNMR用基準物質のひょう量
1H qNMRにおいて,定量値は式(4)(4.2)によって求められるため,精度よく分析するためには,試料
の質量を,次によって精密にはかりとる。
なお,吸湿,揮発又は蒸発しやすい試料の場合は,ひょう量値に偏りが生じないように試料の特性に応
じた対策をとる必要がある。
さらに,試料のひょう量の注意事項を,附属書Dに示す。
a) ひょう量 試料及びqNMR用基準物質の質量をはかりとる場合には,目標とする精確さに応じて決定
する。通常,最小計量値より大きい質量を目安とする。
b) ひょう量の手順 試料及びqNMR用基準物質のひょう量の手順は,次による。
1) ひょう量に使用する器具類の準備及び整理整頓を行う。
2) 天びんの感度調整を行う3)。
注3) 天びんの感度は経時的な変化を生じ,その変化量は良好に管理された分銅の変化よりも大
きいため,感度調整が行われていない天びんを用いてはかりとった質量に偏りが生じる。
そのため,使用前には感度調整を行う。
注記 前回の使用から1時間以上使用しなかった場合は,改めて感度調整を行うことが望ましい。
6.3
基準物質
NMR分析において用いられる基準物質は,化学シフト用とqNMR用に分類される。
6.3.1
化学シフト基準物質
化学シフト基準物質は,測定対象物質の化学シフトを求める際の基準となる周波数をもつ化学物質をい
う。国際純正・応用化学連合(International Union of Pure and Applied Chemistry:IUPAC)では,試料溶液を
有機溶媒で調製する場合は,テトラメチルシラン(TMS)を用いることとされている。一方,試料溶液が
13
K 0138:2018
水溶液の場合は,化学シフト基準物質として3-(トリメチルシリル)-1-プロパンスルホン酸ナトリウム塩
(DSS)を,主に用いる。
6.3.2
qNMR用基準物質
qNMR用基準物質は,定量する際の物質量の基準となる物質をいう。qNMR用基準物質にはSIへのトレ
ーサビリティを確保しているものが供給されており,1H qNMRでは,これを上位標準として用いるとき,
試料中の分析種の純度をSIにトレーサブルな値として間接的に算出することができる。このため,SIに
トレーサブルな値付けをされた認証標準物質(Certified Reference Material:CRM)をqNMR用基準物質と
して,通常用いる。既にCRMとして供給されているものには,3,5-ビス(トリフルオロメチル)安息香酸,
1,4-ビス(トリメチルシリル)ベンゼン-d4(1,4-BTMSB-d4),3-(トリメチルシリル)-1-プロパンスルホン
酸-d6-ナトリウム塩(DSS-d6),マレイン酸,ジメチルスルホンなどがある。これらCRMのうち,取扱い
の容易な固体化合物であり,特異的な化学シフトである0 ppm付近に鋭い1本のシグナルを示す
1,4-BTMSB-d4及びDSS-d6がqNMR用基準物質として主に用いられる。ただし,前者は有機溶媒に,後者
はメタノール,ジメチルスルホキシド,重水などに適している。
いずれのqNMR用基準物質を用いる場合にも,試料溶液中で反応,分解などの相互作用を起こさないこ
と,定量に用いる分析種とqNMR用基準物質のシグナルが異なる化学シフトに完全に分離して観察される
ことが必要である。
6.4
重水素化溶媒の選定
1H qNMR溶媒としては,NMR測定用重水素化溶媒を用いる。溶媒の選択に当たっては,試料のシグナ
ルと重なるシグナルを示さない,試料の溶解性が高い,試料と反応しないなどを考慮して選択する。溶媒
の種類,溶液の濃度,水素イオン濃度(pH)などによって化学シフトが変化する。また,試料溶液の粘度
が高い場合には分解能が低下するので注意する必要がある。
なお,NMR測定用重水素化溶媒中の不純物のシグナルが定量精度に影響を与えるので高純度のものが
よい。
6.5
NMR試料管
NMR試料管は,感度・分解能が良好となる真円度が高く,反りが小さく,かつ,肉厚が薄くて均一な
ほうけい酸ガラス製のもので,そのキャップの材質は,可塑剤などの溶出がないものを使用する。
注記1 NMR試料管として,外径が5 mmのものが多く使用されるが,外径が3 mm,10 mmなどの
ものもある。いずれのサイズのNMR試験管を用いる場合においても,1H qNMR測定に適し
たもの(5 mm管の場合,外経4.965
014
.0005
.0
−
+
mm,反り5 μm以下,全長180 mm〜200 mm)を
使用するのがよい。
注記2 NMR試料管は,NMR試験管とも呼ばれている。
6.6
NMR装置の最適化
NMR測定では,次のa) 及びb) に留意して,試料空間における静磁場の空間的な強度むらを試料部位
周りに取り巻かれた複数のシムコイルに電流を流して補正する磁場を追加するシム調整及び分解能調整を
行い,NMR装置を最適化する。
a) NMR装置は,機器の取扱説明書などに従って,あらかじめ測定に支障のない状態に調整したものを
用いる。
b) NMR装置は,0.1 %エチルベンゼン重水素化クロロホルム溶液などを用いて装置の感度を最適条件に
調整する。また,1 %〜3 %クロロホルム重水素化アセトン溶液などを用いて装置の分解能を最適条件
に調整する。
14
K 0138:2018
注記 感度調整用として封管された0.1 %エチルベンゼン重水素化クロロホルム溶液が,分解能調
整用として封管された1 %〜3 %クロロホルム重水素化アセトン溶液が供給されている。
6.7
試料溶液の調製
試料溶液の調製は,次による。ただし,採取環境の条件,qNMR用基準物質又は試料の調製条件などが
個別規格に規定されている場合は,それに従う。
なお,一般的な試料溶液の調製を附属書Aに示す。
a) 採取環境(温度及び湿度)が,試料及びqNMR用基準物質の取扱いに適していることを確認する。
b) 試料及びqNMR用基準物質は,それぞれの定量に用いるシグナル面積ができるだけ同程度になるよう
に採取する。
c) 試料及びqNMR用基準物質を加えた容器に適量の重水素化溶媒を加えて溶かし,試料溶液を調製す
る。
d) 測定に適切な量の試料溶液をとり,NMR試料管に入れる。
e) 試料及びqNMR用基準物質の基本的なひょう量の手順は,次による。
質量差(試料又はqNMR用基準物質を含めた採取用容器の質量をはかりとった後,採取用容器の質
量を減算した質量)として,はかりとる場合を例として示す。
注記1 採取用容器を天びんの計量皿へ載せた後に,天びんの風袋引き装置を用いて表示値をゼロ
に設定後,採取用容器に試料をはかりとる場合は,試料又はqNMR用基準物質の質量が表
示器に表示される。天びんの風袋引き装置はJIS B 7611-1[11]を参照。
1) ひょう量前の周囲環境(温度,相対湿度及び大気圧)を記録する。
注記2 ひょう量を行う周囲環境の温度変化は天びんに感度変化を起こす。また,気圧が急激に
変化すると試料への浮力効果が異なるため,ひょう量値に誤差を与える可能性がある。
注記3 帯電しやすい試料若しくは採取用容器,又は測定室内の相対湿度が40 %以下の環境では,
天びんと電荷の力の作用によって表示値が上方又は下方に変動し,はかりとった質量に
誤差を与える可能性がある。
2) 天びんの計量皿が清掃されており,ひょう量に不要な物が何も載せられていないかを目視で確認す
る。
3) 天びんの零点設定機能によって,その表示値がゼロを示しているかを確認する。
注記4 零点設定後の表示値の安定性についても確認することが望ましい。天びんの零点設定装
置はJIS B 7611-1を参照。
4) 採取用容器を計量皿へ載せ,一定の読取時間の経過後,表示値を記録する。
注記5 読取時間の時間間隔は,ひょう量の手順又は環境条件によって異なるが,ひょう量作業
全体を通して同じ時間間隔を用いることが望ましい。
5) 試料又はqNMR用基準物質を採取用容器に採取し,計量皿へ載せ,読取時間経過後,表示値を記録
する。
6) 計量皿から全てを取り除いた後,表示値がゼロに戻るかを確認する。
注記6 表示値がゼロに戻らない場合は,再度ひょう量を行う,又は無視できる許容範囲を設定
し質量結果の取扱いとして個別に手順化しておくことが望ましい。
7) はかりとり後の周囲環境(温度,相対湿度及び大気圧)を記録する。
6.8
1H qNMR測定条件の設定
使用するNMR装置に従い,測定対象とする核,デジタル分解能,スペクトル幅,スピニング,パルス
15
K 0138:2018
角,13Cデカップリング,遅延時間,積算回数,ダミースキャン,測定温度などの項目を定量分析に適し
た次の条件に適切に設定する。精確な定量分析には,十分なシグナルの分離及びSN比が必要である。有
効数字2桁以上の精確さで分子構造が明確な有機化合物の純度測定を行う際に初期設定として望ましい測
定条件は,次のa)〜j) による。
なお,分析種及びqNMR用基準物質の両者にCRMなど純度が保証されたものを用いて分析種の純度又
は含有率の測定を行い,目標とする精確さ(不確かさ)が得られていると確認した場合には,各項目のパ
ラメータを変更してもよい。
なお,個別規格に1H qNMR測定条件が規定されている場合は,それに従う。
さらに,1H qNMRスペクトルの測定の詳細な注意事項を,附属書Bに示す。
a) NMR装置 プロトン(1H)共鳴周波数300 MHz以上の分光分析装置。
注記1 目標とする精確さ(不確かさ)が得られる感度及び分解能をもつ装置を用いることが望ま
しい。
b) 分解能 0.25 Hz以下。
注記2 十分なデータポイント数が確保されているとき,定量に用いるシグナル面積を正確に求め
ることができることから,分解能は,通常,0.25 Hz以下にすることが望ましい。
c) スペクトル幅 −5 ppm〜15 ppmを含む20 ppm以上。
注記3
1Hのシグナルが通常検出される範囲は化学シフトとして−5 ppm〜15 ppmの範囲にあるこ
とから,この範囲を満たす20 ppm以上に設定することを推奨する。ただし,スペクトル
の両端付近で観測されるシグナルが認められる場合には,通常より広いスペクトル幅を適
切に設定することが望ましい。
d) スピニング オフ。
注記4 スピニングサイドバンドの存在は不純物のシグナルの存否判定を困難にする場合がある。
また,スピニングサイドバンドが定量対象のシグナルと重なったとき,そのシグナル面積
に影響を及ぼし,結果として正確な定量値を求めることができない。これらの問題を回避
するため,通常はスピニングを行わない設定にすることが望ましい。
e) パルス角 90°。
注記5 精確な定量条件を達成するための遅延時間(少なくともT1の7倍以上)を確保した場合,
パルス角を90°とすることで単位時間当たりのSN比を最も高くすることができるため,
パルス角を90°にすることを推奨する。ただし,シグナルが非常に広帯域に観測される場
合には,パルス角を小さくすることで励起中心から離れたシグナルにおける励起効率の低
下を抑制できる場合もあるため,パルス角を小さくする検討を行うことが望ましい。
f)
13Cデカップリング あり。
注記6
13Cサテライトサイドバンドが定量対象のシグナルと重なったとき,そのシグナル面積に
影響を及ぼし,結果として正確な定量値を求めることができない。また,13Cサテライト
サイドバンドの存在は不純物のシグナルの存否判定を困難にする場合がある。このため,
通常は13Cデカップリングを行う設定で測定することが望ましい。
13Cデカップリングは,MPF8[周波数スイッチングによる広帯域(13C)デカップリング
法の一つ]などの13Cのシグナルが観測される帯域が完全にデカップリングされるものを
用いることが望ましい。
なお,有意な不純物が認められない場合には,13Cサテライトサイドバンドを積分区間
16
K 0138:2018
に含めることを前提に13Cデカップリングを行わずに測定することもできる。
g) 遅延時間 縦緩和時間T1の7倍以上。
注記7 通常の化合物の縦緩和時間T1は長いものでも5秒〜7秒であることから,その7倍を十分
にカバーしている遅延時間(繰返しパルス待ち時間)60秒以上を設定することが望ましい。
ただし,分析種及びqNMR用基準物質の定量に用いるシグナルの縦緩和時間T1が極端に
長いと想定されるとき,縦緩和時間T1の測定を別に行い,その7倍以上になるように遅延
時間を設定することが望ましい。
h) 積算回数 SN比100以上を示す積算回数。
注記8 目標とする精度に応じて,十分なシグナルのSN比が得られる回数を設定することが望ま
しい。
シグナルのSN比とは,求めたいシグナルのベースラインから頂点までの高さをSとし,
そのシグナルの近傍で1H由来のシグナルが認められないベースラインの200 Hz以上の帯
域から得たノイズの二乗平均平方根をノイズNとしたとき,Sを2Nで除したものである
(参考文献[7]を参照)。SN比は積算回数の正の平方根に比例するため,10倍のSN比を得
るためには100倍の積算が必要である。
i)
ダミースキャン 2回以上。
注記9 測定前の空スキャンのことをいう。NMRシグナルの取得を開始する前に,信号を取得せ
ずにパルスシーケンスを繰り返し実行する。測定の際には,RFパルスの出力によって試料
の加熱などが起きるため,装置及び試料が安定化するまで繰り返してから信号の取得を開
始する。安定した環境が必要な測定では,多めに設定する。
j)
測定温度 一定温度。
注記10 分析種及びqNMR用基準物質のシグナルがシャープに得られる一定温度に設定すること
が望ましい。
7
測定
7.1
1H qNMR測定条件
6.8に従って,定量分析に適した測定条件を設定する。
7.2
測定操作
操作は,次のとおりとする。
a) 測定前にシム調整を行い,シグナルの対称性及び裾の立ち上がりが良好であることを確認する。
b) 試料溶液を入れたNMR試料管をNMR装置に導入し,定量分析に最適化した条件で測定する。
7.3
FIDデータの処理及び保存
データ処理装置を用いて,測定されたFIDデータを保存する。FIDデータは,定量分析に適した条件で
1H qNMRスペクトルにフーリエ変換し,定量に用いるシグナルの位相を適切に調整する。また,処理に用
いる窓関数(ウィンドウ関数),ゼロフィリング,ベースライン補正,位相補正,積分範囲の設定などのパ
ラメータは,定量分析に適した条件を用いる。
8
解析
8.1
一般
試料溶液について得られた1H qNMRスペクトル上に検出されるqNMR用基準物質及び分析種のシグナ
17
K 0138:2018
ル面積をそれぞれ評価し,qNMR用基準物質を内標準物質として分析種を定量する。それぞれのシグナル
面積は,システムに附属したデータ処理ソフト又はシステムに後から組み込んだデータ処理ソフトによっ
て評価する。シグナル面積の測定はシグナルの形状に合わせて適切な積分範囲を設定しなければならない。
システムに後から組み込む処理ソフトによる場合は,製造業者などが定めた手順を参考にするなどによっ
て,処理ソフトが正しい結果を与えることを検証した上で適用する。
8.2
計算式
分析種のシグナルiから求められる含有率(Pi)は,式(5)を用いて算出する。
なお,通常十分に分離したシグナルを定量に用いるが,それぞれの官能基に由来するシグナルが近接し
分離して個別に評価できない場合,それらを一区間とみなしてシグナル面積を求めてもよい。このとき,
分析種のシグナルiのプロトン数(Ni)には,設定した積分区間内に観測されるプロトン数の合計を用い
る。
なお,具体的な1H qNMRスペクトル解析の基本的な注意事項を附属書Cに示す。
s
a
s
s
i
i
s
s
i
i
P
m
m
M
M
N
N
S
S
P
×
×
×
×
=
··························································· (5)
ここに,
Pi: 分析種のシグナルiから求められる含有率
Ps: qNMR用基準物質の純度
Si: 分析種のシグナルiの面積
Ss: qNMR用基準物質のシグナル面積
Ni: 分析種のシグナルiのプロトン数
Ns: qNMR用基準物質のプロトン数
Mi: 分析種のモル質量
Ms: qNMR用基準物質のモル質量
ma: 試料の質量
ms: qNMR用基準物質の質量
8.3
分析値の表し方
8.2で得られた計算値は,kg/kg,mg/kg,%(質量分率)などで表す。
8.4
データの整理及び保存
調製及び測定データには,次のうち必要な項目を記載する。
a) 調製及び測定年月日
b) 試料,qNMR用基準物質及び使用した重水素化溶媒の情報(qNMR用基準物質及び重水素化溶媒の名
称,qNMR用基準物質の純度,ロット番号など)
c) 測定条件,測定繰返し数及びデータ処理条件
d) 分析結果(定量に使用したシグナル情報を含む。)
e) シグナルの帰属情報
f)
その他の必要事項
9
データの質の管理
9.1
一般事項
データの質の管理のために,qNMR用基準物質,1H qNMRスペクトルのベースライン及びシグナル形状
の記録,定期的な装置性能の点検などが必要である。評価結果は,生データ(FIDデータ,スペクトルデ
ータ),測定記録,1H qNMRスペクトルとともに電子ファイル及び文書として保管する。
注記 管理については,適切なものを選別し,個別分析方法の標準作業手順書(SOP)などに書き込
18
K 0138:2018
むことが望ましい。
9.2
NMR装置の適格性確認
NMR装置の適格性確認には,次のa),b) で示す装置の適格性及びqNMR測定操作に対する装置の適格
性の二つの確認がある。
a) 定期点検又は日常点検 NMR装置は,6.6に規定するように装置に対してあらかじめ設定された仕様
(性能)に適合していることを定期的又は日常的に行う。
b) qNMR測定操作に対する適格性 使用するNMR装置が,1H qNMR測定操作に対して適格性をもつこ
とを確認するため,装置の性能管理を実施する。その場合,測定対象のシグナル面積が,シグナルが
観測されるスペクトル幅内で,正確に定量できることを確認する。通常,類似構造の不純物を有意に
含まない純度が非常に高い(99.5 %以上が望ましい。)化合物を用いて1H qNMR測定条件で測定した
とき,分子内の複数のシグナルから得られる理論上のシグナル面積の比が正確であることを確認する。
なお,標準物質の純度として認証値が付されている分析種を使用することが望ましい。
注記 NMR装置の適格性確認用として,ビンクロゾリン(CRM)及び1,4-BTMSB-d4(CRM)をジ
メチルスルホキシド-d6に溶解して調製された1H qNMR用の混合溶液などが供給されている。
9.3
システム適合性試験
分析種の純度又は含有率を測定する場合,目標とする定量精度に合わせてa)〜c) に適合することを確認
する。ただし,個別規格にシステム適合性の要件が規定されている場合には,それに従う。
a) システムの再現性 試料溶液について,1H qNMR測定条件で測定を繰り返すとき,分析種の定量に用
いる各シグナル面積とqNMR用基準物質のシグナル面積との比が一定であることを確認する。
注記 分析種の純度を精確に測定する場合,繰返し測定を行ったとき,分析種の定量に用いる各シ
グナル面積とqNMR用基準物質のシグナル面積との比の相対標準偏差が目標とする定量精度
を達成できる水準であることを確認することが望ましい。
例えば,1 %以内の精確さを目標とした場合,その相対標準偏差は0.5 %以下(6回以上繰
返し測定を行った場合)であることが望ましい。
b) システムの性能 試料溶液について,1H qNMR測定条件で測定するとき,分析種のシグナルとqNMR
用基準物質のシグナルとが完全に分離して観測され,かつ,定量に用いるシグナルと不純物のシグナ
ルとが完全に分離していることを確認する。
c) 検出の確認 分析種及びqNMR用基準物質のシグナルが十分なSN比をもつことを確認する。例えば,
1 %以内の精確さを目標とした場合,定量に用いるシグナルのSN比は100以上であることが望ましい。
9.4
トレーサビリティの確保
調製に用いる天びんは定期的な校正を行い,その校正にはSIへのトレーサビリティが保証されている分
銅(JIS B 7609のもの。)を用いる。
SIへのトレーサビリティが確保されているqNMR用基準物質に用いる標準物質は,ISO 17034に適合す
る標準物質生産者からCRM 4) として供給されており,不確かさが明記されている。
なお,適切なCRMが入手できない場合は,SIへのトレーサビリティが確保されたCRMを用いたqNMR
分光法によって純度又は含有率を評価した試料をqNMR用基準物質として用いることができる。
注4) ISO Guide 34[12]に基づいて2019年10月31日までに生産されたCRMは,その有効期間内に
おいて使用することができる。
9.5
分析値の信頼性の確保
分析値の信頼性を確保する場合には,SIへのトレーサビリティが保証された値をもつqNMR用基準物質
19
K 0138:2018
として用いる標準物質及びSIへのトレーサビリティが保証された分銅によって校正された天びんを用い
て試料調製を行い,適切な条件でNMR測定及びデータ解析を行うことが要件であり,分析値に不確かさ
を付与することが求められる。影響のある全ての不確かさを評価し分析値に付与するのが困難な場合でも,
定量に用いたシグナル間での測定値のばらつき(複数のシグナルから測定値を得た場合)及び試料調製間
での測定値のばらつきを求めておき,測定の繰返し性と比較するなどによって異常値がないことを確認し
ておくことは省略しない。さらに,適切な試験所間比較への参加及び適切なCRMによる分析値の検証は,
分析値の信頼性の確保に向けた客観性の高い手段として望ましい。
なお,不確かさの求め方の例を附属書Eに示す。
10
個別規格で1H qNMRとして取り入れる際に記載すべき事項
1H qNMRによる分析を規定するに際しては,8.4及び9.3のうち必要な項目について記載する。
20
K 0138:2018
附属書A
(参考)
試料溶液の調製
A.1 一般
この附属書は,試料溶液の調製に際して,その一般的な手順,溶媒の選択,qNMR用基準物質の選定な
どの注意事項を示す。
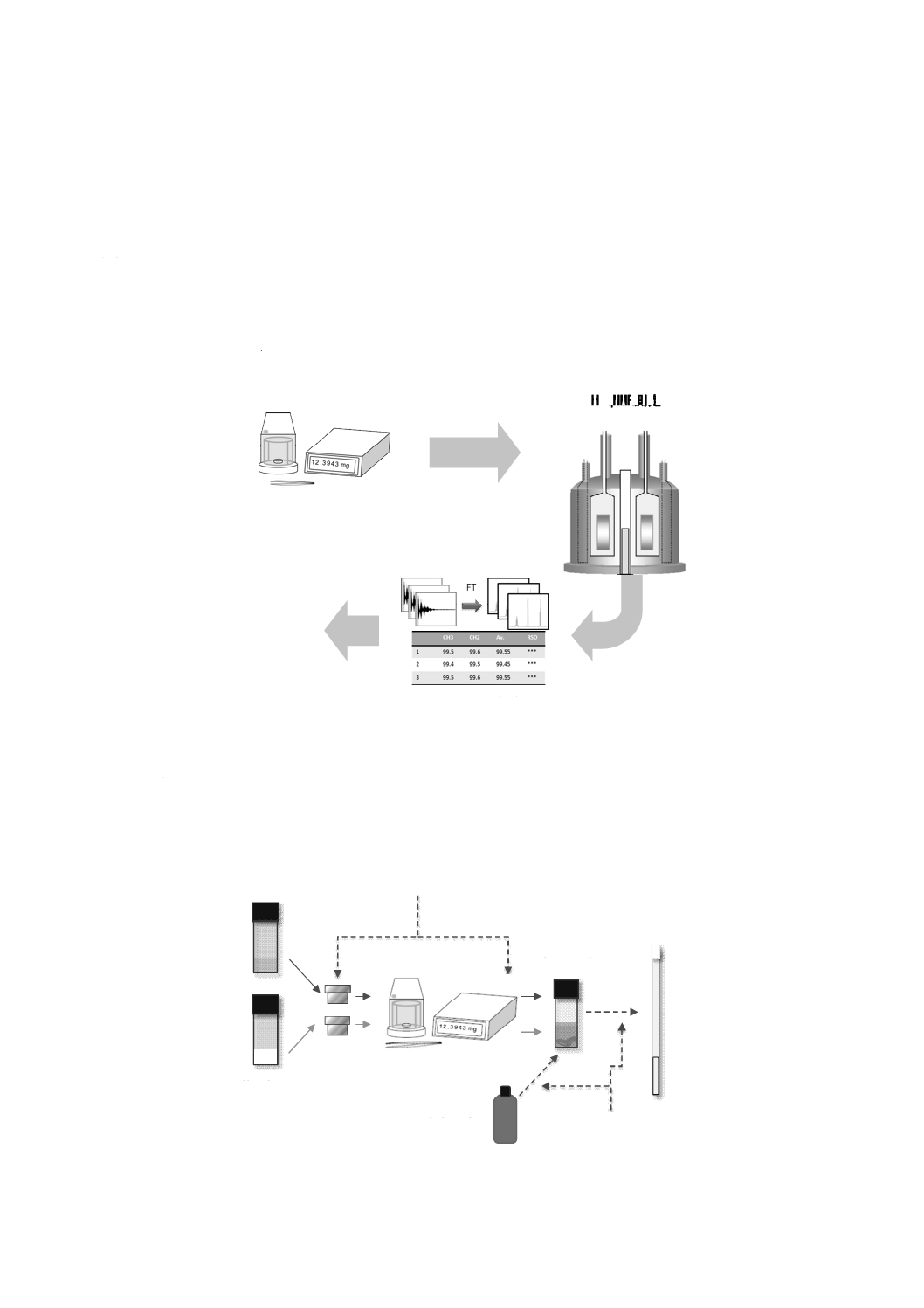
なお,試料調製,1H qNMR測定及びデータ解析の全体フロー図を,参考として図A.1に示す。
定量結果
試料調製
1H qNMR測定
データ解析
図A.1−試料調製,1H qNMR測定及びデータ解析の全体フロー図
A.2 一般的な手順及び注意事項
試料調製に関する一般的な手順は,次による。
なお,その手順の詳細なフローを図A.2に示す。
天びん
基準物質
重水素化溶媒
試料
採取用容器
バイアル瓶
NMR試料管
持ち運びにはピンセット
などを使用する。
パスツールピペットなど
を使用する。
図A.2−試料調製の手順のフロー図
21
K 0138:2018
a) 器具 試料溶液の調製に使用する一般的な器具は,次による。
1) 採取用容器 試料又はqNMR用基準物質のいずれとも反応しない質量が安定した材質で,はかりと
る質量に合わせたものを用いる。
注記1 天びんでの計量に適した帯電しにくいアルミニウム製などの容器を用いることが望まし
い。
2) 採取器 試料又はqNMR用基準物質のいずれとも反応しない材質の薬さじ,パスツールピペットな
どを用いる。
注記2 薬さじには,ミクロスパーテルなどがある。
3) ピンセットなど 採取用容器の持ち運びに使用することができるもの。
なお,はかりとる採取用容器の形状・種類に合わせたものを選択してもよい。
4) パスツールピペットなど 液体を移すことができるもの,又はそれと同等なもの。
5) バイアル瓶 加える重水素化溶媒に合わせた容量のもの。
b) 試料及びqNMR用基準物質の準備 準備は,次による。
1) 試料及びqNMR用基準物質は,指定された保管条件に従って保管する。
2) 試料及びqNMR用基準物質の保管条件によって,次のいずれかの準備を行い,試料溶液の調製を開
始する。
2.1) 保管条件が室温の場合,試料又はqNMR用基準物質を調製環境に1時間以上放置する。
2.2) 保管条件が冷蔵又は冷凍の場合,試料又はqNMR用基準物質を保管庫から取り出した後,直ちに
シリカゲルなどを乾燥剤としたデシケーターに入れ,室温に戻した後,調製環境に1時間以上放
置する。
c) 試料溶液の調製 6.7 e) のひょう量の手順として,試料又はqNMR用基準物質を質量差(試料又は
qNMR用基準物質を含めた採取用容器の質量をはかりとった後,採取用容器の質量を減算した質量)
としてはかりとる場合の調製は,次による。
1) 採取器を用い,採取用容器に試料をはかりとり,バイアル瓶に入れて密栓する。採取用容器の移動
(持ち運び)にはピンセットなどを使用する。
2) 採取器を用い,採取用容器にqNMR用基準物質をはかりとり,1) のバイアル瓶に入れて密栓する。
採取用容器の移動(持ち運び)にはピンセットなどを使用する。
3) パスツールピペットなどを用いて,2) のバイアル瓶に適量の重水素化溶媒を加えて密栓し溶解させ
る。
4) パスツールピペットなどを用いて,3) の試料溶液をNMR試料管へ移して密栓する。
5) 4) で調製した試料溶液を入れたNMR試料管をNMR装置に導入し,速やかにNMR測定する。
なお,個別規格に試料又はqNMR用基準物質の取扱方法及び調製方法が規定されている場合は,
それに従う。
A.3 吸湿性・揮発性(昇華性)の高い化合物を測定する場合
吸湿性及び/又は昇華性(揮発性)をもつ試料を精密にひょう量するためには,作業環境の準備及び調
製操作を考慮する必要がある。
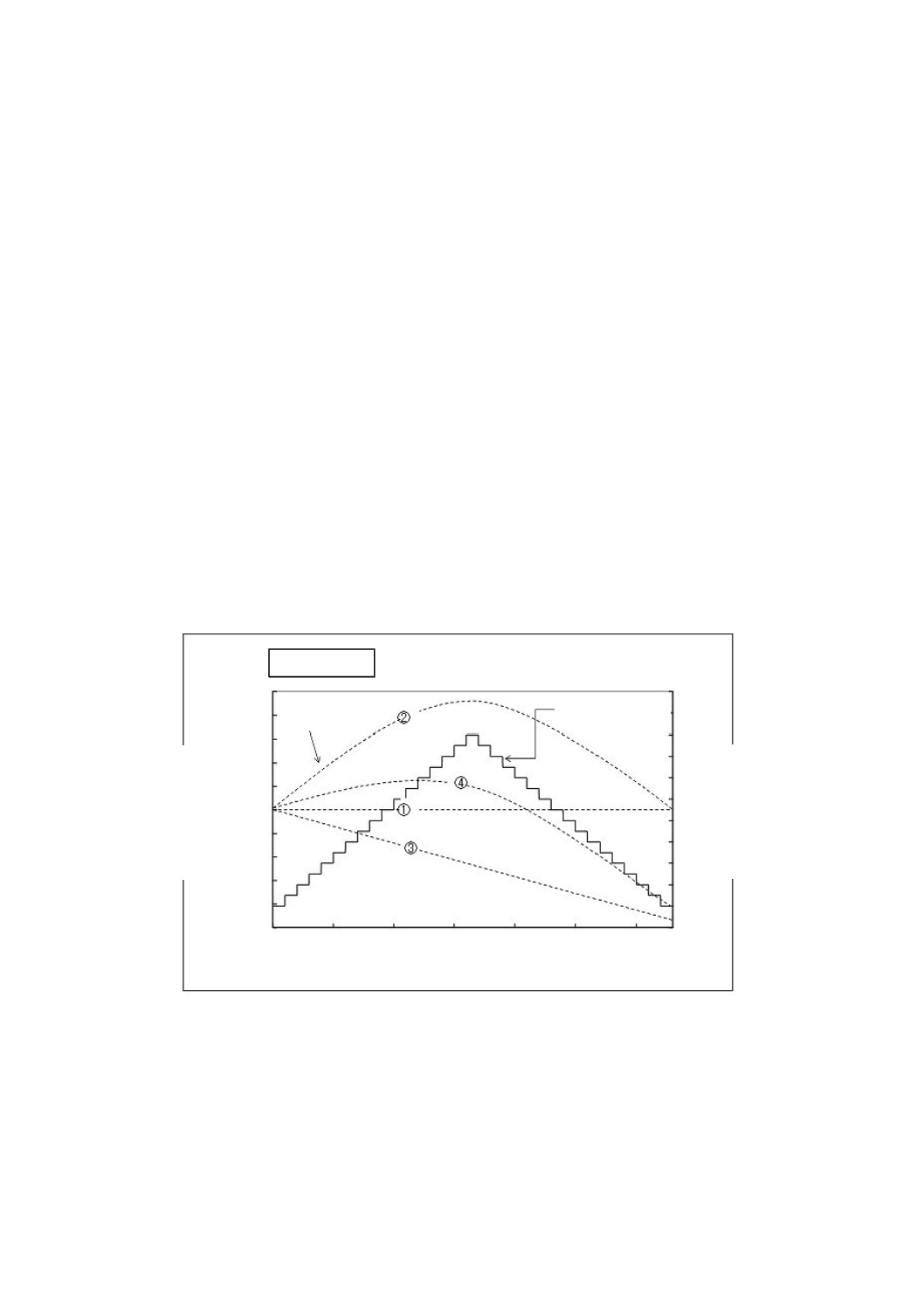
吸湿性及び/又は昇華性(揮発性)が質量変動にどのような影響を与えるのか,参考として蒸気吸脱着
分析チャートの模式図(図A.3)を用いて説明する。蒸気吸脱着分析は,温度を一定にした条件で湿度を
変化させたときの分析試料の質量の変化を測定する分析法である(参考文献[10]参照)。
22
K 0138:2018
蒸気吸脱着分析では相対湿度を経時的に階段状に上昇させ,また,最初の相対湿度まで戻す際の試料の
質量変化を計測する(図A.3参照)。左軸は分析試料の質量分率,右軸は相対湿度,横軸は時間を示してい
る。点線①〜点線④は階段状の実線のように相対湿度を時間ごとに変化させたときの試料の質量の変化
(%)を表している。
①のように相対湿度を変化させても質量変動がないものは,吸湿性なし及び昇華性(揮発性)なしと判
断できる。このような性質をもつ試料の場合,常温常湿の環境において精密なひょう量が可能である。
一方,②,③又は④のように時間経過とともに質量変動が観察される試料は,常温常湿の環境において
精密なひょう量が困難である。
②は,相対湿度の変化に応じて質量変動があるが質量減少はなく,吸湿性あり及び昇華性(揮発性)な
しと判断される。温度一定で相対湿度の上昇に合わせて質量が増加していることから,このような試料は
ひょう量を行う周囲の相対湿度状態を注視することが望ましい。
③は,相対湿度の変化に応じた質量変動はないが質量減少があることから,吸湿性なし及び昇華性(揮
発性)ありと判断され,採取用容器に蓋をするなどひょう量作業に配慮することが望ましい。
④は,相対湿度の変化に応じた質量変動だけでなく質量減少があり,吸湿性あり及び昇華性(揮発性)
ありと判断され,このような試料の場合,周囲環境状態を注視し,ひょう量作業についても配慮すること
が望ましい。
図A.3には示されていないが,吸湿性はあるが,吸湿後,水和物として完全に安定する試料の場合,一
定の湿度環境下で吸湿させ,質量が安定した後ひょう量することが望ましい。
① 吸湿性なし・昇華性(揮発性)なし
② 吸湿性あり・昇華性(揮発性)なし
③ 吸湿性なし・昇華性(揮発性)あり
④ 吸湿性あり・昇華性(揮発性)あり
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図A.3−蒸気吸脱着分析チャートの模式図a)
25 ℃一定温度
相対湿度の変化(%)
質量の変化(%)
質
量
分
率
(
%
)
時間(分)
110
108
106
104
102
100
98
96
94
92
90
0
50
100
150
200
250
300
100
90
80
70
60
50
40
30
20
10
0
相
対
湿
度
(
%
)
23
K 0138:2018
A.4 試料及びqNMR用基準物質に対する溶媒
溶媒の選定及び確認事項は,次による。
a) 溶媒の選定 1H qNMRでは,試料及びqNMR用基準物質を溶媒に溶かした試料溶液について測定す
るため,試料及びqNMR用基準物質の溶媒への溶解性だけでなく溶液中の安定性,すなわち,分解,
異性化などによる経時的変化の有無を考慮して溶媒を選択しなければならない。このため,調製した
試料溶液は速やかにNMR測定を行うとともに繰返し測定を行い,分解,異性化などの経時的変化が
生じていないことを確認する。経時的変化が試料溶液に確認された場合,試料溶液の調製に用いる溶
媒を替えるか,用時調製によって直ちに測定を行うなどの調製から測定までの操作上の検討を行う必
要がある。
注記 溶媒にクロロホルム-dを用いる場合は,保管中に発生した塩化水素,ラジカルなどの影響で
試料及びqNMR用基準物質が分解することがあるので,なるべく新しいものを用いる。この
場合,試料溶液の調製後,可能な限り速やかに測定する。また,試料及びqNMR用基準物質
と反応していないことを確認する。
b) 溶解性の確認 試料及びqNMR用基準物質が溶媒に完全に溶解していない場合は,精確な定量結果を
求めることができない。試料及びqNMR用基準物質は,物質ごとに溶媒の種類,温度,pHなど様々
な条件で溶解性が異なるため,溶解性に関する情報が得られていればそれらを参考にする。
試料及びqNMR用基準物質が溶媒に完全に溶解したかどうかの確認は,通常目視で行う。目視によ
る確認をより慎重に行うためには,例えば,透明なガラス容器を用いて試料溶液を調製し,蛍光灯を
照射して確認する方法がある。このとき試料溶液が僅かに濁っている,又は試料溶液中に浮遊物が観
察される場合は,定量結果に影響を与える場合があるので,細心の注意を払って確認する。試料及び
qNMR用基準物質が溶解したかどうかの判断が困難な場合は,別の溶媒を検討することが望ましい。
c) 溶媒中の不純物の確認 溶媒中の不純物は,試料溶液と同容量の溶媒だけをNMR試料管に入れ,試
料溶液と同じNMR条件で測定して確認する。溶媒中の不純物のシグナルが,分析種及びqNMR用基
準物質のシグナルと重複する場合,そのシグナルから求めた定量値には偏りが生じるため,そのシグ
ナルの棄却又は他の溶媒の利用を検討することが望ましい。
また,溶媒中の不純物とは別にNMR試料管由来のシグナル及び/又はプローブ中の水分のシグナ
ルが観測される場合があるので注意する。前者の場合,高純度石英ガラス製NMR試料管の使用で問
題を解決することができ,後者の場合,メンブレンフィルタなどを使用することで水のシグナルを除
去することができる。
d) qNMR用基準物質中の不純物の確認 qNMR用基準物質中の不純物は,qNMR用基準物質だけをこの
試験と同容量の溶媒に溶解させNMR試料管へ採取し,試料溶液と同じNMR条件で測定して確認す
る。
qNMR用基準物質の種類によっては化合物特有のシグナルが観測される場合がある。例えば,日本
薬局方などの公定法において,qNMR用基準物質として採用されている1,4-ビス(トリメチルシリル)
ベンゼン-d4(1,4-BTMSB-d4)は,重溶媒の種類によっても異なるが0 ppm付近に単一線の定量の基準
となるシグナルを与えるが,このシグナルの両肩部分には,けい素(29Si)とのスピン結合を示す分裂
シグナルが観測される。これは,トリメチルシリル基由来のシグナルであり,不純物由来のシグナル
ではないので定量性に影響を与えることはない。
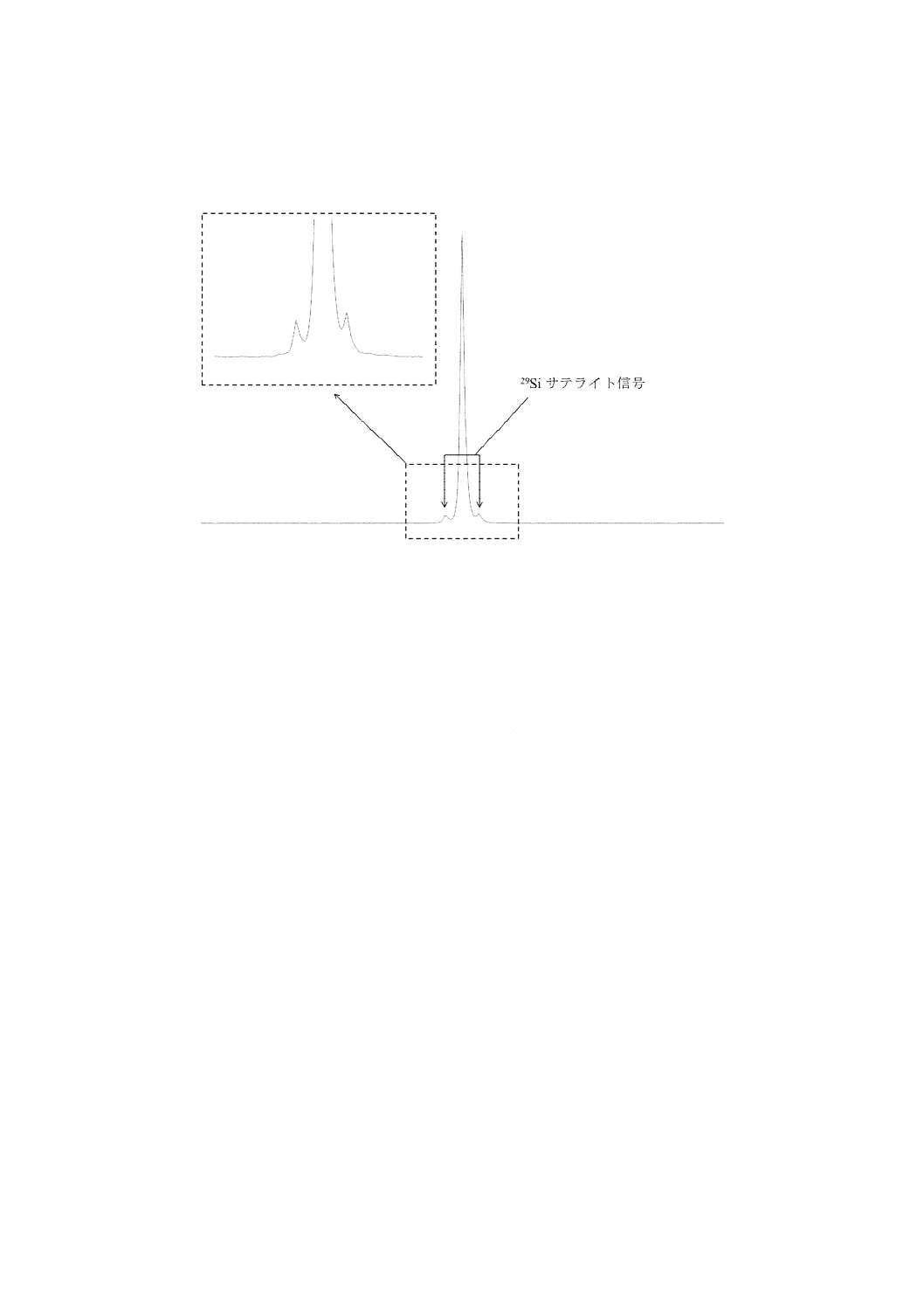
なお,29Siサテライトシグナルの面積は,29Siの天然存在比が4.67 %であることから,主シグナル
に対して4.67 %の割合で観測される(図A.4参照)。
24
K 0138:2018
注記 1,4-BTMSB-d4のシグナルの両肩部分のシグナル(図A.4参照)は,1,4-BTMSB-d4由来のシ
グナルのため,これらのシグナルを含めてシグナル面積とすることができる。
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図A.4−1,4-BTMSB-d4の分裂シグナルa)
一方,1,4-BTMSB-d4はベンゼン環の四つの水素が重水素化されているものであるが,完全な重水素
化が困難であることから,重溶媒の種類によっても異なるが,1,4-BTMSB-d4のトリメチルシリル基
(TMS基)を0.00 ppmとしたときに7.3 ppm付近(アセトン-d6:7.27 ppm付近,メタノール-d4:7.24
ppm付近又はクロロホルム-d:7.26 ppm付近)にベンゼン環の四つの水素に由来する微少なシグナル
が観測される。このため,特にベンゼン環をもつ分析種を定量する場合は,この微小なシグナルが重
複していないことを慎重に確認する必要がある。また,安定剤不含のクロロホルム-d中では,
1,4-BTMSB-d4は,ラジカル反応によって経時的に分解することが確認されている。よって,クロロホ
ルム-dを試料調製の重溶媒に用いる場合には,試料溶液を調製後,直ちにNMR測定を行う必要があ
る。
日本薬局方において同様にqNMR用基準物質として採用されている3-(トリメチルシリル)-1-プロ
パンスルホン酸-d6-ナトリウム塩(DSS-d6)の使用に際しても,次の点に注意するのがよい。この化合
物は,試験室環境(温度15 ℃〜25 ℃,相対湿度20 %〜80 %)で一水和物として完全に安定化する。
したがって,DSS-d6を使用する場合,試験室環境で開封後30分以上放置した後使用しなければなら
ない。また,一水和物として安定したときのDSS-d6の水分含有量は7.4 %程度であることから,DSS-d6
をqNMR用基準物質として用いた試料溶液の1H qNMRスペクトル上に水のシグナルが観察されるた
め,分析種のシグナルとの分離を考慮する必要がある。
A.5 qNMR用基準物質の選定
8.2から,分析種のシグナルiから求められる純度又は含有率(Pi)は,式(A.1)を用いて算出する。
25
K 0138:2018
s
a
s
s
i
i
s
s
i
i
P
m
m
M
M
N
N
S
S
P
×
×
×
×
=
························································ (A.1)
ここに,
Pi: 分析種のシグナルiから求められる純度又は含有率
Ps: qNMR用基準物質の純度
Si: 分析種のシグナルiの面積
Ss: qNMR用基準物質のシグナル面積
Ni: 分析種のシグナルiのプロトン数
Ns: qNMR用基準物質のプロトン数
Mi: 分析種のモル質量
Ms: qNMR用基準物質のモル質量
ma: 試料の質量
ms: qNMR用基準物質の質量
分析種又はqNMR用基準物質のプロトン数(N)及びモル質量(M)は,それぞれの化合物に固有な値
であり,定量結果に偏りを与える要因とはならない。一方,1H qNMRスペクトルから得られるシグナル面
積(S),はかりとった質量(m)及び使用したqNMR用基準物質の純度(Ps)は,化合物又は分析種の固
有値ではなく変数であることから,定量結果に偏りを与える要因となる。すなわち,シグナル面積(Ss),
質量(ms)及びqNMR用基準物質の純度(Ps)は,全てqNMR用基準物質の品質と密接に関係する変数で
あり,qNMR用基準物質には主に次の六つの要件を満たすものを選定することが望ましい。
次のa)〜f) に,精確な定量結果を得るための観点からqNMR用基準物質に一般的に求められる要件に
ついて解説したが,あらゆる試料の定量分析に適用可能なqNMR用基準物質は存在しない。したがって,
供給されている複数のqNMR用基準物質の特性を十分理解し,試料の溶解及び測定に用いる重溶媒の性質
を考慮して,最適なqNMR用基準物質を選択する必要がある。さらに,精確な定量結果を得るためには,
SIトレーサビリティが確保された信頼性の高いqNMR用基準物質を用いることが望ましい。表A.1には
qNMR用基準物質として有用な化合物の性状,可溶溶媒,化学シフトなどを示した。
a) 高純度なもの qNMR用基準物質はできるだけ高純度であることが望ましい。純度が低く不純物が多
く含まれる場合,分析種のシグナルと重複し正確なシグナル面積(S)を測定することができない。
qNMR用基準物質中の不純物と分析種のシグナルが重複した場合,他の変数であるはかりとった質量
(m)がいかに精確であったとしても,定量結果に偏りを与え,最終的に得られる定量結果は精確さ
をもたない。
b) 安定して精確にひょう量できるもの qNMR用基準物質は吸湿性及び/又は昇華性(揮発性)がなく,
質量を安定してひょう量できるものが望ましい。例えば,トルエン,ベンゼンなどの揮発性有機化合
物及び化学シフト用基準物質に使用されるテトラメチルシラン[(CH3)4Si](TMS)は,はかりとり操
作中に揮発するので,はかりとった精密な質量(m)を求めることは困難である。さらに,吸湿性及
び/又は昇華性(揮発性)が非常に高い化合物については,瓶の蓋を開閉するたびに純度が変化する
可能性があることから,純度(Ps)の信頼性が確保できない。このような観点からもqNMR用基準物
質として望ましくない。また,吸湿性が高い化合物もはかりとった精密な質量(m)を求めることは
困難である。ただし,後者に関しては,吸湿によって水和物として質量が安定する化合物であれば問
題ない。
c) シグナルの数とスピン結合によるシグナルの分裂が少ないもの 一種類の単一線(singlet)のシグナ
ルを与えるものがqNMR用基準物質として望ましい。複数のシグナルをもつ場合,基準物質と分析種
のシグナル(又は両者に含まれる不純物のシグナル)が定量に用いるシグナルに重複してしまい,精
確なシグナル面積(S)を測定することが困難になる場合があるためである。スピン結合によるシグナ
26
K 0138:2018
ルの分裂,すなわち,多重線(multiplet)を与えるものは,シグナル範囲が多重度に比例して広くな
り,SN比が多重度に反比例して悪くなり,シグナル面積(S)を精確に測定することが困難となるた
め,qNMR用基準物質には適さない。
d) 特異的な領域に化学シフトを与えるもの 特異的な領域とは,一般的な有機化合物のシグナルが観測
されない範囲をいう。化学シフトの基準物質として利用されるTMSでは,構造中のけい素の磁気遮
蔽効果によってメチル基の1Hシグナルが高磁場側へシフトして0 ppm付近に観察される。TMSのよ
うに構造中にけい素が含まれる有機化合物を除き,0 ppm付近にはシグナルを与えない。このような
通常シグナルが観察されない0 ppm付近の特異的な領域にシグナルをもつ化合物をqNMR用基準物質
とした場合,試料,重溶媒又は不純物のシグナルが重複するという問題はほとんど生じない。ただし,
0 ppm付近に試料又は不純物のシグナルが観測される場合は,この限りでない。このような場合には,
試料又は不純物のシグナルが観測されない他の領域,例えば低磁場側にシグナルを与える化合物を
qNMR用基準物質として選択することが必要となる。
e) 重溶媒中で安定かつ試料と反応しないもの qNMR用基準物質には,重溶媒中において安定で分解し
ない,試料と反応しないものを選択することが重要である。qNMR用基準物質が分解又は反応する場
合,変数であるqNMR用基準物質のシグナル面積(Ss)が精確に測定できないため,定量結果の信頼
性を確保することは困難となる。
f)
重溶媒への溶解性が良好であるもの qNMR用基準物質が重溶媒に完全に溶解していない場合,
qNMR用基準物質のシグナル面積(Ss)が過小評価され,分析種の純度又は含有率が高く見積もられ
る。また,重溶媒に対する溶解性がよくないとNMR測定の安定性が損なわれ,分解能が低下する場
合がある。qNMR用基準物質の種類によって,重溶媒への溶解性が異なるので,分析者は適宜使い分
けることが求められる。この問題は試料に関しても同様である。試料が重溶媒に完全に溶解していな
い場合は,分析種のシグナル面積(Si)が過小評価され,結果として試料の純度又は含有率が低く見
積もられるので,qNMR用基準物質及び試料の重溶媒への溶解性を確認した上でqNMR用基準物質を
選択する。

27
K 0138:2018
表A.1−主なqNMR用基準物質一覧表
基準物質名
性状
可溶溶媒
化学シフトa)
(ppm)
注記
1,4-ビス(トリメチルシリル)ベンゼン-d4
(1,4-BTMSB-d4)
固体
有機溶媒系
0
CRM又は
RM b)
デュロキノン
固体
有機溶媒系
2
CRM
安息香酸ベンジルエステル
固体
有機溶媒系
5〜6
CRM
安息香酸
固体
有機溶媒系
7〜9
CRM c)
1,2,4,5-テトラクロロ-3-ニトロベンゼン
固体
有機溶媒系
7〜9
CRM
3,5-ビス(トリフルオロメチル)安息香酸
固体
有機溶媒系
8〜9
CRM
チモール
固体
有機溶媒系
1〜2,2〜3,3〜4,6〜7
CRM
エチル-4-ジメチルアミノベンゼン
固体
有機溶媒系
1〜2,3,4〜5,6〜7,7〜8
CRM
1,2,4,5-テトラメチルベンゼン
固体
有機溶媒系
2〜3,6〜7
CRM
テレフタル酸ジメチル
固体
有機溶媒系
4,8
CRM
メチル-3,5-ジニトロベンゼン
固体
有機溶媒系
4〜5,9
CRM
ジメチルスルホン
固体
水系及び
有機溶媒系
3
CRM又は
RM b)
ジメチルマロン酸
固体
水系及び
有機溶媒系
1〜2
CRM
3-(トリメチルシリル)-1-プロパン-
1,1,2,2,3,3-d6-スルホン酸ナトリウム
(DSS-d6)
固体
水系
0
CRM
マレイン酸
固体
水系
6〜7
CRM
フタル酸水素カリウム
固体
水系
7〜9
CRM c)
ぎ酸カルシウム
固体
水系
7〜8
CRM
3-(トリメチルシリル)-1-プロパン-
1,1,2,2,3,3-d6-スルホン酸ナトリウム
・重水溶液(DSS-d6・重水溶液)d)
溶液
−
0
RM
ヘキサメチルジシラン
・メタノール-d4溶液d)
溶液
−
0
RM
注a) 大まかな数値を示したが試料濃度などの測定条件の違いによって変わる場合がある。
b) RMは,標準物質の略号で,特性値(濃度)が十分に均一で,適切に値付けされているもの。
c) 用途の異なるCRMとして供給されているが,qNMR用基準物質としても使用可能。
d) 一定量のqNMR用基準物質を含むqNMR用標準液として供給されている。
qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]を参照。

28
K 0138:2018
附属書B
(参考)
1H qNMRスペクトルの測定
B.1
一般
1H qNMR測定条件の最適化は,使用するNMR装置の性能,測定対象分析種の物理的な性質に依存する。
1H qNMR測定によって精確な定量値を得るために,通則の1H qNMR測定条件には各種パラメータについ
て具体的な推奨値を示している。この附属書では,特に注意するのがよい事項を記載する。
なお,NMR測定のパラメータであるパルス幅(フリップ角),遅延時間(繰返しパルス待ち時間),縦
緩和時間(T1)などの用語の具体的な説明は書籍,参考文献などを参考にすることが望ましい。
B.2
装置
1H qNMRでは,分析種とqNMR用基準物質のシグナル面積比から定量値が算出される。すなわち,分
析種とqNMR用基準物質の各シグナルが同一スペクトル上で完全に分離して観測されるとき,1H共鳴周
波数の異なるNMR装置においても,理論上,同一の定量値を求めることができる。ただし,分析種及び
qNMR用基準物質に由来する各プロトンのシグナル面積を精確に測定でき,かつ,共存する不純物に由来
するシグナルを分離して観測できるとき,1H qNMRによって精確な定量値が得られるため,可能な限り1H
共鳴周波数の大きい高分解能のNMR装置を用いることが望ましい。
B.3
測定条件
1H qNMRの測定条件の設定のための,主なパラメータは,次のとおりとする。
a) パルス角と遅延時間 1H qNMR測定において,精確な定量値を得るためには,測定対象分析種及び
qNMR用基準物質を含む試料溶液にパルスを加えた後,定量に用いる全てのプロトンシグナルの磁化
を99.9 %以上まで回復させる必要がある。これは,異なるシグナルの面積比を正確に算出するための
必須条件である。
パルス角90°及び遅延時間(繰返しパルス待ち時間)60秒以上(縦緩和時間T1の7倍以上)を推
奨値とした理由は,次のとおりである。
− 核スピンはパルスによって励起され,その後,熱平衡状態に次第に回復していく。この過程を
緩和といい,その時定数を緩和時間という。
− 縦緩和(スピン−格子緩和)と呼ばれる過程の緩和時間が定量精度に最も関係する。
− 外部磁場方向をzとしたとき,核スピンがもつ磁化のz成分(磁化Mz)は,熱平衡状態のz磁
化,すなわち,初期磁化Mz0に向かって回復していく。
このときの時定数を縦緩和時間T1といい,磁化Mzは,式(B.1)で表される。
]}
1[
)
cos
1(
{cos
)
/
(
z0
z
1T
RD
e
M
M
−
−
×
−
+
×
=
θ
θ
···································· (B.1)
ここに,
Mz: 核スピンがもつ磁化のz成分
Mz0: 初期磁化
RD: 遅延時間(繰返しパルス待ち時間)
T1: 縦緩和時間
29
K 0138:2018
θ: パルス角
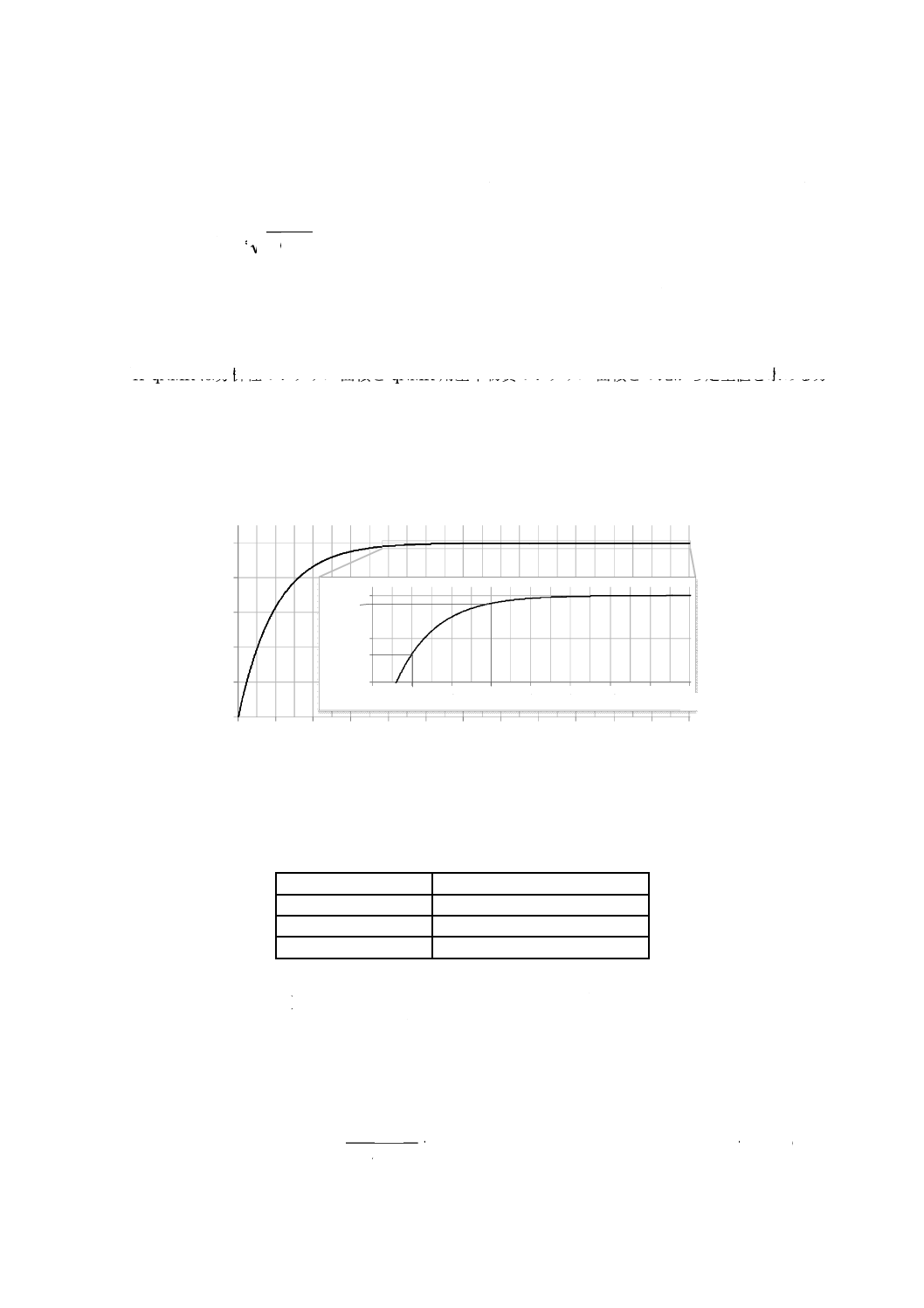
式(B.1)から,磁化回復率(%)Mz/Mz0×100とRD/T1との関係を,図B.1に示す。すなわち,磁化を
99.9 %回復させるためには,パルス角θ=90°の場合はRD/T1=7以上を,パルス角θ=45°の場合は
RD/T1=5.7以上を必要とする。さらに,99.9 %の磁化回復率を前提としたとき,信号の単位時間当た
りのSNは
(
)
RD
T1
sin
θ
に比例するため,パルス角θ=90°と45°との強度比率は0.378:0.296 2
=1:0.783 6となる。したがって,99.9 %の磁化の回復を前提としたとき,パルス角θ=90°で測定す
るほうが信号の単位時間当たりのSN比がよく,測定時間の短縮においても有利である。
さらに,磁化回復率から見積もられる遅延時間(パルス角90°の場合)とシグナル面積の偏りとの
関係を表B.1に示したが,遅延時間がT1の7倍以上のとき,シグナル面積の偏りは0.1 %未満となる。
1H qNMRは分析種のシグナル面積とqNMR用基準物質のシグナル面積との比から定量値を求める方
法であることから,このシグナル面積の偏りを加味すると,有効数字2桁以上の定量精度を目標とす
るとき,遅延時間にはT1の7倍以上を設定する必要がある。通常の化合物のT1が長いものでも5秒
〜7秒であり,その7倍を十分に確保している遅延時間(繰返しパルス待ち時間)60秒以上を奨励値
としている。
0%
20%
40%
60%
80%
100%
0
1
2
3
4
5
6
7
8
9
10
11
12
99.0%
99.5%
100.0%
4
5
6
7
8
9
10
11
12
99.33%
99.91%
磁
化
回
復
率
(%
)
RD/T1
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図B.1−磁化回復率(%)とRD/T1との関係a)
表B.1−遅延時間(パルス角90°の場合)とシグナル面積の偏りとの関係
遅延時間
シグナル面積の偏り
T1の5倍以上
0.7 %未満
T1の7倍以上
0.1 %未満
T1の10倍以上
0.005 %未満
b) パルス角と励起範囲 a) で示したとおり,99.9 %の磁化の回復を前提としたとき,パルス角90°のほ
うが単位時間当たりのSN比がよく,積算効率が高い。さらに,励起範囲においても,パルス角90°
に設定することが次の理由から適切である。
一般にパルス幅twのく(矩)形パルスによるラジオ波(RF)磁場強度のオフセット依存性は,式(B.2)
で与えられる。
off
w
off
w
1
off
1
π
)
π
sin(
v
t
v
t
v
v
×
=
································································ (B.2)
30
K 0138:2018
ここに,
off
1v: 磁場強度のオフセット依存性
v1: 照射中心でのRF強度
voff: 磁場強度のオフセット
tw: く(矩)形パルスのパルス幅
パルス角90°をt90とするとき,
2
π
π
2
90
1
=
t
v
であることから,v1は,
90
1
4
1
t
v=
となる。したがって,
パルス幅twのく(矩)形パルスによって,オフセット位置voffで励起されるシグナル強度Mは,式(B.3)
として表される。
=
=
90
off
w
off
w
off
1
2
)
π
sin(
sin
)
π
2
sin(
t
v
t
v
t
v
M
············································· (B.3)
ここに,
M: オフセット位置で励起されるシグナル強度
off
1v: 磁場強度のオフセット依存性
tw: く(矩)形パルスのパルス幅
voff: オフセット位置
t90: パルス角
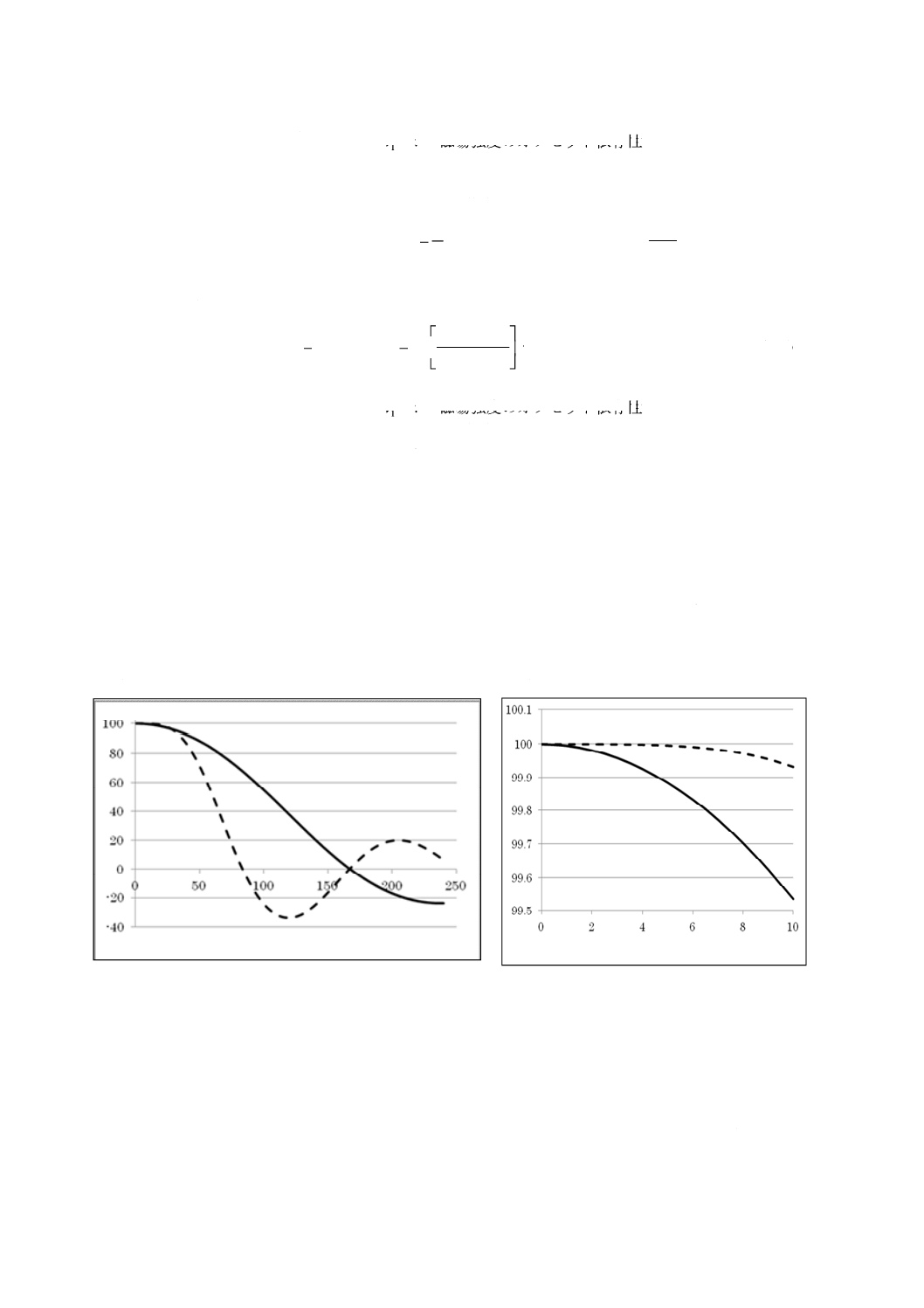
600 MHzのNMR装置によって,90°パルス(ここでは20 μsとする)と45°パルス(10 μs)での
励起範囲を図B.2に示す。ただし,縦軸は観測中心でのシグナル強度で規格化したシグナル強度(%)
で,横軸は600 MHzでの1Hの化学シフト値(ppm)とする。全体の励起範囲は(2/tw Hz)で定義さ
れるように,パルス幅の逆数に比例する。しかし,観測中心付近のシグナル強度99.9 %の領域では,
45°パルスのときと比べ,90°パルスのほうが,パルス幅が長いにもかかわらず励起範囲が広い[20 µs
(90°パルス)が99.9 %ラインでは広い(図B.2参照)]。
シグナル強度(%)
シグナル強度(%)
実線(−) 10 μs(45°パルス)
点線(- -) 20 μs(90°パルス)
実線(−) 10 μs(45°パルス)
点線(- -) 20 μs(90°パルス)
図B.2−600 MHzのNMR装置を用いた励起範囲
このように励起範囲を考慮すると,1H qNMR測定において90°パルスを用いることが適切である
といえる。表B.2には,90°パルス,20 μsにおける各磁場での99.9 %励起範囲を示した。このように,
20 μs以下の90°パルスのとき,現在,主に使用されている1H共鳴周波数300 MHzから1 GHzまで
の磁場において,1H qNMRが支障なく測定可能となる。
化学シフト(ppm)
信号強度(%)
化学シフト(ppm)
31
K 0138:2018
表B.2−各磁場における99.9 %の励起範囲(90°パルス,20 μs)
磁場(1H 共鳴周波数)(MHz)
励起範囲(ppm)
300
44
400
31
500
26
600
22
700
18
800
16
900
14
1 000
12
c) 遅延時間 a) 及びb) から,1H qNMR測定をする場合,パルス角90°,20 μs以下のパルス幅におい
て,分析種及びqNMR用基準物質の定量対象シグナルとなるプロトンの縦緩和時間T1の7倍以上の
遅延時間(繰返しパルス待ち時間)を設定したとき,精確な定量値を求めることが可能となる。
プロトンの縦緩和時間T1は,測定溶媒,測定磁場,測定温度,試料溶液の濃度などに依存して変化
し,また,同じ分子上のプロトンごとに異なる。すなわち,正確な定量性を確保して1H qNMRスペ
クトルを得るためには,試料溶液ごとに全てのプロトンのT1をあらかじめ測定する必要がある。しか
し,T1を求めるNMR測定は,1H NMRスペクトル測定の何倍もの長時間が必要であることから,試
料溶液ごとに全てのプロトンのT1測定を行うとするとあまりにも効率が悪い。このような非効率な測
定を避けるため,1H qNMR測定条件には遅延時間(繰返しパルス待ち時間)として,次の理由から
60秒以上とすることが望ましい。
分子量100〜1 000程度の低中分子の分析種の場合,これらのプロトンのT1は例外的な極端なもの
を除き,長くても7秒程度であり,分子量が大きくなればT1が短くなる傾向がある。よって,1H qNMR
測定条件には,定量精度に影響を及ぼさないように,パルス角90°のとき,遅延時間(繰返しパルス
待ち時間)を通常のプロトンのT1の7倍以上となる60秒を設定している。
なお,分析種及びqNMR用基準物質の定量対象シグナルとなる全てのプロトンの縦緩和時間T1に
ついて測定を行い,そのT1の7倍以上の遅延時間(繰返しパルス待ち時間)を設定できるとき,この
値を変更しても問題はない。また,個別規格に遅延時間(繰返しパルス待ち時間)が示されていると
き,定量精度に関係することから,その値に従うことが必要となる。
d) デジタル分解能,観測スペクトル幅及び取込み時間 試料溶液のNMRシグナルはアナログ信号とし
て検出され,アナログ−デジタル変換器(A/Dコンバータ)によってデジタル化された後,FID信号
がデジタル信号として取得される。FID信号のもつデータポイント数は,フーリエ変換(FT)後のス
ペクトルに反映されるが,このとき周波数で表されるデータ点の間隔をデジタル分解能DR(Hz)と
いい,式(B.4)で表される。
Ndata
Sw
DR=
·········································································· (B.4)
ここに,
DR: デジタル分解能(Hz)
Sw: 観測幅
Ndata: データポイント数
デジタル分解能が小さいとき,ポイント落ちが生じてしまいシグナルの形状が崩れ,正確なシグナ
ルが再現できず,正確なシグナル面積が求められない。したがって,1H qNMRによって正確な定量値
を得るためには,十分なデジタル分解能(Hz)でデータを取得する必要がある。さらに,1H qNMR
32
K 0138:2018
では正確な位相補正とベースライン補正が必要であることを考慮すると,観測スペクトル幅はシグナ
ル存在範囲の少なくとも2倍以上の範囲が望ましい。このため,デジタル分解能は0.25 Hz以下,観
測スペクトル幅は通常プロトンシグナルが観測される範囲より広い−5 ppm〜15 ppmを含む20 ppm以
上を推奨している。
e) レシーバゲインの設定 プローブの検出コイルで検出されたNMR信号は,受信器(レシーバ)でデ
ジタル信号として取り込まれるまでの時間の間に増幅される。このときの増幅の係数がレシーバゲイ
ンである。よって,レシーバゲインの値が小さすぎるとSN比が悪くなる。一方,レシーバゲインの
値を大きくすると増幅率は大きくなりSN比が良くなるが,FID信号がA/Dコンバータの入力電圧範
囲を超えたとき,正確に取り込むことができなくなり,ベースラインにひずみが生じ,1H qNMRスペ
クトルとして用いることができなくなる(図B.3参照)。
2016年時点のNMR装置では,レシーバゲインは自動調整機能(オートゲイン)で求めることがで
きる。したがって,自動調整機能(オートゲイン)を用い,定性的な1H NMRスペクトルをあらかじ
め測定し,レシーバゲインを確認後,1H qNMR測定に適したレシーバゲインを設定することが必要と
なる。
入力範囲
FIDのclipping
FT
FT
入力範囲
FIDのclipping
FT
FT
a)
レシーバゲインが適切
レシーバゲインが大きすぎるb)
注a) clippingしたFID信号をFT(フーリエ変換)するとベースラインにひずみが生じる。
b) 定量測定では定性測定よりも積算1回当たりの信号強度が大きくなることが考えられる。
そのときレシーバゲインが不適切であれば,NMRスペクトル上でベースラインにひずみが生じる。
c) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図B.3−レシーバゲインとNMRスペクトルとの関係c)
33
K 0138:2018
附属書C
(参考)
1H qNMRスペクトル解析
C.1 一般
この附属書は,1H qNMRスペクトル解析において,位相補正,ベースライン補正,積分範囲の設定など
のデータ処理に不適切な操作が含まれるとき,定量に用いるシグナル面積を精確に得ることができない。
このため,データ処理に関する基本的な注意事項を記載する。
C.2 窓関数(ウィンドウ関数)
窓関数(ウィンドウ関数)とは,得られたFID信号をフーリエ変換(FT)するとき,スペクトルのSN
比,線幅などを改善させるために掛け合わせる関数のことをいう。FID信号は時間とともに減衰していく
が,最初のポイントの大きさはシグナル面積に関係している。FID信号の前半部分には雑音が相対的に少
なく,後半部分には相対的に大きくなる。このため,FID信号の後半部分を多く含むか,又は後半部分を
強調するような窓関数を乗じるとスペクトルのSN比が悪くなる。一方,線幅の大きい(横緩和時間T2の
短い)シグナル及びスペクトルの低周波成分は,FIDの初期に含まれており,FID信号の後半部分を強調
する窓関数を乗じるとシグナルの線幅は狭くなり,分解能が向上する。このように,デジタルデータとし
て得られたFID信号は,フーリエ変換によってスペクトルを得るとき,窓関数を設定することによって解
析に適したスペクトルに都合良く加工することができる。一方,不適切な窓関数によってFID信号を加工
するとシグナル面積の精確さを損なうため,精確さを検証している場合を除いて,窓関数によるFID信号
の加工は行わないことが望ましい(図C.1参照)。
34
K 0138:2018
A:信号強度 B:周波数1周期分の時間 C:FID減衰の時間定数(1/T2)
A':シグナル面積 B':化学シフト C':信号の半値幅
注a) A,B及びCの値をもつFID信号をFT(フーリエ変換)すると,A',B'及びC' の周波数軸のシグ
ナル面積に変換される。このことは,時間軸のFID信号を加工することによって,周波数軸のシ
グナルとすることができる。(上図)
b) FID信号に指数型ウィンドウ関数を乗じると信号線幅は太くなるが,シグナル面積は変わらない。
しかし,sin型ウィンドウ関数を乗じると信号線幅は細くなるが,シグナル面積が変わる。このよ
うに,指数型ウィンドウ関数以外を乗じるとシグナル面積が変わるので,qNMRでは基本的にウ
ィンドウ関数を使用しない。もし使用するときは指数型ウィンドウ関数に限られる。(下図)
図C.1−FID信号の特性及び窓関数の効果
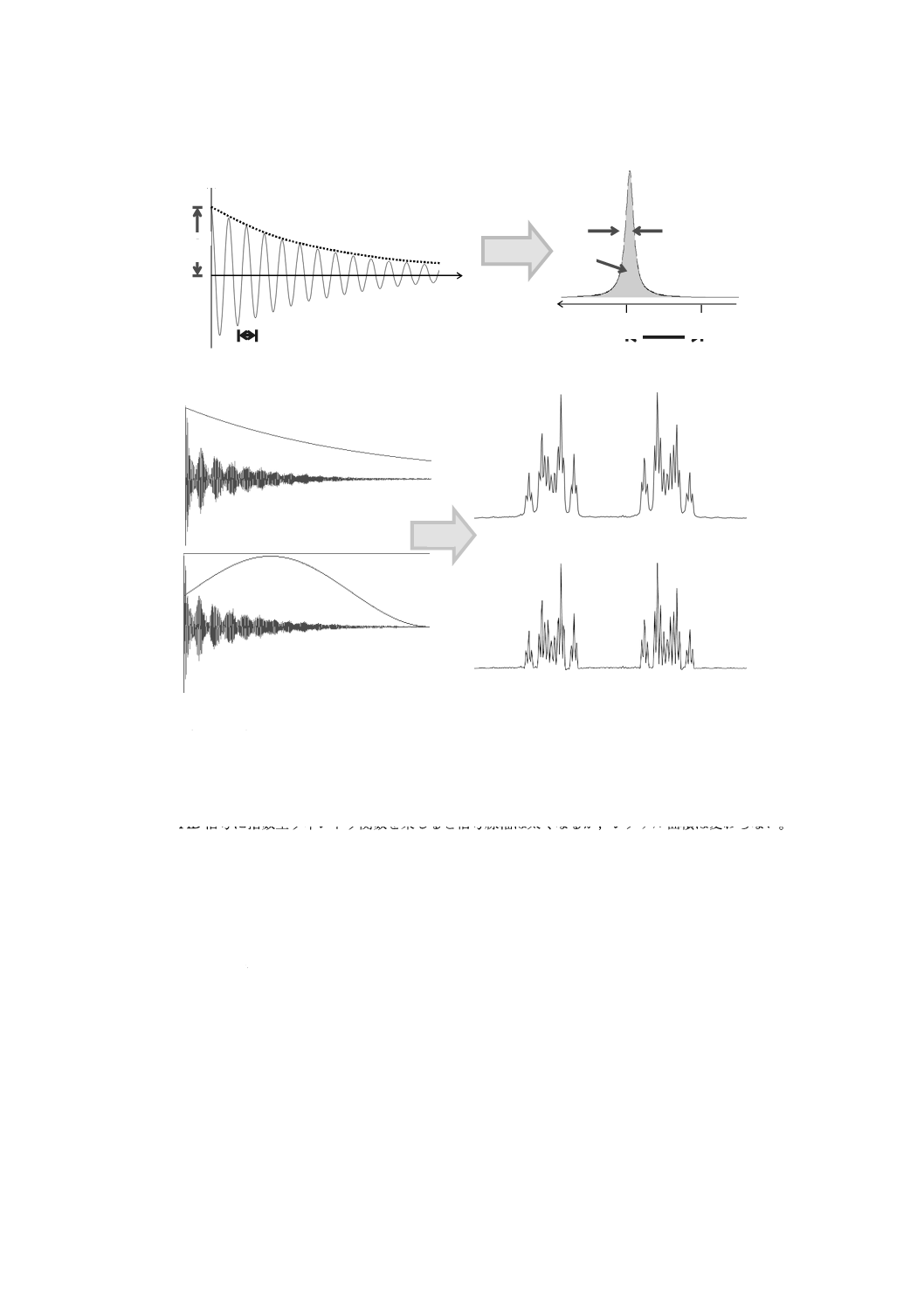
C.3 ゼロフィリング
ゼロフィリングとは,得られたFID信号の後ろに強度ゼロのデータを補完し,見掛け上のデジタル分解
能を向上させる方法である。また,ゼロフィリングで足したデータポイントは,フーリエ変換(FT)する
と,元のポイントの間に入る点となる(図C.2参照)。
FT
周波数
ω0
ω1
強度
時間
Aʼ
A
強度情報
B
Bʼ
周波数情報
Cʼ
C
減衰の速さ
指数型ウィンドウ関数
FT
sin型ウィンドウ関数
a)
a)
a)
b)
b)

35
K 0138:2018
図C.2−ゼロフィリングの効果
なお,挿入するゼロの個数をN個より増やして,ポイント数を元のデータポイントの4倍,8倍…とで
きるが,分解能は2倍より向上することはない。しかし,ゼロフィリング後に得られたデータポイント間
が三角関数で補完されるため,スペクトルが滑らかになる(図C.3参照)。
ゼロフィリングは,デジタル分解能を上げたいが,取込み時間を伸ばせない場合に通常利用する。ゼロ
フィリングによって,データポイントが補完され,スペクトルが滑らかになるため,結果的にシグナル面
積の精度が向上する。
a) ポイント落ちしたデータ
b) デジタル分解能2倍のデータ
c) デジタル分解能8倍のデータ
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図C.3−データポイント数が異なるシグナルから得られる面積a)
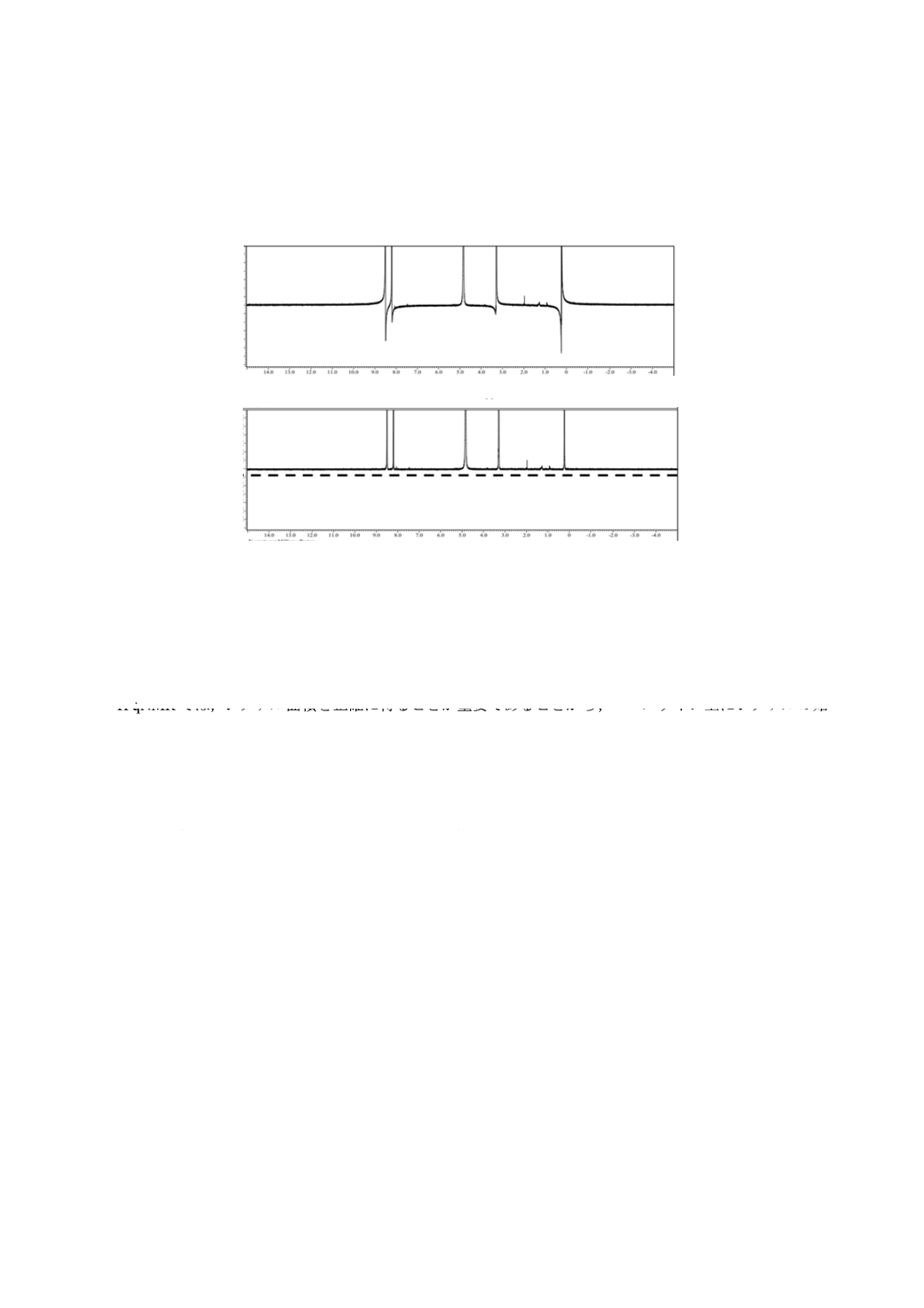
C.4 位相補正
NMRシグナルの位相は測定前に明らかでない。このため,測定後に解析に適した位相に合わせる必要
があり,この補正を位相補正という。2016年時点のNMR装置では,NMRシグナルの位相補正は自動的
に行われるが,シグナルの形状,測定範囲,ノイズなどの影響によって,微小な位相ずれが残ることが多
い。定性分析において,この位相ずれは問題になることはないが,1H qNMRでは,位相ずれはシグナル面
積のばらつきの原因となるため,手動操作で除く必要がある。この際,シグナルの位相だけではなく,ベ
ースラインをできるだけ平たんに合わせるように注意する必要がある。そこで,平たんなベースラインに
位相補正がなされていることを確認するために,定量に用いるシグナルを全て含む十分に広い観測スペク
トル範囲を設定して測定することが必要となる。
36
K 0138:2018
位相補正の具体例として,1H qNMRスペクトルを拡大し,各シグナルが全て上向きに,かつ,各シグナ
ルの両裾の傾きが同じになるように位相を合わせ,更に,ベースライン付近を拡大し,位相を合わせた段
階でベースラインが平たんであることを確認したものを,図C.4に示す。
化学シフト(ppm)
化学シフト(ppm)
上図 補正前
下図 補正後
図C.4−位相補正の例
C.5 積分範囲の設定
1H qNMRでは,シグナル面積を正確に得ることが重要であることから,ベースライン上にシグナルの始
点と終点を正しく設定する必要がある(図C.5参照)。NMR測定によって得られるシグナルはローレンツ
型であるため,厳密にはシグナルの裾の部分はかなりの幅に広がっており,理論的にはシグナルの半値幅
の64倍〜128倍を積分範囲として設定しなければ真に精確なシグナル面積を得ることができない。しかし,
実際には,このように広い範囲を設定した場合,測定対象とした分析種以外のシグナル,すなわち,試料,
溶媒などに含まれる不純物に由来するシグナルを加算し,かえって不確かな結果を与える可能性が高まる。
また,スピン結合によるシグナルの分裂,すなわち,多重線(multiplet)を与えるものは,シグナル範囲
が多重度に比例して広くなり,更に,SN比が多重度に反比例して悪くなり,シグナル面積(S)を精確に
測定することが困難となるため,このようなシグナルを可能な限り選択しないことが望ましい。したがっ
て,シグナル範囲は目的に応じてルールを設定したうえで一定の条件を適用し,かつ,シグナル範囲に不
純物のシグナルを含まないように設定する方が現実的である。
37
K 0138:2018
良い例
悪い例
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図C.5−シグナル面積の取り方の例a)
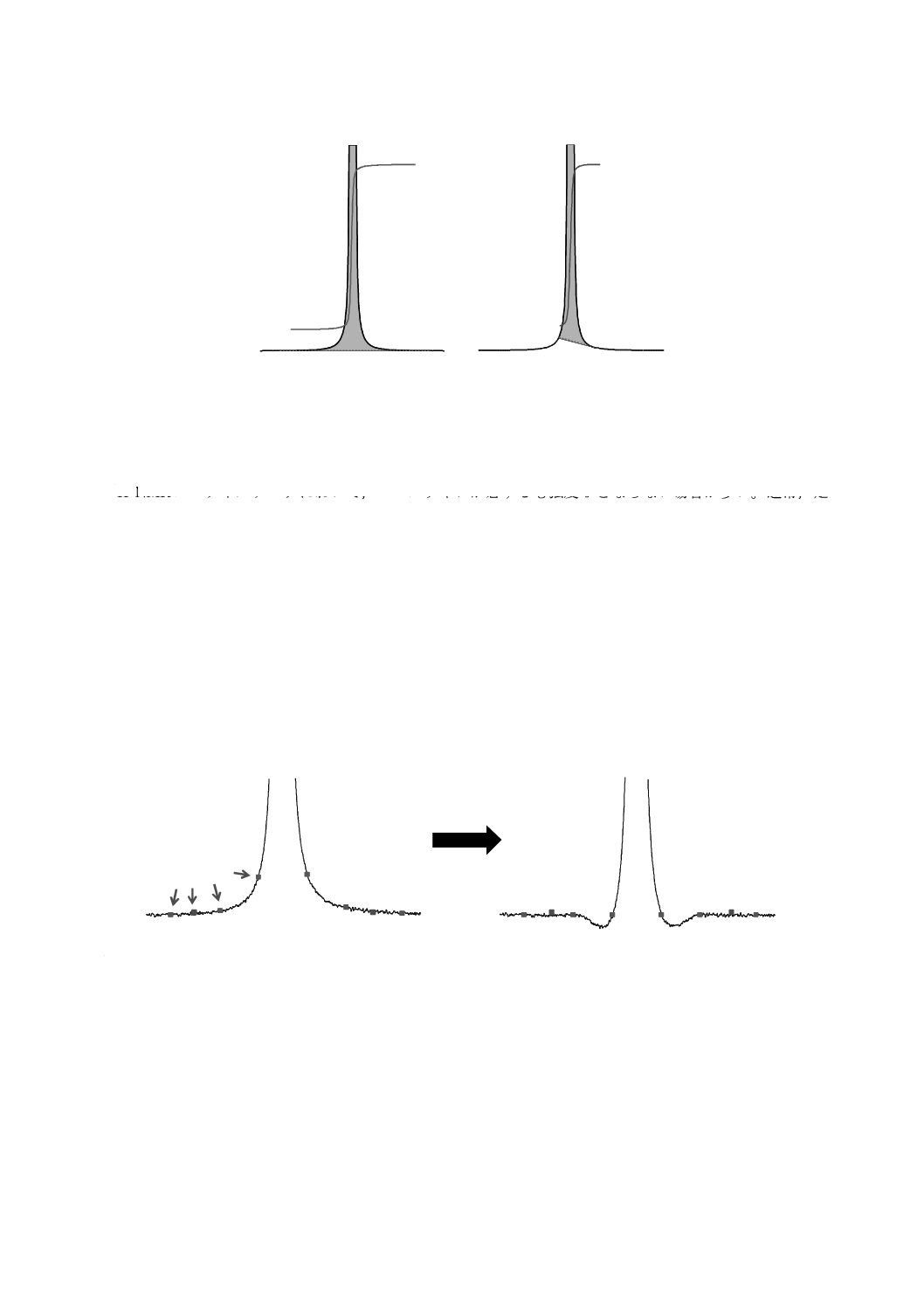
C.6 ベースライン補正
1H NMRスペクトルデータにおいて,ベースラインが必ずしも強度0とならない場合が多い。通常,定
性分析において,このようなさ(些)少なずれは問題ではないが,1H qNMRでは,シグナル面積の取得に
おいてばらつきの要因となる。これを避けるため,1H qNMRではベースライン補正を行う。
ベースライン補正には様々なアルゴリズムがあるが,通常は“ベースラインとみなされるデータポイン
ト”を補正点として利用してひずみを平たんにする処理を行う。補正点が真にベースラインであれば問題
ないが,シグナルが裾の一部でも補正点に使用されてしまえば,シグナルの形状が影響を受けて積分面積
が正しく評価できなくなってしまう(図C.6参照)。特に広幅なシグナルの場合には,シグナルの裾が補正
点となるシグナル形状にひずみが生じてしまうので,ひずみが生じないようにシグナルの外側が補正点と
なるように設定することが重要である。したがって,ベースラインのひずみは,シグナル面積に影響を及
ぼすので一定のルールを決めて慎重に行うことが望ましい。
補正点
ベースライン補正
注a) qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]参照
図C.6−不適切な補正点によるベースライン補正a)
38
K 0138:2018
附属書D
(参考)
試料のひょう量
D.1 一般
1H qNMR測定において,試料の質量のはかりとりは,通常,非常に微量(ミリグラム程度の量)である
と考えられる。ひょう量結果は分析値の精度に影響を与える要因の一つであることから,この附属書では,
ひょう量に関する注意事項について記載する。
D.2 天びんの取扱い
ひょう量に使用する天びんは,次の事項について留意し,精度,関連する外部規制の内容,その他生産
性などの目的に合致した仕様の機器を使用する。
a) 天びんの計量性能 計量性能として代表的な項目は,次による。
1) 最大ひょう量値 天びんでひょう量することが可能な最大許容質量。
2) 最小表示(目量又は表示分解能) 表示器がひょう量した結果として表示可能な最小桁(量)。
3) 繰返し性 同一試料を天びんの計量皿へ複数回のはかりとったひょう量値のまとまり度合で,高い
表示分解能を表示する天びんの性能評価に最も必須な特性である。性能は,標準偏差又は最大値と
最小値との差によって評価される。
4) 直線性誤差(非直線性) 零点から最大ひょう量値の範囲を分割した各荷重点(例えば,0 %,25 %,
50 %及び100 %付近)において,零点から最大ひょう量点までの理想直線からの偏りの程度を示す。
5) 偏置誤差 計量皿を真上方向から見た場合に,中心位置に荷重を加えた際の表示値に対して,中心
から離れた箇所(例えば,四角形の計量皿であれば対角線を四分割した四隅の点)に荷重を加えた
際の表示値の変化の程度。試料及び採取用容器が特殊な形状である場合は配慮するのがよい項目で
ある。
6) 温度ドリフトによる感度変化 温度変化による表示値の変化で,天びん周囲の温度変化量に対して
表示値の変化量を温度係数(1 ℃当たりの偏差の百分率)によって算出される。
7) 最小計量値 繰返し性試験によって得られる要求精度への検証項目であり,要求精度を満たす下限
値。はかりとる質量は,最小計量値より大きいことが目安となる。特に測定範囲の小領域において
は想定できない大きなばらつき(精密さ)又は測定誤差が含まれる可能性があるため,微小の質量
のはかりとりに対して注意を払うべき要因の一つである。測定結果の精密さが読み値の1/1 000
(0.10 %)以下の精度で包含係数2を許容できるひょう量範囲の下限となる質量は式(D.1)で表され,
比較検証で使用される最小計量値の推定は,式(D.2)で算出される(参考文献[8]及び[9]を参照)。
%)
10
.0(
000
1
1
2
sn
≦
w
σ
×
······························································ (D.1)
ここに,
σ: 任意の質量で10回以上の繰返しひょう量から得た表示
値の標準偏差
wsn: はかりとる質量
000
1
2
E
×
×
=
σ
w
···································································· (D.2)
ここに,
wE: 最小計量値の推定値
39
K 0138:2018
σ: 任意の質量で10回以上の繰返しひょう量から得た表示
値の標準偏差
b) 天びんの設置環境 天びんは1階以下の広すぎない部屋で,振動源,通風箇所及び直射日光を受ける
壁面(又は室内電灯の放射熱)を避けた常時周囲環境状態が変化しない測定場所であることが望まし
い。また,振動の影響が低いとされる部屋の隅又は大きな建物柱の傍で使用することが理想的であり,
天びんが据付けされる計量台(除振台,防振台など)はそれ自身に十分な重量があり,計量台へ重量
物などの負荷を加えても上下のひずみがなく堅ろうで,磁性及び帯電性に配慮されていることが望ま
しい。特に表示器の最小表示桁,読取限度桁が0.1 mg以下の天びんは,ヒトの感覚では感じることが
できない微震についても試料自身への伝搬又は天びんの計量センサが反応し表示値に不安定性を起こ
すので,設置又は移設する際には注意を払うことも必要となる。
保全管理の面においても天びんの機器部品への劣化を避けるため結露の要因となる急激な温度変化
がない環境を確保する。また,電子機器である天びんの使用環境として,温度範囲は5 ℃〜40 ℃で,
かつ,相対湿度は20 %〜80 %の範囲であり,静電気などの影響を考慮する場合は45 %〜60 %の範囲
であることが望ましい。
使用場所を移動した場合は,その場所に加わる重力加速度が異なってくるため感度調整が必要とな
ることもある。両皿双方に荷重が加わり平衡状態ではかる機械式天びんとは異なり,電子式天びんは
電磁力と自由落下の加速度(重力)との釣り合いにおいて補正し質量の値として表示させるため,移
動する前の場所で釣り合いを感度調整された天びんは,移動先の環境条件の違いによって実際とは偏
った質量を表示する。
c) 天びんの使用前の動作確認 天びんを使用する前には,次に示す事項について確認を行う。
1) 予熱待機時間の確保 電源供給後,検出器の内部温度を安定化させるために予熱待機時間を確保す
る。例えば,必要な予熱時間は,最小表示が10 mg以上の場合は30分間以上,1 mg以下の場合は
2時間以上,0.01 mg以下の場合は1日間を確保することが望ましい。
2) 据付状態の確認 天びんに装備されている水平器の気泡が中心位置にあるか確認する。水平調整の
際には,天びんががたついていないかの確認及び計量台と接している天びんの足と設置面に隙間が
ないか目視確認することが望ましい。
3) 感度調整の実施 感度調整機能を備えた(調整用内蔵分銅が装備された)天びんの場合,表示器の
零点及び最大ひょう量値付近について変化した周辺温度の状態に応じて理想的な感度調整を行うこ
とが可能である。前回の使用から1時間以上空いた場合は,使用前に感度調整を行うことが望まし
い。
分解能が高いほどこの影響は大きくなり感度変化による測定誤差は,一般的に零点からひょう量
する質量付近まで相対的に大きくなる。感度調整機能を備えていない機器については,分銅(天び
んのひょう量する質量付近又は使用者が決定する測定範囲の最大の質量付近)を用いて,感度調整
を手動で実施することが望ましい。
D.3 ひょう量に関する留意点
ひょう量に関する注意事項などは,次による。
a) 準備 試料及び取扱いジグは,手を伸ばして届く範囲に配置するなど作業しやすい環境を整える。ま
た,測定者の体温及び息は,天びんを囲う風防内又は計量皿周辺へ放出されると温度差による空気対
流を起こす可能性があるため,マスク,手袋など着衣に配慮する。
40
K 0138:2018
b) 試料の取扱い 揮発及び/又は吸湿を起こす試料は,状態変化によって計量値に不安定性を引き起こ
す。そのため,試料測定において不安定性が確認された場合は,測定結果に求める要求精度を見直す,
又は適切な天びん用アクセサリなどを活用し,天びんを使用する環境条件とともに注視して作業を行
う。
c) 表示値の記録 同一試料にもかかわらず測定者によるひょう量誤差を防止するために,読取時間を設
定し,一定の時間間隔後ひょう量値を取得する。
d) ひょう量作業 試料及び採取用容器を計量皿へ載せる際は,余分な力を加えないよう静かに丁寧に行
う。また,体温による計量皿周辺への温度変化に配慮し可能な限り天びんから身体を離して操作する。
e) 環境条件の記録 天びんを使用する場所の周囲環境の記録は,湿度の状態及び物理的浮力量の確認,
温度変化による天びんの感度ドリフトの確認に使用する。
f)
清掃 意図しない計量を避けるため,清掃を定期的に行う。天びんの構造を理解し,簡易的に分解し
て清掃が可能な場合は,ガラスクリーナー又は毛羽立ちのない布を用いて各部をこまめに清掃し,計
量皿及び計量室内は衛生的な状態を保つ。
g) 計量結果に影響する外的要因の排除 計量結果に影響を及ぼす主な外的要因は,可能な限り排除する
ことが望ましい。しかし,測定条件が特異なために完全に外的要因を排除できない場合,又は測定結
果に大きく依存すると考えられる場合には,はかりとりを行った質量を補正することを検討する。代
表的な要因は,次による。
1) 計量皿周辺と試料(採取用容器を含む。)間の温度差 試料の冷蔵保管庫,異なる温度の室外からの
持ち込み,熱処理,物性特性,体温による熱伝導などによって,計量皿周辺と試料間に温度差が生
じる。試料及び採取用容器が計量室内の温度よりも高い場合は,計量皿付近に上向きの微量な風が
発生し,その現象が試料及び採取用容器を押し上げる力となり表示値の減少又は不安定性を生じさ
せる。温度関係が逆の場合は相対して逆の傾向が表れる。これらの現象は計量皿周辺に起こる物理
的現象であるため,天びんに風防が備えられていたとしても避けられない。そのため可能な限り天
びんの風防内部と試料,採取用容器は同等の温度条件ではかりとりを行う。
2) 風(冷暖房空調) はかりとりを行う測定者によって外部からの風が計量皿に対して直接当たること
によって表示値が不安定になるため,空調の風向き,ひょう量作業要領について配慮する。風が直
接的に天びんに吹き当たるような状態で開閉ドアを備えている天びんを使用する際は,必要以上に
ドアを大きく開けない。
3) 静電気 紛体など摩擦によって帯電しやすい試料,採取用容器又は測定室内が相対湿度40 %を下回
る低い湿度状態では,天びんとの電荷の力の作用によって表示値が上方に又は下方に変動するため
測定誤差に影響を与える。はかりとりを行う際には,配慮が必要となる。測定室の湿度を高めに保
つ,蓄積された静電気の消散を待つ,採取用容器を帯電防止加工に変更するなどの対策が取れない
場合は,イオナイザーなどの帯電した電荷を中和させる,又は消散を促進する器具を用いて可能な
限り除電を行った後に,測定を行うことを推奨する。除電は表示値の不安定性を起こす風を計量皿
に直接吹きかけるような器具の使用は避ける。
4) 試料の密度 アルキメデスの法則に従った物理的現象であり,密度8.0 g/cm3の標準分銅であらかじ
め感度調整されている天びんに対して,大きく密度が異なる試料をはかりとりする場合には,浮力
量分の偏りが質量の誤差となって生じる。必要な場合は周囲温度・相対湿度・大気圧による空気密
度を算出するとともに測定結果に浮力効果による補正を加える。
41
K 0138:2018
D.4 天びんの日常点検
日常的に行う自主点検内容(点検に使用する分銅の管理方法も含む。)を含めて作業標準手順書として整
備し,SIトレーサブルな校正証明書付きの分銅を使用して,少なくとも次の点検を行うことが望ましい。
a) 天びんの最大ひょう量値付近(又は使用範囲の最大値付近)に対する正確さ。
b) 天びんの最大ひょう量の約5 %付近(又は使用範囲の下限付近)に対する精密さ(最小計量値)。

42
K 0138:2018
附属書E
(参考)
不確かさの求め方
E.1
一般
分析値がもつ曖昧さは,不確かさという指標を用いて定量的に表現することができる。不確かさを求め,
それを分析値とともに示すことで初めて分析値の信頼性が確保される。
この附属書は,1H qNMR分光法によって内標準法に基づいて化学物質の純度を求める場合を例として,
影響のある主な不確かさ要因を相対標準偏差の計算から評価することで,簡単に分析値の信頼性を確保す
る手順を示したものである。
なお,より厳密な不確かさの求め方は,参考文献[6]などを参照するのがよい。
E.2
不確かさの主な要因
1H qNMR測定における主要な不確かさとして,次の4点が挙げられる。
a) 繰返し測定から得られる純度の不確かさ
b) 複数のシグナルから得られる純度の不確かさ
c) 複数の試料調製から得られる純度の不確かさ
d) qNMR用基準物質の純度の不確かさ
そこで,試料調製を3回行って得た試料溶液それぞれについて繰返し測定を3回ずつ行った後,得られ
た各NMRスペクトルの解析において分析種のシグナルを3個用いて純度を求めた場合を例に,主要な要
因における不確かさの算出方法を示す。
E.3
繰返し測定から得られる純度の不確かさ
表E.1に,繰返し測定から得られる純度の不確かさを算出するための計算表の例を示す。
表E.1−繰返し測定から得られる純度の不確かさを算出するための計算表の例
試料
測定1
測定2
測定3
各シグナル
平均純度(kg/kg)
相対標準偏差(%)
試料溶液1
シグナル1
0.995 5
0.991 3
0.995 3
0.994 0
A
0.236 3
シグナル2
0.999 0
0.996 3
0.998 2
0.997 8
0.140 0
シグナル3
0.993 0
0.993 9
0.996 0
0.994 3
0.155 3
試料溶液2
シグナル1
0.993 8
0.997 5
0.995 1
0.995 5
0.187 7
シグナル2
0.995 0
0.996 4
0.994 8
0.995 4
0.085 6
シグナル3
0.994 3
0.992 9
0.993 6
0.993 6
0.070 0
試料溶液3
シグナル1
0.997 7
1.000 3
0.998 0
0.998 6
0.144 2
シグナル2
1.000 0
1.000 8
1.000 9
1.000 6
0.047 4
シグナル3
0.998 7
0.999 6
1.001 1
0.999 8
0.119 7
まず,試料溶液1について,繰返し測定から得られる3個の純度を,シグナルごとに記載する。試料溶
液2及び試料溶液3についても同様に行い,計27個の純度を記載する。次に,特定の試料溶液における特
定のシグナルに着目し,繰返し測定から得られる純度を平均した各シグナルの平均純度を求めて,その相
43
K 0138:2018
対標準偏差を計算する。表E.1の場合では,9個の相対標準偏差が得られるが,過小評価を避けるために
最も大きな値を示す相対標準偏差を採用する。すなわち,試料溶液1におけるシグナル1の平均純度の相
対標準偏差の値(表中のA)であり,相対標準偏差は約0.24 %となるので,これを繰返し測定から得られ
る純度の相対標準不確かさとする。
E.4
複数のシグナルから得られる純度の不確かさ
表E.2に,複数のシグナルから得られる純度の不確かさを算出するための計算表の例を示す。
なお,表中における各シグナルの平均純度は,表E.1で求めた値である。
表E.2−複数のシグナルから得られる純度の不確かさを算出するための計算表の例
試料
各シグナル
各試料溶液
平均純度(kg/kg)
平均純度(kg/kg)
相対標準偏差(%)
試料溶液1
シグナル1
0.994 0
0.995 4
B
0.212 5
シグナル2
0.997 8
シグナル3
0.994 3
試料溶液2
シグナル1
0.995 5
0.994 8
0.105 8
シグナル2
0.995 4
シグナル3
0.993 6
試料溶液3
シグナル1
0.998 6
0.999 7
0.096 2
シグナル2
1.000 6
シグナル3
0.999 8
特定の試料溶液に着目し,各シグナルの平均純度を平均した各試料溶液の平均純度を求めて,その相対
標準偏差を計算する。表E.2の場合では,3個の相対標準偏差が得られるが,過小評価を避けるために最
も大きな値を示す相対標準偏差を採用する。すなわち,試料溶液1の平均純度の相対標準偏差の値(表中
のB)であり,相対標準偏差は約0.21 %となるので,これを複数のシグナルから得られる純度の相対標準
不確かさとする。
なお,シグナルから得られる純度の相対標準不確かさが他の相対標準不確かさと比べて有意に大きい場
合は,適切なシグナル面積が得られていないシグナルが含まれている可能性があるので,慎重な判断が必
要である。
E.5
複数の試料調製から得られる純度の不確かさ
表E.3に,複数の試料調製から得られる純度の不確かさを算出するための計算表の例を示す。
なお,表中における各試料溶液の平均純度は,表E.2で求めた値である。
44
K 0138:2018
表E.3−複数の試料調製から得られる純度の不確かさを算出すための計算表の例
試料
各試料溶液
全試料溶液
平均純度(kg/kg)
平均純度(kg/kg)
相対標準偏差(%)
試料溶液1
0.995 4
0.996 6
C
0.266 3
試料溶液2
0.994 8
試料溶液3
0.999 7
各試料溶液の平均純度を平均した全試料溶液の平均純度を求めて,その相対標準偏差を計算する。表E.3
の場合では表中のCであり,相対標準偏差は約0.27 %となるので,これを複数の試料調製から得られる純
度の相対標準不確かさとする。
E.6
qNMR用基準物質の純度の不確かさ
qNMR用基準物質として用いた標準物質の認証書に記載されている純度の不確かさを参照する。標準物
質の認証書に拡張不確かさが記載されている場合は,これを併記されている包含係数で除算して標準不確
かさとする。例えば,qNMR用基準物質の純度の拡張不確かさが0.005 0 kg/kgであり,包含係数k=2であ
るとすると,標準不確かさは0.002 5 kg/kgとなる。
得られた標準不確かさをqNMR用基準物質として用いた標準物質の認証書に記載されている純度で除
算することで相対標準不確かさとする。例えば,標準物質の認証書に記載された純度が0.998 0 kg/kgであ
るとすると,qNMR用基準物質の純度の相対標準不確かさは約0.25 %となる。
E.7
不確かさの合成
得られた各要因の不確かさが互いに独立であると仮定し,不確かさをそれぞれ二乗して加算し,正の平
方根とすることで,qNMR測定における総合的な不確かさである相対合成標準不確かさが算出できる。す
なわち,表E.1で示した測定例の場合では,相対合成標準不確かさは約0.49 %となる。
得られた相対合成標準不確かさに包含係数k(データ数が10以上の場合は,通常k=2と考えてよい。)
を乗じることで,信頼性の指標として通常表現される相対拡張不確かさが算出できる。すなわち,表E.1
で示した測定例の場合では,相対拡張不確かさは約1.0 %となる。
上記より得られた分析値の相対拡張不確かさの値を,目標とした不確かさ(精確さ)と比較する。目標
とした分析値の不確かさが1 %であったとすると,この事例の場合は目標が達成できたことになる。一方,
目標が達成できなかった場合は,4点の不確かさのうち,大きな寄与をしている要因から順に不確かさの
低減を検討することが合理的である。このとき,各要因の相対標準不確かさとそれらの相対合成標準不確
かさの関係を視覚的に把握できるグラフを作成すると,不確かさの寄与の割合を容易に把握することがで
きる。
なお,この附属書に記載した計算方法では,次に示す不確かさが統計的には完全に独立しているとはい
えないため,厳密には正しく不確かさの評価ができていない。
a) 繰返し測定から得られる純度の不確かさ
b) 複数のシグナルから得られる純度の不確かさ
c) 複数の試料調製から得られる純度の不確かさ
そこで,より精確にa)〜c) を評価したい場合には,統計的手法である分散分析を利用するのがよい。さ
らに,上記以外の不確かさの要因としては,天びんを使用することによる不確かさ,分析種及びqNMR用
45
K 0138:2018
基準物質のモル質量の不確かさ,分析種及びqNMR用基準物質のシグナル面積に寄与する1H核の個数の
不確かさなどがある。多くの場合は割愛しても分析値の信頼性には影響しないが,一度は計算してみるこ
とが望ましい。
この附属書は,qNMRプライマリーガイド−基礎から実践まで−(共立出版)[6]を参照。
参考文献
[1] 第十七改正日本薬局方(平成28年3月7日厚生労働省告示第64号)
入手先:
<http://www.mhlw.go.jp/file/06-Seisakujouhou-11120000-Iyakushokuhinkyoku/JP17.pdf>
<http://www.mhlw.go.jp/file/06-Seisakujouhou-11120000-Iyakushokuhinkyoku/JP17̲SANKOU.pdf>
[2] Fujiwara, T., Anai, T., Kurihara, N., Nagayama, K. J. Magn. Reson. A 104, 103-105 (2011)
[3] Saito, T., Ihara, T., Koike, M., Kinugasa, S., Fujimine, Y., Nose, K., Hirai, T. Accred. Qual. Assur., 14, 79-86
(2009)
[4] Saito, T., Ihara, T., Miura, T., Yamada, Y., Chiba, K. Accred. Qual. Assur., 16, 421-428 (2011)
[5] Saito, T., Nakaie, S., Kinoshita, M., Ihara, T., Kinugasa, S., Nomura, A., Maeda, T. Metrologia, 41, 213-218
(2004)
[6] “qNMRプライマリーガイド”ワーキング・グループ,(2015),qNMRプライマリーガイド−基礎か
ら実践まで−,(共立出版)
[7] ASTM E2977:2015,Standard Practice for Measuring and Reporting Performance of Fourier-Transform
Nuclear Magnetic Resonance (FT-NMR) Spectrometers for Liquid Samples
[8] USP, “General Chapter 41 Balances”, US Pharmacopeia USP39-NF34, 2016
[9] USP, “General Information 1251 Weighing on an Analytical Balance”, US Pharmacopeia USP39−NF34, 2016
[10] 加藤尚志,鮑新努,鈴木彰子,井原俊英,分析化学,66, 375-380 (2017)
[11] JIS B 7611-1 非自動はかり−性能要件及び試験方法−第1部:一般計量器
[12] ISO Guide 34:2009,General requirements for the competence of reference material producers