K 0128 : 2000K 0128 : 2000K 0128 : 2000
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本工業
用水協会 (JIWA)/財団法人日本規格協会 (JSA) から工業標準原案を具して日本工業規格を改正すべきと
の申出があり,日本工業標準調査会の審議を経て,通商産業大臣が改正した日本工業規格である。これに
よってJIS K 0128 : 1994は改正され,この規格に置き換えられる。
K 0128 : 2000
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 1
3. 共通事項 ························································································································ 1
4. 試料 ······························································································································ 2
4.1 試料の採取 ··················································································································· 3
4.2 試料の取扱い ················································································································ 3
5. 結果の表示 ····················································································································· 3
6. 試料の前処理 ·················································································································· 3
6.1 溶媒抽出法 ··················································································································· 3
6.2 固相抽出法 ··················································································································· 5
6.3 クロマトグラフ分離 ······································································································· 6
7. 多成分同時測定法 ··········································································································· 10
7.1 ガスクロマトグラフ質量分析法 ························································································ 10
7.2 ガスクロマトグラフ法···································································································· 16
7.2.1 熱イオン化検出器 (FTD) を用いたガスクロマトグラフ法 ··················································· 17
7.2.2 炎光光度検出器 (FPD) を用いたガスクロマトグラフ法 ······················································ 20
7.2.3 電子捕獲検出器 (ECD) を用いたガスクロマトグラフ法 ······················································ 23
7.3 高速液体クロマトグラフ法······························································································ 25
8. 1, 3-ジクロロプロペン ····································································································· 29
9. EPN ····························································································································· 29
9.1 ガスクロマトグラフ質量分析法 ························································································ 29
9.2 ガスクロマトグラフ法···································································································· 30
10. アシュラム ·················································································································· 32
10.1 ガスクロマトグラフ法 ·································································································· 32
10.2 高速液体クロマトグラフ法 ···························································································· 36
11. アセフェート················································································································ 38
11.1 ガスクロマトグラフ質量分析法 ······················································································ 38
11.2 ガスクロマトグラ ········································································································ 40
12. イソキサチオン ············································································································ 41
12.1 ガスクロマトグラフ質量分析法 ······················································································ 41
12.2 ガスクロマトグラフ法 ·································································································· 43
13. イソフェンホス ············································································································ 45
13.1 ガスクロマトグラフ質量分析法 ······················································································ 45
13.2 ガスクロマトグラフ法 ·································································································· 47
K 0128 : 2000K 0128 : 2000 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
14. イソプロチオラン ········································································································· 49
14.1 ガスクロマトグラフ質量分析法 ······················································································ 49
14.2 ガスクロマトグラフ法 ·································································································· 50
15. イプロジオン ··············································································································· 52
15.1 ガスクロマトグラフ質量分析法 ······················································································ 52
15.2 ガスクロマトグラフ法 ·································································································· 53
16. イプロベンホス [IBP] ···································································································· 54
16.1 ガスクロマトグラフ質量分析法 ······················································································ 55
16.2 ガスクロマトグラフ法 ·································································································· 56
17. イミダクロプリド ········································································································· 57
17.1 高速液体クロマトグラフ法 ···························································································· 57
18. エスプロカルブ ············································································································ 60
18.1 ガスクロマトグラフ質量分析法 ······················································································ 60
18.2 ガスクロマトグラフ法 ·································································································· 62
19. エジフェンホス [EDDP] ································································································· 63
19.1 ガスクロマトグラフ質量分析法 ······················································································ 63
19.2 ガスクロマトグラフ法 ·································································································· 64
20. エトフェンプロックス···································································································· 66
20.1 高速液体クロマトグラフ法 ···························································································· 66
21. エトリジアゾール[エクロメゾール] ··············································································· 68
21.1 ガスクロマトグラフ質量分析法 ······················································································ 68
21.2 ガスクロマトグラフ法 ·································································································· 69
22. オキシン銅[有機銅]···································································································· 72
22.1 高速液体クロマトグラフ法 ···························································································· 72
22.1.1 紫外吸光光度法 ········································································································· 72
22.1.2 蛍光光度法 ··············································································································· 75
23. カルバリル [NAC] ········································································································· 77
23.1 ガスクロマトグラフ質量分析法 ······················································································ 77
23.2 ガスクロマトグラフ法 ·································································································· 79
24. キャプタン ·················································································································· 80
24.1 ガスクロマトグラフ質量分析法 ······················································································ 80
24.2 ガスクロマトグラフ法 ·································································································· 81
25. クロルニトロフェン [CNP] ···························································································· 83
25.1 ガスクロマトグラフ質量分析法 ······················································································ 83
25.2 ガスクロマトグラフ法 ·································································································· 84
26. クロルピリホス ············································································································ 86
26.1 ガスクロマトグラフ質量分析法 ······················································································ 86
26.2 ガスクロマトグラフ法 ·································································································· 87
27. クロロタロニル [TPN] ··································································································· 89
K 0128 : 2000 K 0128 : 2000 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
27.1 ガスクロマトグラフ質量分析法 ······················································································ 89
27.2 ガスクロマトグラフ法 ·································································································· 90
28. クロロネブ ·················································································································· 92
28.1 ガスクロマトグラフ質量分析法 ······················································································ 92
28.2 ガスクロマトグラフ法 ·································································································· 93
29. ジクロフェンチオン [ECP]······························································································ 95
29.1 ガスクロマトグラフ質量分析法 ······················································································ 95
29.2 ガスクロマトグラフ法 ·································································································· 97
30. ジクロルボス [DDVP] ···································································································· 99
30.1 ガスクロマトグラフ質量分析法 ······················································································ 99
30.2 ガスクロマトグラフ法 ································································································· 100
31. ジチオピル ················································································································· 102
31.1 ガスクロマトグラフ質量分析法 ····················································································· 102
31.2 ガスクロマトグラフ法 ································································································· 103
32. シマジン [CAT] ··········································································································· 106
32.1 ガスクロマトグラフ質量分析法 ····················································································· 106
32.2 ガスクロマトグラフ法 ································································································· 107
33. シメトリン ················································································································· 109
33.1 ガスクロマトグラフ質量分析法 ····················································································· 109
33.2 ガスクロマトグラフ法 ································································································· 111
34. ダイアジノン ·············································································································· 113
34.1 ガスクロマトグラフ質量分析法 ····················································································· 113
34.2 ガスクロマトグラフ法 ································································································· 115
35. チウラム ···················································································································· 116
35.1 高速液体クロマトグラフ法 ··························································································· 116
36. チオベンカルブ[ベンチオカーブ] ················································································· 119
36.1 ガスクロマトグラフ質量分析法 ····················································································· 119
36.2 ガスクロマトグラフ法 ································································································· 120
37. テルブカルブ [MBPMC] ································································································ 122
37.1 ガスクロマトグラフ質量分析法 ····················································································· 122
37.2 ガスクロマトグラフ法 ································································································· 123
38. トリクロピル ·············································································································· 124
38.1 高速液体クロマトグラフ法 ··························································································· 124
38.2 ガスクロマトグラフ質量分析法 ····················································································· 128
39. トリクロルホン [DEP] ·································································································· 130
39.1 ガスクロマトグラフ質量分析法 ····················································································· 130
39.2 ガスクロマトグラフ法 ································································································· 132
40. トリシクラゾール ········································································································ 134
40.1 ガスクロマトグラフ質量分析法 ····················································································· 134
K 0128 : 2000K 0128 : 2000 目次
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
40.2 ガスクロマトグラフ法 ································································································· 137
41. トルクロホスメチル ····································································································· 138
41.1 ガスクロマトグラフ質量分析法 ····················································································· 138
41.2 ガスクロマトグラフ法 ································································································· 140
42. ナプロパミド ·············································································································· 141
42.1 ガスクロマトグラフ質量分析法 ····················································································· 141
42.2 ガスクロマトグラフ法 ································································································· 143
43. ピリダフェンチオン ····································································································· 144
43.1 ガスクロマトグラフ質量分析法 ····················································································· 144
43.2 ガスクロマトグラフ法 ································································································· 147
44. ピリブチカルブ ··········································································································· 148
44.1 ガスクロマトグラフ質量分析法 ····················································································· 148
44.2 ガスクロマトグラフ法 ································································································· 151
45. フェニトロチオン [MEP] ······························································································· 152
45.1 ガスクロマトグラフ質量分析法 ····················································································· 152
45.2 ガスクロマトグラフ法 ································································································· 153
46. フェノブカルブ [BPMC] ································································································ 155
46.1 ガスクロマトグラフ質量分析法 ····················································································· 155
46.2 ガスクロマトグラフ法 ································································································· 157
47. フサライド ················································································································· 158
47.1 ガスクロマトグラフ質量分析法 ····················································································· 158
47.2 ガスクロマトグラフ法 ································································································· 160
48. ブタミホス ················································································································· 161
48.1 ガスクロマトグラフ質量分析法 ····················································································· 161
48.2 ガスクロマトグラフ法 ································································································· 163
49. ブプロフェジン ··········································································································· 164
49.1 ガスクロマトグラフ質量分析法 ····················································································· 164
49.2 ガスクロマトグラフ法 ································································································· 166
50. フルトラニル ·············································································································· 168
50.1 ガスクロマトグラフ質量分析法 ····················································································· 168
50.2 ガスクロマトグラフ法 ································································································· 170
51. プレチラクロール ········································································································ 171
51.1 ガスクロマトグラフ質量分析法 ····················································································· 172
51.2 ガスクロマトグラフ法 ································································································· 173
52. プロピザミド ·············································································································· 175
52.1 ガスクロマトグラフ質量分析法 ····················································································· 175
52.2 ガスクロマトグラフ法 ································································································· 176
53. プロベナゾール ··········································································································· 178
53.1 ガスクロマトグラフ質量分析法 ····················································································· 178
K 0128 : 2000 K 0128 : 2000 目次
(5)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
53.2 ガスクロマトグラフ法 ································································································· 179
54. ブロモブチド[ブロモチド] ·························································································· 180
54.1 ガスクロマトグラフ質量分析法 ····················································································· 180
54.2 ガスクロマトグラフ法 ································································································· 183
55. ペンシクロン ·············································································································· 184
55.1 ガスクロマトグラフ質量分析法 ····················································································· 184
55.2 ガスクロマトグラフ法 ································································································· 187
56. ベンスリド [SAP] ········································································································ 189
56.1 ガスクロマトグラフ質量分析法 ····················································································· 189
56.2 ガスクロマトグラフ法 ································································································· 190
57. ペンジメタリン ··········································································································· 192
57.1 ガスクロマトグラフ質量分析法 ····················································································· 192
57.2 ガスクロマトグラフ法 ································································································· 193
58. ベンフルラリン[ベスロジン] ······················································································· 195
58.1 ガスクロマトグラフ質量分析法 ····················································································· 195
58.2 ガスクロマトグラフ法 ································································································· 196
59. マラチオン (マラソン) ·································································································· 197
59.1 ガスクロマトグラフ質量分析法 ····················································································· 197
59.2 ガスクロマトグラフ法 ································································································· 199
60. メコプロップ [MCPP] ··································································································· 201
60.1 ガスクロマトグラフ質量分析法 ····················································································· 201
60.2 ガスクロマトグラフ法 ································································································· 203
61. メタラキシル ·············································································································· 206
61.1 ガスクロマトグラフ質量分析法 ····················································································· 206
61.2 ガスクロマトグラフ法 ································································································· 208
62. メチルダイムロン ········································································································ 209
62.1 ガスクロマトグラフ質量分析法 ····················································································· 209
62.2 ガスクロマトグラフ法 ································································································· 210
63. メフェナセツト ··········································································································· 212
63.1 ガスクロマトグラフ質量分析法 ····················································································· 212
63.2 ガスクロマトグラフ法 ································································································· 213
64. メプロニル ················································································································· 214
64.1 ガスクロマトグラフ質量分析法 ····················································································· 215
64.2 ガスクロマトグラフ法 ································································································· 216
65. モリネート ················································································································· 217
65.1 ガスクロマトグラフ質量分析法 ····················································································· 217
65.2 ガスクロマトグラフ法 ································································································· 219
K 0128 : 2000K 0128 : 2000 目次
(6)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
付表1 農薬の名称 ············································································································· 221
付表2 試験方法概要一覧 ···································································································· 228
付表3 対象農薬の構造式,分子式及び分子量 ········································································· 231
付表4 引用規格 ················································································································ 237
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 0128 : 2000
用水・排水中の農薬試験方法
Testing methods for pesticides in industrial water and waste water
序文 この規格は,工業用水及び工場排水中の農薬類について1994年に制定されたものであるが,その後
の分析技術の進歩,社会情勢の変化などに伴う社会的要請を勘案して改正したものである。
1. 適用範囲 この規格は,工業用水及び工場排水中の農薬類のうち,付表1に示す農薬の試験方法につ
いて規定する。
備考1. この規格の試験方法の概要一覧を,付表2に示す。
2. 付表1に示す農薬の構造式などを,付表3に示す。
2. 引用規格 付表4に示す規格は,この規格に引用されることによって,この規格の規定の一部を構成
する。これらの引用規格は,その最新版(追補を含む。)を適用する。
3. 共通事項 共通事項は,次による。
a) 通則 化学分析に共通する一般事項は,JIS K 0050による・
b) 定義 この規格で用いる主な用語の定義は,JIS K 0101,JIS K 0102,JIS K 0211及びJIS K 0215に
よる。
c) ガスクロマトグラフ法 ガスクロマトグラフ法に共通する一般事項は,JIS K 0114による。
d) ガスクロマトグラフ質量分析法 ガスクロマトグラフ質量分析法に共通する一般事項は,JIS K 0123
による。
e) 高速液体クロマトグラフ法 高速液体クロマトグラフ法に共通する一般事項は,JIS K 0124による。
f)
項目の名称 項目の名称は,国内だけの登録農薬名であるEPNを除いて,ISO登録農薬名を片仮名で
表示したもので,付表1の国内通称名を用いて表現している。ただし,一部に商標登録名と一致する
ものもある。また,化合物名は通称名を用いたものもある。
なお,各項目における国内通称名の化合物名は,“Chemical Abstracts Index Guide 9th, 10th” で使用さ
れている化合物を,国内通称名に続いて括弧内に併記した。
g) 定量範囲 それぞれの試験方法に示してある定量範囲は,各試験方法に用いられている装置に注入す
る溶媒中の対象農薬の質量 (ng) で示す。
h) 繰返し分析精度 繰返し分析精度は,それぞれの試験方法の定量範囲内において繰返し試験で求めた
変動係数 (%) で示す(1)。
注(1)
100
(%)
×
=xσ
変動係数
ここに,
σ: 標準偏差
2
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
x: 平均値
i)
水 この規格で用いる水は,JIS K 0557に規定するA1〜A4の水とする。試薬の調製,空試験などに
用いる水は,A4(又はA3)の水とするが,使用前に各試験方法で空試験を行い,使用の適否を確認
する。
j)
試薬
1) 試薬は,品目指定されている場合には,JISに規定するものを用いる。JISに規定されるものがない
場合には,試験に支障のないものを用いる。
2) 試薬類の溶液の濃度は,一般に質量濃度はg/L又はmg/L(化合物の場合は無水物としての質量を用
いる。)で,モル濃度はmol/L又はmmol/Lで示す。
標準液の濃度は,1ml中の質量(mg/ml,μg/ml又はng/ml)で表す。
3) 液体試薬の濃度は,水(又は別の液体試薬)との混合比[試薬 (a+b)]で表す。この表し方は,試
薬amlと水(又は別の液体試薬)bmlとを混合したことを示す。ただし,JIS K 0050の表3の液体
試薬を薄めないで用いる場合は,その試薬名だけで示す。
4) 試薬類の溶液名称の後に括弧で示されている濃度は,標準液以外は概略の濃度であることを意味す
る。例えば,水酸化ナトリウム溶液 (0.1mol/L) は約0.1mol/Lの水酸化ナトリウム溶液であること
を示す。
また,溶液名の前に示す濃度は,正確な濃度を意味する。ただし,一般には,端数のない数値で
示し,別にファクターを求めておく。
5) 試薬類の名称は,国際純正及び応用化学連合 (IUPAC) の無機化学命名法及び有機化学命名法を基
にして,社団法人日本化学会が定めた化合物命名法及びJIS試薬の名称にできるだけ整合させた。
6) 標準液の調製に使用する農薬は,純度既知のものを用いるが,この規格では,それを標準品と記述
してある。それらには類似した化合物が不純物として含まれることが多いので,試験に支障のない
ものを使用する。
7) 試薬類及び廃液などの取扱いについては,関係法令規則などに従い十分に注意する。
k) ガラス器具類 ガラス器具類は,特に断らない限りJIS R 3503及びJIS R 3505に規定するものを使用
する。ただし,特殊な器具を必要とする場合には,それぞれの項目に,その一例を図示又は説明する。
また,加熱操作を伴う場合には,JIS R 3503に規定するほうけい酸ガラス-1を用いる。
デシケーターに用いる乾燥剤は,特に断らない限りシリカゲル(2)とする。
試験に用いるガラス器具類は,使用前にあらかじめ水で洗浄した後,更にアセトン(JIS K 8040に
規定する濃縮300以上の品質のもの。)で洗浄しておく。
注(2) JIS Z 0701に規定する包装用シリカゲル乾燥剤A形1種を用いる。
l)
検量線(ガスクロマトグラフ法,ガスクロマトグラフ質量分析法,高速液体クロマトグラフ法) 検
量線の作成に当たっては,試験方法に示される定量範囲内を4〜6段階に分け,これに一致するように
標準液をとり,定量範囲内について作成する。
検量線は,試験に際して新たに作成したものを用い,同一項目を多数の試料について連続して試験
する場合には,試験の途中において,適宜,標準液を用いて指示値の確認を行う。
m) 注及び備考 注及び備考番号は,各項目ごとに一連番号を付けてある。
備考 注番号については,本文中では(1),備考の中では(*1)として区別してある。
4. 試料
3
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1
試料の採取 試料の採取は,次による。
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 JIS K 8180に規定するもの。
2) 水酸化ナトリウム溶液 (100g/L) JIS K 8576に規定する水酸化ナトリウムを用いて調製する。
3) アセトン JIS K 8040に規定する濃縮300以上の品質のもの(1)。
注(1) 使用前に試験に支障のないことを確認する。開封後は汚染のない場所に保管しておく。
b) 器具 器具は,次による。
1) 試料容器 共栓付きガラス瓶200〜1 000mlをあらかじめ水で洗浄した後,更にアセトンで洗浄して
乾燥したもの。
c) 採取操作 採取操作は,次のとおり行う。
1) 表層水の採取 試料をJIS K 0094の4.1.1(試料容器による採取)又は4.1.2(バケツ類による採取)
に従って採取し,試料容器に移し入れ,満たして密栓する。
2) 各深度の水の採取 試料をJIS K 0094の4.1.4(バンドーン採水器による採取)に従って採取し,試
料容器に移し入れ,満たして密栓する。
3) 配管装置からの採取 試料をJIS K 0094の4.3(採取弁を用いる採取)に従って採取し,試料容器
に移し入れ,満たして密栓する。
備考 残留塩素が存在する場合には,共栓付きガラス瓶に泡立てないように静かに採取し,残留塩素
1mg当たりL (+) -アスコルビン酸溶液 (40g/L) (JIS K 9502に規定するもので調製する。)を
0.1mlの割合で添加した後,気泡が残らないように満たして密栓する。
4.2
試料の取扱い 農薬の中には分解しやすいものがあるので,試験は試料採取後直ちに行う。直ちに
行えない場合には,0〜10℃の暗所に保存し,できるだけ早く試験する。
5. 結果の表示 結果の表示には,用いた試験方法を明記する。
6. 試料の前処理 各項目で試験操作を行う前の準備操作は,農薬の種類に関係なく共通するものがほと
んどであるため,多くの農薬に共通して適用できる操作を一括して次に規定する。ただし,項目ごとに規
定してある場合は,それによる。
6.1
溶媒抽出法 試料を塩化ナトリウムの共存下でジクロロメタンで抽出した後,脱水,濃縮して一定
量とする。
なお,揮発性有機化合物を試験している試験室では,ジクロロメタンによる室内の汚染に注意が必要で
ある。また,ジクロロメタンは有害な物質であるから,その取扱いに気を付ける。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム JIS K 8150に規定する塩化ナトリウム(600℃で約1時間加熱し,デシケーター中
で放冷したもの。)。
2) 硫酸ナトリウム JIS K 8987に規定するもの。
3) ジクロロメタン JIS K 8117に規定する濃縮300以上の品質のもの(1)。
4) ヘキサン JIS K 8825に規定する濃縮300以上の品質のもの(1)。
注(1) 4.の注(1)による。
b) 器具 器具は,次による。
1) 分液漏斗 200〜1 000ml
4
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) なす形フラスコ 100〜500mlの共通すり合わせで濃縮器に接続できるもの。
3) 三角フラスコ 300〜500mlの適切な容量
4) 振とう器
5) 濃縮器(2) ロータリーエバポレーター又はクデルナー-ダニッシュ形濃縮器
注(2) 濃縮時に試料溶液と接触する部分は,あらかじめ水及びアセトンで洗浄する。
c) 操作 操作は,次のとおり行う。
1) 4.1c)で採取した試料(3)の適量 (100〜1 000ml) を分液漏斗にとり,液量100mlについて塩化ナトリ
ウム5gとジクロロメタン10ml(4)とを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
ジクロロメタンで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量100mlについ
てジクロロメタン10 mlを加え,再び振とう器を用いて約10分間振り混ぜ,放置する。ジクロロメ
タン層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液20mlについて硫酸ナトリウム約3g(5)を加え,軽く振り混ぜ,約10分間放置し
た後(6),ろ紙5種A(又は5種B)(7)を用いてろ過し,ろ液をなす形フラスコ(8)に受ける。少量の
ジクロロメタンを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙上の硫酸ナトリウ
ムも洗浄し,洗液をジクロロメタン溶液に合わせる。
4) 濃縮器(9)を用いて,約40℃(9)の水浴上でジクロロメタン溶液が約5mlになるまで濃縮する。
5) この濃縮液にヘキサン約50mlを加え,4)の操作を行い,5mlの一定量にする(10)。
6) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gとジクロ
ロメタン10mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。続いて2)〜5)の操作を
行う。
注(3) 試料の酸性又はアルカリ性が強い場合には,水酸化ナトリウム溶液 (40g/L) (JIS K 8576に規
定する水酸化ナトリウムを用いて調製する。)又は塩酸 (1+11) (JIS K 8180に規定する塩酸を
用いて調製する。)を用いて中性にする。
(4) エマルションが生じて分離が困難な場合は,ジクロロメタンの量を増加する。
(5) 溶媒量が200mlの場合,硫酸ナトリウムは約30gでよいが,エマルションが生じた場合には,
更に過剰に加える必要がある。
(6) クデルナー-ダニッシュ形濃縮器による濃縮のとき,脱水が不十分な場合には,突沸することが
ある。十分な脱水を行うには2時間以上を要することもある。
脱水の方法には,このほかに,抽出溶媒を−20℃の暗所に保存し,水分を凍結させる方法も
ある。この方法は,4)の濃縮操作を引き続き行わないときに用いるとよい。
(7) ろ紙は,使用時に抽出に用いる溶媒で洗浄しておく。
(8) クデルナー-ダニッシュ形濃縮器を用いる場合は,なす形フラスコに代え,濃縮管付きの濃縮フ
ラスコ500mlを用いる。
(9) クデルナー-ダニッシュ形濃縮器を用いる場合は,減圧方式ではなく,大気圧で75℃以下で加熱
して濃縮する。濃縮終了後,スニーダーカラムを濃縮部につけたまま装置からとりはずし,ス
ニーダーカラムの上部から少量のヘキサンを加え洗浄し,スニーダーカラムを付けたまま冷却
する。
(10) 引き続いて測定操作を行わない場合には,この濃縮液を0〜10℃の暗所に保管する。
5
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2
固相抽出法 試料中の農薬をシリカゲル系又はポリマーゲル系の基材を充てんした固相カラム又は
固相ディスクに加圧法又は減圧法を用いて吸着させ,アセトンで共栓付き試験管に溶出し,一定量とする。
カラムクロマトグラフ分離を行う場合には,一定量としたアセトン溶液にヘキサンを加え,濃縮後,一定
量とする。
a) 試薬 試薬は,次のものを用いる。
1) アセトン 4.1a)3)による(1)。
2) 窒素 JIS K 1107に規定する高純度窒素2級。
b) 器具 器具は,次による。
1) 共栓付き試験管 5〜20mlの適切な容量のもの。
2) 固相カラム 内径5〜10mm,長さ30〜l00mmのカートリッジで,カラム充てん剤にはスチレンジ
ビニルベンゼン共重合体(ポリスチレン系ゲル)又はシリカゲルにオクタデシル基を化学結合させ
たもの,若しくはこれと同じ性能をもつもの。使用前にアセトン約10ml及び水約10mlで洗浄して
おく。
備考1. 農薬分析用の固相は,市販品でディスク形のものもあり,これを用いてもよい。この際の通
水の速度及び溶出溶媒についてはあらかじめ確認しておく。
参考1. 固相カラム又は固相ディスクには,次のようなものがある。
Sep-Pak PS-2,OasisTMHLBカートリッジ,エムポアディスクSDB-RPSなど。
c) 操作 操作は,次のとおり行う。
1) 4.1c)で採取した試料(11)の適量(通常は,200ml)を固相カラム(12)(13)に加圧法(14)又は減圧法(15)によ
って流量10〜20ml/min(16)で通水する。
2) 固相カラムに水10ml(17)を流し,洗浄した後,約30分間吸引などで水分を除去する(18)。
3) 固相カラムの上端からアセトン約3ml(17)を緩やかに通し(19),対象農薬を溶出させる。溶出液の受器
には,共栓付き試験管を用いる。
4) 溶出液に窒素を緩やかに吹き付け,1mlの一定量にする(10)(20)(21)。
5) 空試験として水200mlについて,固相カラム(12)(13)に加圧法(14)又は減圧法(15)によって流量10〜
20ml/min(16)で通水する。続いて2)〜4)の操作を行う。
6) 6.3のクロマトグラフ分離を行う場合には,4)の操作に引き続き,ヘキサン約50mlを加え,濃縮器
(8)(9)を用いて,6〜7mlになるまで濃縮し,濃縮液に窒素を緩やかに吹き付け,1mlの一定量にする
(21)。
この場合の空試験は,水200mlについて,2)〜4)の操作を行い,引き続きヘキサン約50mlを加え,
濃縮器(8)(9)を用いて,6〜7mlになるまで濃縮し,濃縮液に窒素を緩やかに吹き付け,1mlの一定量
にする(10)(21)。
注(11) 懸濁物の多い試料は,ガラス繊維ろ紙(孔径1μm以下のもの)を用いてろ過する。ろ紙上の懸
濁物に吸着している対象農薬をアセトン又はアセトニトリル[JIS K 8039に規定する濃縮300以
上の品質のもの。注(1)による。]で溶出する。この溶出液は,3)の溶出液に合わせる。
使用するガラス繊維ろ紙は,あらかじめ一連の操作を行って試験に影響のないことを確認し
ておく。
(12) 固相カラムの調製の手順,回収率などをあらかじめ確認して使用する。
(13) 対象農薬又は充てん剤によっては固相への保持率に,pH依存性のあるものがある。
例えば,オキシン銅の場合,充てん剤にスチレンジビニルベンゼン共重合体を用いるときは
6
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
pHを3.5にする。充てん剤にオクタデシル基 (ODS) を化学結合したシリカゲルを用いる場合
は,pH調節の必要はない。ポリスチレン系ゲル及びポリメタクリレートのポリマーゲルを用い
る場合は,製品によってpHを3.5に調節する必要があるものもある。
pHの調節にはJIS K 8180に規定する塩酸を適切な濃度に薄めたものを用いるが,充てん剤
によってはJIS K 8541に規定する硝酸を適切な濃度に薄めたものを用いた方がよい場合もある。
(14) 固相カラムに対して試料を加圧状態で送り込む方法をいう。
装置は,調節部及びポンプ部から成り,調節部は,通水時間及び通水速度を制御する。ポン
プ部は,シリンジを用いる方式とペリスタルティックポンプを用いる方式がある。
シリンジの場合は,チューブは四ふっ化エチレン樹脂製のものを使用する。ペリスタルティ
ックポンプの場合は,使用する溶媒に適したポンプ用チューブを用いなければならない。
(15) 固相カラムの底部側を減圧状態として試料を送り込む方法をいう。
装置は,吸引用配管群と真空ポンプから成る。
減圧法は固相の通液抵抗のばらつき及び試料中の懸濁物などによって流量を一定に制御する
ことが困難であるが,簡易な装置で一度に多数の試料を処理できるなど有効な場合もある。
(16) 通水量が多すぎると対象農薬によっては,保持率が低下するものがある。通水量は,20ml/min
以上にはしない。固相カラムへの吸着のバンド幅は,通水量1ml/min程度で最小となるが,農
薬の場合は吸着速度が非常に速いため,10〜20ml/minでも十分な再現性と回収率が得られ,処
理時間の短縮といった利点もある。しかし,有機物の濃度が高い試料の場合,通水量を小さく
すると(5ml/min以下),再現性と回収率の向上につながる。また,通水量は一定に保つ。
(17) 吸着後の洗浄及び溶出に使用する量は,あらかじめ対象農薬を溶出するために適切な量である
ことを確認しておく。
(18) 溶出液の濃縮を容易にするため及びキャピラリーガスクロマトグラフに注入する溶液への水分
の混入を少なくするための操作である。試料を固相カラムに通水後,アスピレーターなどによ
る吸引を行い,できるだけ水分を除去する。
(19) 流量は,カラムからの溶出液の液滴が連続しない程度とする。
(20) ディスク形を用いた場合は,4)の操作に引き続き,ヘキサン約50mlを加え,脱水した後,濃縮
器を用いて濃縮し,濃縮液に窒素を緩やかに吹き付け,1mlの一定量とする。
(21) 窒素を吹き付ける操作では,濃縮液の表面が動いているのがようやく見える程度に窒素の流量
を調節し,濃縮液が飛散しないように注意する。また,乾固させると窒素の吹き付けで対象農
薬が揮散することもあるので注意する。
6.3
クロマトグラフ分離 6.1又は6.2を行った後,カラムの充てん剤に活性けい酸マグネシウムを用い
たカラムクロマトグラフ分離操作を行う。
妨害物質がないときは,この操作を省略してもよい。
カラムクロマトグラフの選択は妨害物質の内容による。
a) 試薬 試薬は,次のものを用いる。
1) アセトン 4.1a)3)による。
2) ジエチルエーテル JIS K 8357に規定する濃縮300以上の品質のもの(1)。
3) ヘキサン 6.1a)4)による。
4) ジエチルエーテル-ヘキサン溶離液 (7+13) 2)のジエチルエーテルと3)のヘキサンで調製する。
5) 窒素 6.2a)2)による。
7
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具 器具は,次による。
1) なす形フラスコ 6.1b)2)による。
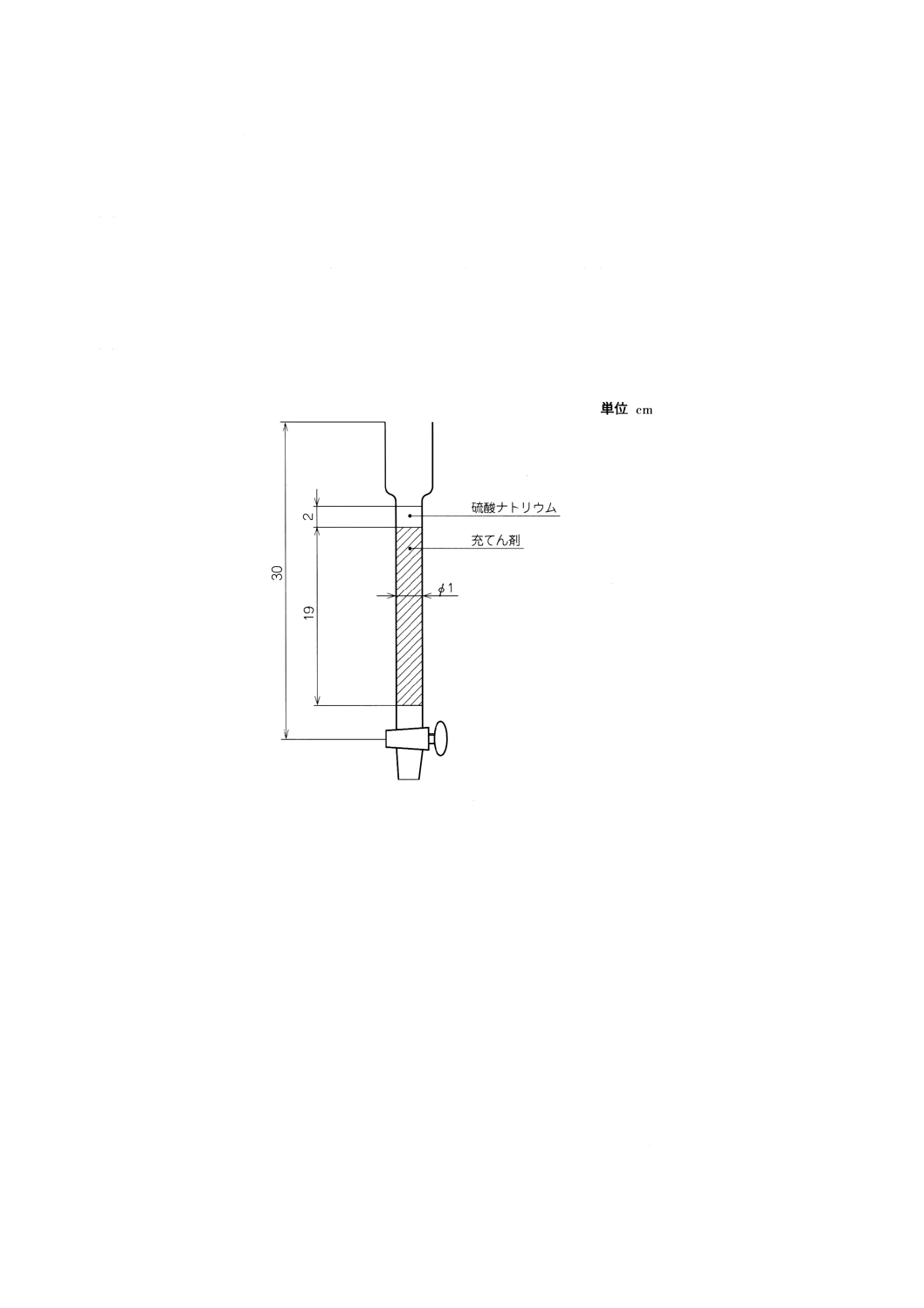
2) クロマトグラフ管 次に掲げる条件を満たすもの。一例を図1に示す。
2.1)
カラム用管 内径約1cm,長さ約30cmのコック付きガラス管。
2.2)
カラム充てん剤 カラムクロマトグラフ用の活性けい酸マグネシウム(22)(粒径75〜150μm,130℃
で約16時間加熱した後,デシケーター中で放冷したもの。)。対象農薬の保持時間にピークを生じな
いもの。
参考2. 活性けい酸マグネシウムは,フロリジルなどの名称で市販されている。
2.3)
クロマトグラフ管の作り方 カラム充てん剤約8gをヘキサンでスラリー状にして気泡が入らない
ようにカラム用管に流し込み(23),その上部に硫酸ナトリウム[6.1a)2)による。]約5gを積層する。
図1 クロマトグラフ管の一例
3) 濃縮器 6.1b)5)による。
注(22) ジクロルボスの測定には,この充てん剤を用いたカラムクロマトグラフ分離は適用できない。
この場合は,備考2.の残留農薬用のシリカゲルを用いる。
(23) カラム用管にカラム充てん剤を均一に充てんするには,流し込んだ後,カラム用管に縦横の振
動を与えるとよい。
備考2. カラム充てん剤として残留農薬用のシリカゲル[(粒径150〜250μm,130℃で約16時間加熱し
た後,デシケーター中で放冷したもの。)。対象農薬の保持時間にピークを生じないもの。]を
用いることができる。
c) 操作 操作は,それぞれ次のとおり行う。
1) 6.1c)5)で一定量 (5ml) にしたへキサン溶液から1ml,又は6.2c)6)で一定量にしたヘキサン溶液の全
量,それぞれをクロマトグラフ管に流し込む。
2) クロマトグラフ管の上部から,ヘキサン100mlを流下させ,ヘキサン流出液は捨てる。
3) 引き続きクロマトグラフ管の上部から,ジエチルエーテル-ヘキサン溶離液 (7+13) (24)100mlを流量
8
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
約1ml/minで流下して対象農薬を溶出させ(25),溶出液をなす形フラスコ(8)に受ける。
4) さらに,アセトン100mlをクロマトグラフ管の上部から流量約1ml/minで流下して,対象農薬を溶
出させ(25),溶出液をなす形フラスコ100〜200ml(8)に受ける。
5) これらの溶出液を,濃縮器(9)を用いて,約40℃(9)の水浴上で溶出液を約10mlになるまで濃縮し,
引き続きそれぞれのなす形フラスコにヘキサン約100mlを加えた後,再び濃縮器(9)を用いて有機溶
媒層を2〜5mlになるまで濃縮し,受器を取り外し,窒素を緩やかに吹き付け,1mlの一定量にする
(10)(21)。
6) 空試験として,6.1c)6)で一定量 (5ml) にしたへキサン溶液から1ml,又は6.2c)6)で一定量にしたヘ
キサン溶液全量,それぞれをクロマトグラフ管に流し込み,続いて2),3),5)又は2),4),5)の操作
を行う。
注(24) ジエチルエーテル-ヘキサン溶離液 (7+13) の代わりに,アセトン-ヘキサン溶離液 (1+19) を
用いることができる。この場合の操作は,備考4.による。
(25) あらかじめ目的成分(対象農薬)が溶出してくる画分を確認しておく。
備考3. 備考2.の残留農薬用のシリカゲルをカラム充てん剤として用いる場合は,次による。
カラム充てん剤にシリカゲルを用いた場合
1) 6.1c)5)で一定量 (5ml) にしたへキサン溶液から1ml又は6.2c)6)で一定量にしたヘキサン溶液
全量をクロマトグラフ管に流し込む。
2) クロマトグラフ管の上部から,ヘキサン80mlを流下させ,ヘキサン流出液は捨てる。
3) c)3)〜5)の操作を行う。
4) 空試験として,c)6)の操作を行う。
備考4. アセトン-ヘキサン溶離液 (1+19) を用いた場合の操作は,次による。
1) 6.1のc)1)〜3)の操作を行う。
2) 濃縮器(*1)を用いて,約40℃(*1)の水浴上でジクロロメタン溶液を1〜2mlになるまで濃縮し,
受器を取り外し,窒素を緩やかに吹き付け,ジクロロメタンを揮散させる(*2)(*3)。
3) 受器の内容物を少量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更に受器を
ヘキサン2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ10mlに移し入れ,ヘキサンを
標線まで加える。
4) 全量フラスコ10mlの内容物全量をクロマトグラフ管(*4)の上部から流し込む。更に全量フラ
スコ10 mlをヘキサン2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ管に流し込む。
5) 引き続き,アセトン-ヘキサン溶離液 (1+19) [6.2a)1)のアセトンと6.1a)4)のヘキサンを用
いて調製する。]50mlを流下して対象農薬を溶出させ(*5),溶出液をなす形フラスコ(*6)に受け
る。
6) これを,濃縮器(*1)を用いて約40℃の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,
窒素を緩やかに吹き付け,溶出液を揮散させ,アセトン1mlを正しく加える(*7)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(*1) 注(9)による。
(*2) ジクロロメタンが残留していると,クロマトグラフ分離に影響するのでジクロロメタンを
揮散させる。ただし,ジクロロメタンが揮散した後も窒素を吹き付けると対象農薬が揮散
することもあるので注意する。
(*3) 注(21)による。
9
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(*4) 6.3のb)2)による。
(*5) 注(25)による。
(*6) 注(8)による。
(*7) 注(10)による。
5. 対象農薬によっては,酢酸エチル-ヘキサン混合の有機溶媒を用いる抽出法を用いてもよい。
抽出操作などは次による。
混合有機溶媒を用いる溶媒抽出法 試料を塩化ナトリウム共存の下で酢酸エチル-ヘキサン
混合溶液で抽出した後,脱水,濃縮して一定量とする。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) 酢酸エチル JIS K 8110に規定する濃縮300以上の品質のもの(*1)。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 備考4.の5)による。
7) 酢酸エチル-ヘキサン混合溶液 (1+1) 4)の酢酸エチルと5)のヘキサンで調製する。
8) 窒素 6.2a)2)による。
注(*1) 注(1)による。
b) 器具 器具は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 次に掲げる条件を満たすもの。
4.1) カラム用管 内径約1.5cm,長さ約30cmのコック付きガラス管。
4.2) カラム充てん剤 6.3b)2.2)による。
4.3) クロマトグラフ管の作り方 カラム充てん剤約5gをヘキサンでスラリー状にして気泡が
入らないようにカラム用管に流し込み(*2),その上部に硫酸ナトリウム約4gを積層する。
5) 振とう器
6) 濃縮器 6.1b)5)による。
注(*2) 注(23)による。
c) 操作 操作は,次のとおり行う。
1) 4.1c)で採取した試料(*3)の適量 (200〜400ml) を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gと酢酸エチル-ヘキサン混合溶液 (1+1) 10ml(*4)とを加え,振とう器を用い
て約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移す。有機溶媒を三角フラスコに移し入れ,分液漏斗を少量の酢酸
エチル-ヘキサン混合溶液 (1+1) で洗い,洗液は先の三角フラスコに合わせる。分液漏斗
の水層に液量100mlについて酢酸エチル-ヘキサン混合溶液 (1+1) 10mlを加え,再び振と
う器を用いて約10分間振り混ぜ,放置する。有機溶媒層を先の三角フラスコに合わせる。
3) 有機溶媒20mlについて硫酸ナトリウム約5g(*5)を加え,軽く振り混ぜ,約10分間放置し
た後(*6),ろ紙5種A(又は5種B)(*7)を用いてろ過し,ろ液をなす形フラスコ(*8)に受け
10
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。少量の酢酸エチル-ヘキサン混合溶液 (1+1) を用いて三角フラスコを2,3回洗浄し,
更にその洗液で先のろ紙上の硫酸ナトリウムも洗浄し,洗液を有機溶媒に合わせる。
4) 濃縮器(*9)を用いて,約40℃(*9)の水浴上で有機溶媒を1〜2mlになるまで濃縮し,受器を取
り外し,窒素を緩やかに吹き付け,有機溶媒を揮散させる(*10)(*11)。
5) 内容物を少量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更に受器をヘキサ
ン2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ10mlに移し入れ,ヘキサンを標線
まで加える(*12)。
6) このヘキサン溶液10mlから,その5mlをクロマトグラフ管に流し込む。
7) クロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+19) 50mlを流量約1ml/min
で流下して対象農薬を溶出させ(*13),溶出液をなす形フラスコ(*8)に受ける。
8) 濃縮器(*9)を用いて,約40℃(*9)の水浴上で溶出液を1〜2mlになるまで濃縮し,受器を取り
外し,窒素を緩やかに吹き付け,溶出液を揮散させ,アセトン4mlを正しく加える(*10)(*11)。
9) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5g
と酢酸エチル-ヘキサン混合溶液 (1+1) 10ml(*4)とを加え,振とう器を用いて約10分間振
り混ぜ,放置する。続いて2)〜8)の操作を行う。
注(*3) 注(3)による。
(*4) 注(4)による。
(*5) 注(5)による。
(*6) 注(6)による。
(*7) 注(7)による。
(*8) 注(8)による。
(*9) 注(9)による。
(*10) 有機溶媒が揮散した後も,窒素を吹き付けると対象農薬が揮散することもあるので注意す
る。
(*11) 注(21)による。
(*12) 注(10)による。
(*13) 注(25)による。
7. 多成分同時測定法 試料中の多成分の農薬類を同時に測定する方法で,ガスクロマトグラフ質量分析
法,ガスクロマトグラフ法及び高速液体クロマトグラフ法を適用する。
備考1. この試験方法は,試料に色及び濁りがなく,かつ,この試験の妨害となる成分を含まない場
合で,6.3の操作を省略できる試料に適用する。
7.1
ガスクロマトグラフ質量分析法 試料中の農薬を,前処理に6.1又は6.2を適用して濃縮した後,そ
の一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法 (SIM) 又は
マスクロマトグラフ法 (MC) を用いて定量する。対象農薬とその定量範囲及び繰返し分析精度(変動係数
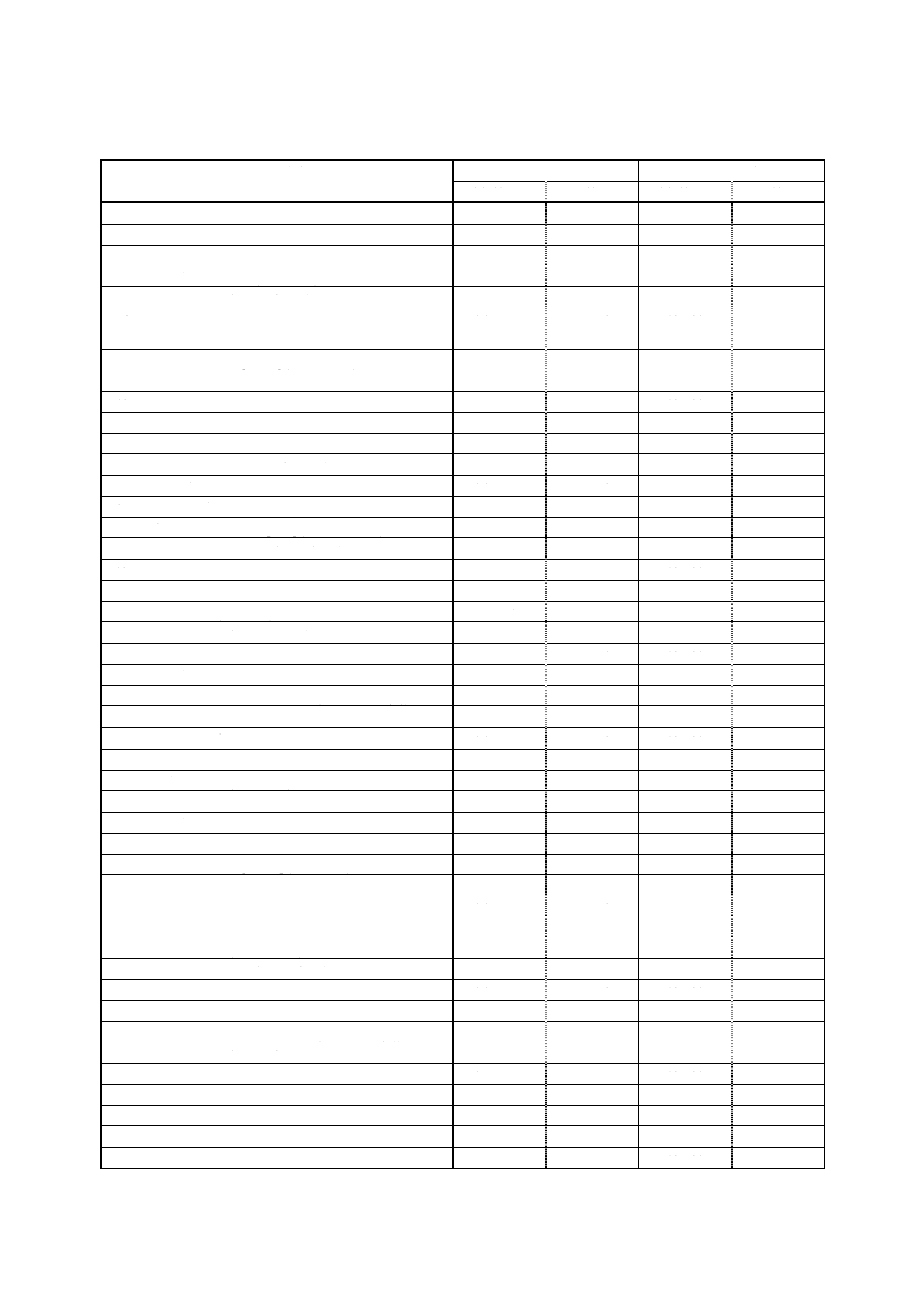
で)は,表1のとおりである(いずれも装置,測定条件によって異なる。)。
11
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
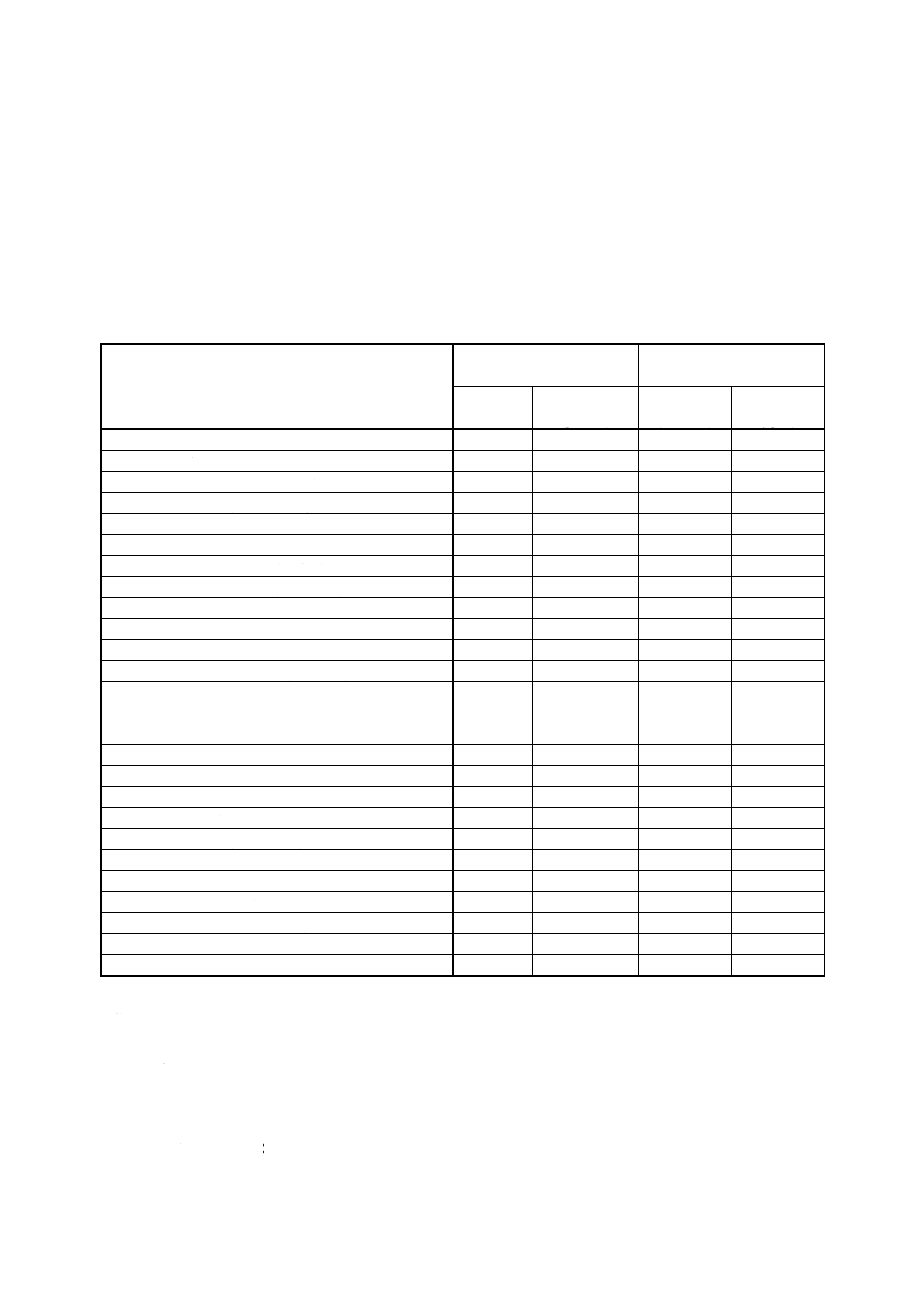
表1 対象農薬とその定量範囲及び繰返し分析精度
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
溶媒抽出法
固相抽出法
溶媒抽出法
固相抽出法
1
EPN (C14H14NO4PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
2
イソキサチオン (C13H16NO4PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
3
イソフェンホス (C15H24NO4PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
4
イソプロチオラン (C12H18O4S2)
0.1 〜2
0.1 〜2
20〜30
20〜30
5
イプロジオン (C13H13Cl2N3O3)
0.2 〜4
0.2 〜4
20〜30
20〜30
6
イプロベンホス [IBP] (C13H21O3PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
7
エスプロカルブ (C15H23NOS)
0.4 〜8
0.4 〜8
10〜20
10〜20
8
エジフェンホス [EDDP] (C14H15O2PS2)
0.4 〜8
0.4 〜8
10〜20
10〜20
9
エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS)
0.3 〜6
0.3 〜6
10〜20
10〜20
10
カルバリル [NAC] (C12H11NO2)
0.4 〜8
0.4 〜8
10〜20
10〜20
11
キャプタン (C9H8Cl3NO2S)
0.1 〜2
0.1 〜2
20〜30
20〜30
12
クロルニトロフェン [CNP] (C12H6Cl3NO3)
0.1 〜2
0.1 〜2
10〜20
10〜20
13
クロルピリホス (C9H11Cl3NO3PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
14
クロロタロニル [TPN] (C8Cl4N2)
0.1 〜2
0.1 〜2
20〜30
20〜30
15
クロロネブ (C8H8Cl2O2)
0.1 〜2
0.1 〜2
10〜20
10〜20
16
ジクロフェンチオン [ECP] (C10H13Cl2O3PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
0.1 〜2
0.1 〜2
10〜20
10〜20
18
ジチオピル (C15H16F5NO2S2)
0.4 〜8
0.4 〜8
10〜20
10〜20
19
シマジン [CAT] (C7H12ClN5)
0.1 〜2
0.1 〜2
10〜20
10〜20
20
シメトリン (C8H15N5S)
0.05 〜1
0.05 〜1
10〜20
10〜20
21
ダイアジノン (C12H21N2O3PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
0.05 〜1
0.05 〜1
10〜20
10〜20
23
テルブカルブ [MBPMC] (C17H27NO2)
0.1 〜2
0.1 〜2
10〜20
10〜20
24
トリクロピルブトキシエチル (C13H16Cl3NO4) (1)
0.4 〜8
0.4 〜8
10〜20
10〜20
25
トリクロルホン [DEP] (C4H8Cl3O4P)
0.5 〜10
0.5 〜10
20〜30
20〜30
26
トリシクラゾール (C9H7N3S)
0.1 〜2
0.1 〜2
10〜20
10〜20
27
トルクロホスメチル (C9H11Cl2O3PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
28
ナプロパミド (C17H21NO2)
0.1 〜2
0.1 〜2
10〜20
10〜20
29
ピリダフェンチオン (C14H17N2O4PS)
0.2 〜4
0.2 〜4
20〜30
20〜30
30
ピリブチカルブ (C18H22N2O2S)
0.1 〜2
0.1 〜2
10〜20
10〜20
31
フェニトロチオン [MEP] (C9H12NO5PS)
0.1 〜2
0.1 〜2
10〜20
10〜20
32
フェノブカルブ [BPMC] (C12H17NO2)
0.1 〜2
0.1 〜2
10〜20
10〜20
33
フサライド (C8H2Cl4O2)
0.1 〜2
0.1 〜2
10〜20
10〜20
34
ブタミホス (C13H21N2O4PS)
0.1 〜2
0.1 〜2
20〜30
20〜30
35
ブプロフェジン (C16H23N3OS)
0.1 〜2
0.1 〜2
10〜20
10〜20
36
フルトラニル (C17H16F3NO2)
0.1 〜2
0.1 〜2
20〜30
20〜30
37
プレチラクロール (C17H26ClNO2)
0.1 〜2
0.1 〜2
10〜20
10〜20
38
プロピザミド (C12H11Cl2NO)
0.1 〜2
0.1 〜2
10〜20
10〜20
39
プロベナゾール (C10H9NO3S)
0.4 〜8
0.4 〜8
10〜20
10〜20
40
ブロモブチド[ブロモチド] (C15H22BrNO) (2)
0.1 〜2
0.1 〜2
10〜20
10〜20
41
ペンシクロン (C19H21ClN2O)
0.1 〜2
0.1 〜2
10〜20
10〜20
42
ベンスリド [SAP] (C14H24NO4PS3)
0.5 〜10
0.5 〜10
10〜20
10〜20
43
ペンジメタリン (C13H19N3O4)
0.1 〜2
0.1 〜2
10〜20
10〜20
44
ベンフルラリン[ベスロジン] (C13H16F3N3O4)
0.1 〜2
0.1 〜2
10〜20
10〜20
45
マラチオン(マラソン) (C10H19O6PS2)
0.1 〜2
0.1 〜2
10〜20
10〜20
46
メタラキシル (C15H21NO4)
0.4 〜8
0.4 〜8
10〜20
10〜20

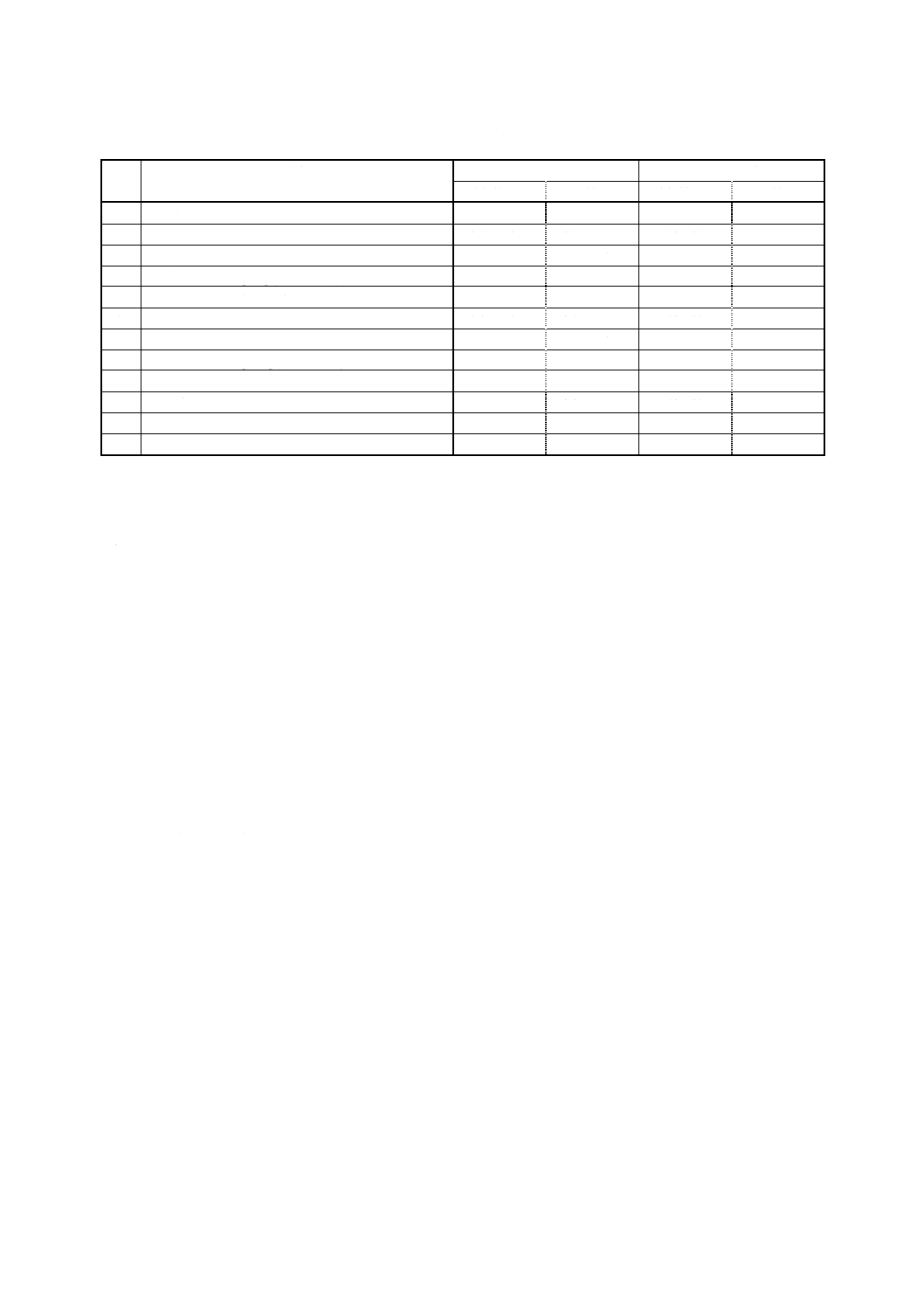
12
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
溶媒抽出法
固相抽出法
溶媒抽出法
固相抽出法
47
メチルダイムロン (C17H20N2O)
0.1 〜2
0.1 〜2
10〜20
10〜20
48
メフェナセット (C16H14N2O2S)
0.4 〜8
0.4 〜8
10〜20
10〜20
49
メプロニル (C17H19NO2)
0.1 〜2
0.1 〜2
10〜20
10〜20
50
モリネート (C9H17NOS)
0.4 〜8
0.4 〜8
10〜20
10〜20
注(1) トリクロピルの定量は,ガスクロマトグラフ質量分析法又は38.1でトリクロピルブトキシエチルを定量し,
これに係数0.719 2を乗じた値と,別に,トリクロピル(遊離)を7.3又は38.1によって定量し,両者の合量を
求めてトリクロピルとする。
(2) ブロモブチドの定量は,ガスクロマトグラフ質量分析法を用いて,ブロモブチドの量 (ng) とブロモブチド
脱臭素体の量 (ng) に係数1.338を乗じてブロモブチドに換算したものとを合計して,d)4)の式によって濃度
を算出して求める。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
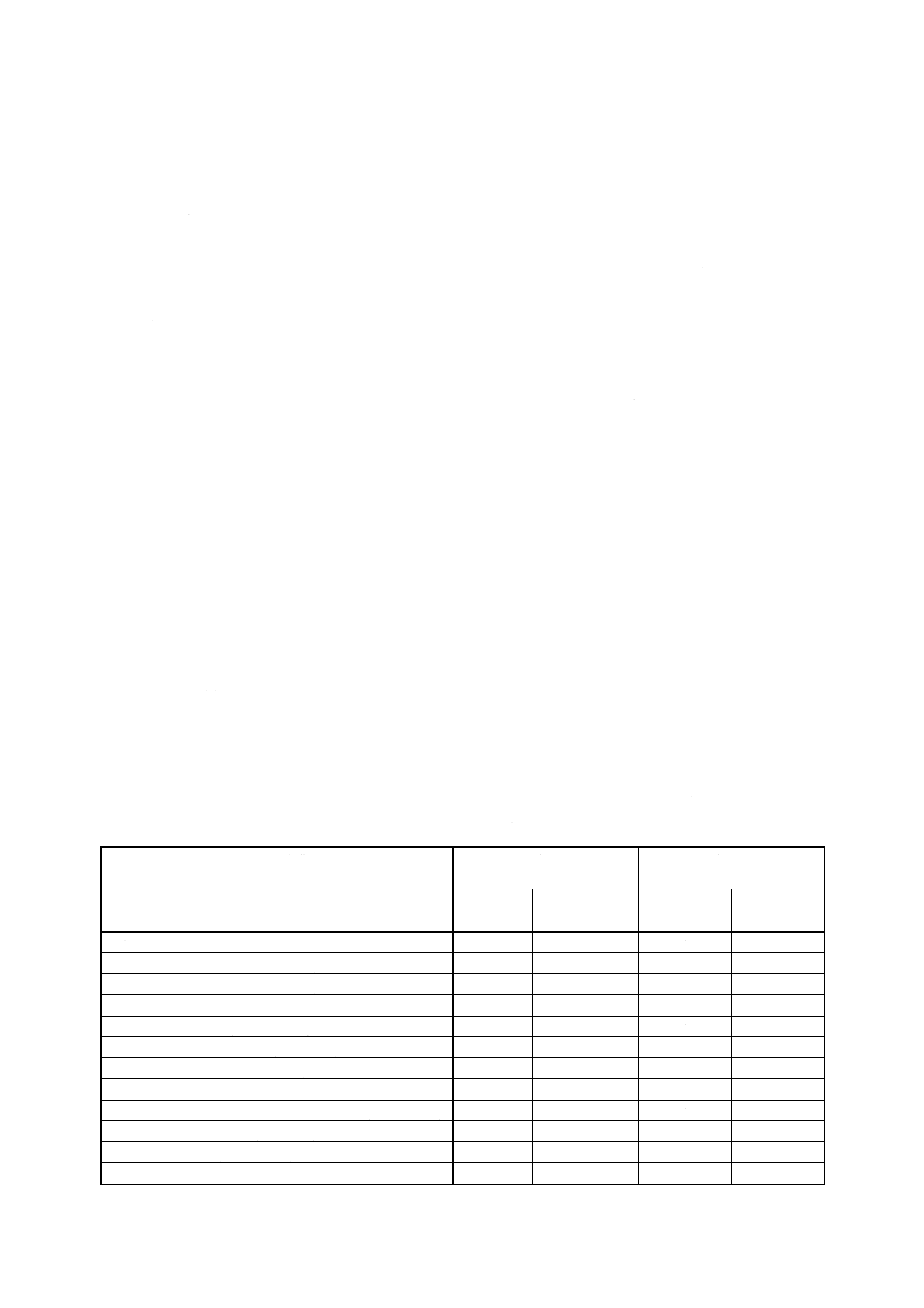
6) 対象農薬標準液 (0.2〜5mg/ml) 対象農薬の標準液の調製に用いる標準品の採取量及び溶解に使
用する溶媒を表2に示す(3)。これら対象農薬の標準液の調製は,次による。
対象農薬の標準品の一定量[採取量は表2の表中の濃度 (A) に示す。]を,対象農薬別に少量の
アセトン又はヘキサンに溶かし,全量フラスコ100mlにそれぞれ移し入れ,アセトン又はヘキサン
をそれぞれ標線まで加える(4)。
7) 混合標準液 (5〜50μg/ml) 6)で個別に調製した対象農薬標準液 (0.2〜5mg/ml) の一定量(採取量は,
表2の表中の混合標準液に示す。)を全量フラスコ100mlにとり,ヘキサンを標線まで加える(5)。保
存する場合は,−20℃の暗所におく。
8) 混合標準液 (0.5〜5μg/ml) 7)で調製した混合標準液 (5〜50μg/ml) 1mlを全量フラスコ10mlにと
り,ヘキサンを標線まで加える(5)。使用時に調製する。
9) 窒素 6.2a)2)による。
注(3) イソキサチオン,クロルニトロフェン,クロロタロニル,ジクロルボス,フェノブカルブ及び
プロピザミドについては,それぞれの標準品の一定量を少量のアセトンに溶かし,全量フラス
コ100mlに移し入れ,ヘキサンを標線まで加える。
(4) 標準液を長期間保存する場合は,調製した標準液を直ちにアンプルに入れ,溶封し,−20℃の
暗所におく。
(5) 6.2の前処理操作を行った場合の混合標準液の調製には,ヘキサンに代えアセトンを用いる。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) マイクロシリンジ 1〜10μlの適切なもの。又は自動注入装置を用いてもよい。
13
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) ガスクロマトグラフ質量分析計 次に掲げる条件を満たすもの(6)。
7.1)
キャピラリーカラム用管 内径0.2〜0.7mm,長さ10〜60mのステンレス鋼,石英ガラス又はほう
けい酸ガラス製のもの。又は同等のもの。
7.2)
キャピラリーカラム キャピラリーカラム用管の内壁にメチルシリコーン系固定相液体(7)0.1〜
1.0μmの厚さで被覆したもの。又は同等の分離性能をもつもの。
参考1. この試験に用いるキャピラリーカラムには,メチルシリコーン系固定相液体として100%メチ
ルシリコーンを用いているDB-1,CBP-1,CP-SIL-5CB,NB-1,SPB-1,Ultra-1などがある。
7.3)
検出器 選択イオン検出法 (SIM) 又はマスクロマトグラフ法 (MC) が行えるもの。
7.4)
キャリヤーガス ヘリウム(99.999 9vol%以上),線速度は20〜40cm/sの範囲に調節して用いる。
7.5)
試料導入方法及び試料導入部温度 試料導入方法は,スプリットレス注入法(非分割導入方式)又
はコールドオンカラム注入法(全量導入方式)による。試料導入部温度は,スプリットレス注入法
の場合は220〜280℃,コールドオンカラム注入法の場合は50〜100℃
7.6)
GC/MS接続部(8)温度 200〜280℃
7.7)
イオン源温度 200〜250℃
7.8)
電子加速電圧 20〜70V
7.9)
昇温プログラム 50〜280℃(2〜30℃/minの昇温)
8) 振とう器
9) 濃縮器 6.1b)5)による。
注(6) これらの条件は,装置,測定条件によって異なる。
(7) メチルシリコーン系固定相液体としては,100%メチルシリコーン,5%フェニル−95%メチルシ
リコーン及び50%フェニル−50%メチルシリコーンなど,又はこれと同等の分離性能をもつも
のを用いる。
(8) 装置によっては,セパレーター部という場合もある。
備考1. ガスクロマトグラフ質量分析計の感度として,混合標準液 (0.05〜0.5μg/ml) [a)8)の混合標
準液 (0.5〜5μg/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入
し,対象農薬の量 (0.05〜0.5ng) が確認できるように検出器の感度を調節する。
表2 対象農薬標準液及び混合標準液
番
号
対象農薬
濃度 (A)
0.2〜5mg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
1
EPN (C14H14NO4PS)
ヘキサン
0.100
1
10
2
イソキサチオン (C13H16NO4PS)
注(3)
0.100
1
10
3
イソフェンホス (C15H24NO4PS)
アセトン
0.100
1
10
4
イソプロチオラン (C12H18O4S2)
ヘキサン
0.100
1
10
5
イプロジオン (C13H13Cl2N3O3)
アセトン
0.200
1
20
6
イプロベンホス [IBP] (C13H21O3PS)
ヘキサン
0.100
1
10
7
エスプロカルブ (C15H23NOS)
アセトン
0.400
1
40
8
エジフェンホス [EDDP] (C14H15O2PS2)
アセトン
0.400
1
40
9
エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS)
アセトン
0.300
1
30
10
カルバリル [NAC] (C12H11NO2)
アセトン
0.400
1
40
11
キャプタン (C9H8Cl3NO2S)
アセトン
0.100
1
10
12
クロルニトロフェン [CNP] (C12H6Cl3NO3)
注(3)
0.100
1
10

14
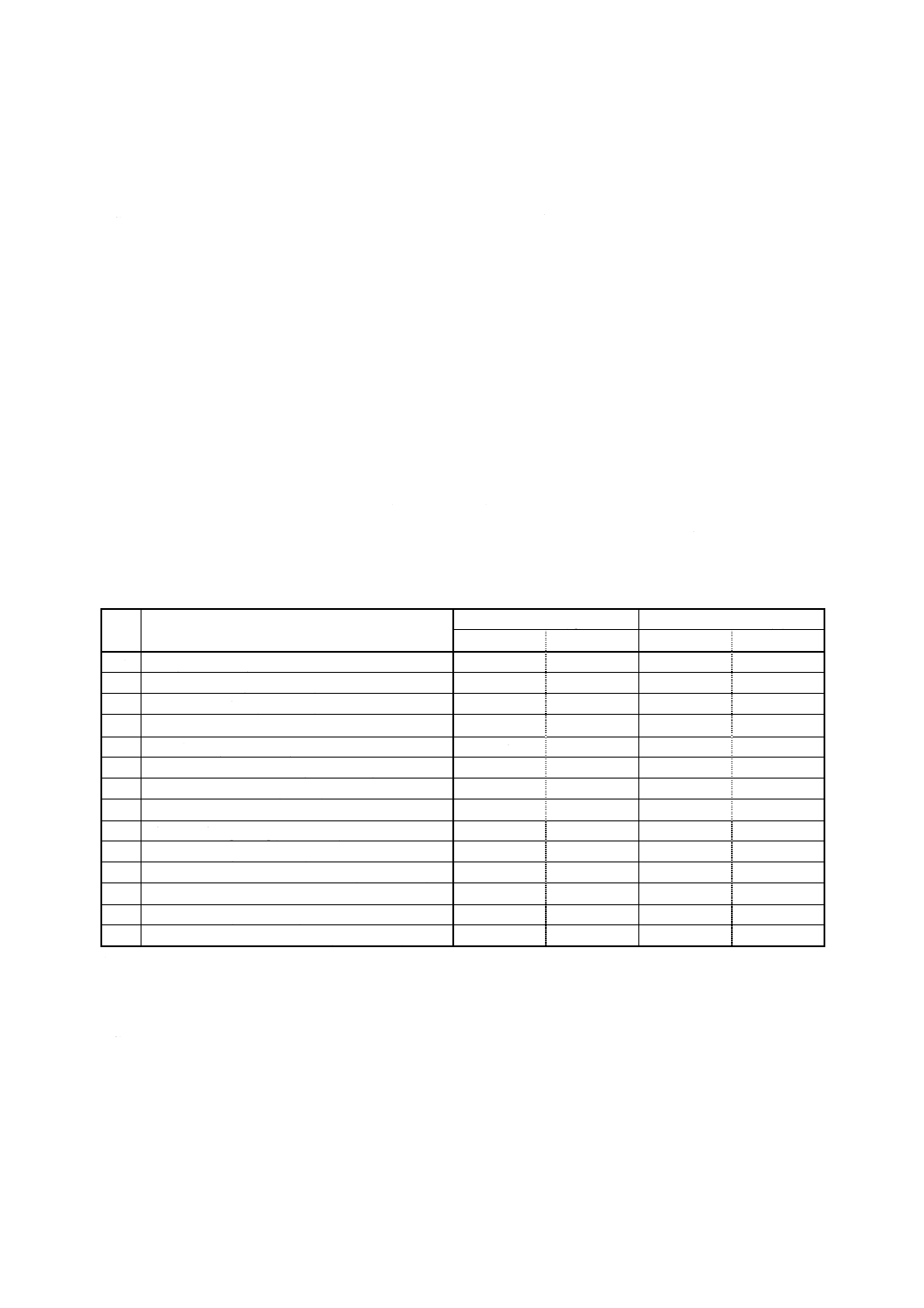
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
番
号
対象農薬
濃度 (A)
0.2〜5mg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
13
クロルピリホス (C9H11Cl3NO3PS)
アセトン
0.100
1
10
14
クロロタロニル [TPN] (C8Cl4N2)
注(3)
0.100
1
10
15
クロロネブ (C8H8Cl2O2)
アセトン
0.100
1
10
16
ジクロフェンチオン [ECP] (C10H13Cl2O3PS)
アセトン
0.100
1
10
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
注(3)
0.100
1
10
18
ジチオピル (C15H16F5NO2S2)
アセトン
0.400
1
10
19
シマジン [CAT] (C7H12ClN5)
アセトン
0.020
2.5
5
20
シメトリン (C8H15N5S)
アセトン
0.100
1
10
21
ダイアジノン (C12H21N2O3PS)
ヘキサン
0.100
1
10
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
ヘキサン
0.050
1
5
23
テルブカルブ [MBPMC] (C17H27NO2)
アセトン
0.100
1
10
24
トリクロピルブトキシエチル (C13H16Cl3NO4)
アセトン
0.400
1
40
25
トリクロルホン [DEP] (C4H8Cl3O4P)
アセトン
0.500
1
50
26
トリシクラゾール (C9H7N3S)
アセトン
0.100
1
10
27
トルクロホスメチル (C9H11Cl2O3PS)
アセトン
0.100
1
10
28
ナプロパミド (C17H21NO2)
アセトン
0.100
1
10
29
ピリダフェンチオン (C14H17N2O4PS)
アセトン
0.200
1
20
30
ピリブチカルブ (C18H22N2O2S)
アセトン
0.100
1
10
31
フェニトロチオン [MEP] (C9H12NO5PS)
ヘキサン
0.100
1
10
32
フェノブカルブ [BPMC] (C12H17NO2)
注(3)
0.100
1
10
33
フサライド (C8H2Cl4O2)
アセトン
0.100
1
10
34
ブタミホス (C13H21N2O4PS)
アセトン
0.100
1
10
35
ブプロフェジン (C16H23N3OS)
アセトン
0.100
1
10
36
フルトラニル (C17H16F3NO2)
アセトン
0.100
1
10
37
プレチラクロール (C17H26ClNO2)
アセトン
0.100
1
10
38
プロピザミド C12H11Cl2NO)
注(3)
0.100
1
10
39
プロベナゾール (C10H9NO3S)
アセトン
0.400
1
40
40
ブロモブチド[ブロモチド] (C15H22BrNO)
アセトン
0.050
1
5
ブロモブチド脱臭素体 (C15H23NO)
アセトン
0.050
1
5
41
ペンシクロン (C19H21ClN2O)
アセトン
0.100
1
10
42
ベンスリド [SAP] (C14H24NO4PS3)
アセトン
0.500
1
50
43
ペンジメタリン (C13H19N3O4)
アセトン
0.100
1
10
44
ベンフルラリン[ベスロジン] (C13H16F3N3O4)
アセトン
0.100
1
10
45
マラチオン(マラソン) (C10H19O6PS2)
アセトン
0.100
1
10
46
メタラキシル (C15H21NO4)
アセトン
0.400
1
40
47
メチルダイムロン (C17H20N2O)
アセトン
0.100
1
10
48
メフェナセット (C16H14N2O2S)
アセトン
0.400
1
40
49
メプロニル (C17H19NO2)
アセトン
0.100
1
10
50
モリネート (C9H17NOS)
アセトン
0.400
1
40
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)の混合標準液 (0.5〜5μg/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,対象農薬特有のフラグメントイオン

15
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(m/z) を設定し(10),選択イオン検出法又はマスクロマトグラフ法によって測定してそのマスフラグ
メントグラムを記録し,対象農薬の保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(9)でとり,1)の操作を行ってマスフラグメントグラムを記録
し,1)の保持時間と一致していることを確認し,保持時間に相当する位置のピークについて,指示
値(11)を読み取る。
3) 空試験として,c)で得た空試験の濃縮液について1)及び2)の操作を行い,対象農薬の保持時間に相
当する位置にピークが検出され,その指示値(11)が定量下限値の指示値の31以上である場合には,準
備操作から再度操作し直す。
4) 検量線から対象農薬の量を求め,次の式によって試料中の対象農薬の濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.1c)5)のヘキサンの量又は6.2c)4)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)7)の混合標準液 (5〜50μg/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,ヘキ
サン(5)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロ
シリンジでとり,1)及び2)の操作を行って対象農薬の量 (ng) と指示値(11)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(9) 検量線作成時と同じものを用いる。
(10) 特有のフラグメントイオンを設定するには表3を参考にするとよい。
(11) ピーク高さ又はピーク面積
備考2. 分析した農薬の確認が必要な場合には,試料を濃縮し,対象農薬のマススペクトルをとる。
試料中の対象農薬の濃度が低く,マススペクトルが得られない場合は,複数のフラグメント
イオンを設定し,測定して得られたクロマトグラムのイオン強度比を求める。対象農薬の標
準品をこれと同様に測定して得られたマススペクトル又はピーク強度比とを比較して判断す
る。
3. ナフタレン-d8 (m/z 136),フェナントレン-d10 (m/z 188),フルオランテン-d10 (m/z 212),クリ
セン-d10 (m/z 240) などの内標準物質を用いた方法によって定量を行ってもよい。
表3 選択イオン検出法におけるフラグメントイオンの一例
番
号
対象農薬
フラグメントイオン (m/z) (12)(13)
1
EPN (C14H14NO4PS)
157
169
185
141
−
2
イソキサチオン (C13H16NO4PS)
105
177
77
313
−
3
イソフェンホス (C15H24NO4PS)
213
121
255
185
−
4
イソプロチオラン (C12H18O4S2)
118
162
189
290
204
5
イプロジオン (C13H13Cl2N3O3)
314
316
187
189
−
6
イプロベンホス [IBP] (C13H21O3PS)
91
204
123
−
−
7
エスプロカルブ (C15H23NOS)
91
222
71
162
−
8
エジフェンホス [EDDP] (C14H15O2PS2)
109
173
310
201
−
9
エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS)
211
183
213
185
140

16
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
番
号
対象農薬
フラグメントイオン (m/z) (12)(13)
10
カルバリル [NAC] (C12H11NO2)
144
115
−
−
−
11
キャプタン (C9H8Cl3NO2S)
79
149
264
−
−
12
クロルニトロフェン [CNP] (C12H6Cl3NO3)
317
319
236
287
289
13
クロルピリホス (C9H11Cl3NO3PS)
197
199
314
316
258
14
クロロタロニル [TPN] (C8Cl4N2)
266
264
268
−
−
15
クロロネブ (C8H8Cl2O2)
191
193
206
208
141
16
ジクロフェンチオン [ECP] (C10H13Cl2O3PS)
279
223
97
251
162
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
109
185
79
−
−
18
ジチオピル (C15H16F5NO2S2)
354
306
−
−
−
19
シマジン [CAT] (C7H12ClN5)
201
186
173
158
−
20
シメトリン (C8H15N5S)
213
170
155
−
−
21
ダイアジノン (C12H21N2O3PS)
179
137
304
152
199
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
100
72
125
257
−
23
テルブカルブ [MBPMC] (C17H27NO2)
205
220
145
−
−
24
トリプロピルブトキシエチル (C13H16Cl3NO4)
57
85
210
212
182
25
トリクロルホン [DEP] (C4H8Cl3O4P)
109
79
145
139
−
26
トリシクラゾール (C9H7N3S)
189
162
161
−
−
27
トルクロホスメチル (C9H11Cl2O3PS)
265
267
125
−
−
28
ナプロパミド (C17H21NO2)
72
128
271
100
115
29
ピリダフェンチオン (C14H17N2O4PS)
340
199
188
97
77
30
ピリブチカルブ (C18H22N2O2S)
165
108
181
−
−
31
フェニトロチオン [MEP] (C9H12NO5PS)
277
125
109
260
79
32
フェノブカルブ [BPMC] (C12H17NO2)
121
150
91
−
−
33
フサライド (C8H2Cl4O2)
243
241
245
272
270
34
ブタミホス (C13H21N2O4PS)
286
200
232
202
258
35
ブプロフェジン (C16H23N3OS)
105
172
106
175
305
36
フルトラニル (C17H16F3NO2)
173
281
145
−
−
37
プレチラクロール (C17H26ClNO2)
162
238
176
202
262
38
プロピザミド (C12H11Cl2NO)
173
175
255
145
254
39
プロベナゾール (C10H9NO3S)
130
103
159
104
132
40
ブロモブチド[ブロモチド] (C15H22BrNO)
119
120
118
232
91
ブロモブチド脱臭素体 (C15H23NO)
119
233
177
−
−
41
ペンシクロン (C19H21ClN2O)
125
180
127
209
182
42
ベンスリド [SAP] (C14H24NO4PS3)
77
131
141
−
−
43
ペンジメタリン (C13H19N3O4)
252
281
162
−
−
44
ベンフルラリン[ベスロジン] (C13H16F3N3O4)
292
264
276
−
−
45
マラチオン(マラソン) (C10H19O6PS2)
173
125
127
93
158
46
メタラキシル (C15H21NO4)
206
249
220
234
−
47
メチルダイムロン (C17H20N2O)
107
119
91
−
−
48
メフェナセット (C16H14N2O2S)
192
120
136
−
−
49
メプロニル (C17H19NO2)
119
269
91
−
−
50
モリネート (C9H17NOS)
126
55
187
−
−
注(12) フラグメントイオン強度の大きいものから順次表示した。
(13) 試料の測定において妨害するフラグメントイオンは避ける。また,フラグメントイオン100以下の使
用は避けることが望ましい。
7.2
ガスクロマトグラフ法 熱イオン化検出器 (FTD),炎光光度検出器 (FPD) 及び電子捕獲検出器
(ECD) をそれぞれ用いたガスクロマトグラフ法を適用する。
17
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2.1
熱イオン化検出器 (FTD) を用いたガスクロマトグラフ法 試料中の農薬を,前処理に6.1又は6.2
を適用して濃縮した後,その一定量を,熱イオン化検出器を用いた方法で定量する。対象農薬とその定量
範囲及び繰返し分析精度(変動係数で)は,表4のとおりである(いずれも装置,測定条件によって異な
る。)。
表4 対象農薬とその定量範囲及び繰返し分析精度 (FTD)
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
溶媒抽出法
固相抽出法
溶媒抽出法
固相抽出法
1
EPN (C14H14NO4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
2
イソキサチオン (C13H16NO4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
3
イソフェンホス (C15H24NO4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
5
イプロジオン (C13H13Cl2N3O3)
0.2 〜 4
0.2 〜 4
20〜30
20〜30
6
イプロベンホス [IBP] (C13H21O3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
13
クロルピリホス (C9H11Cl3NO3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
14
クロロタロニル [TPN] (C8Cl4N2)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
19
シマジン [CAT] (C7H12ClN5)
0.05 〜 1
0.05 〜 1
10〜20
10〜20
21
ダイアジノン (C12H21N2O3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
23
テルブカルブ [MBPMC] (C17H27NO2)
0.4 〜 8
0.4 〜 8
10〜20
10〜20
27
トルクロホスメチル (C9H11Cl2O3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
28
ナプロパミド (C17H21NO2)
0.4 〜 8
0.4 〜 8
10〜20
10〜20
29
ピリダフェンチオン (C14H17N2O4PS)
0.2 〜 4
0.2 〜 4
10〜20
10〜20
30
ピリブチカルブ (C18H22N2O2S)
0.05 〜 1
0.05 〜 1
10〜20
10〜20
31
フェニトロチオン [MEP] (C9H12NO5PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
32
フェノブカルブ [BPMC] (C12H17NO2)
0.6 〜 12
0.6 〜 12
10〜20
10〜20
34
ブタミホス (C13H21N2O4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
36
フルトラニル (C17H16F3NO2)
0.2 〜 4
0.2 〜 4
10〜20
10〜20
38
プロピザミド (C12H11Cl2NO)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
41
ペンシクロン (C19H21ClN2O)
0.4 〜 8
0.4 〜 8
10〜20
10〜20
42
ベンスリド [SAP] (C14H24NO4PS3)
0.5 〜 10
0.5 〜 10
10〜20
10〜20
43
ペンジメタリン (C13H19N3O4)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
47
メチルダイムロン (C17H20N2O)
1.0 〜 20
1.0 〜 20
10〜20
10〜20
49
メプロニル (C17H19NO2)
1.0 〜 20
1.0 〜 20
10〜20
10〜20
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) 対象農薬標準液 (0.5〜6mg/ml) 対象農薬の標準液の調製に使用する標準品の採取量及び溶解に
使用する溶媒を表5に示す(14)。これら対象農薬標準液の調製は,次による。
対象農薬標準品の一定量[採取量は表5の表中の濃度 (A) に示す。]を対象農薬別に少量のアセ
トン又はヘキサンに溶かし,全量フラスコ100mlにそれぞれ移し入れ,アセトン又はヘキサンをそ
れぞれ標線まで加える(4)。
7) 混合標準液 (5〜100μg/ml) 6)で個別に調製した対象農薬標準液 (0.5〜6mg/ml) について,それぞ
18
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
れの一定量(採取量は表5の表中の混合標準液に示す。)を量フラスコ100mlにとり,ヘキサンを
標線まで加える(5)。保存する場合は,−20℃の暗所におく。
8) 混合標準液 (0.5〜10μg/ml) 7)で調製した混合標準液 (5〜100μg/ml) 1mlを全量フラスコ10mlに
とり,ヘキサンを標線まで加える(5)。使用時に調製する。
9) 窒素 6.2a)2)による。
注(14) イソキサチオン,クロロタロニル,ジクロルボス,フェノブカルブ及びプロピザミドについて
は,注(3)による。
表5 対象農薬標準液及び混合標準液 (FTD)
番
号
対象農薬
濃度 (A)
0.5〜6mg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
1
EPN (C14H14NO4PS)
ヘキサン
0.100
1
10
2
イソキサチオン (C13H16NO4PS)
注(14)
0.100
1
10
3
イソフェンホス (C15H24NO4PS)
アセトン
0.100
1
10
5
イプロジオン (C13H13Cl2N3O3)
アセトン
0.200
1
20
6
イプロベンホス [IBP] (C13H21O3PS)
ヘキサン
0.100
1
10
13
クロルピリホス (C9H11Cl3NO3PS)
アセトン
0.100
1
10
14
クロロタロニル [TPN] (C8Cl4N2)
注(14)
0.100
1
10
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
注(14)
0.100
1
10
19
シマジン [CAT] (C7H12ClN5)
アセトン
0.020
2.5
5
21
ダイアジノン C12H21N2O3PS)
ヘキサン
0.100
1
10
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
ヘキサン
0.100
1
10
23
テルブカルブ [MBPMC] (C17H27NO2)
アセトン
0.400
1
40
27
トルクロホスメチル (C9H11Cl2O3PS)
アセトン
0.100
1
10
28
ナプロパミド (C17H21NO2)
アセトン
0.400
1
40
29
ピリダフェンチオン (C14H17N2O4PS)
アセトン
0.200
1
20
30
ピリブチカルブ (C18H22N2O2S)
アセトン
0.050
1
5
31
フェニトロチオン [MEP] (C9H12NO5PS)
ヘキサン
0.100
1
10
32
フェノブカルブ [BPMC] (C12H17NO2)
注(14)
0.600
1
60
34
ブタミホス (C13H21N2O4PS)
アセトン
0.100
1
10
36
フルトラニル (C17H16F3NO2)
アセトン
0.200
1
20
38
プロピザミド (C12H11Cl2NO)
注(14)
0.100
1
10
41
ペンシクロン (C19H21ClN2O)
アセトン
0.400
1
40
42
ベンスリド [SAP] (C14H24NO4PS3)
アセトン
0.500
1
50
43
ペンジメタリン (C13H19N3O4)
アセトン
0.100
1
10
47
メチルダイムロン (C17H20N2O)
アセトン
0.200
5
100
49
メプロニル (C17H19NO2)
アセトン
0.200
5
100
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) マイクロシリンジ 7.1b)6)による。
19
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 熱イオン化検出器(15)
7.4)
キャリヤーガス ヘリウム(99.999 9vol%以上)を用い,線速度は20〜40cm/sの範囲に調節して用
いる。カラム出口には付加ガス(16)としてキャリヤーガスと同じガスを接続し,流量は30〜60ml/min
に調節して用いる。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
7.6)
燃料ガス及び助燃ガス 燃料ガスにはJIS K 0512に規定する水素4級,助燃ガスには精製した空気
を用いる。流量は,燃料ガスは2〜10ml/min,助燃ガスは100〜180ml/minに設定する。
7.7)
カラム槽温度 50〜60℃で2分間保ち,50(又は60)〜260℃の範囲で,2〜20℃/minの昇温を行う
(17)。
7.8)
検出器槽温度 250〜280℃
8) 振とう器
9) 濃縮器 6.1b)5)による。
注(15) 窒素及びりんに対して高感度に検出できる高感度窒素りん検出器 (NPD) を用いてもよい。
(16) 内径0.53mm以上の充てんキャピラリーカラムを用いた場合には,付加ガスを用いなくてもよ
い。
(17) 最終溶媒にヘキサンを用いた場合のカラム槽温度である。最終溶媒にアセトンを用いる場合の
カラム槽の温度調節は,次による。
最終溶媒にアセトンを用いた場合 40〜50℃で2分間保ち,40(又は50)〜280℃の範囲で,2
〜20℃/minの昇温を行う。
備考4. ガスクロマトグラフの感度として,混合標準液 (0.05〜1μg/ml) [a)8)の混合標準液 (0.5〜
10μg/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入し,対象農薬の量 (0.05
〜1ng) が確認できるように検出器の感度を調節する。
5. c)の準備操作に引き続き,6.3の操作を行った場合は,ガスクロマトグラフの分離カラムとし
て充てんカラムを用いることができる。この場合のガスクロマトグラフの条件は,次のとお
りである。
ガスクロマトグラフ 次に掲げる条件を満たすもの。
1) カラム用管 内径2〜3mm,長さ1 000〜1 500mmのガラス管。
2) カラム充てん剤 耐火れんが(*1)(粒径150〜250μmのもの)を塩酸 (6mol/L) (JIS K 8180
に規定する塩酸を用いて調製したもの。)で約2時間加熱・還流して洗い,水で洗液が中性に
なるまで洗った後,乾燥する。次に,メチルシラザン処理した後,シリコーン系固定相液体
約5%を含浸させたもの。
3) 検出器 熱イオン化検出器(*2)
4) キャリヤーガス ヘリウム(99.999 9vol%以上)を用い,流量は40〜50ml/minに調節して用
いる。
5) 燃料ガス及び助燃ガス b)7.6)による。
6) 試料気化室温度 250〜300℃
7) カラム槽温度(*3) 180〜220℃
20
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) 検出器槽温度 250〜280℃
注(*1) けい藻土を主成分とした耐火度1 100℃のれんが。
(*2) 注(15)による。
(*3) 農薬によってカラム槽温度が異なることがあるので,対象農薬によってカラム槽温度を設
定する。
参考2. カラム充てん剤の市販品には,耐火れんがとしてクロモゾルブW,クロモゾルムG又はこれ
と同等の性能をもつものを担体とし,これにシリコーン系固定相液体としてDC-200,OV-1,
V-17などを含浸させたものがある。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)の混合標準液 (0.5〜10μg/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,対象農薬特有の保持時間に相当する
ピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(9)でとり,1)の操作を行ってガスクロマトグラムを記録し,
保持時間が1)の保持時間と一致していることを確認し,保持時間に相当する位置のピークについて,
指示値(11)を読み取る。
3) 空試験として,c)で得た空試験の濃縮液について1)及び2)の操作を行って,対象農薬の保持時間に
相当する位置にピークが検出され,その指示値(11)が定量下限値の指示値の31以上である場合には,
準備操作から再度操作し直す。
4) 検量線から対象農薬の量を求め,7.1d)4)の式によって試料中の対象農薬の濃度 (μg/L) を算出する。
検量線 a)7)の混合標準液 (5〜100μg/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,ヘキサン
(5)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロシリンジ
でとり,1)及び2)の操作を行って対象農薬の量 (ng) と指示値(11)との関係線を作成する。検量線
の作成は,試料測定時に行う。
備考6. 備考2.による。
7.2.2
炎光光度検出器 (FPD) を用いたガスクロマトグラフ法 試料中の農薬を,前処理に6.1又は6.2
を適用して濃縮した後,その一定量を,炎光光度検出器を用いた方法で定量する。対象農薬とその定量範
囲及び繰返し分析精度(変動係数で)は,表6のとおりである(いずれも装置,測定条件によって異なる。)。
21
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表6 対象農薬とその定量範囲及び繰返し分析精度 (FPD)
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
溶媒抽出法
固相抽出法
溶媒抽出法
固相抽出法
1
EPN (C14H14NO4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
2
イソキサチオン (C13H16NO4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
3
イソフェンホス (C15H24NO4PS)
0.05 〜 1
0.05 〜 1
10〜20
10〜20
6
イプロベンホス [IBP] (C13H21O3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
13
クロルピリホス (C9H11Cl3NO3PS)
0.05 〜 1
0.05 〜 1
10〜20
10〜20
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
21
ダイアジノン (C12H21N2O3PS)
0.05 〜 1
0.05 〜 1
10〜20
10〜20
25
トリクロルホン [DEP] (C4H8Cl3O4P)
1.0 〜20
1.0 〜20
10〜20
10〜20
27
トルクロホスメチル (C9H11Cl2O3PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
29
ピリダフェンチオン (C14H17N2O4PS)
0.2 〜 4
0.2 〜 4
10〜20
10〜20
31
フェニトロチオン [MEP] (C9H12NO5PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
34
ブタミホス (C13H21N2O4PS)
0.1 〜 2
0.1 〜 2
10〜20
10〜20
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) 対象農薬標準液 (0.5〜2mg/ml) 対象農薬の標準液の調製に使用する標準品の採取量及び溶解に
使用する溶媒を表7に示す(18)。これら対象農薬標準液の調製は,次による。
対象農薬標準品の一定量[採取量は表7の表中の濃度 (A) に示す。]を対象農薬別に少量のアセ
トン又はヘキサンに溶かし,全量フラスコ100mlにそれぞれ移し入れ,アセトン又はヘキサンをそ
れぞれ標線まで加える(4)。
7) 混合標準液 (5〜100μg/ml) 6)で個別に調製した対象農薬標準液 (1〜2mg/ml) について,それぞれ
の一定量(採取量は表7の表中の混合標準液に示す。)を全量フラスコ100mlにとり,ヘキサンを
標線まで加える(5)。
保存する場合は,−20℃の暗所におく。
8) 混合標準液 (0.5〜10μg/ml) 7)で調製した混合標準液 (5〜100μg/ml) 1mlを全量フラスコ10mlに
とり,ヘキサンを標線まで加える(5)。使用時に調製する。
9) 窒素 6.2a)2)による。
注(18) イソキサチオン及びジクロルボスについては,注(3)による。
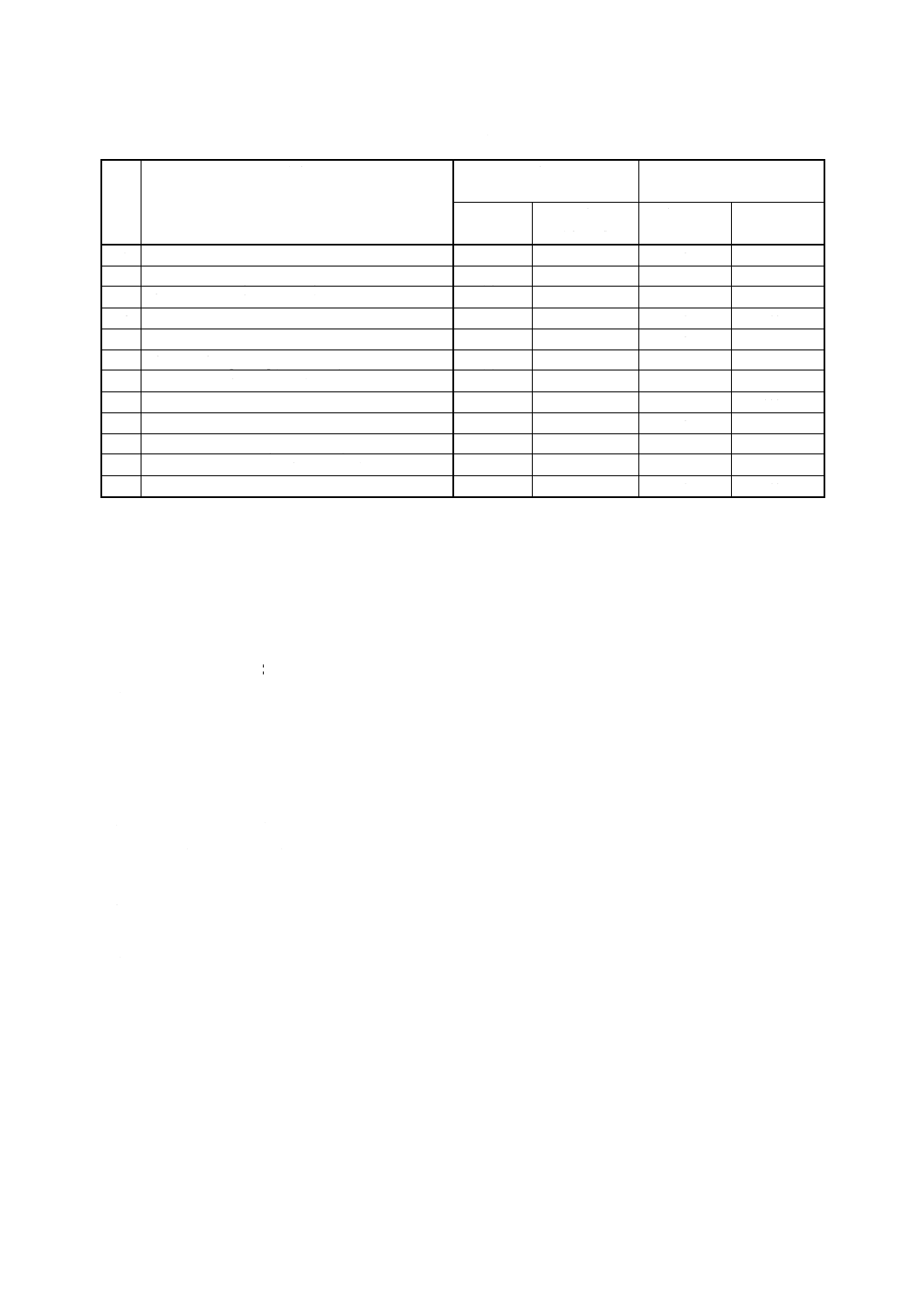
22
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7 対象農薬標準液及び混合標準液 (FPD)
番
号
対象農薬
濃度 (A)
0.5〜2mg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
1
EPN (C14H14NO4PS)
ヘキサン
0.100
1
10
2
イソキサチオン (C13H16NO4PS)
注(18)
0.100
1
10
3
イソフェンホス (C15H24NO4PS)
アセトン
0.050
1
5
6
イプロベンホス [IBP] (C13H21O3PS)
ヘキサン
0.100
1
10
13
クロルピリホス (C9H11Cl3NO3PS)
アセトン
0.050
1
5
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
注(18)
0.100
1
10
21
ダイアジノン (C12H21N2O3PS)
ヘキサン
0.050
1
5
25
トリクロルホン [DEP] (C4H8Cl3O4P)
アセトン
0.200
5
100
27
トルクロホスメチル (C9H11Cl2O3PS)
アセトン
0.100
1
10
29
ピリダフェンチオン (C14H17N2O4PS)
アセトン
0.200
1
20
31
フェニトロチオン [MEP] (C9H12NO5PS)
ヘキサン
0.100
1
10
34
ブタミホス (C13H21N2O4PS)
アセトン
0.100
1
10
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) マイクロシリンジ 7.1b)6)による。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 炎光光度検出器。干渉フィルターとしてりんフィルターを用い,測定波長は526nmとする。
7.4)
キャリヤーガス 7.2.1b)7.4)による。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
7.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。ただし,燃料ガスは50〜100ml/min,助燃ガスは80〜
150ml/minの流量に設定する。
7.7)
カラム槽温度 7.2.1b)7.7)による。
7.8)
検出器槽温度 7.2.1b)7.8)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
備考7. 備考4.による。
8. c)の準備操作に引き続き,6.3のカラムクロマトグラフ分離を行った場合は,ガスクロマトグ
ラフの分離カラムとして充てんカラムを用いることができる。この場合のガスクロマトグラ
フの条件は,備考5.によるが,検出器,燃料ガス及び助燃ガスの条件は,次による。
1) 検出器 b)7.3)による。
2) 燃料ガス及び助燃ガス b)7.6)による。
23
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)の混合標準液 (0.5〜10μg/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,対象農薬特有の保持時間に相当する
ピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(9)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線から対象農薬の量を求め,7.1d)4)の式によって試料中の対象農薬の濃度 (mg/L) を算出する。
検量線 a)7)の混合標準液 (5〜100μg/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,ヘキサン
(5)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロシリンジ
でとり,1)及び2)の操作を行って対象農薬の量 (ng) と指示値(11)との関係線を作成する。検量線
の作成は,試料測定時に行う。
備考9. 備考2.による。
7.2.3
電子捕獲検出器 (ECD) を用いたガスクロマトグラフ法 試料中の農薬を,前処理に6.1又は6.2
を適用して濃縮した後,その一定量を,電子捕獲検出器を用いた方法で定量する。対象農薬とその定量範
囲及び繰返し分析精度(変動係数で)は,表8のとおりである(いずれも装置,測定条件によって異なる。)。
表8 対象農薬とその定量範囲及び繰返し分析精度 (ECD)
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
溶媒抽出法
固相抽出法
溶媒抽出法
固相抽出法
1
EPN (C14H14NO4PS)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
2
イソキサチオン (C13H16NO4PS)
0.05 〜1
0.05 〜1
10〜20
10〜20
4
イソプロチオラン (C12H18O4S2)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
9
エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS)
0.02 〜0.4
0.02 〜0.4
20〜30
20〜30
11
キャプタン (C9H8Cl3NO2S)
0.05 〜1
0.05 〜1
10〜20
10〜20
12
クロルニトロフェン [CNP] (C12H6Cl3NO3)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
14
クロロタロニル [TPN] (C8Cl4N2)
0.02 〜0.4
0.02 〜0.4
20〜30
20〜30
15
クロロネブ (C8H8Cl2O2)
0.1 〜2
0.05 〜1
10〜20
10〜20
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
21
ダイアジノン (C12H21N2O3PS)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
0.1 〜2
0.05 〜1
10〜20
10〜20
31
フェニトロチオン [MEP] (C9H12NO5PS)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
38
プロピザミド (C12H11Cl2NO)
0.1 〜2
0.1 〜2
10〜20
10〜20
44
ベンフルラリン[ベスロジン] (C13H16F3N3O4)
0.02 〜0.4
0.02 〜0.4
10〜20
10〜20
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) 対象農薬標準液 (0.2〜1mg/ml) 対象農薬の標準液の調製に使用する標準品の採取量及び溶解に
使用する溶媒を表9に示す(19)。これら対象農薬標準液の調製は,次による。
対象農薬標準品の一定量[採取量は表9の表中の濃度 (A) に示す。]を対象農薬別に少量のアセ

24
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
トン又はヘキサンに溶かし,全量フラスコ100mlにそれぞれ移し入れ,アセトン又はヘキサンをそ
れぞれ標線まで加える(4)。
7) 混合標準液 (2〜10μg/ml) 6)で個別に調製した対象農薬の標準液 (0.2〜1μg/ml) について,それぞ
れの一定量(採取量は表9の表中の混合標準液に示す。)を全量フラスコ100mlにとり,ヘキサン
を標線まで加える(5)。保存する場合は,−20℃の暗所におく。
8) 混合標準液 (0.2〜1μg/ml) 7)で調製した混合標準液 (2〜10μg/ml) 1mlを全量フラスコ10mlにと
り,ヘキサンを標線まで加える(5)。使用時に調製する。
9) 窒素 6.2a)2)による。
注(19) イソキサチオン,クロルニトロフェン,クロロタロニル,ジクロルボス及びプロピザミドにつ
いては,注(3)による。
表9 対象農薬標準液及び混合標準液 (ECD)
番
号
対象農薬
濃度 (A)
0.2〜1μg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
1
EPN (C14H14NO4PS)
ヘキサン
0.020
1
2
2
イソキサチオン (C13H16NO4PS)
注(19)
0.050
1
5
4
イソプロチオラン (C12H18O4S2)
ヘキサン
0.020
1
2
9
エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS)
アセトン
0.020
1
2
11
キャプタン (C9H8Cl3NO2S)
アセトン
0.050
1
5
12
クロルニトロフェン [CNP] (C12H6Cl3NO3)
注(19)
0.020
1
2
14
クロロタロニル [TPN] (C8Cl4N2)
注(19)
0.020
1
2
15
クロロネブ (C8H8Cl2O2)
アセトン
0.100
1
10
17
ジクロルボス [DDVP] (C4H7Cl2O4P)
注(19)
0.020
1
2
21
ダイアジノン (C12H21N2O3PS)
ヘキサン
0.020
1
2
22
チオベンカルブ[ベンチオカーブ] (C12H16ClNOS)
アセトン
0.100
1
10
31
フェニトロチオン [MEP] (C9H12NO5PS)
ヘキサン
0.020
1
2
38
プロピザミド (C12H11Cl2NO)
注(19)
0.100
1
10
44
ベンフルラリン[ベスロジン] (C13H16F3N3O4)
アセトン
0.020
1
2
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) マイクロシリンジ 7.1b)6)による。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 電子捕獲検出器
7.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,カラム出口には付加ガス(20)としてJIS K 1107に規定
する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用いる。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
25
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.6)
カラム槽温度 7.2.1b)7.7)による。
7.7)
検出器槽温度 7.2.1b)7.8)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
注(20) 注(16)による。
なお,付加ガスにはメタン5vol%を含むアルゴン混合ガスを用いてもよい。
備考10. ガスクロマトグラフの感度として,混合標準液 (0.02〜0.1μg/ml) [a)8)の混合標準液 (0.2〜
1μg/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入し,対象農薬の量 (0.02
〜0.1ng) が確認できるように検出器の感度を調節する。
11. c)の準備操作に引き続き,6.3のカラムクロマトグラフ分離を行った場合は,ガスクロマトグ
ラフの分離カラムとして充てんカラムを用いることができる。この場合のガスクロマトグラ
フの条件は,備考5.によるが,検出器,キャリヤーガスの条件は,次による。
1) 検出器 b)7.3)による。
2) キャリヤーガス b)7.4)による。
3) 試料気化室温度 250〜300℃
4) カラム槽温度 200〜240℃
5) 検出器槽温度 250〜300℃
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)の混合標準液 (0.2〜1μg/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,対象農薬特有の保持時間に相当する
ピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(9)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線から対象農薬の量を求め,7.1d)4)の式によって試料中の対象農薬の濃度 (μg/L) を算出する。
検量線 a)7)の混合標準液 (2〜10μg/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,ヘキサン(5)
を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロシリンジで
とり,1)及び2)の操作を行って対象農薬の量 (ng) と指示値(11)との関係線を作成する。検量線の
作成は,試料測定時に行う。
備考12. 備考2.による。
7.3
高速液体クロマトグラフ法 試料に塩酸を加えて,pHを3.5に調節し,スチレンビニルベンゼン共
重合体を充てんした固相カラムに流し入れ,アセトニトリルで展開し,溶出液に2, 2'-オキシビスエタノー
ル(ジエチレングリコール)溶液を加えて,濃縮後,アセトニトリル-水混液 (7+13) を加えて一定量とす
る。その一定量を高速液体クロマトグラフ (HPLC) に注入し,吸光光度検出器を用いて吸光度を測定して
定量する。対象農薬とその定量範囲及び繰返し分析精度(変動係数で)は,表10のとおりである(いずれ
も装置,測定条件によって異なる。)。

26
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表10 対象農薬とその定量範囲及び繰返し分析精度 (HPLC)
番
号
対象農薬
定量範囲 (ng)
繰返し分析精度 (%)
1
アシュラム (C8H10N2O4S)
4〜 80
10〜20
2
オキシン銅 (C18H12CuN2O2)
20〜200
10〜20
3
チウラム (C6H12N2S4)
2〜 40
10〜20
4
メコプロップ [MCPP] (C10H11ClO3)
20〜200
10〜20
5
トリクロピル(遊離) (C7H4Cl3NO3) (21)
5〜100
10〜20
注(21) 7.1.1のトリクロピルの定量のために測定する。
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) JIS K 8180に規定する塩酸を用いて調製する。
2) アセトニトリル JIS K 8039に規定する濃縮300以上の品質のもの(22)。
3) アセトン 4.1a)3)による。
4) クロロホルム JIS K 8322に規定する濃縮300以上の品質のもの(22)。
5) 2, 2'-オキシビスエタノール(ジエチレングリコール) (22)(23)
6) ヘキサン 6.1a)4)による。
7) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 5)の2, 2'-オキシビスエタノールと3)のアセト
ンを用いて調製する。
8) アセトニトリル-水混液 (7+13) 2)のアセトニトリルと水を用いて調製する。
9) 対象農薬標準液 (0.2〜1μg/ml) 対象農薬の標準液の調製に使用する標準品の採取量及び溶解に使
用する溶媒を表11に示す。これら対象農薬標準液の調製は,次による。
対象農薬標準品の一定量[採取量は表11の表中の濃度 (A) に示す。]を対象農薬別に少量のアセ
トニトリル又はクロロホルムに溶かし,全量フラスコ100mlにそれぞれ移し入れ,アセトニトリル
又はクロロホルムをそれぞれ標線まで加える(4)。
10) 混合標準液 (5〜50μg/ml) 9)で個別に調製した対象農薬標準液 (0.2〜1μg/ml) について,それぞれ
の一定量(採取量は表11の表中の混合標準液に示す。)を全量フラスコ100mlにとり,アセトニト
リルを標線まで加える。保存する場合は,−20℃の暗所におく。
11) 混合標準液 (0.5〜5μg/ml) 10)の混合標準液 (5〜50μg/ml) 1mlを全量フラスコ10mlにとり,アセ
トニトリルを標線まで加える。使用時に調製する。
注(22) 4.の注(1)による。
(23) 300mlを約1mlに濃縮し,その40μlについてd)において対象農薬に相当する保持時間にピーク
を生じないもの。
27
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表11 対象農薬の標準液及び混合標準液 (HPLC)
番
号
対象農薬
濃度 (A)
0.2〜1μg/ml
混合標準液
使用溶媒
標準品採取量
採取量
濃度
(g/100ml)
(ml/100ml)
(μg/ml)
1
アシュラム (C8H10N2O4S)
アセトニトリル
0.100
1
10
2
オキシン銅 (C18H12CuN2O2)
クロロホルム
0.020
25
50
3
チウラム (C6H12N2S4)
クロロホルム
0.100
0.5
5
4
メコプロップ [MCPP] (C10H11ClO3)
アセトニトリル
0.100
5
50
5
トリクロピル(遊離) (C7H4Cl3NO3)
アセトニトリル
0.050
2.5
12.5
b) 器具及び装置 器具及び装置は,次による。
1) なす形フラスコ 100mlの共通すり合わせで,濃縮器に接続できるもの。
2) 三角フラスコ 6.1b)3)による。
3) 共栓付き試験管 6.2b)1)による。
4) 固相カラム 6.2b)2)による。
5) マイクロシリンジ 100μl以下の適切なもの。又は自動注入装置を用いてもよい。
6) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
6.1)
分離管 内径2〜6mm,長さ15〜30cmのステンレス鋼製のもの。
6.2)
充てん剤 ポリウレタン系中極性ゲル又はシリカゲルにオクタデシル基 (ODS) を化学的に結合さ
せたもの。若しくはこれと同等の分離性能をもつもの。
6.3)
溶離液 溶離液の調製は,次による。
6.3.1) りん酸塩緩衝液 (pH3.3) りん酸 (1mol/L) (JIS K 9005に規定するりん酸69mlを水に溶かして
1Lにする。)10mlに水約950mlを加えた後,水酸化ナトリウム溶液 (10mol/L) (JIS K 8576に規定
する水酸化ナトリウムを用いて調製したもの。)を加えてpHを3.3に調節し,水を加えて1Lとす
る。
6.3.2) 溶離液 a)2)のアセトニトリルとりん酸塩緩衝液 (pH3.3) を体積比7 : 13で混合する。
6.4)
流量 約1ml/min
6.5)
検出器 吸光光度検出器で230〜280nmが測定できるもの。対象農薬別の測定波長は,次による。
アシュラムは279nm,オキシン銅は240nm,チウラム,メコプロップ及びトリクロピル酸は230nm。
6.6)
カラム槽温度 40〜45℃
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考13. 高速液体クロマトグラフの感度として,混合標準液 (0.05〜0.5μg/ml) [a)11)の混合標準液
(0.5〜5μg/ml) 1mlを10mlに薄めたもの。]40μlを高速液体クロマトグラフに注入し,対象農
薬の量 (2〜20ng) が確認できるように検出器の感度を調節する。
14. 6.の備考1.による。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(24)250mlを三角フラスコにとり,塩酸 (2mol/L) を加えてpHを3.5に調節する。
2) この溶液を固相カラムに加圧法(25)又は減圧法(26)によって,流量10〜20ml/min(27)で通水する。
3) 固相カラムに水10ml(28)を流し,カラムを洗浄した後,約10分間吸引を続け,水分を分離除去する
(29)。
28
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 固相カラムの上端からアセトニトリル約5ml(28)を緩やかに通して(30),対象農薬を溶出させる。溶出
液をなす形フラスコに受ける。
5) このなす形フラスコに2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 0.5mlを加える。
6) 濃縮器(31)を用いて,40℃以下で溶媒を留去する。
7) 残留物にアセトニトリル-水混液 (7+13) を滴加して溶かし,アセトニトリル-水混液 (7+13) で
2mlの一定量とする(32)。
8) 空試験として水250mlについて,2)〜7)の操作を行う。
注(24) 6.の注(11)による。
(25) 6.の注(14)による。
(26) 6.の注(15)による。
(27) 6.の注(16)による。
(28) 6.の注(17)による。
(29) 6.の注(18)による。
(30) 6.の注(19)による。
(31) 6.の注(9)による。
(32) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)11)の混合標準液 (0.5〜5μg/ml) 40μlをマイクロシリンジ(9)でとり,高速液体クロマトグラフに注
入し,対象農薬の保持時間に相当するピークの位置を確認しておく。
2) c)7)で得たアセトニトリル-水混液 (7+13) 40μlをマイクロシリンジ(9)でとり,1)の操作を行ってク
ロマトグラムを記録し,1)の保持時間と一致していることを確認し,保持時間に相当する位置のピ
ークについて,指示値(11)を読み取る。
3) 空試験として,c)8)で得たアセトニトリル-水混液 (7+13) について2)の操作を行って,対象農薬の
保持時間に相当する位置にピークが検出され,その指示値(11)が定量下限値の指示値の31以上である
場合には,準備操作から再度操作し直す。
4) 検量線から対象農薬の量を求め,次の式によって試料中の対象農薬の濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)6)のアセトニトリル-水混液 (7+13) の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)10)の混合標準液 (5〜50μg/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,アセトニ
トリル-水混液 (7+13) を標線まで加える。これらの溶液の一定量[試料と同量(例えば,40μl)]
をマイクロシリンジでとり,1)及び2)の操作を行って対象農薬の量 (ng) と指示値(11)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考15. 備考2.による。ガスクロマトグラフ質量分析法によっても判断が困難な場合には,高速液体
クロマトグラフ質量分析法を用いるとよい。
29
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8. 1, 3-ジクロロプロペン (C3H4Cl2) JIS K 0125による。
9. EPN (C14H14NO4PS) EPN[フェニルホスホノチオ酸O-エチルO-(4-ニトロフェニル)エステル]
の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
9.1
ガスクロマトグラフ質量分析法 試料を6.1又は6.2による抽出操作を行い,引き続き6.3の操作を
行って濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオ
ン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C14H14NO4PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) EPN標準液 (1mgC14H14NO4PS/ml) EPNの標準品0.100gをとり,少量のヘキサンに溶かし,全量
フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) EPN標準液 (10μgC14H14NO4PS/ml) EPN標準液 (1mgC14H14NO4PS/ml) 1mlを全量フラスコ100ml
にとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) EPN標準液 (1μgC14H14NO4PS/ml) EPN標準液 (10μgC14H14NO4PS/ml) 1mlを全量フラスコ10ml
にとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) c)の準備操作において,6.2を用い,6.3の操作を省略する場合の混合標準液の調製には,ヘキ
サンに代えてアセトンを用いる。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計は,EPN標準液 (0.1μgC14H14NO4PS/ml) [a)9)のEPN標準液
(1μgC14H14NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフ質量分析計に注
入し,対象農薬の量 (0.1ng) が確認できるように検出器の感度を調節する。
30
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作に引き続き,6.3の操作を行う(3)(4)。
注(3) 試料に色及び濁りがなく,かつ,この試験の妨害となる成分を含まない場合は,6.3の操作を省
略できる。
(4) EPNだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 6.3のカラムクロマトグラフによる分離操作において,カラム充てん剤に6.の備考2.による残
留農薬用のシリカゲルを用いた場合は,6.の備考3.の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)9)のEPN標準液 (1μgC14H14NO4PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス注入法
又はコールドオンカラム注入法によってガスクロマトグラフに注入し,EPNのフラグメントイオン
(m/z157,169,185など)を設定し(6),選択イオン検出法又はマスクロマトグラフ法によって測定
してそのマスフラグメントグラムを記録し,EPNの保持時間に相当するピークの位置を確認してお
く。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からEPNの量を求め,次の式によって試料中のEPNの濃度 (μg/L) を算出する。
V
C
a
N
1000
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.1c)5),6.2c)4)又は6)のいずれかにおける有機溶媒の量 (ml)
υ2: 6.3c)1),6.備考3.1)のいずれかにおける有機溶媒の量 (ml)
υ3: 6.3c)5),6.備考3.3)のいずれかにおける有機溶媒の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のEPN標準液 (10μgC14H14NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロ
シリンジでとり,1)及び2)の操作を行ってEPNの量 (ng) と指示値(7)との関係線を作成する。検
量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
9.2
ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器,熱イオン化検出器又は炎光光度検出器をそれぞれ用いた方法で定量する。
定量範囲:C14H14NO4PS 0.02〜0.4ng(8) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によ
って異なる。)
注(8) 熱イオン化検出器又は炎光光度検出器を用いた場合は,0.1〜2ng
a) 試薬 試薬は,次のものを用いる。
31
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) EPN標準液 (10μgC14H14NO4PS/ml) 9.1a)8)による。
8) EPN標準液 (2μgC14H14NO4PS/ml) EPN標準液 (10μgC14H14NO4PS/ml) 2mlを全量フラスコ10ml
にとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) EPN標準液 (0.2μgC14H14NO4PS/ml) EPN標準液 (2μgC14H14NO4PS/ml) 1mlを全量フラスコ10ml
にとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。電子捕獲検出器を用いる場合は,7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(9)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。電子捕獲検出器では用いない。
8.7)
カラム槽温度 7.2.1b) 7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注(9) 7.の注(20)による。
備考5. ガスクロマトグラフの感度として,EPN標準液 (20ngC14H14NO4PS/ml) [a)9)のEPN標準液
(0.2μgC14H14NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入してEPN
の量 (0.02ng) が十分に確認できるように検出器の感度を調節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
32
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ができる。この場合のガスクロマトグラフの条件などは,7.の備考5.,7.の備考8.又は7.の備
考11.による。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
備考7. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のEPN標準液 (0.2μgC14H14NO4PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス注入法
又はコールドオンカラム注入法によってガスクロマトグラフに注入し,EPNの保持時間に相当する
ピークの位置を確認しておく。
2) c)で得た濃縮液1plをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からEPNの量を求め,9.1d)4)の式によって試料中のEPNの濃度 (μg/L) を算出する。
検量線 a)8)のEPN標準液 (2μgC14H14NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロ
シリンジでとり,1)及び2)の操作を行ってEPNの量 (ng) と指示値(7)との関係線を作成する。検
量線の作成は,試料測定時に行う。
備考8. 備考3.による。
10. アシュラム (C8H10N2O4S) アシュラム〔[(4-アミノフェニル)スルホニル]カルバミン酸メチルエ
ステル〕の定量にはガスクロマトグラフ法及び高速液体クロマトグラフ法を適用する。
10.1 ガスクロマトグラフ法 試料中のアシュラムを塩酸でpHを2〜3とし,酢酸エチルで抽出した後,
脱水,濃縮し,残留物にジアゾメタン-アセトン溶液を加えてアシュラムをメチル化する。引き続きその溶
液をカラムクロマトグラフ分離・濃縮する。その一定量を,ガスクロマトグラフに注入し,熱イオン化検
出器を用いた方法で定量する。
定量範囲:C8H10N2O4S 0.2〜4ng,繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) 7.3a)1)による。
2) 塩化ナトリウム 6.1a)1)による。
3) 硫酸ナトリウム 6.1a)2)による。
4) アセトン 4.1a)3)による。
5) 酢酸エチル 6.備考5.のa)4)による(1)。
6) メタノール JIS K 8891に規定するもの(1)。
7) ヘキサン 6.1a)4)による。
8) アセトン-ヘキサン溶離液 (3+7) 4)のアセトンと7)のヘキサンで調製する。
9) アセトン-ヘキサン溶離液 (3+17) 4)のアセトンと7)のヘキサンで調製する。
10) 1, 2-エタンジオール-アセトン混合液 (1+49) JIS K 8105に規定するエチレングリコール(1, 2-エ
タンジオール)と4)のアセトンで調製する。
11) ジアゾメタン-アセトン溶液 図2のジアゾメタン発生装置の容器 (A1) にジエチルエーテル[JIS K
8357に規定する濃縮300以上のもの(1)。]50mlを入れ,容器 (A2) には2-(2-エトキシエトキシ)-
エタノール(2)6ml,水酸化カリウム溶液 (560g/L) (JIS K 8574に規定する水酸化カリウムで調製し
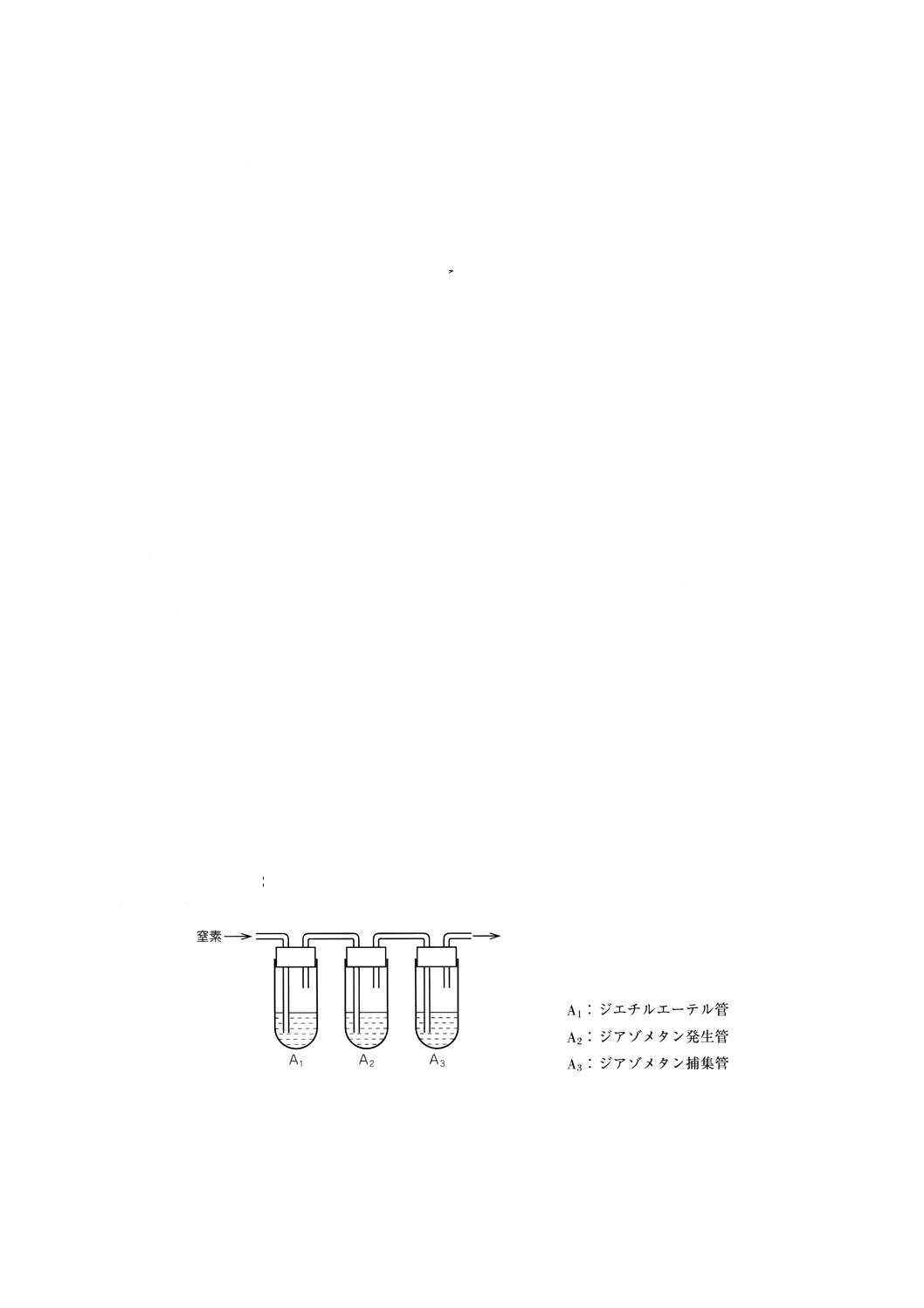
33
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
たもの。)3ml及びN, 4-ジメチル-N-ニトロソベンゼンスルホンアミドのジエチルエーテル溶液(3)
(20g/L) 10mlを加える。また,容器 (A3) に4)のアセトン50mlを加える。
次に,容器 (A1),容器 (A2) 及び容器 (A3) を図2のように接続した後,容器 (A1) の方から窒素
を通気し,容器 (A3) のアセトンに生成したジアゾメタンを捕集する。このときアセトン溶液の色
が濃い黄色になるまで窒素を通気する。この溶液は,使用時に調製する。
12) アシュラム標準液 (1mgC8H10N2O4S/ml) アシュラムの標準品0.100gをとり,少量のメタノールに
溶かし,全量フラスコ100mlに移し入れ,メタノールを標線まで加える(4)。
13) アシュラム標準液 (10μgC8H10N2O4S/ml) (5) アシュラム標準液 (1mgC8H10N2O4S/ml) 1mlをなす形
フラスコ200mlにとり,窒素を緩やかに吹き付けメタノールを揮散させ,ジアゾメタン-アセトン溶
液20mlを加えて,c)の5)及び6)の操作を行ってメチル化を行う。次に残留物を少量のメタノールで
溶かし,全量フラスコ100mlに移し入れ,更にこのなす形フラスコを少量のメタノールで数回よく
洗浄し,洗液も全量フラスコ100mlに合わせ,メタノールを標線まで加える。使用時に調製する。
14) アシュラム標準液 (2μgC8H10N2O4S/ml) (5) アシュラム標準液 (10μgC8H10N2O4S/ml) (5)2mlを全量
フラスコ10mlにとり,メタノールを標線まで加える。使用時に調製する。
15) 窒素 6.2a)2)による。
注(1) 4.の注(1)による。
(2) 慣用名は,ジエチレングリコールモノエチルエーテル又はカルビトール
(3) N, 4-ジメチル-N-ニトロソベンゼンスルホンアミド-ジエチルエーテル溶液 (20g/L) の調製は,
次による。
N, 4-ジメチル-N-ニトロソベンゼンスルホンアミド(p-トルエンスルホニル-N-メチルニトロソ
アミド)1gをジエチルエーテル[JIS K 8357に規定する濃縮300以上の品質のもの(1)。]50ml
に溶かす。使用時に調製する。
(4) 7.の注(4)による。
(5) ここでいうアシュラム標準液は,メチル化したものである。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ジアゾメタン発生装置 図2にジアゾメタン発生装置の一例を示す。
図2 ジアゾメタン発生装置の一例
6) クロマトグラフ管 次に掲げる条件を満たすもの。
6.1)
カラム用管 6.3b)2.1)による。
34
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2)
カラム充てん剤 活性酸化アルミニウム(酸性)[粒径63〜212(又は180)μm]約50gに水2ml
を加え振り混ぜた後,16時間放置したもの。
参考 市販品には,クロマトグラフ用アルミナ(酸性)がある。
6.3)
クロマトグラフ管の作り方 カラム充てん剤約5gをヘキサンでスラリー状にしてカラム用管に流
し込み,上部に硫酸ナトリウム約4gを積層する。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 7.2.1b)7.3)による。
7.4)
キャリヤーガス 7.2.1b)7.4)による。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
7.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
7.7)
カラム槽温度 7.2.1b)7.7)による。
7.8)
検出器槽温度 7.2.1b)7.8)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフの感度として,アシュラム標準液 (0.2μgC8H10N2O4S/ml) (5)[a)14)のアシ
ュラム標準液 (2μgC8H10N2O4S/ml) (5)1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに
注入してアシュラムの量 (0.2ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料の適量 (100〜1 000ml) を分液漏斗にとり,試料100mlについて塩化ナトリウ
ム5gを加えて溶かし,塩酸 (2mol/L) を加えてpHを2〜3に調節し,酢酸エチル10ml(6)を加え,
振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。酢酸エチルを三角フラスコに移し入れ,分液漏斗を少量の酢酸
エチルで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に酢酸エチル10mlを加え,
振とう器を用いて約5分間振り混ぜ,放置する。水層を別の分液漏斗に移し入れる。酢酸エチル層
を先の三角フラスコに合わせる。分液漏斗の水層に酢酸エチル10mlを加え,この抽出操作をもう
一度繰り返し,酢酸エチル層を先の三角フラスコに合わせる。
3) 酢酸エチル溶液30mlについて硫酸ナトリウム約1g(7)を加え,軽く振り混ぜて,約10分間放置した
後(8),ろ紙5種A(又は5種B)(9)を用いてろ過し,ろ液をなす形フラスコ(10)に受ける。少量の酢
酸エチルを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙上の硫酸ナトリウムを洗
浄し,この洗液も酢酸エチル溶液に合わせる。この溶液に1, 2-エタンジオール-アセトン混合液 (1
+49) 数滴を加える。
4) 濃縮器(11)を用いて約40℃(11)の水浴で,酢酸エチル溶液を約1mlになるまで濃縮し,受器を取り外
し,窒素を緩やかに吹き付け,酢酸エチルを揮散させる(12)(13)。
5) この受器にジアゾメタン-アセトン溶液20mlを加え密栓して,ときどき軽く振り混ぜて,室温で約
1時間放置する。
6) 濃縮器(11)を用いて約40℃(11)の水浴で,ジアゾメタン-アセトン溶液を1〜2mlになるまで濃縮し,
受器を取り外し,窒素を緩やかに吹き付け,ジアゾメタン-アセトン溶液を揮散させる(12)(13)。
7) 受器の内容物を少量のアセトン-ヘキサン溶離液 (3+17) で溶かした後,全量フラスコ20mlに移し
35
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
入れ,更に2〜3mlのアセトン-ヘキサン溶離液 (3+17) で2,3回洗い,洗液も全量フラスコ20ml
に合わせ,アセトン-ヘキサン溶離液 (3+17) を標線まで加える。
8) 活性酸化アルミニウム(酸性)を充てんしたクロマトグラフ管の上部から,7)の全量フラスコ20ml
の内容物全量を注ぎ,流下させる。
9) 次に,アセトン-ヘキサン溶離液 (3+17) 40ml,更にアセトン-ヘキサン溶離液 (3+7) 50mlを流下し
て初めの流出液約40mlは捨て,次の溶出液50mlをなす形フラスコに受ける(14)。
10) これを,濃縮器(11)を用いて約40℃(11)の水浴で約1mlになるまで濃縮し,受器を取り外し,窒素を
緩やかに吹き付け,溶出液を揮散させた後,メタノール1mlを正しく加える(12)(13)(15)。
11) 空試験として試料と同量の水を分液漏斗にとり,溶液100mlについて塩化ナトリウム5gを加えて
溶かし,塩酸 (2mol/L) を加えてpHを2〜3に調節し,酢酸エチル10ml(6)を加え,振とう器を用い
て約5分間振り混ぜ,放置する。続いて2)〜10)の操作を行う。
注(6) 6.の注(4)による。ただし,酢酸エチルの量を増加する。
(7) 6.の注(5)による。
(8) 6.の注(6)による。
(9) 6.の注(7)による。
(10) 6.の注(8)による。
(ll) 6.の注(9)による。
(12) 6.の注(21)による。
(13) 6.備考5.の注(*10)による。
(14) 6.の注(25)による。
(15) 6.の注(10)による。
備考2. 試料に濁りがなく,かつ,この試験に妨害となる成分を含まない場合には,c)7)〜10)の操作
を省略してもよい。この場合は,c)6)の操作に引き続きメタノール1mlを正しく加える。また,
この場合の対象農薬の濃度はd)4)の式によって算出する。
d) 操作 操作は,次のとおり行う。
1) a)14)のアシュラム標準液 (2μgC8H10N2O4S/ml) 1μlをマイクロシリンジ(16)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,アシュラムの保持時間
に相当するピークの位置を確認しておく。
2) c)10)で得たメタノール溶液1μlをマイクロシリンジ(16)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)11)で得たメタノール溶液について7.2.1d)3)の操作を行う。
4) 検量線からアシュラムの量を求め,次の式によって試料中のアシュラムの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)10)のメタノールの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)13)のアシュラム標準液 (10μgC8H10N2O4S/ml) 0.2〜4mlを全量フラスコ10mlに段階的に
とり,メタノールを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
36
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
イクロシリンジでとり,1)及び2)の操作を行ってアシュラムの量 (ng) と指示値(17)との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(16) 7.の注(9)による。
(17) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 試料中の対象農薬の濃度が高い場合は,c)7)のアセトン-ヘキサン溶離液 (3+17) の適量をと
り,c)8)のクロマトグラフ管の上部から流下させる。この場合の対象農薬の濃度は,次の式
によって算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めたアシュラムの量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)7)のアセトン-ヘキサン溶離液 (3+17) の量 (ml)
υ2: c)8)のアセトン-ヘキサン溶離液 (3+17) の量 (ml)
υ3: c)10)で用いたメタノールの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
10.2 高速液体クロマトグラフ法 試料中のアシュラムを,塩化ナトリウムの共存のもとで塩酸でpHを2
〜3とし,酢酸エチルで抽出した後,脱水及び濃縮する。その一定量を高速液体クロマトグラフに注入し,
吸光光度検出器で波長270nmの吸光度を測定して定量する。
定量範囲:C8H10N2O4S 4〜80ng,繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (1+5) 10.1a)1)による。
2) 塩化ナトリウム 6.1a)1)による。
3) 硫酸ナトリウム 6.1a)2)による。
4) 酢酸エチル 6.備考5.のa)4)による(18)。
5) メタノール 10.1a)6)による(18)。
6) アシュラム標準液 (1mgC8H10N2O4S/ml) 10.1a)12)による。
7) アシュラム標準液 (10μgC8H10N2O4S/ml) アシュラム標準液 (1mgC8H10N2O4S/ml) 1mlを全量フラ
スコ100mlにとり,メタノールを標線まで加える。使用時に調製する。
8) アシュラム標準液 (2μgC8H10N2O4S/ml) アシュラム標準液 (10μgC8H10N2O4S/ml) 2mlを全量フラ
スコ10mlにとり,メタノールを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(18) 7.の注(23)による。ただし,高速液体クロマトグラフに注入する量は20μlとする。
b) 器具及び装置 器具及び装置は,次による。
1) 三角フラスコ 6.1b)3)による。
2) 共栓付き試験管 6.2b)1)による。
3) マイクロシリンジ 50μl以下の適切なもの。又は自動注入装置を用いてもよい。
4) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
37
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1)
分離管 7.3b)6.1)による。
4.2)
充てん剤 7.3b)6.2)による。
4.3)
溶離液 水と5)のメタノールとJIS K 8355に規定する酢酸をそれぞれ体積比18 : 1 : 1で混合したも
の。
4.4)
流量 約1ml/min
4.5)
検出器 吸光光度検出器 波長270nmで測定できるもの。
4.6)
カラム槽温度 40〜45℃
5) 振とう器
6) 濃縮器 6.1b)5)による。
備考5. 高速液体クロマトグラフの感度として,アシュラム標準液 (0.2μgC8H10N2O4S/ml) [a)8)のア
シュラム標準液 (2μgC8H10N2O4S/ml) 1mlを10mlに薄めたもの。]20μlを高速液体クロマトグ
ラフに注入してアシュラムの量 (4ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 10.1c)1)及び2)の操作を行う。
2) 酢酸エチル溶液30mlについて硫酸ナトリウム約1g(7)を加え,軽く振り混ぜ,約10分間放置した後
(8),ろ紙5種A(又は5種B)(9)を用いてろ過し,ろ液をなす形フラスコ(10)に受ける。少量の酢酸
エチルを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙上の硫酸ナトリウムを洗浄
し,この洗液も酢酸エチル溶液に合わせる。
3) 濃縮器(19)を用い,酢酸エチル溶液を約1mlまで濃縮し,さらに窒素を緩やかに吹き付け,酢酸エチ
ルを完全に揮散させた後,メタノール1mlを正しく加える(12)(13)(15)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(19) 6.の注(9)による。ただし,大気圧で95℃以下で加熱する。濃縮後,スニーダーカラムを少量の
酢酸エチルで洗い,スニーダーカラムを付けたまま冷却する。
d) 操作 操作は,次のとおり行う。
1) a)8)のアシュラム標準液 (2μgC8H10N2O4S/ml) 20μlをマイクロシリンジ(16)でとり,高速液体クロマト
グラフに注入し,アシュラムの保持時間に相当するピークの位置を確認しておく。
2) c)3)で得たメタノール溶液20μlをマイクロシリンジ(16)でとり,1)の操作を行ってクロマトグラムを
記録し,保持時間が1)の保持時間と一致していることを確認し,保持時間に相当する位置のピーク
について指示値(17)を読み取る。
3) 空試験として,c)4)で得たメタノール溶液について2)の操作を行って,1)の保持時間に相当する位置
のピークが検出され,その指示値(17)が定量下限値の指示値の31以上の場合には,準備操作から再度
操作をやり直す。
4) 検量線からアシュラムの量を求め,次の式によって試料中のアシュラムの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)3)のメタノールの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
38
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検量線 a)7)のアシュラム標準液 (10μgC8H10N2O4S/ml) 0.4〜8mlを全量フラスコ10mlに段階的にと
り,メタノールを標線まで加える。これらの溶液の一定量[試料と同量(例えば,20μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってアシュラムの量 (ng) と指示値(17)との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考6. 7.の備考15.による。
11. アセフェート (C4H10NO3PS) アセフェート(アセチルホスホロアミドチオ酸O, S-ジメチルエステ
ル)の定量にはガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
定量に当たっては,これらの試験方法によって,アセフェート及びメタミドホス[ホスホロアミドチオ
酸O, S-ジメチルエステル, (C2H8NO2PS)]の量 (ng) を求め,メタミドホスの量に係数1.298を乗じてア
セフェートの量 (ng) に換算したものの合量 (ng) をもとに算出する。
11.1 ガスクロマトグラフ質量分析法 試料を濃縮器を用いて50℃以下で20mlまで濃縮する。続いて塩
化ナトリウムを加え,クロマトグラフ管に移し,15分間放置する。これを酢酸エチルで展開し,溶出液を
40℃以下で再び濃縮する。その一定量をガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選
択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C4H10NO3PS 0.2〜4ng,繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) アセトン 4.1a)3)による。
3) 酢酸エチル 6.備考5.のa)4)による。
4) アセフェート標準液 (2mgC4H10NO3PS/ml) アセフェートの標準品0.200gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
5) メタミドホス標準液 (2mgC2H8NO2PS/ml) メタミドホスの標準品0.200gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) 混合標準液 [(20μgC4H10NO3PS, 20μgC2H8NO2PS) /ml] アセフェート標準液 (2mgC4H10NO3PS/ml)
及びメタミドホス標準液 (2mgC2H8NO2PS/ml) それぞれ1mlを全量フラスコ100mlにとり,アセト
ンを標線まで加える。使用時に調製する。
7) 混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 混合標準液 [(20μgC4H10NO3PS,
20μgC2H8NO2PS) /ml] 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製
する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) なす形フラスコ 6.1b)2)による。
2) マイクロシリンジ 7.1b)6)による。
3) クロマトグラフ管 次に掲げる条件を満たすもの(図1を参照。)。
3.1)
カラム用管 内径約2cm,長さ約15cmのガラス管。
3.2)
カラム充てん剤 カラムクロマトグラフ用の粒状多孔性けい藻土(粒径70〜150μm,130℃で約16
時間加熱した後,デシケーター中で放冷したもの。)。対象農薬の保持時間にピークを生じないもの
39
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
又は同等の性能をもつもの。
3.3)
クロマトグラフ管の作り方 カラム充てん剤約20mlを気泡が入らないようにカラム用管に流し込
む(2)。
4) ガスクロマトグラフ質量分析計 7.1b)7)による。
5) 濃縮器 6.1b)5)のロータリーエバポレーター
注(2) 6.の注(23)による。
備考1. ガスクロマトグラフ質量分析計の感度として,混合標準液 [(0.2μgC4H10NO3PS,
0.2μgC2H8NO2PS) /ml] 〔a)7)の混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 1mlを10ml
に薄めたもの。〕1μlをガスクロマトグラフ質量分析計に注入してアセフェート及びメタミド
ホスの量 (0.2ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(3)の適量(20〜250mlの一定量)をなす形フラスコ(4)にとり,濃縮器(5)を用い
て,50℃以下の水浴上で約20mlになるまで濃縮する。
2) この濃縮液に塩化ナトリウム5gを加えて溶かす。
3) この溶液をクロマトグラフ管の上部から流し入れ,約15分間室温で放置する。
4) クロマトグラフ管の上部から,酢酸エチル200mlを流量約1ml/minで流下して対象農薬を溶出させ
(6),溶出液をなす形フラスコ(4)に受ける。
5) 濃縮器(5)を用いて,40℃(5)以下の水浴上で溶出液を2〜5mlになるまで濃縮し,受器を取り外し,
窒素を緩やかに吹き付け,溶出液を揮散させる(7)(8)。
6) 残留物にアセトン1mlを正しく加える(9)。
7) 空試験として試料と同量の水をとり,濃縮器を用いて,50℃以下の水浴上で約20mlになるまで濃
縮する。続いて2)〜6)の操作を行う。
注(3) 6.の注(3)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。ただし,スニーダーカラムの洗浄には,少量の酢酸エチルを用いる。
(6) 6.の注(25)による。
(7) 6.の注(21)による。
(8) 6.備考5.の注(*10)による。
(9) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)7)の混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 1μlをマイクロシリンジ(10)でとり,スプリ
ットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,アセフェー
ト及びメタミドホス特有のフラグメントイオン (m/z) を設定し(11),選択イオン検出法又はマスクロ
マトグラフ法によって測定してそのマスフラグメントグラムを記録し,アセフェート及びメタミド
ホスの保持時間に相当するピークの位置を確認しておく。
2) c)6)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からアセフェート及びメタミドホスの量をそれぞれ求め,次の式によって試料中のアセフェ
ートの濃度 (μg/L) を算出する。
40
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V
C
b
a
N
000
1
10
10
)]
298
.1
(
[
3
1
3
×
×
×
×
×
+
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めたアセフェートの量 (ng)
b: 検量線から求めたメタミドホスの量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)6)のアセトンの量 (ml)
1.298: アセフェートの量 (ng) に換算する係数
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)6)の混合標準液 [(20μgC4H10NO3PS, 20μgC2H8NO2PS) /ml] 0.1〜2mlを全量フラスコ10ml
に段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってアセフェート及びメタミドホスの量 (ng) と
指示値(12)との関係線をそれぞれ作成する。検量線の作成は,試料測定時に行う。
注(10) 7.の注(9)による。
(11) それぞれのフラグメントイオンは,次を参考(イオン強度の大きいものから順に表示してある。)
とするとよい。
アセフェート 136,94,125,183
メタミドホス 94,95,141
(12) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
11.2 ガスクロマトグラフ法 試料について11.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,炎光光度検出器を用いた方法で定量する。
定量範囲:C4H10NO3PS 0.2〜4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) アセトン 4.1a)3)による。
3) 酢酸エチル 6.備考5.のa)4)による。
4) 混合標準液 [(20μgC4H10NO3PS, 20μgC2H8NO2PS) /ml] 11.1a)6)による。
5) 混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 11.1a)7)による。
6) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) なす形フラスコ 6.1b)2)による。
2) マイクロシリンジ 7.1b)1)による。
3) クロマトグラフ管 11.1b)3)による。
4) ガスクロマトグラフ 次に掲げる条件を満たすもの。
4.1)
キャピラリーカラム用管 7.1b)7.1)による。
4.2)
キャピラリーカラム 7.1b)7.2)による。
4.3)
検出器 7.2.2b)7.3)による。
41
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.4)
キャリヤーガス 7.2.1b)7.4)による。
4.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
4.6)
燃料ガス及び助燃ガス 7.2.2b)7.6)による。
4.7)
カラム槽温度 7.2.1b)7.7)による。
4.8)
検出器槽温度 260〜300℃
5) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,混合標準液 [(0.2μgC4H10NO3PS, 0.2μgC2H8NO2PS) /ml]
〔a)5)の混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 1mlを10mlに薄めたもの。〕1μl
をガスクロマトグラフに注入してアセフェート及びメタミドホスの量 (0.2ng) が十分に確認
できるように検出器の感度を調節する。
5. 7.の備考5.による。
c) 準備操作 準備操作は,11.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)5)の混合標準液 [(2μgC4H10NO3PS, 2μgC2H8NO2PS) /ml] 1μlをマイクロシリンジ(10)でとり,スプリ
ットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,アセフェー
ト及びメタミドホスの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からアセフェート及びメタミドホスの量をそれぞれ求め,11.1d)の式によって試料中のアセ
フェートの濃度 (μg/L) を算出する。
検量線 a)4)の混合標準液 [(20μgC4H10NO3PS,20μgC2H8NO2PS) /ml] 0.1〜2mlを全量フラスコ10ml
に段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってアセフェート及びメタミドホスの量 (ng) と
指示値(12)との関係線をそれぞれ作成する。検量線の作成は,試料測定時に行う。
備考6. 備考2.による。
12. イソキサチオン (C13H16NO4PS) イソキサチオン[ホスホロチオ酸O, O-ジエチルO-(5-フェニル-3-
イソキサゾリル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適
用する。
12.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C13H16NO4PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
42
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イソキサチオン標準液 (1mgC13H16NO4PS/ml) イソキサチオンの標準品0.100gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) イソキサチオン標準液 (10μgC13H16NO4PS/ml) イソキサチオン標準液 (1mgC13H16NO4PS/ml) 1ml
を全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所にお
く。
9) イソキサチオン標準液 (1μgC13H16NO4PS/ml) イソキサチオン標準液 (10μgC13H16NO4PS/ml) 1ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,イソキサチオン標準液 (0.1μgC13H16NO4PS/ml)
[a)9)のイソキサチオン標準液 (1μgC13H16NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガス
クロマトグラフ質量分析計に注入してイソキサチオンの量 (0.1ng) が十分に確認できるよう
に検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
注(3) 9.の注(3)による。
(4) イソキサチオンだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 9.の備考2.による。
3. 6.3の操作において,ジエチルエーテル-ヘキサン溶離液 (7+13) に代えて,アセトン-ヘキサ
ン溶離液 (1+19) を用いることができる。この場合の準備操作は,6.の備考4.による。この
操作を行った場合は,検量線の作成に用いる標準液の調製にはアセトンを用いる。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソキサチオン標準液 (1μgC13H16NO4PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソキサチオンの
フラグメントイオン(m/z 105,177,77など)を設定し(6),選択イオン検出法又はマスクロマトグ
ラフ法によって測定してそのマスフラグメントグラムを記録し,イソキサチオンの保持時間に相当
するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
43
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からイソキサチオンの量を求め,次の式によって試料中のイソキサチオンの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.1c)5),6.2c)4)又は6),6.備考4.の3)のいずれかにおける有
機溶媒の量 (ml)
υ2: 6.3c)1),6.備考3.の1),6.備考4.の4)のいずれかにおける有
機溶媒の量 (ml)
υ3: 6.3c)5),6.備考3.の3),6.備考4.の6)のいずれかにおける有
機溶媒の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のイソキサチオン標準液 (10μgC13H16NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってイソキサチオンの量 (ng) と指示値(7)との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
12.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器,熱イオン化検出器又は炎光光度検出器をそれぞれ用いた方法で定量する。
定量範囲:C13H16NO4PS 0.05〜1ng(8) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によっ
て異なる。)
注(8) 9.の注(8)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イソキサチオン標準液 (10μgC13H16NO4PS/ml) 12.1a)8)による。
8) イソキサチオン標準液 (5μgC13H16NO4PS/ml) イソキサチオン標準液 (10μgC13H16NO4PS/ml) 5ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) イソキサチオン標準液 (0.5μgC13H16NO4PS/ml) イソキサチオン標準液 (5μgC13H16NO4PS/ml) 1ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
44
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。電子捕獲検出器を用いる場合は,7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(9)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。ただし,電子捕獲検出器では用いない。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注 (9) 7.の注 (20)による。
備考6. ガスクロマトグラフの感度として,イソキサチオン標準液 (50ngC13H16NO4PS/ml) [a)9)のイ
ンキサチオン標準液 (0.5μgC13H16NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマト
グラフに注入してイソキサチオンの量 (0.05ng) が十分に確認できるように検出器の感度を
調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.,7.の備考8.又は7.の備
考11.による。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
備考8. 備考2.による。
9. 備考3.による。
10. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作は6.の備考5.に
よる。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソキサチオン標準液 (0.5μgC13H16NO4PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソキサチオン
45
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からイソキサチオンの量を求め,次の式によって試料中のイソキサチオンの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.1c)5),6.2c)4)又は6),6.備考4.の3),6.備考5.のc)5)の
いずれかにおける有機溶媒の量 (ml)
υ2: 6.3c)1),6.備考3.の1),6.備考4.の4),6.備考5.のc)6)のい
ずれかにおける有機溶媒の量 (ml)
υ3: 6.3c)5),6.備考3.の3),6.備考4.の6),6.備考5.のc)8)のい
ずれかにおける有機溶媒の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のイソキサチオン標準液 (5μgC13H16NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってイソキサチオンの量 (ng) と指示値(7)との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考11. 備考4.による。
13. イソフェンホス (C15H24NO4PS) イソフェンホス〔2-{<エトキシ[(1-メチルエチル)アミノ]ホ
スフィノチオイル>オキシ}安息香酸1-メチルエチルエステル〕の定量には,ガスクロマトグラフ質量分
析法及びガスクロマトグラフ法を適用する。
13.1 ガスクロマトグラフ質量分析法 試料を6.の備考4.による抽出・分離・濃縮を行う。その一定量を,
ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ
法を用いて定量する。
定量範囲:C15H24NO4PS 0.1〜2ng,繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) イソフェンホス標準液 (1mgC15H24NO4PS/ml) イソフェンホスの標準品0.100gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) イソフェンホス標準液 (10μgC15H24NO4PS/ml) イソフェンホス標準液 (1mgC15H24NO4PS/ml) 1ml
46
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を全量フラスコl00mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) イソフェンホス標準液 (1μgC15H24NO4PS/ml) イソフェンホス標準液 (10μgC15H24NO4PS/ml) 1ml
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,イソフェンホス標準液 (0.1μgC15H24NO4PS/ml)
[a)9)のイソフェンホス標準液 (1μgC15H24NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガス
クロマトグラフ質量分析計に注入してイソフェンホスの量 (0.1ng) が十分に確認できるよう
に検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜4)の操作を行う(2)。
2) 次に,アセトン-ヘキサン溶離液 (3+17) 50mlを流下してイソフェンホスを溶出させ(3),溶出液を
なす形フラスコ(4)に受ける。
3) これを,濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を完全に揮散させた後,アセトン1mlを正しく加える(6)(7)(8)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(2) 試料に色及び濁りがなく,かつ,この試験の妨害となる成分を含まない場合は,6.備考4.の1)及
び2)の操作までを行い,ジクロロメタンを揮散させた後,アセトン1mlを正しく加える。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
(8) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソフェンホス標準液 (1μgC15H24NO4PS/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソフェンホスの
フラグメントイオン(m/z 213,121,255など)を設定し(10),選択イオン検出法又はマスクロマト
グラフ法によって測定してそのマスフラグメントグラムを記録し,イソフェンホスの保持時間に相
当するピークの位置を確認しておく。
2) c)3)で得たアセトン溶液1μlをマイクロシリンジ(9)でとり,7.1d)2)の操作を行う。
47
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 空試験として,c)4)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からイソフェンホスの量を求め,次の式によって試料中のイソフェンホスの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.備考4.の3)の有機溶媒の量 (ml)
υ2: 6.備考4.の4)の有機溶媒の量 (ml)
υ3: c)3)の有機溶媒の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のイソフェンホス標準液 (10μgC15H24NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってイソフェンホスの量 (ng) と指示値(11)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(9) 7.の注(9)による。
(10) 7.の注(10)による。
(11) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
13.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,炎光光度検出器又は熱イオン化検出器をそれぞれ用いた方法で定量する。
定量範囲:C15H24NO4PO 0.05〜1ng(12) 繰返し分析精度:変動係数で10〜20% (装置,測定条件に
よって異なる。)
注(12) 熱イオン化検出器を用いた場合の定量範囲は,0.1〜2ngである。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) イソフェンホス標準液 (10μgC15H24NO4PS/ml) 13.1a)8)による。
8) イソフェンホス標準液 (5μgC15H24NO4PS/ml) イソフェンホス標準液 (10μgC15H24NO4PS/ml) 5ml
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) イソフェンホス標準液 (0.5μgC15H24NO4PS/ml) イソフェンホス標準液 (5μgC15H24NO4PS/ml) 1ml
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
48
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,イソフェンホス標準液 (50ngC15H24NO4PS/ml) [a)9)のイ
ソフェンホス標準液 (0.5μgC15H24NO4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマト
グラフに注入してイソフェンホスの量 (0.05ng) が十分に確認できるように検出器の感度を
調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフ条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,13.1c)による(2)。
備考6. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作は6.備考5.のc)1)
〜3)及び8)の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソフェンホス標準液 (0.5μgC15H24NO4PS/ml) 1μlをマイクロシリンジ(9)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソフェンホス
の保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(9)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からイソフェンホスの量を求め,13.1d)4)の式によって試料中のイソフェンホスの濃度
(μg/L) を算出する。
検量線 a)8)のイソフェンホス標準液 (5μgC15H24NO4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってイソフェンホスの量 (ng) と指示値(11)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
49
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14. イソプロチオラン (C12H18O4S2) イソプロチオラン[1, 3-ジチオラン-2-イリデンプロパン二酸ビス
(1-メチルエチル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を
適用する。
14.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H18O4S2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)4)による。
5) ヘキサン 6.1a)5)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イソプロチオラン標準液 (1mgC12H18O4S2/ml) イソプロチオランの標準品0.100gをとり,少量の
ヘキサンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) イソプロチオラン標準液 (10μgC12H18O4S2/ml) イソプロチオラン標準液 (1mgC12H18O4S2/ml) 1ml
を全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所にお
く。
9) イソプロチオラン標準液 (1μgC12H18O4S2/ml) イソプロチオラン標準液 (10μgC12H18O4S2/ml) 1ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)7)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,イソプロチオラン標準液 (0.1μgC12H18O4S2/ml)
[a)9)のイソプロチオラン標準液 (1μgC12H18O4S2/ml) 1mlを10mlに薄めたもの。]1μlをガス
クロマトグラフ質量分析計に注入してイソプロチオランの量 (0.1ng) が十分に確認できるよ
50
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
うに検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
3. 6.3の操作において,ジエチルエーテル-ヘキサン溶離液 (7+13) に代えて,アセトン-ヘキサ
ン溶離液 (3+17) を用いることができる。この場合の準備操作は,13.1c)による。この操作
を行った場合は,検量線の作成に用いる標準液の調製にはアセトンを用いる。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソプロチオラン標準液 (1μgC12H18O4S2/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソプロチオラン
のフラグメントイオン(m/z 118,162,189など)を設定し(5),選択イオン検出法又はマスクロマト
グラフ法によって測定してそのマスフラグメントグラムを記録し,イソプロチオランの保持時間に
相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からイソプロチオランの量を求め,12.1d)4)の式によって試料中のイソプロチオランの濃度
(μg/L) を算出する。
検量線 a)8)のイソプロチオラン標準液 (10μgC12H18O4S2/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってイソプロチオランの量 (ng) と指示値(6)と
の関係線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
14.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C12H18O4S2 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イソプロチオラン標準液 (10μgC12H18O4S2/ml) 14.1a)8)による。
8) イソプロチオラン標準液 (5μgC12H18O4S2/ml) イソプロチオラン標準液 (10μgC12H18O4S2/ml) 5ml
を全量フラスコ10mlにとり,ヘキサンを標線まで加える。保存する場合は,−20℃の暗所におく。
51
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9) イソプロチオラン標準液 (0.5μgC12H18O4S2/ml) イソプロチオラン標準液 (5μgC12H18O4S2/ml) 1ml
を全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.3b)7.4)による。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
カラム槽温度 7.2.1b)7.7)による。
8.7)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考6. ガスクロマトグラフの感度として,イソプロチオラン標準液 (50ngC12H18O4S2/ml) [a)9)のイ
ソプロチオラン標準液 (0.5μgC12H18O4S2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマト
グラフに注入してイソプロチオランの量 (0.05ng) が十分に確認できるように検出器の感度
を調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,9.1c)による(3)。
備考8. 備考2.による。
9. 12.の備考3.による。
10. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作などは6.備考5.
のc)1)〜5)の操作に引き続き,ヘキサン溶液全量を用いて,13.1c)の2)及び3)の操作を行う。
ただし,これらの操作を行った場合の最終溶媒量はヘキサン20mlとする。
d) 操作 操作は,次のとおり行う。
1) a)9)のイソプロチオラン標準液 (0.5μgC12H18O4S2/ml) 1μlをマイクロシリンジ(4)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イソプロチオラ
ンの保持時間に相当するピークの位置を確認しておく。
2) c)で得たヘキサン溶液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からイソプロチオランの量を求め,12.2d)4)の式によって試料中のイソプロチオランの濃度
52
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(μg/L) を算出する。
検量線 a)8)のイソプロチオラン標準液 (5μgC12H18O4S2/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってイソプロチオランの量 (ng) と指示値(6)との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考9. 備考4.による。
15. イプロジオン (C13H13Cl2N3O3) イプロジオン[3-(3, 5-ジクロロフェニル)-N-(1-メチルエチル)
-2, 4-ジオキソ-1-イミダゾリジンカルボキサミド]の定量には,ガスクロマトグラフ質量分析法及びガスク
ロマトグラフ法を適用する。
15.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C13H13Cl2N3O3 0.2〜4ng 繰返し分析精度:変動係数で30〜40%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液(3+17) 10.1a)9)による。
7) イプロジオン標準液 (2mgC13H13Cl2N3O3/ml) イプロジオンの標準品0.200gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) イプロジオン標準液 (20μgC13H13Cl2N3O3/ml) イプロジオン標準液 (2mgC13H13Cl2N3O3/ml) 1mlを
全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) イプロジオン標準液 (2μgC13H13Cl2N3O3/ml) イプロジオン標準液 (20μgC13H13Cl2N3O3/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,イプロジオン標準液 (0.2μgC13H13Cl2N3O3/ml)
53
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[a)9)のイプロジオン標準液 (2μgC13H13Cl2N3O3/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してイプロジオンの量 (0.2ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,13.1c)による(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のイプロジオン標準液 (2μgC13H13Cl2N3O3/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イプロジオンのフ
ラグメントイオン(m/z 314,316,187など)を設定し(4),選択イオン検出法又はマスクロマトグラ
フ法によって測定してそのマスフラグメントグラムを記録し,イプロジオンの保持時間に相当する
ピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からイプロジオンの量を求め,13.1d)4)の式によって試料中のイプロジオンの濃度 (μg/L) を
算出する。
検量線 a)8)のイプロジオン標準液 (20μgC13H13Cl2N3O3/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってイプロジオンの量 (ng) と指示値(5)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
15.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C13H13Cl2N3O3 0.2〜4ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液(3+17) 10.1a)9)による。
7) イプロジオン標準液 (20μgC13H13Cl2N3O3/ml) 15.1a)8)による。
8) イプロジオン標準液 (2μgC13H13Cl2N3O3/ml) 15.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
54
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,イプロジオン標準液 (0.2μgC13H13Cl2N3O33/ml) [a)8)のイ
プロジオン標準液 (2μgC13H13Cl2N3O3/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフに注入してイプロジオンの量 (0.2ng) が十分に確認できるように検出器の感度を調節す
る。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフ条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,13.1c)による(2)。
備考6. 14.の備考10.による。ただし,最終溶媒量はアセトン4mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のイプロジオン標準液 (2μgC13H13Cl2N3O3/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イプロジオンの保
持時間に相当するピークの位置を確認しておく。
2) c)2)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からイプロジオンの量を求め,13.1d)4)の式によって試料中のイプロジオンの濃度 (μg/L) を
算出する。
検量線 a)7)のイプロジオン標準液 (20μgC13H13Cl2N3O3/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってイプロジオンの量 (ng) と指示値(5)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
16. イプロベンホス [IBP] (C13H21O3PS) イプロベンホス [IBP] [ホスホルチオ酸O, O-ビス(1-メチル
エチル)-S-(フェニルメチル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマト
グラフ法を適用する。
55
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
16.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C13H21O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イプロベンホス標準液 (1mgC13H21O3PS/ml) イプロベンホスの標準品0.100gをとり,少量のヘキ
サンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) イプロベンホス標準液 (10μgC13H21O3PS/ml) イプロベンホス標準液 (1mgC13H21O3PS/ml) 1mlを
全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) イプロベンホス標準液 (1μgC13H21O3PS/ml) イプロベンホス標準液 (10μgC13H21O3PS/ml) 1mlを
全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,イプロベンホス標準液 (0.1μgC13H21O3PS/ml)
[a)9)のイプロベンホス標準液 (1μgC13H21O3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してイプロベンホスの量 (0.1ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
d) 操作 操作は,次のとおり行う。
56
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) a)9)のイプロベンホス標準液 (1μgC13H21O3PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イプロベンホスのフ
ラグメントイオン(m/z 91,204,123など)を設定し(5),選択イオン検出法又はマスクロマトグラ
フ法によって測定してそのマスフラグメントグラムを記録し,イプロベンホスの保持時間に相当す
るピークの位置を確認しておく。
2) c)で得た濃縮液1plをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からイプロベンホスの量を求め,9.1d)4)の式によって試料中のイプロベンホスの濃度 (μg/L)
を算出する。
検量線 a)8)のイプロベンホス標準液 (10μgC13H21O3PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってイプロベンホスの量 (ng) と指示値(6)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
16.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器又は炎光光度検出器をそれぞれ用いた方法で定量する。
定量範囲:C13H21O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) イプロベンホス標準液 (10μgC13H21O3PS/ml) 16.1a)8)による。
8) イプロベンホス標準液 (1μgC13H21O3PS/ml) 16.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
57
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考5. ガスクロマトグラフの感度として,イプロベンホス標準液 (0.1μgC13H21O3PS/ml) [a)8)のイ
プロベンホス標準液 (1μgC13H21O3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フに注入してイプロベンホスの量 (0.1ng) が十分に確認できるように検出器の感度を調節す
る。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,9.1c)による(3)。
備考7. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)8)のイプロベンホス標準液 (1μgC13H21O3PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,イプロベンホスの保
持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からイプロベンホスの量を求め,9.1d)4)の式によって試料中のイプロベンホスの濃度 (μg/L)
を算出する。
検量線 a)7)のイプロベンホス標準液 (10μgC13H21O3PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってイプロベンホスの量 (ng) と指示値(6)との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考8. 備考3.による。
17. イミダクロプリド (C9H10ClN5O2) イミダクロプリド〔1-[-(6-クロロ-3-ピリジニル)メチル]-4, 5-
ジヒドロ-N-ニトロ-1H-イミダゾール-2-アミン〕の定量には,高速液体クロマトグラフ法を適用する。
17.1 高速液体クロマトグラフ法 試料中のイミダクロプリドを塩化ナトリウムの共存下で,ジクロロメ
タン抽出を行い,濃縮,脱水後,アセトニトリル-水混液で溶かし一定量とする。その一定量を高速液体ク
ロマトグラフ (HPLC) に注入し,吸光光度検出器を用いて波長270nmの吸光度を測定して定量する。
58
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C9H10ClN5O2 1〜20ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトニトリル 7.3a)2)による。
4) ジクロロメタン 6.1a)3)による。
5) アセトニトリル-水混液 (1+3) 3)のアセトニトリルと水で調製する。
6) イミダクロプリド標準液 (0.5mgC9H10ClN5O2/ml) イミダクロプリドの標準品0.05gをとり,少量
のアセトニトリルに溶かし,全量フラスコ100mlに移し入れ,アセトニトリルを標線まで加える(1)。
7) イミダクロプリド標準液 (10μgC9H10ClN5O2/ml) イミダクロプリド標準液
(0.5mgC9H10ClN5O2/ml) 2mlを全量フラスコ100mlにとり,アセトニトリル-水混液 (1+3) を標線ま
で加える。保存する場合は,−20℃の暗所におく。
8) イミダクロプリド標準液 (1μgC9H10ClN5O2/ml) イミダクロプリド標準液 (10μgC9H10ClN5O2/ml)
1mlを全量フラスコ10mlにとり,アセトニトリル-水混液 (1+3) を標線まで加える。使用時に調製
する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200〜500mlの適切な容量
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 50μl以下の適切なもの。又は自動注入装置を用いてもよい。
5) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
5.1)
分離管 7.3b)6.1)による。
5.2)
充てん剤 7.3b)6.2)による。
5.3)
溶離液 a)5)アセトニトリル-水混液 (1+3) を用いる。
5.4)
流量 約1ml/min
5.5)
検出器 吸光光度検出器 波長270nmで測定できるもの。
5.6)
カラム槽温度 40〜45℃
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. 高速液体クロマトグラフの感度として,イミダクロプリド標準液 (0.1μgC9H10ClN5O2/ml)
[a)8)のイミダクロプリド標準液 (1μgC9H10ClN5O2/ml) 1mlを10mlに薄めたもの。]10μlを高
速液体クロマトグラフに注入してイミダクロプリドの量 (1ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gとジクロロメタン50ml(3)を加え,振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
59
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ジクロロメタンで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量100mlについ
てジクロロメタン50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。ジクロロメタン
層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液50mlについて硫酸ナトリウム約10g(4)を加え,軽く振り混ぜて,約30分間放
置した後(5),ろ紙5種A(又は5種B)(6)を用いてろ過し,ろ液をなす形フラスコ(7)に受ける。少
量のジクロロメタンを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙上の硫酸ナト
リウムを洗浄し,この洗液もジクロロメタン溶液に合わせる。
4) 濃縮器(8)を用いて約40℃(8)の水浴で,ジクロロメタン溶液を約1mlになるまで濃縮し,受器を取り
外し,窒素を緩やかに吹き付け,ジクロロメタンを完全に揮散させる(9)(10)。
5) 残留物にアセトニトリル-水混液 (1+3) 2mlを正しく加える(11)。
6) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gを溶かし,
ジクロロメタン50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。続いて2)〜5)の操作
を行う。
注(2) 6.の注(3)による。
(3) 6.の注(4)による。
(4) 6.の注(5)による。
(5) 6.の注(6)による。
(6) 6.の注(7)による。
(7) 6.の注(8)による。
(8) 6.の注(9)による。ただし,大気圧で95℃以下で加熱する。
(9) 6.の注(21)による。
(10) 6.備考5.の注(*10)による。
(11) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のイミダクロプリド標準液 (1μgC9H10ClN5O2/ml) 10μlをマイクロシリンジ(12)でとり,高速液体
クロマトグラフに注入し,イミダクロプリドの保持時間に相当するピークの位置を確認しておく。
2) c)5)で得たアセトニトリル-水混液 (1+3) 10μlをマイクロシリンジ(12)でとり,1)の操作を行ってク
ロマトグラムを記録し,保持時間が1)の保持時間と一致していることを確認し,保持時間に相当す
る位置のピークについて指示値(13)を読み取る。
3) 空試験として,c)6)で得たアセトニトリル-水混液 (1+3) について2)の操作を行って1)の保持時間
に相当する位置のピークが検出され,その指示値(13)が定量下限値の指示値の31以上の場合には,準
備操作から再度操作をやり直す。
4) 検量線からイミダクロプリドの量を求め,次の式によって試料中のイミダクロプリドの濃度 (μg/L)
を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)5)のアセトニトリル-水混液 (1+3) の量 (ml)
60
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)7)のイミダクロプリド標準液 (10μgC9H10ClN5O2/ml) 0.1〜2mlを全量フラスコ10mlに段
階的にとり,アセトニトリル-水混液 (1+3) を標線まで加える。これらの溶液の一定量[試料と
同量(例えば,10μl)]をマイクロシリンジでとり,1)及び2)の操作を行ってイミダクロプリドの
量 (ng) と指示値(13)との関係線を作成する。検量線の作成は,試料測定時に行う。
注(12) 7.の注(9)による。
(13) 7.の注(11)による。
備考2. 7.の備考15.による。
18. エスプロカルブ (C15H23NOS) エスプロカルブ[S-ベンジル-N-(1, 2-ジメチルプロピル)-N-エチル
カーバーマート]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
18.1 ガスクロマトグラフ質量分析法 試料中のエスプロカルブを塩化ナトリウムの共存下で,ジクロロ
メタン抽出を行い濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法
に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C15H23NOS 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) エスプロカルブ標準液 (2mgC15H23NOS/ml) エスプロカルブの標準品0.200gをとり,少量のアセ
トンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) エスプロカルブ標準液 (20μgC15H23NOS/ml) エスプロカルブ標準液 (2mgC15H23NOS/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) エスプロカルブ標準液 (2μgC15H23NOS/ml) エスプロカルブ標準液 (20μgC15H23NOS/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,エスプロカルブ標準液 (0.2μgC15H23NOS/ml)
[a)7)のエスプロカルブ標準液 (2μgC15H23NOS/ml) 1mlを10mlに薄めたもの。]2μlをガスク
ロマトグラフ質量分析計に注入してエスプロカルブの量 (0.4ng) が十分に確認できるように
61
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gとジクロロメタン25ml(3)とを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
ジクロロメタンで洗い,洗液は先の三角フラスコに合せる。分液漏斗の水層に液量100mlについて
ジクロロメタン25mlを加え,再び振とう器を用いて約10分間振り混ぜ,放置する。ジクロロメタ
ン層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液25mlについて硫酸ナトリウム約10g(4)を加え,軽く振り混ぜ,約30分間放置
した後(5),ろ紙5種A(又は5種B)(6)を用いてろ過し,ろ液をなす形フラスコ(7)に受ける。少量
のジクロロメタンを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙上の硫酸ナトリ
ウムも洗浄し,洗液をジクロロメタン溶液に合わせる。
4) 濃縮器(8)を用いて,約40℃(8)の水浴上でジクロロメタン溶液を約1mlになるまで濃縮する。受器を
取り外し,窒素を緩やかに吹き付け,ジクロロメタンを揮散させる(9)。
5) 残留物にアセトン1mlを正しく加える(10)。
6) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gとジクロ
ロメタン25mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。続いて2)〜5)の操作を
行う。
注(2) 6.の注(3)による。
(3) 6.の注(4)による。
(4) 6.の注(5)による。
(5) 6.の注(6)による。
(6) 6.の注(7)による。
(7) 6.の注(8)による。
(8) 6.の注(9)による。
(9) 6.の注(21)による。
(10) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のエスプロカルブ標準液 (2μgC15H23NOS/ml) 2μlをマイクロシリンジ(11)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エスプロカルブの
フラグメントイオン(m/z 91,222,71など)を設定し(12),選択イオン検出法又はマスクロマトグ
ラフ法によって測定してそのマスフラグメントグラムを記録し,エスプロカルブの保持時間に相当
するピークの位置を確認しておく。
2) c)5)で得たアセトン溶液2μlをマイクロシリンジ(11)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)6)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からエスプロカルブの量を求め,次の式によって試料中のエスプロカルブの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
62
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)5)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)6)のエスプロカルブ標準液 (20μgC15H23NOS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってエスプロカルブの量 (ng) と指示値(13)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(11) 7.の注(9)による。
(12) 7.の注(10)による。
(13) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
18.2 ガスクロマトグラフ法 試料について18.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C15H23NOS 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) エスプロカルブ標準液 (20μgC15H23NOS/ml) 18.1a)6)による。
6) エスプロカルブ標準液 (2μgC15H23NOS/ml) 18.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
63
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,エスプロカルブ標準液 (0.2μgC15H23NOS/ml) [a)6)のエ
スプロカルブ標準液 (2μgC15H23NOS/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フに注入してエスプロカルブの量 (0.4ng) が十分に確認できるように検出器の感度を調節す
る。
c) 準備操作 準備操作は,18.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のエスプロカルブ標準液 (2μgC15H23NOS/ml) 2μlをマイクロシリンジ(11)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エスプロカルブの
保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(11)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からエスプロカルブの量を求め,18.1d)4)の式によって試料中のエスプロカルブの濃度
(μg/L) を算出する。
検量線 a)5)のエスプロカルブ標準液 (20μgC15H23NOS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってエスプロカルブの量 (ng) と指示値(13)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
19. エジフェンホス [EDDP] (C14H15O2PS2) エジフェンホス [EDDP] [ホスホロジチオ酸O-エチルS, S-
ジフェニルエステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
19.1 ガスクロマトグラフ質量分析法 試料中のエジフェンホスを塩化ナトリウムの共存下で,ジクロロ
メタン抽出を行い濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法
に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C14H15O2PS2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) エジフェンホス標準液 (2mgC14H15O2PS2/ml) エジフェンホスの標準品0.200gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) エジフェンホス標準液 (20μgC14H15O2PS2/ml) エジフェンホス標準液 (2mgC14H15O2PS2/ml) 1mlを
全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) エジフェンホス標準液 (2μgC14H15O2PS2/ml) エジフェンホス標準液 (20μgC14H15O2PS2/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
64
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,エジフェンホス標準液 (0.2μgC14H15O2PS2/ml)
[a)7)のエジフェンホス標準液 (2μgC14H15O2PS2/ml) 1mlを10mlに薄めたもの。]2μlをガスク
ロマトグラフ質量分析計に注入してエジフェンホスの量 (0.4ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(400ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gとジクロロメタン10ml(3)とを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 18.1c)2)〜5)の操作を行う。ただし,ジクロロメタンは液量100mlについて10mlを加える。
3) 空試験として試料と同量の水を分液漏斗にとり,1)及び2)の操作を行う。
注(2) 6.の注(3)による。
(3) 6.の注(4)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のエジフェンホス標準液 (2μgC14H15O2PS2/ml) 2μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エジフェンホスの
フラグメントイオン(m/z 109,173,310など)を設定し(5),選択イオン検出法又はマスクロマトグ
ラフ法によって測定してそのマスフラグメントグラムを記録し,エジフェンホスの保持時間に相当
するピークの位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からエジフェンホスの量を求め,18.1d)4)の式によって試料中のエジフェンホスの濃度
(μg/L) を算出する。
検量線 a)6)のエジフェンホス標準液 (20μgC14H15O2PS2/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってエジフェンホスの量 (ng) と指示値(6)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
19.2 ガスクロマトグラフ法 試料について19.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器又は炎光光度検出器を用いた方法で定量する。
65
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C14H15O2PS2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) エジフェンホス標準液 (20μgC14H15O2PS2/ml) 19.1a)6)による。
6) エジフェンホス標準液 (2μgC14H15O2PS2/ml) 19.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,エジフェンホス標準液 (0.2μgC14H15O2PS2/ml) [a)6)のエ
ジフェンホス標準液 (2μgC14H15O2PS2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグ
ラフに注入してエジフェンホスの量 (0.4ng) が十分に確認できるように検出器の感度を調節
する。
c) 準備操作 準備操作は,19.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のエジフェンホス標準液 (2μgC14H15O2PS2/ml) 2μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エジフェンホスの
保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からエジフェンホスの量を求め,18.1d)4)の式によって試料中のエジフェンホスの濃度
66
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(μg/L) を算出する。
検量線 a)5)のエジフェンホス標準液 (20μgC14H15O2PS2/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってエジフェンホスの量 (ng) と指示値(6)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
20. エトフェンプロックス (C25H28O3) エトフェンプロックス[2-(4-エトキシフェニル)-2-メチルプロ
ピル-3-フェノキシベンジルエーテル]の定量には,高速液体クロマトグラフ法を適用する。
20.1 高速液体クロマトグラフ法 試料中のエトフェンプロックスを塩化ナトリウムの共存下で,ジクロ
ロメタン抽出を行い,濃縮,脱水後,メタノール-水混液で溶かし一定量とする。その一定量を高速液体ク
ロマトグラフ (HPLC) に注入し,吸光光度検出器を用いて波長225nmの吸光度を測定して定量する。
定量範囲:C25H28O3 1〜20ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) メタノール 10.1)6)による。
4) ジクロロメタン 6.1a)3)による。
5) メタノール-水混液 (9+1) メタノールと水で調製する。
6) エトフェンプロックス標準液 (0.5mgC25H28O3/ml) エトフェンプロックスの標準品0.05gをとり,
少量のメタノールに溶かし,全量フラスコ100mlに移し入れ,メタノールを標線まで加える(1)。
7) エトフェンプロックス標準液 (10μgC25H28O3/ml) エトフェンプロックス標準液
(0.5mgC25H28O3/ml) 2mlを全量フラスコ100mlにとり,メタノール-水混液 (9+1) を標線まで加える。
保存する場合は,−20℃の暗所におく。
8) エトフェンプロックス標準液 (1μgC25H28O3/ml) エトフェンプロックス標準液 (10μgC25H28O3/ml)
1mlを全量フラスコ10mlにとり,メタノール-水混液 (9+1) を標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 17.1b)4)による。
5) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
5.1)
分離管 7.3b)6.1)による。
5.2)
充てん剤 7.3b)6.2)による。
5.3)
溶離液 a)5)のメタノール-水混液 (9+1) を用いる。
5.4)
流量 約1ml/min
5.5)
検出器 吸光光度検出器 波長225nmで測定できるもの。
67
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.6)
カラム槽温度 40〜45℃
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. 高速液体クロマトグラフの感度として,エトフェンプロックス標準液 (0.1μgC25H28O3/ml)
[a)8)のエトフェンプロックス標準液 (1μgC25H28O3/ml) 1mlを10mlに薄めたもの。]10μlを高
速液体クロマトグラフに注入してエトフェンプロックスの量 (1ng) が十分に確認できるよ
うに検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 17.1c)1)〜4)の操作を行う。
2) 残留物にメタノール-水混液 (9+1) 2mlを正しく加える(2)。
3) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gを溶かし,
ジクロロメタン50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。続いて2)の操作を
行う。
注(2) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のエトフェンプロックス標準液 (1μgC25H28O3/ml) 10μlをマイクロシリンジ(3)でとり,高速液体
クロマトグラフに注入し,エトフェンプロックスの保持時間に相当するピークの位置を確認してお
く。
2) c)2)で得たメタノール-水混液 (9+1) 10μlをマイクロシリンジ(3)でとり,1)の操作を行ってクロマト
グラムを記録し,保持時間が1)の保持時間と一致していることを確認し,保持時間に相当する位置
のピークについて指示値(4)を読み取る。
3) 空試験として,c)3)で得たメタノール-水混液 (9+1) について2)の操作を行って1)の保持時間に相
当する位置のピークが検出され,その指示値(4)が定量下限値の指示値の31以上の場合には,準備操
作から再度操作をやり直す。
4) 検量線からエトフェンプロックスの量を求め,次の式によって試料中のエトフェンプロックスの濃
度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)3)のメタノール-水混液 (9+1) の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)7)のエトフェンプロックス標準液 (10μgC25H28O3/ml) 0.1〜2mlを全量フラスコ10mlに段
階的にとり,メタノール-水混液 (9+1) を標線まで加える。これらの溶液の一定量[試料と同量
(例えば,10μl)]をマイクロシリンジでとり,1)及び2)の操作を行ってエトフェンプロックスの
量 (ng) と指示値(4)との関係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(11)による。
備考2. 7.の備考15.による。
68
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
21. エトリジアゾール[エクロメゾール] (C5H5Cl3N2OS) エトリジアゾール[エクロメゾール][5-エ
トキシ-3-(トリクロロメチル)-1, 2, 4-チアジアゾール]の定量には,ガスクロマトグラフ質量分析法及び
ガスクロマトグラフ法を適用する。
21.1 ガスクロマトグラフ質量分析法 試料を6.の備考4.による抽出を行い,続いて分離・濃縮を行う。
その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマス
クロマトグラフ法を用いて定量する。
定量範囲:C5H5Cl3N2OS 0.3〜6ng,繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジエチルエーテル 6.3a)2)による。
5) ジクロロメタン 6.1a)3)による。
6) ヘキサン 6.1a)4)による。
7) ジエチルエーテル-ヘキサン溶離液 (1+9) 4)のジエチルエーテルと6)のヘキサンで調製する。
8) エトリジアゾール標準液 (3mgC5H5Cl3N2OS/ml) エトリジアゾールの標準品0.300gをとり,少量
のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
9) エトリジアゾール標準液 (30μgC5H5Cl3N2OS/ml) エトリジアゾール標準液 (3mgC5H5Cl3N2OS/ml)
1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所に
おく。
10) エトリジアゾール標準液 (3μgC5H5Cl3N2OS/ml) エトリジアゾール標準液 (30μgC5H5Cl3N2OS/ml)
1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
11) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,エトリジアゾール標準液
(0.3μgC5H5Cl3N2OS/ml) [a)10)のエトリジアゾール標準液 (3μgC5H5Cl3N2OS/ml) 1mlを10ml
に薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入してエトリジアゾールの量
(0.3ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
69
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 6.備考4.の1)〜4)の操作を行う(2)。
2) 次に,クロマトグラフ管の上部から,ジエチルエーテル-ヘキサン溶離液 (1+9) 50mlを流下してエ
トリジアゾールを溶出させ(3),溶出液をなす形フラスコ(4)に受ける。
3) これを,濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を完全に揮散させた後,アセトン1mlを正しく加える(6)(7)(8)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(2) 13.の注(2)による。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
(8) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)10)のエトリジアゾール標準液 (3μgC5H5Cl3N2OS/ml) 1μlをマイクロシリンジ(9)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エトリジアゾ
ールのフラグメントイオン(m/z 211,183,213など)を設定し(10),選択イオン検出法又はマスク
ロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,エトリジアゾールの保持
時間に相当するピークの位置を確認しておく。
2) c)3)で得たアセトン溶液1μlをマイクロシリンジ(9)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)4)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からエトリジアゾールの量を求め,13.1d)4)の式によって試料中のエトリジアゾールの濃度
(μg/L) を算出する。
検量線 a)9)のエトリジアゾール標準液 (30μgC5H5Cl3N2OS/ml) 0.1〜2mlを全量フラスコ10mlに段
階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってエトリジアゾールの量 (ng) と指示値(11)と
の関係線を作成する。検量線の作成は,試料測定時に行う。
注(9) 7.の注(9)による。
(10) 7.の注(10)による。
(11) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
21.2 ガスクロマトグラフ法 試料について21.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C5H5Cl3N2OS 0.02〜0.4ng,繰返し分析精度:変動係数で20〜30%(装置,測定条件によっ
て異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
70
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (1+9) 21.1a)7)による。
7) エトリジアゾール標準液 (2mgC5H5Cl3N2OS/ml) エトリジアゾールの標準品0.200gをとり,少量
のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) エトリジアゾール標準液 (20μgC5H5Cl3N2OS/ml) エトリジアゾール標準液 (2mgC5H5Cl3N2OS/ml)
1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所に
おく。
9) エトリジアゾール標準液 (2μgC5H5Cl3N2OS/ml) エトリジアゾール標準液 (20μgC5H5Cl3N2OS/ml)
1mlを全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
10) エトリジアゾール標準液 (0.2μgC5H5Cl3N2OS/ml) エトリジアゾール標準液 (2μgC5H5Cl3N2OS/ml)
1mlを全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
11) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.3b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
カラム槽温度 7.2.1b)7.7)による。
6.7)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,エトリジアゾール標準液 (20ngC5H5Cl3N2OS/ml) [a)10)
のエトリジアゾール標準液 (0.2μgC5H5Cl3N2OS/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフに注入してエトリジアゾールの量 (0.02ng) が十分に確認できるように検出器
の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフ条件などは7.の備考11.による。
c) 準備操作 準備操作は,21.1c)による(12)。ただし,溶出液を揮散させた後,ヘキサンを正しく加える
(13)。
注(12) 抽出溶媒をジクロロメタンに代えて酢酸エチルを用いてもよい。この場合の準備操作などは備
考6.による。
(13) 6.の注(10)による。
71
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考6. c)準備操作において,抽出溶媒に酢酸エチルを用いた場合の準備操作は,次による。
1) 4.1c)で採取した試料(*1)の適量(400ml以下の一定量)を分液漏斗にとり,液量100mlについ
て塩化ナトリウム5gを加えて溶かし,酢酸エチル[6.備考5.のa)4)による。]10ml(*2)を加え,
振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。酢酸エチルを三角フラスコに移し入れ,分液漏斗を少量
の酢酸エチルで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の液量100mlについて
酢酸エチル10mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。酢酸エチル層を
先の三角フラスコに合わせる。
3) 酢酸エチル溶液20mlについて硫酸ナトリウム約10g(*3)を加え,軽く振り混ぜて,約30分間
放置した後(*4),ろ紙5種A(又は5種B)(*5)を用いてろ過し,ろ液をなす形フラスコ(*6)に
受ける。少量の酢酸エチルを用いて三角フラスコを2,3回洗浄し,更にその洗液で先のろ紙
上の硫酸ナトリウムを洗浄し,この洗液も酢酸エチル溶液に合わせる。
4) 濃縮器(*7)を用いて約40℃(*7)の水浴で,酢酸エチル溶液が約1mlになるまで濃縮し,受器を
取り外し,窒素を緩やかに吹き付け,酢酸エチルを揮散させる(*8)(*9)。
5) 残留物にヘキサン10mlを正しく加える(*10)。
6) このヘキサン溶液5mlをクロマトグラフ管の上部から流し込む。
7) クロマトグラフ管(*11)の上部から,アセトン-ヘキサン溶離液 (1+19) [6.備考4.の5)による。]
50mlを流下して対象農薬を溶出させ(*12),溶出液をなす形フラスコ(*7)に受ける。
8) 4)の操作を行い,残留物にヘキサン20mlを正しく加える(*10)。
9) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gを
加えて溶かし,酢酸エチル10mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。
引き続き,2)〜8)の操作を行う。
注(*1) 6.の注(3)による。
(*2) 6.の注(4)による。ただし,酢酸エチルの量を増加する。
(*3) 6.の注(5)による。
(*4) 6.の注(6)による。
(*5) 6.の注(7)による。
(*6) 6.の注(8)による。
(*7) 6.の注(9)による。
(*8) 6.の注(21)による。
(*9) 6.備考5.の注(*10)による。
(*10) 6.の注(10)による。
(*11) 6.の備考5.b)4)による。
(*12) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)10)のエトリジアゾール標準液 (0.2μgC5H5Cl3N2OS/ml) 1μlをマイクロシリンジ(9)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,エトリジアゾ
ールの保持時間に相当するピークの位置を確認しておく。
2) c)で得たヘキサン溶液1μlをマイクロシリンジ(9)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)3)で得た空試験のヘキサン溶液について7.2.1d)3)の操作を行う。
72
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 検量線からエトリジアゾールの量を求め,13.1d)4)の式によって試料中のエトリジアゾールの濃度
(μg/L) を算出する。
検量線 a)9)のエトリジアゾール標準液 (2μgC5H5Cl3N2OS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってエトリジアゾールの量 (ng) と指示値(11)との
関係線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
22. オキシン銅[有機銅] (C18H12CuN2O2) オキシン銅[有機銅][ビス(8-キノリノラト-N1,O8)銅]
の定量には高速液体クロマトグラフ法を適用する。
22.1 高速液体クロマトグラフ法 紫外吸光光度法及び蛍光光度法を適用する。
22.1.1 紫外吸光光度法 試料のpHを2以下にし,8-キノリノールと銅を解離させ,水溶性とし,妨害物
質をジクロロメタンで抽出,除去する。水層を水酸化ナトリウム溶液で中和し,ジクロロメタンでオキシ
ン銅を抽出する。脱水・濃縮後,アセトニトリルで一定量とする。又は試料のpHを3.5とした後,6.2に
よる固相抽出した後,脱水し,アセトニトリルを用いて対象農薬を溶出し,濃縮後,一定量とする。これ
らアセトニトリル溶液の一定量を高速液体クロマトグラフに注入し,吸光光度検出器で波長240nmの吸光
度を測定してオキシン銅を定量する。
定量範囲:C18H12CuN2O2 20〜200ng 繰返し分析精度:変動係数で5〜10%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) 7.1a)1)による。
2) 水酸化ナトリウム溶液 (2mol/L) JIS K 8576に規定する水酸化ナトリウム80gを水に溶かして1L
とする。
3) 四ほう酸ナトリウム溶液 (50mmol/L) JIS K 8866に規定する四ほう酸ナトリウム十水和物19.1g
を水に溶かして1Lとする。
4) 塩化ナトリウム 6.1a)1)による。
5) 硫酸ナトリウム 6.1a)2)による。
6) アセトニトリル 7.3a)2)による。
7) ジクロロメタン 6.1a)3)による(1)。
8) オキシン銅標準液 (0.5mgC18H12CuN2O2/ml) オキシン銅の標準品0.050gをとり,少量のアセトニ
トリルに溶かし,全量フラスコ100mlに移し入れ,アセトニトリルを標線まで加える(2)。
9) オキシン銅標準液 (50μgC18H12CuN2O2/ml) オキシン銅標準液 (0.5mgC18H12CuN2O2/ml) 1mlを全
量フラスコ10mlにとり,アセトニトリルを標線まで加える。保存する場合は,−20℃の暗所にお
く。
10) オキシン銅標準液 (10μgC18H12CuN2O2/ml) オキシン銅標準液 (50μgC18H12CuN2O2/ml) 2mlを全量
フラスコ10mlにとり,アセトニトリルを標線まで加える。使用時に調製する。
11) オキシン銅標準液 (1μgC18H12CuN2O2/ml) オキシン銅標準液 (10μgC18H12CuN2O2/ml) 1mlを全量
フラスコ10mlにとり,アセトニトリルを標線まで加える。使用時に調製する。
12) 窒素 6.2a)2)による。
注(1) 7.の注(23)による。ただし,高速液体クロマトグラフへ注入する量は20μlとする。
73
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。ただし,充てん剤にはスチレンジビニルベンゼン重合体(ポリスチレ
ン系ゲル)又はこれと同等の性能をもつもの。
6) マイクロシリンジ 7.1b)6)による。
7) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
7.1)
分離管 7.3b)6.1)による。
7.2)
充てん剤 7.3b)6.2)による。
7.3)
溶離液 溶離液の調製は,次による。
7.3.1) EDTA-りん酸塩緩衝液 (pH3.5) JIS K 9007に規定するりん酸二水素カリウム6.8gを水約900ml
に溶かし,これにJIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物2g
を加え,JIS K 9005に規定するりん酸を加えてpHを3.5に調節し,水を加えて1Lとする。
7.3.2) 溶離液 a)6)のアセトニトリルとEDTA-りん酸塩緩衝液 (pH3.5) をそれぞれ体積比2 : 3で混合した
もの。
7.4)
流量 約1ml/min
7.5)
検出器 吸光光度検出器。波長240nmで測定できるもの。
7.6)
カラム槽温度 40〜45℃
8) 振とう器
9) 濃縮器 6.1b)5)による。
備考1. 高速液体クロマトグラフの感度として,a)11)のオキシン銅標準液 (1μgC18H12CuN2O2/ml) 20μl
を高速液体クロマトグラフに注入してオキシン銅の量 (20ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,ジクロロメタン抽出法又は固相抽出法を適用する。
c.1)
ジクロロメタン抽出法 ジクロロメタン抽出法の操作は,次のとおり行う。
1) 4.1c)で採取した試料の適量(例えば,1 000ml)を分液漏斗にとり,塩酸 (2mol/L) を用いて,pHを
2以下(3)に調節し,液量100ml(4)についてジクロロメタン10mlを加えて振とう器を用いて約5分間
振り混ぜ,放置する。
2) 分離したジクロロメタン層は捨てる。分液漏斗の液量100mlについてジクロロメタン10mlを加え,
再び振とう器を用いて約5分間振り混ぜ,放置後,ジクロロメタン層は捨てる。
3) 分液漏斗の水層に水酸化ナトリウム溶液 (2mol/L) を加え中和した後,液量100mlについて四ほう
酸ナトリウム溶液 (50mmol/L) 2ml,ジクロロメタン10ml及び塩化ナトリウム5gとを加え,約5分
間振とう器を用いて振り混ぜる。放置後,ジクロロメタン層を三角フラスコに移す。分液漏斗の液
量100mlについてジクロロメタン10mlを加え,再び振とう器を用いて約5分間振り混ぜ,放置後,
ジクロロメタン層を先の三角フラスコに合わせる。
4) ジクロロメタン溶液10mlについて硫酸ナトリウム2g(5)を加え,軽く振り混ぜ,約10分間放置した
後(6),ろ紙5種A(又は5種B)(7)を用いてろ過し,ろ液をなす形フラスコ(8)に受ける。少量のジ
74
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
クロロメタンを用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリウ
ムも洗浄し,洗液をジクロロメタン溶液に合わせる。
5) 濃縮器(9)を用いて,ジクロロメタン溶液を約7mlまで濃縮し,受器を取り外し,なす形フラスコに
アセトニトリル(10)約50mlを加える。
6) 再び濃縮器(9)を用いて,約1〜2mlまで濃縮し,窒素を緩やかに吹き付け,アセトニトリル溶液と
して1mlまで正しく濃縮する(11)(12)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(3) pHを2以下に調節しない場合は,オキシン銅の一部がジクロロメタン層に移行し,回収率が低
下する。
(4) 6.の注(4)による。
(5) 6.の注(5)による。
(6) 6.の注(6)による。
(7) 6.の注(7)による。
(8) 6.の注(8)による。
(9) 6.の注(9)による。ただし,大気圧で95℃以下で加熱する。
(10) メタノールを用いてもよい。その場合の準備操作は,備考2.による。
(11) 6.の注(21)による。
(12) 6.の注(10)による。
備考2. アセトニトリルに代えて,メタノールを用いてもよい。この場合の準備操作は,次による。
a) 試薬 試薬は,次のもの以外はa)と同じものを用いる。
1) メタノール 10.1a)6)による(*1)(*2)。
注(*1) 4.の注(1)による。
(*2) 注(1)による。
b) 準備操作 準備操作は,次のとおり行う。
1) c.1)1)〜4)の操作を行う。
2) 濃縮器(*3)を用いて,約40℃(*3)の水浴上で約1〜2mlまで濃縮し,受器を取り外し窒素を緩
やかに吹き付け,ジクロロメタンを完全に揮散させた後,メタノール1mlを正しく加える
(*4)(*5)。
3) 空試験として試料と同量の水を分液漏斗にとり,1)及び2)の操作を行う。
注(*3) 6.の注(9)による。ただし,大気圧で95℃以下で加熱する。
(*4) 6.の注(21)による。
(*5) 6.の注(10)による。
c.2)
固相抽出法 固相抽出法の操作は,次のとおり行う。
1) 4.1c)で採取した試料(13)を塩酸 (2mol/L) を用いて,pHを3.5に調節し,その適量(通常は,500ml)
を固相カラム(14)(15)に加圧法(16)又は減圧法(17)によって,流量10〜20ml/min(18)で通水する。
2) 固相カラムに水10ml(19)を流し,洗浄した後,約30分間吸引などで水分を分離除去する(20)。
3) 固相カラムの上端からアセトニトリル約3ml(19)を緩やかに通し(21),対象農薬を溶出させる。溶出液
の受器には,共栓付き試験管を用いる。
4) 溶出液に窒素を緩やかに吹き付け,1mlの一定量にする(11)(12)(22)。
5) 空試験としてあらかじめ,塩酸 (2mol/L) を用いて,pHを3.5に調節した水500mlについて固相カ
75
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ラム(14)(15)に加圧法(16)又は減圧法(17)によって,流量10〜20ml/min(18)で通水する。続いて2)〜4)の
操作を行う。
注(13) 6.の注(11)による。ただし,ろ紙上の懸濁物に吸着している対象農薬についてはアセトニトリル
だけで溶出する。
(14) 6.の注(12)による。
(15) 6.の注(13)による。
(16) 6.の注(14)による。
(17) 6.の注(15)による。
(18) 6.の注(16)による。
(19) 6.の注(17)による。
(20) 6.の注(18)による。
(21) 6.の注(19)による。
(22) 6.の注(20)による。
d) 操作 操作は,次のとおり行う。
1) a)11)のオキシン銅標準液 (1μgC18H12CuN2O2/ml) 20μlをマイクロシリンジ(23)でとり,高速液体クロ
マトグラフに注入し,オキシン銅の保持時間に相当するピークの位置を確認しておく。
2) c.1)6)又はc.2)4)で得たアセトニトリル溶液20μlをマイクロシリンジ(23)でとり,10.2d)2)の操作を行
う。
3) 空試験として,c.1)7)又はc.2)5)で得たアセトニトリル溶液について10.2d)3)の操作を行う。
4) 検量線からオキシン銅の量を求め,次の式によって試料中のオキシン銅の濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c.1)6)又はc.2)4)のアセトニトリルの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)10)のオキシン銅標準液 (10μgC18H12CuN202/ml) 1〜10mlを全量フラスコ10mlに段階的に
とり,アセトニトリルを標線まで加える。これらの溶液の一定量[試料と同量(例えば,20μl)]
をマイクロシリンジでとり1)及び2)の操作を行ってオキシン銅の量 (ng) と指示値(24)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(23) 7.の注(9)による。
(24) 7.の注(11)による。
備考3. 7.の備考15.による。
22.1.2 蛍光光度法 試料を減圧濃縮し,硫酸銅(II),塩化ナトリウムの共存下で塩酸酸性とした試料から
妨害物質をジクロロメタンで抽出,除去する。水層を水酸化ナトリウム溶液で中和し,ジクロロメタンで
オキシン銅を抽出する。脱水,濃縮する。その一定量を高速液体クロマトグラフに注入し,蛍光光度検出
器を用いて波長380nmで励起させ,波長520nmの蛍光強度を測定してオキシン銅を定量する。
備考4. この試験方法は,試料中のオキシン銅及び8-キノリノール系化合物の全量を試験する場合に
適用する。
76
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C18H12CuN2O2 10〜200ng,繰返し分析精度:変動係数で5〜10%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (1mol/L) 22.1.1a)1)の塩酸 (2mol/L) を用いて調製する。
2) 水酸化ナトリウム溶液 (1mol/L) 22.1.1a)2)の水酸化ナトリウム溶液 (2mol/L) を用いて調製する。
3) 塩化ナトリウム 6.1a)1)による。
4) 硫酸ナトリウム 6.1a)2)による。
5) 硫酸銅(II)溶液 JIS K 8983に規定する硫酸銅(II)五水和物1.6gを水に溶かして100mlとする。
6) メタノール 10.1a)6)による(1)。
7) ジクロロメタン 6.1a)3)による。
8) オキシン銅標準液 (0.5mgC18H12CuN2O2/ml) 22.1.1a)8)による(2)。ただし,メタノールを用いて調
製する。
9) オキシン銅標準液 (50μgC18H12CuN2O2/ml) 22.1.1a)9)による。ただし,メタノールを用いて調製
する。
10) オキシン銅標準液 (10μgC18H12CuN2O2/ml) 22.1.1a)10)による。ただし,メタノールを用いて調製
する。
11) オキシン銅標準液 (1μgC18H12CuN2O2/ml) 22.1.1a)11)による。ただし,メタノールを用いて調製す
る。
12) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 容量300ml
2) なす形フラスコ 6.1b)2)による又は容量2 000ml
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
5.1)
分離管 7.3b)6.1)による。
5.2)
充てん剤 多孔性ポリマー又は高純度のシリカゲル(平均粒径5〜10μm)にオクタデシル基 (ODS)
を化学的に結合したもの。又はこれと同等の分離性能をもつもの。
5.3)
溶離液 JIS K 8544に規定する硝酸アルミニウム九水和物10gをa)6)のメタノール1Lに溶かしたも
の。
5.4)
流量 約1ml/min
5.5)
検出器 蛍光光度検出器で励起波長380nm及び蛍光波長520nmが測定できるもの。
5.6)
カラム槽温度 40〜45℃
6) 振とう器
7) 濃縮器 6.1b)5)のロータリーエバポレーター
備考5. 高速液体クロマトグラフの感度として,オキシン銅標準液 (0.5μgC18H12CuN2O2/ml) [a)11)
のオキシン銅標準液 (1μgC18H12CuN2O2/ml) 5mlを10mlに薄めたもの。]20μlを高速液体クロ
マトグラフに注入してオキシン銅の量 (10ng) が十分に確認できるように検出器の感度を調
節する。
c) 準備操作 準備操作は,次のとおり行う。
77
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 4.1c)で採取した試料(25)の適量(例えば,1 000ml)をなす形フラスコにとり,濃縮器を用いて約40℃
の水浴上で約20mlまで濃縮する。
2) 濃縮液を分液漏斗に移す。濃縮に使用した容器も水約80mlを用いて洗浄し,この洗液も先の分液
漏斗に合わせる。
3) 塩酸 (1mol/L) 2ml,硫酸銅(II)溶液0.5ml,塩化ナトリウム30g及びジクロロメタン100ml(26)を加え,
振とう器を用いて約5分間振り混ぜ,放置後,ジクロロエタン層を捨てる。
4) 分液漏斗の水層に水酸化ナトリウム溶液 (1mol/L) を加え,pHを7〜8に調節し,ジクロロメタン
100ml(25)を加えて,振とう器を用いて約5分間振り混ぜ,放置後,ジクロロメタン層を三角フラス
コに移す。分液漏斗の水層にジクロロメタン100mlを加え,再び振とう器を用いて約5分間振り混
ぜ,放置後,ジクロロメタン層を先の三角フラスコに合わせる。
5) 22.1.1c)1)4)の操作を行う(12)。
6) 濃縮器を用いて,約40℃の水浴上で約1〜2mlまで濃縮し,受器を取り外し,窒素を緩やかに吹き
付け,ジクロロメタンを揮散させた後,メタノール1mlを正しく加える(11)(12)。
7) 空試験として試料と同量の水を,なす形フラスコにとり,1)〜6)の操作を行う。
注(25) 6.の注(3)による。
(26) ジクロロメタンに代え酢酸エチルを用いて,洗浄,抽出した準備操作を行ってもよい。
d) 操作 操作は,次のとおり行う。
1) 22.1.1d)1)の操作を行う。
2) c)6)で得たメタノール溶液20μlをマイクロシリンジ(23)でとり,10.2d)2)の操作を行う。
3) 空試験として,c)7)で得たメタノール溶液について10.2d)3)の操作を行う。
4) 検量線からオキシン銅の量を求め,22.1.1d)4)の式によって試料中のオキシン銅の濃度 (μg/L) を算
出する。
検量線 a)10)のオキシン銅標準液 (10μgC18H12CuN2O2/ml) 0.5〜10mlを全量フラスコ10mlに段階的
にとり,メタノールを標線まで加える。これらの溶液の一定量[試料と同量(例えば,20μl)]を
マイクロシリンジでとり1)及び2)の操作を行ってオキシン銅の量 (ng) と指示値(24)との関係線を
作成する。検量線の作成は,試料測定時に行う。
備考6. 備考3.による。
23. カルバリル [NAC] (C12H11NO2) カルバリル [NAC] (1-ナフタレノールメチルカーバメート)の定
量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
23.1 ガスクロマトグラフ質量分析法 試料中のカルバリルを塩化ナトリウムの共存下でジクロロメタン
抽出を行い濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択
イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C12H11NO2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1)3)による。
4) ジクロロメタン 6.1a)3)による。
78
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) カルバリル標準液 (2mgC12H11NO2/ml) カルバリルの標準品0.200gをとり,少量のアセトンに溶
かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) カルバリル標準液 (20μgC12H11NO2/ml) カルバリル標準液 (2mgC12H11NO2/ml) 1mlを全量フラス
コ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) カルバリル標準液 (2μgC12H11NO2/ml) カルバリル標準液 (20μgC12H11NO2/ml) 1mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,カルバリル標準液 (0.2μgC12H11NO2/ml) [a)7)
のカルバリル標準液 (2μgC12H11NO2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フ質量分析計に注入してカルバリルの量 (0.4ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,18.1c)による。ただし,塩化ナトリウムの添加量は,液量100mlにつき10g
とする。
d) 操作 操作は,次のとおり行う。
1) a)7)のカルバリル標準液 (2μgC12H11NO2/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,カルバリルのフラグメン
トイオン (m/z 144, 115) を設定し(3),選択イオン検出法又はマスクロマトグラフ法によって測定し
てそのマスフラグメントグラムを記録し,カルバリルの保持時間に相当するピークの位置を確認し
ておく。
2) c)で得たアセトン溶液2μをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からカルバリルの量を求め,18.1d)4)の式によって試料中のカルバリルの濃度 (μg/L) を算出
する。
検量線 a)6)のカルバリル標準液 (20μgC12H11NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってカルバリルの量 (ng) と指示値(4)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(2) 7.の注(8)による。
(3) 7.の注(9)による。
(4) 7.の注(11)による。
備考2. 7.の備考2.による。
79
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 7.の備考3.による。
23.2 ガスクロマトグラフ法 試料について23.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C12H11NO2 0.4〜8ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) カルバリル標準液 (20μgC12H11NO2/ml) 23.1a)6)による。
6) カルバリル標準液 (2μgC12H11NO2/ml) 23.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,カルバリル標準液 (0.2μgC12H11NO2/ml) [a)6)のカルバリ
ル標準液 (2μgC12H11NO2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフに注入し
てカルバリルの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,23.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のカルバリル標準液 (2μgC12H11NO2/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,カルバリルの保持時間に
相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(2)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からカルバリルの量を求め,18.1d)4)の式によって試料中のカルバリルの濃度 (μg/L) を算出
80
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。
検量線 a)5)のカルバリル標準液 (20μgC12H11NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってカルバリルの量 (ng) と指示値(4)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
24. キャプタン (C9H8Cl3NO2S) キャプタン〔3a, 4, 7, 7a-テトラヒドロ-2-[(トリクロロメチル)チオ]
-1H-イソインドール-1, 3- (2H) -ジオン〕の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグ
ラフ法を適用する。
24.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C9H8Cl3NO2S 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) キャプタン標準液 (1mgC9H8Cl3NO2S/ml) キャプタンの標準品0.100gをとり,少量のアセトンに
溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) キャプタン標準液 (10μgC9H8Cl3NO2S/ml) キャプタン標準液 (1mgC9H8Cl3NO2S/ml) 1mlを全量フ
ラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) キャプタン標準液 (1μgC9H8Cl3NO2S/ml) キャプタン標準液 (10μgC9H8Cl3NO2S/ml) 1mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,キャプタン標準液 (0.1μgC9H8Cl3NO2S/ml)
81
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[a)9)のキャプタン標準液 (1μgC9H8Cl3NO2S/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してキャプタンの量 (0.1ng) が十分に確認できるように検出器
の感度を調節する。
c) 準備操作 準備操作は,13.1c)による(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のキャプタン標準液 (1μgC9H8Cl3NO2S/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,キャプタンのフラグメ
ントイオン(m/z 79,149,264など)を設定し(4),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,キャプタンの保持時間に相当するピークの
位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からキャプタンの量を求め,13.1d)4)の式によって試料中のキャプタンの濃度 (μg/L) を算出
する。
検量線 a)8)のキャプタン標準液 (10μgC9H8Cl3NO2S/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってキャプタンの量 (ng) と指示値(5)との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
24.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C9H8Cl3NO2S 0.05〜1ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) キャプタン標準液 (10μgC9H8Cl3NO2S/ml) 24.1a)8)による。
8) キャプタン標準液 (5μgC9H8Cl3NO2S/ml) キャプタン標準液 (10μgC9H8Cl3NO2S/ml) 5mlを全量フ
ラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
9) キャプタン標準液 (0.5μgC9H8Cl3NO2S/ml) キャプタン標準液 (5μgC9H8Cl3NO2S/ml) 1mlを全量フ
ラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
82
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.3b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
カラム槽温度 7.2.1b)7.7)による。
6.7)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,キャプタン標準液 (50ngC9H8Cl3NO2S/ml) [a)9)のキャプ
タン標準液 (0.5μgC9H8Cl3NO2S/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに
注入してキャプタンの量 (0.05ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,13.1c)による(2)。ただし,溶出液を揮散させた後,ヘキサン1mlを正しく加
える(6)。
注(6) 6.の注(10)による。
備考6. 14.の備考10.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のキャプタン標準液 (0.5μgC9H8Cl3NO2S/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,キャプタンの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たヘキサン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からキャプタンの量を求め,13.1d)4)の式によって試料中のキャプタンの濃度 (μg/L) を算出
する。
検量線 a)8)のキャプタン標準液 (5μgC9H8Cl3NO2S/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってキャプタンの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
83
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
25. クロルニトロフェン [CNP] (C12H6Cl3NO3) クロルニトロフェン [CNP] [1, 3, 5-トリクロロ-2-(4-
ニトロフェノキシ)ベンゼン]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を
適用する。
25.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H6Cl3NO3 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) クロルニトロフェン標準液 (1mgC12H6Cl3NO3/ml) クロルニトロフェンの標準品0.100gをとり,
少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) クロルニトロフェン標準液 (10μgC12H6Cl3NO3/ml) クロルニトロフェン標準液
(1mgC12H6Cl3NO3/ml) 1mlを全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場
合は,−20℃の暗所におく。
9) クロルニトロフェン標準液 (1μgC12H6Cl3NO3/ml) クロルニトロフェン標準液
(10μgC12H6Cl3NO3/ml) 1mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調
製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,クロルニトロフェン標準液
(0.1μgC12H6Cl3NO3/ml) [a)9)のクロルニトロフェン標準液 (1μgC12H6Cl3NO3/ml) 1mlを10ml
に薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入してクロルニトロフェンの量
84
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(0.1ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
注(3) 9.の注(3)による。
(4) クロルニトロフェンだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 9.の備考2.による。
3. 12.の備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロルニトロフェン標準液 (1μgC12H6Cl3NO3/ml) 1μlをマイクロシリンジ(5)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロルニトロ
フェンのフラグメントイオン(m/z 317,319,236など)を設定し(6),選択イオン検出法又はマスク
ロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,クロルニトロフェンの保
持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からクロルニトロフェンの量を求め,12.1d)4)の式によって試料中のクロルニトロフェンの濃
度 (μg/L) を算出する。
検量線 a)8)のクロルニトロフェン標準液 (10μgC12H6Cl3NO3/ml) 0.1〜2mlを全量フラスコ10mlに
段階的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってクロルニトロフェンの量 (ng) と指示値(7)
との関係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
25.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C12H6Cl3NO3 0.02〜0.4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によっ
て異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) クロルニトロフェン標準液 (10μgC12H6Cl3NO3/ml) 25.1a)8)による。
8) クロルニトロフェン標準液 (2μgC12H6Cl3NO3/ml) クロルニトロフェン標準液
(10μgC12H6Cl3NO3/ml) 2mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調
製する。
85
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9) クロルニトロフェン標準液 (0.2μgC12H6Cl3NO3/ml) クロルニトロフェン標準液
(2μgC12H6Cl3NO3/ml) 1mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調
製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.3b)7.4)による。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
カラム槽温度 7.2.1b)7.7)による。
8.7)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考6. ガスクロマトグラフの感度として,クロルニトロフェン標準液 (20ngC12H6Cl3NO3/ml) [a)9)
のクロルニトロフェン標準液 (0.2μgC12H6Cl3NO3/ml) 1mlを10mlに薄めたもの。]1μlをガス
クロマトグラフに注入してクロルニトロフェンの量 (0.02ng) が十分に確認できるように検
出器の感度を調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,9.1c)による(3)。
備考8. 備考2.による。
9. 備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロルニトロフェン標準液 (0.2μgC12H6Cl3NO3/ml) 1μlをマイクロシリンジ(5)でとり,スプリ
ットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロルニト
ロフェンの保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からクロルニトロフェンの量を求め,12.1d)4)の式によって試料中のクロルニトロフェンの濃
度 (μg/L) を算出する。
検量線 a)8)のクロルニトロフェン標準液 (2μgC12H6Cl3NO3/ml) 0.1〜2mlを全量フラスコ10mlに段
86
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
階的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってクロルニトロフェンの量 (ng) と指示値(7)
との関係線を作成する。検量線の作成は,試料測定時に行う。
備考10. 備考4.による。
26. クロルピリホス (C9H11Cl3NO3PS) クロルピリホス[ホスホロチオ酸O, O-ジエチルO-(3, 5, 6-トリ
クロロ-2-ピリジニル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法
を適用する。
26.1 ガスクロマトグラフ質量分析法 試料について6.備考4.の操作を行う。濃縮液の一定量を,ガスク
ロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用
いて定量する。
定量範囲:C9H11Cl3NO3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によっ
て異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) クロルピリホス標準液 (1mgC9H11Cl3NO3PS/ml) クロルピリホスの標準品0.100gをとり,少量の
アセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) クロルピリホス標準液 (10μgC9H11Cl3NO3PS/ml) クロルピリホス標準液 (1mgC9H11Cl3NO3PS/ml)
1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所に
おく。
9) クロルピリホス標準液 (1μgC9H11Cl3NO3PS/ml) クロルピリホス標準液 (10μgC9H11Cl3NO3PS/ml)
1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,クロルピリホス標準液
(0.1μgC9H11Cl3NO3PS/ml) [a)9)のクロルピリホス標準液 (1μgC9H11Cl3NO3PS/ml) 1mlを10ml
87
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入してクロルピリホスの量 (0.1ng)
が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,6.備考4.による(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロルピリホス標準液 (1μgC9H11Cl3NO3PS/ml) 1μlをマイクロシリンジ(3)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロルピリホス
のフラグメントイオン(m/z 197,199,314など)を設定し(4),選択イオン検出法又はマスクロマト
グラフ法によって測定してそのマスフラグメントグラムを記録し,クロルピリホスの保持時間に相
当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からクロルピリホスの量を求め,次の式によって試料中のクロルピリホスの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: 6.備考4.の3)の有機溶媒の量 (ml)
υ2: 6.備考4.の4)の有機溶媒の量 (ml)
υ3: 6.備考4.の6)の有機溶媒の量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のクロルピリホス標準液 (10μgC9H11Cl3NO3PS/ml) 0.1〜2mlを全量フラスコ10mlに段
階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってクロルピリホスの量 (ng) と指示値(5)との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
26.2 ガスクロマトグラフ法 試料について6.備考4.の操作を行う。濃縮液の一定量を,ガスクロマトグ
ラフに注入し,炎光光度検出器又は熱イオン化検出器を用いた方法で定量する。
定量範囲:C9H11Cl3NO3PS 0.05〜1ng(6) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によ
って異なる。)
注(6) 13.の注(12)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
88
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) クロルピリホス標準液 (10μgC9H11Cl3NO3PS/ml) 26.1a)8)による。
8) クロルピリホス標準液 (5μgC9H11Cl3NO3PS/ml) クロルピリホス標準液 (10μgC9H11Cl3NO3PS/ml)
5mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) クロルピリホス標準液 (0.5μgC9H11Cl3NO3PS/ml) クロルピリホス標準液 (5μgC9H11Cl3NO3PS/ml)
1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,クロルピリホス標準液 (50ngC9H11Cl3NO3PS/ml) [a)9)の
クロルピリホス標準液 (0.5μgC9H11Cl3NO3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフに注入してクロルピリホスの量 (0.05ng) が十分に確認できるように検出器の感
度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフ条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,6.の備考4.による(2)。
備考6. 13.の備考6.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロルピリホス標準液 (0.5μgC9H11Cl3NO3PS/ml) 1μlをマイクロシリンジ(3)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロルピリホ
89
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
スの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からクロルピリホスの量を求め,26.1d)4)の式によって試料中のクロルピリホスの濃度
(μg/L) を算出する。
検量線 a)8)のクロルピリホス標準液 (5μgC9H11Cl3NO3PS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってクロルピリホスの量 (ng) と指示値(5)との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
27. クロロタロニル [TPN] (C8Cl4N2) クロロタロニル [TPN] (2, 4, 5, 6-テトラクロロ-1, 3-ベンゼンジ
カルボニトリル)の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
27.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C8Cl4N2 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) クロロタロニル標準液 (1mgC8Cl4N2/ml) クロロタロニルの標準品0.100gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) クロロタロニル標準液 (10μgC8Cl4N2/ml) クロロタロニル標準液 (1mgC8Cl4N2/ml) 1mlを全量フ
ラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) クロロタロニル標準液 (1μgC8Cl4N2/ml) クロロタロニル標準液 (10μgC8Cl4N2/ml) 1mlを全量フラ
スコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
90
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,クロロタロニル標準液 (1μgC8Cl4N2/ml) [a)9)
のクロロタロニル標準液 (1μgC8Cl4N2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフ質量分析計に注入してクロロタロニルの量 (0.1ng) が十分に確認できるように検出器の
感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
注(3) 9.の注(3)による。
(4) クロロタロニルだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 9.の備考2.による。
3. 6.3の操作において,ジエチルエーテル-ヘキサン溶離液 (7+13) に代えて,ジエチルエーテ
ル-ヘキサン溶離液 (1+9) を用いることができる。この場合の準備操作は,21.2c)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロロタロニル標準液 (1μgC8Cl4N2/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロロタロニルのフラ
グメントイオン(m/z 266,264,268など)を設定し(6),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,クロロタロニルの保持時間に相当する
ピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からクロロタロニルの量を求め,12.1d)4)の式によって試料中のクロロタロニルの濃度
(μg/L) を算出する。
検量線 a)8)のクロロタロニル標準液 (10μgC8Cl4N2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってクロロタロニルの量 (ng) と指示値(7)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
27.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器又は熱イオン化検出器を用いた方法で定量する。
定量範囲:C8Cl4N2 0.02〜0.4ng(8) 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
注(8) 13.の注(12)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
91
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) クロロタロニル標準液 (10μgC8Cl4N2/ml) 27.1a)8)による。
8) クロロタロニル標準液 (2μgC8Cl4N2/ml) クロロタロニル標準液 (10μgC8Cl4N2/ml) 2mlを全量フラ
スコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) クロロタロニル標準液 (0.2μgC8Cl4N2/ml) クロロタロニル標準液 (2μgC8Cl4N2/ml) 1mlを全量フ
ラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。電子捕獲検出器を用いる場合は,
7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(9)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。電子捕獲検出器では
用いない。
8.6)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注 (9) 7.の注(20)による。
備考6. ガスクロマトグラフの感度として,クロロタロニル標準液 (20ngC8Cl4N2/ml) [a)9)のクロロ
タロニル標準液 (0.2μgC8Cl4N2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに
注入してクロロタロニルの量 (0.02ng) が十分に確認できるように検出器の感度を調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考11.による。
92
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
備考8. 備考2.による。
9. 備考3.による。
10. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作などは6.備考5.
のc)1)〜5)の操作に引き続き,ヘキサン溶液全量を用いて,21.1c)の2)及び3)の操作を行う。
ただし,これらの操作を行った場合の最終溶媒量はヘキサン20mlとする。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロロタロニル標準液 (0.2μgC8Cl4N2/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロロタロニルの保
持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からクロロタロニルの量を求め,12.1d)4)の式によって試料中のクロロタロニルの濃度
(μg/L) を算出する。
検量線 a)8)のクロロタロニル標準液 (2μgC8Cl4N2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってクロロタロニルの量 (ng) と指示値(7)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考11. 備考4.による。
28. クロロネブ (C8H8Cl2O2) クロロネブ(1, 4-ジクロロ-2, 5-ジメトキシベンゼン)の定量には,ガスク
ロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
28.1 ガスクロマトグラフ質量分析法 試料について21.2c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C8H8Cl2O2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (1+9) 21.1a)7)による。
7) クロロネブ標準液 (1mgC8H8Cl2O2/ml) クロロネブの標準品0.100gをとり,少量のアセトンに溶
かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) クロロネブ標準液 (10μgC8H8Cl2O2/ml) クロロネブ標準液 (1mgC8H8Cl2O2/ml) 1mlを全量フラス
コ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) クロロネブ標準液 (1μgC8H8Cl2O2/ml) クロロネブ標準液 (10μgC8H8Cl2O2/ml) 1mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
93
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,クロロネブ標準液 (0.1μgC8H8Cl2O2/ml) [a)9)
のクロロネブ標準液 (1μgC8H8Cl2O2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フ質量分析計に注入してクロロネブの量 (0.1ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,21.1c)による(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のクロロネブ標準液 (1μgC8H8Cl2O2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロロネブのフラグメン
トイオン(m/z 191,193,206など)を設定し(4),選択イオン検出法又はマスクロマトグラフ法によ
って測定してそのマスフラグメントグラムを記録し,クロロネブの保持時間に相当するピークの位
置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からクロロネブの量を求め,13.1d)4)の式によって試料中のクロロネブの濃度 (μg/L) を算出
する。
検量線 a)8)のクロロネブ標準液 (10μgC8H8Cl2O2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってクロロネブの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
28.2 ガスクロマトグラフ法 試料について21.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C8H8Cl2O2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
94
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (1+9) 21.1a)7)による。
7) クロロネブ標準液 (10μgC8H8Cl2O2/m1) 28.1a)8)による。ただし,溶媒はヘキサンを用いる。
8) クロロネブ標準液 (1μgC8H8Cl2O2/m1) 28.1a)9)による。ただし,溶媒はヘキサンを用いる。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.3b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
カラム槽温度 7.2.1b)7.7)による。
6.7)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,クロロネブ標準液 (0.1μgC8H8C12O2/ml) [a)8)のクロロ
ネブ標準液 (1μgC8H8Cl2O2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入
してクロロネブの量 (0.1ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,21.1c)による(2)。ただし,溶出液を揮散させた後,ヘキサン1mlを正しく加
える(6)。
注(6) 6.の注(10)による。
備考6. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作などは6.備考5.
の操作を行う。ただし,溶離液にはジエチルエーテル-ヘキサン溶離液 (1+19) [a)4)のジエ
チルエーテルとa)5)のヘキサンを用いて調製する。]を用いる。また,これらの操作を行った
場合の最終溶媒量はヘキサン20mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のクロロネブ標準液 (1μgC8H8Cl2O2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注入
95
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,クロロネブの保持時間に
相当するピークの位置を確認しておく。
2) c)2)で得たヘキサン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)3)で得たヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からクロロネブの量を求め,13.1d)4)の式によって試料中のクロロネブの濃度 (μg/L) を算出
する。
検量線 a)7)のクロロネブ標準液 (10μgC8H8Cl2O2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってクロロネブの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
29. ジクロフェンチオン [ECP] (C10H13Cl2O3PS) ジクロフェンチオン [ECP] [ホスホロチオ酸O-(2, 4-
ジクロロフェニル)O, O-ジエチルエステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマ
トグラフ法を適用する。
29.1 ガスクロマトグラフ質量分析法 試料中のジクロフェンチオンを塩化ナトリウムの共存下でヘキサ
ン抽出を行い,引き続きカラムクロマトグラフ分離を行った後,濃縮する。その一定量を,ガスクロマト
グラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定
量する。
定量範囲:C10H13Cl2O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジエチルエーテル 6.3a)2)による。
5) ヘキサン 6.1a)3)による。
6) ジエチルエーテル-ヘキサン溶離液 (1+49) 4)のジエチルエーテルと5)のヘキサンを用いて調製
する。
7) ジクロフェンチオン標準液 (1mgC10H13Cl2O3PS/ml) ジクロフェンチオンの標準品0.100gをとり,
少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ジクロフェンチオン標準液 (10μgC10H13Cl2O3PS/ml) ジクロフェンチオン標準液
(1mgC10H13Cl2O3PS/ml) 1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場
合は,−20℃の暗所におく。
9) ジクロフェンチオン標準液 (5μgC10H13Cl2O3PS/ml) ジクロフェンチオン標準液
(10μgC10H13Cl2O3PS/ml) 5mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調
製する。
10) ジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) ジクロフェンチオン標準液
(5μgC10H13Cl2O3PS/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製
する。
96
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ジクロフェンチオン標準液
(50ngC10H13Cl2O3PS/ml) [a)7)のジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) 1mlを
10mlに薄めたもの。]2μlをガスクロマトグラフ質量分析計に注入してジクロフェンチオンの
量 (0.1ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gとヘキサン25ml(3)とを加え,振とう器を用いて約30分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ヘキサンを三角フラスコに移し入れ,分液漏斗を少量のヘキサ
ンで洗い,洗液は先の三角フラスコに合せる。分液漏斗の水層に液量100mlについてヘキサン25ml
を加え,再び振とう器を用いて約30分間振り混ぜ,放置する。ヘキサン層を先の三角フラスコに合
わせる。
3) ヘキサン溶液25mlについて硫酸ナトリウム10g(4)を加え,軽く振り混ぜ,約30分間放置した後(5),
ろ紙5種A(又は5種B)(6)を用いてろ過し,ろ液をなす形フラスコ(7)に受ける。少量のヘキサン
を用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリウムも洗浄し,
洗液をヘキサン溶液に合わせる。
4) 濃縮器(8)を用いて,約40℃(8)の水浴上でヘキサン溶液を約1mlになるまで濃縮する。受器を取り外
し,窒素を緩やかに吹き付け,ヘキサンを完全に揮散させる(9)(10)。
5) 受器の内容物を少量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更に受器をヘキサ
ン2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ10mlに移し入れ,ヘキサンを標線まで加え
る(11)。
6) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から流し込む。更に全量フラスコ10ml
もヘキサン2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ管に流し込む。
7) クロマトグラフ管の上部からヘキサン50mlを流下する。この流出液は捨てる。
8) ジエチルエーテル-ヘキサン溶離液 (1+49) 100mlを流下してジクロフェンチオンを溶出させ(12),溶
出液をなす形フラスコ(7)に受ける。
9) 濃縮器(8)を用いて約40℃(8)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,アセトン1mlを正しく加える(9)(10)(11)。
10) 空試験として試料と同量の水を分液漏斗にとり,1)〜9)の操作を行う。
注(2) 6.の注(3)による。
97
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 6.の注(4)による。ただし,ヘキサンの量を増加する。
(4) 6.の注(5)による。
(5) 6.の注(6)による。
(6) 6.の注(7)による。
(7) 6.の注(8)による。
(8) 6.の注(9)による。
(9) 6.の注(21)による。
(10) 6.備考5.の注(*10)による。
(11) 6.の注(10)による。
(12) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)10)のジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) 2μlをマイクロシリンジ(13)でとり,スプ
リットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジクロフ
ェンチオンのフラグメントイオン(m/z 279,223,97など)を設定し(14),選択イオン検出法又はマ
スクロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,ジクロフェンチオン
の保持時間に相当するピークの位置を確認しておく。
2) c)9)で得たアセトン溶液2μlをマイクロシリンジ(13)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)10)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からジクロフェンチオンの量を求め,次の式によって試料中のジクロフェンチオンの濃度
(μg/L) を算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)5)のヘキサンの量 (ml)
υ2: c)6)のヘキサンの量 (ml)
υ3: c)9)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)9)のジクロフェンチオン標準液 (5μgC10H13Cl2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってジクロフェンチオンの量 (ng) と指示値(15)
との関係線を作成する。検量線の作成は,試料測定時に行う。
注(13) 7.の注(9)による。
(14) 7.の注(10)による。
(15) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
29.2 ガスクロマトグラフ法 試料について29.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
98
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C10H13Cl2O3PS 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)3)による。
5) ジエチルエーテル-ヘキサン溶離液(1+49) 29.1a)6)による。
6) ジクロフェンチオン標準液 (5μgC10H13Cl2O3PS/ml) 29.1a)9)による。
7) ジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) 29.1a)10)による。
8) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ジクロフェンチオン標準液 (50ngC10H13Cl2O3PS/ml) [a)7)
のジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) 1mlを10mlに薄めたもの。]2μlをガス
クロマトグラフに注入してジクロフェンチオンの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
c) 準備操作 準備操作は,29.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のジクロフェンチオン標準液 (0.5μgC10H13Cl2O3PS/ml) 2μlをマイクロシリンジ(13)でとり,スプ
リットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジクロフ
ェンチオンの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(13)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からジクロフェンチオンの量を求め,29.1d)4)の式によって試料中のジクロフェンチオンの濃
99
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
度 (μg/L)を算出する。
検量線 a)6)のジクロフェンチオン標準液 (5μgC10H13Cl2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってジクロフェンチオンの量 (ng) と指示値(15)
との関係線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
30. ジクロルボス [DDVP] (C4H7Cl2O4P) ジクロルボス [DDVP] (りん酸2, 2-ジクロロエテニルジメチ
ルエステル)の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
30.1 ガスクロマトグラフ質量分析法 試料を6.1又は6.2による抽出操作を行い,引き続き6.の備考3.
の操作を行って濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に
選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C4H7Cl2O4P 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液(7+13) 6.3a)4)による。
7) ジクロルボス標準液 (1mgC4H7Cl2O4P/ml) ジクロルボスの標準品0.100gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) ジクロルボス標準液 (10μgC4H7Cl2O4P/ml) ジクロルボス標準液 (1mgC4H7Cl2O4P/ml) 1mlを全量
フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) ジクロルボス標準液 (1μgC4H7Cl2O4P/ml) ジクロルボス標準液 (10μgC4H7Cl2O4P/ml) 1mlを全量
フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。ただし,カラム充てん剤は6.の備考2.による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
100
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ジクロルボス標準液 (0.1μgC4H7Cl2O4P/ml)
[a)9)のジクロルボス標準液 (1μgC4H7Cl2O4P/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してジクロルボスの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.1又は6.2の操作に引き続き,6.の備考3.の操作を行う(3)。
注(3) 試料に濁りがなく,かつ,この試験に妨害となる成分を含まない場合は,6.の備考3.の操作は省
略できる。
d) 操作 操作は,次のとおり行う。
1) a)9)のジクロルボス標準液 (1μgC4H7Cl2O4P/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジクロルボスのフラグ
メントイオン (m/z 109, 185, 79) を設定し(5),選択イオン検出法又はマスクロマトグラフ法によって
測定してそのマスフラグメントグラムを記録し,ジクロルボスの保持時間に相当するピークの位置
を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からジクロルボスの量を求め,12.1d)4)の式によって試料中のジクロルボスの濃度 (μg/L) を
算出する。
検量線 a)8)のジクロルボス標準液 (10μgC4H7Cl2O4p/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってジクロルボスの量 (ng) と指示値(6)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
30.2 ガスクロマトグラフ法 試料について30.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器,熱イオン化検出器又は炎光光度検出器をそれぞれ用いた方法で定量する。
定量範囲:C4H7Cl2O4P 0.02〜0.4ng(7) 繰返し分析精度:変動係数で20〜30%(装置,測定条件によっ
て異なる。)
注(7) 9.の注(8)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
101
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) ジクロルボス標準液 (10μgC4H7Cl2O4P/ml) 30.1a)8)による。
8) ジクロルボス標準液 (2μgC4H7Cl2O4P/ml) ジクロルボス標準液 (10μgC4H7Cl2O4P/ml) 2mlを全量
フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) ジクロルボス標準液 (0.2μgC4H7Cl2O4P/ml) ジクロルボス標準液 (2μgC4H7Cl2O4P/ml) 1mlを全量
フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) クロマトグラフ管 30.1b)6)による。
6) マイクロシリンジ 7.1b)6)による。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。電子捕獲検出器を用いる場合は,7.2.3b)7.3)による。
7.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(8)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
7.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。電子捕獲検出器では用いない。
7.7)
カラム槽温度 7.2.1b)7.7)による。
7.8)
検出器槽温度 7.2.1b)7.8)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
注(8) 7.の注(20)による。
備考4. ガスクロマトグラフの感度として,ジクロルボス標準液 (20ngC4H7Cl2O4P/ml) [a)9)のジク
ロルボス標準液 (0.2μgC4H7Cl2O4P/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフ
に注入してジクロルボスの量 (0.02ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは,7.の備考5.,7.の備考8.又は7.の備
考11.による。
c) 準備操作 準備操作は,30.1c)による(3)。
d) 操作 操作は,次のとおり行う。
1) a)9)のジクロルボス標準液 (0.2μgC4H7Cl2O4P/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジクロルボスの保持
時間に相当するピークの位置を確認しておく。
102
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からジクロルボスの量を求め,12.1d)4)の式によって試料中のジクロルボスの濃度 (μg/L) を
算出する。
検量線 a)8)のジクロルボス標準液 (2μgC4H7Cl2O4P/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってジクロルボスの量 (ng) と指示値(6)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考6. 備考2.による。
31. ジチオピル (C15H16F5NO2S2) ジチオピル[2-(ジフルオロメチル)-4-(2-メチルプロピル)-6-(ト
リフルオロメチル)-3, 5-ピリジンカルボチオ酸S, S-ジメチルエステル]の定量には,ガスクロマトグラフ
質量分析法及びガスクロマトグラフ法を適用する。
31.1 ガスクロマトグラフ質量分析法 試料中のジチオピルを塩化ナトリウムの共存下でヘキサン抽出を
行い,脱水・濃縮後,2, 2'-オキシビスエタノール-アセトン溶液を加え,さらに濃縮を繰り返す。その一定
量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマト
グラフ法を用いて定量する。
定量範囲:C15H16F5NO2S2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)4)による。
5) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
6) ジチオピル標準液 (2mgC15H16F5NO2S2/ml) ジチオピルの標準品0.200gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
7) ジチオピル標準液 (20μgC15H16F5NO2S2/ml) ジチオピル標準液 (2mgC15H16F5NO2S2/ml) 1mlを全量
フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
8) ジチオピル標準液 (2μgC15H16F5NO2S2/ml) ジチオピル標準液 (20μgC15H16F5NO2S2/ml) 1mlを全量
フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
103
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ジチオピル標準液 (0.2μgC15H16F5NO2S2/ml)
[a)8)のジチオピル標準液 (2μgC15H16F5NO2S2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロ
マトグラフ質量分析計に注入してジチオピルの量 (0.4ng) が十分に確認できるように検出器
の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlにつき塩化ナ
トリウム5gとヘキサン25ml(3)とを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 29.1c)2)及び3)の操作を行う。
3) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 0.5mlを加える。
4) 濃縮器(4)を用いて,約40℃(4)の水浴上でヘキサン溶液を約1mlになるまで濃縮する。受器を取り外
し,窒素を緩やかに吹き付け,ヘキサンを揮散させ,アセトン1mlを正しく加える(5)(6)(7)。
5) 空試験として試料と同量の水を分液漏斗にとり,1)〜4)の操作を行う。
注(2) 6.の注(3)による。
(3) 6.の注(4)による。ただし,ヘキサンの量を増加する。
(4) 6.の注(9)による。
(5) 6.の注(21)による。
(6) 6.備考5.の注(*10)による。
(7) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のジチオピル標準液 (2μgC15H16F5NO2S2/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジチオピルのフラグ
メントイオン (m/z 354, 306) を設定し(9),選択イオン検出法又はマスクロマトグラフ法によって測
定してそのマスフラグメントグラムを記録し,ジチオピルの保持時間に相当するピークの位置を確
認しておく。
2) c)4)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)5)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からジチオピルの量を求め,29.1d)4)の式によって試料中のジチオピルの濃度 (μg/L) を算出
する。
検量線 a)7)のジチオピル標準液 (20μgC15H16F5NO2S2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってジチオピルの量 (ng) と指示値(10)との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) 7.の注(10)による。
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
31.2 ガスクロマトグラフ法 試料について31.1c)の操作を行う。濃縮した溶液の一定量を,ガスクロマト
グラフに注入し,熱イオン化検出器を用いた方法で定量する。
104
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C15H16F5NO2S2 0.4〜8ng(11) 繰返し分析精度:変動係数で20〜30%(装置,測定条件によ
って異なる。)
注(11) 検出器に電子捕獲検出器を用いてもよい。その場合の定量範囲,試薬,準備操作などは備考6.
による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)4)による。
5) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
6) ジチオピル標準液 (20μgC15H16F5NO2S2/ml) 31.1a)7)による。
7) ジチオピル標準液 (2μgC15H16F5NO2S2/ml) 31.1a)8)による。
8) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ジチオピル標準液 (0.2μgC15H16F5NO2S2/ml) [a)7)のジチ
オピル標準液 (2μgC15H16F5NO2S2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフ
に注入してジチオピルの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,31.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のジチオピル標準液 (2μgC15H16F5NO2S2/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ジチオピルの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からジチオピルの量を求め,29.1d)4)の式によって試料中のジチオピルの濃度 (μg/L) を算出
105
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。
検量線 a)6)のジチオピル標準液 (20μgC15H16F5NO2S2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってジチオピルの量 (ng) と指示値(10)との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
6. 電子捕獲検出器を用いてもよい。この場合の操作は次による。
定量範囲:C15H16F5NO2S2 4〜80pg
繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による(*1)。
4) ヘキサン 6.1a)4)による(*1)。
5) ジエチルエーテル-ヘキサン溶離液 (1+49) 29.1a)6)による。
6) ジチオピル標準液 (1mgC15H16F5NO2S2/ml) ジチオピルの標準品0.100gをとり,少量の
アセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(*2)。
7) ジチオピル標準液 (20μgC15H16F5NO2S2/ml) ジチオピル標準液 (1mgC15H16F5NO2S2/ml)
2mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃
の暗所におく。
8) ジチオピル標準液 (0.2μgC15H16F5NO2S2/ml) ジチオピル標準液 (20μgC15H16F5NO2S2/ml)
1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。使用時に調製する。
9) ジチオピル標準液 (0.02μgC15H16F5NO2S2/ml)
ジチオピル標準液
(0.2μgC15H16F5NO2S2/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使
用時に調製する。
10) 窒素 6.2a)2)による。
注(*1) 試験前に,次によって品質を確認する。
濃縮器を用いてその300mlを5mlに濃縮する。その5μlをとり,d)の操作を行い,ガスク
ロマトグラム上のγ-BHCに相当するピーク高さが,γ-BHC 20pgが示すピーク高さより低いも
のを用いる。
(*2) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,28.2b)による。
備考*1. ガスクロマトグラフの感度として,ジチオピル標準液 (2ngC15H16F5NO2S2/ml) [a)9)のジ
チオピル標準液 (0.02μgC15H16F5NO2S2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマ
トグラフに注入してジチオピルの量 (4pg) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 31.1c)1)〜3)の操作を行う。
2) 31.1c)4)の操作を行う。ただし,最終溶媒量はアセトン100mlを正しく加える。
3) 空試験として試料と同量の水を分液漏斗にとり,1)及び2)の操作を行う。
106
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 操作 操作は,次のとおり行う。
1) a)9)のジチオピル標準液 (0.02μgC15H16F5NO2S2/ml) 2μlをマイクロシリンジ(*3)でとり,スプ
リットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,
ジチオピルの保持時間に相当するピークの位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(*3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)3)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からジチオピルの量を求め,29.1d)4)の式によって試料中のジチオピルの濃度
(μg/L) を算出する。
検量線 a)8)のジチオピル標準液 (0.2μgC15H16F5NO2S2/ml) 0.1〜2mlを全量フラスコ10ml
に段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例
えば,2μl)]をマイクロシリンジでとり,1)及び2)の操作を行ってジチオピルの量 (pg) と
指示値(*4)との関係線を作成する。検量線の作成は,試料測定時に行う。
注(*3) 7.の注(9)による。
(*4) 7.の注(11)による。
備考*2. 備考2.による。
32. シマジン [CAT] (C7H12ClN5) シマジン [CAT] (6-クロロ-N, N-ジエチル-1, 3, 5-トリアジン-2, 4-ジア
ミン)の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
32.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C7H12ClN5 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) シマジン標準液 (0.2mgC7H12ClN5/ml) シマジンの標準品0.020gをとり,少量のアセトンに溶かし,
全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) シマジン標準液 (10μgC7H12ClN5/ml) シマジン標準液 (0.2mgC7H12ClN5/ml) 5mlを全量フラスコ
100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) シマジン標準液 (5μgC7H12ClN5/ml) シマジン標準液 (10μgC7H12ClN5/ml) 5mlを全量フラスコ
10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) シマジン標準液 (0.5μgC7H12ClN5/ml) シマジン標準液 (5μgC7H12ClN5/ml) 1mlを全量フラスコ
10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
11) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
107
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,シマジン標準液 (50ngC7H12ClN5/ml) [a)10)
のシマジン標準液 (0.5μgC7H12ClN5/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フ質量分析計に注入してシマジンの量 (0.05ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,9.1c)による(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
3. 14.の備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)10)のシマジン標準液 (0.5μgC7H12ClN5/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,シマジンのフラグメント
イオン(m/z 201,186,173など)を設定し(5),選択イオン検出法又はマスクロマトグラフ法によっ
て測定してそのマスフラグメントグラムを記録し,シマジンの保持時間に相当するピークの位置を
確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からシマジンの量を求め,12.1d)4)の式によって試料中のシマジンの濃度 (μg/L) を算出する。
検量線 a)9)のシマジン標準液 (5μgC7H12ClN5/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
ヘキサン(1)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロ
シリンジでとり,1)及び2)の操作を行ってシマジンの量 (ng) と指示値(6)との関係線を作成する。
検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
32.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
108
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C7H12ClN5 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) シマジン標準液 (5μgC7H12ClN5/ml) 32.1a)9)による。
8) シマジン標準液 (0.5μgC7H12ClN5/ml) 32.1a)10)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 7.2.1b)7.6)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
カラム槽温度 7.2.1b)7.7)による。
8.7)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考6. ガスクロマトグラフの感度として,シマジン標準液 (50ngC7H12ClN5/ml) [a)8)のシマジン標
準液 (0.5μgC7H12ClN5/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入して
シマジンの量 (0.05ng) が十分に確認できるように検出器の感度を調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,9.1c)による(3)。
備考8. 備考2.による。
9. 備考3.による。
10. 14.の備考10.による。
d) 操作 操作は,次のとおり行う。
109
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) a)8)のシマジン標準液 (0.5μgC7H12ClN5/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,シマジンの保持時間に相
当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からシマジンの量を求め,12.1d)4)の式によって試料中のシマジンの濃度 (μg/L) を算出する。
検量線 a)7)のシマジン標準液 (5μgC7H12ClN5/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイクロ
シリンジでとり,1)及び2)の操作を行ってシマジンの量 (ng) と指示値(6)との関係線を作成する。
検量線の作成は,試料測定時に行う。
備考11. 備考4.による。
33. シメトリン (C8H15N5S) シメトリン[N, N'-ジエチル-6-(メチルチオ)-1, 3, 5-トリアジン-2, 4-ジア
ミン]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
33.1 ガスクロマトグラフ質量分析法 試料中のシメトリンを塩化ナトリウムの共存下でジクロロメタン
抽出を行い,引き続きカラムクロマトグラフ分離を行った後,濃縮する。その一定量を,ガスクロマトグ
ラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量
する。
定量範囲:C8H15N5S 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジエチルエーテル 6.3a)2)による。
5) ジクロロメタン 6.1a)3)による。
6) ヘキサン 6.1a)4)による。
7) アセトン-ヘキサン溶離液 (1+5) 3)のアセトンと6)のヘキサンを用いて調製する。
8) ジエチルエーテル-ヘキサン混液 (1+1) 4)のジエチルエーテルと6)のヘキサンを用いて調製する。
9) シメトリン標準液 (1mgC8H15N5S/ml) シメトリンの標準品0.100gをとり,少量のアセトンに溶か
し,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
10) シメトリン標準液 (10μgC8H15N5S/ml) シメトリン標準液 (1mgC8H15N5S/ml) 1mlを全量フラスコ
100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
11) シメトリン標準液 (5μgC8H15N5S/ml) シメトリン標準液 (10μgC8H15N5S/ml) 5mlを全量フラスコ
10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) シメトリン標準液 (0.5μgC8H15N5S/ml) シメトリン標準液 (5μgC8H15N5S/ml) 1mlを全量フラスコ
10mlにとり,アセトンを標線まで加える。使用時に調製する。
13) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
110
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,シメトリン標準液 (50ngC8H15N5S/ml) [a)8)
のシメトリン標準液 (0.5μgC8H15N5S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フ質量分析計に注入してシメトリンの量 (0.1ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム5gとジクロロメタン25ml(3)とを加え,振とう器を用いて約30分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
ジクロロメタンで洗い,洗液は先の三角フラスコに合せる。分液漏斗の水層に液量100mlについて
ジクロロメタン25mlを加え,再び振とう器を用いて約30分間振り混ぜ,放置する。ジクロロメタ
ン層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液25mlについて硫酸ナトリウム10g(4)を加え,軽く振り混ぜ,約30分間放置し
た後(5),ろ紙5種A(又は5種B)(6)を用いてろ過し,ろ液をなす形フラスコ(7)に受ける。ジクロ
ロメタン5mlを用いて三角フラスコを数回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリウム
も洗浄し,洗液をジクロロメタン溶液に合わせる。
4) 濃縮器(8)を用いて,約40℃(8)の水浴上でジクロロメタン溶液を約1mlになるまで濃縮する。受器を
取り外し,窒素を緩やかに吹き付け,ジクロロメタンを完全に揮散させる(9)(10)。
5) 受器の内容物を少量のジエチルエーテル-ヘキサン混液 (1+1) で溶かした後,全量フラスコ10ml
に移し入れ,更に受器をジエチルエーテル-ヘキサン混液 (1+1) 2〜3mlで2,3回よく洗浄し溶か
した後,洗液も全量フラスコ10mlに移し入れ,ジエチルエーテル-ヘキサン混液 (1+1) を標線ま
で加える(11)。
6) このジエチルエーテル-ヘキサン混液 (1+1) 全量をクロマトグラフ管の上部から注ぎ,流下させる。
更に全量フラスコ10mlもジエチルエーテル-ヘキサン混液 (1+1) 2〜3mlで2,3回よく洗浄し,洗
液もクロマトグラフ管に流し込む。
7) クロマトグラフ管の上部から,ジエチルエーテル-ヘキサン混液 (1+1) 100mlを流下させる。この
流出液は捨てる。
8) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+5) 100mlを流下し,シメト
リンを溶出させ(12),溶出液をなす形フラスコ(7)に受ける。
9) 濃縮器(8)を用いて約40℃(8)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,アセトン4mlを正しく加える(9)(10)(11)。
10) 空試験として試料と同量の水を分液漏斗にとり,1)〜9)の操作を行う。
注(2) 6.の注(3)による。
111
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 6.の注(4)による。
(4) 6.の注(5)による。
(5) 6.の注(6)による。
(6) 6.の注(7)による。
(7) 6.の注(8)による。
(8) 6.の注(9)による。
(9) 6.の注(21)による。
(10) 6.備考5.の注(*10)による。
(11) 6.の注(10)による。
(12) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)12)のシメトリン標準液 (0.5μgC8H15N5S/ml) 2μlをマイクロシリンジ(13)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,シメトリンのフラグメ
ントイオン (m/z 213, 170, 155) を設定し(14),選択イオン検出法又はマスクロマトグラフ法によって
測定してそのマスフラグメントグラムを記録し,シメトリンの保持時間に相当するピークの位置を
確認しておく。
2) c)9)で得たアセトン溶液2μlをマイクロシリンジ(13)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)10)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からシメトリンの量を求め,次の式によって試料中のシメトリンの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)5)のジエチルエーテル-ヘキサン混液 (1+1) の量 (ml)
υ2: c)6)のジエチルエーテル-ヘキサン混液 (1+1) の量 (ml)
υ3: c)9)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)11)のシメトリン標準液 (5μgC8H15N5S/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってシメトリンの量 (ng) と指示値(15)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(13) 7.の注(9)による。
(14) 7.の注(10)による。
(15) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
33.2 ガスクロマトグラフ法 試料について33.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C8H15N5S 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異な
112
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) アセトン-ヘキサン溶離液 (1+5) 33.1a)7)による。
6) ジエチルエーテル-ヘキサン混液 (1+1) 33.1a)8)による。
7) シメトリン標準液 (5μgC8H15N5S/ml) 33.1a)11)による。
8) シメトリン標準液 (0.5μgC8H15N5S/ml) 33.1a)12)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,シメトリン標準液 (50ngC8H15N5S/ml) [a)8)のシメトリ
ン標準液 (0.5μgC8H15N5S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフに注入し
てシメトリンの量 (0.1ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,33.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のシメトリン標準液 (0.5μgC8H15N5S/ml) 2μlをマイクロシリンジ(13)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,シメトリンの保持時間
に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(13)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からシメトリンの量を求め,33.1d)4)の式によって試料中のシメトリンの濃度 (μg/L) を算出
する。
113
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検量線 a)7)のシメトリン標準液 (5μgC8H15N5S/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイクロシ
リンジでとり,1)及び2)の操作を行ってシメトリンの量 (ng) と指示値(15)との関係線を作成する。
検量線の作成は,試料測定時に行う。
備考4. 備考2.による。
34. ダイアジノン (C12H21N2O3PS) ダイアジノン〔ホスホロチオ酸O, O-ジメチルO-[6-メチル-2-(1-
メチルエチル)-4-ピリミジニル]エステル〕の定量には,ガスクロマトグラフ質量分析法及びガスクロマ
トグラフ法を適用する。
34.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H21N2O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) ダイアジノン標準液 (1mgC12H21N2O3PS/ml) ダイアジノンの標準品0.100gをとり,少量のヘキサ
ンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) ダイアジノン標準液 (10μgC12H21N2O3PS/ml) ダイアジノン標準液 (1mgC12H21N2O3PS/ml) 1mlを
全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) ダイアジノン標準液 (1μgC12H21N2O3PS/ml) ダイアジノン標準液 (10μgC12H21N2O3PS/ml) 1mlを
全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
114
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ダイアジノン標準液 (0.1μgC12H21N2O3PS/ml)
[a)9)のダイアジノン標準液 (1μgC12H21N2O3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してダイアジノンの量 (0.1ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
注(3) 9.の注(3)による。
(4) ダイアジノンだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 9.の備考2.による。
3. 6.3の操作において,ジエチルエーテル-ヘキサン溶離液 (7+13) に代えて,アセトン-ヘキサ
ン溶離液 (1+49) [4.1a)3)のアセトン及び6.1a)4)のヘキサンを用いて調製する。]を用いる
ことができる。この場合の準備操作は,備考4.による。
4. 溶離液にアセトン-ヘキサン溶離液 (1+49) を用いた場合の準備操作は,次による。
1) 6.備考4.の1)〜4)の操作を行う。
2) 次に,アセトン-ヘキサン溶離液 (1+49) 70mlを流下してダイアジノンを溶出させ(*1),溶出
液をなす形フラスコ(*2)に受ける。
3) これを,濃縮器(*3)を用いて約40℃(*3)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,
窒素を緩やかに吹き付け,溶出液を揮散させた後,アセトン又はヘキサン1mlを正しく加え
る(*4)(*5)(*6)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(*1) 6.の注(25)による。
(*2) 6.の注(8)による。
(*3) 6.の注(9)による。
(*4) 6.の注(21)による。
(*5) 6.備考5.の注(*10)による。
(*6) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のダイアジノン標準液 (1μgC12H21N2O3PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ダイアジノンのフラ
グメントイオン(m/z 179,137,304など)を設定し(6),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,ダイアジノンの保持時間に相当するピ
ークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からダイアジノンの量を求め,12.1d)4)の式によって試料中のダイアジノンの濃度 (μg/L) を
算出する。
検量線 a)8)のダイアジノン標準液 (10μgC12H21N2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってダイアジノンの量 (ng) と指示値(7)との関係
線を作成する。検量線の作成は,試料測定時に行う。
115
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考5. 7.の備考2.による。
6. 7.の備考3.による。
34.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器,熱イオン化検出器又は炎光光度検出器を用いた方法で定量する。
定量範囲:C12H21N2O3PS 0.02〜0.4ng(8) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によ
って異なる。)
注(8) 熱イオン化検出器を用いた場合は,0.1〜2ngである。炎光光度検出器を用いた場合は,0.05〜
1ngである。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) ダイアジノン標準液 (10μgC12H21N2O3PS/ml) 34.1a)8)による。
8) ダイアジノン標準液 (2μgC12H21N2O3PS/ml) ダイアジノン標準液 (10μgC12H21N2O3PS/ml) 2mlを
全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。
9) ダイアジノン標準液 (0.2μgC12H21N2O3PS/ml) ダイアジノン標準液 (2μgC12H21N2O3PS/ml) 1mlを
全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。電子捕獲検出器を用いる場合は,7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(9)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
116
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。電子捕獲検出器では用いない。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注(9) 7.の注(20)による。
備考7. ガスクロマトグラフの感度として,ダイアジノン標準液 (20ngC12H21N2O3PS/ml) [a)9)のダ
イアジノン標準液 (0.2μgC12H21N2O3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフに注入してダイアジノンの量 (0.02ng) が十分に確認できるように検出器の感度を調節
する。
8. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.,7.の備考8.又は7.の備
考11.による。
c) 準備操作 準備操作は,9.1c)による(3)(10)。
注(10) 6.の備考5.の抽出操作を用いることができる。
備考9. 備考2.による。
10. 備考4.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のダイアジノン標準液 (0.2μgC12H21N2O3PS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ダイアジノンの保
持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からダイアジノンの量を求め,12.1d)4)の式によって試料中のダイアジノンの濃度 (μg/L) を
算出する。
検量線 a)8)のダイアジノン標準液 (2μgC12H21N2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってダイアジノンの量 (ng) と指示値(7)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考11. 備考5.による。
35. チウラム (C6H12N2S4) チウラム(テトラメチルチオペルオキシジカルボンジアミド)の定量には,
高速液体クロマトグラフ (HPLC) 法を適用する。
35.1 高速液体クロマトグラフ法 試料中のチウラムを塩化ナトリウムの共存下でジクロロメタン抽出を
行い,脱水・濃縮後,アセトニトリルで一定量にする。又は試料のpHを3.5とした後,6.2による固相抽
出した後,脱水し,アセトニトリルを用いて対象農薬を溶出し,濃縮後,一定量とする。これらアセトニ
トリル溶液の一定量を高速液体クロマトグラフに注入し,吸光光度検出器で波長272nmの吸光度を測定し,
チウラムを定量する。
117
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C6H12N2S4 2〜40ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) 22.1.1a)1)による。
2) 塩化ナトリウム 6.1a)1)による。
3) 硫酸ナトリウム 6.1a)2)による。
4) アセトニトリル 7.3a)2)による。
5) ジクロロメタン 6.1a)3)による(1)。
6) メタノール 10.1a)6)による(1)。
7) チウラム標準液 (1mgC6H12N2S4/ml) チウラムの標準品0.100gをとり,少量のアセトニトリルに
溶かし,全量フラスコ100mlに移し入れ,アセトニトリルを標線まで加える(2)。
8) チウラム標準液 (10μgC6H12N2S4/ml) チウラム標準液 (1mgC6H12N2S4/ml) 1mlを全量フラスコ
100mlにとり,アセトニトリルを標線まで加える。保存する場合は,−20℃の暗所におく。
9) チウラム標準液 (1μgC6H12N2S4/ml) チウラム標準液 (10μgC6H12N2S4/ml) 1mlを全量フラスコ10ml
にとり,アセトニトリルを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(23)による。ただし,高速液体クロマトグラフへ注入する量は20μlとする。
(2) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 22.1.1b)4)による。
6) マイクロシリンジ 7.1b)5)による。
7) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
7.1)
分離管 7.3b)6.1)による。
7.2)
充てん剤 7.3b)6.2)による。
7.3)
溶離液(移動相) 溶離液の調製は,次による(3)。
7.3.1) りん酸塩緩衝液 (pH3.0) JIS K 9007に規定するりん酸二水素カリウム6.8gを水1Lに溶かし,JIS
K 9005に規定するりん酸を加えてpHを3.0に調節したもの。
7.3.2) 溶離液 a)4)のアセトニトリルとりん酸塩緩衝液 (pH3.0) をそれぞれ体積比55 : 45で混合し,超音
波処理などで十分に脱気したもの。
7.4)
流量 約1ml/min
7.5)
検出器 吸光光度検出器 波長272nmで測定できるもの。
7.6)
カラム槽温度 40〜45℃
8) 振とう器
9) 濃縮器 6.1b)5)による。
注(3) 溶離液にアセトニトリル-水混液 (1+1) を用いてもよい。
備考1. 6.の備考1.による。
118
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. 高速液体クロマトグラフの感度として,チウラム標準液 (0.1μgC6H12N2S4/ml) [a)9)のチウラ
ム標準液 (1μgC6H12N2S4/ml) 1mlを10mlに薄めたもの。]20μlを高速液体クロマトグラフに
注入してチウラムの量 (2ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,ジクロロメタン抽出法(4)及び固相抽出法を適用する。
注(4) チウラムはジクロロメタン中で分解するので,直ちにアセトニトリルを加える操作 [c.1)4)] ま
で完了させる。アセトニトリル中では分解しない。
c.1)
ジクロロメタン抽出法 ジクロロメタン抽出法の操作は,次のとおり行う。
1) 4.1c)で採取した試料(5)の適量 (例えば,1 000ml)を分液漏斗にとり,液量1 000mlについて塩化ナト
リウム40gとジクロロメタン100mlとを加えて振とう器を用いて約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
ジクロロメタンで洗い,洗液は先の三角フラスコに合せる。分液漏斗の水層に液量1 000mlについ
てジクロロメタン50mlを加え,再び振とう器を用いて約10分間振り混ぜ,放置後,ジクロロメタ
ン層を先の三角フラスコに合わせる。
3) 6.1c)3)及び4)の操作を行う。
4) 濃縮液にアセトニトリル約50mlを加え,再び濃縮器(6)を用いて,5mlの一定量にする(7)。
5) 空試験として試料と同量の水を分液漏斗にとり,1)〜4)の操作を行う。
注(5) 6.の注(3)による。
(6) 6.の注(9)による。
(7) 6.の注(10)による。
c.2)
固相抽出法 固相抽出法の操作は,次のとおり行う。
1) 22.1.1c.2)1)〜3)の操作を行う。
2) 溶出液に窒素を緩やかに吹き付け,1mlの一定量とする(7)(8)。
3) 空試験としてあらかじめ,塩酸 (2mol/L) を用いて,pHを3.5に調節した試料と同量の水について,
1)及び2)の操作を行う。
注(8) 6.の注(21)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のチウラム標準液 (1μgC6H12N2S4/ml) 20μlをマイクロシリンジ(9)でとり,高速液体クロマトグラ
フに注入し,チウラムの保持時間に相当するピークの位置を確認しておく。
2) c.1)4)又はc.2)2)で得たアセトニトリル溶液20μlをマイクロシリンジ(9)でとり,10.2d)2)の操作を行
う。
3) 空試験として,c.1)5)又はc.2)3)で得たアセトニトリル溶液について10.2d)3)の操作を行う。
4) 検量線からチウラムの量を求め,22.1.1d)の式によって試料中のチウラムの濃度 (μg/L) を算出する。
検量線 a)8)のチウラム標準液 (10μgC6H12N2S4/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,
アセトニトリルを標線まで加える。これらの溶液の一定量[試料と同量(例えば,20μl)]をマイ
クロシリンジでとり1)及び2)の操作を行ってチウラムの量 (ng) と指示値(10)との関係線を作成す
る。検量線の作成は,試料測定時に行う。
注(9) 7.の注(9)による。
(10) 7.の注(11)による。
備考3. 7.の備考15.による。
119
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
36. チオベンカルブ[ベンチオカーブ] (C12H16ClNOS) チオベンカルブ[ベンチオカーブ]〔ジエチル
カルバモチオ酸S-[(4-クロロフェニル)メチル]エステル〕の定量には,ガスクロマトグラフ質量分析法
及びガスクロマトグラフ法を適用する。
36.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H16ClNOS 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) チオベンカルブ標準液 (1mgC12H16ClNOS/ml) チオベンカルブの標準品0.100gをとり,少量のヘ
キサンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) チオベンカルブ標準液 (10μgC12H16ClNOS/ml) チオベンカルブ標準液 (1mgC12H16ClNOS/ml) 1ml
を全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所にお
く。
9) チオベンカルブ標準液 (5μgC12H16ClNOS/ml) チオベンカルブ標準液 (10μgC12H16ClNOS/ml) 5ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) チオベンカルブ標準液 (0.5μgC12H16ClNOS/ml) チオベンカルブ標準液 (5μgC12H16ClNOS/ml) 1ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
11) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,チオベンカルブ標準液 (50ngC12H16ClNOS/ml)
[a)10)のチオベンカルブ標準液 (0.5μgC12H16ClNOS/ml) 1mlを10mlに薄めたもの。]1μlをガ
120
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
スクロマトグラフ質量分析計に注入してチオベンカルブの量 (0.05ng) が十分に確認できる
ように検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
注(3) 9.の注(3)による。
(4) チオベンカルブだけの定量を行うので,6.3のc)4)の操作は省略してよい。
備考2. 9.の備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)10)のチオベンカルブ標準液 (0.5μgC12H16ClNOS/ml) 1μlをマイクロシリンジ(5)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,チオベンカルブ
のフラグメントイオン(m/z 100,72,125など)を設定し(6),選択イオン検出法又はマスクロマト
グラフ法によって測定してそのマスフラグメントグラムを記録し,チオベンカルブの保持時間に相
当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からチオベンカルブの量を求め,9.1d)4)の式によって試料中のチオベンカルブの濃度 (μg/L)
を算出する。
検量線 a)9)のチオベンカルブ標準液 (5μgC12H16ClNOS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってチオベンカルブの量 (ng) と指示値(7)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(10)による。
(7) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
36.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器又は電子捕獲検出器を用いた方法で定量する。
定量範囲:C12H16ClNOS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) チオベンカルブ標準液 (10μgC12H16ClNOS/ml) 36.1a)8)による。
8) チオベンカルブ標準液 (1μgC12H16ClNOS/ml) チオベンカルブ標準液 (10μgC12H16ClNOS/ml) 1ml
を全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
121
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。電子捕獲検出器を用いる場合は,
7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(8)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
カラム槽温度 7.2.1b)7.7)による。
8.7)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注(8) 7.の注(20)による。
備考5. ガスクロマトグラフの感度として,チオベンカルブ標準液 (0.1μgC12H16ClNOS/ml) [a)8)の
チオベンカルブ標準液 (1μgC12H16ClNOS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマト
グラフに注入してチオベンカルブの量 (0.1ng) が十分に確認できるように検出器の感度を調
節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考11.による。
c) 準備操作 準備操作は,9.1c)による(3)(4)。
備考7. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)8)のチオベンカルブ標準液 (1μgC12H16ClNOS/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,チオベンカルブの
保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からチオベンカルブの量を求め,9.1d)4)の式によって試料中のチオベンカルブの濃度 (μg/L)
を算出する。
検量線 a)7)のチオベンカルブ標準液 (10μgC12H16ClNOS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
122
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
マイクロシリンジでとり,1)及び2)の操作を行ってチオベンカルブの量 (ng) と指示値(7)との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考8. 備考3.による。
37. テルブカルブ [MBPMC] (C17H27NO2) テルブカルブ [MBPMC] [2, 6-ビス(1, 1-ジメチルエチル)-4-
メチルフェノールメチルカーバマート]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグ
ラフ法を適用する。
37.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C17H27NO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) テルブカルブ標準液 (1mgC17H27NO2/ml) テルブカルブの標準品0.100gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) テルブカルブ標準液 (10μgC17H27NO2/ml) テルブカルブ標準液 (1mgC17H27NO2/ml) 1mlを全量フ
ラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) テルブカルブ標準液 (1μgC17H27NO2/ml) テルブカルブ標準液 (10μgC17H27NO2/ml) 1mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,テルブカルブ標準液 (0.1μgC17H27NO2/ml)
[a)9)のテルブカルブ標準液 (1μgC17H27NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してテルブカルブの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
123
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のテルブカルブ標準液 (1μgC17H27NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,テルブカルブのフラグ
メントイオン(m/z 205,220,145など)を設定し(4),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,テルブカルブの保持時間に相当するピー
クの位置を確認しておく。
2) c)2)で得たアセトン1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得た空試験のアセトンについて7.1d)3)の操作を行う。
4) 検量線からテルブカルブの量を求め,13.1d)4)の式によって試料中のテルブカルブの濃度 (μg/L) を
算出する。
検量線 a)8)のテルブカルブ標準液 (10μgC17H27NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってテルブカルブの量 (ng) と指示値(5)との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
37.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H27NO2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) テルブカルブ標準液 (10μgC17H27NO2/ml) 37.1a)8)による。
8) テルブカルブ標準液 (4μgC17H27NO2/ml) テルブカルブ標準液 (10μgC17H27NO2/ml) 4mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
124
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,テルブカルブ標準液 (0.4μgC17H27NO2/ml) [a)8)のテルブ
カルブ標準液 (4μgC17H27NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注
入してテルブカルブの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
備考6. 13.の備考6.による。
d) 操作 操作は,次のとおり行う。
1) a)8)のテルブカルブ標準液 (4μgC17H27NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,テルブカルブの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からテルブカルブの量を求め,13.1d)4)の式によって試料中のテルブカルブの濃度 (μg/L) を
算出する。
検量線 a)7)のテルブカルブ標準液 (10μgC17H27NO2/ml) 0.4〜8mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってテルブカルブの量 (ng) と指示値(5)との関係線を
作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
38. トリクロピル (C7H4Cl3NO3) トリクロピル〔[(3, 5, 6-トリクロロ-2-ピリジニル)オキシ]酢酸〕の
定量は,高速液体クロマトグラフ (HPLC) 法によって,トリクロピル酸を定量する。別に,高速液体クロ
マトグラフ法又はガスクロマトグラフ質量分析法によって,トリクロピルブトキシエチルを定量し,係数
0.719 2を乗じた値と,先に求めたトリクロピル酸の値との合量を求めてトリクロピルとする。
38.1 高速液体クロマトグラフ法 トリクロピル酸及びトリクロピルブトキシエチルの定量に適用する。
試料中のトリクロピルを塩酸酸性として,酢酸エチルによる抽出を行い,脱水後,2, 2'-オキシビスエタ
125
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ノール-アセトン溶液 (1+49) を加え,一定量まで濃縮する。
この濃縮液の一定量について,さらに濃縮揮散し,一定量のアセトニトリル-水混液 (2+3) を加え,ト
リクロピル酸の測定溶液とする。
また,残りの濃縮液の一定量について,さらに濃縮揮散し,アセトニトリル-水混液 (4+1) を加え,ト
リクロピルブトキシエチルの測定溶液とする。
トリクロピル酸の測定溶液の一部及びトリクロピルブトキシエチルの測定溶液の一部をそれぞれ高速液
体クロマトグラフ (HPLC) を用い,吸光光度検出器で波長295nmの吸光度を測定し,それぞれ定量する。
なお,トリクロピルブトキシエチルの定量には,ガスクロマトグラフ質量分析法も適用できる。
トリクロピルの定量は,トリクロピルブトキシエチルに係数0.719 2を乗じた値とトリクロピル酸の値と
の合量を求める。
定量範囲:C7H4Cl3NO3(トリクロピル酸)0.2〜4ng
C13H16Cl3NO4(トリクロピルブトキシエチル)0.2〜4ng
繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) 22.1.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトニトリル 22.1.1a)6)による。
4) 酢酸エチル 6.備考5.a)4)による。
5) アセトニトリル-水混液 (2+3) 4)のアセトニトリルと水で調製する。
6) アセトニトリル-水混液 (4+1) 4)のアセトニトリルと水で調製する。
7) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
8) トリクロピル酸標準液 (0.5mgC7H4Cl3NO3/ml) トリクロピル酸の標準品0.050gをとり,少量のア
セトニトリルに溶かし,全量フラスコ100mlに移し入れ,アセトニトリルを標線まで加える(1)。
9) トリクロピル酸標準液 (10μgC7H4Cl3NO3/ml) トリクロピル酸標準液 (0.5mgC7H4Cl3NO3/ml) 2ml
を全量フラスコ100mlにとり,アセトニトリル-水混液 (2+3) を標線まで加える。保存する場合は,
−20℃の暗所におく。
10) トリクロピル酸標準液 (1μgC7H4Cl3NO3/ml) トリクロピル酸標準液 (10μgC7H4Cl3NO3/ml) 1mlを
全量フラスコ10mlにとり,アセトニトリル-水混液 (2+3) を標線まで加える。使用時に調製する。
11) トリクロピルブトキシエチル標準液 (0.5mgC13H16Cl3NO4/ml) トリクロピルブトキシエチルの標
準品0.050gをとり,少量のアセトニトリルに溶かし,全量フラスコ100mlに移し入れ,アセトンを
標線まで加える(1)。
12) トリクロピルブトキシエチル標準液 (10μgC13H16Cl3NO4/ml) トリクロピルブトキシエチル標準
液 (0.5mgC13H16Cl3NO4/ml) 2mlを全量フラスコ100mlにとり,アセトニトリル-水混液 (4+1) を標
線まで加える。保存する場合は,−20℃の暗所におく。
13) トリクロピルブトキシエチル標準液 (1μgC13H16Cl3NO4/ml) トリクロピルブトキシエチル標準液
(10μgC13H16Cl3NO4/ml) 1mlを全量フラスコ10mlにとり,アセトニトリル-水混液 (4+1) を標線まで
加える。使用時に調製する。
14) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
126
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 50〜100μlの適切なもの。
5) 高速液体クロマトグラフ 次に掲げる条件を満たすもの。
5.1)
分離管 7.3b)6.1)による。
5.2)
充てん剤 7.3b)6.2)による。
5.3)
溶離液 溶離液の調製は,次による。
5.3.1) トリクロピル酸用溶離液 りん酸塩緩衝液 (pH3.0) (JIS K 9007に規定するりん酸二水素カリウム
1.4gを水1Lに溶かし,JIS K 9005に規定するりん酸を加えてpHを3.0に調節したもの。)とa)3)
のアセトニトリルとを体積比で3 : 2の割合で混合する。
5.3.2) トリクロピルブトキシエチル用溶離液 りん酸塩緩衝液 (pH3.0) [5.3.1)のりん酸塩緩衝液 (pH3.0)
による。]とa)3)のアセトニトリルとを体積比で1 : 4の割合で混合する。
5.4)
流量 約1ml/min
5.5)
検出器 吸光光度検出器で295nmが測定できるもの。
5.6)
カラム槽温度 40〜45℃
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. 高速液体クロマトグラフの感度として,トリクロピル酸標準液 (0.01μgC7H4Cl3NO3/ml)
[a)10)のトリクロピル酸標準液 (1μgC7H4Cl3NO3/ml) 1mlを100mlに薄めたもの。]20μlを高
速液体クロマトグラフに注入してトリクロピル酸の量 (0.2ng) が十分に確認できるように検
出器の感度を調節する。
トリクロピルブトキシエチル標準液 (0.01μgC13H16Cl3NO4/ml) [a)13)のトリクロピルブト
キシエチル標準液 (1μgC13H16Cl3NO4/ml) 1mlを100mlに薄めたもの。]20μlを高速液体クロマ
トグラフに注入してトリクロピルブトキシエチルの量 (0.2ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(250ml以下の一定量)を分液漏斗にとり,液量250mlについて塩酸
(2mol/L) 5ml及び酢酸エチル50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。酢酸エチルを三角フラスコに移し入れ,分液漏斗を少量の酢酸
エチルで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に,液量250mlについて酢酸
エチル50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。酢酸エチル層を先の三角フ
ラスコに合わせる。
3) 酢酸エチル溶液100mlについて硫酸ナトリウム20〜30g(3)を加え,軽く振り混ぜて,約30分間放置
した後(4),ろ紙5種A(又は5種B)(5)を用いてろ過し,ろ液をなす形フラスコ(6)に受ける。酢酸
エチル約15mlを用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリ
ウムを洗浄し,この洗液も酢酸エチル溶液に合わせる。
4) 酢酸エチル溶液に2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 0.5mlを加える。
5) 濃縮器(7)を用いて約40℃(7)の水浴上で,酢酸エチル溶液が正しく25mlになるまで濃縮する。
6) 濃縮した酢酸エチル溶液25mlから10mlをなす形フラスコに分取し,濃縮器(7)を用いて約40℃(7)
127
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の水浴で約1mlまで濃縮する。受器を取り外し,窒素を緩やかに吹き付け,酢酸エチルを揮散させ
る(8)(9)。
7) 残留物にアセトニトリル-水混液 (2+3) 1mlを正しく加え,トリクロピル酸定量用の溶液とする(10)。
8) 5)で濃縮した酢酸エチル溶液25mlから10mlについて6)の操作を行い,酢酸エチルを揮散させる(8)(9)。
9) 残留物にアセトニトリル-水混液 (4+1) 1mlを正しく加え,トリクロピルブトキシエチル定量用の
溶液とする(10)。
10) 空試験として,試料と同量の水を分液漏斗にとり,液量250mlについて塩酸 (2mol/L) 5ml及び酢酸
エチル50mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。引き続き,2)〜9)の操作を行
う。
注(2) 6.の注(3)による。
(3) 6.の注(5)による。
(4) 6.の注(6)による。
(5) 6.の注(7)による。
(6) 6.の注(8)による。
(7) 6.の注(9)による。
(8) 6.の注(21)による。
(9) 6.備考5.の注(*10)による。
(10) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)10)のトリクロピル酸標準液 (1μgC7H4Cl3NO3/ml) 又はa)13)のトリクロピルブトキシエチル標準
液 (1μgC13H16Cl3NO4/ml) 20μlをそれぞれマイクロシリンジ(11)でとり,高速液体クロマトグラフに注
入し,トリクロピル酸及びトリクロピルブトキシエチルのそれぞれの保持時間に相当するピークの
位置を確認しておく。
2) c)7)で得たアセトニトリル-水混液 (2+3) 又はc)9)で得たアセトニトリル-水混液 (4+1) のそれぞ
れ20μlをマイクロシリンジ(11)でとり,1)の操作を行ってクロマトグラムを記録し,それぞれの保持
時間が1)の保持時間と一致していることを確認し,それぞれの保持時間に相当する位置のピークに
ついて指示値(12)を読み取る。
3) 空試験として,c)10)で得たアセトニトリル-水混液 (2+3) 又はアセトニトリル-水混液 (4+1) のそ
れぞれについて1)及び2)の操作を行う。1)の保持時間に相当する位置のピークが検出され,それぞ
れのその指示値(12)が定量下限値の指示値の1/3以上の場合には,準備操作から再度操作をやり直す。
4) 検量線からトリクロピル酸及びトリクロピルブトキシエチルの量を求め,次の式によって試料中の
トリクロピル酸及びトリクロピルブトキシエチルのそれぞれの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
2
3
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)7)のアセトニトリル-水混液 (2+3) の量又はc)9)のアセト
ニトリル-水混液 (4+1) の量 (ml)
υ2: c)5)の酢酸エチルの量 (ml)
128
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
υ3: c)6)又はc)8)の酢酸エチルの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
5) トリクロピル酸及びトリクロピルブトキシエチルのそれぞれの濃度 (μg/L) から,次の式によって
試料中のトリクロピルの濃度 (μg/L) を求める。
トリクロピルの濃度 (μg/L) =トリクロピル酸の濃度 (μg/L) +トリクロピルブトキシエチルの
濃度 (μg/L) ×0.719 2
検量線 a)9)のトリクロピル酸標準液 (10μgC7H4Cl3NO3/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトニトリル-水混液 (2+3) を標線まで加える。別に,a)12)のトリクロピルブトキシ
エチル標準液 (10μgC13H16Cl3NO4/ml) 0.1〜2mlを全量フラスコ10mlに段階的にとり,アセトニト
リル-水混液 (4+1) を標線まで加える。それぞれの溶液の一定量[試料と同量(例えば,20μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってトリクロピル酸及びトリクロピルブトキシ
エチルのそれぞれの量 (ng) とそれぞれの指示値(12)との関係線を作成する。検量線の作成は,試
料測定時に行う。
注(11) 7.の注(9)による。
(12) 7.の注(11)による。
備考2. 7.の備考15.による。
38.2 ガスクロマトグラフ質量分析法 トリクロピルブトキシエチルの定量に適用する。
試料中のトリクロピルを塩酸酸性として,酢酸エチルによる抽出を行い,脱水後,2, 2'-オキシビスエタ
ノール-アセトン溶液 (1+49) を加え,一定量まで濃縮する。その一定量について,更に濃縮し,アセトニ
トリル-水混液 (4+1) を加え,トリクロピルブトキシエチルの測定溶液とする。その溶液の一定量を,ガ
スクロマトグラフ質量分析計に注入し,検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定
量する。
定量範囲:C13H16Cl3NO4(トリクロピルブトキシエチル)0.2〜4ng
繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (2mol/L) 22.1.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) 酢酸エチル 6.備考5.のa)4)による。
5) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
6) トリクロピルブトキシエチル標準液 (1mgC13H16Cl3NO4/ml) トリクロピルブトキシエチルの標準
品0.100gをとり,少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで
加える(1)。
7) トリクロピルブトキシエチル標準液 (10μgC13H16Cl3NO4/ml) トリクロピルブトキシエチル標準
液 (1mgC13H16Cl3NO4/ml) 1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する
場合は,−20℃の暗所におく。
8) トリクロピルブトキシエチル標準液 (1μgC13H16Cl3NO4/ml) トリクロピルブトキシエチル標準液
(10μgC13H16Cl3NO4/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製
する。
9) 窒素 6.2a)2)による。
129
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考3. ガスクロマトグラフ質量分析計の感度として,トリクロピルブトキシエチル標準液
(0.1μgC13H16Cl3NO4/ml) [a)8)のトリクロピルブトキシエチル標準液 (1μgC13H16Cl3NO4/ml)
1mlを10mlに薄めたもの。]2μlをガスクロマトグラフ質量分析計に注入してトリクロピルブ
トキシエチルの量 (0.2ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 38.1c)1)〜6)の操作を行う。
2) 残留物にアセトン1mlを正しく加える(10)。
3) 空試験として,試料と同量の水を分液漏斗にとり,1)及び2)の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)のトリクロピルブトキシエチル標準液 (1μgC13H16Cl3NO4/ml) 2μlをマイクロシリンジ(11)でとり,
スプリットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,トリ
クロピルブトキシエチルのフラグメントイオン(m/z 210,212,182など)を設定し(13),選択イオ
ン検出法又はマスクロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,トリ
クロピルブトキシエチルの保持時間に相当するピークの位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(11)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について,7.1d)3)の操作を行う。
4) 検量線からトリクロピルブトキシエチルの量を求め,次の式によって試料中のトリクロピルブトキ
シエチルの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
2
3
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: C)のアセトンの量 (ml)
υ2: 38.1c)5)の酢酸エチルの量 (ml)
υ3: 38.1c)8)の酢酸エチルの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
5) トリクロピルブトキシエチルの濃度 (μg/L) 及び38.1d)によって求めたトリクロピル酸の濃度
(μg/L) から,38.1d)5)の式によって試料中のトリクロピルの濃度 (μg/L) を求める。
検量線 a)7)のトリクロピルブトキシエチル標準液 (10μgC13H16Cl3NO4/ml) 0.1〜2mlを全量フラス
コ10mlに段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例
えば,2μl)]をマイクロシリンジでとり,1)及び2)の操作を行ってトリクロピルブトキシエチル
130
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のそれぞれの量 (ng) とそれぞれの指示値(12)との関係線を作成する。検量線の作成は,試料測定
時に行う。
注(13) 7.の注(10)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
39. トリクロルホン [DEP] (C4H8Cl3O4P) トリクロルホン [DEP] [(2, 2, 2-トリクロロ-1-ヒドロキシエ
チル)ホスホン酸ジメチルエステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラ
フ法を適用する。
39.1 ガスクロマトグラフ質量分析法 試料中のトリクロルホンを塩化ナトリウムの共存下でジクロロメ
タン抽出を行い,引き続きカラムクロマトグラフ分離を行った後,その一定量を,ガスクロマトグラフ質
量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C4H8Cl3O4P 0.5〜10ng,繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+1) 3)のアセトンと5)のヘキサンで調製する。
7) トリクロルホン標準液 (1mgC4H8Cl3O4P/ml) トリクロルホンの標準品0.100gをとり,少量のアセ
トンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) トリクロルホン標準液 (50μgC4H8Cl3O4P/ml) トリクロルホン標準液 (1mgC4H8Cl3O4P/ml) 5mlを
全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) トリクロルホン標準液 (5μgC4H8Cl3O4P/ml) トリクロルホン標準液 (50μgC4H8Cl3O4P/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。ただし,試料導入方法及び試料導入部温度は,次
による。
6.1)
試料導入方法及び試料導入部温度 コールドオンカラム方式による。試料導入部温度は,50〜100℃。
7) 振とう器
8) 濃縮器 6.1b)5)による。
131
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考1. ガスクロマトグラフ質量分析計の感度として,トリクロルホン標準液 (0.5μgC4H8Cl3O4P/ml)
[a)9)のトリクロルホン標準液 (5μgC4H8Cl3O4P/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してトリクロルホンの量 (0.5ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(1 000ml以下の一定量)を分液漏斗にとり,液量1 000mlについて塩
化ナトリウム360gとジクロロメタン100ml(3)とを加え,振とう器を用いて約5分間振り混ぜ,放置
する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を少量の
ジクロロメタンで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量1 000mlにつ
いてジクロロメタン100mlを加え,再び振とう器を用いて約5分間振り混ぜ,放置する。ジクロロ
メタン層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液20mlについて硫酸ナトリウム約3g(4)を加え,軽く振り混ぜ,約10分間放置し
た後(5),ろ紙5種A(又は5種B)(6)を用いてろ過し,ろ液をなす形フラスコ(7)に受ける。少量の
ジクロロメタンを用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリ
ウムも洗浄し,洗液をジクロロメタン溶液に合わせる。
4) 濃縮器(8)を用いて約40℃(8)の水浴上でジクロロメタン溶液を1〜2mlになるまで濃縮し,受器を取
り外し,窒素を緩やかに吹き付け,揮散させる(9)(10)。
5) 受器の内容物を少量のジクロロメタンで溶かした後,全量フラスコ10mlに移し入れ,更に受器を
ジクロロメタン2〜3mlで,2,3回よく洗浄し,洗液も全量フラスコ10mlに移し入れ,ジクロロメ
タンを標線まで加える(11)。
6) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から流し込む。さらに,全量フラスコ
10mlもジクロロメタン2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ管に流し込む。
7) アセトン-ヘキサン溶離液 (1+1) 50mlを流下してトリクロルホン (DEP) を溶出させ(12),溶出液を
なす形フラスコ(7)に受ける。
8) これを,濃縮器(8)を用いて約40℃(8)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を完全に揮散させた後,アセトン1mlを正しく加える(9)(10)(11)。
9) 空試験として試料と同量の水を分液漏斗にとり,液量1 000mlについて塩化ナトリウム360gとジク
ロロメタン100ml(3)とを加え,振とう器を用いて約5分間振り混ぜ,放置する。続いて,2)〜8)の操
作を行う。
注(2) 6.の注(3)による。
(3) 6.の注(4)による。
(4) 6.の注(5)による。
(5) 6.の注(6)による。
(6) 6.の注(7)による。
(7) 6.の注(8)による。
(8) 6.の注(9)による。
(9) 6.の注(21)による。
(10) 6.備考5.の注(*10)による。
(11) 6.の注(10)による。
132
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(12) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のトリクロルホン標準液 (5μgC4H8Cl3O4p/ml) 1μlをマイクロシリンジ(13)でとり,コールドオン
カラム注入法によってガスクロマトグラフに注入し,トリクロルホンのフラグメントイオン(m/z
109,79,145など)を設定し(14),選択イオン検出法又はマスクロマトグラフ法によって測定して
そのマスフラグメントグラムを記録し,トリクロルホンの保持時間に相当するピークの位置を確認
しておく。
2) c)8)で得たアセトン溶液1μlをマイクロシリンジ(13)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)9)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からトリクロルホンの量を求め,次の式によって試料中のトリクロルホンの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
2
3
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)のアセトンの量 (ml)
υ2: c)5)のジクロロメタンの量 (ml)
υ3: c)6)のジクロロメタンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のトリクロルホン標準液 (50μgC4H8Cl3O4P/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってトリクロルホンの量 (ng) と指示値(15)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(13) 7.の注(9)による。
(14) 7.の注(10)による。
(15) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
39.2 ガスクロマトグラフ法 試料について39.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,炎光光度検出器を用いた方法で定量する。
定量範囲:C4H8Cl3O4P 1〜20ng,繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+1) 39.1a)6)による。
133
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) トリクロルホン標準液 (50μgC4H8Cl3O4P/ml) 39.1a)8)による。
8) トリクロルホン標準液 (5μgC4H8Cl3O4P/ml) 39.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 39.1b)6.1)による。
6.6)
燃料ガス及び助燃ガス 7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,トリクロルホン標準液 (1μgC4H8Cl3O4P/ml) [a)8)のトリ
クロルホン標準液 (5μgC4H8Cl3O4P/ml) 2mlを10mlに薄めたもの。]1μlをガスクロマトグラフ
に注入してトリクロルホンの量 (1ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフ条件などは7.の備考8.による。
c) 準備操作 準備操作は,39.1c)の操作を行う(16)。
注(16) 抽出溶媒をジクロロメタンに代えて,酢酸エチルを用いてもよい。この場合の準備操作は,備
考6.による。
備考6. 抽出溶媒に酢酸エチルを用いてもよい。この場合の準備操作は,次による。
1) 4.1c)で採取した試料(*1)の適量(500ml以下の一定量)を分液漏斗にとり,液量100mlについ
て塩化ナトリウム12gを加えて溶かし,酢酸エチル[6.備考5.のa)4)による。]10ml(*2)を加え,
振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。酢酸エチルを三角フラスコに移し入れ,分液漏斗を少量
の酢酸エチルで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に,液量100ml
について酢酸エチル10mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。酢酸エ
チル層を先の三角フラスコに合わせる。
3) 酢酸エチル溶液20mlについて硫酸ナトリウム5g(*3)を加え,軽く振り混ぜて,約30分間放
置した後(*4),ろ紙5種A(又は5種B)(*5)を用いてろ過し,ろ液をなす形フラスコ(*6)に受
ける。酢酸エチル約3mlを用いて三角フラスコを数回洗浄し,さらにその洗液で先のろ紙上
の硫酸ナトリウムを洗浄し,この洗液も酢酸エチル溶液に合わせる。
134
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 濃縮器(*7)を用いて約40℃(*7)の水浴で,酢酸エチル溶液を約1mlになるまで濃縮し,受器を
取り外し,窒素を緩やかに吹き付け,酢酸エチルを揮散させる(*8)(*9)。
5) 残留物にアセトン2mlを正しく加える(*10)。
6) 空試験として,試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム12g
を加えて溶かし,酢酸エチル10mlを加え,振とう器を用いて約5分間振り混ぜ,放置する。
引き続き,2)〜5)の操作を行う。
注(*1) 6.の注(3)による。
(*2) 6.の注(4)による。ただし,酢酸エチルの量を増加する。
(*3) 6.の注(5)による。
(*4) 6.の注(6)による。
(*5) 6.の注(7)による。
(*6) 6.の注(8)による。
(*7) 6.の注(9)による。
(*8) 6.の注(21)による。
(*9) 6.備考5.の注(*10)による。
(*10) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のトリクロルホン標準液 (5μgC4H8Cl3O4P/ml) 1μlをマイクロシリンジ(13)でとり,コールドオン
カラム注入法によってガスクロマトグラフに注入し,トリクロルホンの保持時間に相当するピーク
の位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(13)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からトリクロルホンの量を求め,39.1d)4)の式によって試料中のトリクロルホンの濃度
(μg/L) を算出する。
検量線 a)7)のトリクロルホン標準液 (50μgC4H8Cl3O4P/ml) 0.2〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってトリクロルホンの量 (ng) と指示値(15)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
40. トリシクラゾール (C9H7N3S) トリシクラゾール[5-メチル-1, 2, 4-トリアゾロ (3, 4-b) ベンゾチアゾ
ール]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
40.1 ガスクロマトグラフ質量分析法 試料中のトリシクラゾールを塩化ナトリウムの共存下でジクロロ
メタン抽出を行い,引き続きカラムクロマトグラフ分離を行う。濃縮した溶液の一定量を,ガスクロマト
グラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定
量する。
定量範囲:C9H7N3S 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
135
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+9) 3)のアセトンと5)のヘキサンで調製する。
7) アセトン-ヘキサン溶離液 (3+7) 10.1a)8)による。
8) トリシクラゾール標準液 (1mgC9H7N3S/ml) トリシクラゾールの標準品0.100gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
9) トリシクラゾール標準液 (10μgC9H7N3S/ml) トリシクラゾール標準液 (1mgC9H7N3S/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
10) トリシクラゾール標準液 (5μgC9H7N3S/ml) トリシクラゾール標準液 (10μgC9H7N3S/ml) 5mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
11) トリシクラゾール標準液 (0.5μgC9H7N3S/ml) トリシクラゾール標準液 (5μgC9H7N3S/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。ただし,クロマトグラフ管にカラムクロマトグラフ用の活性け
い酸マグネシウムを充てんする場合には,a)6)のアセトン-ヘキサン溶離液 (1+9) を用いる。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,トリシクラゾール標準液 (50ngC9H7N3S/ml)
[a)11)のトリシクラゾール標準液 (0.5μgC9H7N3S/ml) 1mlを10mlに薄めたもの。]2μlをガス
クロマトグラフ質量分析計に注入してトリシクラゾールの量 (0.1ng) が十分に確認できるよ
うに検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 33.1c)1)〜4)の操作を行う。
2) 受器の内容物を少量のアセトン-ヘキサン溶離液 (1+9) で溶かした後,全量フラスコ10mlに移し
入れ,更に受器をアセトン-ヘキサン溶離液 (1+9) 2〜3mlで2,3回よく洗浄し,洗液も全量フラ
スコ10mlに移し入れ,アセトン-ヘキサン溶離液 (1+9) を標線まで加える(2)。
3) 全量フラスコ10mlの内容物全量をクロマトグラフ管上部から注ぎ,流下させる。更に全量フラス
コ10mlもアセトン-ヘキサン溶離液 (1+9) 2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ管
に流し込む。
4) クロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+9) 80mlを流下させる。この流出液は
捨てる。
136
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (3+7) 100mlを流下し,トリシ
クラゾールを溶出させ(3),溶出液をなす形フラスコ(4)に受ける。
6) 濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を揮散させた後,アセトン4mlを正しく加える(2)(6)(7)。
7) 空試験として試料と同量の水を分液漏斗にとり,液量100mlについて塩化ナトリウム5gとジクロ
ロメタン25mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。続いて2)〜6)の操作を
行う。
注(2) 6.の注(10)による。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
d) 操作 操作は,次のとおり行う。
1) a)11)のトリシクラゾール標準液 (0.5μgC9H7N3S/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,トリシクラゾール
のフラグメントイオン (m/z 189, 162, 161) を設定し(9),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,トリシクラゾールの保持時間に相当す
るピークの位置を確認しておく。
2) c)6)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について,7.1d)3)の操作を行う。
4) 検量線からトリシクラゾールの量を求め,次の式によって試料中のトリシクラゾールの濃度 (μg/L)
を算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)2)のアセトン-ヘキサン溶離液 (1+9) の量 (ml)
υ2: c)3)のアセトン-ヘキサン溶離液 (1+9) の量 (ml)
υ3: c)6)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)10)のトリシクラゾール標準液 (5μgC9H7N3S/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってトリシクラゾールの量 (ng) と指示値(10)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) 7.の注(10)による。
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
137
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 7.の備考3.による。
40.2 ガスクロマトグラフ法 試料について40.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C9H7N3S 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) アセトン-ヘキサン溶離液 (1+9) 40.1a)6)による。
6) アセトン-ヘキサン溶離液 (3+7) 10.1a)8)による。
7) トリシクラゾール標準液 (5μgC9H7N3S/ml) 40.1a)10)による。
8) トリシクラゾール標準液 (0.5μgC9H7N3S/ml) 40.1a)11)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 40.1b)4)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,トリシクラゾール標準液 (50ngC9H7N3S/ml) [a)8)のトリ
シクラゾール標準液 (0.5μgC9H7N3S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フに注入してトリシクラゾールの量 (0.1ng) が十分に確認できるように検出器の感度を調節
する。
c) 準備操作 準備操作は,40.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のトリシクラゾール標準液 (0.5μgC9H7N3S/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,トリシクラゾール
138
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からトリシクラゾールの量を求め,40.1d)4)の式によって試料中のトリシクラゾールの濃度
(μg/L) を算出する。
検量線 a)7)のトリシクラゾール標準液 (5μgC9H7N3S/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってトリシクラゾールの量 (ng) と指示値(10)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
41. トルクロホスメチル (C9H11Cl2O3PS) トルクロホスメチル[O, O-ジメチルO-(2, 6-ジクロロ-4-メチ
ルフェニル)ホスホロチオ酸エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグ
ラフ法を適用する。
41.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C9H11Cl2O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) トルクロホスメチル標準液 (1mgC9H11Cl2O3PS/ml) トルクロホスメチルの標準品0.100gをとり,
少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) トルクロホスメチル標準液 (10μgC9H11Cl2O3PS/ml) トルクロホスメチル標準液
(1mgC9H11Cl2O3PS/ml) 1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場
合は,−20℃の暗所におく。
9) トルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) トルクロホスメチル標準液
(10μgC9H11Cl2O3PS/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製
する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
139
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,トルクロホスメチル標準液
(0.1μgC9H11Cl2O3PS/ml) [a)9)のトルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) 1mlを10ml
に薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入してトルクロホスメチルの量
(0.1ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)(3)。
注(2) 13.の注(2)による。
(3) 抽出溶媒をジクロロメタンに代えて,ヘキサンを用いてもよい。この場合の準備操作は,備考
2.による。
備考2. 抽出溶媒にヘキサンを用いてもよい。この場合の準備操作などは,次による。
1) 29.1c)1)〜6)の操作を行う。
2) 次に,クロマトグラフ管の上部からヘキサン70mlを流下する。この流出液は捨てる。
3) ジエチルエーテル-ヘキサン溶離液 (1+4) [6.3a)2)のジエチルエーテル及び6.1a)4)のヘキサ
ンを用いて調製する。]70mlを流下してトルクロホスメチルを溶出させ(*1),溶出液をなす形
フラスコ(*2)に受ける。
4) 濃縮器(*3)を用いて約40℃(*3)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を
緩やかに吹き付け,溶出液を揮散させた後,ヘキサン4mlを正しく加える(*4)(*5)(*6)。
5) 空試験として試料と同量の水を分液漏斗にとり,1)〜4)の操作を行う。
注(*1) 6.の注(25)による。
(*2) 6.の注(8)による。
(*3) 6.の注(9)による。
(*4) 6.の注(21)による。
(*5) 6.備考5.の注(*10)による。
(*6) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のトルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,トルクロホス
メチルのフラグメントイオン(m/z 265,267,125など)を設定し(5),選択イオン検出法又はマスク
ロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,トルクロホスメチルの保
持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からトルクロホスメチルの量を求め,13.1d)4)の式によって試料中のトルクロホスメチルの濃
度 (μg/L) を算出する。
検量線 a)8)のトルクロホスメチル標準液 (10μgC9H11Cl2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
140
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
をマイクロシリンジでとり,1)及び2)の操作を行ってトルクロホスメチルの量 (ng) と指示値(6)
との関係線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
41.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器又は炎光光度検出器を用いた方法で定量する。
定量範囲:C9H11Cl2O3PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) トルクロホスメチル標準液 (10μgC9H11Cl2O3PS/ml) 41.1a)8)による。
8) トルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) 41.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
141
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考5. ガスクロマトグラフの感度として,トルクロホスメチル標準液 (0.1μgC9H11Cl2O3PS/ml) [a)8)
のトルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフに注入してトルクロホスメチルの量 (0.1ng) が十分に確認できるように検出器
の感度を調節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)(3)。
備考7. 備考2.による。
8. 13.の備考6.による。ただし,最終溶媒量はアセトン2mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のトルクロホスメチル標準液 (1μgC9H11Cl2O3PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,トルクロホス
メチルの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からトルクロホスメチルの量を求め,13.1d)4)の式によって試料中のトルクロホスメチルの濃
度 (μg/L) を算出する。
検量線 a)7)のトルクロホスメチル標準液 (10μgC9H11Cl2O3PS/ml) 0.1〜2mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってトルクロホスメチルの量 (ng) と指示値(6)
との関係線を作成する。検量線の作成は,試料測定時に行う。
備考9. 7.の備考3.による。
42. ナプロパミド (C17H21NO2) ナプロパミド[N, N-ジエチル-2-(1-ナフタレニルオキシ)プロパンアミ
ド]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
42.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C17H21NO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ナプロパミド標準液 (1mgC17H21NO2/ml) ナプロパミドの標準品0.100gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ナプロパミド標準液 (10μgC17H21NO2/ml) ナプロパミド標準液 (1mgC17H21NO2/ml) 1mlを全量フ
142
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ナプロパミド標準液 (1μgC17H21NO2/ml) ナプロパミド標準液 (10μgC17H21NO2/ml) 1mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ナプロパミド標準液 (0.1μgC17H21NO2/ml)
[a)9)のナプロパミド標準液 (1μgC17H21NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してナプロパミドの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のナプロパミド標準液 (1μgC17H21NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ナプロパミドのフラグ
メントイオン(m/z 72,128,271など)を設定し(4),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,ナプロパミドの保持時間に相当するピー
クの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からナプロパミドの量を求め,13.1d)4)の式によって試料中のナプロパミドの濃度 (μg/L) を
算出する。
検量線 a)8)のナプロパミド標準液 (10μgC17H21NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってナプロパミドの量 (ng) と指示値(5)との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
143
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
42.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H21NO2 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ナプロパミド標準液 (10μgC17H21NO2/ml) 41.1a)8)による。
8) ナプロパミド標準液 (4μgC17H21NO2/ml) ナプロパミド標準液 (10μgC17H21NO2/ml) 4mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ナプロパミド標準液 (0.4μgC17H21NO2/ml) [a)8)のナプロ
パミド標準液 (4μgC17H21NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注
入してナプロパミドの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
備考6. 14.の備考10.による。ただし,最終溶媒量はアセトン2mlとする。
d) 操作 操作は,次のとおり行う。
144
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) a)8)のナプロパミド標準液 (4μgC17H21NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ナプロパミドの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からナプロパミドの量を求め,13.1d)4)の式によって試料中のナプロパミドの濃度 (μg/L) を
算出する。
検量線 a)7)のナプロパミド標準液 (10μgC17H21NO2/ml) 0.4〜8mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってナプロパミドの量 (ng) と指示値(5)との関係線を
作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
43. ピリダフェンチオン (C14H17N2O4PS) ピリダフェンチオン[ホスホロチオ酸O-(1, 6-ジヒドロ-6-オ
キソ-1-フェニル-3-ピリダジニル)O, O-ジエチルエステル]の定量には,ガスクロマトグラフ質量分析法
及びガスクロマトグラフ法を適用する。
43.1 ガスクロマトグラフ質量分析法 試料中のピリダフェンチオンについて6.の備考4.による抽出操作
を行い,引き続きカラムクロマトグラフ分離を行う。濃縮液の一定量を,ガスクロマトグラフ質量分析計
に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C14H17N2O4PS 0.2〜4ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ピリダフェンチオン標準液 (1mgC14H17N2O4PS/ml) ピリダフェンチオンの標準品0.100gをとり,
少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ピリダフェンチオン標準液 (10μgC14H17N2O4PS/ml) ピリダフェンチオン標準液
(1mgC14H17N2O4PS/ml) 1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場
合は,−20℃の暗所におく。
9) ピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) ピリダフェンチオン標準液
(10μgC14H17N2O4PS/ml) 2mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調
製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
145
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ピリダフェンチオン標準液
(0.2μgC14H17N2O4PS/ml) [a)9)のピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) 1mlを10ml
に薄めたもの。]1μlをガスクロマトグラフ質量分析計に注入してピリダフェンチオンの量
(0.2ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜4)の操作を行う(2)。
2) 次に,アセトン-ヘキサン溶離液 (3+17) (3)120mlを流下してピリダフェンチオンを溶出させ(4),溶
出液をなす形フラスコ(5)に受ける。
3) これを,濃縮器(6)を用いて約40℃(6)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を完全に揮散させた後,アセトン1mlを正しく加える(7)(8)(9)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(2) 13.の注(2)による。
(3) アセトン-ヘキサン溶離液 (3+17) に代えて,アセトン-ヘキサン溶離液 (1+19) [6.備考4.の
5)による。]及びアセトン-ヘキサン溶離液 (3+17) とを用いてもよい。この場合の準備操作は
備考2.による。
(4) 6.の注(25)による。
(5) 6.の注(8)による。
(6) 6.の注(9)による。
(7) 6.の注(21)による。
(8) 6.備考5.の注(*10)による。
(9) 6.の注(10)による。
備考2. アセトン-ヘキサン溶離液 (3+17) に代えて,アセトン-ヘキサン溶離液 (1+19) 及びアセト
ン-ヘキサン溶離液 (3+17) とを用いた場合の準備操作は,次による。
1) 4.1c)で採取した試料(*1)の適量(1 000ml以下の一定量)を分液漏斗にとり,液量1 000mlに
ついて塩化ナトリウム50gとジクロロメタン50mlとを加え,振とう器を用いて約10分間振
り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジクロロメタンを三角フラスコに移し入れ,分液漏斗を
少量のジクロロメタンで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量
1 000mlについてジクロロメタン50mlを加え,再び振とう器を用いて5〜10分間振り混ぜ,
放置する。ジクロロメタン層を先の三角フラスコに合わせる。
3) ジクロロメタン溶液100mlについて硫酸ナトリウム20〜30g(*2)を加え,軽く振り混ぜ,約30
分間放置した後(*3),ろ紙5種A(又は5種B)(*4)を用いてろ過し,ろ液をなす形フラスコ(*5)
に受ける。ジクロロメタン10〜20mlを用いて三角フラスコを数回洗浄し,さらにその洗液
146
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で先のろ紙上の硫酸ナトリウムも洗浄し,洗液をジクロロメタン溶液に合わせる。
4) 濃縮器(*6)を用いて,約40℃(*6)の水浴上でジクロロメタン溶液を約1mlになるまで濃縮する。
受器を取り外し,窒素を緩やかに吹き付け,ジクロロメタンを完全に揮散させ,ヘキサン2ml
を正しく加える(*7)(*8)(*9)。
5) このヘキサン溶液全量をクロマトグラフ管(*10)に注ぎ,流下させる。さらに,受器をヘキサ
ン2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ管に流し込む。
6) クロマトグラフ管(*10)の上部から,アセトン-ヘキサン溶離液 (1+19) 20mlを流下させる。こ
の流出液は捨てる。
7) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (3+17) 20mlを流下し,
ピリダフェンチオンを流出させ(*11),溶出液をなす形フラスコ(*5)に受ける。
8) 濃縮器(*6)を用いて約40℃(*6)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を
緩やかに吹き付け,溶出液を完全に揮散させた後,アセトン1mlを正しく加える(*7)(*8)(*9)。
9) 空試験として試料と同量の水を分液漏斗にとり,1)〜8)の操作を行う。
注(*1) 6.の注(3)による。
(*2) 6.の注(5)による。
(*3) 6.の注(6)による。
(*4) 6.の注(7)による。
(*5) 6.の注(8)による。
(*6) 6.の注(9)による。
(*7) 6.の注(21)による。
(*8) 6.備考5.の注(*10)による。
(*9) 6.の注(10)による。
(*10) 用いるクロマトグラフ管は,次による。
けい酸マグネシウムミニカラム 内径約10mm,長さ約25mmでコック付きのカラムに,カ
ラムクロマトグラフ用の合成けい酸マグネシウム(又はこれと同等の性能をもつもの。)約
900mgを充てんし,使用前にヘキサンで洗浄しておく。
(*11) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) 1μlをマイクロシリンジ(10)でとり,スプリ
ットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ピリダフェ
ンチオンのフラグメントイオン(m/z 340,199,188など)を設定し(11),選択イオン検出法又はマ
スクロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,ピリダフェンチオン
の保持時間に相当するピークの位置を確認しておく。
2) c)3)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)4)で得たアセトン溶液について7.1d) 3)の操作を行う。
4) 検量線からピリダフェンチオンの量を求め,13.1d)4)の式によって試料中のピリダフェンチオンの濃
度 (μg/L) を算出する。
検量線 a)8)のピリダフェンチオン標準液 (10μgC14H17N2O4PS/ml) 0.2〜4mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える,これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってピリダフェンチオンの量 (ng) と指示値(12)
147
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
との関係線を作成する。検量線の作成は,試料測定時に行う。
注(10) 7.の注 (9)による。
(11) 7.の注(10)による。
(12) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
43.2 ガスクロマトグラフ法 試料について43.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器又は炎光光度検出器を用いた方法で定量する。
定量範囲:C14H17N2O4PS 0.2〜4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ピリダフェンチオン標準液 (10μgC14H17N2O4PS/ml) 43.1a)8)による。
8) ピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) 43.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.2b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1)7.5)による。
6.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考5. ガスクロマトグラフの感度として,ピリダフェンチオン標準液 (0.2μgC14H17N2O4PS/ml) [a)8)
148
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフに注入してピリダフェンチオンの量 (0.2ng) が十分に確認できるように検出器
の感度を調節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,43.1c)の操作を行う(2)。
備考7. 備考2.による。
8. 13.の備考6.による。ただし,最終溶媒量はアセトン2mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のピリダフェンチオン標準液 (2μgC14H17N2O4PS/ml) 1μlをマイクロシリンジ(10)でとり,スプリ
ットレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ピリダフェ
ンチオンの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からピリダフェンチオンの量を求め,13.1d)4)の式によって試料中のピリダフェンチオンの濃
度 (μg/L)を算出する。
検量線 a)7)のピリダフェンチオン標準液 (10μgC14H17N2O4PS/ml) 0.2〜4mlを全量フラスコ10mlに
段階的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってピリダフェンチオンの量 (ng) と指示値(12)
との関係線を作成する。検量線の作成は,試料測定時に行う。
備考9. 備考3.による。
44. ピリブチカルブ (C18H22N2O2S) ピリブチカルブ[O-3-(1, 1-ジメチルエチル)-N-(6-メトキシ-2-
ピリジル)-N-メチル(チオカーバマート)]の定量には,ガスクロマトグラフ質量分析法及びガスクロマ
トグラフ法を適用する。
44.1 ガスクロマトグラフ質量分析法 試料中のピリブチカルブを塩化ナトリウムの共存下でヘキサン抽
出を行い,脱水・濃縮後,けい酸マグネシウムを用いたカラムクロマトグラフ分離を行い,アセトン-ヘキ
サン溶離液 (1+4) を用いて,ピリブチカルブを溶出させ,さらに濃縮を繰り返す。その一定量を,ガス
クロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を
用いて定量する。
定量範囲:C18H22N2O2S 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)4)による。
5) アセトン-ヘキサン溶離液 (1+4) 3)のアセトンと4)のヘキサンで調製する。
6) ピリブチカルブ標準液 (1mgC18H22N2O2S/ml) ピリブチカルブの標準品0.100gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
149
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) ピリブチカルブ標準液 (10μgC18H22N2O2S/ml) ピリブチカルブ標準液 (1mgC18H22N2O2S/ml) 1ml
を全量フラスコ100mlにとり,ヘキサンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ピリブチカルブ標準液 (5μgC18H22N2O2S/ml) ピリブチカルブ標準液 (10μgC18H22N2O2S/ml) 5mlを
全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
10) ピリブチカルブ標準液 (0.5μgC18H22N2O2S/ml) ピリブチカルブ標準液 (5μgC18H22N2O2S/ml) 1ml
を全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
11) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 次に掲げる条件を満たすもの。
4.1)
カラム用管 内径約10mm,長さ約25mmのもの。
4.2)
カラム充てん剤 カラムクロマトグラフ用の合成けい酸マグネシウム又はこれと同等の性能をもつ
もの。
4.3)
カラム用管 カラム充てん剤900mgをカラム用管に充てんし,使用前にヘキサンで洗浄しておく。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ピリブチカルブ標準液 (50ngC18H22N2O2S/ml)
[a)10)のピリブチカルブ標準液 (0.5μgC18H22N2O2S/ml) 1mlを10mlに薄めたもの。]2μlをガ
スクロマトグラフ質量分析計に注入してピリブチカルブの量 (0.1ng) が十分に確認できるよ
うに検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(100ml以下の一定量)を分液漏斗にとり,液量100mlについて塩化
ナトリウム10gとヘキサン100mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ヘキサンを三角フラスコに移し入れ,分液漏斗を少量のヘキサ
ンで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量100mlについてヘキサン50ml
を加え,再び振とう器を用いて約10分間振り混ぜ,放置する。ヘキサン層を先の三角フラスコに合
わせる。
3) ヘキサン溶液100mlについて硫酸ナトリウム10〜15g(3)を加え,軽く振り混ぜ,約30分間放置した
後(4),ろ紙5種A(又は5種B)(5)を用いてろ過し,ろ液をなす形フラスコ(6)に受ける。ヘキサン
10〜20mlを用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリウム
も洗浄し,洗液をヘキサン溶液に合わせる。
4) 濃縮器(7)を用いて,約40℃(7)の水浴上でヘキサン溶液を約1mlになるまで濃縮する。受器を取り外
し,窒素を緩やかに吹き付け,ヘキサンを揮散させる(8)(9)。
5) 受器の内容物を少量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更に受器をヘキサ
ン2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ10mlに移し入れ,ヘキサンを標線まで加え
150
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る(10)。
6) このヘキサン全量をクロマトグラフ管の上部から注ぎ,流下させる。さらに,全量フラスコ10ml
もヘキサン2〜3mlで数回よく洗浄し,洗液もクロマトグラフ管に流し込む。
7) クロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+4) 20mlを流下し,ピリブチカルブを
溶出させ(11),溶出液をなす形フラスコ(6)に受ける。
8) 濃縮器(7)を用いて約40℃(7)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を揮散させた後,ヘキサン4mlを正しく加える(8)(9)(10)。
9) 空試験として試料と同量の水を分液漏斗にとり,1)〜8)の操作を行う。
注(2) 6.の注(3)による。
(3) 6.の注(5)による。
(4) 6.の注(6)による。
(5) 6.の注(7)による。
(6) 6.の注(8)による。
(7) 6.の注(9)による。
(8) 6.の注(21)による。
(9) 6.備考5.の注(*10)による。
(10) 6.の注(10)による。
(11) 6.の注(25)による。
d) 操作 操作は,次のとおり行う。
1) a)10)のピリブチカルブ標準液 (0.5μgC18H22N2O2S/ml) 2μlをマイクロシリンジ(12)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ピリブチカルブ
のフラグメントイオン (m/z 165, 108, 181) を設定し(13),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,ピリブチカルブの保持時間に相当する
ピークの位置を確認しておく。
2) c)8)で得たヘキサン溶液2μlをマイクロシリンジ(12)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)9)で得たヘキサン溶液について7.1d)3)の操作を行う。
4) 検量線からピリブチカルブの量を求め,次の式によって試料中のピリブチカルブの濃度 (μg/L) を
算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)5)のヘキサンの量 (ml)
υ2: c)6)のヘキサンの量 (ml)
υ3: c)8)のヘキサンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)9)のピリブチカルブ標準液 (5μgC18H22N2O2S/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってピリブチカルブの量 (ng) と指示値(14)との関係
151
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
線を作成する。検量線の作成は,試料測定時に行う。
注(12) 7.の注(9)による。
(13) 7.の注(10)による。
(14) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
44.2 ガスクロマトグラフ法 試料について44.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C18H22N2O2S 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) ヘキサン 6.1a)4)による。
4) アセトン-ヘキサン溶離液 (1+4) 44.1a)5)による。
5) ピリブチカルブ標準液 (5μgC18H22N2O2S/ml) 44.1a)9)による。
6) ピリブチカルブ標準液 (0.5μgC18H22N2O2S/ml) 44.1a)10)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 44.1b)4)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ピリブチカルブ標準液 (50ngC18H22N2O2S/ml) [a)6)のピ
リブチカルブ標準液 (0.5μgC18H22N2O2S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマト
グラフに注入してピリブチカルブの量 (0.1ng) が十分に確認できるように検出器の感度を調
節する。
c) 準備操作 準備操作は,44.1c)による。
152
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 操作 操作は,次のとおり行う。
1) a)6)のピリブチカルブ標準液 (0.5μgC18H22N2O2S/ml) 2μlをマイクロシリンジ(12)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ピリブチカルブ
の保持時間に相当するピークの位置を確認しておく。
2) c)で得たヘキサン溶液2μlをマイクロシリンジ(12)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からピリブチカルブの量を求め,44.1d)4)の式によって試料中のピリブチカルブの濃度
(μg/L) を算出する。
検量線 a)5)のピリブチカルブ標準液 (5μgC18H22N2O2S/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってピリブチカルブの量 (ng) と指示値(14)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
45. フェニトロチオン [MEP] (C9H12NO5PS) フェニトロチオン [MEP] [ホスホロチオ酸O, O-ジメチ
ルO-(3-メチル-4-ニトロフェニル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロ
マトグラフ法を適用する。
45.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C9H12NO5PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) フェニトロチオン標準液 (1mgC9H12NO5PS/ml) フェニトロチオンの標準品0.100gをとり,少量
のヘキサンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) フェニトロチオン標準液 (10μgC9H12NO5PS/ml) フェニトロチオン標準液 (1mgC9H12NO5PS/ml)
1mlを全量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所
におく。
9) フェニトロチオン標準液 (1μgC9H12NO5PS/ml) フェニトロチオン標準液 (10μgC9H12NO5PS/ml)
1mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
153
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,フェニトロチオン標準液
(0.1μgC9H12NO5PS/ml) [a)9)のフェニトロチオン標準液 (1μgC9H12NO5PS/ml) 1mlを10mlに薄
めたもの。]1μlをガスクロマトグラフ質量分析計に注入してフェニトロチオンの量 (0.1ng)
が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
3. 12.の備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のフェニトロチオン標準液 (1μgC9H12NO5PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フェニトロチオ
ンのフラグメントイオン(m/z 277,125,109など)を設定し(5),選択イオン検出法又はマスクロマ
トグラフ法によって測定してそのマスフラグメントグラムを記録し,フェニトロチオンの保持時間
に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からフェニトロチオンの量を求め,12.1d)4)の式によって試料中のフェニトロチオンの濃度
(μg/L) を算出する。
検量線 a)8)のフェニトロチオン標準液 (10μgC9H12NO5PS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってフェニトロチオンの量 (ng) と指示値(6)と
の関係線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
45.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,炎光光度検出器,電子捕獲検出器又は熱イオン化検出器を用いた方法で定量する。
定量範囲:C9H12NO5PS 0.05〜1ng(7) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によっ
154
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
て異なる。)
注(7) 13.の注(12)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) フェニトロチオン標準液 (10μgC9H12NO5PS/ml) 45.1a)8)による。
8) フェニトロチオン標準液 (5μgC9H12NO5PS/ml) フェニトロチオン標準液 (10μgC9H12NO5PS/ml)
5mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) フェニトロチオン標準液 (0.5μgC9H12NO5PS/ml) フェニトロチオン標準液 (5μgC9H12NO5PS/ml)
1mlを全量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。電子捕獲検出器を用いる場合は,7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(8)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。電子捕獲検出器では用いない。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注(8) 7.の注(20)による。
備考6. ガスクロマトグラフの感度として,フェニトロチオン標準液 (50ngC9H12NO5PS/ml) [a)9)の
155
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フェニトロチオン標準液 (0.5μgC9H12NO5PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマ
トグラフに注入してフェニトロチオンの量 (0.05ng) が十分に確認できるように検出器の感
度を調節する。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.,7.の備考8.又は7.の備
考11.による。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)(9)。
注(9) 6.の備考5.の抽出操作を行ってもよい。
備考8. 備考2.による。
9. 備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のフェニトロチオン標準液 (0.5μgC9H12NO5PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フェニトロチ
オンの保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からフェニトロチオンの量を求め,12.1d)4)の式によって試料中のフェニトロチオンの濃度
(μg/L)を算出する。
検量線 a)8)のフェニトロチオン標準液 (5μgC9H12NO5PS/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]
をマイクロシリンジでとり,1)及び2)の操作を行ってフェニトロチオンの量 (ng) と指示値(6)と
の関係線を作成する。検量線の作成は,試料測定時に行う。
備考10. 備考4.による。
46. フェノブカルブ [BPMC] (C12H17NO2) フェノブカルブ [BPMC] [2-(1-メチルプロピル)-フェノ
ールメチルカーバマート]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用
する。
46.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H17NO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) フェノブカルブ標準液 (1mgC12H17NO2/ml) フェノブカルブの標準品0.100gをとり,少量のアセ
156
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
トンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) フェノブカルブ標準液 (10μgC12H17NO2/ml) フェノブカルブ標準液 (1mgC12H17NO2/ml) 1mlを全
量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) フェノブカルブ標準液 (1μgC12H17NO2/ml) フェノブカルブ標準液 (10μgC12H17NO2/ml) 1mlを全
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,フェノブカルブ標準液 (0.1μgC12H17NO2/ml)
[a)9)のフェノブカルブ標準液 (1μgC12H17NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してフェノブカルブの量 (0.1ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
3. 12.の備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のフェノブカルブ標準液 (10μgC12H17NO2/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フェノブカルブの
フラグメントイオン (m/z 121, 150, 91) を設定し(5),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,フェノブカルブの保持時間に相当するピー
クの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からフェノブカルブの量を求め,12.1d)4)の式によって試料中のフェノブカルブの濃度
(μg/L) を算出する。
検量線 a)8)のフェノブカルブ標準液 (10μgC12H17NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってフェノブカルブの量 (ng) と指示値(6)との関
157
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
係線を作成する。検量線の作成は,試料測定時に行う
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
46.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C12H17NO2 0.6〜12ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) フェノブカルブ標準液 (10μgC12H17NO2/ml) 46.1a)8)による。
8) フェノブカルブ標準液 (6μgC12H17NO2/ml) フェノブカルブ標準液 (10μgC12H17NO2/ml) 6mlを全
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。
9) フェノブカルブ標準液 (0.6μgC12H17NO2/ml) フェノブカルブ標準液 (6μgC12H17NO2/ml) 1mlを全
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 7.2.1b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
158
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考6. ガスクロマトグラフの感度として,フェノブカルブ標準液 (60ngC12H17NO2/ml) [a)9)のフェ
ノブカルブ標準液 (0.6μgC12H17NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フに注入してフェノブカルブの量 (0.6ng) が十分に確認できるように検出器の感度を調節す
る。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)。
備考8. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のフェノブカルブ標準液 (0.6μgC12H17NO2/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フェノブカルブの
保持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からフェノブカルブの量を求め,12.1d)4)の式によって試料中のフェノブカルブの濃度
(μg/L) を算出する。
検量線 a)8)のフェノブカルブ標準液 (6μgC12H17NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってフェノブカルブの量 (ng) と指示値(6)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考9. 備考4.による。
47. フサライド (C8H2Cl4O2) フサライド[4, 5, 6, 7-テトラクロロ-1 (3H) -イソベンゾフラノン]の定量
には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
47.1 ガスクロマトグラフ質量分析法 試料中のフサライドを塩化ナトリウムの共存下でヘキサン抽出を
行い,脱水・濃縮を行う。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に
選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C8H2Cl4O2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)4)による。
5) フサライド標準液 (1mgC8H2Cl4O2/ml) フサライドの標準品0.100gをとり,少量のアセトンに溶
かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) フサライド標準液 (10μgC8H2Cl4O2/ml) フサライド標準液 (1mgC8H2Cl4O2/ml) 1mlを全量フラス
コ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
159
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) フサライド標準液 (5μgC8H2Cl4O2/ml) フサライド標準液 (10μgC8H2Cl4O2/ml) 5mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) フサライド標準液 (0.5μgC8H2Cl4O2/ml) フサライド標準液 (5μgC8H2Cl4O2/ml) 1mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,フサライド標準液 (50ngC8H2Cl4O2/ml) [a)6)
のフサライド標準液 (0.5μgC8H2Cl4O2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグ
ラフ質量分析計に注入してフサライドの量 (0.1ng) が十分に確認できるように検出器の感度
を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 29.1c)1)〜4)の操作を行う。
2) 受器の内容物を少量のアセトンで溶かした後,全量フラスコ20mlに移し入れ,更に受器をアセト
ン2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ20mlに移し入れ,アセトンを標線まで加え
る。
3) 空試験として試料と同量の水を分液漏斗にとり,1)及び2)の操作を行う。
d) 操作 操作は,次のとおり行う。
1) a)8)のフサライド標準液 (0.5μgC8H2Cl4O2/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フサライドのフラグメ
ントイオン(m/z 243,241,245など)を設定し(3),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,フサライドの保持時間に相当するピークの
位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(2)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からフサライドの量を求め,次の式によって試料中のフサライドの濃度 (μg/L) を算出する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)2)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
160
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検量線 a)7)のフサライド標準液 (5μgC8H2Cl4O2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってフサライドの量 (ng) と指示値(4)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(2) 7.の注(9)による。
(3) 7.の注(10)による。
(4) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
47.2 ガスクロマトグラフ法 試料について47.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C8H2Cl4O2 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ヘキサン 6.1a)4)による。
5) フサライド標準液 (5μgC8H2Cl4O2/ml) 47.1a)7)による。
6) フサライド標準液 (0.5μgC8H2Cl4O2/ml) 47.1a)8)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.3b)7.4)による。
5.4)
キャリヤーガス 7.2.3b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
カラム槽温度 7.2.1b)7.7)による。
5.7)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,フサライド標準液 (50ngC8H2Cl4O2/ml) [a)6)のフサライ
ド標準液 (0.5μgC8H2Cl4O2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフに注入
してフサライドの量 (0.1ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,47.1c)による。
161
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 操作 操作は,次のとおり行う。
1) a)6)のフサライド標準液 (0.5μgC8H2Cl4O2/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フサライドの保持時間
に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(2)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からフサライドの量を求め,47.1d)4)の式によって試料中のフサライドの濃度 (μg/L) を算出
する。
検量線 a)5)のフサライド標準液 (5μgC8H2Cl4O2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってフサライドの量 (ng) と指示値(4)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
48. ブタミホス (C13H21N2O4PS) ブタミホス[N-(1-メチルプロピル)ホスファミドチオ酸O-エチルO-
(5-メチル-2-ニトロフェニル)エステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマト
グラフ法を適用する。
48.1 ガスクロマトグラフ質量分析法 試料を6.の備考4.による抽出・分離・濃縮を行う。その一定量を,
ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ
法を用いて定量する。
定量範囲:C13H21N2O4PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) ブタミホス標準液 (1mgC13H21N2O4PS/ml) ブタミホスの標準品0.100gをとり,少量のアセトンに
溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ブタミホス標準液 (10μgC13H21N2O4PS/ml) ブタミホス標準液 (1mgC13H21N2O4PS/ml) 1mlを全量
フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ブタミホス標準液 (1μgC13H21N2O4PS/ml) ブタミホス標準液 (10μgC13H21N2O4PS/ml) 1mlを全量
フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
162
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ブタミホス標準液 (0.1μgC13H21N2O4PS/ml)
[a)9)のブタミホス標準液 (1μgC13H21N2O4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してブタミホスの量 (0.1ng) が十分に確認できるように検出器
の感度を調節する。
c) 準備操作 準備操作は,6.備考4.による(2)(3)。
注(2) 13.の注(2)による。
(3) ジクロロメタンに代えて,酢酸エチルを用いてもよい。この場合の操作などは備考2.による。
備考2. 抽出溶媒をジクロロメタンに代えて,酢酸エチルを用いた場合の準備操作は,次による。
1) 4.1c)で採取した試料(*1)の適量(200ml以下の一定量)を分液漏斗にとり,液量100mlについ
て塩化ナトリウム10gと酢酸エチル[6.備考5.のa)4)による。]25ml(*2)を加え,振とう器を用
いて約10分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。酢酸エチルを三角フラスコに移し入れ,分液漏斗を少量
の酢酸エチルで洗い,洗液は先の三角フラスコに合わせる。分液漏斗の水層に液量100mlに
ついて酢酸エチル25mlを加え,再び振とう器を用いて約10分間振り混ぜ,放置する。酢酸
エチル層を先の三角フラスコに合わせる。
3) 酢酸エチル溶液50mlについて硫酸ナトリウム10g(*3)を加え,軽く振り混ぜ,約30分間放置
した後(*4),ろ紙5種A(又は5種B)(*5)を用いてろ過し,ろ液をなす形フラスコ(*6)に受け
る。少量の酢酸エチルを用いて三角フラスコを2,3回洗浄し,さらにその洗液で先のろ紙上
の硫酸ナトリウムも洗浄し,洗液を酢酸エチル溶液に合わせる。
4) 濃縮器(*7)を用いて,約40℃(*7)の水浴上で酢酸エチル溶液を約1mlになるまで濃縮する。受
器を取り外し,窒素を緩やかに吹き付け,酢酸エチルを揮散させ,アセトン2mlを正しく加
える(*8)(*9)(*10)。
5) 空試験として試料と同量の水を分液漏斗にとり,1)〜4)の操作を行う。
注(*1) 6.の注(3)による。
(*2) 6.の注(4)による。ただし,酢酸エチルの量を増加する。
(*3) 6.の注(5)による。
(*4) 6.の注(6)による。
(*5) 6.の注(7)による。
(*6) 6.の注(8)による。
(*7) 6.の注(9)による。
(*8) 6.の注(21)による。
(*9) 6.備考5.の注(*10)による。
(*10) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
163
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) a)9)のブタミホス標準液 (1μgC13H21N2O4PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ブタミホスのフラグメ
ントイオン(m/z 286,200,232など)を設定し(5),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,ブタミホスの保持時間に相当するピークの
位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からブタミホスの量を求め,12.1d)4)の式によって試料中のブタミホスの濃度 (μg/L) を算出
する。
検量線 a)8)のブタミホス標準液 (10μgC13H21N2O4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってブタミホスの量 (ng) と指示値(6)との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
48.2 ガスクロマトグラフ法 試料を6.の備考4.による抽出・分離・濃縮を行う。濃縮液の一定量を,ガ
スクロマトグラフに注入し,熱イオン化検出器又は炎光光度検出器を用いた方法で定量する。
定量範囲:C13H21N2O4PS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) ブタミホス標準液 (10μgC13H21N2O4PS/ml) 48.1a)8)による。
8) ブタミホス標準液 (1μgC13H21N2O4PS/ml) 48.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
164
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。炎光光度検出器を用いる場合は,
7.2.2b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。炎光光度検出器を用
いる場合は,7.2.2b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考5. ガスクロマトグラフの感度として,ブタミホス標準液 (0.1μgC13H21N2O4PS/ml) [a)8)のブタ
ミホス標準液 (1μgC13H21N2O4PS/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに
注入してブタミホスの量 (0.1ng) が十分に確認できるように検出器の感度を調節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考8.による。
c) 準備操作 準備操作は,6.の備考4.による(2)(3)。
備考7. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)8)のブタミホス標準液 (1μgC13H21N2O4PS/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ブタミホスの保持時間
に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からブタミホスの量を求め,12.1d)4)の式によって試料中のブタミホスの濃度 (μg/L) を算出
する。
検量線 a)7)のブタミホス標準液 (10μgC13H21N2O4PS/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってブタミホスの量 (ng) と指示値(6)との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考8. 備考3.による。
49. ブプロフェジン (C16H23N3OS) ブプロフェジン[2-(1, 1-ジメチルエチルイミノ)-3-(1-メチルエ
チル)-5-フェニル-1, 3, 5-チアジアジン-4-オン]の定量には,ガスクロマトグラフ質量分析法及びガスクロ
マトグラフ法を適用する。
49.1 ガスクロマトグラフ質量分析法 試料中のブプロフェジンを塩化ナトリウムの共存下でジクロロメ
タン抽出を行い,引き続きカラムクロマトグラフ分離を行った後,濃縮する。その一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C16H23N3OS 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
165
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジエチルエーテル 6.3a)2)による。
5) ジクロロメタン 6.1a)3)による。
6) ヘキサン 6.1a)4)による。
7) ジエチルエーテル-ヘキサン混液 (3+7) 4)のジエチルエーテルと5)のヘキサンを用いて調製する。
8) アセトン-ヘキサン溶離液 (1+1) 39.1a)6)による。
9) ブプロフェジン標準液 (1mgC16H23N3OS/ml) ブプロフェジンの標準品0.100gをとり,少量のアセ
トンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
10) ブプロフェジン標準液 (10μgC16H23N3OS/ml) ブプロフェジン標準液 (1mgC16H23N3OS/ml) 1mlを
全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
11) ブプロフェジン標準液 (5μgC16H23N3OS/ml) ブプロフェジン標準液 (10μgC16H23N3OS/ml) 5mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) ブプロフェジン標準液 (0.5μgC16H23N3OS/ml) ブプロフェジン標準液 (5μgC16H23N3OS/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
13) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。ただし,充てん剤の流し込みにはジエチルエーテル-ヘキサン混
液 (3+7) を用いる。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ブプロフェジン標準液 (50ngC16H23N3OS/ml)
[a)12)のブプロフェジン標準液 (0.5μgC16H23N3OS/ml) 1mlを10mlに薄めたもの。]2μlをガス
クロマトグラフ質量分析計に注入してブプロフェジンの量 (0.1ng) が十分に確認できるよう
に検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 33.1c)1)〜4)の操作を行う。
2) 受器の内容物を少量のジエチルエーテル-ヘキサン混液 (3+7) で溶かした後,全量フラスコ20ml
に移し入れ,更にジエチルエーテル-ヘキサン混液 (3+7) 2〜3mlで2,3回よく洗浄し,洗液も全
量フラスコ20mlに移し入れ,ジエチルエーテル-ヘキサン混液 (3+7) を標線まで加える。
3) 全量フラスコ20mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。更に全量フラ
166
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
スコ10mlもジエチルエーテル-ヘキサン混液 (3+7) 2〜3mlで2,3回よく洗浄し,洗液もクロマト
グラフ管に流し込む。
4) クロマトグラフ管の上部から,ジエチルエーテル-ヘキサン混液 (3+7) 50mlを流下させる。この流
出液は捨てる。
5) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+1) 150mlを流下し,ブプロ
フェジンを溶出させ(2),溶出液をなす形フラスコ(3)に受ける。
6) 濃縮器(4)を用いて約40℃(4)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,アセトン2mlを正しく加える(5)(6)(7)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜5)の操作を行う。
注(2) 6.の注(25)による。
(3) 6.の注(8)による。
(4) 6.の注(9)による。
(5) 6.の注(21)による。
(6) 6.備考5.の注(*10)による。
(7) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)12)のブプロフェジン標準液 (0.5μgC16H23N3OS/ml) 2μlをマイクロシリンジ(8)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ブプロフェジン
のフラグメントイオン (m/z 105, 172, 106) を設定し(9),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,ブプロフェジンの保持時間に相当する
ピークの位置を確認しておく。
2) c)6)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からブプロフェジンの量を求め,33.1d)4)の式によって試料中のブプロフェジンの濃度
(μg/L) を算出する。
検量線 a)11)のブプロフェジン標準液 (5μgC16H23N3OS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってブプロフェジンの量 (ng) と指示値(10)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) 7.の注(10)による。
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
49.2 ガスクロマトグラフ法 試料について49.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C16H23N3OS 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
167
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1)3)による。
5) ジエチルエーテル-ヘキサン混液 (3+7) 49.1a)7)による。
6) アセトン-ヘキサン溶離液 (1+1) 39.1a)6)による。
7) ブプロフェジン標準液 (5μgC16H23N3OS/ml) 49.1a)11)による。
8) ブプロフェジン標準液 (0.5μgC16H23N3OS/ml) 49.1a)12)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 49.1b)4)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ブプロフェジン標準液 (50ngC16H23N3OS/ml) [a)8)のブ
プロフェジン標準液 (0.5μgC16H23N3OS/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグ
ラフに注入してブプロフェジンの量 (0.1ng) が十分に確認できるように検出器の感度を調節
する。
c) 準備操作 準備操作は,49.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のブプロフェジン標準液 (0.5μgC16H23N3OS/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ブプロフェジンの
保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からブプロフェジンの量を求め,33.1d)4)の式によって試料中のブプロフェジンの濃度
(μg/L) を算出する。
検量線 a)7)のブプロフェジン標準液 (5μgC16H23N3OS/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
168
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
イクロシリンジでとり,1)及び2)の操作を行ってブプロフェジンの量 (ng) と指示値(10)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
50. フルトラニル (C17H16F3NO2) フルトラニル〔2, 2, 2-トリフルオロメチル-N-[3-(1-メチルエトキシ)
フェニル]ベンザミド〕の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用す
る。
50.1 ガスクロマトグラフ質量分析法 試料中のフルトラニルについて6.の備考4.による抽出操作を行い,
引き続きカラムクロマトグラフ分離を行う。濃縮液の一定量を,ガスクロマトグラフ質量分析計に注入し,
対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C17H16F3NO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+4) 44.1a)5)による。
7) フルトラニル標準液 (1mgC17H16F3NO2/ml) フルトラニルの標準品0.100gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) フルトラニル標準液 (10μgC17H16F3NO2/ml) フルトラニル標準液 (1mgC17H16F3NO2/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) フルトラニル標準液 (1μgC17H16F3NO2/ml) フルトラニル標準液 (10μgC17H16F3NO2/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,フルトラニル標準液 (0.1μgC17H16F3NO2/ml)
[a)9)のフルトラニル標準液 (1μgC17H16F3NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してフルトラニルの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
169
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜4)の操作を行う(2)。
2) 次に,アセトン-ヘキサン溶離液 (1+4) (3)50mlを流下してフルトラニルを溶出させ(4),溶出液をな
す形フラスコ(5)に受ける。
3) これを,濃縮器(6)を用いて約40℃(6)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を揮散させた後,アセトン1mlを正しく加える(7)(8)(9)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(2) 13.の注(2)による。
(3) アセトン-ヘキサン溶離液 (1+4) に代えて,アセトン-ヘキサン混液 (1+9) 及びアセトン-ヘキ
サン溶離液 (1+4) とを用いてもよい。この場合の準備操作は備考2.による。
(4) 6.の注(25)による。
(5) 6.の注(8)による。
(6) 6.の注(9)による。
(7) 6.の注(21)による。
(8) 6.備考5.の注(*10)による。
(9) 6.の注(10)による。
備考2. アセトン-ヘキサン溶離液 (1+4) に代え,アセトン-ヘキサン混液 (1+9) [6.備考4.の5]に
よる。]及びアセトン-ヘキサン溶離液 (1+4) とを用いた場合の準備操作は,次による。
1) 33.1c)1)〜3)の操作を行う。
2) 濃縮器(*1)を用いて,約40℃(*1)の水浴上でジクロロメタン溶液を約1mlになるまで濃縮する。
受器を取り外し,窒素を緩やかに吹き付け,ジクロロメタンを揮散させる。内容物を少量の
アセトン-ヘキサン混液 (1+9) で溶かした後,全量フラスコ10mlに移し入れ,更に受器を
アセトン-ヘキサン混液 (1+9) 2〜3mlで2,3回洗浄し,この洗液も全量フラスコ10mlに移
す。続いてアセトン-ヘキサン混液 (1+9) を標線まで加える。
3) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。さらに
全量フラスコ10mlもアセトン-ヘキサン混液 (1+9) 2〜3mlで2,3回洗浄し,洗液もクロマ
トグラフ管(*2)に流し込む。
4) クロマトグラフ管の上部から,アセトン-ヘキサン混液 (1+9) 70mlを流下させる。この流出
液は捨てる。
5) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+4) 70mlを流下し,フ
ルトラニルを溶出させ(*3),溶出液をなす形フラスコ(*4)に受ける。
6) 濃縮器(*1)を用いて約40℃(*1)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を
緩やかに吹き付け,溶出液を揮散させた後,アセトン4mlを正しく加える(*5)(*6)(*7)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(*1) 6.の注(9)による。
(*2) 49.1b)5)による。
(*3) 6.の注(25)による。
(*4) 6.の注(8)による。
(*5) 6.の注(21)による。
(*6) 6.備考5.の注(*10)による。
170
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(*7) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のフルトラニル標準液 (1μgC17H16F3NO2/ml) 1μlをマイクロシリンジ(10)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フルトラニルのフラ
グメントイオン (m/z 173, 281, 145) を設定し(11),選択イオン検出法又はマスクロマトグラフ法によ
って測定してそのマスフラグメントグラムを記録し,フルトラニルの保持時間に相当するピークの
位置を確認しておく。
2) c)3)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)4)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からフルトラニルの量を求め,13.1d)4)の式によって試料中のフルトラニルの濃度 (μg/L) を
算出する。
検量線 a)8)のフルトラニル標準液 (10μgC17H16F3NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってフルトラニルの量 (ng) と指示値(12)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(10) 7.の注(9)による。
(11) 7.の注(10)による。
(12) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
50.2 ガスクロマトグラフ法 試料について50.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H16F3NO2 0.2〜4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+4) 44.1a)5)による。
7) フルトラニル標準液 (10μgC17H16F3NO2/ml) 50.1a)8)による。
8) フルトラニル標準液 (2μgC17H16F3NO2/ml) フルトラニル標準液 (10μgC17H16F3NO2/ml) 2mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
171
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考5. ガスクロマトグラフの感度として,フルトラニル標準液 (0.2μgC17H16F3NO2/ml) [a)8)のフル
トラニル標準液 (2μgC17H16F3NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフ
に注入してフルトラニルの量 (0.2ng) が十分に確認できるように検出器の感度を調節する。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,50.1c)の操作を行う。
備考7. 備考2.による。
8. 13.の備考6.による。
d) 操作 操作は,次のとおり行う。
1) a)8)のフルトラニル標準液 (2μgC17H16F3NO2/ml) 1μlをマイクロシリンジ(10)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,フルトラニルの保持
時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からフルトラニルの量を求め,13.1d)4)の式によって試料中のフルトラニルの濃度 (μg/L) を
算出する。
検量線 a)7)のフルトラニル標準液 (10μgC17H16F3NO2/ml) 0.2〜4mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってフルトラニルの量 (ng) と指示値(12)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考9. 備考3.による。
51. プレチラクロール (C17H26ClNO2) プレチラクロール[2-クロロ-N-(2, 6-ジエチルフェニル)-N-(2-
プロポキシエチル)アセトアミド]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ
法を適用する。
172
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
51.1 ガスクロマトグラフ質量分析法 試料中のプレチラクロールを塩化ナトリウムの共存下でヘキサン
抽出を行い,脱水・濃縮後,カラムクロマトグラフ分離を行い,濃縮する。その一定量を,ガスクロマト
グラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定
量する。
定量範囲:C17H26ClNO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) ヘキサン 6.1a)4)による。
4) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
5) プレチラクロール標準液 (1mgC17H26ClNO2/ml) プレチラクロールの標準品0.100gをとり,少量
のアセトンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
6) プレチラクロール標準液 (10μgC17H26ClNO2/ml) プレチラクロール標準液 (1mgC17H26ClNO2/ml)
1mlを全量フラスコ100mlにとり,ヘキサンを標線まで加える。保存する場合は,−20℃の暗所に
おく。
7) プレチラクロール標準液 (5μgC17H26ClNO2/ml) プレチラクロール標準液 (10μgC17H26ClNO2/ml)
5mlを全量フラスコ10mlにとり,ヘキサンを標線まで加える。
8) プレチラクロール標準液 (0.5μgC17H26ClNO2/ml) プレチラクロール標準液 (5μgC17H26ClNO2/ml)
1mlを全量フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,プレチラクロール標準液
(50ngC17H26ClNO2/ml) [a)8)のプレチラクロール標準液 (0.5μgC17H26ClNO2/ml) 1mlを10mlに
薄めたもの。]2μlガスクロマトグラフ質量分析計に注入してプレチラクロールの量 (0.1ng)
が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 29.1c)1)〜6)の操作を行う。
2) クロマトグラフ管の上部からヘキサン100mlを流下する。この流出液は捨てる。
3) アセトン-キサン溶離液 (1+19) 100mlを流下してプレチラクロールを溶出させ(2),溶出液をなす形
フラスコ(3)に受ける。
173
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 濃縮器(4)を用いて約40℃(4)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,ヘキサン4mlを正しく加える(5)(6)(7)。
5) 空試験として試料と同量の水を分液漏斗にとり,1)〜4)の操作を行う。
注(2) 6.の注(25)による。
(3) 6.の注(8)による。
(4) 6.の注(9)による。
(5) 6.の注(21)による。
(6) 6.備考5.の注(*10)による。
(7) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のプレチラクロール標準液 (0.5μgC17H26ClNO2/ml) 2μlをマイクロシリンジ(8)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プレチラクロ
ールのフラグメントイオン(m/z 162,238,176など)を設定し(9),選択イオン検出法又はマスクロ
マトグラフ法によって測定してそのマスフラグメントグラムを記録し,プレチラクロールの保持時
間に相当するピークの位置を確認しておく。
2) c)4)で得たヘキサン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)5)で得たヘキサン溶液について7.1d)3)の操作を行う。
4) 検量線からプレチラクロールの量を求め,次の式によって試料中のプレチラクロールの濃度 (μg/L)
を算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めた対象農薬の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)2)のヘキサンの量 (ml)
υ2: c)3)のヘキサンの量 (ml)
υ3: c)4)のヘキサンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)7)のプレチラクロール標準液 (5μgC17H26ClNO2/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってプレチラクロールの量 (ng) と指示値(10)との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) 7.の注(10)による。
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
51.2 ガスクロマトグラフ法 試料について51.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H26ClNO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
174
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) ヘキサン 6.1a)4)による。
4) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
5) プレチラクロール標準液 (5μC17H26ClNO2/ml) 51.1a)7)による。
6) プレチラクロール標準液 (0.5μgC17H26ClNO2/ml) 51.1a)8)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,プレチラクロール標準液 (50ngC17H26ClNO2/ml) [a)6)の
プレチラクロール標準液 (0.5μgC17H26ClNO2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロ
マトグラフに注入してプレチラクロールの量 (0.1ng) が十分に確認できるように検出器の感
度を調節する。
c) 準備操作 準備操作は,51.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のプレチラクロール標準液 (0.5μgC17H26ClNO2/ml) 2μlをマイクロシリンジ(8)でとり,スプリッ
トレス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プレチラクロ
ールの保持時間に相当するピークの位置を確認しておく。
2) c)で得たヘキサン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からプレチラクロールの量を求め,51.1d)4)の式によって試料中のプレチラクロールの濃度
(μg/L) を算出する。
検量線 a)5)のプレチラクロール標準液 (5μgC17H26ClNO2/ml) 0.1〜2mlを全量フラスコ10mlに段階
175
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
的にとり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってプレチラクロールの量 (ng) と指示値(10)との
関係線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
52. プロピザミド (C12H11Cl2NO) プロピザミド[3, 5-ジクロロ-N-(1, 1-ジメチル-2-プロピニル)ベンズ
アミド]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
52.1 ガスクロマトグラフ質量分析法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C12H11Cl2NO 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) プロピザミド標準液 (1mgC12H11Cl2NO/ml) プロピザミドの標準品0.100gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,ヘキサンを標線まで加える(1)。
8) プロピザミド標準液 (10μgC12H11Cl2NO/ml) プロピザミド標準液 (1mgC12H11Cl2NO/ml) 1mlを全
量フラスコ100mlにとり,ヘキサン(2)を標線まで加える。保存する場合は,−20℃の暗所におく。
9) プロピザミド標準液 (1μgC12H11Cl2NO/ml) プロピザミド標準液 (10μgC12H11Cl2NO/ml) 1mlを全
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
(2) 9.の注(2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ質量分析計 7.1b)7)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,プロピザミド標準液 (0.1μgC12H11Cl2NO/ml)
176
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[a)9)のプロピザミド標準液 (1μgC12H11Cl2NO/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してプロピザミドの量 (0.1ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)。
注(3) 9.の注(3)による。
備考2. 9.の備考2.による。
3. 12.の備考3.による。
d) 操作 操作は,次のとおり行う。
1) a)9)のプロピザミド標準液 (1μgC12H11Cl2NO/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プロピザミドのフラ
グメントイオン(m/z 173,175,255など)を設定し(5),選択イオン検出法又はマスクロマトグラフ
法によって測定してそのマスフラグメントグラムを記録し,プロピザミドの保持時間に相当するピ
ークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.1d)3)の操作を行う。
4) 検量線からプロピザミドの量を求め,12.1d)4)の式によって試料中のプロピザミドの濃度 (μg/L) を
算出する。
検量線 a)8)のプロピザミド標準液 (10μgC12H11Cl2NO/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってプロピザミドの量 (ng) と指示値(6)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(4) 7.の注(9)による。
(5) 7.の注(10)による。
(6) 7.の注(11)による。
備考4. 7.の備考2.による。
5. 7.の備考3.による。
52.2 ガスクロマトグラフ法 試料について9.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,電子捕獲検出器又は熱イオン化検出器を用いた方法で定量する。
定量範囲:C12H11Cl2NO 0.01〜0.2ng(7) 繰返し分析精度:変動係数で10〜20%(装置,測定条件によ
って異なる。)
注(7) 13.の注(10)による。
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン溶離液 (7+13) 6.3a)4)による。
7) プロピザミド標準液 (10μgC12H11Cl2NO/ml) 52.1a)8)による。
8) プロピザミド標準液 (1μgC12H11C12NO/ml) プロピザミド標準液 (10μgC12H11Cl2NO/ml) 1mlを全
177
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
9) プロピザミド標準液 (0.1μgC12H11Cl2NO/ml) プロピザミド標準液 (1μgC12H11Cl2NO/ml) 1mlを全
量フラスコ10mlにとり,ヘキサン(2)を標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) 固相カラム 6.2b)2)による。
6) クロマトグラフ管 6.3b)2)による。
7) マイクロシリンジ 7.1b)6)による。
8) ガスクロマトグラフ 次に掲げる条件を満たすもの。
8.1)
キャピラリーカラム用管 7.1b)7.1)による。
8.2)
キャピラリーカラム 7.1b)7.2)による。
8.3)
検出器 熱イオン化検出器を用いる場合は,7.2.1b)7.3)による。電子捕獲検出器を用いる場合は,
7.2.3b)7.3)による。
8.4)
キャリヤーガス 7.2.1b)7.4)による。ただし,電子捕獲検出器を用いる場合は,カラム出口には付加
ガス(8)としてJIS K 1107に規定する高純度窒素1級を接続し,流量は30〜60ml/minに調節して用
いる。
8.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
8.6)
燃料ガス及び助燃ガス 熱イオン化検出器を用いる場合は,7.2.1b)7.6)による。電子捕獲検出器では
用いない。
8.7)
カラム槽温度 7.2.1b)7.7)による。
8.8)
検出器槽温度 7.2.1b)7.8)による。
9) 振とう器
10) 濃縮器 6.1b)5)による。
注(8) 7.の注(20)による。
備考6. ガスクロマトグラフの感度として,プロピザミド標準液 (10ngC12H11Cl2NO/ml) [a)9)のプロ
ピザミド標準液 (0.1μgC12H11Cl2NO/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フに注入してプロピザミドの量 (0.01ng) が十分に確認できるように検出器の感度を調節す
る。
7. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.又は7.の備考11.による。
c) 準備操作 準備操作は,9.1c)の操作を行う(3)。
備考8. 備考2.による。
9. 備考3.による。
10. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作は6.の備考5.に
よる。ただし,最終溶媒量はヘキサン20mlとする。
d) 操作 操作は,次のとおり行う。
178
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) a)9)のプロピザミド標準液 (0.1μgC12H11Cl2NO/ml) 1μlをマイクロシリンジ(4)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プロピザミドの保
持時間に相当するピークの位置を確認しておく。
2) c)で得た濃縮液1μlをマイクロシリンジ(4)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験の濃縮液について7.2.1d)3)の操作を行う。
4) 検量線からプロピザミドの量を求め,12.1d)4)の式によって試料中のプロピザミドの濃度 (μg/L) を
算出する。
検量線 a)8)のプロピザミド標準液 (1μgC12H11Cl2NO/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,ヘキサン(2)を標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってプロピザミドの量 (ng) と指示値(6)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考11. 備考4.による。
53. プロベナゾール (C10H9NO3S) プロベナゾール[3-(2-プロペニルオキシ)-1, 2-ベンズイソチアゾー
ル-1, 1-ジオキシド]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
53.1 ガスクロマトグラフ質量分析法 試料について18.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C10H9NO3S 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) プロベナゾール標準液 (2mgC10H9NO3S/ml) プロベナゾールの標準品0.200gをとり,少量のアセ
トンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) プロベナゾール標準液 (20μgC10H9NO3S/ml) プロベナゾール標準液 (2mgC10H9NO3S/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) プロベナゾール標準液 (2μgC10H9NO3S/ml) プロベナゾール標準液 (20μgC10H9NO3S/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
179
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,プロベナゾール標準液 (0.2μgC10H9NO3S/ml)
[a)7)のプロベナゾール標準液 (2μgC10H9NO3S/ml) 1mlを10mlに薄めたもの。]2μlをガスク
ロマトグラフ質量分析計に注入してプロベナゾールの量 (0.4ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,18.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のプロベナゾール標準液 (2μgC10H9NO3S/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プロベナゾールのフ
ラグメントイオン (m/z 130, 103, 159) を設定し(3),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,プロベナゾールの保持時間に相当するピー
クの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(2)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からプロベナゾールの量を求め,18.1d)4)の式によって試料中のプロベナゾールの濃度
(μg/L) を算出する。
検量線 a)6)のプロベナゾール標準液 (20μgC10H9NO3S/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってプロベナゾールの量 (ng) と指示値(4)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(2) 7.の注(9)による。
(3) 7.の注(10)による。
(4) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
53.2 ガスクロマトグラフ法 試料について18.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C10H9NO3S 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) プロベナゾール標準液 (20μgC10H9NO3S/ml) 53.1a)6)による。
6) プロベナゾール標準液 (2μgC10H9NO3S/ml) 53.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
180
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,プロベナゾール標準液 (0.2μgC10H9NO3S/ml) [a)6)のプ
ロベナゾール標準液 (2μgC10H9NO3S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フに注入してプロベナゾールの量 (0.4ng) が十分に確認できるように検出器の感度を調節す
る。
c) 準備操作 準備操作は,18.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のプロベナゾール標準液 (2μgC10H9NO3S/ml) 2μlをマイクロシリンジ(2)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,プロベナゾールの保
持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(2)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からプロベナゾールの量を求め,18.1d)4)の式によって試料中のプロベナゾールの濃度
(μg/L) を算出する。
検量線 a)5)のプロベナゾール標準液 (20μgC10H9NO3S/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってプロベナゾールの量 (ng) と指示値(4)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
54. ブロモブチド[ブロモチド] (C15H22BrNO) ブロモブチド[ブロモチド][2-ブロモ-3, 3-ジメチル-N-
(1-メチル-1-フェニルエチル)ブタンアミド]の定量には,ガスクロマトグラフ質量分析法及びガスクロ
マトグラフ法を適用する。
ブロモブチドの定量は,ブロモブチドとブロモブチド脱臭素体[(RS) 3, 3-ジメチル-N-(1-メチル-1-フェ
ニルエチル)ブタンアミド]に係数1.338を乗じてブロモブチドに換算したものとを合計して算出する。
181
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
54.1 ガスクロマトグラフ質量分析法 試料中のブロモブチドを塩化ナトリウムの共存下でジクロロメタ
ン抽出を行い,脱水・濃縮後,引き続きカラムクロマトグラフ分離を行った後,再び濃縮を行う。その一
定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマ
トグラフ法を用いて定量する。
定量範囲:C15H22BrNO 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン混液 (1+30) 3)のアセトンと5)のヘキサンで調製する。
7) アセトン-ヘキサン溶離液 (1+5) 33.1a)7)による。
8) ブロモブチド標準液 (2.5mgC15H22BrNO/ml) ブロモブチドの標準品0.250gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
9) ブロモブチド標準液 (25μgC15H22BrNO/ml) ブロモブチド標準液 (2.5mgC15H22BrNO/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
10) ブロモブチド標準液 (2.5μgC15H22BrNO/ml) ブロモブチド標準液 (25μgC15H22BrNO/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
11) ブロモブチド標準液 (0.25μgC15H22BrNO/ml) ブロモブチド標準液 (2.5μgC15H22BrNO/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) ブロモブチド脱臭素体標準液 (2.5μgC15H23NO/ml) ブロモブチド脱臭素体の標準品0.250gをとり,
少量のアセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
13) ブロモブチド脱臭素体標準液 (25μgC15H23NO/ml) ブロモブチド脱臭素体標準液
(2.5mgC15H23NO/ml) 1mlを全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合
は,−20℃の暗所におく。
14) ブロモブチド脱臭素体標準液 (2.5μgC15H23NO/ml) ブロモブチド脱臭素体標準液
(25μgC15H23NO/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製す
る。
15) ブロモブチド脱臭素体標準液 (0.25μgC15H23NO/ml) ブロモブチド脱臭素体標準液
(2.5μgC15H23NO/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製す
る。
16) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
182
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ブロモブチド標準液 (25ngC15H22BrNO/ml) 及
びブロモブチド脱臭素体標準液 (25ngC15H23NO/ml) [a)11)のブロモブチド標準液
(0.25μgC15H22BrNO/ml) 及びa)15)のブロモブチド脱臭素体標準液 (0.25μgC15H23NO/ml),それ
ぞれ1mlを10mlに薄めたもの。],それぞれ2μlをガスクロマトグラフ質量分析計に注入して
ブロモブチド及びブロモブチド脱臭素体のそれぞれの量 (0.05ng) が十分に確認できるよう
に検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 33.1c)1)〜4)の操作を行う。
2) 受器の内容物を少量のアセトン-ヘキサン混液 (1+30) で溶かした後,全量フラスコ10mlに移し入
れ,更に受器をアセトン-ヘキサン混液 (1+30) 2〜3mlで2,3回よく洗浄し,洗液も全量フラスコ
10mlに移し入れ,アセトン-ヘキサン混液 (1+30) を標線まで加える(2)。
3) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。更に全量フラ
スコ10mlもアセトン-ヘキサン混液 (1+30) 2〜3mlで2,3回よく洗浄し,洗液もクロマトグラフ
管に流し込む。
4) クロマトグラフ管の上部から,アセトン-ヘキサン混液 (1+30) 110mlを流下させる。はじめの流出
液50mlは捨てる。
5) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+5) 40mlを流下し,対象農薬
を溶出させ(3),溶出液をなす形フラスコ(4)に受ける。
6) 濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,アセトン2mlを正しく加える(2)(6)(7)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(2) 6.の注(10)による。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
d) 操作 操作は,次のとおり行う。
1) a)11)のブロモブチド標準液 (0.25μgC15H22BrNO/ml) 及びa)15)のブロモブチド脱臭素体標準液
(0.25μgC15H23NO/ml),それぞれ2μlをマイクロシリンジ(8)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,ブロモブチド及びブロモブチド脱臭
素体のフラグメントイオンを設定し(9),選択イオン検出法又はマスクロマトグラフ法によって測定
してそのマスフラグメントグラムを記録し,ブロモブチド及びブロモブチド脱臭素体の保持時間に
相当するピークの位置を確認しておく。
2) c)6)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について7.1d)3)の操作を行う。
183
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 検量線からブロモブチドの量を求め,次の式によって試料中のブロモブチドの濃度 (μg/L) を算出
する。
V
C
b
a
N
000
1
10
10
)]
338
.1
(
[
3
3
2
1
3
×
×
×
×
×
×
+
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めたブロモブチドの量 (ng)
b: 検量線から求めたブロモブチド脱臭素体の量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)2)のアセトン-ヘキサン混液 (1+30) の量 (ml)
υ2: c)3)のアセトン-ヘキサン混液 (1+30) の量 (ml)
υ3: c)6)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)10)のブロモブチド標準液 (2.5μgC15H22BrNO/ml) 及びa)14)のブロモブチド脱臭素体標準
液 (2.5μgC15H23NO/ml) それぞれ0.1〜2mlを全量フラスコ10mlに段階的にとり,アセトンを標線
まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイクロシリンジでとり,
1)及び2)の操作を行ってブロモブチドの量 (ng) と指示値(10)との関係線を作成する。検量線の作
成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) それぞれのフラグメントイオンは,次を参考(イオン強度の大きいものから順に表示している。)
にするとよい。
ブロモブチド
119,120,118,232
ブロモブチド脱臭素体
119,233,177
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
54.2 ガスクロマトグラフ法 試料について54.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C15H22BrNO 0.1〜2ng 繰返し分析精度:変動係数で20〜30%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) アセトン-ヘキサン混液 (1+30) 54.1a)6)による。
6) アセトン-ヘキサン溶離液 (1+5) 33.1a)7)による。
7) ブロモブチド標準液 (2.5μgC15H22BrNO/ml) 54.1a)10)による。
8) ブロモブチド標準液 (0.25μgC15H22BrNO/ml 54.1a)11)による。
9) ブロモブチド脱臭素体標準液 (2.5μgC15H23NO/ml) 54.1a)14)による。
10) ブロモブチド脱臭素体標準液 (0.25μgC15H23NO/ml) 54.1a)15)による。
11) 窒素 6.2a)2)による。
184
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) クロマトグラフ管 6.3b)2)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ブロモブチド標準液 (25ngC15H22BrNO/ml) 及びブロモブ
チド脱臭素体標準液 (25ngC15H23NO/ml) [a)8)のブロモブチド標準液 (0.25μgC15H22BrNO/ml)
及びa)10)のブロモブチド脱臭素体標準液 (0.25μgC15H23NO/ml),それぞれ1mlを10mlに薄め
たもの。],それぞれ2μlをガスクロマトグラフに注入してブロモブチド及びブロモブチド脱
臭素体のそれぞれの量 (0.05ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,54.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のブロモブチド標準液 (0.25μgC15H22BrNO/ml) 及びa)10)のブロモブチド脱臭素体標準液
(0.25μgC15H23NO/ml),それぞれ2μlをマイクロシリンジ(8)でとり,スプリットレス注入法又はコー
ルドオンカラム注入法によってガスクロマトグラフに注入し,ブロモブチド及びブロモブチド脱臭
素体の保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からブロモブチドの量を求め,54.1d)4)の式によって試料中のブロモブチドの濃度 (μg/L) を
算出する。
検量線 a)7)のブロモブチド標準液 (2.5μgC15H22BrNO/ml) 及びa)9)のブロモブチド脱臭素体標準液
(2.5μgC15H23NO/ml),それぞれ0.1〜2mlを全量フラスコ10mlに段階的にとり,アセトンを標線ま
で加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイクロシリンジでとり,1)
及び2)の操作を行ってブロモブチド及びブロモブチド脱臭素体の量 (ng) と指示値(10)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
185
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
55. ペンシクロン (C19H21ClN2O) ペンシクロン[1-(4-クロロベンジル)-1-シクロペンチル-3-フェニル
尿素]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
55.1 ガスクロマトグラフ質量分析法 試料中のペンシクロンについて6.の備考4.による抽出操作を行い,
引き続きカラムクロマトグラフ分離を行う。濃縮液の一定量を,ガスクロマトグラフ質量分析計に注入し,
対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C19H21ClN2O 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ペンシクロン標準液 (1mgC19H21ClN2O/ml) ペンシクロンの標準品0.100gをとり,少量のアセト
ンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ペンシクロン標準液 (10μgC19H21ClN2O/ml) ペンシクロン標準液 (1mgC19H21ClN2O/ml) 1mlを全
量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ペンシクロン標準液 (1μgC19H21ClN2O/ml) ペンシクロン標準液 (10μgC19H21ClN2O/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) クロマトグラフ管 6.3b)2)による。
6) マイクロシリンジ 7.1b)6)による。
7) ガスクロマトグラフ質量分析計 7.1b)7)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ペンシクロン標準液 (0.1μgC19H21ClN2O/ml)
[a)9)のペンシクロン標準液 (1μgC19H21ClN2O/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してペンシクロンの量 (0.1ng) が十分に確認できるように検
出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜2)の操作を行う(2)(3)。
2) 受器の内容物を少量のアセトン-ヘキサン溶離液 (3+17) で溶かした後,全量フラスコ10mlに移し
入れ,更にアセトン-ヘキサン溶離液 (3+17) 2〜3mlで2,3回よく洗浄し,洗液を全量フラスコ10ml
186
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に移し入れ,アセトン-ヘキサン溶離液 (3+17) を標線まで加える。
3) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。更に全量フラ
スコ10mlもアセトン-ヘキサン溶離液 (3+17) 2〜3mlで2,3回洗浄し,洗液もクロマトグラフ管
に流し込む。
4) 次に,アセトン-ヘキサン溶離液 (3+17) 100mlを流下してペンシクロンを溶出させ(4),溶出液をな
す形フラスコ(5)に受ける。
5) これを,濃縮器(6)を用いて約40℃(6)の水浴上で約10mlになるまで濃縮し,この溶液を共栓付き試
験管20mlに少量のヘキサンで洗い移す。
6) 共栓付き試験管20mlに濃縮器を取り付け,約40℃の水浴上で1〜2mlになるまで濃縮し,受器を取
り外し,窒素を緩やかに吹き付け,溶液を完全に揮散させた後,アセトン1mlを正しく加える(7)(8)(9)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(2) 13.の注(2)による。
(3) 抽出溶媒にヘキサンを用いてもよい。この場合の準備操作は備考1.による。
(4) 6.の注(25)による。
(5) 6.の注(8)による。
(6) 6.の注(9)による。
(7) 6.の注(21)による。
(8) 6.備考5.の注(*10)による。
(9) 6.の注(10)による。
備考1. 抽出溶媒に,ヘキサンを用いた場合の準備操作は,次による
1) 29.1c)1)〜3)の操作を行う。
2) 濃縮器(*1)を用いて,約40℃(*1)の水浴上でヘキサン溶液を約1mlになるまで濃縮する。受器
を取り外し,窒素を緩やかに吹き付け,ヘキサンを完全に揮散させる(*2)(*3)。
3) メチル化の操作を55.2c)1)〜7)によって行う。
4) このろ液と洗液を,2)の操作を行ってヘキサンを揮散させた後,アセトン1mlを正しく加え
る(*2)(*3)(*4)。
5) 空試験として水200mlを分液漏斗にとり,1)〜4)の操作を行う。
注(*1) 6.の注(9)による。
(*2) 6.の注(21)による。
(*3) 6.備考5.の注(*10)による。
(*4) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のペンシクロン標準液 (1μgC19H21ClN2O/ml) 1μlをマイクロシリンジ(10)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ペンシクロンのフラ
グメントイオン(m/z 125,180,127など)を設定し(11),選択イオン検出法又はマスクロマトグラ
フ法によって測定してそのマスフラグメントグラムを記録し,ペンシクロンの保持時間に相当する
ピークの位置を確認しておく。
2) c)6)で得たアセトン溶液1μlをマイクロシリンジ(10)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について,7.1d)3)の操作を行う。
4) 検量線からペンシクロンの量を求め,12.1d)4)の式によって試料中のペンシクロンの濃度 (μg/L) を
187
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
算出する。
検量線 a)8)のペンシクロン標準液 (10μgC19H21ClN2O/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってペンシクロンの量 (ng) と指示値(12)との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(10) 7.の注(9)による。
(11) 7.の注(10)による。
(12) 7.の注(11)による。
備考3. 7.の備考2.による。
4. 7.の備考3.による。
55.2 ガスクロマトグラフ法 試料中のペンシクロンについて6.の備考4.による抽出操作を行い,引き続
きカラムクロマトグラフ分離を行った後,濃縮し,アセトン-ヘキサン溶離液で一定量にし,カラムクロマ
トグラフ分離で精製し,濃縮する。残留物をベンゼンで溶かし,ジメチルスルホキシド,ヨードメタン及
び水素化ナトリウムを加えてメチル化し,この溶液をヘキサンで抽出し,抽出液の一定量をガスクロマト
グラフに注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C19H21ClN2O 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 水素化ナトリウム あらかじめ8)のヘキサンを用いて洗浄し,ヘキサンに浸して保存しておく。使
用時はろ紙5種A(又は5種B)(13)を用いてろ過し,直ちに使用する。
3) ヨードメタン JIS K 8919に規定するもの(14)。
4) 硫酸ナトリウム 6.1a)2)による。
5) アセトン 4.1a)3)による。
6) ジクロロメタン 6.1a)3)による。
7) ジメチルスルホキシド JIS K 9702に規定するもの(14)。
8) ヘキサン 6.1a)4)による。
9) ベンゼン JIS K 8858に規定するもの(14)。
10) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
11) ペンシクロン標準液 (10μgC19H21ClN2O/ml) (15) 55.1a)7)のペンシクロン標準液
(1mgC19H21ClN2O/ml) 1mlを全量フラスコ20mlにとり,アセトンを標線まで加える。この溶液10ml
を共栓付き試験管20ml[c)3)で用いたものと同じものを用いる。]に少量のヘキサンを用いて洗い移
し,c)3)の操作を行い,溶媒を揮散させ,続いてc)4)〜7)の操作を行う。再びc)3)の操作を行い,溶
媒を揮散させ,メチル化を行う。残留物を少量のヘキサンで2,3回洗浄し,洗液を全量フラスコ
50mlに移し入れ,アセトンを標線まで加える。
12) ペンシクロン標準液 (4μgC19H21ClN2O/ml) (15) ペンシクロン標準液 (10μgC19H21ClN2O/ml) 4mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。
13) 窒素 6.2a)2)による。
注(13) 6.の注(7)による。
(14) 4.の注(1)による。
188
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(15) ここでいうペンシクロン標準液は,メチル化したものである。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) 共栓付き試験管 6.2b)1)による。
5) クロマトグラフ管 6.3b)2)による。
6) マイクロシリンジ 7.1b)6)による。
7) ガスクロマトグラフ 次に掲げる条件を満たすもの。
7.1)
キャピラリーカラム用管 7.1b)7.1)による。
7.2)
キャピラリーカラム 7.1b)7.2)による。
7.3)
検出器 7.2.1b)7.3)による。
7.4)
キャリヤーガス 7.2.1b)7.4)による。
7.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
7.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
7.7)
カラム槽温度 7.2.1b)7.7)による。
7.8)
検出器槽温度 7.2.1b)7.8)による。
8) 振とう器
9) 濃縮器 6.1b)5)による。
備考5. ガスクロマトグラフの感度として,ペンシクロン標準液 (0.4μgC19H21ClN2O/ml) (15)[a)12)の
ペンシクロン標準液 (4μgC19H21ClN2O/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフに注入してペンシクロンの量 (0.4ng) が十分に確認できるように検出器の感度を調節す
る。
6. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜3)の操作を行う(2)(3)。
2) 55.1の2)〜5)の操作を行う。
3) 共栓付き試験管20mlに濃縮器(6)を取り付け,約40℃(6)の水浴上で1〜2mlになるまで濃縮し,受器
を取り外し,窒素を緩やかに吹き付け,ヘキサンを揮散させる(7)(8)(9)。
4) 濃縮した残留物にベンゼン0.5mlを加えて溶かし,ジメチルスルホキシド0.5ml,ヨードメタン0.5ml
及び水素化ナトリウム0.2gを加え,30±1℃に調節してある恒温槽に移し,栓をして時々振り混ぜ
ながら約30分間放置する。
5) この溶液にヘキサン5mlを加え,約1分間振り混ぜた後,水10mlを徐々に滴加する。
6) 少量のヘキサンを用いて,分液漏斗50mlに移し入れ,振とう器を用いて約5分間振り混ぜる。
7) 放置後,ヘキサン層を三角フラスコに移し入れ,硫酸ナトリウム5〜10gを加え,軽く振り混ぜ,約
10分間放置した後,ろ紙5種A(又は5種B)(13)を用いてろ過し,ろ液をなす形フラスコに受ける。
ヘキサン5〜6mlを用いて三角フラスコを数回洗浄し,さらにその洗液で先のろ紙上の硫酸ナトリ
ウムも洗浄し,洗液をヘキサン溶液に合わせる。
8) このヘキサン溶液について3)の操作を行って,ヘキサンを揮散させた後,アセトン1mlを正しく加
189
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
える(7)(8)(9)。
9) 空試験として試料と同量の水を分液漏斗にとり,1)〜8)の操作を行う。
備考7. 備考2.による。
d) 操作 操作は,次のとおり行う。
1) a)12)のペンシクロン標準液 (4μgC19H21ClN2O/ml) 1μlをマイクロシリンジ(10)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ペンシクロンの保
持時間に相当するピークの位置を確認しておく。
2) c)8)で得たヘキサン溶液1μlをマイクロシリンジ(10)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)9)で得たヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からペンシクロンの量を求め,12.1d)4)の式によって試料中のペンシクロンの濃度 (μg/L) を
算出する。
検量線 a)11)のペンシクロン標準液 (10μgC19H21ClN2O/ml) 0.4〜8mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってペンシクロンの量 (ng) と指示値(12)との関係線
を作成する。検量線の作成は,試料測定時に行う。
備考8. 備考3.による。
56. ベンスリド [SAP] (C14H24NO4PS3) ベンスリド [SAP] 〔ホスホロジチオ酸O, O-ビス(1-メチルエチ
ル)S-{2-[(フェニルスルホニル)アミノ]-エチル}エステル〕の定量には,ガスクロマトグラフ質量分
析法及びガスクロマトグラフ法を適用する。
56.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C14H24NO4PS3 0.5〜10ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ベンスリド標準液 (1mgC14H24NO4PS3/ml) ベンスリドの標準品0.100gをとり,少量のアセトンに
溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ベンスリド標準液 (50μgC14H24NO4PS3/ml) ベンスリド標準液 (1mgC14H24NO4PS3/ml) 5mlを全量
フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ベンスリド標準液 (5μgC14H24NO4PS3/ml) ベンスリド標準液 (50μgC14H24NO4PS3/ml) 1mlを全量
フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
190
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ベンスリド標準液 (0.5μgC14H24NO4PS3/ml)
[a)9)のベンスリド標準液 (5μgC14H24NO4PS3/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してベンスリドの量 (0.5ng) が十分に確認できるように検出器
の感度を調節する。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のベンスリド標準液 (5μgC14H24NO4PS3/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ベンスリドのフラグメ
ントイオン(m/z 77,131,141など)を設定し(4),選択イオン検出法又はマスクロマトグラフ法に
よって測定してそのマスフラグメントグラムを記録し,ベンスリドの保持時間に相当するピークの
位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からベンスリドの量を求め,13.1d)4)の式によって試料中のベンスリドの濃度 (μg/L) を算出
する。
検量線 a)8)のベンスリド標準液 (50μgC14H24NO4PS3/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってベンスリドの量 (ng) と指示値(5)との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
56.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C14H24NO4PS3 0.5〜10ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
191
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) ベンスリド標準液 (50μgC14H24NO4PS3/ml) 56.1a)8)による。
8) ベンスリド標準液 (5μgC14H24NO4PS3/ml) 56.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ベンスリド標準液 (0.5μgC14H24NO4PS3/ml) [a)8)のベン
スリド標準液 (5μgC14H24NO4PS3/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに
注入してベンスリドの量 (0.5ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
備考6. 14.の備考10.による。ただし,最終溶媒量はアセトン4mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のベンスリド標準液 (5μgC14H24NO4PS3/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ベンスリドの保持時間
に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からベンスリドの量を求め,13.1d)4)の式によって試料中のベンスリドの濃度 (μg/L) を算出
する。
192
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検量線 a)7)のベンスリド標準液 (50μgC14H24NO4PS3/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってベンスリドの量 (ng) と指示値(5)との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
57. ペンジメタリン (C13H19N3O4) ペンジメタリン[N-(1-エチルプロピル)-3, 4-ジメチル-2, 6-ジニト
ロベンゼンアミン]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
57.1 ガスクロマトグラフ質量分析法 試料中のペンジメタリンについて6.の備考4.による抽出操作を行
い,引き続きカラムクロマトグラフ分離を行う。濃縮液の一定量を,ガスクロマトグラフ質量分析計に注
入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C13H19N3O4 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4. の5)による。
7) ペンジメタリン標準液 (1mgC13H19N3O4/ml) ペンジメタリンの標準品0.100gをとり,少量のアセ
トンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ペンジメタリン標準液 (10μgC13H19N3O4/ml) ペンジメタリン標準液 (1mgC13H19N3O4/ml) 1mlを
全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ペンジメタリン標準液 (1μgC13H19N3O4/ml) ペンジメタリン標準液 (10μgC13H19N3O4/ml) 1mlを全
量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ペンジメタリン標準液 (0.1μgC13H19N3O4/ml)
[a)9)のペンジメタリン標準液 (1μgC13H19N3O4/ml) 1mlを10mlに薄めたもの。]1μlをガスク
ロマトグラフ質量分析計に注入してペンジメタリンの量 (0.1ng) が十分に確認できるように
193
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 6.備考4.の1)〜4)の操作を行う(2)。
2) 次に,アセトン-ヘキサン溶離液 (1+19) 60mlを流下してペンジメタリンを溶出させ(3),溶出液を
なす形フラスコ(4)に受ける。
3) これを,濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を揮散させた後,アセトン1mlを正しく加える(6)(7)(8)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(2) 13.の注(2)による。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
(8) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のペンジメタリン標準液 (1μgC13H19N3O4/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ペンジメタリンのフ
ラグメントイオン(m/z 252,281,162など)を設定し(10),選択イオン検出法又はマスクロマトグ
ラフ法によって測定してそのマスフラグメントグラムを記録し,ペンジメタリンの保持時間に相当
するピークの位置を確認しておく。
2) c)3)で得たアセトン溶液1μlをマイクロシリンジ(9)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)4)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からペンジメタリンの量を求め,13.1d)4)の式によって試料中のペンジメタリンの濃度
(μg/L) を算出する。
検量線 a)8)のペンジメタリン標準液 (10μgC13H19N3O4/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってペンジメタリンの量 (ng) と指示値(11)との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(9) 7.の注(9)による。
(10) 7.の注(10)による。
(11) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
57.2 ガスクロマトグラフ法 試料につき57.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフに
注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C13H19N3O4 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
194
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) ペンジメタリン標準液 (10μgC13H19N3O4/ml) 57.1a)8)による。
8) ペンジメタリン標準液 (1μgC13H19N3O4/ml) 57.1a)9)による。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ペンジメタリン標準液 (0.1μgC13H19N3O4/ml) [a)8)のペ
ンジメタリン標準液 (1μgC13H19N3O4/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フに注入してペンジメタリンの量 (0.1ng) が十分に確認できるように検出器の感度を調節す
る。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,57.1c)の操作を行う。
備考6. ジクロロメタンに代えて混合有機溶媒を用いてもよい。この場合の抽出操作などは6.備考5.
のc)1)〜5)の操作に引き続き,ヘキサン溶液全量を用いて,57.1c)の2)及び3)の操作[この場
合の溶離液はジエチルエーテル-ヘキサン溶離液 (1+19) (28.1の備考6.による。)を用いる。]
を行う。ただし,これらの操作を行った場合の最終溶媒量はアセトン4mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のペンジメタリン標準液 (1μgC13H19N3O4/ml) 1μlをマイクロシリンジ(9)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ペンジメタリンの保
持時間に相当するピークの位置を確認しておく。
195
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) c)2)で得たアセトン溶液1μlをマイクロシリンジ(9)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からペンジメタリンの量を求め,13.1d)4)の式によって試料中のペンジメタリンの濃度
(μg/L) を算出する。
検量線 a)7)のペンジメタリン標準液 (10μgC13H19N3O4/ml) 0.1〜2mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
ィクロシリンジでとり,1)及び2)の操作を行ってペンジメタリンの量 (ng) と指示値(11)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
58. ベンフルラリン[ベスロジン] (C13H16F3N3O4) ベンフルラリン[ベスロジン][N-ブチル-N-エチル
-2, 6-ジニトロ-4-(トリフルオロメチル)ベンゼンアミン]の定量には,ガスクロマトグラフ質量分析法及
びガスクロマトグラフ法を適用する。
58.1 ガスクロマトグラフ質量分析法 試料を6.の備考4.による抽出・分離・濃縮を行う。濃縮液の一定
量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマト
グラフ法を用いて定量する。
定量範囲:C13H16F3N3O4 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) ベンフルラリン標準液 (1mgC13H16F3N3O4/ml) ベンフルラリンの標準品0.100gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) ベンフルラリン標準液 (10μgC13H16F3N3O4/ml) ベンフルラリン標準液 (1mgC13H16F3N3O4/ml) 1ml
を全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) ベンフルラリン標準液 (1μgC13H16F3N3O4/ml) ベンフルラリン標準液 (10μgC13H16F3N3O4/ml) 1ml
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
196
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,ベンフルラリン標準液
(0.1μgC13H16F3N3O4/ml) [a)9)のベンフルラリン標準液 (1μgC13H16F3N3O4/ml) 1mlを10mlに薄
めたもの。]1μlをガスクロマトグラフ質量分析計に注入してベンフルラリンの量 (0.1ng) が
十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,6.備考4.による(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のベンフルラリン標準液 (1μgC13H16F3N3O4/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ベンフルラリンの
フラグメントイオン(m/z 292,264,276など)を設定し(4),選択イオン検出法又はマスクロマトグ
ラフ法によって測定してそのマスフラグメントグラムを記録し,ベンフルラリンの保持時間に相当
するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からベンフルラリンの量を求め,13.1d)4)の式によって試料中のベンフルラリンの濃度
(μg/L) を算出する。
検量線 a)8)のベンフルラリン標準液 (10μgC13H16F3N3O4/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってベンフルラリンの量 (ng) と指示値(5)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
58.2 ガスクロマトグラフ法 試料を6.の備考4.による抽出・分離・濃縮を行う。濃縮液の一定量を,ガ
スクロマトグラフに注入し,電子捕獲検出器を用いた方法で定量する。
定量範囲:C13H16F3N3O4 0.02〜0.4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によっ
て異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
7) ベンフルラリン標準液 (1μgC13H16F3N3O4/ml) 58.1a)9)による。
8) ベンフルラリン標準液 (0.2μgC13H16F3N3O4/ml) ベンフルラリン標準液 (1μgC13H16F3N3O4/ml) 2ml
197
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.3b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
カラム槽温度 7.2.1b)7.7)による。
6.7)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,ベンフルラリン標準液 (20ngC13H16F3N3O4/ml) [a)8)のベ
ンフルラリン標準液 (0.2μgC13H16F3N3O4/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマト
グラフに注入してベンフルラリンの量 (0.02ng) が十分に確認できるように検出器の感度を
調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,6.備考4.による(2)。
備考6. 57.の備考6.による。ただし,最終溶媒量はヘキサン20mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のベンフルラリン標準液 (0.2μgC13H16F3N3O4/ml) 1μlをマイクロシリンジ(3)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,ベンフルラリン
の保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からベンフルラリンの量を求め,13.1d)4)の式によって試料中のベンフルラリンの濃度
(μg/L) を算出する。
検量線 a)7)のベンフルラリン標準液 (1μgC13H16F3N3O4/ml) 0.2〜4mlを全量フラスコ10mlに段階的
にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマ
イクロシリンジでとり,1)及び2)の操作を行ってベンフルラリンの量 (ng) と指示値(5)との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
198
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
59. マラチオン (マラソン) (C10H19O6PS2) マラチオン(マラソン)[(ジメトキシホスフィノチオイル)
チオブタン二酸ジエチルエステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ
法を適用する。
59.1 ガスクロマトグラフ質量分析法 試料を33.1c)の抽出操作を行い,引き続きカラムクロマトグラフ分
離を行った後濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法に選
択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C10H19O6PS2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジエチルエーテル 6.3a)2)による。
5) ジクロロメタン 6.1a)3)による。
6) ヘキサン 6.1a)4)による。
7) ジエチルエーテル-ヘキサン混液 (3+17) 4)のジエチルエーテルと6)のヘキサンで調製する。
8) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
9) マラチオン標準液 (1mgC10H19O6PS2/ml) マラチオンの標準品0.100gをとり,少量のアセトンに
溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
10) マラチオン標準液 (10μgC10H19O6PS2/ml) マラチオン標準液 (1mgC10H19O6PS2/ml) 1mlを全量フラ
スコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
11) マラチオン標準液 (5μgC10H19O6PS2/ml) マラチオン標準液 (10μgC10H19O6PS2/ml) 5mlを全量フラ
スコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) マラチオン標準液 (0.5μgC10H19O6PS2/ml) マラチオン標準液 (5μgC10H19O6PS2/ml) 1mlを全量フラ
スコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
13) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。ただし,カラム充てん剤を流し込むときは,a)7)のジエチルエ
ーテル-ヘキサン混液 (3+17) を用いる。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,マラチオン標準液 (50ngC10H19O6PS2/ml)
[a)12)のマラチオン標準液 (0.5μgC10H19O6PS2/ml) 1mlを10mlに薄めたもの。]2μlをガスク
ロマトグラフ質量分析計に注入してマラチオンの量 (0.1ng) が十分に確認できるように検出
199
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 33.1c)1)〜4)の操作を行う。
2) 受器の内容物を少量のジエチルエーテル-ヘキサン混液 (3+17) で溶かした後,全量フラスコ10ml
に移し入れ,更に受器をジエチルエーテル-ヘキサン混液 (3+17) 2〜3mlで2,3回よく洗浄し,洗
液も全量フラスコ10mlに移し入れ,ジエチルエーテル-ヘキサン混液 (3+17) を標線まで加える(2)。
3) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。更に全量フラ
スコ10mlをジエチルエーテル-ヘキサン混液 (3+17) 2〜3mlで2,3回よく洗浄し,洗液もクロマ
トグラフ管に流し込む。
4) クロマトグラフ管の上部から,ジエチルエーテル-ヘキサン混液 (3+17) 100mlを流下させ,この流
出液は捨てる。
5) 引き続きクロマトグラフ管の上部から,アセトン-ヘキサン溶離液 (1+19) 100mlを流下し,マラチ
オンを溶出させ(3),溶出液をなす形フラスコ(4)に受ける。
6) 濃縮器(5)を用いて約40℃(5)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素を緩やか
に吹き付け,溶出液を完全に揮散させ,アセトン4mlを正しく加える(2)(6)(7)。
7) 空試験として試料と同量の水を分液漏斗にとり,1)〜6)の操作を行う。
注(2) 6.の注(10)による。
(3) 6.の注(25)による。
(4) 6.の注(8)による。
(5) 6.の注(9)による。
(6) 6.の注(21)による。
(7) 6.備考5.の注(*10)による。
d) 操作 操作は,次のとおり行う。
1) a)12)のマラチオン標準液 (0.5μgC10H19O6PS2/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,マラチオンのフラグ
メントイオン(m/z 173,125,127など)を設定し(9),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,マラチオンの保持時間に相当するピーク
の位置を確認しておく。
2) c)6)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)7)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からマラチオンの量を求め,33.1d)4)の式によって試料中のマラチオンの濃度 (μg/L) を算出
する。
検量線 a)11)のマラチオン標準液 (5μgC10H19O6PS2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってマラチオンの量 (ng) と指示値(10)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(8) 7.の注(9)による。
(9) 7.の注(10)による。
(10) 7.の注(11)による。
備考2. 7.の備考2.による。
200
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 7.の備考3.による。
59.2 ガスクロマトグラフ法 試料について59.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C10H19O6PS2 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって
異なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジエチルエーテル-ヘキサン混液 (3+17) 59.1a)7)による。
7) アセトン-ヘキサン溶離液 (1+19) 6.備考4.の5)による。
8) マラチオン標準液 (2.5mgC10H19O6PS2/ml) マラチオンの標準品0.250gをとり,少量のアセトンに
溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
9) マラチオン標準液 (25μgC10H19O6PS2/ml) マラチオン標準液 (2.5mgC10H19O6PS2/ml) 1mlを全量フ
ラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
10) マラチオン標準液 (2.5μgC10H19O6PS2/ml) マラチオン標準液 (25μgC10H19O6PS2/ml) 1mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
11) マラチオン標準液 (0.25μgC10H19O6PS2/ml) マラチオン標準液 (2.5μgC10H19O6PS2/ml) 1mlを全量
フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
12) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 59.1b)4)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,マラチオン標準液 (25ngC10H19O6PS2/ml) [a)11)のマラチ
201
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
オン標準液 (0.25μgC10H19O6PS2/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフに
注入してマラチオンの量 (0.05ng) が十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,59.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)11)のマラチオン標準液 (0.25μgC10H19O6PS2/ml) 2μlをマイクロシリンジ(8)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,マラチオンの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(8)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からマラチオンの量を求め,33.1d)4)の式によって試料中のマラチオンの濃度 (μg/L) を算出
する。
検量線 a)10)のマラチオン標準液 (2.5μgC10H19O6PS2/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってマラチオンの量 (ng) と指示値(10)との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考5. 備考2.による。
60. メコプロップ [MCPP] (C10H11C1O3) メコプロップ [MCPP] [2-(4-クロロ-2-メチルフェノキシ)
プロパン酸]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
60.1 ガスクロマトグラフ質量分析法 試料に塩酸を加えて,pHを2以下とし,ジクロロメタンで抽出し
た後,脱水・濃縮し,残留物にジアゾメタン-アセトン溶液を加えてメチル化し,溶媒を揮散させた後,ア
セトンに溶かし,その溶液の一定量を,ガスクロマトグラフ質量分析計に注入し,検出法に選択イオン検
出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C10H11ClO3 0.2〜4ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 JIS K 8180に規定するもの。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) ジアゾメタン-アセトン溶液 10.1a)11)による。
7) メコプロップ標準液 (1mgC10H11ClO3/ml) メコプロップの標準品0.100gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) メコプロップ標準液 (10μgC10H11ClO3/ml) (2) メコプロップ標準液 (1mgC10H11ClO3/ml) 1mlをなす
形フラスコにとり,窒素を緩やかに吹き付け,アセトンを完全に揮散させ,ジアゾメタン-アセトン
溶液20mlを加えて,10.1のc)5)及び6)の操作を行って,メチル化を行う。残留物を少量のアセトン
で溶かし,全量フラスコ100mlに移し入れ,さらにこのなす形フラスコを少量のアセトンで数回よ
く洗い,洗液も全量フラスコ100mlに合わせ,アセトンを標線まで加える。使用時に調製する。
9) メコプロップ標準液 (1μgC10H11C103/ml) (2) メコプロップ標準液 (10μgC10H11ClO3/ml) 1mlを全量
202
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(2)による。
(2) ここでいうメコプロップ標準液は,メチル化したものである。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ジアゾメタン発生装置 10.1b)5)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,メコプロップ標準液 (0.1μgC10H11ClO3/ml)
[a)9)のメコプロップ標準液 (1μgC10H11ClO3/ml) 1mlを10mlに薄めたもの。]1μlをガスクロ
マトグラフ質量分析計に注入してメコプロップの量 (0.1ng) が十分に確認できるように検出
器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(3)の適量(1 000ml以下の一定量)を分液漏斗にとり,塩酸を加えてpHを2以
下に調節し,液量1 000mlにつきジクロロメタン100mlを加え,振とう器を用いて約10分間振り混
ぜ,放置する。
2) 6.1c)2)〜5)の操作を行う。
3) 10.1のc)5)及び6)の操作を行い,ジアゾメタン-アセトン溶液を完全に揮散させ,アセトン1mlを正
しく加える(4)。
4) 空試験として試料と同量の水を分液漏斗にとり,1)〜3)の操作を行う。
注(3) 6.の注(3)による。
(4) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のメコプロップ標準液 (1μgC10H11ClO3/ml) (2)2μlをマイクロシリンジ(5)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メコプロップのフラ
グメントイオン[m/z(メチル化誘導体として)169,228など]を設定し,選択イオン検出法又は
マスクロマトグラフ法によって測定してそのマスフラグメントグラムを記録し,メコプロップの保
持時間に相当するピークの位置を確認しておく。
2) c)3)で得たアセトン溶液2μlをマイクロシリンジ(5)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)4)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からメコプロップの量を求め,次の式によって試料中のメコプロップの濃度 (μg/L) を算出
する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
203
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a: 検量線から求めたメコプロップの量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)3)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)8)のメコプロップ標準液 (10μgC10H11ClO3/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってメコプロップの量 (ng) と指示値(6)との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(5) 7.の注(9)による。
(6) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
60.2 ガスクロマトグラフ法 試料中のメコプロップを塩酸酸性として,ジエチルエーテルで抽出した後,
脱水・濃縮し,残留物にN, N'-ジシクロヘキシルカルボジイミドピリジン溶液,2, 2, 2-トリクロロエタノー
ルを加えて,恒温槽中で約1時間反応させ,エステル化を行う。この溶液に炭酸水素ナトリウム溶液を加
えて,ヘキサンで抽出し,再び脱水,濃縮を行い,カラムクロマトグラフ分離で精製した溶液について,
電子捕獲検出器を用いたガスクロマトグラフに注入して定量する。
定量範囲:C10H11ClO3 0.05〜1ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩酸 (1mol/L) 22.1.1a)の塩酸を用いて調製する。
2) 炭酸水素ナトリウム溶液 (20g/L) JIS K 8622に規定する炭酸水素ナトリウム2gを水に加えて
100mlとする。
3) 硫酸ナトリウム 6.1a)2)による。
4) ジエチルエーテル 6.3a)2)による。
5) N, N'-ジシクロヘキシルカルボジイミド(7)
6) 2, 2, 2-トリクロロエタノール(7)
7) ヘキサン 6.1a)4)による。
8) ピリジン JIS K 8777に規定するもの(7)。
9) ジエチルエーテル-ヘキサン溶離液 (1+29) 4)のジエチルエーテルと7)のヘキサンを用いて調製
する。
10) N, Nʼ-ジシクロヘキシルカルボジイミド-ピリジン溶液 (2vol%) 5)のN, N'-ジシクロヘキシルカル
ボジイミド1mlをとり,8)のピリジンに溶かして50mlとする。使用時に調製する。
12) メコプロップ標準液 (1mgC10H11ClO3/ml) 60.1a)7)による。
13) メコプロップ標準液 (10μgC10H11ClO3/ml) (8) メコプロップ標準液 (1mgC10H11ClO3ml) 1mlを全量
フラスコ10mlにとり,アセトンを標線まで加える。この溶液1mlをなす形フラスコ200mlにとり,
窒素を緩やかに吹き付けアセトンを揮散させ,残留物にN, N'-ジシクロヘキシルカルボジイミド-ピ
リジン溶液 (2vol%) 0.2mlと2, 2, 2-トリクロロエタノール0.5mlとを加え,c)の5)〜7)の操作を行い,
ヘキサンを完全に揮散させ,メコプロップトリクロロエチルエステル化物を生成させる。これを少
量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更にこのなす形フラスコをヘキサン2
204
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
〜3mlで2,3回よく洗い,洗液も全量フラスコ10mlに合わせ,ヘキサンを標線まで加える。使用
時に調製する。
14) メコプロップ標準液 (5μgC10H11ClO3/ml) (8) メコプロップ標準液 (10μgC10H11ClO3/ml) 5mlを全量
フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
15) メコプロップ標準液 (0.5μgC10H11ClO3/ml) (8) メコプロップ標準液 (5μgC10H11ClO3/ml) 1mlを全量
フラスコ10mlにとり,ヘキサンを標線まで加える。使用時に調製する。
16) 窒素 6.2a)2)による。
注(7) 4.の注(1)による。
(8) ここでいうメコプロップ標準液は,エステル化したものである。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) クロマトグラフ管 6.3b)2)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.3b)7.3)による。
6.4)
キャリヤーガス 7.2.3b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
カラム槽温度 7.2.1b)7.7)による。
6.7)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,メコプロップ標準液 (50ngC10H11ClO3/ml) (8)[a)15)のメ
コプロップ標準液 (0.5μgC10H11ClO3/ml) (8)1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフに注入してメコプロップの量 (0.05ng) が十分に確認できるように検出器の感度を調節
する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考11.による。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(3)200mlを分液漏斗500mlにとり,塩酸 (1mol/L) 10ml,ジエチルエーテル50ml
を加え,振とう器を用いて約5分間振り混ぜ,放置する。
2) 水層を別の分液漏斗に移し入れる。ジエチルエーテルを三角フラスコ200mlに移し入れ,分液漏斗
を少量のジエチルエーテルで洗い,洗液も先の三角フラスコに合わせる。分液漏斗の水層にジエチ
ルエーテル50mlを加えて,再び振とう器を用いて約5分間振り混ぜ,放置後,ジエチルエーテル
層を先の三角フラスコ200mlに合わせる。同じ操作を繰り返し,放置後,ジエチルエーテル層を先
の三角フラスコ200mlに合わせる。
3) ジエチルエーテル溶液50mlについて硫酸ナトリウム10〜15g(9)を加え,軽く振り混ぜて,約10分
205
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
間放置した後(10),ろ紙5種A(又は5種B)(11)を用いてろ過し,ろ液をなす形フラスコ200ml(12)
に受ける。ジエチルエーテル約15mlを用いて三角フラスコを数回洗浄し,さらにその洗液で先の
ろ紙上の硫酸ナトリウムを洗浄し,この洗液もジエチルエーテル溶液に合わせる。
4) これを,濃縮器(13)を用いて約40℃(13)の水浴上でジエチルエーテル溶液が1〜2mlになるまで濃縮し,
受器を取り外し,窒素を緩やかに吹き付け,ジエチルエーテルを揮散させる(14)(15)。
5) 残留物にN, N'-ジシクロヘキシルカルボジイミド-ピリジン溶液 (2vol%) 0.2mlと2, 2, 2-トリクロロ
エタノール0.5mlとを加え,60±1℃の恒温槽に移し入れ,栓をして時々軽く振り混ぜて,約1時間
放置する。
6) 放冷後,ヘキサン50mlを用いて分液漏斗200mlに移し入れ,炭酸水素ナトリウム溶液 (20g/L) 50ml
を加え,振とう器を用いて約1分間軽く振り混ぜて放置する。分離した水層は捨て,ヘキサン層に
塩酸 (1mol/L) 50mlを加え,再び振とう器を用いて約5分間振り混ぜて放置する。分離した水層は
捨て,ヘキサン層に水50mlを加え,振とう器を用いて約5分間振り混ぜて放置する。分離した水
層を捨て,ヘキサン層を三角フラスコ100mlに移す。
7) このヘキサン溶液に硫酸ナトリウム20gを加え,3)及び4)の操作を行い,ヘキサンを揮散させる。
8) 受器の内容物を少量のヘキサンで溶かした後,全量フラスコ10mlに移し入れ,更に受器を2〜3ml
のヘキサンで2,3回洗い,洗液も全量フラスコ10mlに移し入れ,ヘキサンを標線まで加える。
9) 全量フラスコ10mlの内容物全量をクロマトグラフ管の上部から注ぎ,流下させる。更に全量フラ
スコ10mlを2〜3mlのヘキサンで2,3回よく洗浄し,洗液もクロマトグラフ管に流下させる。
10) 次に,ジエチルエーテル-ヘキサン溶離液 (1+29) 50mlを流下してエステル化物を溶出させ(16),溶
出液をなす形フラスコ200ml(12)に受ける。
11) これを,濃縮器(13)を用いて約40℃(13)の水浴上で1〜2mlになるまで濃縮し,受器を取り外し,窒素
を緩やかに吹き付け,溶出液を揮散させた後,ヘキサン1mlを正しく加える(4)(14)(15)。
12) 空試験として試料と同量の水を分液漏斗500mlにとり,1)〜11)の操作を行う。
注(9) 6.の注(5)による。
(10) 6.の注(6)による。
(11) 6.の注(7)による。
(12) 6.の注(8)による。
(13) 6.の注(9)による。
(14) 6.の注(21)による。
(15) 6.備考5.の注(*10)による。
(16) あらかじめメコプロップトリクロロエチルエステル化物が溶出してくる画分を確認しておく。
備考6. 試料に濁りがなく,かつ,この試験に妨害となる成分を含まない場合には,c)9)及び10)の操
作を省略してもよい。この場合は,c)7)の操作に引き続きヘキサンを正しく加える。
d) 操作 操作は,次のとおり行う。
1) a)15)のメコプロップ標準液 (0.5μgC10H11ClO3/ml) 1μlをマイクロシリンジ(5)でとり,スプリットレス
注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メコプロップの保持
時間に相当するピークの位置を確認しておく。
2) c)11)で得たヘキサン溶液1μlをマイクロシリンジ(5)でとり,7.2.1d)2)の操作を行う。
3) 空試験としてc)12)で得たヘキサン溶液について7.2.1d)3)の操作を行う。
4) 検量線からメコプロップの量を求め,次の式によって試料中のメコプロップの濃度 (μg/L) を算出
206
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: メコプロップの濃度 (μg/L)
a: 検量線から求めたメコプロップの量 (ng)
C: 注入量 (μl)
υ1: c)11)で用いたヘキサンの量 (ml)
V: 試料 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)14)のメコプロップ標準液 (5μgC10H11ClO3/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,ヘキサンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってメコプロップの量 (ng) と指示値(6)との関係線を
作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
8. 試料中の対象農薬の濃度が高い場合は,c)8)のヘキサンから適量をとり,c)9)のクロマトグラ
フ管に流下させる。この場合の対象農薬の濃度は,次の式によって算出する。
V
C
a
N
000
1
10
10
3
3
2
1
3
×
×
×
×
×
=
−
−
υ
υ
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めたメコプロップの量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)8)のヘキサンの量 (ml)
υ2: c)9)のヘキサンの量 (ml)
υ3: c)11)のヘキサンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
61. メタラキシル (C15H21NO4) メタラキシル[N-(2, 6-ジメチルフェニル)-N-(メトキシアセチル)-DL-
アラニンメチルエステル]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用
する。
61.1 ガスクロマトグラフ質量分析法 試料について6.の備考5.の操作を行う。濃縮液の一定量を,ガス
クロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を
用いて定量する。
定量範囲:C15H21NO4 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) 酢酸エチル-ヘキサン混合溶液 (1+1) 6.備考5.のa)7)による。
5) メタラキシル標準液 (2mgC15H21NO4/ml) メタラキシルの標準品0.200gをとり,少量のアセトン
に溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) メタラキシル標準液 (20μgC15H21NO4/ml) メタラキシル標準液 (2mgC15H21NO4/ml) 1mlを全量フ
207
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) メタラキシル標準液 (2μgC15H21NO4/ml) メタラキシル標準液 (20μgC15H21NO4/ml) 1mlを全量フ
ラスコ10mlにとり,アセトンを標線まで加える。
8) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,メタラキシル標準液 (0.2μgC15H21NO4/ml)
[a)7)のメタラキシル標準液 (2μgC15H21NO4/ml) 1mlを10mlに薄めたもの。]2μlをガスクロ
マトグラフ質量分析計に注入してメタラキシルの量 (0.4ng) が十分に確認できるように検出
器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(200ml以下の一定量)を分液漏斗にとり,液量200mlについて塩化
ナトリウム10gと酢酸エチル-ヘキサン混合溶液 (1+1) 50mlとを加え,振とう器を用いて約10分
間振り混ぜ,放置する。
2) 6.備考5.のc)2)〜4)と同じ操作を行い,アセトン1mlを正しく加える(3)(4)(5)。
3) 空試験として試料と同量の水を分液漏斗にとり,液量200mlについて塩化ナトリウム10gと酢酸エ
チル-ヘキサン混合溶液 (1+1) 50mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。
引き続き2)の操作を行う。
注(2) 6.の注(3)による。
(3) 6.の注(21)による。
(4) 6.備考5.の注(*10)による。
(5) 6.の注(10)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のメタラキシル標準液 (2μgC15H21NO4/ml) 2μlをマイクロシリンジ(6)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メタラキシルのフラグ
メントイオン(m/z 206,248,220など)を設定し(7),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,メタラキシルの保持時間に相当するピー
クの位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(6)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からメタラキシルの量を求め,次の式によって試料中のメタラキシルの濃度 (μg/L) を算出
する。
208
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V
C
a
N
000
1
10
10
3
1
3
×
×
×
×
=
−
−
υ
ここに,
N: 対象農薬の濃度 (μg/L)
a: 検量線から求めたメタラキシルの量 (ng)
C: 注入量 (μl)
V: 試料 (ml)
υ1: c)2)のアセトンの量 (ml)
10−3: ngをμgに,μlをmlに換算する係数
検量線 a)6)のメタラキシル標準液 (20μgC15H21NO4/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってメタラキシルの量 (ng) と指示値(8)との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(6) 7.の注(9)による。
(7) 7.の注(10)による。
(8) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
61.2 ガスクロマトグラフ法 試料について61.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C15H21NO4 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) 酢酸エチル-ヘキサン混合溶液 (1+1) 6.備考5.のa)7)による。
5) メタラキシル標準液 (20μgC15H21NO4/ml) 61.1a)6)による。
6) メタラキシル標準液 (2μgC15H21NO4/ml) 61.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
209
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,メタラキシル標準液 (0.2μgC15H21NO4/ml) [a)6)のメタラ
キシル標準液 (2μgC15H21NO4/ml) 1mlを10mlに薄めたもの。]2μ1をガスクロマトグラフに注
入してメタラキシルの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,61.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のメタラキシル標準液 (2μgC15H21NO4/ml) 2μlをマイクロシリンジ(6)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メタラキシルの保持時
間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(6)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からメタラキシルの量を求め,61.1d)4)の式によって試料中のメタラキシルの濃度 (μg/L) を
算出する。
検量線 a)5)のメタラキシル標準液 (20μgC15H21NO4/ml) 0.1〜2mlを全量フラスコ10mlに段階的に
とり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイ
クロシリンジでとり,1)及び2)の操作を行ってメタラキシルの量 (ng) と指示値(8)との関係線を
作成する。検量線の作成は,試料測定時に行う。
備考6. 備考2.による。
62. メチルダイムロン (C17H20N2O) メチルダイムロン[N-メチル-N'-(1-メチル-1-フェニルエチル)-N-
フェニル尿素]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
62.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C17H20N2O 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) メチルダイムロン標準液 (1mgC17H20N2O/ml) メチルダイムロンの標準品0.100gをとり,少量の
アセトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
210
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) メチルダイムロン標準液 (10μgC17H20N2O/ml) メチルダイムロン標準液 (1mgC17H20N2O/ml) 1ml
を全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) メチルダイムロン標準液 (1μgC17H20N2O/ml) メチルダイムロン標準液 (10μgC17H20N2O/ml) 1ml
を全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,メチルダイムロン標準液
(0.1μgC17H20N2O/ml) [a)9)のメチルダイムロン標準液 (1μgC17H20N2O/ml) 1mlを10mlに薄め
たもの。]1μlをガスクロマトグラフ質量分析計に注入してメチルダイムロンの量 (0.1ng) が
十分に確認できるように検出器の感度を調節する。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
注(2) 13.の注(2)による。
d) 操作 操作は,次のとおり行う。
1) a)9)のメチルダイムロン標準液 (1μgC17H20N2O/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メチルダイムロン
のフラグメントイオン (m/z 107, 119, 91) を設定し(4),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,メチルダイムロンの保持時間に相当する
ピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からメチルダイムロンの量を求め,13.1d)4)の式によって試料中のメチルダイムロンの濃度
(μg/L)を算出する。
検量線 a)8)のメチルダイムロン標準液 (10μgC17H20N2O/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってメチルダイムロンの量 (ng) と指示値(5)との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
211
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
62.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H20N2O 1〜20ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) メチルダイムロン標準液 (100μgC17H20N2O/ml) 62.1a)7)のメチルダイムロン標準液
(1mgC17H20N2O/ml) 1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。保存する場合は,
−20℃の暗所におく。
8) メチルダイムロン標準液 (10μgC17H20N2O/ml) メチルダイムロン標準液 (100μgC17H20N2O/ml)
1mlを全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,メチルダイムロン標準液 (1μgC17H20N2O/ml) [a)8)のメ
チルダイムロン標準液 (10μgC17H20N2O/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグ
ラフに注入してメチルダイムロンの量 (1ng) が十分に確認できるように検出器の感度を調
節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
212
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
備考6. 14.の備考10.による。ただし,最終溶媒量はアセトン4mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のメチルダイムロン標準液 (10μgC17H20N2O/ml) 1μlをマイクロシリンジ(3)でとり,スプリット
レス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メチルダイムロ
ンの保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からメチルダイムロンの量を求め,13.1d)4)の式によって試料中のメチルダイムロンの濃度
(μg/L) を算出する。
検量線 a)7)のメチルダイムロン標準液 (100μgC17H20N2O/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってメチルダイムロンの量 (ng) と指示値(5)との
関係線を作成する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
63. メフェナセツト (C16H14N2O2S) メフェナセット[2-ベンゾチアゾール-2-イルオキシ-N-メチル(フェ
ニルアセトアミド)]の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
63.1 ガスクロマトグラフ質量分析法 試料中のメフェナセットを塩化ナトリウムの共存下でジクロロメ
タン抽出を行い,濃縮する。その一定量を,ガスクロマトグラフ質量分析計に注入し,対象農薬の検出法
に選択イオン検出法又はマスクロマトグラフ法を用いて定量する。
定量範囲:C16H14N2O2S 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) メフェナセット標準液 (2mgC16H14N2O2S/ml) メフェナセットの標準品0.200gをとり,少量のア
セトンに溶かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
6) メフェナセット標準液 (20μgC16H14N2O2S/ml) メフェナセット標準液 (2mgC16H14N2O2S/ml) 1ml
を全量フラスコ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
7) メフェナセット標準液 (2μgC16H14N2O2S/ml) メフェナセット標準液 (20μgC16H14N2O2S/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
8) 窒素 6.2a)2)による。
注(1) 7.の注(5)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
213
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,メフェナセット標準液 (0.2μgC16H14N2O2S/ml)
[a)7)のメフェナセット標準液 (2μgC16H14N2O2S/ml) 1mlを10mlに薄めたもの。]2μlをガスク
ロマトグラフ質量分析計に注入してメフェナセットの量 (0.4ng) が十分に確認できるように
検出器の感度を調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(400ml以下の一定量)を分液漏斗にとり,液量400mlについて塩化
ナトリウム20gとジクロロメタン50mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 18.1c)2)〜5)の操作を行う。ただし,ジクロロメタンの量は液量400mlについて50mlとする。
3) 空試験として試料と同量の水を分液漏斗にとり,液量400mlについて塩化ナトリウム20gとジクロ
ロメタン50mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。続いて2)の操作を行う。
注(2) 6.の注(3)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のメフェナセット標準液 (2μgC16H14N2O2S/ml) 2μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メフェナセットの
フラグメントイオン (m/z 192, 120, 136) を設定し(4),選択イオン検出法又はマスクロマトグラフ法
によって測定してそのマスフラグメントグラムを記録し,メフェナセットの保持時間に相当するピ
ークの位置を確認しておく。
2) c)2)で得たアセトン溶液2μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)3)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からメフェナセットの量を求め,18.1d)4)の式によって試料中のメフェナセットの濃度
(μg/L) を算出する。
検量線 a)6)のメフェナセット標準液 (20μgC16H14N2O2S/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってメフェナセットの量 (ng) と指示値(5)との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
63.2 ガスクロマトグラフ法 試料について63.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C16H14N2O2S 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
214
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) メフェナセット標準液 (20μgC16H14N2O2S/ml) 63.1a)6)による。
6) メフェナセット標準液 (2μgC16H14N2O2S/ml) 63.1a)7)による。
7) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,メフェナセット標準液 (0.2μgC16H14N2O2S/ml) [a)6)のメ
フェナセット標準液 (2μgC16H14N2O2S/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグ
ラフに注入してメフェナセットの量 (0.4ng) が十分に確認できるように検出器の感度を調節
する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,63.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)6)のメフェナセット標準液 (2μgC16H14N2O2S/ml) 2μlをマイクロシリンジ(3)でとり,スプリットレ
ス注入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メフェナセットの
保持時間に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からメフェナセットの量を求め,18.1d)4)の式によって試料中のメフェナセットの濃度
(μg/L) を算出する。
検量線 a)5)のメフェナセット標準液 (20μgC16H14N2O2S/ml) 0.1〜2mlを全量フラスコ10mlに段階
的にとり,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]を
マイクロシリンジでとり,1)及び2)の操作を行ってメフェナセットの量 (ng) と指示値(5)との関
215
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
係線を作成する。検量線の作成は,試料測定時に行う。
備考6. 備考2.による。
64. メプロニル (C17H19NO2) メプロニル〔2-メチル-N-[3-(1-メチルエトキシ)フェニル]ベンザミド〕
の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
64.1 ガスクロマトグラフ質量分析法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマ
トグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法を用いて
定量する。
定量範囲:C17H19NO2 0.1〜2ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異
なる。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) メプロニル標準液 (1mgC17H19NO2/ml) メプロニルの標準品0.100gをとり,少量のアセトンに溶
かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
8) メプロニル標準液 (10μgC17H19NO2/ml) メプロニル標準液 (1mgC17H19NO2/ml) 1mlを全量フラス
コ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
9) メプロニル標準液 (1μgC17H19NO2/ml) メプロニル標準液 (10μgC17H19NO2/ml) 1mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
10) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ質量分析計 7.1b)7)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,メプロニル標準液 (0.1μgC17H19NO2/ml) [a)9)
のメプロニル標準液 (1μgC17H19NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラ
フ質量分析計に注入してメプロニルの量 (0.1ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
注(2) 13.の注(2)による。
216
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 操作 操作は,次のとおり行う。
1) a)9)のメプロニル標準液 (1μgC17H19NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メプロニルのフラグメン
トイオン (m/z 119, 269, 91) を設定し(4),選択イオン検出法又はマスクロマトグラフ法によって測定
してそのマスフラグメントグラムを記録し,メプロニルの保持時間に相当するピークの位置を確認
しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からメプロニルの量を求め,13.1d)4)の式によって試料中のメプロニルの濃度 (μg/L) を算出
する。
検量線 a)8)のメプロニル標準液 (10μgC17H19NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってメプロニルの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
64.2 ガスクロマトグラフ法 試料について13.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C17H19NO2 1〜20ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) ヘキサン 6.1a)4)による。
6) アセトン-ヘキサン溶離液 (3+17) 10.1a)9)による。
7) メプロニル標準液 (100μgC17H19NO2/ml) 64.1a)7)のメプロニル標準液 (1mgC17H19NO2/ml) 1mlを
全量フラスコ10mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
8) メプロニル標準液 (10μgC17H19NO2/ml) メプロニル標準液 (100μgC17H19NO2/ml) 1mlを全量フラ
スコ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 6.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) クロマトグラフ管 6.3b)2)による。
217
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) マイクロシリンジ 7.1b)6)による。
6) ガスクロマトグラフ 次に掲げる条件を満たすもの。
6.1)
キャピラリーカラム用管 7.1b)7.1)による。
6.2)
キャピラリーカラム 7.1b)7.2)による。
6.3)
検出器 7.2.1b)7.3)による。
6.4)
キャリヤーガス 7.2.1b)7.4)による。
6.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
6.6)
燃料ガス及び助燃ガス 7.2.1b)7.6)による。
6.7)
カラム槽温度 7.2.1b)7.7)による。
6.8)
検出器槽温度 7.2.1b)7.8)による。
7) 振とう器
8) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,メプロニル標準液 (1μgC17H19NO2/ml) [a)8)のメプロニ
ル標準液 (10μgC17H19NO2/ml) 1mlを10mlに薄めたもの。]1μlをガスクロマトグラフに注入し
てメプロニルの量 (1ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,13.1c)の操作を行う(2)。
備考6. 14.の備考10.による。ただし,最終溶媒量はアセトン4mlとする。
d) 操作 操作は,次のとおり行う。
1) a)8)のメプロニル標準液 (10μgC17H19NO2/ml) 1μlをマイクロシリンジ(3)でとり,スプリットレス注
入法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,メプロニルの保持時間
に相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液1μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からメプロニルの量を求め,13.1d)4)の式によって試料中のメプロニルの濃度 (μg/L) を算出
する。
検量線 a)7)のメプロニル標準液 (100μgC17H19NO2/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,1μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってメプロニルの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考7. 備考2.による。
65. モリネート (C9H17NOS) モリネート{ヘキサヒドロ-1H-アゼピン-1-カルボチオ酸S-エチルエステ
ル)の定量には,ガスクロマトグラフ質量分析法及びガスクロマトグラフ法を適用する。
65.1 ガスクロマトグラフ質量分析法 試料中のモリネートを塩化ナトリウムの共存下でジクロロメタン
抽出を行い,脱水後,2, 2'-オキシビスエタノール-アセトン溶液を添加し,濃縮を行う。その一定量を,ガ
スクロマトグラフ質量分析計に注入し,対象農薬の検出法に選択イオン検出法又はマスクロマトグラフ法
を用いて定量する。
定量範囲:C9H17NOS 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
218
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
6) モリネート標準液 (2mgC9H17NOS/ml) モリネートの標準品0.200gをとり,少量のアセトンに溶
かし,全量フラスコ100mlに移し入れ,アセトンを標線まで加える(1)。
7) モリネート標準液 (20μgC9H17NOS/ml) モリネート標準液 (2mgC9H17NOS/ml) 1mlを全量フラス
コ100mlにとり,アセトンを標線まで加える。保存する場合は,−20℃の暗所におく。
8) モリネート標準液 (2μgC9H17NOS/ml) モリネート標準液 (20μgC9H17NOS/ml) 1mlを全量フラス
コ10mlにとり,アセトンを標線まで加える。使用時に調製する。
9) 窒素 6.2a)2)による。
注(1) 7.の注(4)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ質量分析計 7.1b)7)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考1. ガスクロマトグラフ質量分析計の感度として,モリネート標準液 (0.2μgC9H17NOS/ml) [a)8)
のモリネート標準液 (2μgC9H17NOS/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラ
フ質量分析計に注入してモリネートの量 (0.4ng) が十分に確認できるように検出器の感度を
調節する。
c) 準備操作 準備操作は,次のとおり行う。
1) 4.1c)で採取した試料(2)の適量(400ml以下の一定量)を分液漏斗にとり,液量400mlについて塩化
ナトリウム20gとジクロロメタン50mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。
2) 18.1c)2)及び3)の操作を行う。
3) 脱水したジクロロメタン層に2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 0.1mlを添加する。
4) 18.1c)4)及び5)の操作を行う。
5) 空試験として試料と同量の水を分液漏斗にとり,液量400mlについて塩化ナトリウム20gとジクロ
ロメタン50mlとを加え,振とう器を用いて約10分間振り混ぜ,放置する。続いて2)〜4)の操作を
行う。
注(2) 6.の注(3)による。
d) 操作 操作は,次のとおり行う。
1) a)8)のモリネート標準液 (2μgC9H17NOS/ml) 2μlをマイクロシリンジ(3)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,モリネートのフラグメン
219
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
トイオン (m/z 126, 55, 187) を設定し(4),選択イオン検出法又はマスクロマトグラフ法によって測定
してそのマスフラグメントグラムを記録し,モリネートの保持時間に相当するピークの位置を確認
しておく。
2) c)4)で得たアセトン溶液2μlをマイクロシリンジ(3)でとり,7.1d)2)の操作を行う。
3) 空試験として,c)5)で得たアセトン溶液について7.1d)3)の操作を行う。
4) 検量線からモリネートの量を求め,18.1d)4)の式によって試料中のモリネートの濃度 (μg/L) を算出
する。
検量線 a)7)のモリネート標準液 (20μgC9H17NOS/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってモリネートの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(3) 7.の注(9)による。
(4) 7.の注(10)による。
(5) 7.の注(11)による。
備考2. 7.の備考2.による。
3. 7.の備考3.による。
65.2 ガスクロマトグラフ法 試料について65.1c)の操作を行う。濃縮液の一定量を,ガスクロマトグラフ
に注入し,熱イオン化検出器を用いた方法で定量する。
定量範囲:C9H17NOS 0.4〜8ng 繰返し分析精度:変動係数で10〜20%(装置,測定条件によって異な
る。)
a) 試薬 試薬は,次のものを用いる。
1) 塩化ナトリウム 6.1a)1)による。
2) 硫酸ナトリウム 6.1a)2)による。
3) アセトン 4.1a)3)による。
4) ジクロロメタン 6.1a)3)による。
5) 2, 2'-オキシビスエタノール-アセトン溶液 (1+49) 7.3a)7)による。
6) モリネート標準液 (20μgC9H17NOS/ml) 65.1a)7)による。
7) モリネート標準液 (2μgC9H17NOS/ml) 65.1a)8)による。
8) 窒素 6.2a)2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 17.1b)1)による。
2) なす形フラスコ 6.1b)2)による。
3) 三角フラスコ 6.1b)3)による。
4) マイクロシリンジ 7.1b)6)による。
5) ガスクロマトグラフ 次に掲げる条件を満たすもの。
5.1)
キャピラリーカラム用管 7.1b)7.1)による。
5.2)
キャピラリーカラム 7.1b)7.2)による。
5.3)
検出器 7.2.1b)7.3)による。
5.4)
キャリヤーガス 7.2.1b)7.4)による。
5.5)
試料導入方法及び試料導入部温度 7.1b)7.5)による。
220
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.6)
燃料ガス及び助燃ガス 7.2.1b)7.6による。
5.7)
カラム槽温度 7.2.1b)7.7)による。
5.8)
検出器槽温度 7.2.1b)7.8)による。
6) 振とう器
7) 濃縮器 6.1b)5)による。
備考4. ガスクロマトグラフの感度として,モリネート標準液 (0.2μgC9H17NOS/ml) [a)7)のモリネー
ト標準液 (2μgC9H17NOS/ml) 1mlを10mlに薄めたもの。]2μlをガスクロマトグラフに注入し
てモリネートの量 (0.4ng) が十分に確認できるように検出器の感度を調節する。
5. c)準備操作においてカラムクロマトグラフ分離を行った場合は,充てんカラムを用いること
ができる。この場合のガスクロマトグラフの条件などは7.の備考5.による。
c) 準備操作 準備操作は,65.1c)による。
d) 操作 操作は,次のとおり行う。
1) a)7)のモリネート標準液 (2μgC9H17NOS/ml) 2μlをマイクロシリンジ(3)でとり,スプリットレス注入
法又はコールドオンカラム注入法によってガスクロマトグラフに注入し,モリネートの保持時間に
相当するピークの位置を確認しておく。
2) c)で得たアセトン溶液2μlをマイクロシリンジ(3)でとり,7.2.1d)2)の操作を行う。
3) 空試験として,c)で得た空試験のアセトン溶液について7.2.1d)3)の操作を行う。
4) 検量線からモリネートの量を求め,18.1d)4)の式によって試料中のモリネートの濃度 (μg/L) を算出
する。
検量線 a)6)のモリネート標準液 (20μgC9H17NOS/ml) 0.1〜2mlを全量フラスコ10mlに段階的にと
り,アセトンを標線まで加える。これらの溶液の一定量[試料と同量(例えば,2μl)]をマイク
ロシリンジでとり,1)及び2)の操作を行ってモリネートの量 (ng) と指示値(5)との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考6. 備考2.による。
2
2
1
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 農薬の名称
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
1
1, 3-ジクロロプロペン
dichloropropene
542-75-6
1, 3-ジクロロプロペン[1, 3-ジクロロ-1-プロペン]
1, 3-dichloropropene [1, 3-dichloro-1-propene]
−
−
2
EPN
−
2104-64-5
フェニルホスホノチオ酸O-エチルO-(4-ニトロフェニ
ル)エステル
phenylphosphonothioic acid O-ethylO- (4-
nitrophenyl) ester
O-エチルO-p-ニトロフェニルフェニルホスホノチオ
エート
O-ethylO-p-nitrophenyl phenylphosphonothioate
3
アシュラム
asulam
2302-17-2
[(4-アミノフェニル)スルホニル]カルバミン酸メチ
ルエスチル
[(4-aminophenyl) sulfonyl] carbamic acid
methylester
p-スルファニリルカルバミン酸メチル
p-sulfanilycarbamate methyl
4
アセフェート
acephate
(30560-19-1)
アセチルホスホロアミドチオ酸O, S-ジメチルエステ
ル
acetylphosphoramidothioic acid O, S-dimethyl
ester
O, S-ジメチアセチルホスホスホロアミドエート
O, S-dimethyl acetylphosphosphoramidethioate
5
イソキサチオン
isoxathion
18854-01-8
ホスホロチオ酸O, O-ジエチルO-(5-フェニル-3-イソ
キサゾリル)エステル
phosphorothioic acid O, O-diethylO- (5-phenyl-3-
isoxazolyl) ester
O, O-ジエチルO-5-フェニルイソオキサゾ-3-イルホス
ホロチオエート
O, O-diethylO-5-phenylisoxazol-3-yl
phosphorothioate
6
イソフェンホス
isofenphos
25311-71-1
2-{〈エトキシ[(1-メチルエチル)アミノ]ホスフィ
ノチオイル〉オキシ}安息香酸1-メチルエチルエステ
ル
2- {<ethoxy [(1-methylethyl) amino]
phosphinothioyl>oxy} benzonic acid 1-
methylethylester
O-エチルO-2-イソプロポキシカルボニルフェニルイ
ソプロピルホスホロアミドチオエート
O-ethylO-2-isopropoxycarbonylphenyl
isopropylphosphoramidothioate
7
イソプロチオラン
isoprothiolane
50512-35-1
1, 3-ジチオラン-2-イリデンプロパン二酸ビス(1-メチ
ルエチル)エステル
1, 3-dithiolane-2-ylidenpropanedioic acid bis (1-
methylethyl) ester
ジイソプロピル1, 3-ジチオラン-2-イリデンマロネー
ト
diisopropyl 1, 3-dithiolan-2-yldenemalonate
8
イプロジオン
iprodione
36734-19-7
3-(3, 5-ジクロロフェニル)-N-(1-メチルエチル)-2, 4-
ジオキソ-1-イミダゾリジンカルボキサミド
3- (3, 5-dichlorophenyl) -N- (1-methylethyl-2, 4-
dioxo-1-imidazolidine carboxamide
3-(3, 5-ジクロロフェニル)-N-イソプロピル-2, 4-ジオ
キソー1-イミダゾリジニルカルボキサミド
3- (3, 5-dichlorophenyl) -N-isopropyl-2, 4-
dioxoimidazolidin-1-yl-carboxamid
2
2
2
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
9
イプロベンホス [IBP]
iprobenfos
26087-47-8
ホスホルチオ酸O, O-ビス(1-メチルエチル)-S-(フ
ェニルメチル)エステル
phosphorothioic acid O, O-bis (1-methyl ethyl) -S-
(phenyl methyl) ester
S-ベンジル O, O-ジイソプロピルホスホロチオエート
S-benzylO, O-diisopropyl phosphorothioate
10
イミダクロプリド
imidacloprid
(138261-41-3)
1-[(6-クロロ-3-ピリジニル)メチル]-4, 5-ジヒドロ-N-
ニトロ-1H-イミダゾール-2-アミン
1-
[
(6-chloro-3-pyridinyl)
methyl]
-4,
5-dihydro-N-nitro-1H-imidazol-2-amine
1-(6-クロロ-3-ピリジルメチル)-N-ニトロイミダゾリ
ジン-2-イリデンアミン
1- (6-chloro-3-pyridylmethyl) -N-
nitroimidazolidin-2-ylideneamine
11
エスプロカルブ
esprocarb
85785-20-2
S-ベンジル-N-1,(2-ジメチルプロピル)-N-エチルチオ
カルバマート
S-benzyl-N- (1, 2-dimethylpropyl) -N-ethyl
thiocarbamate
S-ベンジル-N-(1, 2-ジメチルプロピル)-N-エチルチオ
カルバマート
S-benzyl-N- (1, 2-dimethylpropyl) -N-ethyl
thiocarbamate
12
エジフェンホス [EDDP]
edifenphos
17109-49-8
ホスホロジチオ酸O-エチルS, S-ジフェニルエステル
phosphorodithioic acid O-ethyl S, S-diphenyl ester
O-エチルS, S-ジフェニルホスホロジチオエート
O-ethyl S, S-diphenyl phosphorodithioate
13
エトフェンプロックス
etofenprox
80844-07-1
2-(4-エトキシフェニル)-2-メチルプロピル 3-フェノ
キシベンジルエーテル
2- (4-ethoxyphenyl) -2-methylpropyl3-
phenoxybenzyl ether
2-(4-エトキシフェニル)-2-メチルプロピル3-フェノ
キシベンジルエーテル
2- (4-ethoxyphenyl) -2-methylpropyl3-
phenoxybenzyl ether
14
エトリジアゾール[エク
ロメゾール]
etridiazole
2593-15-9
5-エトキシ-3-(トリクロロメチル)-1, 2, 4-チアジアゾ
ール
5-ethoxy-3- (trichlorometyl) -1, 2, 4-thiadiazole
エチル3-トリクロロメチル-1, 2, 4-チアゾール-5-イル
エーテル
ethyl3-trichloromethyl-1, 2, 4-thiazol-5-ylether
15
オキシン銅[有機銅]
oxine-copper
10380-28-6
ビス(8-キノリノラト-N1, O8)銅
bis (8-quinolinolato-N1, O8) copper
ビス(キノリン-8-オラト)銅
bis (quinolin-8-olato) copper
16
カルバリル [NAC]
carbaryl
63-25-2
1-ナフタレノールメチルカーバマート
1-Naphthalenol methylcarbamate
1-ナフチルメチルカーバマート
1-Naphthyl methylcarbamate
17
キャプタン
captan
133-06-2
3a, 4, 7, 7a-テトラヒドロ-2-[(トリクロロメチル)チオ]
-1H-イソインドール-1, 3- (2H) -ジオン
3a, 4, 7, 7a-tetrahydro-2- [(trichloromethyl) thio]
-1H-isoindole-1, 3- (2H) -dione
N-(トリクロロメチルチオ)-4-シクロヘキセン-1, 2-
ジカルボキシイミド
N- (trichloromethylthio) cyclohex-4-ene-1, 2-
dicarboximide
18
クロルニトロフェン
[CNP]
chlornitrofen
1836-77-7
1, 3, 5-トリクロロ-2-(4-ニトロフェノキシ)ベンゼン
1, 3, 5-trichloro-2- (4-nitrophenoxy) benzene
2, 4, 6-トリクロロフェニル4-ニトロフェニルエーテル
2, 4, 6-trichlorophenyl 4-nitrophenyl ether
2
2
3
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
19
クロルピリホス
chlorpyrifos
2921-88-2
ホスホロチオ酸O, O-ジエチルO-(3, 5, 6-トリクロロ
-2-ピリジニル)エステル
phosphorothioic acid O, O-diethylO- (3, 5, 6-
trichloro-2-pyridinyl) ester
O, O-ジエチルO-3, 5, 6-トリクロロ-2-ピリジルホスホ
ロチオエート
O, O-diethylO-3, 5, 6-trichloro-2-pyridyl
phosphorothioate
20
クロロタロニル [TPN]
chlorothalonil
1897-45-6
2, 4, 5, 6-テトラクロロ-1, 3-ベンゼンジカルボニトリ
ル
2, 4, 5, 6-tetrachloro-1, 3-benzenedicarbonitrile
テトラクロロイソフタロニトリル
tetrachloroisophthalonitrile
21
クロロネブ
chloroneb
2675-77-6
1, 4-ジクロロ-2, 5-ジメトキシベンゼン
1, 4-dichloro-2, 5-dimethoxybenzene
1, 4-ジクロロ-2, 5-ジメトキシベンゼン
1, 4-dichloro-2, 5-dimethoxybenzene
22
ジクロフェンチオン
[ECP]
dichlofenthion
97-17-6
ホスホロチオ酸O-(2, 4-ジクロロフェニル)O, O-ジエ
チルエステル
phosphorothioic acid O- (2, 4-dichlorophenyl) O,
O-diethylester
O, -2, 4-ジクロロフェニルO, O-ジエチルホスホロチオ
エート
O, -2, 4-dichlorophenyl O, O-diethyl
phophorothioate
23
ジクロルボス [DDVP]
dichlorvos
62-73-7
りん酸2, 2-ジクロロエテニルジメチルエステル
phosphoric acid2, 2-dichloroethenyl dimethyl
ester
2, 2-ジクロロビニルジメチルホスフェート
2, 2-dicohlorovinyl dimethyl phosphate
24
ジチオピル
dithiopyr
97886-45-8
2-(ジフルオロメチル)-4-(2-メチルプロピル)-6-(ト
リフルオロメチル)-3, 5-ピリジンジカルボチオ酸S, S-
ジメチルエステル
2- (Difluoro methyl) -4- (2-methylpropyl) -6-
(trifluoromethyl) -3, 5-pyridinedicarbothioic acid S,
S-dimethyl ester
S, S-ジメチル2-(ジフルオロメチル)-4-イソブチル-6-
(トリフルオロメチル)-3, 5-ピリジンジカルボチオエ
ート
S, S-dimetyl 2- (difluoromethyl) -4-isobutyl-6-
(trifluoromethyl) -3, 5-pyridinedicarbothioate
25
シマジン [CAT]
simazine
(122-34-9)
6-クロロ-N, N-ジエチル-1, 3, 5-トリアジン-2, 4-ジアミ
ン
6-chloro-N, N-diethyl-1, 3, 5-triazine-2,.4-diamine
2-クロロ-4, 6-ビス(エチルアミノ)-1, 3, 5-トリアジン
2-chloro-4, 6-bis (ethylamino) -1, 3, 5-triazine
26
シメトリン
simetlyn
1014-70-6
N, N'-ジエチル-6-(メチルチオ)-1, 3, 5-トリアジン-2, 4-
ジアミン
N, N'-diethyl-6 (methylthio) -1,.3, 5-triazine-2, 4-
diamine
2, 4-ビス(エチルアミノ)-6-メチルチオ-1, 3, 5-トリア
ジン
2, 4-bis (ethylamino) -6-mlethylthio-1, 3, 5-triazine
27
ダイアジノン
diazinon
(333-41-5)
ホスホロチオ酸O, O-ジエチルO-[6-メチル-2-(1-メ
チルエチル)-4-ピリミジニル]エステル
phosphorothioic acid O, O-diethylO- [6-methyl-
2- (1-methylethyl) -4-pyrimidinyl] ester
O, O-ジエチルO-2-イソプロピル-6-メチル-ピリミジン
-4-イルホスホロチオエート
O, O-diethylO-2-isopropyl-6-methylpyrimidin-
4-yl phosphorothioate
2
2
4
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
28
チウラム
thiram
137-26-8
テトラメチルチオペルオキシジカルボン酸ジアミド
tetramethylthioperoxydicarbonic diamide
テトラメチルチウラムジスルフィド
tetramethylthiuram disulfide
29
チオベンカルブ[ベンチ
オカーブ]
thiobencarb
[benthiocarb]
28249-77-6
ジエチルカルバモチオ酸S-[(4-クロロフェニル)メチ
ル]エステル
diethylcarbamothioic acid S- [(4-chlorophenyl)
methyl] ester
S-4-クロロベンジルジエチルカルバマート
S-4-chlorobenzyl diethylcarbamate
30
テルブカルブ [MBPMC]
terbucarb
1918-11-2
2, 6-ビス(1, 1-ジメチルエチル)-4-メチルフェノール
メチルカルバマート
2, 6-bis (1, 1-dimethylethyl) -4-methylpenol
methylcarbamate
2, 6-ジ-tert-ブチル-p-トリルメチルカルバマート
2, 6-di-tert-buthyl-p-toly methylcarbamate
31
トリクロピル(トリクロ
ピルブトキシエチル)
triclopyr
55335-06-3
[(3, 5, 6-トリクロロ-2-ピリジニル)オキシ]酢酸
[(3, 5, 6-trichloro-2-pyridinyl) oxy] acetic acid
3, 5, 6-トリクロロ-2-ピリジルオキシ酢酸
3, 5, 6-trichloro-2-pyridyloxyacetic acid
32
トリクロルホン [DEP]
trichlorfon
52-68-6
(2, 2, 2-トリクロロ-1-ヒドロキシエチル)ホスホン酸
ジメチルエステル
(2, 2, 2-trichloro-1-hydroxyethyl) phosphonic
acid dimethyl ester
ジメチル2, 2, 2-トリクロロ-1-ヒドロキシエチルホス
ホネート
dimethyl 2, 2, 2-trichloro-1-hydroxyethyl
phosphonate
33
トリシクラゾール
tricyclazole
41814-78-2
5-メチル-1, 2, 4-トリアゾロ [3, 4-b] ベンゾチアゾー
ル
5-methyl-1, 2, 4-triazolo [3, 4-b] benzothiazole
5-メチル-1, 2, 4-トリアゾロ [3, 4-b] ベンゾチアゾー
ル
5-methyl-1, 2, 4-triazolo [3, 4-b] benzothiazole
34
トルクロホスメチル
tolclophos-methyl
57018-04-9
O, O-ジメチルO-(2, 6-ジクロロ-4-メチルフェニル)
ホスホロチオ酸エステル
phosphorothioic acid O, O-dimethylO- (2, 6-
dichloro-4-methylphenyl) ester
O-2, 6-ジクロロ-p-トリルO, O-ジメチルホスホロチオ
エート
O-2, 6-dichloro-p-tolylO, O-dimethyl
phosphorothioate
35
ナプロパミド
napropamide
15299-99-7
N, N-ジエチル2-(1-ナフタレニルオキシ)プロパンア
ミド
N, N-diethyl-2- (1-naphthalenyloxy) propanamide
N, N-ジエチル-2-(1-ナフチルオキシ)プロピオンアミ
ド
N, N-diethyl-2- (1-naphthyloxy) propionamide
36
ピリダフェンチオン
pyridaphenthion
119-12-0
ホスホロチオ酸O-(1, 6-ジヒドロ-6-オキソ-1-フェニ
ル-3-ピリダジニル)O, O-ジエチルエステル
phosphorothioic acid O- (1, 6-dihydoro-6-oxo-1-
phenyl-3-pyridazinyl) O, O-diethyl ester
O, O-ジエチルO-(2, 3-ジヒドロ-3-オキソ-2-フェニル
ピリダジン-6-イル)ホスホロチオエート
O, O-diethylO- (2, 3-dihydro-3-oxo-2-phenyl
pyradazin-6-yl) phosphorotioate
2
2
5
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
37
ピリブチカルブ
pyributicarb
(88678-67-5)
O-3-(1, 1-ジメチルエチル)-N-(6-メトシキ-2-ピリジル)
-N-メチル(チオカーバメート)
O-3- (1, 1-dimethyl ethyl) -N- (6-methoxy-2-
pridyl) -N-methyl (thiocarbamate)
O-3-tert-ブチルフェニル-N-(6-メトキシ-2-ピリジル)
-N-メチル(チオカルバマート)
O-3-tert-butylphenyl N- (6-methoxy-2-pyridyl)
-N-methyl (thiocarbamate)
38
フェニトロチオン [MEP]
fenitrothion
(122-14-5)
ホスホロチオ酸O, O-ジメチルO-(3-メチル-4-ニトロ
フェニル)エステル
phosphorothioic
acid
O,
O-dimethylO-
(3-methyl-4-nitrophenyl) ester
O, O-ジメチルO-4-ニトロ-m-トリルホスホロチオエー
ト
O, O-dimethyl O-4-nitro-m-tolyl
phosphorothioate
39
フェノブカルブ [BPMC]
fenobucarb
3766-81-2
2-(1-メチルプロピル)-フェノールメチルカルバマー
ト
2- (1-Methylpropyl) -phenol methyl carbamate
o-sec-ブチルフェニルメチルカルバマート
o-sec-butylphenyl methylcarbamate
40
フサライド
fthalide
27355-22-2
4, 5, 6, 7-テトラクロロ-1 (3H) -イソベンゾフラノン
4, 5, 6, 7-tetrachloro-1 (3H) -isobenzofuranone
4, 5, 6, 7-テトラクロロフタリド
4, 5, 6. 7-tetrachlorophthalide
41
ブタミホス
butamifos
36335-67-8
N-(1-メチルプロピル)ホスファミドチオ酸O-エチル
O-(5-メチル-2-ニトロフェニル)エステル
N- (1-methylpropyl) phosphamidothioic acid O-
ethyl O- (5-methyl-2-nitrophenyl) ester
O-エチルO-6-ニトロ-m-トリルsec-ブチルホスホロア
ミドチオエート
O-ethyl O-6-nitro-m-tolyl-sec-
butylphosphoramidothioate
42
ブプロフェジン
buprofezin
69327-76-0
2-(1, 1-ジメチルエチルイミノ)-3-(1-メチルエチル)
-5-フェニル-1, 3, 5-チアジアジン-4-オン
2- (1, 1-dimethyl ethylimino) -3- (1-methyl ethyl)
-5-phenyl-1, 3, 5-thiadiazine-4-one
2-tert-ブチルイミノ-3-イソプロピル-5-フェニル-1, 3,
5-チアジアジン-4-オン
2-tert-butylimino-3-isopropyl-5-phenyl-1, 3, 5-
thiadiazinan-4-one
43
フルトラニル
flutolanil
66332-96-5
2, 2, 2-トリフルオロメチル-N-[3-(1-メチルエトキシ)
フェニル]ベンザミド
2, 2, 2-trifluoromethyl-N- [3- (1-methylethoxy)
phenyl] benzamide
α,α,α-トリフルオロ-3'-イソプロポキシ-o-トルアニ
リド
α,α,α-trifluoro-3-isopropoxy-o-toluanilide
44
プレチラクロール
pretilachlor
51218-49-6
2-クロロ-N-(2, 6-ジエチルフェニル)-N-(2-プロポキ
シエチル)アセトアミド
2-chloro-N- (2, 6-diethylphenyl) -N- (2-
propoxyethyl) acetamide
2-クロロ-2', 6'-ジエチル-N-(2-プロポキシエチル)-ア
セタニリド
2-chloro2', 6'-diethyl-N- (2-propoxyethyl) -
acetanilide
45
プロピザミド
propyzamide
23950-58-5
3, 5-ジクロロ-N-(1, 1-ジメチル-2-プロピニル)ベンズ
アミド
3, 5-dichloro-N- (1, 1-dimethyl-2-propynyl)
benzamide
3, 5-ジクロロ-N-(1, 1-ジメチルプロピニル)ベンズア
ミド
3, 5-dichloro-N- (1, 1-dimethylpropynyl)
benzamide
2
2
6
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
46
プロベナゾール
probenazole
27605-76-1
3-(2-プロペニルオキシ)-1, 2-ベンズイソチアゾール
-1, 1-ジオキシド
3- (2-propenyloxy) -1, 2-benzisothiazole-1, 1-
dioxide
3-アリルオキシ-1, 2-ベンゾイソチアゾール1, 1-ジオ
キシド
3-allyloxy-1, 2-benzoisothiazole1, 1-dioxide
47
ブロモブチド[ブロモチ
ド]
bromobutide
74712-19-9
2-ブロモ-3, 3-ジメチル-N-(1-メチル-1-フェニルエチ
ル)ブタンアミド
2-bromo-3, 3-dimethy-N- (1-methyl-1-phenylethyl)
butanamide
2-ブロモ-3, 3-ジメチル-N-(1-メチル-1-フェニルエチ
ル)ブチルアミド
2-bromo-3, 3-dimethyl-N- (1-methyl-1-
phenylethyl) butylamide
48
ペンシクロン
pencycuron
(66063-05-6)
1-(4-クロロベンジル)-1-シクロペンチル-3-フェニル
尿素
1- (4-chlorobenzyl) -1-cyclopentyl-3-phenylurea
1-(4-クロロベンジル)-1-シクロペンチル-3-フェニル
尿素
1- (4-chlorobenzyl) -1-cyclopentyl-3-phenylurea
49
ベンスリド [SAP]
bensulide
741-58-2
ホスホロジチオ酸O, O-ビス(1-メチルエチル)S-<2-
[(フェニルスルホニル)アミノ]-エチル>エステル
phosphorodithioic acid O, O-bis (1-methylethyl)
S-<2- [(phenylsulfonyl) amino] -ethyl>ester
O, O-ジイソプロピルS-2-ベンゼンスルホンアミノエ
チルホスホロジチオエート
O, O-diisopropyl S-2-
benzenesulfonaminoethylphosphorodithioate
50
ペンジメタリン
pendimethalin
40487-42-1
N-(1-エチルプロピル)-3, 4-ジメチル-2, 6-ジニトロベ
ンゼンアミン
N- (1-ethylpropyl) -3, 4-dimethyl-2, 6-
dinitrobenzenamine
N-(1-エチルプロピル)-2, 6-ジニトロ-3, 4-キシリジン
N- (1-ethylpropyl) -2, 6-dinitro-3, 4-xylidine
51
ベンフルラリン[ベスロ
ジン]
benfluralin
732-11-6
N-ブチル-N-エチル-2, 6-ジニトロ-4-(トリフルオロメ
チル)ベンゼンアミン
N-butyl-N-ethyl-2, 6-dinitro-4- (trifluoromethyl)
benzenamine
N-ブチル-N-エチル-α,α,α-トリフルオロ-2, 6-ジニト
ロ-p-トルイジイン
N-butyl-N-ethyl-α,α,α-trifluoro-2, 6-dinitro-p-
toluidine
52
マラチオン(マラソン)
malathion
121-75-5
(ジメトキシホスフィノチオイル)チオブタン二酸ジ
エチルエステル
(dimethoxyphosphinothioyl) thiobutanedioic acid
diethyl ester
S-1, 2-ビス(エトキシカルボニル)エチルO, O-ジメチ
ルホスホロジチオエート
S-1, 2-bis (ethoxycarbonyl) ethyl O, O-dimethyl
phosphorodithioate
53
メコプロップ [MCPP]
mecoprop
7085-19-0
2-(4-クロロ-2-メチルフェノキシ)プロパン酸
2- (4-chloro-2-methylphenoxy) propanoictacid
(R) or (RS) -2-(4-クロロ-2-トリルオキシ)プロピオン
酸
(R) or (RS) -2- (4-chloro-2-tolyloxy) propionic acid
2
2
7
K
0
1
2
8
:
2
0
0
0
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名(1)[登録名]
ISO登録農薬名
Chemical Abstractsの
Registry Number(2)
化合物名(3)
IUPAC規則による化合物名(4)
54
メタラキシル
metalaxyl
57837-19-1
N-(2, 6-ジメチルフェニル)-N-(メトキシアセチル)
-DL-アラニンメチルエステル
N- (2, 6-dimethylphenyl) -N- (methoxyacetyl)
-DL-alanine methyl ester
メチルN-(2-メトキシアセチル)-N-(2, 6-キシリル)-DL-
アラネート
methylN- (2-methoxyacetyl) -N- (2, 6-xylyl)
-DL-alaninate
55
メチルダイムロン
methyldymron
42609-73-4
N-メチル-N'-(1-メチル-1-フェニルエチル)-N-フェニ
ル尿素
N-methyl-N'- (1-methyl-1-phenylethyl) -N-
Phenylurea
1-(α,α-ジメチルベンジル)-3-メチル-3-フェニル尿
素
1- (α,α-dimethylbenzyl) -3-methyl-3-phenylurea
56
メフェナセット
mefenacet
73250-68-7
2-ベンゾチアゾール-2-イルオキシ-N-メチル(フェニル
アセトアミド)
2-benzothiazol-2-yloxy-N-methyl-phenylacetamide
2-ベンゾチアゾール-2-イルオキシ-N-メチルアセトア
ニリド
2-benzothiazol-2-yloxy-N-methylacetanilide
57
メプロニル
mepronil
55814-41-0
2-メチル-N-[3-(1-メチルエトキシ)フェニル]ベン
ズアミド
2-methyl-N- [3- (1-methylethoxy) phenyl]
benzamide
3'-イソプロポキシ-o-トルアニリド
3'-isopropoxy-o-toluanilide
58
モリネート
molinate
2212-67-1
ヘキサヒドロ-1H-アゼピン-1-カルボチオ酸S-エチル
エステル
hexahydiro-1H-azepine-1-carbothioic acid S-ethyl
ester
S-エチルペルヒドロアジピン-1-カルボチオエート
S-ethyl perhydroazepin-1-carbothoate
注(1) 農林水産省への登録名と国内通称名が一致していない場合に,別に登録名を[ ]で表示してある。
(2) 国内通称名と同じ呼称で商標登録が確認されたもの(平成12年7月現在の化学品,薬剤,医療補助品の商品分類)については,Chemical AbstractsのRegistry Number
を ( ) で示した。
(3) IUPAC規則による化合物名を字訳による日本語で表示したものである。
(4) 上段,下段ともIUPAC規則による化合物名であるが,上段が“Chemical Abstracts Index Guide 9th, 10th”で使用されている化合物名で,下段は“Pesticides and other
agrochemicals−Common names字訳対照表”(ISO農薬部会1981)によった化合物名である。規格本文では上段の化合物名を国内通称名の後に ( ) で表示してあ
る。

228
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表2 試験方法概要一覧
No
.
国内通称名
[登録名]
ISO登録農薬名
多成分同時測定法
個別測定方法
GC/MS
ガスクロマトグラフ法
(キャピラリーカラム用管使用)
HPLC GC/
MS
ガスクロマト
グラフ法
HPLC
(SIM又は
MC)
(FTD)
(FPD)
(ECD)
本文備考
で充てん
カラム使
用
(UV)
FTD FPD ECD UV 蛍光
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
固相
抽出
1 1, 3-ジクロロプ
ロペン
JIS K 0125を引用
2 EPN
EPN
この場合,
対象農薬
ごとの検
出器を選
択し,カラ
ム槽温度
を対象農
薬ごとに
設定する
ことを明
示してあ
る。
○ ○ ○ ○
3 アシュラム
asulam
○
○
4 アセフェート
acephate
○ ○
5 イソキサチオン isoxathion
○ ○ ○ ○
6 イソフェンホス isofenphos
○ ○ ○
7 イソプロチオラ
ン
isoprothiolane
○
○
8 イプロジオン
iprodione
○ ○
9 イプロベンホス
[IBP]
iprobenfos
○
10 イミダクロプリ
ド
imidacloprid
○
11 エスプロカルブ esprocarb
○ ○
12 エジフェンホス
[EDDP]
edifenphos
○ ○ ○
13 エトフェンプロ
ックス
etofenprox
○
14 エトリジアゾー
ル[エクロメゾ
ール]
etridiazole
○
○
15 オキシン銅[有
機銅]
oxine-copper
○ ○
16 カルバリル
[NAC]
carbaryl
○ ○
17 キャプタン
captan
○
○
18 クロルニトロフ
ェン [CNP]
chlornitrofen
○
○
19 クロルピリホス chlorpyrifos
○ ○ ○
20 クロロタロニル
[TPN]
chlorothalonil
○ ○
○
21 クロロネブ
chloroneb
○
○
22 ジクロフェンチ
オン [ECP]
dichlofenthion
○ ○
23 ジクロルボス
[DDVP]
dichlorvos
○ ○ ○ ○
24 ジチオピル
dithiopyr
○ ○
25 シマジン [CAT] simazine
○ ○
26 シメトリン
simetryn
○ ○
27 ダイアジノン
diazinon
○ ○ ○ ○
28 チウラム
thiram
○
229
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No
.
国内通称名
[登録名]
ISO登録農薬名
多成分同時測定法
個別測定方法
GC/MS
ガスクロマトグラフ法
(キャピラリーカラム用管使用)
HPLC GC/
MS
ガスクロマト
グラフ法
HPLC
(SIM又は
MC)
(FTD)
(FPD)
(ECD)
本文備考
で充てん
カラム使
用
(UV)
FTD FPD ECD UV 蛍光
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
固相
抽出
29 チオベンカルブ
[ベンチオカー
ブ]
thiobencarb
[benthiocarb]
この場合,
対象農薬
ごとの検
出器を選
択し,カラ
ム槽温度
を対象農
薬ごとに
設定する
ことを明
示してあ
る。
○ ○
○
30 テルブカルブ
[MBPMC]
terbucarb
○ ○
31 トリクロピル
(トリクロピルブ
トキシエチル)
triclopyr
○
○
32 トリクロルホン
[DEP]
trichlorfon
○
○
33 トリシクラゾー
ル
tricyclazole
○ ○
34 トルクロホスメ
チル
tolclophos-
methyl
○ ○ ○
35 ナプロパミド
napropamide
○ ○
36 ピリダフェンチ
オン
pyridaphenthion
○ ○ ○
37 ピリブチカルブ pyributicarb
○ ○
38 フェニトロチオ
ン [MEP]
fenitrothion
○ ○ ○ ○
39 フェノブカルブ
[BPMC]
fenobucarb
○ ○
40 フサライド
fthalide
○
○
41 ブタミホス
butamifos
○ ○ ○
42 ブプロフェジン buprofezin
○ ○
43 フルトラニル
flutolanil
○ ○
44 プレチラクロー
ル
pretilachlor
○ ○
45 プロピザミド
propyzamide
○ ○
○
46 プロベナゾール probenazole
○ ○
47 ブロモブチド
[ブロモチド]
bromobutide
○ ○
48 ペンシクロン
pencycuron
○ ○
49 ベンスリド
[SAP]
bensulide
○ ○
50 ペンジメタリン pendimethalin
○ ○
51 ベンフルラリン
[ベスロジン]
benfluralin
○
○
52 マラチオン(マ
ラソン)
malathion
○ ○
53 メコプロップ
[MCPP]
mecoprop
○
○
54 メタラキシル
metalaxyl
○ ○
230
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No
.
国内通称名
[登録名]
ISO登録農薬名
多成分同時測定法
個別測定方法
GC/MS
ガスクロマトグラフ法
(キャピラリーカラム用管使用)
HPLC GC/
MS
ガスクロマト
グラフ法
HPLC
(SIM又は
MC)
(FTD)
(FPD)
(ECD)
本文備考
で充てん
カラム使
用
(UV)
FTD FPD ECD UV 蛍光
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
溶媒
抽出
固相
抽出
固相
抽出
55 メチルダイムロ
ン
methyldymron
この場合,
対象農薬
ごとの検
出器を選
択し,カラ
ム槽温度
を対象農
薬ごとに
設定する
ことを明
示してあ
る。
○ ○
56 メフェナセット mefenacet
○ ○
57 メプロニル
mepronil
○ ○
58 モリネート
molinate
○ ○
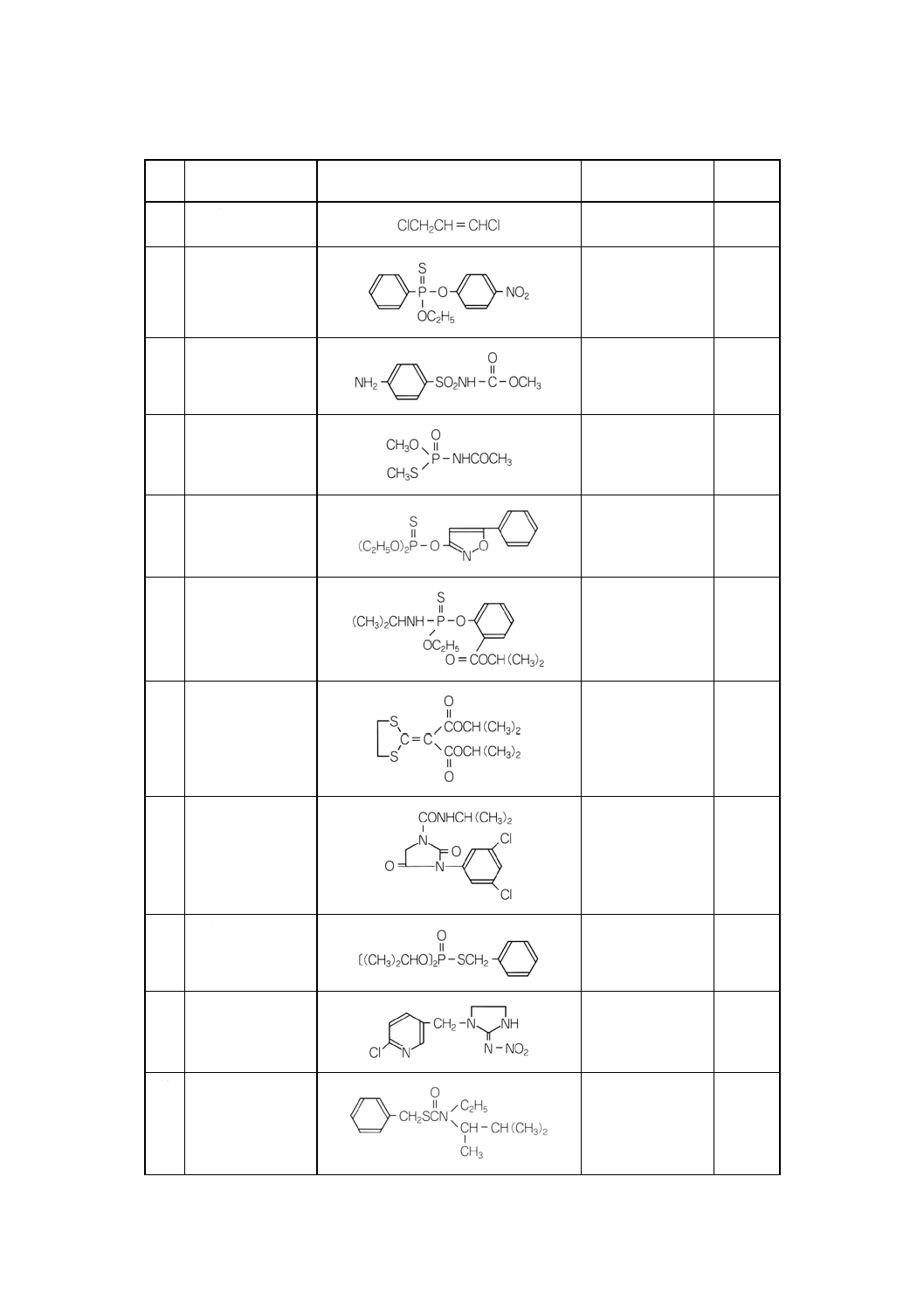
231
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表3 対象農薬の構造式,分子式及び分子量
No.
国内通称名
[登録名]
構造式
分子式
分子量
1
1, 3-ジクロロプロペン
C3H4Cl2
111.0
2
EPN
C14H14NO4PS
323.3
3
アシュラム
C8H10N2O4S
230.2
4
アセフェート
C4H10NO3PS
183.2
5
イソキサチオン
C13H16NO4PS
313.3
6
イソフェンホス
C15H24NO4PS
345.4
7
イソプロチオラン
C12H18O4S2
290.4
8
イプロジオン
C13H13Cl2N3O3
330.2
9
イプロベンホス
[IBP]
C13H21O3PS
288.4
10
イミダクロプリド
C9H10ClN5O2
255.7
11
エスプロカルブ
C15H23NOS
265.4
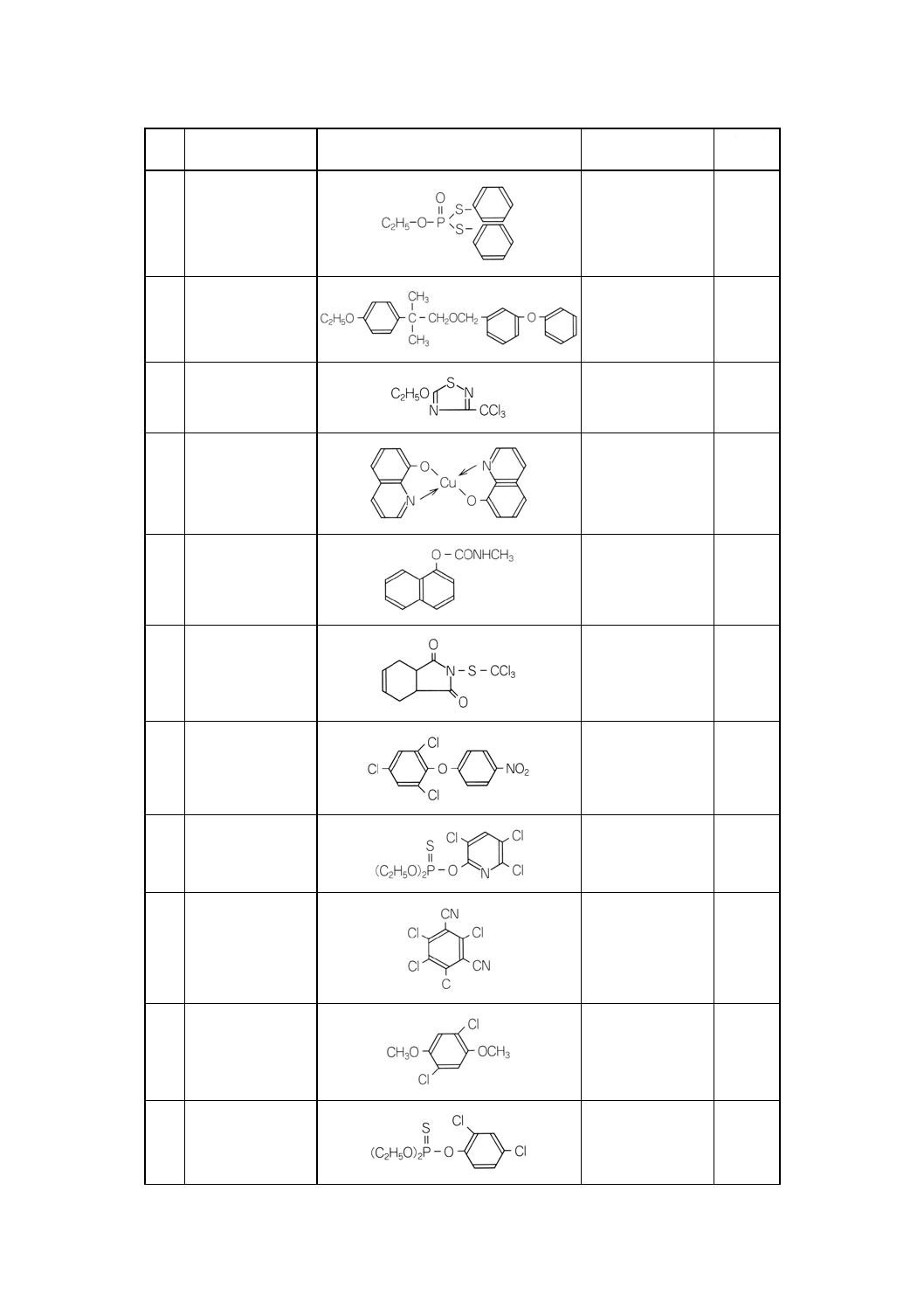
232
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名
[登録名]
構造式
分子式
分子量
12
エジフェンホス
[EDDP]
C14H15O2PS2
310.4
13
エトフェンプロックス
C25H28O3
376.5
14
エトリジアゾール
[エクロメゾール]
C5H5Cl3N2OS
247.5
15
オキシン銅[有機銅]
C18H12CuN2O2
351.9
16
カルバリル [NAC]
C12H11NO2
201.2
17
キャプタン
C9H8Cl3NO2S
300.6
18
クロルニトロフェン
[CNP]
C12H6Cl3NO3
318.6
19
クロルピリホス
C9H11Cl3NO3PS
350.6
20
クロロタロニル
[TPN]
C8Cl4N2
265.9
21
クロロネブ
C8H8Cl2O2
207.1
22
ジクロフェンチオン
[ECP]
C10H13Cl2O3PS
315.2
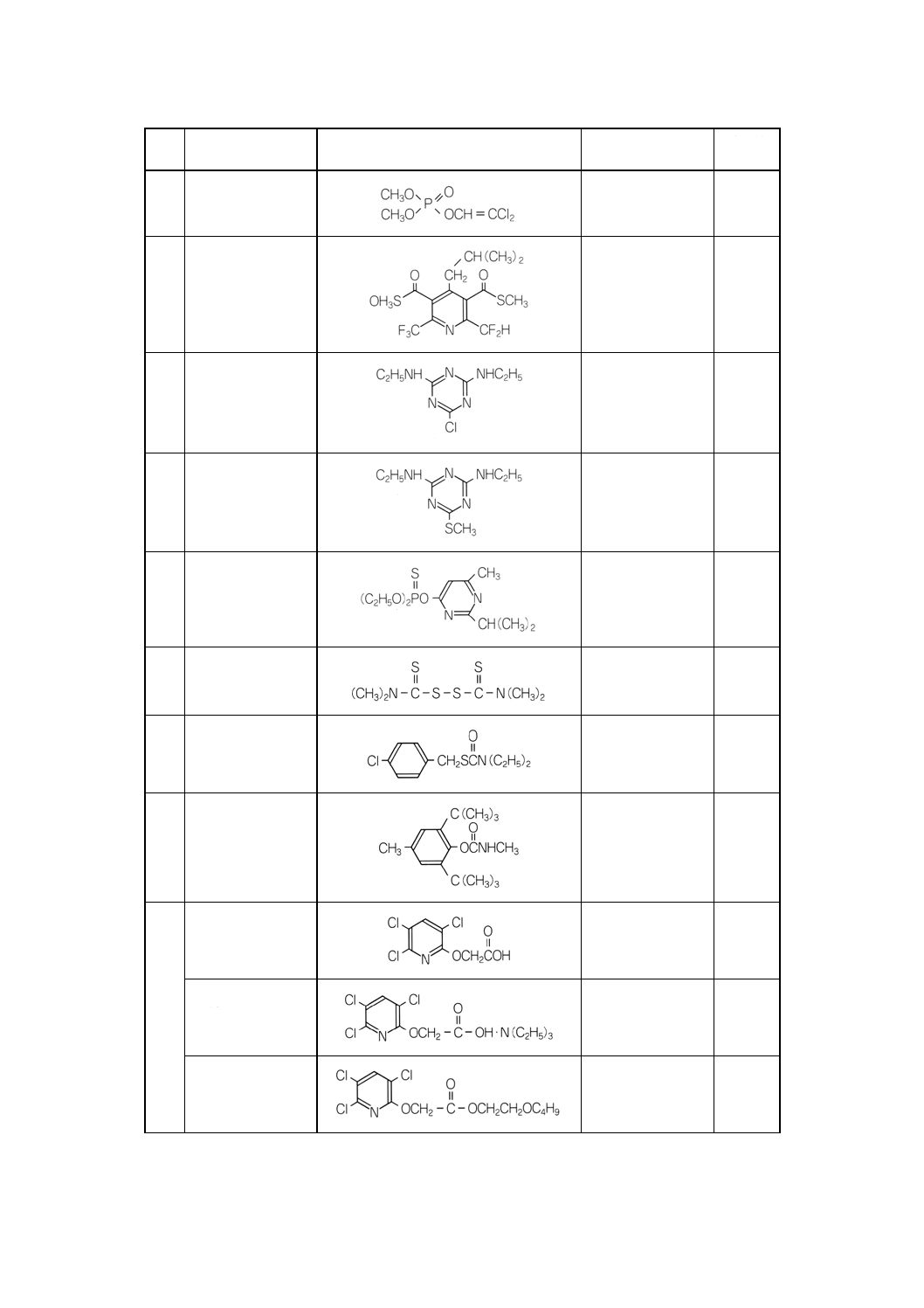
233
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名
[登録名]
構造式
分子式
分子量
23
ジクロルボス
[DDVP]
C4H7Cl2O4p
221.0
24
ジチオピル
C15H16F5NO2S2
401.4
25
シマジン [CAT]
C7H12ClN5
201.7
26
シメトリン
C8H15N5S
213.3
27
ダイアジノン
C12H21N2O3PS
304.4
28
チウラム
C6H12N2S4
240.4
29
チオベンカルブ[ベン
チオカーブ]
C12H16ClNOS
257.8
30
テルブカルブ
[MBPMC]
C17H27NO2
277.4
31
トリクロピル(有離)
C7H4Cl3NO3
256.5
トリクロピルトリエチ
ルアミン
C13H19Cl3N2O3
357.7
トリクロピルブトキシ
エチルエステル
C13H16Cl3NO4
356.6
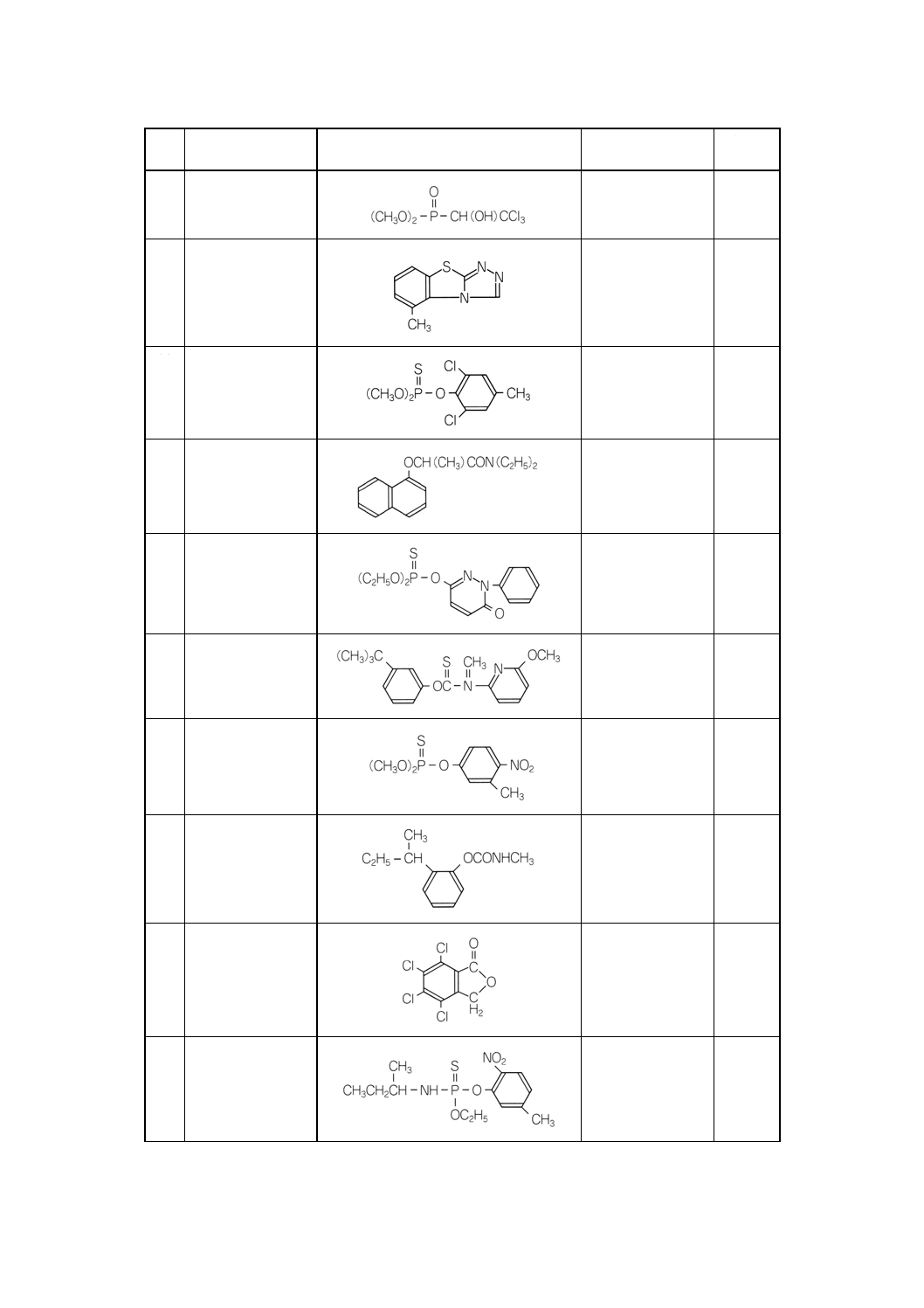
234
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名
[登録名]
構造式
分子式
分子量
32
トリクロルホン[DEP]
C4H8Cl3O4p
257.4
33
トリシクラゾール
C9H7N3S
189.2
34
トルクロホスメチル
C9H11Cl2O3PS
301.1
35
ナプロパミド
C17H21NO2
271.4
36
ピリダフェンチオン
C14H17N2O4PS
340.3
37
ピリブチカルブ
C18H22N2O2S
330.4
38
フェニトロチオン
[MEP]
C9H12NO5PS
277.2
39
フェノブカルブ
[BPMC]
C12H17NO2
207.3
40
フサライド
C8H2Cl4O2
271.9
41
ブタミホス
C13H21N2O4PS
332.4
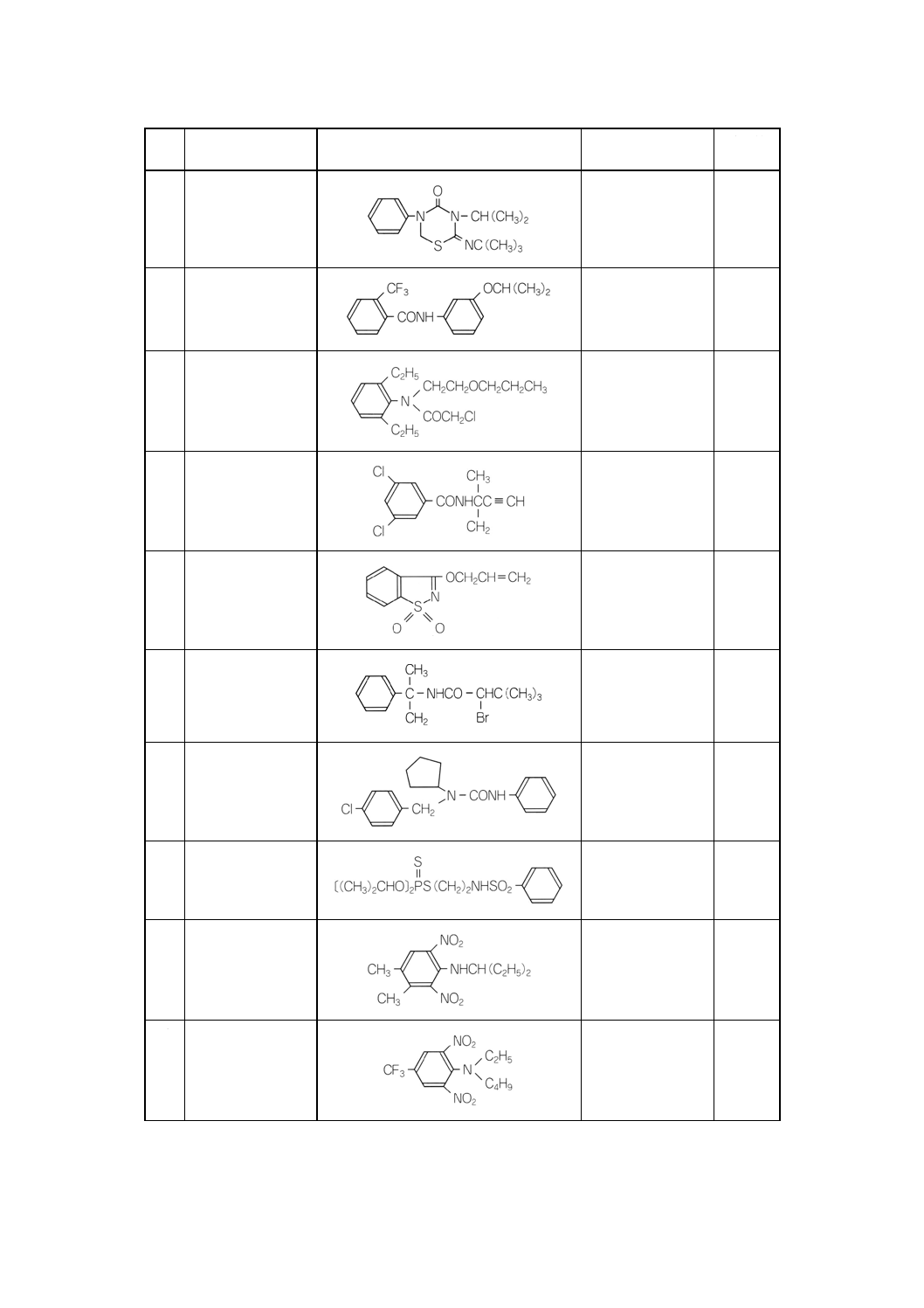
235
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名
[登録名]
構造式
分子式
分子量
42
ブプロフェジン
C16H23N3OS
305.4
43
フルトラニル
C17H16F3NO2
323.3
44
プレチラクロール
C17H26ClNO2
311.9
45
プロピザミド
C12H11Cl2NO
256.1
46
プロベナゾール
C10H9NO3S
223.3
47
ブロモブチド[ブロモ
チド]
C15H22BrNO
312.2
48
ペンシクロン
C19H21ClN2O
328.8
49
ベンスリド [SAP]
C14H24NO4PS3
397.5
50
ペンジメタリン
C13H19N3O4
281.3
51
ベンフルラリン[ベス
ロジン]
C13H16F3N3O4
335.3
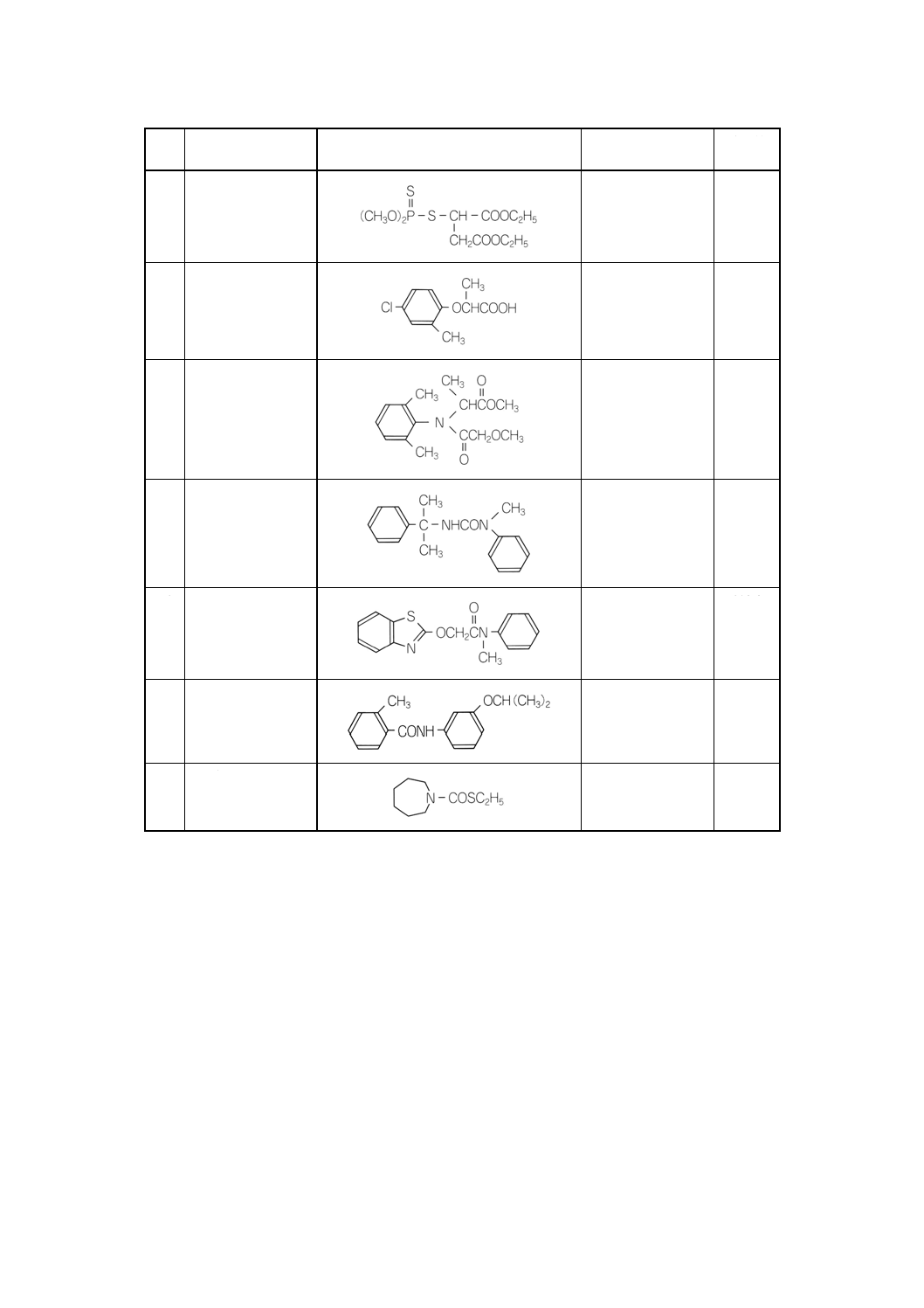
236
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
No.
国内通称名
[登録名]
構造式
分子式
分子量
52
マラチオン(マラソン)
C10H19O6PS2
330.4
53
メコプロップ
[MCPP]
C10H11ClO3
214.6
54
メタラキシル
C15H21NO4
279.3
55
メチルダイムロン
C17H20N2O
268.4
56
メフェナセット
C16H14N2O2S
298.4
57
メプロニル
C17H19NO2
269.4
58
モリネート
C9H17NOS
187.3
237
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表4 引用規格
JIS K 0050 化学分析方法通則
JIS K 0094 工業用水・工場排水の試料採取方法
JIS K 0101 工業用水試験方法
JIS K 0102 工場排水試験方法
JIS K 0114 ガスクロマトグラフ分析通則
JIS K 0123 ガスクロマトグラフ質量分析通則
JIS K 0124 高速液体クロマトグラフ分析通則
JIS K 0125 用水・排水中の揮発性有機化合物試験方法
JIS K 0211 分析化学用語(基礎部門)
JIS K 0215 分析化学用語(分析機器部門)
JIS K 0512 水素
JIS K 0557 用水・排水の試験に用いる水
JIS K 1107 高純度窒素
JIS K 8039 アセトニトリル(残留農薬・PCB試験用)(試薬)
JIS K 8040 アセトン(残留農薬・PCB試験用)(試薬)
JIS K 8105 エチレングリコール(試薬)
JIS K 8110 酢酸エチル(残留農薬試験用)(試薬)
JIS K 8117 ジクロロメタン(残留農薬試験用)(試薬)
JIS K 8150 塩化ナトリウム(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8357 ジエチルエーテル(残留農薬試験用)(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8544 硝酸アルミニウム九水和物(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8622 炭酸水素ナトリウム(試薬)
JIS K 8777 ピリジン(試薬)
JIS K 8825 ヘキサン(残留農薬・PCB試験用)(試薬)
JIS K 8858 ベンゼン(試薬)
JIS K 8866 四ほう酸ナトリウム十水和物(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8919 ヨードメタン(試薬)
JIS K 8983 硫酸銅(II)五水和物(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9007 りん酸二水素カリウム(試薬)
238
K 0128 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 9502 L (+) ‐アスコルビン酸(試薬)
JIS K 9702 ジメチルスルホキシド(試薬)
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS Z 0701 包装用シリカゲル乾燥剤
JIS K 0128改正原案作成委員会 構成表(平成11年3月現在)
氏名
所属
(委員長)
○ 梅 崎 芳 美
社団法人産業環境管理協会名誉参与
○ 土 屋 悦 輝
東京都立衛生研究所環境保健部
○ 宮 崎 正 浩
工業技術院標準部消費生活規格課
谷 重 男
通商産業省環境立地局産業施設課
後 藤 芳 一1)
通商産業省環境立地局環境政策課
野 村 明
工業技術院物質工学工業技術研究所計測化学部
○ 山 下 信 義
工業技術院資源環境技術総合研究所水圏環境保全部
○ 畑 野 浩
環境庁水質保全局水質規制課
○ 高 橋 明 宏
建設省土木研究所下水道部
○ 中 川 順 一
東京都立衛生研究所環境保健部
○ 日 野 隆 信
千葉県衛生研究所
飯 田 勝 彦
神奈川県環境科学センター水質環境部
○ 坂 本 勉
財団法人日本規格協会技術部
橋 本 進
財団法人日本規格協会技術部
○ 伊 藤 尚 美
社団法人日本分析機器工業会(株式会社島津製作所)
○ 久 本 泰 秀
社団法人日本分析機器工業会(株式会社日立製作所)
○ 池 田 久 幸
社団法人日本分析機器工業会(横河アナリティカルシステムズ
株式会社)
○ 鈴 木 弘 七
社団法人日本環境測定分析協会(新日本気象海洋株式会社)
○ 田 中 良 雄
社団法人日本環境測定分析協会(東邦化研株式会社)
○ 橋 場 常 雄
株式会社環境管理センター
岩 﨑 岩 次
社団法人日本工業用水協会
(事務局)
山 本 功
社団法人日本工業用水協会
本 郷 秀 昭
社団法人日本工業用水協会
備考 1):発足当初は,林明夫(通商産業省環境立地局環境指導課)
○は小委員兼任
(文責 土屋 悦輝)