K 0127:2013
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 イオンクロマトグラフィー概説 ··························································································· 4
5 装置の構成 ······················································································································ 5
5.1 構成 ···························································································································· 5
5.2 溶離液槽 ······················································································································ 5
5.3 送液部 ························································································································· 5
5.4 試料導入部(インジェクター) ························································································ 6
5.5 カラム部 ······················································································································ 7
5.6 検出部 ························································································································· 7
5.7 データ処理部 ················································································································ 8
5.8 附属装置 ······················································································································ 8
5.9 溶離液 ························································································································· 8
5.10 分離カラム及び充塡剤 ··································································································· 9
6 装置の設置及び操作方法 ··································································································· 10
6.1 装置の設置 ·················································································································· 10
6.2 安全についての注意事項 ································································································ 10
6.3 測定用試料溶液の準備及び前処理····················································································· 10
6.4 操作 ··························································································································· 13
7 検量線用溶液 ·················································································································· 15
8 定性分析························································································································ 19
9 定量分析························································································································ 19
9.1 定量法 ························································································································ 19
9.2 ピーク面積 ·················································································································· 19
9.3 ピーク高さ ·················································································································· 20
9.4 重複ピーク ·················································································································· 20
9.5 検量線 ························································································································ 21
9.6 標準添加法(作図法) ··································································································· 22
10 試験結果の表示 ············································································································· 23
11 データの質の管理 ·········································································································· 23
11.1 分析法バリデーション ·································································································· 23
11.2 データの質の管理 ········································································································ 23
11.3 定期的な装置性能の点検 ······························································································· 24
11.4 空試験の実施 ·············································································································· 24
K 0127:2013 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
11.5 検出下限の求め方 ········································································································ 24
11.6 定量下限の求め方 ········································································································ 25
12 個別JISでイオンクロマトグラフィーを分析法として取り入れる際の記載事項 ·························· 26
K 0127:2013
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本分析
機器工業会(JAIMA)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格
を改正すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格
である。
これによって,JIS K 0127:2001は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 0127:2013
イオンクロマトグラフィー通則
General rules for ion chromatography
1
適用範囲
この規格は,イオンクロマトグラフィーを用いて,分析種の定性及び定量分析を行う場合の通則につい
て規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0211 分析化学用語(基礎部門)
JIS K 0214 分析化学用語(クロマトグラフィー部門)
JIS K 0215 分析化学用語(分析機器部門)
JIS K 0557 用水・排水の試験に用いる水
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8019 亜硝酸ナトリウム(試薬)
JIS K 8073 安息香酸(試薬)
JIS K 8116 塩化アンモニウム(試薬)
JIS K 8121 塩化カリウム(試薬)
JIS K 8267 ぎ酸ナトリウム(試薬)
JIS K 8372 酢酸ナトリウム(試薬)
JIS K 8443 シアン化カリウム(試薬)
JIS K 8495 p-ジメチルアミノベンジリデンロダニン(試薬)
JIS K 8506 臭化カリウム(試薬)
JIS K 8528 しゅう酸ナトリウム(試薬)
JIS K 8530 臭素酸カリウム(試薬)
JIS K 8548 硝酸カリウム(試薬)
JIS K 8617 炭酸カルシウム(試薬)
JIS K 8875 マグネシウム(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 9001 チオシアン酸カリウム(試薬)
2
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 9007 りん酸二水素カリウム(試薬)
JIS K 9501 アジ化ナトリウム(試薬)
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS K 0050,JIS K 0211,JIS K 0214及びJIS K 0215によるほ
か,次による。
注記 必要に応じてJIS K 0211:2005,JIS K 0214:2006及びJIS K 0215の定義を補足,補強などの変
更を行っている。変更を行った用語には,* を付した。
3.1
イオンクロマトグラフィー*(ion chromatography)
溶離液を移動相として,イオン交換体などを固定相とした分離カラム内で試料溶液中のイオン種成分を
展開溶離させ,電気伝導度検出器,電気化学検出器,分光光度検出器,蛍光検出器などによって測定する
クロマトグラフを用いた分析方法。
3.2
イオンクロマトグラフ*(ion chromatograph)
イオンクロマトグラフィー用装置。
3.3
溶離液*(eluent)
カラムに保持されている試料中のイオン種成分を展開,溶出させるための液体。
3.4
グラジエント溶離法(gradient elution method)
溶離液の組成を連続的に変化させながら試料中のイオン種成分を展開溶離する方法。溶離液の流量を変
化させる場合もある。
3.5
試料導入部(sample injector, injection port)
測定用試料溶液を流路内に導入する機能をもつもの。
3.6
カラム槽(column oven)
カラムを収容する槽。
3.7
プレカラム*(pre-column)
測定するイオン種成分の濃縮,予備分離,異物除去などのために分離カラムの前に配置したカラム。
3.8
分離カラム*(separation column)
イオン種成分の分離を行うため,分離目的に応じた性能をもつカラム。
3.9
イオン交換体*(ion exchanger)
表面にイオン交換基をもつ充塡剤。
3.10
イオン交換容量*(ion exchange capacity)
3
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
イオン交換体の単位質量又は単位体積当たりのイオン交換基の当量。meq/g又はmeq/mLで表す。
3.11
溶出液(eluate)
移動相(溶離液)によって展開したときの分離カラムから流出する液体。
3.12
サプレッサー(suppressor)
電気伝導度検出器を用いる場合,測定するイオン種成分の検出を損なうことなくバックグラウンドとな
る電気伝導度を低減する装置。
3.13
サプレッサー法(suppressor method)
サプレッサーを用いて測定する方法。
3.14
ノンサプレッサー法*(non-suppressor method)
サプレッサーを用いずに,低電気伝導度の溶離液を用いてイオン種成分を分離し,電気伝導度検出器で
測定する方法。
3.15
再生液*(regenerant)
サプレッサーの機能を再生,又は継続的に維持するために用いる液体。除去液ともいう。
3.16
電気伝導度検出器(conductivity detector)
溶出液中の分析する対象成分の電気伝導度を測定する検出器。
3.17
電気化学検出器(electrochemical detector)
作用電極に定電位を印加し,溶出液中の分析する対象成分の電気化学反応によって流れる電流又は電量
を測定する検出器。
3.18
紫外可視吸光光度検出器*(ultraviolet-visible absorption detector)
紫外可視光線領域で吸収される分析種の吸光度を測定する検出器。
3.19
蛍光検出器*(fluorometric detector)
溶出液中の分析する対象成分の蛍光を測定する検出器。
3.20
廃液槽(waste reservoir)
装置から排出される液体を貯留するもの。
3.21
標準液*(standard solution)
標準物質,標準試薬などを用いて,測定するイオン種成分の濃度を1 000 mg/Lなどに調製した液。
3.22
希釈標準液(diluted standard solution)
標準液を所定の濃度に希釈した液。
4
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.23
混合希釈標準液(mixed diluted standard solution)
複数の標準液又は希釈標準液を混合して各成分を所定の濃度に調製した液。
3.24
分離度*(resolution)
二つのイオンのピークの相互分離を示す指標。
3.25
感度*(sensitivity, sensitiveness)
イオン種成分を測定するとき,検出定量できる量又は濃度の変化の最小量(値)。
3.26
検量線*(working curve)
測定するイオン種成分の量又は濃度と測定値との関係を表した線。
3.27
絶対検量線法*(external standard method)
測定するイオン種成分の量又は濃度と,ピーク面積又はピーク高さとの関係を示す検量線を作成し,試
料を同一条件の下で測定したピーク面積又はピーク高さから作成した検量線によって量又は濃度を求める
方法。外標準法ともいう。
3.28
空試験*(blank test)
空試験値を求める試験。
3.29
空試験値*(blank value)
測定するイオン種成分を含まない溶液を用いて,試料を用いたときと同様な操作をして求めた値。
3.30
検出下限*(minimum limit of detection, minimum limit of identification)
測定するイオン種成分を検出できる最小量(値)又は最小濃度。
3.31
定量下限*(minimum limit of determination)
測定するイオン種成分を定量できる最小量(値)又は最小濃度。
3.32
固定相*(stationary phase)
イオンクロマトグラフィーが行われる場の要素の一つで,移動相と平衡状態にあり分析種と相互作用す
る相。
3.33
移動相*(mobile phase)
イオンクロマトグラフィーが行われる場の要素の一つで,固定相に接して流れる流体。
4
イオンクロマトグラフィー概説
イオンクロマトグラフィーは,溶離液を移動相にして試料を導入し,固定相との相互作用(イオン交換
など)の差を利用してイオン種成分を測定する方法で,高速液体クロマトグラフィーの一種である。イオ
5
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ン種は,主にイオン交換,イオン排除,逆相イオンペアなどの分離機構によって分離される。溶液中の無
機イオン,有機酸,低分子量アミン,ハロゲンなどの測定に利用される。大気・水・土壌などの環境モニ
タリング,食品の管理,飲料水,排水などの水質の管理,排ガスの管理,生体試料の解析,樹脂製品中の
臭素系難燃剤など無機・有機材料に含まれるハロゲン及び硫黄元素の定量など,多くの分野で使用される。
試料は,試料溶液の一定量を計量してカラムへ導入する。
カラムには,イオン交換体などを,合成樹脂,金属などの管に充塡したものが用いられる。
検出器には,電気伝導度検出器,紫外可視吸光光度検出器,電気化学検出器,蛍光検出器,質量分析計,
ICP発光分光分析計などが用いられる。測定するイオン種に対する感度及び/又は選択性を高めるための
検出前処理部を設けることができる。
微量に含まれている不純物の測定では,溶媒抽出,固相抽出などの前処理,質量分析装置及び/又は誘
導体化試薬による高感度化を図る必要がある。
5
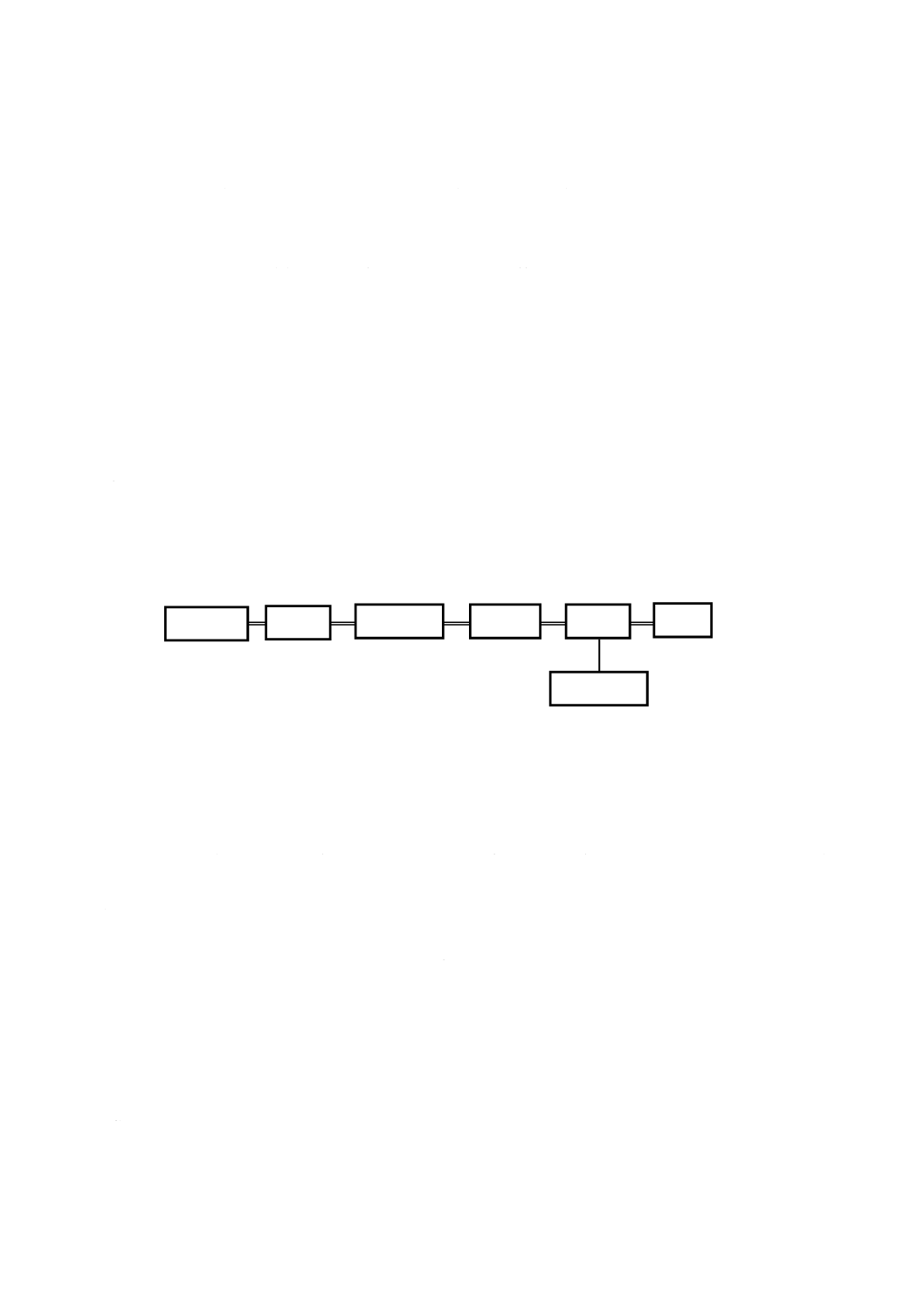
装置の構成
5.1
構成
イオンクロマトグラフは,溶離液槽,送液部,試料導入部,カラム部,検出部,データ処理部及び廃液
槽で構成する。必要があれば,グラジエント装置,脱気装置などの附属装置を備えてもよい。基本構成の
一例を図1に示す。
図1−イオンクロマトグラフの基本構成の一例
5.2
溶離液槽
溶離液槽の材質は,溶離液によって侵されたり,溶離液を汚染したりすることのないものを用いる。
5.3
送液部
脱気装置及び送液ポンプで構成される。グラジエント溶離を行う場合は,グラジエント機能を備えた送
液ポンプを用いる。
a) 脱気装置 溶離液に溶解している空気を連続的に取り除き,装置内で温度変化及び圧力変化に伴い発
生する気泡のトラブルを未然に防ぎ,安定した流量及びバックグラウンドが得られるようにするため
に用いられる。オフラインで脱気した水で調製した溶離液を用いても問題ない場合は,脱気装置はな
くてもよい。
b) 送液ポンプ 溶離液を正確かつ精密な流量でカラムに送液するために,次の条件を満たすことが望ま
しい。
1) 定流量精度が高いものとする。
2) 必要な送液圧力が得られるものとする。
3) 脈流が小さいものとする。
4) 流量の調節が可能なものとする。
溶離液槽
送液部
試料導入部
カラム部
廃液槽
検出部
データ処理部
6
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) 接液部の材質が溶離液によって侵されたり,溶離液を汚染したりすることのないものとする。
6) 溶離液の交換が容易であるものとする。
c) グラジエント装置 複数の溶離液を用い,時間的にその組成を変化させるための制御部と混合溶液を
均一にするためのミキサー部とから成る。送液ポンプの前にミキサー部がある低圧グラジエント装置
及び複数の送液ポンプを用いて送液ポンプの後にミキサー部を設ける高圧グラジエント装置がある。
いずれの装置も設定できる混合比の範囲が広く,濃度が精確であることが望ましい。
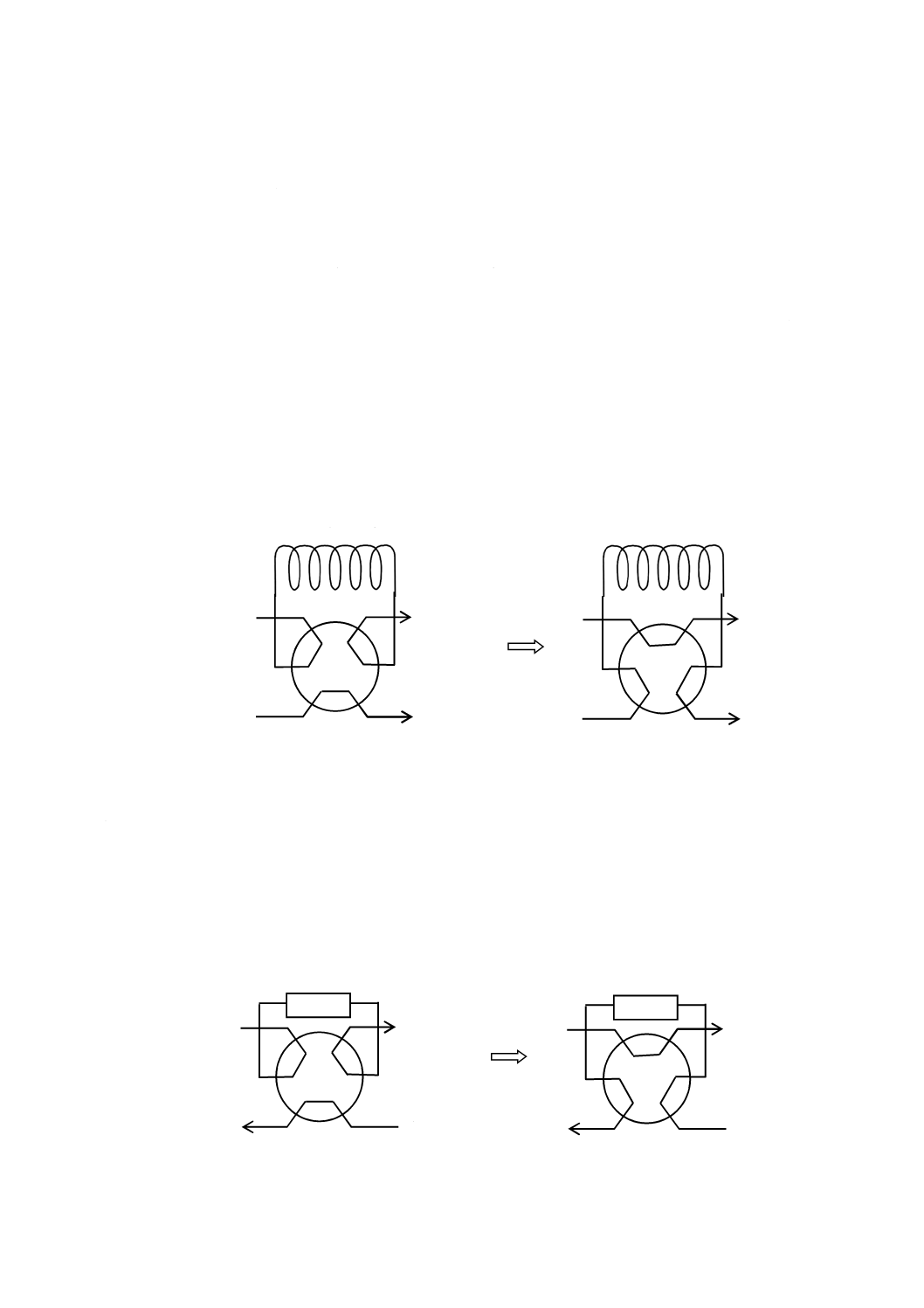
5.4
試料導入部(インジェクター)
試料導入部は,次のいずれかの方法によって試料溶液の一定量を再現性よく流路内に導入する。多数の
試料溶液を自動的に導入するために,自動試料導入装置を用いてもよい。
a) 方法1 被検試料溶液をためておくためのサンプルループを備え,そのループ内に被検試料溶液を注
入した後,6方バルブなどを用いた溶離液流路の切換えによってループ内の被検試料溶液を分離カラ
ムへの流路内に導入する。この方法による試料導入部の一例を図2に記載する。ループ内に試料を全
量満たす方法及びループ内の一部に一定量を導入する方法がある。
図2−試料導入部の一例
b) 方法2 イオン種成分を捕集濃縮するための濃縮カラムを備え,その濃縮カラムに一定量の被検試料
溶液を導入した後,6方バルブなどを用いた溶離液流路の切換えによって,濃縮カラム内に濃縮され
たイオン種成分を分離カラムへの流路内に導入する。この方法による試料導入部の一例を図3に記載
する。
図3−方法2の場合の試料導入部の一例
サンプルループ
試料溶液導入時の状態
分析時の状態
排出
試料導入
ポンプから
分離カラムへ
バルブ
試料溶液濃縮時の状態
分析時の状態
バルブ
濃縮カラム
排出
試料導入
ポンプから
分離カラムへ
7
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.5
カラム部
カラム部は,カラム及びカラム槽からなる。分析目的によっては,分析性能に支障がなければ,カラム
槽を設けなくてもよい。
a) カラム カラムにはプレカラム1) 及び分離カラムがあり,分析目的に応じた性能をもつものとする。
注1) プレカラムは,カラムの保護を目的として,必要に応じて用いるものとし,必ずしも設けな
くてもよい。
b) カラム槽 カラム槽は,必要な長さのカラムを収容できる内容積をもつもの。任意の一定温度に保つ
ための温度制御機能をもつ恒温槽を兼ね備えたものもある。
5.6
検出部
検出部は,カラムからの溶出液を微量フローセルに導き,溶離液をバックグラウンドとしイオン種成分
の濃度又は量を種々の原理によって検出するものである。検出部は,検出器及び溶出液の前処理を行う部
分からなる。分析目的によっては,分析性能に支障がなければ,次に示す溶出液の前処理を行う部分を設
けなくてもよい。
a) 検出器 検出器は,溶離液及び/又は被検試料中の成分によって侵されない材質をもつもので,被検
イオン種成分を検出するのに適した次のものなどを用いる。
1) 電気伝導度検出器
2) 紫外可視吸光光度検出器
3) 電気化学検出器
4) 蛍光検出器
5) 質量分析計
6) ICP発光分光分析計
7) ICP質量分析計
b) 溶出液の前処理を行う部分 被検イオン種成分に対する検出器の感度又は選択性を高めるために,次
の装置を検出器の前に設ける。
1) サプレッサー 電気伝導度検出器を用いる場合に,サプレッサーはイオン交換部位(膜又は樹脂)
を介したイオン交換によって溶離液の電気伝導度を低下し,測定イオンの対イオンをより伝導度の
高いイオンに交換することでSN(シグナルノイズ)比を改善し,測定感度を高める。陰イオン分
析には陽イオン交換膜又は樹脂を用い,溶離液及び測定イオンの陽イオン部分をH+に交換する。
一方,陽イオン分析には陰イオン交換膜又は樹脂を用い,溶離液及び測定イオンの陰イオン部分を
OH−に交換する。イオン交換部位の形状によって,表1に示す膜形,カラム形,ゲル形,ファイバ
ー形又はこれらを組み合わせて用いたものがある。サプレッサーのイオン交換部位を連続使用する
場合は再生が必要であり,その再生方法として,酸性水溶液(陰イオン測定時)又はアルカリ性水
溶液(陽イオン測定時)を用いる化学的再生方式及び水又は検出器からの排出液を電気分解してH+
又はOH−を供給する電気的再生方式がある。

8
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−サプレッサーの構造及びサプレッション原理
種類
構造と原理
膜形
2枚のイオン交換膜間に分離カラムからの溶出液を通過させ,サプレッションを
行う。膜の外側に再生液を供給することで,測定しながら再生ができる。イオン
交換膜は,電気的又は化学的に再生される。
カラム形
イオン交換樹脂を充塡したサプレッサーカラムに分離カラムからの溶出液を通
過させ,サプレッションを行う。定期的にサプレッサーカラムの再生が必要とな
る。複数のサプレッサーカラムを再生しながら交互に使用すれば,連続的な効果
が得られる。サプレッサーカラムを再生する場合は電気的又は化学的に行う。
ゲル形
少量のイオン交換樹脂(ゲル)を流路内に充塡し分離カラムからの溶出液を通過
させ,サプレッションを行う。イオン交換樹脂は一定頻度で交換し,再生は行わ
ない。
ファイバー形
イオン交換ファイバーの内側に分離カラムからの溶出液を通過させ,サプレッシ
ョンを行う。ファイバーの外側に再生液を供給することで,測定しながら再生が
できる。イオン交換ファイバーは化学的に再生される。
2) 誘導体化装置 誘導体化装置は,カラム溶出液に誘導体化試薬溶液,pH調整液などを混合し,必要
に応じて加熱などの処理をして被検イオン種成分を化学的に変化させ,検出器による測定を容易に
するものである。
5.7
データ処理部
データ処理部は次に示す,データ処理装置又は記録計とする。
a) データ処理装置 データ処理装置は,検出器からの信号を処理し,クロマトグラム,保持時間,ピー
ク面積,ピーク高さ,定量値などを記録又は表示できるもの。
b) 記録計 記録計は,検出器からの信号を経時的に記録できるもの。
5.8
附属装置
イオンクロマトグラフは,必要であれば,次の附属装置などを備えてもよい。
a) 自動試料導入装置
b) 溶離液・カラム切換えバルブ
5.9
溶離液
溶離液は,被検イオン種成分,用いる分離カラム及び分離方法(分離機構),検出器,カラム分離後の処
理方法などによって異なるため,カラムの取扱説明書及び各種データを参考にして,適切な溶離液を選択
することが重要である。溶離液は,次による。
a) 基本的な必要事項 溶離液は,次の条件を満たさなければならない。
1) 充塡剤に対して不活性である。
2) 測定するイオン種成分の分離に適切である。
3) 検出器での検出に適している。
4) サプレッサー及び誘導体化装置を用いる場合はその機能を十分に満足しなければならない。
5) 測定するイオン種成分を不純物として含まない。
6) 長時間化学的に安定である。
b) 溶離液の調製に用いる水 溶離液に用いる水は,逆浸透法,蒸留法,イオン交換法,紫外線照射,ろ
過などを組み合わせた方法によって精製した水で,分析に干渉しない水質のものとする。水質は,比
抵抗値(又は電気伝導度),全有機炭素(TOC)などを指標とし評価する。使用直後から容器・環境か

9
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ら汚染を受け劣化するので,分析に影響を与えない材質の容器を選定し,適切な方法で洗浄後使用す
る。分析目的に応じてJIS K 0557に規定するA2〜A4又はこれと同等以上の水を使用する。
c) 溶離液の調製 溶離液はあらかじめ脱気をした水を用いて調製する。保存は細菌又は藻類の生育を避
けるために,冷暗所において貯蔵し,2〜3日ごとに更新する。溶離液の一例を次に示す。
1) サプレッサー法による無機陰イオンの分析 溶離液には通常炭酸塩緩衝液,水酸化カリウム,ほう
酸塩緩衝液などの塩基性溶液を用いる。
2) ノンサプレッサー法による無機陰イオンの分析 溶離液にはモル電気伝導率が比較的低いフタル酸,
p-ヒドロキシ安息香酸などの溶液を用いる。
3) アルカリ金属,アンモニウム,アルカリ土類金属イオンの分析 溶離液には通常強酸(硝酸,硫酸
など),有機酸(メタンスルホン酸,くえん酸,しゅう酸など)などを用いる。
4) イオン排除法による有機酸の分析 溶離液には有機酸の解離を抑制するために,りん酸,過塩素酸,
硫酸などの検出の妨害とならない酸を用いる。
5.10 分離カラム及び充塡剤
分離カラム及び充塡剤は,次による。
a) 分離カラム 分離カラムは,内径0.2〜9 mm,長さ10〜500 mmの不活性な合成樹脂製又はステンレ
ス鋼などの管に充塡剤を詰めたものとする。
b) 充塡剤 イオン種成分は,主に三つの分離手法(イオン交換,イオン排除及びイオン対)を単独又は
複合的に作用させて分離する。イオン交換及びイオン排除に用いられるカラムの充塡剤には,ポリス
チレン,ポリメタクリレート,ポリビニルアルコールなどの有機高分子及びシリカゲルの基材などに
イオン交換基が導入されたものを用いる。イオン対の場合には,ODS(オクタデシルシリル基で修飾
したシリカゲル)のような疎水性充塡剤などを用いる。また,陰イオン及び陽イオンを同時に分離す
る場合には,イオン排除による陰イオンの分離及び陽イオン交換による陽イオンの分離をカラム内で
同時に進行させる方法を用いる。充塡剤の基材,官能基,孔径などは選択性の発現に影響するので,
試料の特性に応じて用いる溶離液及び適切な充塡剤のカラムを選ぶ。表2に分離手法及び主な測定イ
オン種成分例を示す。
表2−主な分離手法及び測定イオン種成分の例
分離手法
充塡剤官能
基
測定イオン種
陽イオン交換
−SO3−
Li+,Na+,K+,NH4+,Rb+,Cs+,Ca2+,Mg2+,Sr2+,Ba2+,低級アミン類,遷
移金属類a)
陽イオン交換
−COO−
Li+,Na+,K+,NH4+,Rb+,Cs+,Ca2+,Mg2+,Sr2+,Ba2+,低級アミン類
陰イオン交換
−N+R3
F−,Cl−,NO2−,Br−,NO3−,SO42−,PO43−,I−,S2O32−,SCN−,CO32−,BrO3
−,ClO4−,ClO3−,ClO2−,有機酸類,糖類,アミノ酸類
イオン排除
−SO3H
有機酸類,CN−,NO2−,PO43−,けい酸,亜ひ酸,ひ酸,炭酸
イオン排除
−COOH
有機酸類,F−,Cl−,NO3−,SO42−,I−,NO2−,PO43−,けい酸,炭酸,硫化物,
ほう酸
イオン排除・
イオン交換
−COOH
(−COO−)
有機酸類,F−,Cl−,NO3−,SO42−,I−,NO2−,PO43−,けい酸,炭酸,硫化物,
ほう酸,Li,Na+,K+,NH4+,Ca2+,Mg2+
イオン対
ODSなど
I−,SCN−,有機酸類,H+,OH−,HCO3−,F−,Cl−,NO2−,Br−,NO3−,SO42−,
ClO4−,Na+,K+,NH4+,Ca2+,Mg2+,Ba2+
注a) 遷移金属はPAR[4-(2-ピリジルアゾ)レゾルシノール]によるポストカラム誘導体化法によって発色させ,
可視域で吸光光度検出することができる。
10
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6
装置の設置及び操作方法
6.1
装置の設置
附属装置を含め,設置場所は,通常,次の条件を備えた室内が望ましい。
a) 結露せず,温度,湿度及び気圧が装置に定められた範囲内にあり,急激な変化を生じないところ。
b) 振動がなく,直射日光が当たらないところ。
c) 空調器具などからの風が直接当たらないところ。
d) 火気が近くにないところ。
e) 腐食性ガス及びほこりが少なく,換気のよいところ。
f)
強い磁界を発生する装置(電気溶接機,高周波電気炉,柱上変圧器など)に近接していないところ。
g) 供給電源は,装置の仕様に指定された電圧,電気容量及び周波数の下で,電圧変動は10 %以下,周波
数の変動がないところ。
h) 接地抵抗100 Ω以下の接地点があるところ。
i)
実験台は,装置の総質量に十分耐えられる強度をもつ。
j)
装置の放熱及びメンテナンスを考慮した設置スペースを確保する。
k) 有機溶媒などによる汚染を防ぐための室内換気設備又は強制排気設備を必要に応じて設ける。
6.2
安全についての注意事項
安全のために,次の事項に十分注意しなければならない。
a) 試料及び分析に使用する薬品の取扱いは,毒性又は有害性に十分注意して行い,それらの廃棄につい
ても安全化,無害化などに配慮する。毒物,劇物の取扱いについては,“毒物及び劇物取締法”の諸規
定に従う。毒性,有害性のある薬品の取扱いには,必要に応じて保護具(保護めがね,ゴム手袋,防
毒マスクなど)を着用する。
b) 装置を接地点に接地する。
c) イオンクロマトグラフの運転に先立ち,配管の接続部,流路などからの液漏れ及びガス漏れがないか
十分に点検する。
d) 装置内部に直接触れると感電のおそれがあるので,装置の点検及び修理は,通常電源プラグを抜いた
状態で行う。
警告 装置にリチウム電池が内蔵されている場合は,取扱い方を誤ると破裂する危険があるため注
意する。
e) 装置が他の機器に電磁波障害を与えたり,他の機器から電磁波妨害を受ける場合があるので注意する。
f)
生物学的試料[血清,血しょう(漿),組織,尿など]を素手で取り扱うと,感染のおそれがあるので,
必ずゴム手袋などを使用して慎重に取り扱う。試料の皮膚への直接接触,ピペッティングによる誤飲,
針刺しなどに注意し,必要に応じて保護具(保護めがね,ゴム手袋,マスクなど)を着用する。
g) 有機溶媒を使用する場合には,発火を防ぐために廃液チューブ及び廃液容器の静電気対策を行う。
h) 感電,火災,故障の原因となるため,ぬれた手で電源プラグの抜き差しを行わない。
6.3
測定用試料溶液の準備及び前処理
6.3.1
一般事項
イオンクロマトグラフィーにおいては,あらかじめその分析目的によって,試料に合わせた試料採取方
法を選択する必要がある。採取した試料は,測定するイオン種の定性又は定量を精度よく行うために,環
境からの汚染,成分の経時変化などが起こらないように試料溶液の保存及び調製に十分注意する。また,
採取した試料は,そのままイオンクロマトグラフに導入できる場合,又は導入前に前処理操作が必要な場
11
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
合があり,分析目的に応じて選択する必要がある。
6.3.2
試料採取
分析目的,試料の性質及び試験項目に最も適した方法で,試料母集団を代表できる又は各試料の特性の
差を明らかにできるような試料採取方法を選択する。試料によっては,採取の時期,時刻によって量的な
変動を生じる場合があるので,分析対象の個体差,内部での不均一性を考慮した採取を行う。個別に規格
がある場合はそれに準じる。また,外的要因,経時変化などに対する変質を防ぐため,なるべく速やかに
前処理操作,イオンクロマトグラフへの試料導入を行う。試料は生物学的,化学的,又は物理的変化を保
存中に受ける可能性がある。これらの変性を防止できない分析対象成分の場合は,採取後直ちに測定を行
わなくてはならない。
なお,液体試料の採取容器は,試料成分の吸着が生じず,妨害成分の溶出のないものを用いる。一般的
には,ガラス製よりもポリエチレン又は四ふっ化エチレン,ポリプロピレンなどの樹脂製容器を使用した
方がよい。試料採取は,次による。
a) 環境試料 環境試料の分析値は,採取条件及び採取方法によって影響を受けるため,事前に十分に考
慮した上で試料を採取する。また,試料の採取の際には,採取場所,日時,気象条件などを記録する。
1) 水試料 試料の種類に応じて採取方法を工夫する。雨水は少ない雨量に対応した容器の形状,回収
までの蒸発又は変質防止,不溶物の混入に対し配慮し,排水は油分,有機溶媒,界面活性剤などが
含まれる可能性を考慮し,必要に応じて対策を取る。不溶物はろ過によって除去する。
2) 降下物 湿性降下物と乾性降下物とを含めた一括採取法(バルク方式)及び湿性降下物だけを採取
する方式がある。一括採取法の場合は常時開放形ろ過式採取装置などによって,湿性降下物だけを
採取する場合は降水時開放形の捕集装置などによって試料を採取する。
3) 底質・土壌 粉砕後,均一に混合するか,又はふるい分けによって粒度分画を行い,一定時間溶媒
に浸せきしたり,超音波をかけて抽出する。
なお,必要に応じて抽出前に乾燥,脱水などの操作を行う。
4) 大気試料 気象条件(気温,湿度,風速,風向など),採取時刻などを十分に考慮し事前に調査した
上で,イオン種を吸収液などによって吸着し採取する。
5) 浮遊物 ろ過剤(セルロースエステル,ポリカーボネート,石英繊維及びテフロン)で捕集後,ろ
過剤を一定時間溶媒に浸せきしたり,超音波をかけて抽出する。
6) 排ガス試料 試料ガスの採取法としては,吸収瓶又は真空フラスコによる捕集があり,測定対象ご
とに定められた吸収液(過酸化水素,水,ほう酸,水酸化ナトリウムなど)を用いる。
b) 食品及び生物学的試料 食品試料及び生物学的試料は,種々の成分を含有する複雑な試料であり,カ
ラムを劣化させる成分が多く含まれている。特に,タンパク質及び脂質は,前処理によって十分に取
り除く必要がある。生物学的試料の全ての検体は感染性をもつものとみなして慎重に取り扱わなけれ
ばならない。
c) 医薬品 医薬品主成分の測定は,目的成分の濃度が高いため,希釈,ろ過などの前処理を行い測定す
る。微量に含まれている不純物の測定では,溶媒抽出,固相抽出などの前処理,質量分析装置及び誘
導体化試薬の使用による高感度化を図る必要がある。
6.3.3
試料保存
採取した試料の測定を直ちに行えない場合は,0〜10 ℃の暗所に保存し,できるだけ早く測定を行う。
0 ℃付近に保存する場合には,凍結させないようにする。また,試験項目に保存方法が示されている場合
には,それに従う。
12
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試料を保存する必要がある場合は,事前にその保存及び運搬方法で試料の変質に問題がないことを確認
する。変質が懸念される場合は,その防止策を施す。試料採取及び保存に使用する容器及び器具は,試料
の吸着,又は試料中へ溶出する成分を含まない材質のものを用い,気密性が高く丈夫な容器が望ましい。
また,保存容器には必ず必要事項を記入したラベルを貼るようにする。データの信頼性を確保するため,
二重測定が義務づけられている場合には,同一試料を同時期に最低2点以上の試料採取を行う必要がある。
6.3.4
試料溶液の前処理
イオンクロマトグラフに導入する前に,場合によって分析目的,試料の性質,試験項目などに最も適し
た方法で,試料の前処理操作を行う。前処理操作は,特異性,精度,感度などの向上,測定妨害物質の除
去,カラム及び分析機器の保護並びに劣化の防止,測定操作,手順の簡易化,イオン種の安定化などを目
的として行われる。試料溶液の前処理は,次による。
a) 試料溶液の希釈又は濃縮 試料溶液は,測定するイオン種があらかじめ適正な濃度範囲内となるよう
に希釈又は濃縮(溶媒抽出,固相抽出など)し,測定用試料溶液を調製してからカラムに導入する。
測定用試料溶液は,溶離液(移動相)に溶けることが必要である。
b) 試料溶液中妨害成分と目的成分との分離 試料溶液中に固形成分,カラムに悪影響を与える成分又は
定性・定量を妨害する成分がある場合,次の方法で除去する。
1) ろ過膜を用いる方法 試料溶液のろ過は,孔径0.45 μm以下のフィルターを用いる。採取した試料
溶液に浮遊物がある場合及び懸濁状態の場合には,ろ過による除去が必要となる。孔径0.45 μmで
のろ過が難しい場合には,あらかじめ孔径の大きなろ過膜を用いてろ過又は遠心分離などの処理後,
孔径0.45 μmでろ過してもよい。また,試料溶液中の高分子化合物及びタンパク質の除去に限外ろ
過膜を用いることができる。限外ろ過膜には,シリンジなどによる加圧でろ過できるもの及び遠心
分離機に装着して使用できるカートリッジタイプがある。
2) 樹脂などを用いる方法 粒子状の無機物質又は樹脂を充塡した小さなカラム(カートリッジ)を用
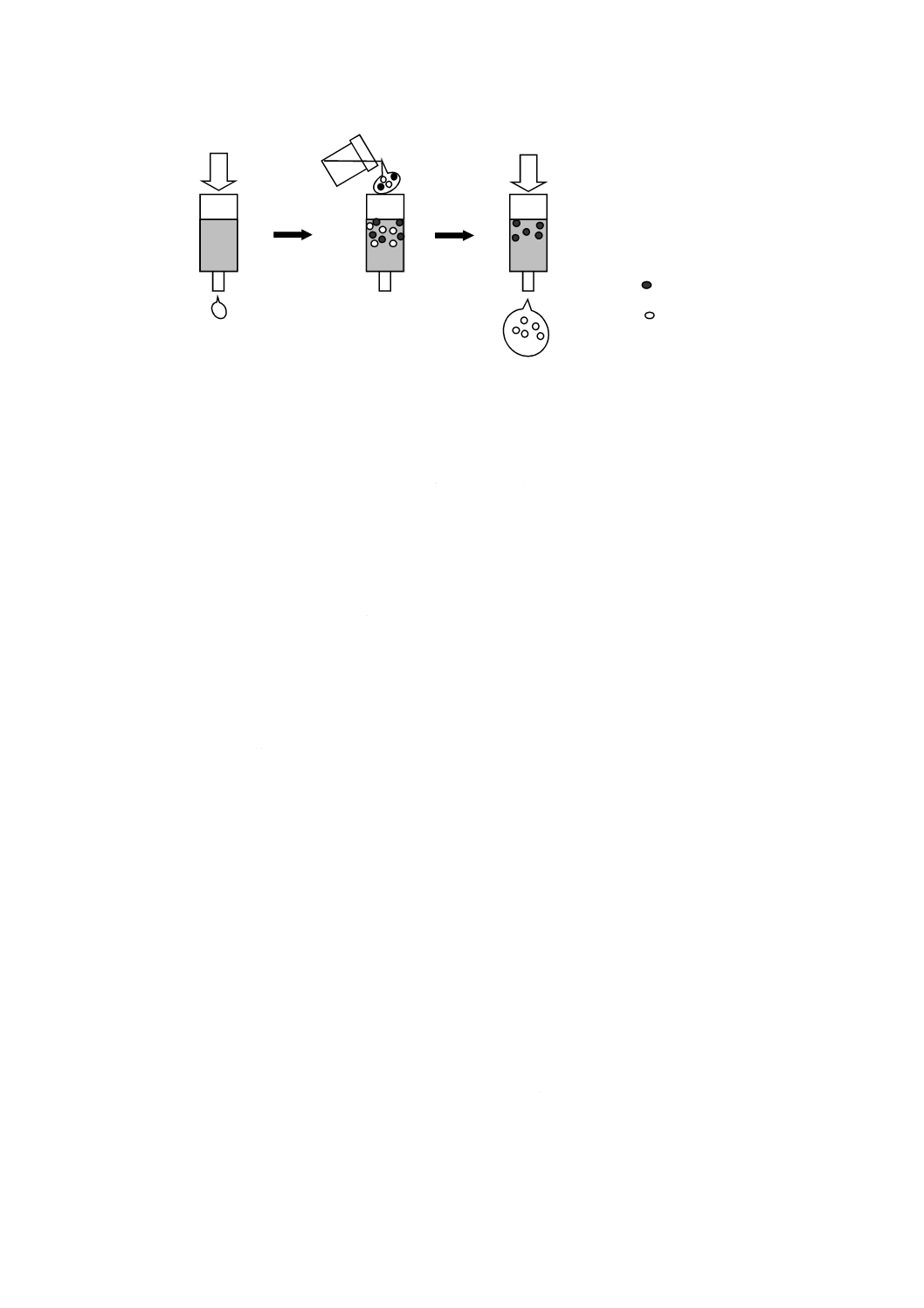
い,特定のイオン種及び疎水性物質を除去するのに有効である。樹脂などを用いる前処理法の例を
表3に,妨害成分を吸着させ,目的成分だけ溶出する固相抽出法の一例を図4に示す。イオン交換
基をもつ機能膜又は透析膜をカラム(カートリッジ)の代わりに用いることもできる。
表3−樹脂などを用いる前処理法の例
種類
目的
Ag+形
ハロゲン化物イオンの除去
H+形
アルカリ金属イオン,アルカリ土類金属イオンなどの除去
アルカリ性試料溶液の中和
OH−形
強酸性イオンの除去
酸性試料溶液の中和
Ba2+形
硫酸イオンの除去
疎水性樹脂
疎水性官能基結合形
疎水性物質の除去
色素などの除去
キレート樹脂
遷移金属イオンの除去
13
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4−目的成分だけ溶出する固相抽出法の一例
3) 液−液抽出を用いる方法 試料溶液と混ざり合わない有機溶媒を用いて,疎水性の高い妨害成分を
抽出除去する方法である。有機溶媒としては,ヘキサン,酢酸エチル,ジエチルエーテル,クロロ
ホルム,ジクロロメタンなどを用いる。妨害成分を有機溶媒相に移動させた後,有機溶媒を取り除
くことによって,分析対象成分溶液を得る。水溶液に不溶な疎水性の高い成分に含まれる無機イオ
ンの測定に有効な手法である。また,水及び溶離液と混ざり合わない試料溶液中のイオン種成分に
ついては,水抽出を行い分析対象成分溶液を得る。
6.3.5
有機化合物の燃焼前処理
有機化合物を酸素フラスコ燃焼法,酸素ボンベ燃焼法又は石英管燃焼法によって燃焼分解し,発生ガス
を吸収液に吸収させ,イオンクロマトグラフ法によって定量する方法である。ふっ素,塩素,臭素及びよ
う素の4種ハロゲン及び硫黄が測定対象元素となる。
燃焼前処理時の検量線用溶液には,箇条7に規定する容量分析用標準物質又は各種陰イオン標準液を用
いて調製する方法(無機検量線法)及びハロゲン及び硫黄を含む有機標準物質又は標準試料を段階的に燃
焼させて検量線を作成する方法(有機検量線法)がある。測定目的によって,いずれかの方法を用いて検
量線を作成し定量計算を行う。装置及び燃焼条件の評価として,ハロゲン及び硫黄濃度が既知である認証
物質(例,EU認証のBCR-680,BCR681,ERM-EC681K及びEC680K)を用いて,回収率,再現性などを
指標として燃焼条件を前もって確認しておくことが望ましい。
吸収液には,水,薄い過酸化水素水,薄いヒドラジン水及びアルカリ溶液などがあり,臭素及びよう素
の定量には還元剤の添加が,硫黄の定量には酸化剤の添加が必要である。
注記 有機物の燃焼前処理については,日本薬局方(第16改正),個々のJIS,JEITA規格などを参
照するとよい。
6.4
操作
イオンクロマトグラフの本体,データ処理装置,附属装置などをそれぞれ操作条件に合わせて設定を行
い,作動させる。その操作は,装置の構成及びデータ処理装置の個別規格で定めた条件の範囲内で設定し
なければならない。箇条5に規定する装置の構成(附属装置も含む。)を基本として,操作する。操作は,
次による。
a) 分析条件の設定 個別規格で定めた条件に従い,次の項目についてそれぞれの値に調節する。
妨害成分
分析対象成分
a) コンディショニング
(洗浄・活性化)
b) 試料溶液添加
c) 妨害成分の吸着
分析対象成分の溶出
14
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 溶離液(移動相)の種類及び流量(グラジエント溶離法を用いる場合は,溶離液の初期組成,組成
変化率,最終組成などの設定)
2) 試料の注入量
3) カラムの種類
4) カラムの温度
5) サプレッサーの設定
6) 検出部の感度
7) データ処理部(データ処理装置)の設定
b) システムの作動,ベースラインの安定度及びノイズレベルの確認 a) で規定した条件で作動させたと
き,ベースラインの変動が測定に支障のないことを確認する。ベースラインの安定度は,ドリフトの
大きさで表し,ノイズレベルは,安定したベースラインの状態で確認する(箇条11参照)。
また,誤差の要因などを特定するために,空試験の測定(箇条11参照)を行うことが望ましい。
c) 標準液の導入 希釈標準液又は混合希釈標準液をシリンジ又は自動試料導入装置を用いて注入し,試
料導入部から導入する。精度管理に必要な測定回数,及び複数の濃度の標準液を測定する(箇条8参
照)。
d) 測定用試料溶液の導入 測定用試料溶液をシリンジ又は自動試料導入装置を用いて注入し,試料導入
部から導入する。
e) クロマトグラムの記録 クロマトグラムの記録は,データ処理装置又は記録計を用いて行う。データ
処理装置を用いる場合,正確なデータを得るために,データの取込みに関するパラメータの数値を状
況に応じ設定する。データの取込み及びピークの計算処理に関するパラメータには,サンプリング周
期,時定数,ピーク検出パラメータなどがある。記録計を用いる場合,測定用試料溶液導入と同時に
記録紙に試料導入点を記入する。測定するイオン種に基づくピークが記録紙上を振り切れることなく,
できるだけ大きなピークを描くように測定するイオン種の濃度に応じて減衰器を調節する。
f)
クロマトグラムの整理 クロマトグラムの整理に当たり,次の事項をクロマトグラムとともに記録す
る。記載内容が分析手順書などに記載されている場合は一部を省略してもよい。単位は一例として示
す。
1) 測定日及び測定者名
2) イオンクロマトグラフの製造業者名又はその形式記号
3) 試料名
4) 測定用試料溶液の調製方法及び導入量(μL又はmL)
5) カラム充塡剤の種類又は充塡カラムの形式名
6) カラム管の材質,内径(mm)及び長さ(mm)
7) カラム温度(℃)
8) 溶離液(移動相)の種類
9) 溶離液(移動相)の流量(mL/min)及びカラム入口圧力(MPa)
10) グラジエント溶離法を採用した場合は,グラジエントの条件
11) サプレッサーを採用した場合は,サプレッサーの方式及び条件
12) 検出器の種類及び設定条件
13) ポストカラム誘導体化法などの化学反応を利用した場合は,反応液及び誘導体化試薬の種類,移動
相及び反応液の混合比並びに反応条件
15
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14) データ処理部の名称及びデータ処理条件
15) その他必要事項
7
検量線用溶液
検量線用溶液の調製には,トレーサビリティが確保された標準液2) 又はそれを一定濃度に希釈したもの
のほか,a)〜aj) に示す方法によって調製した標準液又は市販されているイオンクロマトグラフ用標準液を
用いる。検量線用溶液には,標準液3) を水で希釈した希釈標準液4) 又は混合希釈標準液5) を用いる。検量
線用溶液を調製する水は,JIS K 0557に規定するA2〜A4の水又はこれと同等以上の水を使用する。また,
あらかじめ空試験を行って使用の適否を確認しなければならない。標準液及び希釈標準液の調製例を次に
示す。
注2) トレーサビリティが確保された標準液に相当するものとして,国家計量標準(計量法第134条)
に規定するJCSS(計量法校正事業者登録制度)マーク付き証明書を付したものがある。市販さ
れているイオンクロマトグラフ用標準液にも,値付けされているものがある。
3) 標準液の保存には,ポリエチレン製又は四ふっ化エチレン樹脂(PTFE)製などの気密容器を用
いるとよい。冷暗所に保存すると数か月間安定である。ただし,シアン化物イオン標準液,亜
硝酸イオン標準液,炭酸イオン標準液,塩素酸イオン標準液及び亜塩素酸イオン標準液は,使
用時に調製することが望ましい。
4) 希釈標準液は,使用時に調製することが望ましい。
5) 混合希釈標準液は,使用時に調製することが望ましい。ただし,混合によって沈殿及び/又は
濁りを生じない組合せに限る。
a) ふっ化物イオン標準液(1 000 mg/L) JIS K 8005に規定する容量分析用標準物質のふっ化ナトリウ
ムをあらかじめ約500 ℃で1時間加熱し,デシケーター中で放冷する。その2.210 gをとり,水に溶
かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
b) 塩化物イオン標準液(1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを
あらかじめ約600 ℃で1時間加熱し,デシケーター中で放冷する。その1.648 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
c) 臭化物イオン標準液(1 000 mg/L) JIS K 8506に規定する臭化カリウムをあらかじめ110 ℃で4時
間加熱し,デシケーター中で放冷する。その1.489 gをとり,水に溶かし,全量フラスコ1 000 mLに
移し入れ,水を標線まで加える。
d) よう化物イオン標準液(1 000 mg/L) JIS K 8913に規定するよう化カリウムをあらかじめ110 ℃で
4時間加熱し,デシケーター中で放冷する。その1.308 gをとり,水に溶かし,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。
e) シアン化物イオン標準液(1 000 mg/L) JIS K 8443に規定するシアン化カリウム0.63 gを少量の水
に溶かし,水酸化ナトリウム溶液(20 g/L)2.5 mLを加え,水で250 mLとする。この溶液は使用時に
調製する。
シアン化物イオン標準液の濃度は,次の方法によって求める。
シアン化物イオン標準液100 mLを正しくとり,水酸化ナトリウム溶液(20 g/L)1 mLと,指示薬
として,JIS K 8495に規定するp-ジメチルアミノベンジリデンロダニンのアセトン溶液(0.2 g/L)0.5
mLを加え,0.1 mol/L硝酸銀溶液6) で滴定し,黄色から赤色になった点を終点とする。ここに要した
0.1 mol/L硝酸銀溶液のmL数(a)から,次の式によってこの溶液のシアン化物イオン濃度(mg/L)
16
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を算出する。
000
1
100
1
204
.5
×
×
×
×
=
f
a
C
ここに,
C: シアン化物イオン標準液の濃度(mg/L)
a: 0.1 mol/L硝酸銀溶液の滴定量(mL)
f: 0.1 mol/L硝酸銀溶液のファクター
5.204: 0.1 mol/L硝酸銀溶液1 mLに対応するシアン化物イオン量
(mg)
注6) 0.1 mol/L硝酸銀溶液の調製法は,JIS K 8001のJA.5.2 n) を参照。
f)
炭酸イオン標準液(1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムをあ
らかじめ600 ℃で1時間加熱し,デシケーター中で放冷する。その1.766 gをとり,炭酸を含まない
水に溶かし,全量フラスコ1 000 mLに移し入れ,炭酸を含まない水を標線まで加える。
g) 亜硝酸イオン標準液(1 000 mg/L) JIS K 8019に規定する亜硝酸ナトリウムをあらかじめ110 ℃で
4時間加熱し,デシケーター中で放冷する。NaNO2 100 %に対してその1.500 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
h) 硝酸イオン標準液(1 000 mg/L) JIS K 8548に規定する硝酸カリウムをあらかじめ110 ℃で4時間
加熱し,デシケーター中で放冷する。その1.631 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
i)
りん酸イオン標準液(1 000 mg/L) JIS K 9007に規定するりん酸二水素カリウムをあらかじめ110 ℃
で4時間加熱し,デシケーター中で放冷する。その1.433 gをとり,水に溶かし,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。
j)
硫酸イオン標準液(1 000 mg/L) JIS K 8987に規定する硫酸ナトリウムをあらかじめ110 ℃で2時
間加熱し,デシケーター中で放冷する。その1.479 gをとり,水に溶かし,全量フラスコ1 000 mLに
移し入れ,水を標線まで加える。
k) ナトリウム標準液(1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムをあ
らかじめ約600 ℃で1時間加熱し,デシケーター中で放冷する。その2.542 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
l)
カリウム標準液(1 000 mg/L) JIS K 8121に規定する塩化カリウムをあらかじめ約500 ℃で1時間
加熱し,デシケーター中で放冷する。その1.907 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
m) アンモニウムイオン標準液(1 000 mg/L) JIS K 8116に規定する塩化アンモニウムをあらかじめシ
リカゲルを入れたデシケーター中で16時間以上乾燥する。その2.965 gをとり,水に溶かし,全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。
n) マグネシウム標準液(1 000 mg/L) JIS K 8875に規定するリボン状又は薄片状のマグネシウム1.000
gを最少量の塩酸(1+1)に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
o) カルシウム標準液(1 000 mg/L) JIS K 8617に規定する炭酸カルシウムをあらかじめ180 ℃で1時
間加熱し,デシケーター中で放冷する。その2.497 gをとり,水約600 mLが入った全量フラスコ1 000
mLに移し入れ,塩酸を加えて固形物を消失させ,水を標線まで加える。
p) 混合希釈標準液 混合希釈標準液の一例を,次に示す。
例1 陰イオン混合希釈標準液[(F−10 mg,Cl−10 mg,Br−10 mg,NO2−10 mg,NO3−10 mg,
PO43−10 mg,SO42−10 mg) / L] ふっ化物イオン標準液(1 000 mg/L)10 mL,塩化物イオン
17
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
標準液(1 000 mg/L)10 mL,臭化物イオン標準液(1 000 mg/L)10 mL,亜硝酸イオン標準
液(1 000 mg/L)10 mL,硝酸イオン標準液(1 000 mg/L)10mL,りん酸イオン標準液(1 000
mg/L)10 mL,硫酸イオン標準液(1 000 mg/L)10 mLをそれぞれ全量フラスコ1 000 mLに
とり,水を標線まで加える。
例2 陽イオン混合希釈標準液[(Na+10 mg,K+10 mg,NH4+10 mg,Mg2+10 mg,Ca2+10 mg) / L]
ナトリウム標準液(1 000 mg/L)10 mL,カリウム標準液(1 000 mg/L)10 mL,アンモニウ
ムイオン標準液(1 000 mg/L)10 mL,マグネシウム標準液(1 000 mg/L)10 mL,カルシウム
標準液(1 000 mg/L)10 mLをそれぞれ全量フラスコ1 000 mLにとり,水を標線まで加える。
q) ひ素標準液(1 000 mg/L) ひ素標準液は,JIS K 8005に規定する標準液に相当するものとして,JCSS
の標章付き証明書を付した標準液(1 000 mg/L,100 mg/L)が供給されている。
r) 塩素酸イオン標準液(1 000 mg/L) 塩素酸ナトリウム(NaClO3)1.4 gをとり,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。この溶液は使用時に調製する。
塩素酸イオン標準液の濃度は,次の方法で求める。
共栓付三角フラスコ100 mLに臭化カリウム溶液(5 g/L)5 mL,塩酸10 mL及び水5 mLをとり,
これに塩素酸イオン標準液10 mLを加えて直ちに栓をし,暗所で20分間静置後,よう化カリウム1 g
を加えてよう素を遊離させる。この試験溶液をあらかじめりん酸一水素ナトリウム飽和溶液25 mLを
入れてある共栓付三角フラスコ100 mLに移す。先の共栓付三角フラスコ100 mLを少量の水で洗い,
洗液も共栓付三角フラスコ100 mLに移し,でんぷん溶液5 mLを加えて生じた青色が消えるまで0.1
mol/Lチオ硫酸ナトリウム溶液7) で滴定し,この滴定に要した0.1 mol/Lチオ硫酸ナトリウム溶液の
mL数(a)を求め,次の式によってこの溶液の塩素酸イオンの濃度(mg/L)を算出する。
000
1
1
391
.1
×
×
×
×
=
S
f
a
C
ここに,
C: 塩素酸イオン標準液の濃度(mg/L)
a: 0.1 mol/Lチオ硫酸ナトリウム溶液の滴定量(mL)
f: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
S: 滴定に用いた塩素酸イオン標準液のmL数
1.391: 0.1 mol/Lチオ硫酸ナトリウム1 mLに相当する塩素酸イオン
の質量を示す換算係数(mg/mL)
注7) チオ硫酸ナトリウム溶液0.1 mol/Lの調製法は,JIS K 8001:2009のJA.5.2 t) 2)を参照。
s)
亜塩素酸イオン標準液(1 000 mg/L) 亜塩素酸ナトリウム(NaClO2)1.5 gをとり,全量フラスコ1 000
mLに移し入れ,水を標線まで加える。この溶液は使用時に調製する。
亜塩素酸イオン標準液の濃度は,次の方法で求める。
三角フラスコ300 mLによう化カリウム溶液(100 g/L)30 mL及び塩酸(1+24)50 mLをとり,こ
れに亜塩素酸イオン標準液20 mLを加えて0.1 mol/Lチオ硫酸ナトリウム溶液8) で滴定し,溶液の色
が褐色から淡黄色に変化してからでんぷん溶液3 mLを加え,生じた青色が消えるまで滴定を続け,
要した0.1 mol/Lチオ硫酸ナトリウム溶液のmL数(a)を求め,次の式によってこの溶液の亜塩素酸
イオンの濃度(mg/L)を算出する。
000
1
1
686
.1
×
×
×
×
=
S
f
a
C
ここに,
C: 亜塩素酸イオン標準液の濃度(mg/L)
a: 0.1 mol/Lチオ硫酸ナトリウム溶液の滴定量(mL)
f: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
18
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 滴定に用いた亜塩素酸イオン標準液のmL数
1.686: 0.1 mol/Lチオ硫酸ナトリウム1 mLに相当する亜塩素酸イオ
ンの質量を示す換算係数(mg/mL)
注8) チオ硫酸ナトリウム溶液0.1 mol/Lの調製法は,JIS K 8001:2009のJA.5.2 t) 2)を参照。
t)
臭素酸イオン標準液 JIS K 8530に規定する臭素酸カリウム1.306 gをとり,水に溶かし,全量フラス
コ1 000 mLに移し入れ,水を標線まで加える。
u) チオシアン酸イオン標準液(1 000 mg/L) JIS K 9001に規定するチオシアン酸カリウムをあらかじ
め105 ℃で4時間加熱し,デシケーター中で放冷する。その1.673 gをとり,水に溶かし,全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。
v) アジ化物標準液(1 000 mg/L) JIS K 9501に規定するアジ化ナトリウム1.547 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
w) ぎ酸標準液(1 000 mg/L) JIS K 8267に規定するぎ酸ナトリウム1.511 gをとり,水に溶かし,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
x) 酢酸標準液(1 000 mg/L) JIS K 8372に規定する酢酸ナトリウム1.389 gをとり,水に溶かし,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
y) マロン酸標準液(1 000 mg/L) マロン酸1.020 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
z) しゅう酸標準液(1 000 mg/L) JIS K 8528に規定するしゅう酸ナトリウム1.522 gをとり,水に溶か
し,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
aa) こはく酸標準液(1 000 mg/L) こはく酸1.017 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
ab) 安息香酸標準液(1 000 mg/L) JIS K 8073に規定する安息香酸1.008 gをとり,水に溶かし,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
ac) くえん酸標準液(1 000 mg/L) くえん酸1.016 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
ad) りんご酸標準液(1 000 mg/L) りんご酸1.015 gをとり,水に溶かし,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
ae) メチルアミン標準液(1 000 mg/L) 塩化メチルアンモニウム2.174 gをとり,水に溶かし,全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。
af) ジメチルアミン標準液(1 000 mg/L) 塩化ジメチルアンモニウム1.809 gをとり,水に溶かし,全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。
ag) トリメチルアミン標準液(1 000 mg/L) 塩化トリメチルアンモニウム1.617 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
ah) エチルアミン標準液(1 000 mg/L) 塩化エチルアンモニウム1.809 gをとり,水に溶かし,全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。
ai)
ジエチルアミン標準液(1 000 mg/L) 塩化ジエチルアンモニウム1.499 gをとり,水に溶かし,全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。
aj) トリエチルアミン標準液(1 000 mg/L) 塩化トリエチルアンモニウム1.359 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。

19
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8
定性分析
定性分析は,希釈標準液又は混合希釈標準液を同一条件下の測定で得られたクロマトグラムを用いて,
これら標準液と試料溶液中の未知成分との保持値9), 10) を比較して行う。この場合,一つのピークは必ずし
も一つの成分に対応するとは限らないので,カラムの種類又は溶離液を変えるなど分離条件を変えて測定
するか,又は次の定性手法と併用して確かめる。
注9) 保持値には,保持時間,保持容量又は保持比がある。
10) 同一分析条件における,ある成分の繰返し分析での保持値のばらつきは,相対標準偏差3 %以
下とすることが一般的である。
a) 異なる検出器の使用
b) 化学反応の利用
c) 原子吸光法,フレーム光度法,質量分析法,発光分光分析法,イオン電極法など
9
定量分析
9.1
定量法
定量は,得られたクロマトグラムからデータ処理装置などを用いてピーク面積又はピーク高さを測定し,
絶対検量線法,内標準法,標準添加法のいずれかによって行う11) 12)。
注11) ピーク面積又はピーク高さを求めるに当たっては,ピークの形状に合わせ,適切なベースライ
ンを引くように注意する。
12) 適切な内標準物質が得られる場合は,内標準法を用いることができる。この場合,導入誤差の
補正,前処理誤差の補正を行うことができる。また,標準添加法は,試料マトリックスの影響
が補正できる特長がある。
9.2
ピーク面積
ピークの始まりから終わりにわたってピークの信号値とベースラインの信号値との差を積算したもの,
又はピーク高さ(h)の中点(h/2)から時間軸に平行線を描き,ピークによって切られる線分を半値幅(w0.5h)
とする。これにピーク高さ(h)を乗じたものをピーク面積(A)とする。ただし,この方法は著しいベー
スラインの変動,又はリーディング若しくはテーリングが認められるピークには適用しない。
なお,データ処理装置を用いる場合は,表示された記録値又は指示値による。データ処理装置にはピー
クの形状に合わせ,適切な設定条件を選ばなければならない。
図5−半値幅法によるピーク面積測定
20
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.3
ピーク高さ
ピーク頂点の信号値からピーク頂点の保持時間と同一の保持時間におけるベースラインの信号値を差し
引いたもの,又はピークの頂点から時間軸に垂線を下ろし,これがベースラインと交わる点と頂点との距
離をピーク高さとする。
9.4
重複ピーク
重複ピークの処理方法には,次の方法がある。
a) 垂線法 図6のように,二つのピークの大きさがほぼ等しい場合,ピークの谷から時間軸に下ろした
垂線によってベースライン上のピークを二つに分割し,それぞれの面積又は高さを求める。
図6−垂線法によるピークの分割
b) 谷−谷(valley to valley)法 図7のように,バックグラウンドの上の重複したピークに対して適用す
る。隣接する谷と谷とを結ぶ線分及びクロマトグラムによって囲まれた面積又は高さを求める。
図7−谷−谷法によるピークの分割
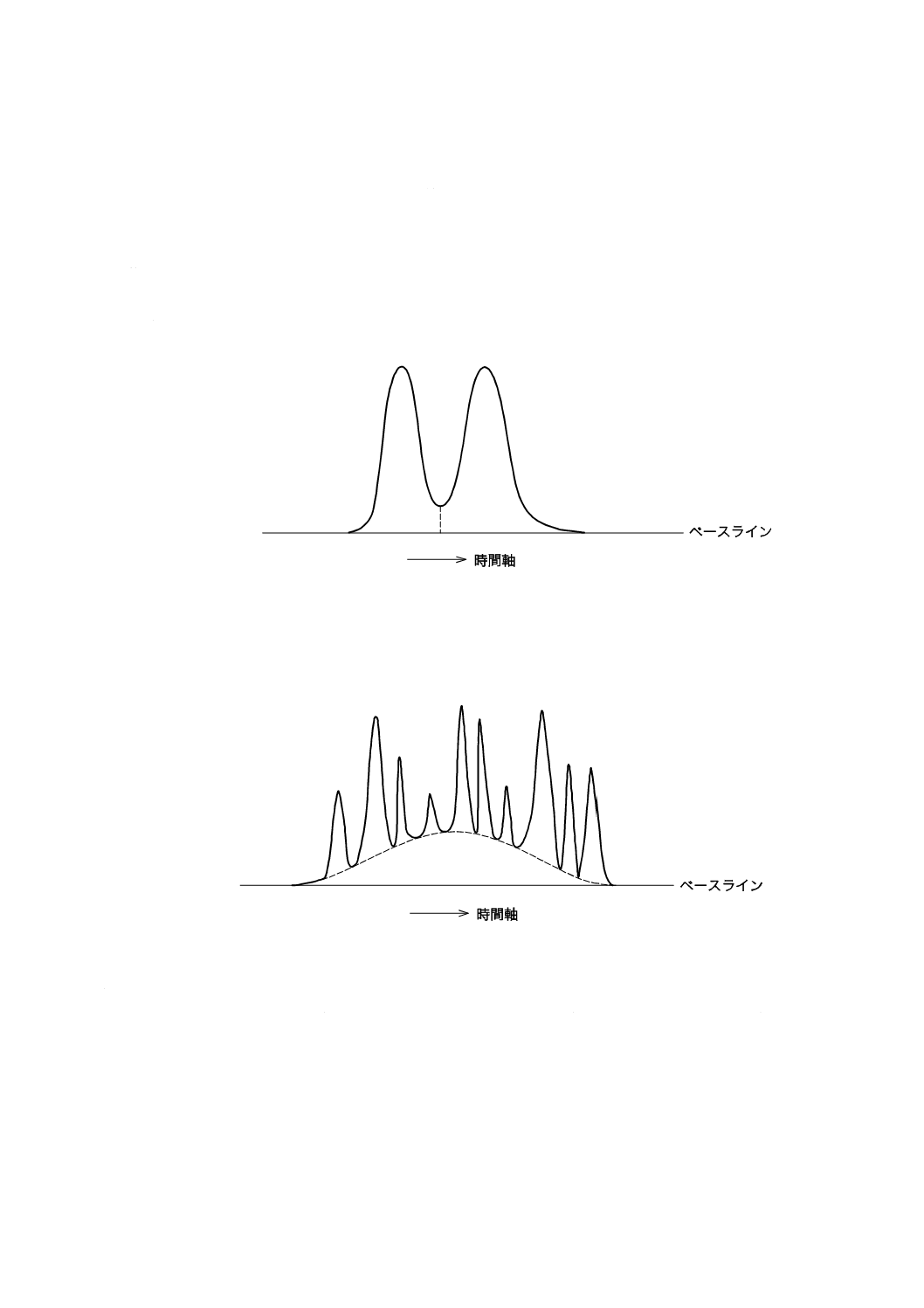
c) 接線法 図8のように,大きなピークのテーリングに重なった小さなピークの場合,ピークの谷と大
きなピークの裾とを結ぶ接線上の部分をピーク面積とする。接線の代わりに,指数関数曲線によるピ
ーク分割もできる。
21
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図8−接線法によるピークの分割
9.5
検量線
9.5.1
一般事項
定量に使用する検量線については,使用する検出器の種類及び分析条件によって,直線又は多次関数を
適切に使い分けて作成する必要がある。
9.5.2
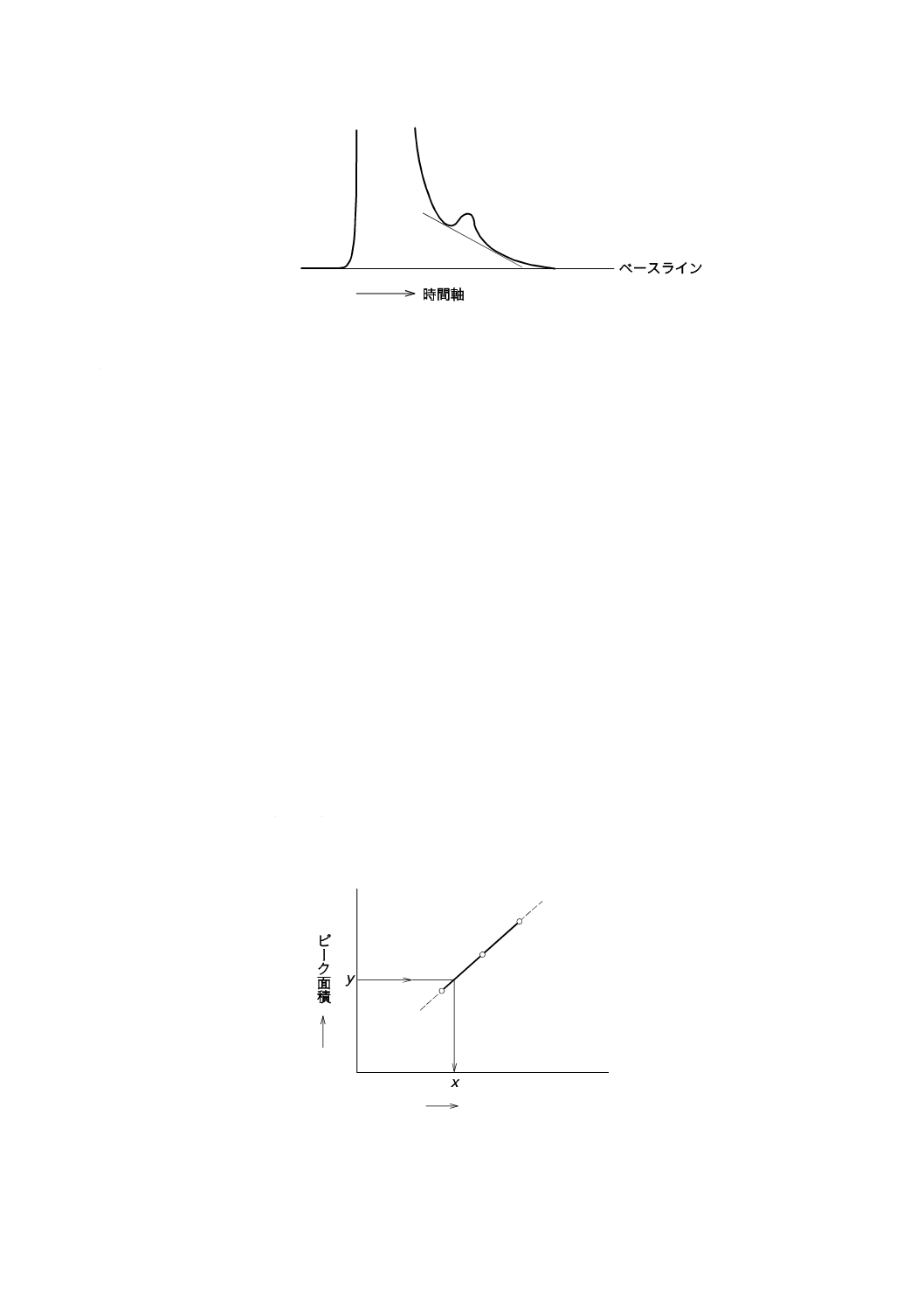
絶対検量線法
測定するイオン種成分の標準液を3段階以上の濃度に調製し,各希釈標準液を一定量導入し,クロマト
グラムを記録してピーク面積又はピーク高さを測定する。次に,導入された希釈標準液中の測定するイオ
ン種成分の量を横軸に,そのピーク面積又はピーク高さを縦軸にし,検量線を作成する13)。同一条件の下
で測定用試料溶液を導入し,クロマトグラムを記録し,ピーク面積又はピーク高さから検量線によって測
定するイオン種成分の量を求め,試料中の濃度を算出する。ピーク面積を用いた場合の例を図9に示す。
注13) 一般には数点を取り,図9のような検量線を作成して定量を行うが,あらかじめ,原点を通る
直線性が確かめられていれば,既知量の導入を一点だけとし,単位ピーク面積当たりの成分量
を求めてもよい。このとき,希釈標準液濃度は,試料濃度より高濃度であるとする。
また,希釈標準液濃度とピーク面積又はピーク高さとの関係が曲線になる場合は,各点を結
ぶ多次関数を検量線とする。このとき,濃度によっては測定精度が悪くなるおそれがあるため,
4段階以上の濃度に調製された希釈標準液を用いる。最小二乗法によって得られた決定係数だ
けを直線性の判断基準としないよう注意する。
曲がる検量線を直線近似する必要があるときには,適切な濃度範囲に分割し,それぞれの濃
度範囲において直線検量線を作成する。
図9−絶対検量線法による検量線
イオン量
22
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
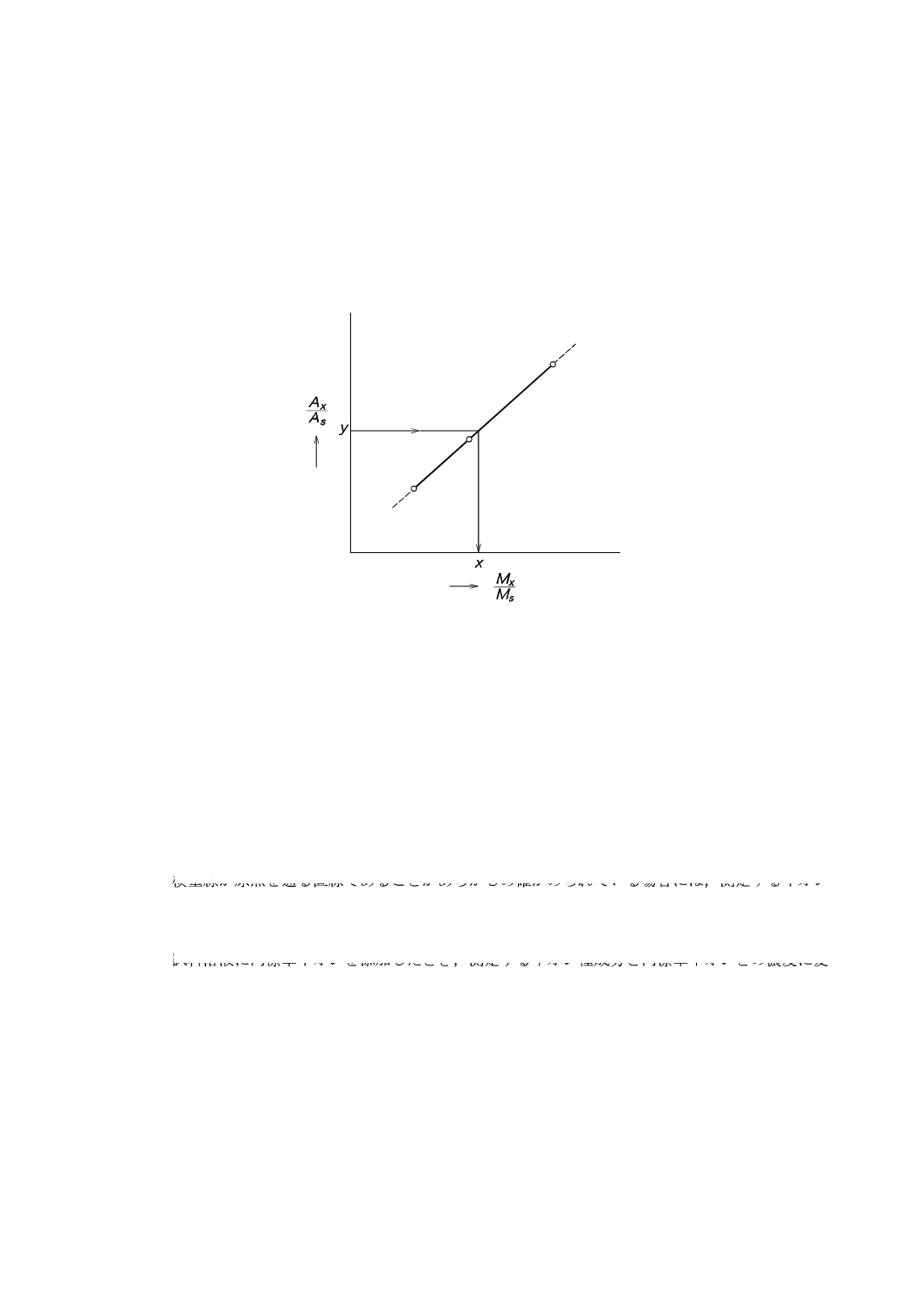
9.5.3
内標準法
一定濃度の内標準イオン14) を含む,3段階以上の濃度15) の希釈標準液を調製する。各希釈標準液を一
定量導入し,クロマトグラムを記録してピーク面積又はピーク高さを測定する。次に,導入された測定す
るイオン種成分の量(Mx)と内標準イオンの量(Ms)との比(Mx/Ms)を横軸に,測定するイオン種成分
のピーク面積又はピーク高さ(Ax)と内標準イオンのピーク面積,又はピーク高さ(As)との比(Ax/As)
を縦軸にして図10に示すような検量線を作成する。
図10−内標準法による検量線
次に,試料溶液に希釈標準液と同じ濃度になるよう内標準イオンを添加16) した測定用試料溶液を調製し,
希釈標準液と同一条件の下で導入してクロマトグラムを記録する。クロマトグラムから測定するイオン種
成分のピーク面積又はピーク高さ(A'x)と内標準イオンのピーク面積又はピーク高さ(A's)との比(A'x/A's)
を算出し,検量線から測定するイオン種成分の量と内標準イオン量との比を求め,導入された内標準イオ
ンの量から測定するイオン種成分の量を算出する。これから試料中の測定するイオン種成分の濃度を算出
する。
注14) 内標準イオンには,そのピークが,測定するイオン種成分のピークの位置になるべく近く,試
料中の他の成分ピークとは完全に分離する安定したものを選択することが望ましい。
15) 検量線が原点を通る直線であることがあらかじめ確かめられている場合には,測定するイオン
種成分の濃度を一点だけとして,これを導入した場合の(Ax/As)を測定し,これに基づいて検
量線を求めてもよい。このときの(Ax/As)は,(A'x/A's)より大きいことが望ましい。
16) 試料溶液に内標準イオンを添加したとき,測定するイオン種成分と内標準イオンとの濃度に変
化を生じさせる沈殿などの化学変化がないよう注意する。
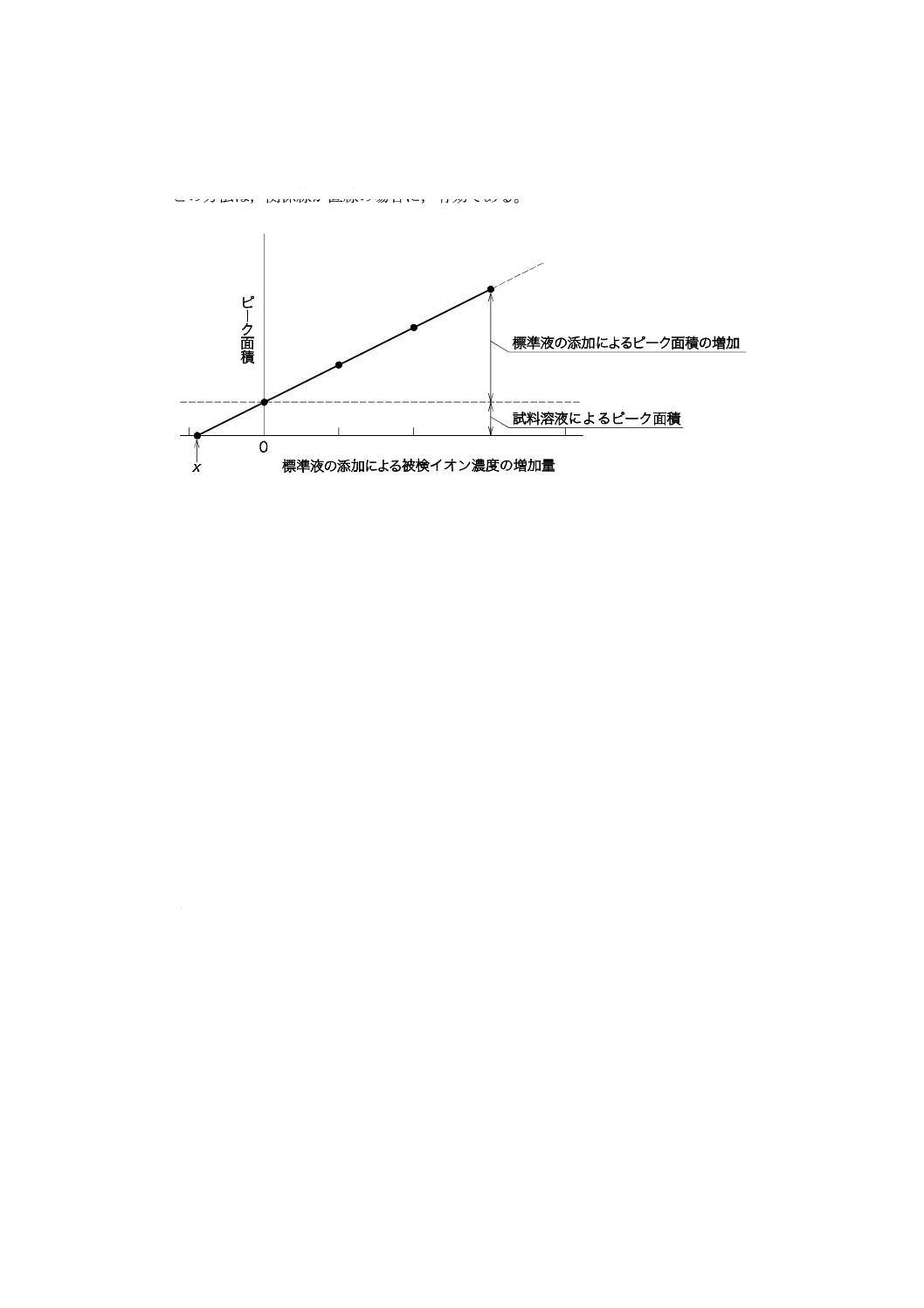
9.6
標準添加法(作図法)
試料溶液から一定量の溶液を4個以上分取する。1個を除き,これらの分取した溶液に,測定するイオ
ン種成分の標準液の濃度が異なるように段階的に加える。これらの溶液及び先に除いた1個の試料溶液を
それぞれ一定量に希釈して測定用試料溶液を調製して,クロマトグラムを記録し,測定するイオン種成分
のピーク面積又はピーク高さを測定する。それぞれに加えた測定するイオン種成分の濃度を算出し,標準
液の添加による目的成分濃度の増加量を横軸17) と,ピーク面積又はピーク高さを縦軸にとり関係線を作成
する。関係線と横軸との交点(図11に示す点X)から測定するイオン種成分の濃度を求める18)。
23
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注17) 測定するイオン種成分の濃度と標準液の添加量又は一定量の添加回数との間に比例関係が成立
するため,横軸に添加量又は添加回数をとることができる。
18) この方法は,関係線が直線の場合に,有効である。
図11−標準添加法(作図法)
10 試験結果の表示
試験結果は,測定の対象としたイオン名及び濃度を併記する。濃度は,mg/mL,mg/L,μg/L,mol/L,
mg/kgなどで表す。
11 データの質の管理
11.1 分析法バリデーション
データの質を保証するために,新たに開発した分析法(分析条件の変更を含む。)については,バリデー
ションを実施する19)。公定法(個別JISなど)を採用する場合でも,その試験室で要求される項目につい
てのバリデーションを行うことが望ましい。その試験室で開発した方法(インハウスメソッド)による場
合は,その方法を適用する前に必ずバリデーションを実施し,少なくともその分析法が適用される間は,
その分析条件を維持する。
注19) 実際に分析を開始する際には,カラム,溶離液などを選定し,その最適化を図ることが望まし
い。すなわち,開発した方法が分析の目的にかなう方法であることを確認するために分析法を
開発又は改良する。バリデーションの項目には,精確さ,精度,真度又は正確さ(回収率),直
線性,検出下限,定量下限,分析法の適用範囲,堅ろう性試験などがある。
11.2 データの質の管理
データの質は,それぞれの分析条件における定量値の“不確かさ”をもって表示する。この規格では,
包含係数k=2とする拡張不確かさUを用いることとする。
また,品質管理用の試料20) を準備し,これを定期的に分析することによって,データの質を監視する。
ルーチン分析では,試料20個に1個の品質管理用の試料及び1個のブランク試料を含め,並行分析を実施
してデータの質の管理を行うのが望ましい。
注20) 品質管理用の試料には,通常インハウス標準物質(試験室で値付けした標準物質),購入した標
準物質,認証標準物質のいずれかを用いるが,均質で安定な試料であれば,大量に残っている
分析試料を用いてもよい。
24
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.3 定期的な装置性能の点検
装置性能の確認には,6.4 a) で規定した条件で作動させたとき,ベースラインの安定度,ノイズレベル
の安定度の確認を行うとともに,一定濃度の混合希釈標準液を用いた場合の分離度の確認などを,一定期
間ごとに行う。
a) ベースラインの安定度の確認 装置が測定可能な状態(ポンプ圧力,カラムオーブン,セル温度など)
であることを確認する。装置が安定したことを確認後,ベースラインを記録し,そのドリフト具合か
ら状態を判断する。
b) ノイズレベルの安定度の確認 測定装置の検出下限が確認できるベースラインノイズで安定している
ことを確認する。
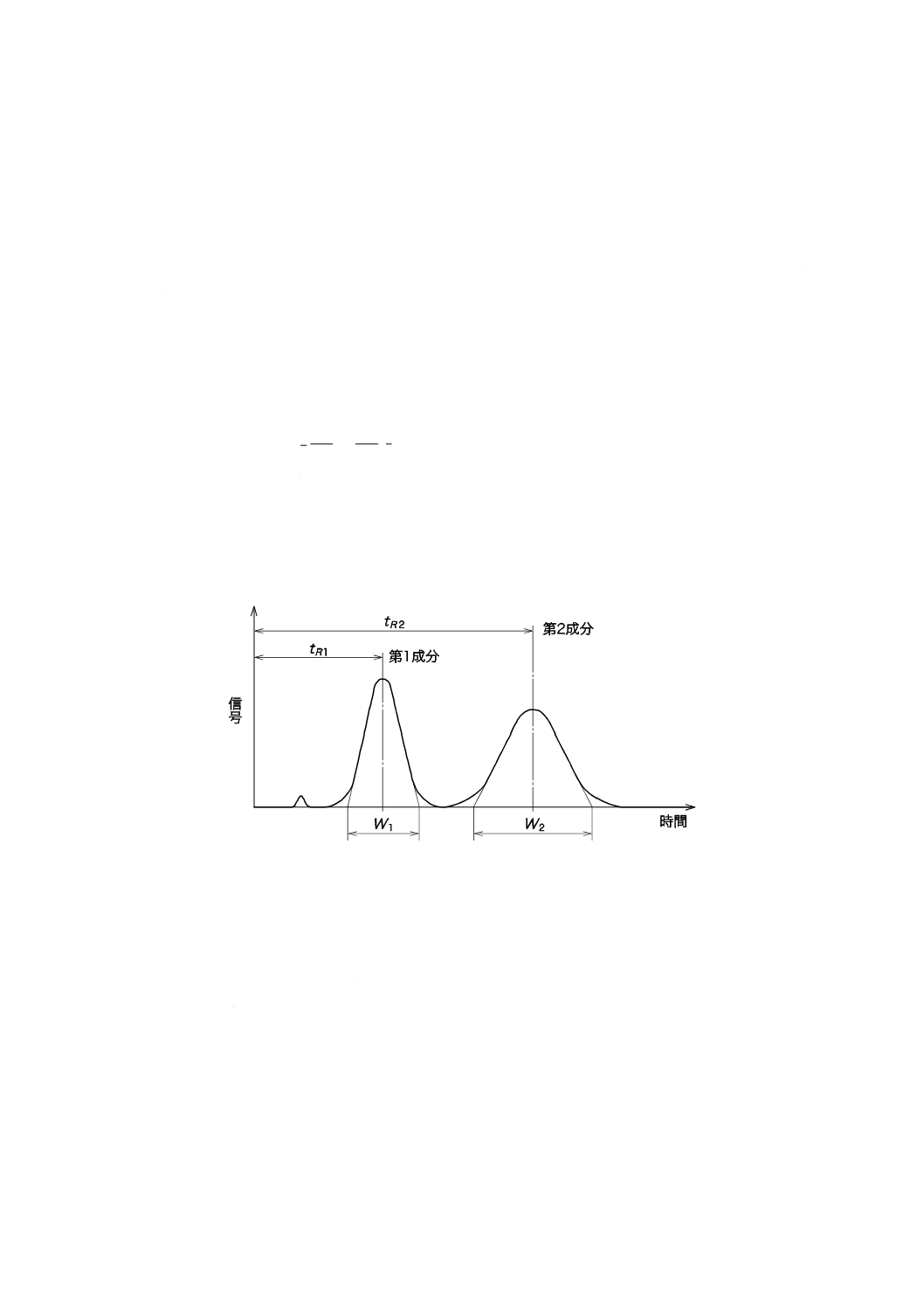
c) 分離度の確認 混合希釈標準液(mg/L)を用いて,各イオン間の分離度を確認する。分離度(R)は,
次の式によって求める(図12参照)。
2
1
1
2
)
(
2
W
W
t
t
R
R
R
+
−
×
=
ここに,
tR1: 最初のピークの保持時間(s)
tR2: 二番目のピークの保持時間(s)
W1: 最初のピークのピーク幅(s)
W2: 二番目のピークのピーク幅(s)
注記 陰イオン混合希釈標準液(mg/L)を用いた場合は,分離度1.3以上を目安とする。
図12−クロマトグラフ分離の模式図
d) 定期的な装置性能の点検 分析機器製造業者の取扱説明書に従い,各装置構成部の点検を定められた
頻度で実施する。点検の記録は保管する。
11.4 空試験の実施
ブランク試料[例えば,分析種を含まないことが明らかな試料を分析種と並行して処理した測定用試料
溶液,又は溶離液21)]を用いてブランクのクロマトグラムを得る。
注21) 分析種を含まないことが明らかな試料を分析種と並行して処理した測定用試料溶液を測定する
と,試料マトリックス及び全分析操作にわたる影響を明らかにできる。溶離液をブランクとし
て測定すれば,溶離液ブランク,試料導入時の汚染,クロスコンタミネーションなど,装置か
らの影響を分離して求められる。
11.5 検出下限の求め方
検出下限を報告する場合は,統計的手法で誤りの確率を説明できる検出下限の求め方を定め,それを記
25
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
述する。
検出下限は,ブランク試料及びそれに一定量スパイクした試料を各々少なくとも20回以上測定して,正
味の(ブランク値を差し引いた)測定値の標準偏差σの3.3倍の値を濃度(量)換算した値を検出下限と
する。測定回数が20回以下の場合には表4のt値(n=5では4.26)を乗じることによって求めた値を濃
度(量)換算した値を検出下限とする。すなわち,検出下限は“95 %の確率[誤りの確率(危険率)5 %]
でブランク信号と区別できる信号を与える分析種の濃度(量)”とする。また,シグナルSとノイズNと
の比,S/Nの値が3のときの目的成分量又は濃度を検出下限としてもよい。この方法で検出下限を述べる
場合は,必ずS/Nの値と,記録計の応答時間又はデータ処理装置のパラメータ(積分時間など)を併記す
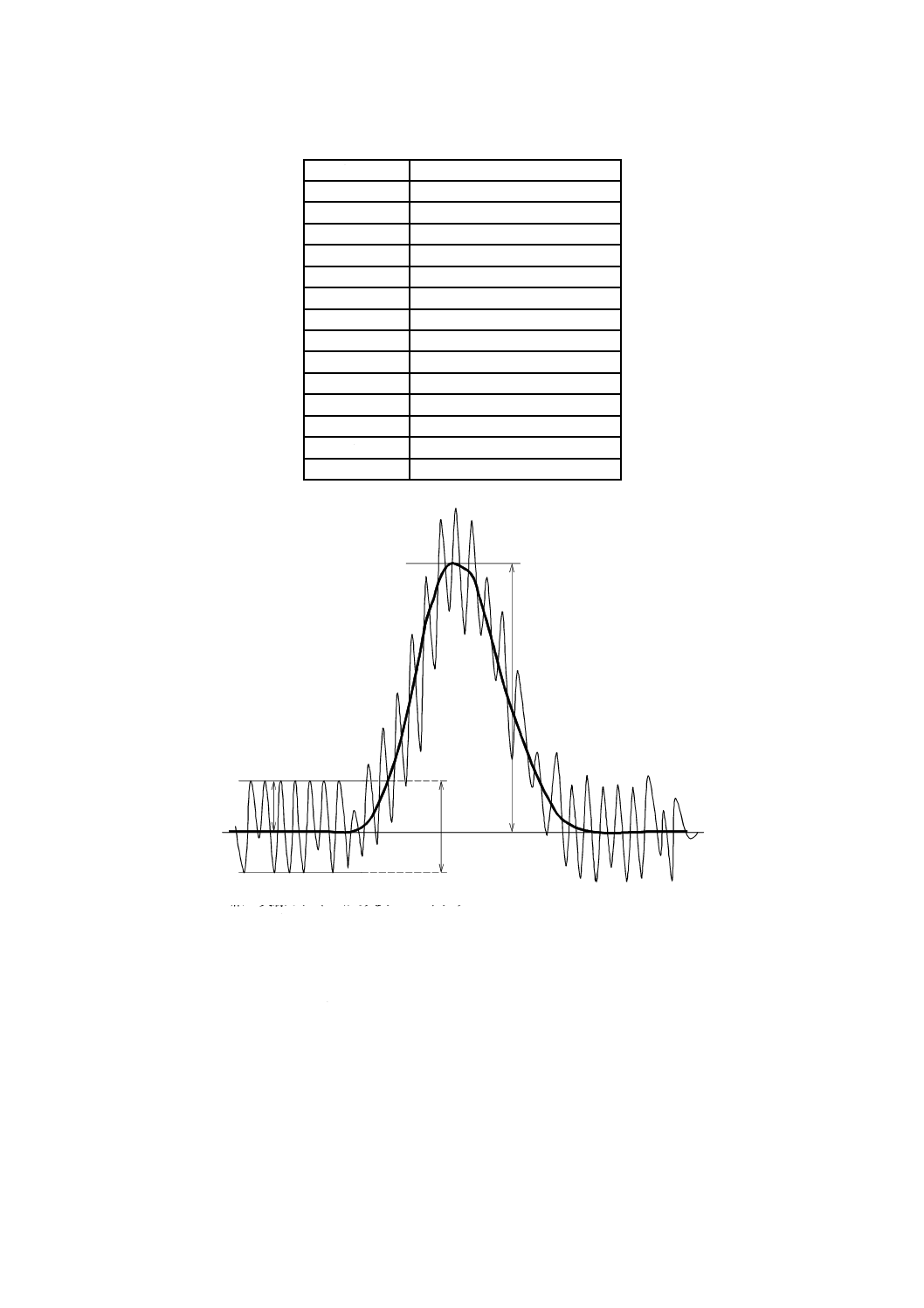
る。これは,単一の分析種を極めて低濃度に含む試料のクロマトグラムを図13に示すように,ノイズレ
ベルが十分に測定できるように高感度に記録して描き(図13の細い実線),次のようにして求める。
a) シグナル 検出器出力の平均値を線で結びノイズを含まないクロマトグラム(図13の太い実線)を得
て,ベースラインからピークの頂点までのピーク高さhをシグナルSとする。
S=h ······················································································· (1)
b) ノイズ ピークの前後におけるベースラインの,ピーク半値幅の20倍の間における出力信号の最大値
と最小値の差の振れ幅の1/2をノイズNとする。
2
h
N
N
=
··················································································· (2)
注記1 検出下限は,ISO 11843-1:1997“Capability of detection”第1部(用語及び定義)では“minimum
detectable value of the net state variable”“検出可能な最小正味状態変数値”と呼び,JIS Z
8462-1:2001としてJIS化され,“検出下限”という用語は使用しない方向にある。
注記2 検出下限の定義には,次に示すように数種類ある。
a) 確率的手法 ブランク試料及びそれに一定量スパイクした試料を繰り返し測定し,そ
のときの測定値の標準偏差から算出する。繰返し回数が多い場合は表4から3.29倍す
ることになるが,繰返し回数が少ない場合にはその回数に応じた倍率tを乗じる。
b) S/Nを用いる手法 クロマトグラフィーにおいては,従来から,シグナルSとノイズ
Nとの比から検出下限を算出する方法が用いられている。
S/N=3を検出下限として定めるのは,S/N=2では理論的には誤りの確率が0.1 %で
あるとはいえ,ドリフト,ふらつき,汚染などの影響で,経験的には2〜3にするのが
妥当であると判断されているからである。
11.6 定量下限の求め方
定量下限を報告する場合は,定量下限の定義を定め,それに従って定量下限の値を報告する。定量下限
の求め方には,検出下限の3〜5倍の値を採る方法,S/N=10のときの分析種量又は濃度とする方法,分析
種量を下げていったときに,定量値の精度が低下し,変動係数が10 %以上になる値を定量下限とする方法
などがある。
26
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表4−測定回数nを減らしたときの倍率t
測定回数n
倍率t
3
5.84
4
4.71
5
4.26
6
4.03
7
3.89
8
3.79
9
3.72
10
3.67
11
3.62
12
3.59
13
3.56
14
3.54
15
3.52
∞
3.29
細い実線:ノイズがあるクロマトグラム
太い実線:ノイズがない場合のクロマトグラム
図13−分析種が検出下限付近の濃度の高感度記録クロマトグラム概念図
12 個別JISでイオンクロマトグラフィーを分析法として取り入れる際の記載事項
イオンクロマトグラフによる分析方法を規定するに当たっては,次のうち必要な項目について記述する。
個別のクロマトグラムについては,6.4 f) を参照。
a) 一般的な事項
1) 測定日付
2) イオンクロマトグラフの製造業者名及び型式記号,附属装置を用いる場合は製造業者名,及び型式
記号
27
K 0127:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 試料名,対象成分及びその濃度範囲
4) 試料採取場所,採集方法及び試料前処理,並びに保存方法
5) 検出器の種類
6) 定性及び定量方法
b) 操作条件
1) カラムの種類,内径,長さ
2) カラムの温度
3) 溶離液の組成,流量
4) 試料量,試料導入方法
5) サプレッサーの有無
c) 成分の確認方法 代表的なクロマトグラム例を示す。
なお,クロマトグラムには操作条件,ピークの成分名及び保持時間を記入する。
d) 定量法
1) ピーク面積又は高さの測定方法
2) 定量方法の種別22) 及び分析回数
3) 分析種の純物質,内標準物質(種類,純度),分析種純物質の混合物の場合には,組成,濃度範囲及
び調製方法
注22) 相対補正係数を用いる場合には,その値を例示することが望ましい。
e) 分析結果の表示
f)
附属装置の操作条件 附属装置固有の操作条件を明記する。
参考文献 ISO 11843-1,Capability of detection−Part 1: Terms and definitions
JIS Z 8462-1 測定方法の検出能力−第1部:用語及び定義
JEITA ET-7304 ハロゲンフリーはんだ材料の定義