K 0124:2011
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 1
4 高速液体クロマトグラフィー概説 ························································································ 5
5 装置······························································································································· 8
5.1 装置の構成 ··················································································································· 8
5.2 移動相送液部 ················································································································ 9
5.3 試料導入部 ··················································································································· 9
5.4 分離部 ························································································································ 10
5.5 検出部 ························································································································ 10
5.6 データ処理部 ··············································································································· 11
5.7 附属装置 ····················································································································· 11
6 水,試薬及び溶媒 ············································································································ 11
7 カラム及びカラム充塡剤 ··································································································· 11
7.1 カラム ························································································································ 11
7.2 カラム充塡剤 ··············································································································· 12
7.3 分離モード及びカラム充塡剤 ·························································································· 12
7.4 カラム性能評価 ············································································································ 12
8 操作······························································································································ 12
8.1 試料の準備 ·················································································································· 12
8.2 溶離液の準備 ··············································································································· 23
8.3 分析種による検出器の選定 ····························································································· 26
8.4 ポストカラム誘導体化法の選択 ······················································································· 27
8.5 溶離液の選択 ··············································································································· 29
8.6 測定操作 ····················································································································· 31
9 定性分析························································································································ 32
10 定量分析 ······················································································································ 33
10.1 定量法 ······················································································································· 33
10.2 ピーク高さの測定 ········································································································ 33
10.3 ピーク面積の測定 ········································································································ 33
10.4 絶対検量線法 ·············································································································· 34
10.5 内標準法 ···················································································································· 35
10.6 標準添加法 ················································································································· 36
10.7 定量値の表し方 ··········································································································· 36
11 分子量分布の測定 ·········································································································· 36
K 0124:2011 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
11.1 校正曲線を用いて平均分子量を測定する方法 ···································································· 37
11.2 パターンによる分子量分布の測定 ··················································································· 38
12 分取液体クロマトグラフィー ··························································································· 38
12.1 分取の準備 ················································································································· 38
12.2 分取法 ······················································································································· 39
12.3 操作 ·························································································································· 40
13 データの質の保証 ·········································································································· 40
13.1 分析法バリデーション ·································································································· 40
13.2 データの質の管理 ········································································································ 40
13.3 検出下限の求め方 ········································································································ 40
13.4 ブランクの測定 ··········································································································· 43
13.5 定期的な装置性能の点検 ······························································································· 43
14 装置の設置 ··················································································································· 43
15 安全 ···························································································································· 44
16 個別規格に記載すべき事項 ······························································································ 44
K 0124:2011
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本分析
機器工業会(JAIMA)及び財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改
正すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格であ
る。これによって,JIS K 0124:2002は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 0124:2011
高速液体クロマトグラフィー通則
General rules for high performance liquid chromatography
1
適用範囲
この規格は,高速液体クロマトグラフを用いて分析種の定性又は定量分析を行う場合及び分析のための
精製を目的とした分取を行う場合の通則について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0127 イオンクロマトグラフ分析通則
JIS K 0211 分析化学用語(基礎部門)
JIS K 0214 分析化学用語(クロマトグラフィー部門)
JIS K 0215 分析化学用語(分析機器部門)
3
用語及び定義
この規格で用いる主な用語の定義は,JIS K 0050,JIS K 0127,JIS K 0211,JIS K 0214及びJIS K 0215
によるほか,次による。
なお,括弧内の対応英語は参考のために示す。
3.1
高速液体クロマトグラフィー(high performance liquid chromatography)
液体の移動相をポンプなどによって加圧してカラムを通過させ,分析種を固定相及び移動相との相互作
用(吸着,分配,イオン交換,サイズ排除など)の差を利用して高性能に分離して検出する方法。
3.2
高速液体クロマトグラフ(high performance liquid chromatograph)
高速液体クロマトグラフィーを行うための装置。
3.3
分析種(analyte)
試料又は試料溶液中の被検成分。
3.4
試料溶媒(sample solvent)
カラムに導入できるよう試料を溶かすために用いられる溶媒。
2
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.5
測定用試料溶液(test sample solution)
測定を行うための試料溶液又は試料溶液に何らかの前処理を行った溶液。
3.6
固定相(stationary phase)
液体クロマトグラフィーが行われる場の要素の一つで,移動相と平衡状態にあり分析種と相互作用する
相。
3.7
移動相(mobile phase)
液体クロマトグラフィーが行われる場の要素の一つで,固定相に接して流れる液体で固定相と平衡状態
にあり,分析種と相互作用しながら移動し,分析種と固定相との相互作用を繰り返させる役目をする相。
3.8
溶離液(eluent, eluant)
カラムに保持されている分析種を展開,溶出させる移動相として用いる液体。
3.9
溶離液槽(eluent reservoir, eluant reservoir)
溶離液を貯留する槽。
3.10
ポンプ(pump)
溶離液(移動相)を送液する装置。
3.11
試料導入装置(sample injector)
測定用試料溶液を高速液体クロマトグラフのカラムに導入するための装置。
3.12
オートサンプラー(automatic sample injector, auto sampler)
測定用試料溶液を連続的に高速液体クロマトグラフのカラムに導入するための装置。自動試料導入装置
ともいう。
3.13
カラム(column)
分析種の分離が行われる管で,クロマト管に充塡剤が充塡されたもの,又は毛管などの管内壁に固定相
となるものが保持されたもの。
3.14
カラム槽(column compartment)
温度制御を目的としてカラムを収容する槽。
3.15
検出器(detector)
カラムにおいて分離された分析種の特性(吸光度,屈折率など)を検知し,電圧などの電気信号に変化
させる装置。
3
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.16
カラム充塡剤(column packings)
カラムに充塡される主に粒状の固定相,固定相を保持させた粒子,及びモノリス状の固定相。
3.17
モノリス(monolith)
全多孔性のシリカゲル又はポリマーを基材とした骨格構造をもつ固定相。ロッド形のもの,フューズド
シリカ等のキャピラリー内で合成したものなどがある。
3.18
クロマトグラフィー管(chromatographic tube)
充塡剤を充填するための管で,円筒状管の両端にフィルターなどを付したもの。
3.19
溶出液(eluate, effluent)
溶離液(移動相)によって展開したときのカラムから流出する液体。
3.20
理論段数(theoretical plate number)
カラムの効率を表す指標の一つで,カラムを理論的に小区画の不連続な段のつながりと考えた場合のカ
ラム全体での段の総数。ピーク幅の求め方によって,接線法,半値幅法,面積高さ法などの方法がある。
次の式によって求める。
2
R
16 ×W
t
N=
····································································· 接線法
2
h
5.0
R
54
.5
×Wt
N=
····························································· 半値幅法
2
R
π
2 ×
A
H
t
N=
····························································· 面積高さ法
ここに,
N: 理論段数
tR: 保持時間
W: ピーク幅
W0.5h: ピーク半値幅
A: ピーク面積
H: ピーク高さ
ただし,tRとW,tRとW0.5hとは同じ単位を用いる。
3.21
分離度(resolution)
目的成分のピークが隣接するピークからどの程度分離しているかを示す尺度。次の式によって求める。
+
−
×
2
h
5.0
1h
5.0
1
R
2
R
18
.1
W
W
t
t
R=
ここに,
R: 分離度
tR1,tR2: 隣接する各ピークの保持時間(tR1<tR2)
W0.5h1,W0.5h2: 隣接する各ピークの高さの中点におけるピーク幅
ただし,tR1,tR2,W0.5h1,W0.5h2は同じ単位を用いる。
4
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.22
シンメトリー係数(symmetry factor)
クロマトグラム上のピークの対称性の度合いを示す係数。次の式によって求める。
S=W0.05h/2f
ここに,
S: シンメトリー係数
W0.05h: ピークのベースラインからピーク高さの1/20の高さにお
けるピーク幅
f: W0.05hのピーク幅をピーク頂点から記録紙の横軸へ下ろ
した垂線で二分したときのピークの立ち上がり側の距離
ただし,W0.05h,fは同じ単位を用いる。
3.23
プレカラム誘導体化(precolumn derivatization, precolumn labeling)
分析種をカラムに流入する前に誘導体化試薬などと反応させることによって誘導体化する操作。
3.24
ポストカラム誘導体化(postcolumn derivatization, postcolumn labeling)
分析種をカラムで分離後,溶出液に誘導体化試薬などを加えて反応させ誘導体化する操作。
3.25
イソクラティック溶離(isocratic elution)
単一組成の溶離液(移動相)によって分析種を展開,溶出させる操作。
3.26
グラジエント溶離(gradient elution)
溶離液(移動相)の組成を変化させながら分析種を展開,溶出させる操作。
3.27
標準液(reference solution)
分析の際の標準となる濃度,分子量(式量)などの特性値が既知の溶液で,その特性値が高速液体クロ
マトグラフィーに使用するのに必要かつ十分な程度で確定されているもの。
3.28
分離モード(separation mode)
高速液体クロマトグラフィーにおいて,主に分離を支配する作用様式に基づいて分類した様式。
3.29
ベースライン(base line)
クロマトグラムにおいて,分析種ピークがなく溶離液だけが検出器を通過している部分の信号を記録し
たもの。基線ともいう。
3.30
希釈標準液(diluted reference solution)
標準液を所定の濃度に希釈した溶液。
3.31
混合希釈標準液(mixed reference solution)
複数の標準液又は希釈標準液を混合して各成分を所定の濃度に調製した溶液。
5
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.32
内標準物質(internal standard)
内標準法において標準として用いる物質。
3.33
分子量分布(molecular weight distribution)
通常の高分子化合物などのような分子量に多分散性がある化合物について,分子量の関数として,それ
ぞれの分子種の量を表したもの。
3.34
分取液体クロマトグラフィー(preparative liquid chromatography)
分析種を含む画分を集めることを目的とした液体クロマトグラフィー。
3.35
分画(fractionation)
カラムで分離され,溶出してきた成分を分け取る操作。
3.36
画分(fraction)
分画操作によって分け取られた溶液。フラクションともいう。
3.37
デッドボリューム(dead volume)
カラムに不活性な成分に対する保持容量。カラム中の遊び空間体積(空隙容量),試料導入装置,配管,
検出器の実効体積などが関係する。死容量,ホールドアップボリュームともいう。
3.38
分析法バリデーション(method validation)
高速液体クロマトグラフィーを用いる分析において,分析方法が分析の目的にかなっていることを実証
するための各種試験。妥当性確認ともいう。
4
高速液体クロマトグラフィー概説
液体クロマトグラフィー(LC)は,移動相として液体を用いるクロマトグラフィーで,カラムの固定相
と移動相との間で生じる各分析種の相互作用の差によって混合物の分離を行う物理化学的分離分析法の一
つである。移動相をポンプを用いて高圧で送液することによって,短時間で高性能の分離を得るようにし
て分析する方法を高速液体クロマトグラフィー(HPLC)と呼ぶ。
液体クロマトグラフィーは,ガスクロマトグラフィーでは測定が困難な不揮発性,熱的に不安定な化合
物の測定などに適用できる。液体中での拡散速度が遅いため,ガスクロマトグラフィーに比べ分離能力は
高くないが,定量性に優れる,分離した化合物を容易に分取できるなどの特長も備えている。応用範囲は,
一般的な有機物だけでなく,イオン性化合物,天然物,高分子化合物などと非常に広く,種々の分野で利
用されている。
高速液体クロマトグラフィーでは,使用する固定相と移動相との組合せによって多種多様な分離機構を
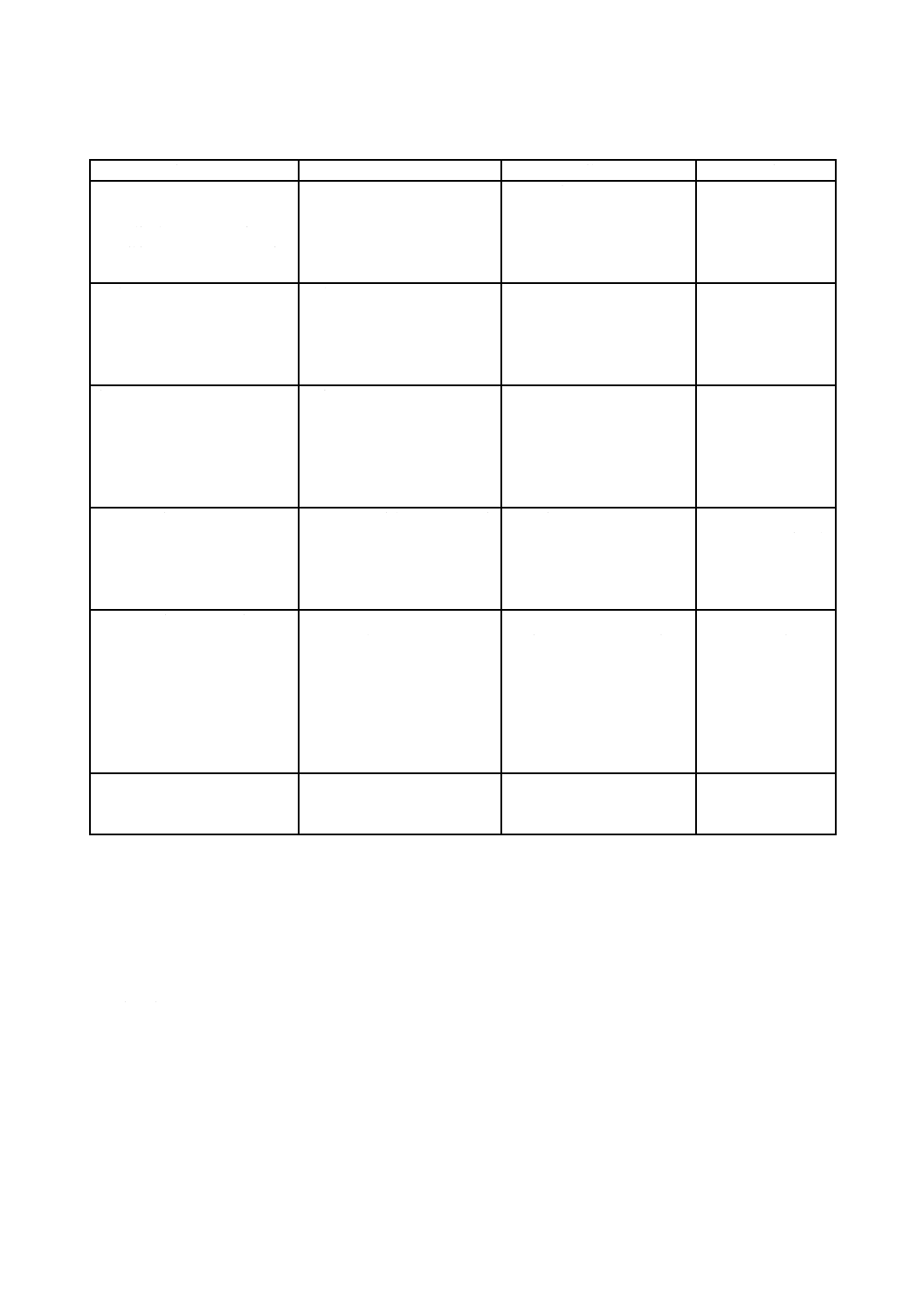
得ることができる。表1に,分離モード,特徴,代表的なカラム充填剤及びその用途を示す。
6
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−高速液体クロマトグラフィーの分離モード,特徴,代表的カラム充塡剤及び用途
分離モード
特徴
代表的充塡剤
用途
分配クロマトグラフィー
(逆相分配クロマトグラフィ
ー,順相分配クロマトグラフィ
ー,逆相イオン対クロマトグラ
フィーを含む。)
・固定相と移動相間との分配
平衡に基づく分離。
・多種類の充塡剤と多様な移
動相の組合せとによって広
範囲の対象に適用可能。
オクタデシル基,アミノ基な
どを導入したシリカゲル,有
機シリカゲル,ポーラスポリ
マーなど。
・低分子から高分子
まで広範な対象
物の分離。
・同族体の分離。
吸着クロマトグラフィー
・無機酸化物固定相による溶
質の吸着平衡に基づく分
離。
・移動相として非極性有機溶
媒を使用。
シリカゲル,アルミナ,チタ
ニア,カーボンなど。
・極性物質,異性体
の分離。
親水性相互作用クロマトグラ
フィー(HILIC)
・親水性相互作用に基づく分
離。
・極性の高い固定相を使用。
移動相には水に混和可能な
有機溶媒を使用。
極性の高いシリカゲル,アミ
ノ基,ジオール基,両性イオ
ン基などを導入した化学結
合形シリカゲル,親水基を含
有するポーラスポリマーな
ど。
・糖,アミノ酸など
の親水性化合物。
イオン交換クロマトグラフィ
ー
(イオン排除クロマトグラフ
ィー,イオンクロマトグラフィ
ーを含む。)
・イオン交換体とイオン性溶
質との静電的相互作用によ
る分離。
・広範囲の対象に適用が可能。
カルボキシメチル基,ジエチ
ルアミノメチル基などのイ
オン交換基を導入したシリ
カゲル,合成ポリマー。
・イオン性物質の分
離分析,脱塩,塩
交換。
サイズ排除クロマトグラフィ
ー
(ゲルろ過クロマトグラフィ
ー,分子ふるいクロマトグラフ
ィー)
・高分子充塡剤のネットワー
ク又は細孔による分子ふる
い作用に基づく分離。
・温和な処理条件。
・親水性高分子充塡剤と合成
高分子充塡剤とによって使
用目的が異なる。
デキストランゲル,アガロー
スゲル,ポリスチレンゲル,
ポリメタクリレートゲル,シ
リカゲルなど。
・たんぱく質,酵素
などの分離及び
精製,脱塩(親水
性高分子充塡
剤)。
・合成高分子の分子
量分画(合成高分
子充塡剤)。
アフィニティークロマトグラ
フィー
・生物由来の分子識別能によ
る分離。
・選択性が極めて高い。
酵素,ペプチド,糖類などを
結合させた担体。
・生理活性物質の濃
縮,分離,精製。
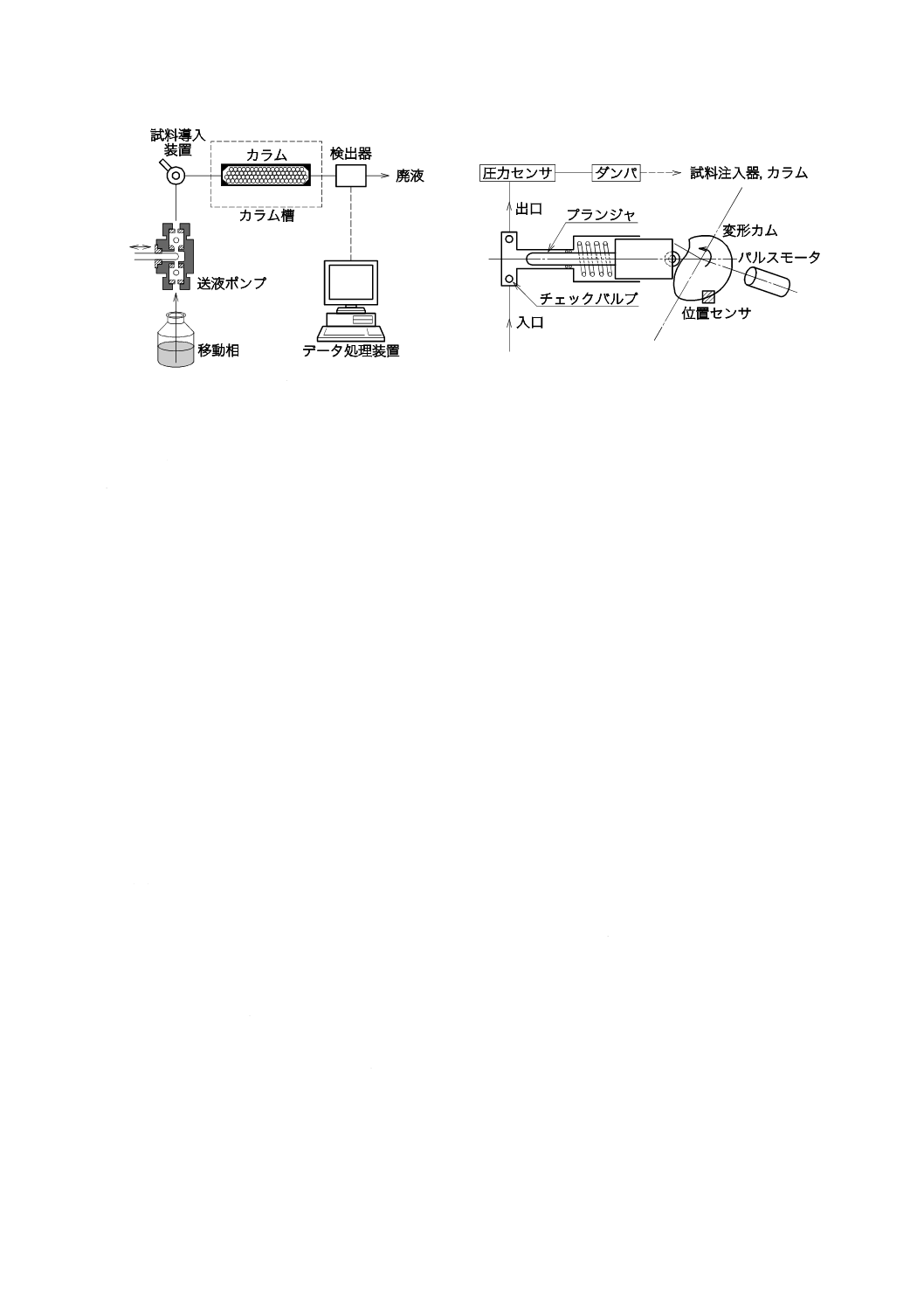
高速液体クロマトグラフの基本構成例を図1に示す。構成は,移動相送液部(送液ポンプ),試料導入部
(試料導入装置),カラム・カラム槽部,検出部(検出器)及びデータ処理部(データ処理装置,記録計)
から成る。移動相の流量はポンプで設定され,一定の流量で試料導入部,カラムへと送液される。ポンプ
は,通常0.01〜10 mL/minの送液流量範囲と,40 MPa程度の吐出圧力とが要求され,図2に示すような往
復動形プランジャポンプが主に用いられている。グラジエント溶離法を用いる場合には1〜4台のポンプ又
はグラジエント装置が用いられる。
7
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1−液体クロマトグラフの基本構成例
図2−往復動形プランジャポンプの基本構成例
試料溶液はマイクロシリンジなどで計量され,試料導入装置によって一定量の試料溶液がカラムに導入
される。多数の検体を順次自動で導入する自動試料導入装置(オートサンプラー)が用いられることもあ
る。
高速液体クロマトグラフィーに用いられるカラム充塡剤は,通常,直径1〜20 µmの比較的均一な粒度
分布をもち,内径1〜12 mm,長さ数十cm以下のステンレススチール又はプラスチック製のクロマト管に
充塡され使用される。カラム充塡剤の粒子径,クロマト管のサイズは,使用目的によって異なる。ロッド
形に成型した連続体であるモノリス形のもの,内径1 mm未満のミクロカラム,内径12 mm以上のセミ分
取カラムも用いられる。カラムの性能は,理論段数,分離度,シンメトリー係数などによって示される。
カラムで分離された分析種は,各成分の物理的,光学的,電気的,化学的などの特性を基に検出器で検出
される。高速液体クロマトグラフィーにおける代表的な検出器は,紫外可視吸光光度検出器である。通常,
4〜20 μLのフローセルが装着され,分離された分析種の特定波長における吸光度(吸収強度)を測定する。
その他の検出器としては,蛍光検出器,示差屈折率検出器,電気化学検出器,電気伝導度検出器,質量分
析計,蒸発光散乱検出器,荷電化粒子検出器,ICP発光分光分析計,ICP質量分析計などがある。高速液
体クロマトグラフィーに用いられる代表的な検出器及びその特徴を表2に示す。選択的な検出,µg/L以下
の高感度検出が要求される場合には,分析種の特性に応じて検出器を使い分ける。
分析種が直接検出できない,又は高感度検出ができないなどの場合には,カラムに導入する前に誘導体
化するプレカラム誘導体化,カラムで分離後,溶出液に誘導体化試薬を混合し誘導体化を行うポストカラ
ム誘導体化が用いられる。
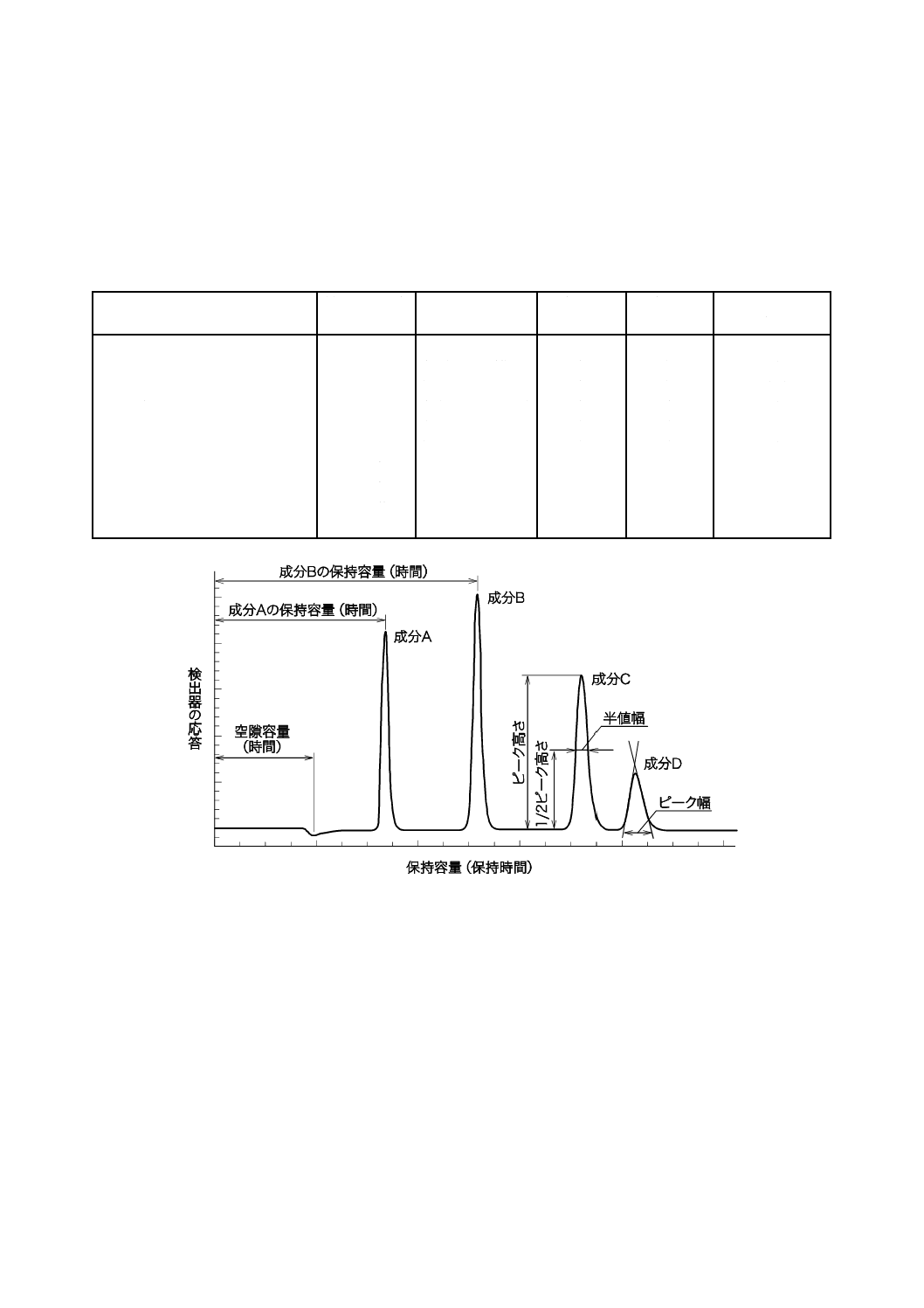
検出器で得られた各分析種の応答は,電気信号に変換されてデータ処理装置に送られ,図3に示すよう
な時間軸に対して検出器の応答を記録したクロマトグラムが得られる。データ処理装置は,このクロマト
グラムを基に種々のパラメーターを計算し,分析種の定性・定量を行う。分離された各分析種の定性は,
同一条件下で得られた標準液のクロマトグラムにおける保持値(保持時間など)との比較によって行う。
質量分析計,フォトダイオードアレイ検出器などを用いる場合には標準液のスペクトルと比較して定性を
行うことができる。定量は,クロマトグラムからピーク面積値又はピーク高さ値を求め,あらかじめ分析
種を含む標準液によって作成した検量線に当てはめ,その量を求める。
一方,得られるクロマトグラムのピーク部分に相当する溶出液を採取することによって,分離された分
析種の分取・精製を行うことができる。
8
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分析の生産性を向上させるために,従来に比べて分析時間を大幅に短縮させた超高速分析を行う場合が
ある。超高速分析は,試料をより短い時間で分析できる,分析条件の検討に要する検討時間が少なくて済
むなどの利点があり,高い圧力で移動相を送液する,専用のカラムを用いるなどの方法で実現することが
できる。
表2−高速液体クロマトグラフィーに用いられる代表的な検出器及びその特徴
検出器
最小検出感度
g
選択性
温度の影響
流速の影響
グラジエント
溶離
紫外可視吸光光度検出器
10−11
有(吸光物質)
小
無
可
蛍光検出器
10−13
有(蛍光物質)
有
無
可
示差屈折率検出器
10−7
無
有
無
不可
電気化学検出器
10−12
有(酸化還元物質)
有
有
可
電気伝導度検出器
10−10
有(イオン)
有
有
困難
質量分析計
10−14
無
有
有
可
蒸発光散乱検出器
10−8
無
無
無
可
荷電化粒子検出器
10−9
無
無
無
可
ICP発光分光分析計
10−11
有
無
無
可
ICP質量分析計
10−14
有
無
無
可
図3−クロマトグラム及び各パラメーターの名称
5
装置
5.1
装置の構成
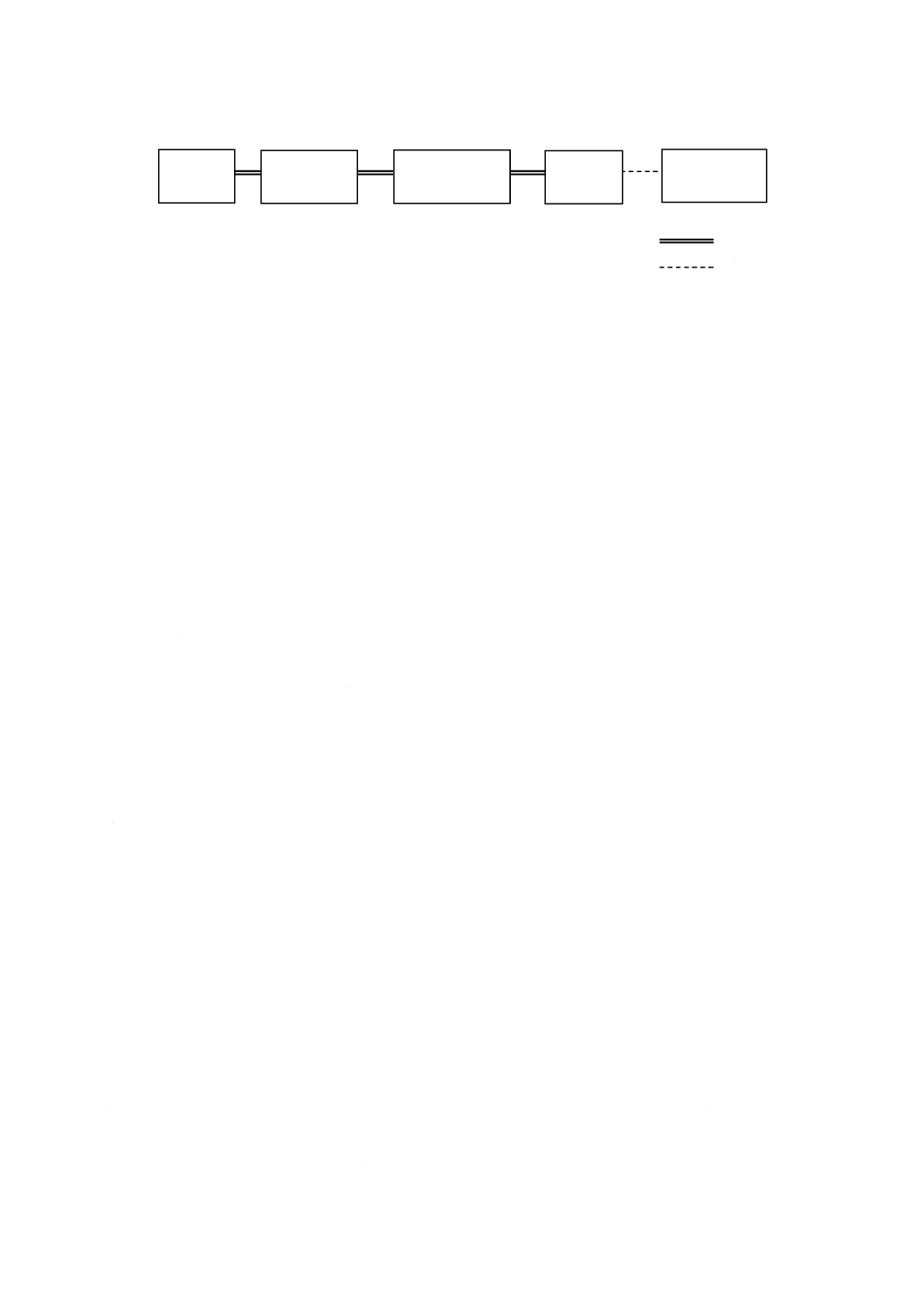
高速液体クロマトグラフは,移動相送液部,試料導入部,分離部,検出部,データ処理部などで構成す
る。その基本構成の一例を図4に示す。試料,試薬及びキャリヤーは,細管の中で連続的な流れ系を形成
する。
9
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4−高速液体クロマトグラフ(一例)
5.2
移動相送液部
溶離液(移動相)をカラムに送液するために構成された装置部で,主に次のものから構成される。
a) 溶離液槽 溶離液槽は,溶離液(移動相)の化学的な組成を変化させないような材質を選択し,使用
中に環境からの汚染,移動相の蒸散・腐敗などが起きないようにする。
b) 脱気装置 溶離液(移動相)に溶解している空気を連続的に取り除き,装置内で温度変化及び圧力変
化に伴い発生する気泡のトラブルを未然に防ぎ,安定した流量及びバックグラウンドが得られるよう
にするために用いられる。
c) 送液ポンプ 設定した溶離液(移動相)を正確かつ精密な流量でカラムに送液するためのもので,更
に次の項目を満足することが望ましい。
1) 広範囲な流量設定を可能とする。
2) 高精度な設定流量を可能とする。
3) 低脈流である。
4) 幅広い送液圧力に対し安定な吐出量が得られる。
5) 接液部の材質は,耐薬品性に優れている。
6) 移動相の交換が容易にできる。
d) グラジエント溶離部 複数の溶離液(移動相)を用い,時間的にその組成を変化させるための制御部
と混合溶液とを均一にするためのミキサー部から成り,設定できる混合比の範囲が広く,濃度が精確
であることが望ましい。
5.3
試料導入部
マイクロシリンジなどを用いて手動で導入するマニュアルインジェクター(手動試料導入装置)及び多
数の検体を順次自動で導入するオートサンプラー(自動試料導入装置)がある。いずれにおいても,測定
用試料溶液を一定量,再現性よく系内に導入するため,導入した試料の残存が少なく,吸着などが生じな
い材質・構造が望ましい。
マニュアルインジェクターでは,試料溶液をマイクロシリンジで必要量計量する方法及び装置に取り付
けられた一定容量の試料ループで計量する方法がある。オートサンプラーでは,種々の方式があり,試料
の計量方法で分けると一定容量の試料ループで計量する方式,シリンジ又はポンプの吸引量で計量する方
式などがある。また,カラムへの導入方法で分けると試料ループだけがカラム流路につながる方式,試料
ループに加えてニードルも直接カラム流路につながる方式などがある。多数の試料溶液を自動で導入する
ためにオートサンプラーを用いる場合は,その仕様として導入量の再現精度,正確さ,残存量の少なさ,
試料最少必要量が少ないなどの基本性能を備えることが望ましい。さらに,分析種の分解,揮散を少なく
する冷却機能の付加など用途にかなったものが望ましい。
なお,試料前処理又はプレカラム誘導体化機能を内蔵するオートサンプラーを使用してもよい。
移動相
送液部
試料導入部
分離部
データ処理部
検出部
細管
信号
10
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.4
分離部
分離部は,次による。
a) カラム 分析目的に応じて箇条7に規定したカラムを用い,試料導入部からの入口流路及び検出部へ
の出口流路との配管には,試料バンドの拡散を防ぐためデッドボリュームをできるだけ少なくするよ
う留意する。
なお,分析カラムの性能劣化を防ぐためガードカラムを使用することがある。
b) カラム槽 試料中の分析種の分離能を一定に保ち,定量精度を確保するためカラム槽を使用する。カ
ラム槽は,温度制御機能をもつことが望ましく,カラム性能の最適化及び分離選択性を高め,環境温
度変化などにカラム温度が影響されることのないように,温度を設定する。
5.5
検出部
検出部は,カラムからの溶出液を微量フローセルに導き,移動相をバックグラウンドとし分析種の濃度
又は量を種々の原理によって検出するものであり,次による。
a) 紫外可視吸光光度検出器 紫外・可視部で吸収される分析種の吸光度を検出するもの。分析種の吸収
特性から吸収波長を設定する。分光された光をフローセルに通す吸光光度検出器が多く利用されてい
る。フォトダイオードアレイ検出器の場合には,フローセルの後で光を分光させ,それぞれの波長に
おける吸光度変化を測定し,各波長でのクロマトグラム及び各保持時間によって分析対象成分の定性,
ピーク成分の純度確認などが行われる。
b) 蛍光検出器 励起光によって励起され特定波長の蛍光を発する分析種について,その蛍光強度を検出
するもの。最適波長の設定,ピーク成分の定性確認などのため,励起,蛍光の波長スキャンをして励
起,蛍光スペクトルを採取できるものも利用されている。
c) 示差屈折率検出器 分析種の溶出による移動相溶液の屈折率の変化を屈折率の差として検出するも
の。
d) 電気化学検出器 一定の電圧を印加した作用電極において,分析種の酸化及び還元反応によって生じ
た電流を検出するもの。
e) 電気伝導度検出器 溶離液(移動相)と分析種との電気伝導度の差を検出するもの。
f)
質量分析計 カラムから溶離された分析種をイオン化し,m/z(質量電荷比)に応じて検出するもの。
g) 蒸発光散乱検出器 溶離液(移動相)を蒸発させて分析種を球状微粒子とし,これに光照射を行うこ
とによって生じる散乱光の強度を検出するもの。
h) 荷電化粒子検出器 溶離液(移動相)を蒸発させて分析種を球状微粒子とし,コロナ電極によってプ
ラス荷電した窒素イオンと衝突させてプラス帯電させ,その電荷量を検出するもの。
i)
ICP発光分光分析計 高周波を用いて発生させた高温アルゴンプラズマ中に分析種(元素)を導入し
て気化励起させ,得られる原子スペクトル線の発光強度を検出するもの。
j)
ICP質量分析計 高周波誘導結合プラズマを用いて分析種(元素)をイオン化し,生成したイオンを
質量分析計に導入して分析種(元素)のm/z(質量電荷比)に応じて検出するもの。
k) その他の検出器 その他の検出器として,次に示すものがある。
1) 赤外分光光度計
2) 旋光度検出器,円二色性検出器
3) 水素炎イオン化検出器
4) 放射線検出器
5) 誘電率検出器
11
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6) 化学発光検出器
7) 原子吸光分光分析装置
8) 熱検出器
9) 光散乱検出器
10) 粘度検出器
11) イオン電極
12) 超音波検出器
13) 核磁気共鳴装置
5.6
データ処理部
データ処理装置は,クロマトグラムを表示させ,保持時間,ピーク高さ・面積などから,定量に必要な
波形処理・検量線作成機能などによって,定量値などを算定し表示できるもの。また,スペクトル解析機
能をもつものもある。
5.7
附属装置
分析の目的によって,必要であれば次の附属装置を備える。
a) コンピューター(ワークステーション用,データ処理・管理用)
b) 流路切換バルブ(カラムスイッチング,リサイクル,バックフラッシュなど)
c) フラクションコレクター
d) ポストカラム反応器
e) 溶離液回収装置
6
水,試薬及び溶媒
水,試薬及び溶媒は,次による。
a) 水 この規格で用いる水は,逆浸透法,蒸留法,イオン交換法,紫外線照射,ろ過などを組み合わせ
た方法によって精製した水で,分析に干渉しない水質のものとする。水質は比抵抗値,総有機物(TOC),
吸光度などを指標とし評価する。使用直後から容器・環境から汚染を受け劣化するので,分析に影響
を与えない材質の容器を選定し,適切な方法で洗浄後使用する。
b) 試薬及び溶媒 試薬及び溶媒は,本来の性質以外で分析に影響を与えない品質のものとする。多くの
等級が存在する場合があるので,目的にあったものを選択する。開封後は環境から汚染を受け劣化す
るので,できるだけ早く使用する。また,製造後時間が経過すると品質が変化する場合があるので,
極力新しいものを使用する。
7
カラム及びカラム充塡剤
7.1
カラム
カラムは,次による。
a) カラム種の表示方法 組合せによって種々のカラムがあるので,カラム仕様を表示する場合は,カラ
ム管の材質,長さ,内径及び充塡剤名を記載する。
b) クロマトグラフィー管の材質 十分な強度をもち,内面が平滑で溶離液及び分析種に対して不活性な
材質を用いる。ステンレス鋼,ポリエーテルエーテルケトン(PEEK),ガラスライニングステンレス
鋼,PEEKライニングステンレス鋼,フューズドシリカ(溶融シリカ)チューブなどが用いられる。
12
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) カラムの分類 カラムは,分析試料の種類,量,特性,分析上の諸条件などによって,種々のサイズ
のものが使用され,内径によって次のように分類する。
1) セミ分取カラム :内径10 mm以上,50 mm未満
2) 汎用カラム :内径 3 mm以上,10 mm未満
3) セミミクロカラム :内径 1 mm以上,3 mm未満
4) ミクロカラム :内径 1 mm未満
7.2
カラム充塡剤
カラム充塡剤は,1〜20 μm程度の比較的均一な粒子径をもつもの,連続体であるモノリス形のものなど
が用いられる。操作条件下で物理的,化学的に安定なもの。
カラム充塡剤の材質としては,シリカゲルなどの無機化合物,シリカゲル骨格にメチル基又はエチレン
基を導入した有機シリカゲル,ポリスチレンゲルなどの高分子化合物,これらを母体として表面に有機分
子を化学結合したものなどがある。
7.3
分離モード及びカラム充塡剤
高速液体クロマトグラフィーには,カラム充塡剤と溶離液との組合せによって種々の分離モードがある
が,分析対象となる試料の物理的・化学的特性によって,適切な分離モードを選択する必要がある。
7.4
カラム性能評価
カラム性能は,ピーク形状,理論段数,シンメトリー係数,カラム寿命,分離度,塩基性化合物の吸着
性,選択性,最大試料負荷量などを評価することがある。
カラム性能評価をする場合には,目的に応じて環境及び装置の影響が少なく,再現性が得られやすい条
件で行う。
8
操作
8.1
試料の準備
高速液体クロマトグラフ分析においては,あらかじめその分析目的によって,試料に合わせた試料採取
方法を選択する必要がある。また,採取した試料は,その形態及び分析目的によって,そのまま高速液体
クロマトグラフに導入できる場合,及び分析種の抽出操作,誘導体化,濃縮操作などの前処理操作を必要
とする場合に分けることができる。
8.1.1
試料採取
分析目的,試料の性質及び試験項目に最も適した方法で,試料母集団を代表できる,又は各試料の特性
の差を明らかにできるような試料採取を行う。目的物質によっては,試料採取の時期,時刻によって量的
に変動する場合,分析対象の個体差,内部での不均一性が高い場合がある。全体の平均値として時間,空
間,個体によらない平均値が欲しい場合,試料の時間変化,試料採取の場所による差,試料の特性をみた
い場合など,目的に応じて試料採取対象物の状態,部位,時間及び方法を選択する。個別に規格がある場
合はそれに準じる。また,外的要因,経時変化などに対する変質を防ぐため,なるべく速やかに前処理操
作,高速液体クロマトグラフへの試料導入を行う。試料は生物学的,化学的,又は物理的変化を保存中に
受ける可能性がある。変性を防止できない分析対象成分の場合は,採取後直ちに測定を行わなくてはなら
ない。
8.1.2
試料保存
試料を保存する必要性がある場合は,事前にその保存及び運搬方法で試料の変質に問題がないことを確
認する。予備実験で取り扱う試料量又は分析種濃度がこの測定のレベルとかけ離れていると,設定される
13
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
条件が効果的ではなくなるので留意が必要である。変質が懸念される場合は,その防止策を施す1)。試料
採取,保存に使用する容器,器具は,試料の吸着,又は試料中へ溶出する成分を含まない材質のものを用
いる。また,保存容器には必ず必要事項を書いたラベルを貼るようにする。データの信頼性を確保するた
め,二重測定が義務づけられている場合は,同一試料を同時に最低2点以上採取する必要がある。
注1) 例えば,生体試料では酵素及び微生物による分析種の分解,代謝産物及び繁殖による汚染など
の可能性がある。このような場合は防腐剤などの保存処理剤の添加,低温保存,pH調整,ろ過,
熱処理,乾燥などの対策を講じる。保存処理剤の添加の分析種への影響及びクロマトグラムへ
の影響については十分注意する必要がある。化学的変化には酸化,変性などがあり,防止には
抗酸化剤の添加,pH調整,低温保存が有効である。低温で試料を保存する場合,保存温度は試
料の種類及び保存期間を考慮して決定する。短期間の保存なら冷蔵保存が可能な場合がある。
長期間保存する場合,冷凍庫,液体窒素中などで凍結保存を行う場合がある。いずれの場合も
冷蔵庫,冷凍庫などの温度が設定どおり保たれているか確認を行う。凍結保存した試料の解凍
は,試料によって適切な方法が異なるので注意する。解凍のとき,局部的に水分の量が異なり,
試料濃度に分布ができるのでよく混和する必要がある。また,解凍が終わる前に保存容器の蓋
を開けると空気中の水分が凝結するので注意する。凍結解凍の繰返しは試料の分解,失活が起
こる可能性があるので避ける。保存の最適条件を設定するためには,この測定に先立ち目的試
料を用いてあらかじめ試しておくとよい。保存容器は,目的成分が反応したり吸着することな
く,また,試料中へ溶出する成分を含まない材質からなり気密で丈夫な容器がよい。容器の密
閉のために,蓋の部分をフィルム,テープなどでシールすることもある。保存容器には必ず必
要事項を書いたラベルを貼るようにする。
8.1.3
前処理操作
高速液体クロマトグラフに導入する前に,場合によって分析目的,試料の性質,試験項目などに最も適
した方法で,試料の前処理操作を行う。前処理操作は,特異性,精度,感度などの向上,測定妨害物質の
除去,カラム及び分析機器の保護並びに劣化の防止,測定操作,手順の簡易化,分析種の安定化などを目
的として行われる。
前処理にはひょう量,除たんぱく,抽出(液液,固相,溶媒蒸気蒸留,限外ろ過膜分離,超臨界流体な
ど),クリーンアップ(液液抽出,固相抽出など),脱水,脱塩,濃縮,定容,誘導体化,標準物質添加,
溶媒の調製(誘導体化,カラム導入に適した溶媒に溶解)などの操作がある。個別に規格がある場合はそ
れに準じる。内標準物質を添加する場合,クロマトグラム上で試料マトリックスと十分に分離され,かつ
目的成分とは必要以上に保持時間の差が大きくならないよう考慮する。ただし,質量分析計を用いる場合
は必ずしも内部標準物質と分離する必要がないことがある。また,質量分析計を用いる場合,見かけ上,
分析種が単一ピークであっても試料マトリックス成分などによるイオン化効率の変動(マトリックス効果)
が起こる場合がある。マトリックス効果が起こるとイオン化抑制又はイオン化促進によって定量値が真値
からずれる危険性があるため注意する。試料は一般には移動相と同一又は移動相より溶出力の小さな少量
の溶媒に溶解するのが望ましいが,目的成分の安定性及びその分離能に影響を与えるので,考慮して溶媒
を選択する。必要に応じ,不溶解成分除去のため孔径0.2〜0.5 μmのメンブランフィルターで試料のろ過
を行う。これらの試料前処理操作において,試薬,容器,環境などからの汚染,不純物の混入,分析種の
損失などがあるので注意する。
14
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.1.3.1
具体的な前処理操作
8.1.3.1.1
除たんぱく
a) 沈殿法 酸,重金属及び中性塩,有機溶媒などの沈殿剤(変性剤,除たんぱく剤)を試料に添加する
ことによってたんぱく質を変成させ沈殿除去する。
b) 限外ろ過法 限外ろ過膜でたんぱく成分を取り除く。
8.1.3.1.2
抽出
抽出は,次による。
a) 有機溶媒抽出(液液抽出) 疎水性物質の分析に適用される方法で,試料水溶液と混ざり合わないク
ロロホルム,酢酸エチルなど有機溶媒を用いて,分析種を有機溶媒層に抽出する。
b) 固相抽出 選択性のある充塡剤を詰めたカラムに試料溶液を通し,目的物質を充塡剤に捕捉し,次い
で,溶離液を通して溶出する。一般にカラムのコンディショニング,試料添加,洗浄,分析種の溶出
という手順で行う。
c) 超臨界流体抽出 超臨界流体を溶剤(移動相)として用い,分析種を抽出する。
8.1.3.1.3
脱塩
試料中に塩が存在することが不都合な場合(例えば,イオン交換クロマトグラフィーにおいて試料溶液
が初期溶離液のイオン濃度よりも濃い場合など),試料の脱塩操作を行う必要がある。透析法,限外ろ過法,
ゲルろ過クロマトグラフィー,イオン交換膜法などが用いられる。
8.1.3.1.4
濃縮
測定法の感度に比較して試料の濃度が低い場合は,試料の濃縮を行う。減圧濃縮,窒素,空気などを吹
き付ける気流濃縮,凍結乾燥,目的物質を小さなカラムなどに吸着させ少量の溶離液で溶出させる方法,
沈殿させ少量の液に溶解する方法,少量の液相に抽出する方法などがある。
8.1.3.1.5
誘導体化
誘導体化は,次による。
a) 誘導体化の目的 分析対象成分が使用する検出器に応答するような化学構造をもっていなかったり,
たとえそのような構造をもっていても,それを利用したのでは十分な感度が得られない場合,検出器
への応答性を向上させる目的で誘導体化を行う。また,単に検出感度が向上するだけでなく,検出に
おける選択性も向上する。その他,固定相への保持を増大させる,固定相への非特異的な吸着を防ぐ,
分離に対する選択性を高める(光学異性体分離など)といった目的で誘導体化を行う。
b) 誘導体化の反応の種類 検出器への応答性の高い官能基をもった試薬を反応・結合させる場合と,分
析対象成分自体を,酸化,還元,分解などの反応によって応答性の高い構造物に変換させる場合があ
る。
8.1.3.2
主な試料分析における試料採取・前処理操作
8.1.3.2.1
生体試料
生体試料は,次による。
a) 試料採取 試料の採取及び情報の利用にはインフォームドコンセントによる被験者の同意及び所属機
関における倫理委員会相当会議の承認が必要となる。採取には,医師,看護師,臨床検査技師などの
有資格者だけが行うことができるなどの制約がある。生体試料の全ての検体は感染性をもつものとみ
なして慎重に取り扱わなければならない。保護着,保護具(手袋・めがね・マスクなど)の使用,手
洗いの励行,実験室での喫煙・飲食・化粧行為の禁止などの対策を講じる。
15
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分析の目的,成分によって,採血の部位,時間,方法などを決定する。抗凝固剤,血液分離剤を用
いる場合には,それらの分析系への影響の有無を考慮する。蓄尿試料の場合に添加する防腐剤の分析
系への影響を考慮する。
b) 前処理操作 血清又は培養ろ液を精製する場合,適切な方法で目的とする物質を予備的に分画して濃
縮しておくことが望ましい。可溶性たんぱく質の予備的分画としては,硫安分画が最も簡便かつ安全
で,脂質成分もかなり取り除ける。
高分子化合物の濃縮に限外ろ過膜が使用されているが,濃度の低いたんぱく質試料(10 μg/mL以下)
の場合にはたんぱく質が膜に吸着されて回収率が低下することがある。このような場合,小さなイオ
ン交換カラムに捕集・溶出させたり,硫酸アンモニウムを添加して疎水性カラムで捕集すると有効な
場合がある。
たんぱく質を含む試料を乾固すると,変性・不溶化して回収が困難になるので注意する。
8.1.3.2.2
医薬品
前処理操作として,以前は,有機溶媒抽出が広く行われていたが,近年,固相抽出を行うことが増えて
いる。分析対象成分の性質によって各種充塡剤を使い分ける。誘導体化を行う場合,特異性及び高感度検
出が可能な蛍光誘導体化試薬を使用することが多い。
8.1.3.2.3
環境試料
環境試料は,次による。
a) 試料の採取
環境試料の分析値は,採取条件及び採取方法によって影響を受けるため,事前に十分に考慮した上
で試料を採取する。また,試料の採取の際には,採取場所,日時,気象条件などを記録する。
1) 大気試料 気象条件(気温,湿度,風速,風向など),採取時刻などを十分に考慮し事前に調査した
うえで,分析種を吸着する捕集管などにより,試料を採取する。
2) 浮遊粒子状物質 ろ過剤(セルロースエステル,ポリカーボネート,石英繊維及びテフロン)で捕
集後,ろ過剤を一定時間溶媒に浸せきしたり,超音波をかけて抽出する。
3) 酸性雨 湿性降下物と乾性降下物を含めた一括採取法(バルク方式)と湿性降下物だけを採取する
方式があり,一括採取法の場合は常時開放形ろ過式採取装置など,湿性降下物だけを採取する場合
は降水時開放形の捕集装置などにより,試料を採取する。
4) 水試料 試料の種類に応じて採取方法を工夫する。雨水は少ない雨量に対応した容器の形状,回収
までの蒸発又は変質防止,不溶物の混入に対し配慮し,排水は油分,有機溶媒,界面活性剤などが
含まれる可能性を考慮し,必要に応じて対策を取る。不溶物はろ過によって除去する。
なお,ガラス製採取容器は無機系分析種に吸着性を示すことがあるが,その場合にはポリエチレ
ン又は四ふっ化エチレン樹脂製のプラスチック容器を使用した方がよい。これらは水で洗浄し,酸
洗いなどコンタミネーションの誘因になるようなことは避ける。
5) 底質・土壌 粉砕後,均一に混合するか,又はふるい分けによって粒度分画を行う。
なお,乾燥,脱水などの操作も必要となるが,揮発性成分,熱分解性成分の分析の場合には十分
注意を払う。
b) 前処理操作
1) 水試料 不溶解成分の除去のため0.2〜0.5 μmのメンブランフィルターでろ過を行う。場合によっ
て抽出,濃縮などの操作を行う。
16
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 底質・土壌 分析対象成分に応じ前処理操作を検討する。無機イオン種の場合,水,pHを調整した
塩水溶液などによって抽出後,酸分解,ろ過,固相抽出による有機化合物の除去を行う。有機化合
物の場合,液液抽出後,固相抽出によって濃縮,クリーンアップを行う。超臨界流体抽出法(SFE)
が用いられる場合がある。
8.1.3.2.4
界面活性剤
原料を移動相に直接溶解させてカラムに導入可能な場合がある。界面活性剤を含む製品が試料の場合,
乾燥後エタノールに溶解,界面活性剤を含む可溶分をイオン交換樹脂を充塡したカラムで分画し必要に応
じ中和した後カラムに導入する。
8.1.3.3
マトリックス効果
質量分析計を用いる場合,見かけ上分析種が単一ピークであっても試料マトリックス成分などによって
イオン化効率の変動(マトリックス効果)が起こる場合がある。マトリックス効果が起こるとイオン化抑
制又はイオン化促進によって定量値が真値からずれる危険性があるため,次の方法などによって評価及び
対策をする。
a) マトリックス効果評価法 分析種の標準液及び後添加標準液の質量分析計によるレスポンスの比をマ
トリックス効果(Matrix Factor: MF)とする評価方法(参考文献[1]参照)がある。ここで,後添加標準
液とは分析種を含まないサンプルを所定の方法で前処理し,後から分析種標準を加えたものをいう。
次の式でマトリックス効果を求めた場合,MF=1はマトリックス効果が起こっていないことを表す。
1以下がイオン化抑制,1以上がイオン化促進が起こっていることを意味する。
MF=後添加標準液のレスポンス/標準液のレスポンス
六つの異なるロットの試料のマトリックス効果を上の式で計算した場合の標準偏差が15 %以下で
あることが要求される場合がある(参考文献[1]参照)。
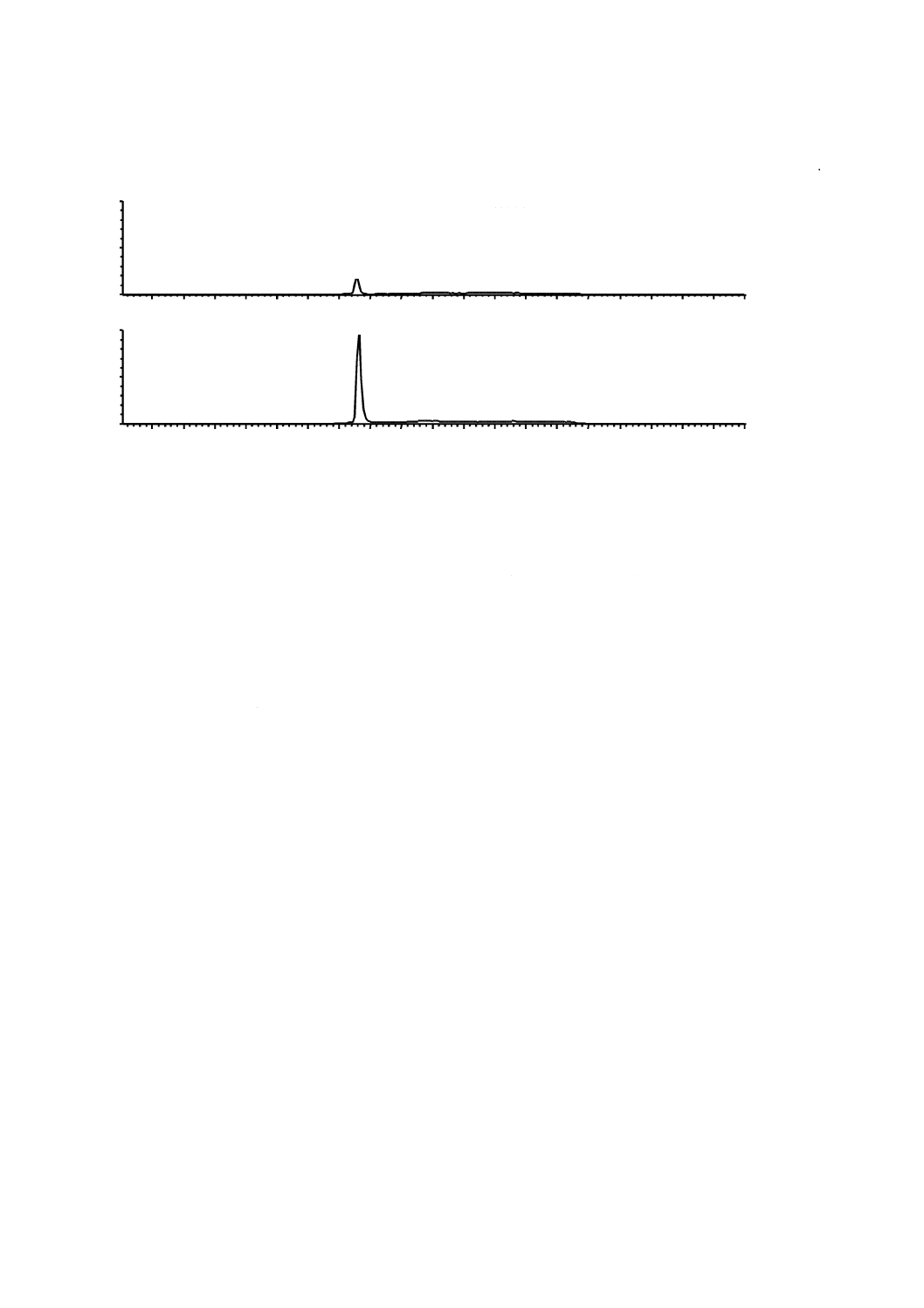
b) マトリックス効果の例 図5に質量分析計を用いた分析におけるマトリックス効果の影響例を示す。
血しょう(漿)を異なる方法で前処理し,抗アレルギー薬として使用されるテルフェナジンを同一濃
度で後添加したものをESI+MRMモードで検出している。理論的には同じ面積値が得られるはずで
あるが,沈殿によるたんぱく質除去を行った試料ではマトリックス効果によるイオン化抑制が顕著
(MF=0.2)に起こり,MSシグナルが減少している。定量分析においてこのような現象が起こると,
定量感度及び信頼性が損なわれる。
17
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
マトリックス効果あり
MF=0.2
マトリックス効果無し
MF=1
0.50
1.00
1.50
2.00
2.50
3.00
3.50
4.00
4.50
5.00
%
0
100
0.50
1.00
1.50
2.00
2.50
3.00
3.50
4.00
4.50
5.00
%
0
100
MRM
472.2 > 436.4
1.27e6
1.89
MRM
472.2 > 436.4
1.27e6
1.91
固相抽出
逆相−陽イオン交換ミックスモード
沈澱法によるタンパク質除去
Terfenadine
Terfenadine
tg= 1.5 min
試料:塩基性化合物(テルフェナジン)を 1 ng/mL 添加した血しょう(漿)
検出:タンデム四重極形質量分析計,ESI+MRMモード
図5−マトリックス効果の影響例
c) マトリックス効果の削減方法 マトリックス効果を削減するためには,前処理でマトリックス成分を
排除するか又は分析カラムでマトリックスと分析種を分離する方法がある。また,内標準に分析種の
安定同位体を用いて補正する方法,検量線作成時にマトリックスを添加して補正する方法なども使用
される。
前処理でマトリックス成分を排除する場合,分析カラムの分離モードと異なるモードの分離技術を
使用することで精製効率が向上することがあり,例えば,血中のマトリックス成分であるりん脂質を
除くために逆相−イオン交換ミックスモード固相が効果的な場合がある(参考文献[2]参照)。
8.1.4
試料調製
試料調製は,分析種の定性又は定量の精度向上,成分の経時変化を抑えるなどの分析種の安定化,カラ
ム,装置保護などの目的のために行われる。試料調製には,試料溶液の希釈・濃縮,試料溶液中の妨害成
分の除去,試料溶液への標準液の添加,分析種の誘導体化などがある。
これらの試料調製は,分析精度及び試料調製作業の効率の向上を目的とし,自動試料導入装置,高圧切
換バルブを用いたカラムスイッチングなどを利用することによって,自動化してもよい。
なお,個別規格で定められたものについては,その条件に従って試料調製を行う。
8.1.4.1
試料溶液の希釈又は濃縮
試料溶液は,分析種があらかじめ適正な濃度範囲となるよう希釈又は濃縮(溶媒抽出,固相抽出など)
し,測定用試料溶液を調製してからカラムに導入する。測定用試料溶液は,移動相に溶けることが必要で
ある。
8.1.4.2
試料溶液中妨害成分の除去
試料溶液中に固形成分,カラムに悪影響を与える成分又は定性・定量を妨害する成分がある場合は,ろ
過膜(孔径0.5 μm以下のメンブランフィルター)を用いる方法,機能膜(限外ろ過膜など)を用いる方法,
固相抽出,溶媒抽出などによって,あらかじめ妨害成分を除去する。
tg=1.5 min
沈殿法によるたんぱく質除去
マトリックス効果あり
MF=0.2
Terfenadine
固相抽出
逆相−陽イオン交換
ミックスモード
マトリックス効果なし
MF=1
Terfenadine
MRM
472.2>436.4
1.27e6
MRM
472.2>436.4
1.27e6
18
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 溶媒抽出(液−液抽出) 試料溶液と混ざり合わない溶媒を用いて,分析対象成分を抽出する方法で
ある。試料溶液が水溶液の場合は,ヘキサン,酢酸エチル,ジエチルエーテル,ベンゼン,クロロホ
ルム,ジクロロメタンなどの水溶液と混ざり合わない有機溶媒,又はこれらの溶媒とエタノール,メ
タノールなどの混合溶媒を用いて,疎水性の分析対象成分を有機溶媒層に移動させ抽出する。抽出後,
有機溶媒を取り除くことによって,濃縮操作なども行うことができる。また,試料中に脂溶性の高い
妨害成分が含まれている場合は,このような妨害成分の抽出除去に利用してもよい。
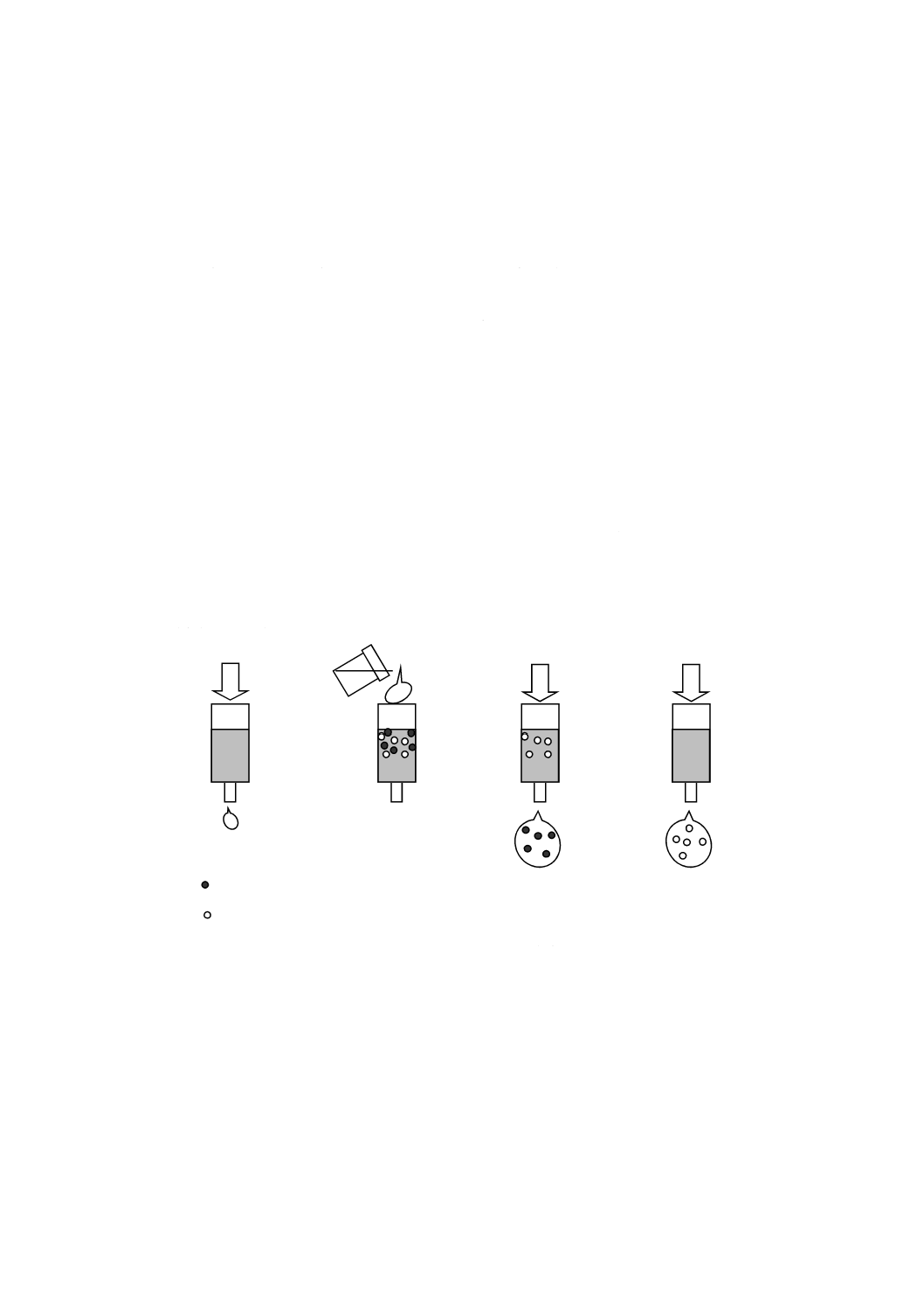
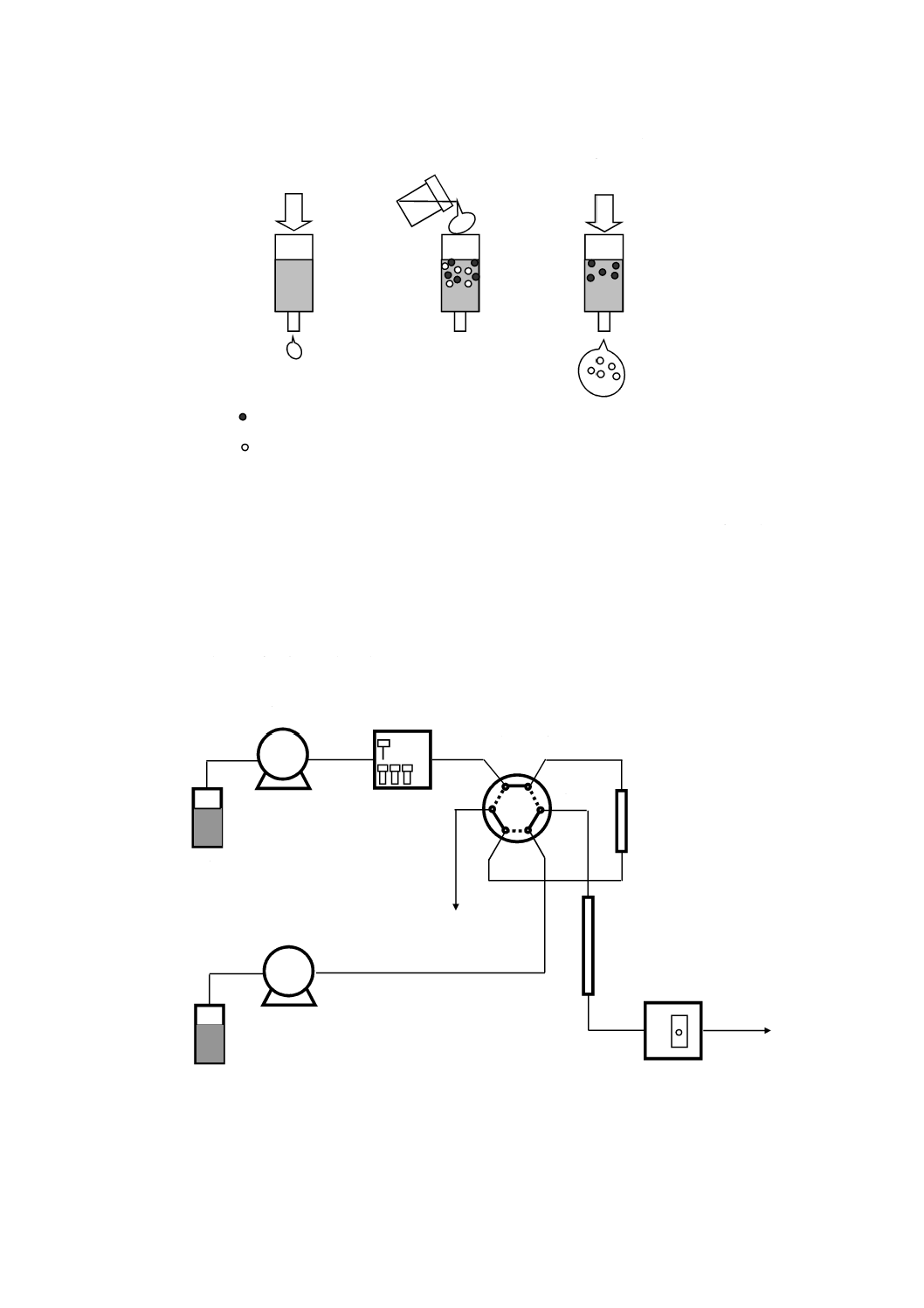
b) 固相抽出 樹脂,シリカゲルなどの充塡剤を充塡した小さなカラム又は膜を利用した方法である。試
料溶液をこの小さなカラム(一般的にカラム又はディスク)に通し,分析対象成分を充塡剤(固相),
膜に抽出(吸着)させた後,溶出液を用いて溶出させる方法(図6),又は妨害成分を充塡剤(又は膜)
に抽出(吸着)させ除去し,溶出してきた分析対象成分溶液を測定用試料溶液とする方法(図7)な
どがある。
この方法の固相抽出用の小さなカラムに充塡される充塡剤には,オクタデシル基結合シリカゲルな
どの化学結合形充塡剤,シリカゲル,アニオン交換,カチオン交換,ポリスチレン系樹脂,ポリメタ
アクリレート系樹脂など多くの種類が利用されている。分析対象成分の前処理の目的に適した充塡剤
を選択することができる。
この方法では,吸引装置又は加圧装置を用いることによって同時に多検体の前処理を行うこともで
きる。
図6−固相抽出法1 分析対象成分を吸着後溶出させる方法
コンディショニング
(洗浄・活性化)
試料溶液添加
洗浄,妨害成
分の溶出
分析対象成分の
溶出
妨害成分
分析対象成分
19
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図7−固相抽出法2 妨害成分を吸着させる方法
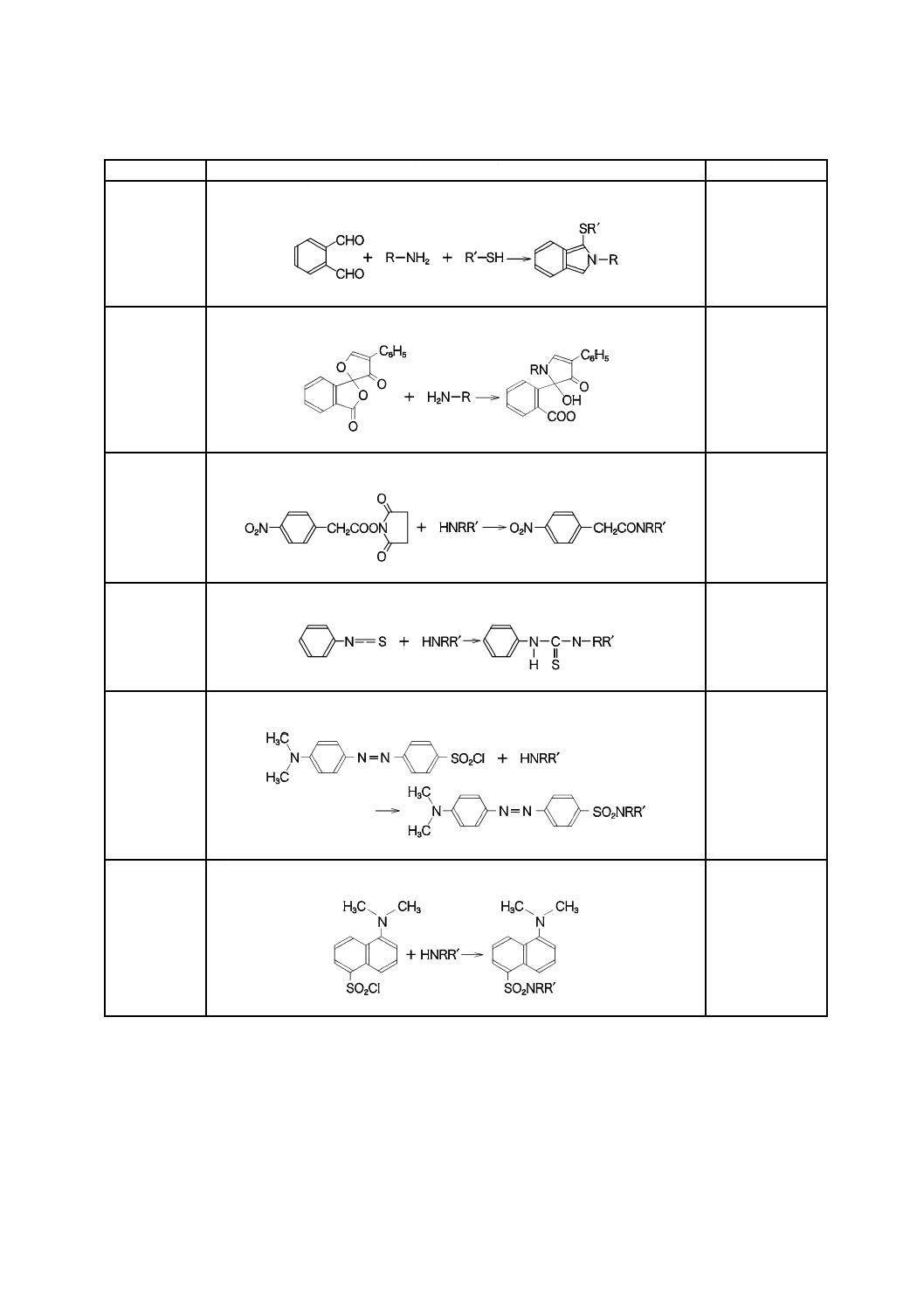
c) 高圧切換バルブを用いたカラムスイッチング法による試料調製(前処理)法 試料調製(前処理)の
一つの方法として,高速液体クロマトグラフの構成に流路切換バルブと妨害成分除去(分析対象成分
を保持し,妨害成分を排出させてしまう方法など)又は分析対象成分の濃縮のためのカラムを接続し,
バルブを切り替えて試料調製(前処理)を自動化2)する方法がある。図8に流路例を示す。これによ
る試料調製の方法例を次に示す。
注2) 切換バルブと自動交換式前処理カラムとを組み合わせた装置も使用することができる。
図8−カラムスイッチング法による試料調製(前処理)法の流路例
コンディショニング
(洗浄・活性化)
試料溶液添加
妨害成分の吸着
分析対象成分の溶出
試料調製液又は
前処理液
自動試料導入装置
ポンプ
溶離液送液ポンプ
溶離液
切換バルブ
検出器
濃縮又は前処
理用カラム
分離カラム
1
2
妨害成分
分析対象成分
20
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 切換バルブは,実線の流路が選択され,分離カラムは溶離液を用いて,前処理用カラムは濃縮用移
動相又は前処理液によって,平衡化される。
2) 試料導入装置から測定用試料溶液が導入され,前処理用カラムにて妨害成分除去(排出),分析対象
成分の濃縮などの操作が行われる。
3) 切換バルブが点線の流路に切り換わり,前処理用カラムに溶離液が流れ3),濃縮又は妨害成分除去
(排出)後の保持されている分析対象成分が溶出され,分離カラムに導かれる。
4) 分離カラムにて分離された分析対象成分を検出器にて検出する。
注3) 前処理用カラムの送液方向は,試料の導入と同じ流れの方向と逆の流れの方向を切換バル
ブのポート1,2の接続配管の位置を替えることによって変更することができる。
試料の導入と同じ流れの方向で溶離液が流れた場合は,前処理カラム内でのピークバン
ドの広がりへの影響などを考慮する必要がある。また,逆の方向で流れた場合は,前処理
カラム上部のフィルターに付着した物質が分離カラムへ流れてしまうことを考慮する必要
がある。
8.1.4.3
試料溶液への標準液の添加
内標準法による場合は内標準物質を,また,標準添加法による場合は標準液又は希釈標準液を添加して
測定用試料溶液を準備する4)。混合希釈標準液を用いてもよい。
注4) 標準液の添加による測定用試料溶液中の沈殿,錯形成反応など化学反応が生じないことをあら
かじめ確認する。
8.1.4.4
プレカラム誘導体化
分析種が直接検出できない場合,高感度,かつ,選択的な検出を行いたい場合,又は最適な分離を得る
ために測定試料溶液をカラムに導入する前に目的成分の誘導体化を行うことがある。プレカラム誘導体化
には,測定試料溶液中の分析種に対して,誘導体化試薬を加える場合と,酸化,還元,分解などを行わせ
る試薬を加える場合とがある。表3に主なプレカラム誘導体化試薬及びその検出条件を示す。
プレカラム誘導体化は,自動試料導入装置に組み込まれた試薬添加,反応液混合,反応時間,反応液の
導入機能などを用いて,自動化してもよい5)。
注5) 試薬の数,添加量,混合回数,反応温度,反応時間などの反応条件によって,利用できるプレ
カラム誘導体化試薬は制限される。
21
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−主なプレカラム誘導体化試薬及び検出条件
対象成分
試薬及び反応式
検出条件
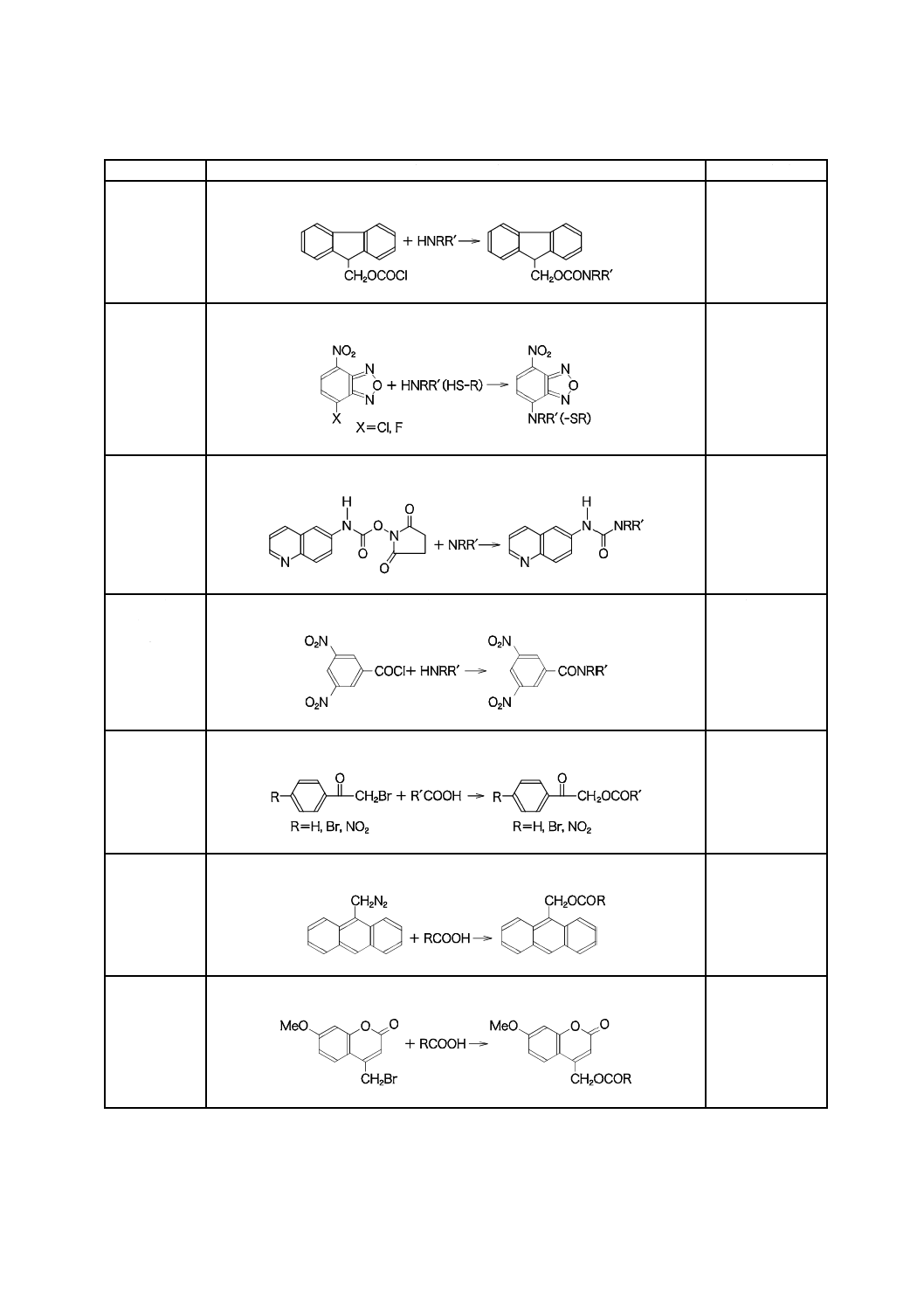
1級アミン
o-フタルアルデヒド(OPA)+チオール化合物
蛍光検出
Ex=340 nm
Em=455 nm
1級アミン
フルオレサミン
蛍光検出
Ex=390 nm
Em=475 nm
アミン(1級,
2級)
N-Succinimidyl p-nitrophenylacetate(SNPA)
UV検出
266 nm
アミン(1級,
2級)
Phenylisothiocyanate(PITC)
UV検出
240〜260 nm
アミン(1級,
2級)
ダブシルクロリド(DABC-Cl)
VIS検出
420〜450 nm
アミン(1級,
2級)
ダンシルクロリド(DNS-Cl)
蛍光検出
Ex=360〜375
nm
Em=510〜540
nm
22
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−主なプレカラム誘導体化試薬及び検出条件(続き)
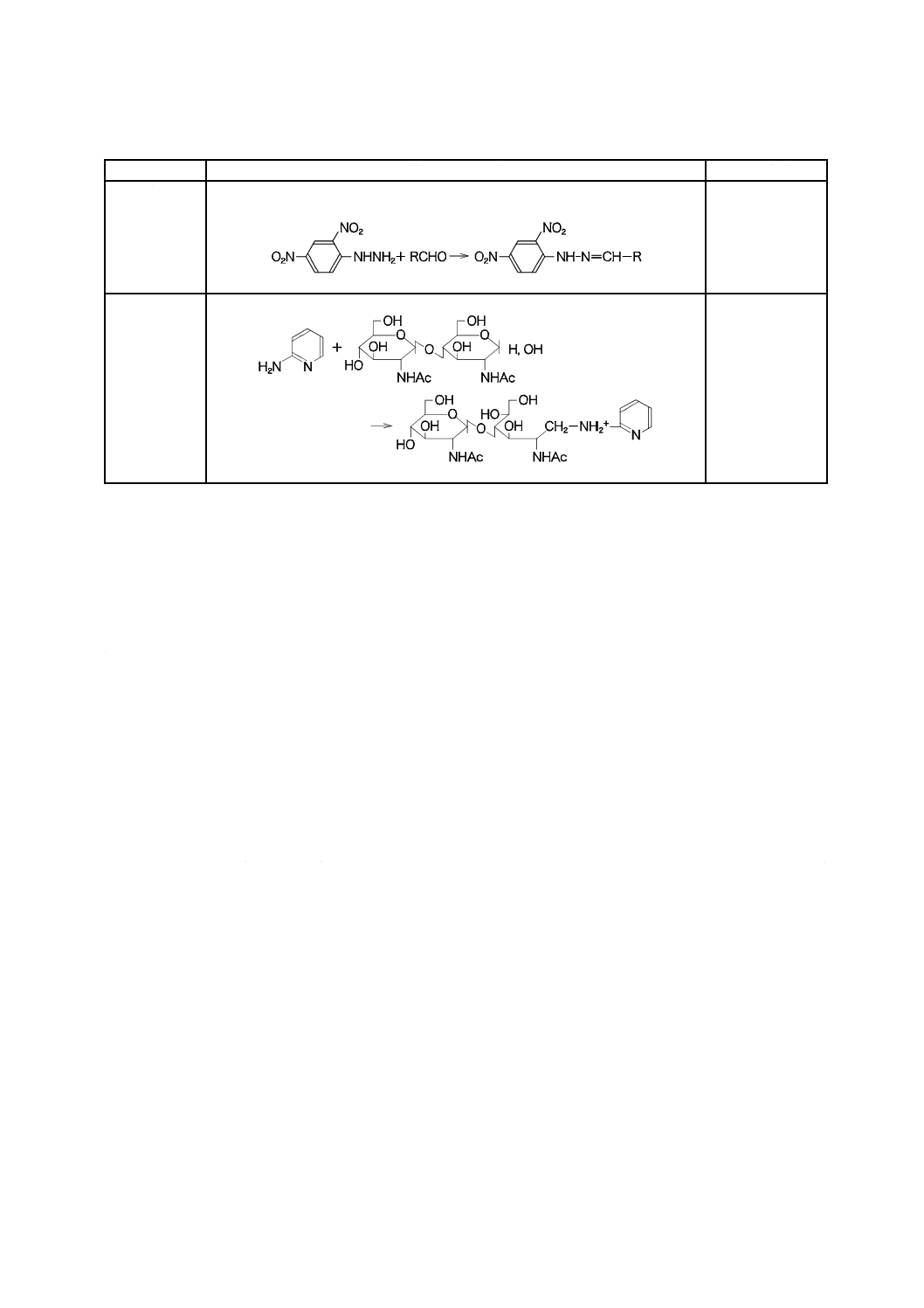
対象成分
試薬及び反応式
検出条件
アミン(1級,
2級)
9-Fluorenylmethyl chloroformate(FMOC-Cl)
蛍光検出
Ex=260〜270
nm
Em=305〜315
nm
アミン(1級,
2級)
4-Harogeno-7-nitrobenzofurazan(NBD-X)(X=Cl, F)
蛍光検出
Ex=425〜475
nm
Em=515〜545
nm
アミン(1級,
2級)
6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate(AQC)
蛍光検出
Ex=250 nm
Em=395 nm
アミン(1級,
2級)
フェノール,
アルコール
3,5-Dinitrobenzoyl Chloride(DNBC)
UV検出
300 nm
カルボン酸
p-Bromophenacylbromide
UV検出
250〜260 nm
カルボン酸
9-Anthryldiazomethane(ADAM)
蛍光検出
Ex=365 nm
Em=412 nm
カルボン酸
4-Bromomethyl-7-methoxycoumarin(Br-MmC)
蛍光検出
Ex=340 nm
Em=420 nm
23
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−主なプレカラム誘導体化試薬及び検出条件(続き)
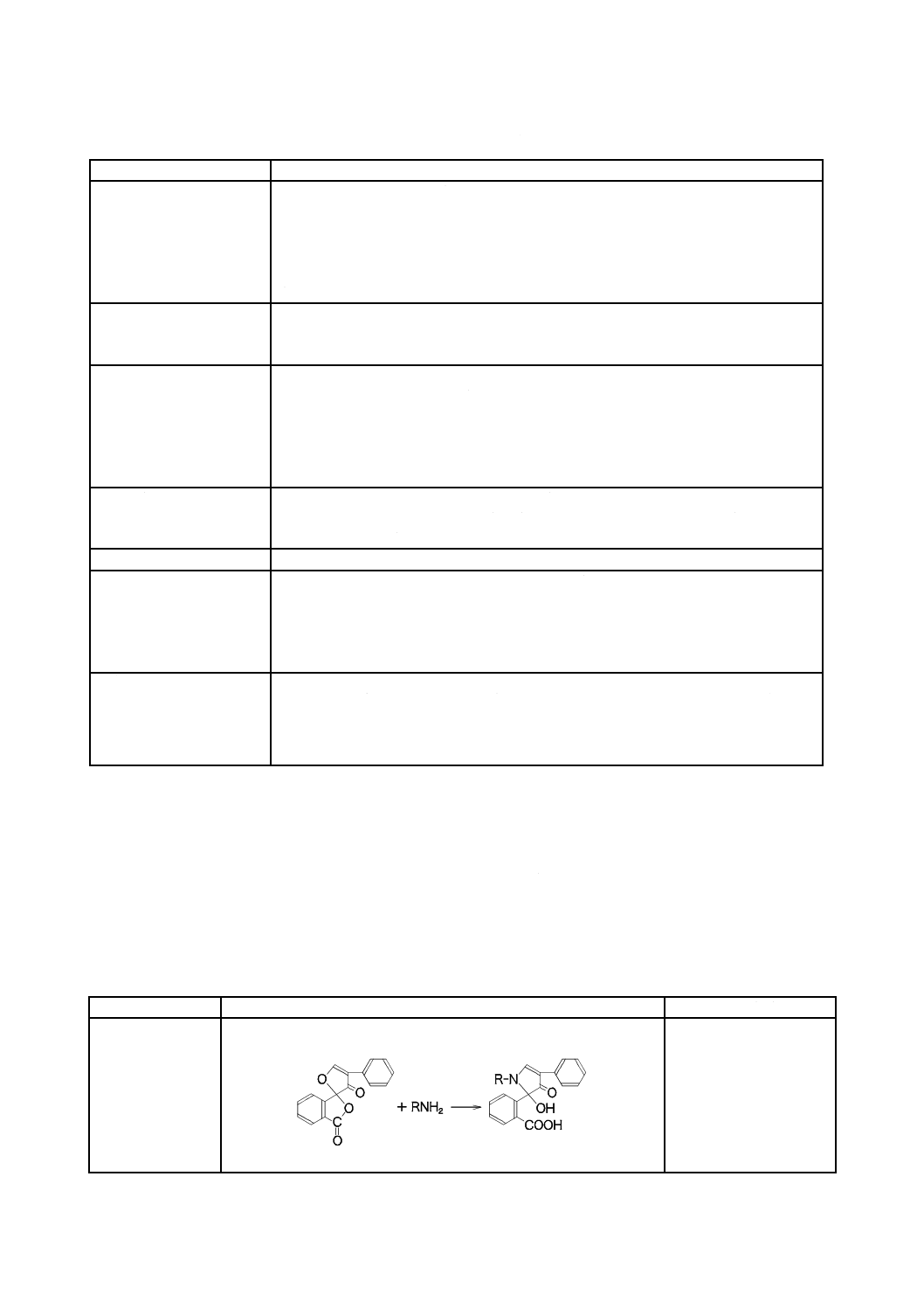
対象成分
試薬及び反応式
検出条件
アルデヒド/
ケトン
2,4-ジニトロフェニルヒドラジン(2,4-DNPH)
UV検出
250〜260 nm
糖
2-アミノピリジン
蛍光検出
Ex=320 nm
Em=400 nm
8.2
溶離液の準備
8.2.1
溶離液の選択
溶離液は,必要な条件を満たし,分離モード又は検出器によって選択する。
8.2.1.1
必要事項
溶離液は次の条件を満たすことが望ましい。
a) 分析種を適切に分離できるもの。
b) 充塡剤に対して化学的,物理的に損傷を与えないもの。
c) 試料を変質することなく溶解できるもの。
d) 検出器の作動に適したもの。
e) 固形物を除去したもの6)。
f)
脱気したもの7)。
g) 混和性がよい。
h) 引火・爆発性が弱い,毒性が低いなど安全性が高い。
注6) 固形物除去は,通常孔径0.5 μm以下のメンブランフィルターを通過させればよい。3 μm以
下の粒子径の充塡剤を使用する場合は,粒子径の1/10の孔径のメンブランフィルターを用い
た方がよい。
7) 溶存ガス除去装置が組み込まれた高速液体クロマトグラフを使用する場合は前もって脱気す
る必要はない。
8.2.1.2
分離モードに基づく選択
分離モードに基づく選択溶離液の選択例を表4に示す。

24
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表4−分離モードに基づく選択の例
分離モード
溶離液
備考
分
配
ク
ロ
マ
ト
グ
ラ
フ
ィ
ー
順相分配クロマト
グラフィー
へキサン,ペンタン,2-プロパノール,
メタノールなど極性の異なる有機溶媒の
混合溶媒を使用する。
溶出力は溶離液の極性が高いほど強くなる。分
離選択性は溶離液と固定相への分析種の溶解
度に支配され,更に溶離液の水素結合供与性,
受容性及び双極子相互作用の違いによっても
変化する。
逆相分配クロマト
グラフィー
メタノール,アセトニトリル,THFa)など
の有機溶媒と水又は緩衝液との混合溶液
を使用する。
溶出力は極性の低い溶離液ほど強くなる。分離
選択性は溶離液及び固定相への分析種の溶解
度に支配され,更に溶離液の水素結合供与性,
受容性及び双極子相互作用の違いによっても
変化する。イオン性化合物を分析種とする場合
は,溶離液pHが保持・分離に大きな影響を与
える。イオン性化合物がイオン化するpHでは
保持が弱くなり,非イオン化状態になるpHで
は保持が強くなる。
逆相イオン対クロ
マトグラフィー
分析対象イオンとイオン対を組むことが
でき,かつ疎水性をもつ試薬を対イオン
として添加した緩衝液と,メタノール,
アセトニトリル,THFa)などの有機溶媒と
の混合溶液を使用する。
溶出力は有機溶媒の混合比が大きいほど強く
なる。イオン対クロマトグラフィーの溶離液で
はイオン対試薬濃度とpHとの調節が重要であ
る。
親水性相互作用ク
ロマトグラフィー
(HILIC)
アセトニトリル,アセトン,THFa)などの
有機溶媒と水又は緩衝液との混合溶液を
使用する。
溶出力は溶離液の極性が高いほど強くなる。分
離選択性は溶離液及び固定相への分離対象成
分溶解度に支配され,更に溶離液のpH,水素
結合供与性,受容性及び双極子相互作用によっ
ても変化する。
吸着クロマトグラフィー へキサン,ペンタン,2-プロパノール,
メタノールなど極性の異なる有機溶媒の
混合溶媒を使用する。
溶出力は溶離液の極性が高いほど強くなる。分
離選択性は溶離液の極性だけではなく,水素結
合供与性,受容性及び双極子相互作用の違いに
よっても変化する。
イオン交換クロマトグラ
フィー
分析種の荷電の差が最も大きくなるpH
において十分な緩衝能をもつ緩衝液が使
用される。通常初期溶離液濃度として20
〜50 mmol/Lを使用する。
分析種がイオン化するpHの緩衝液を初期溶離
液とし,分析種を保持させた後,緩衝液に塩化
ナトリウムなどの塩を添加し,添加濃度を段階
的若しくは直線的に増加させるか,又は緩衝液
のpHを分析種が非イオン化する方向へ変化さ
せることによって溶離させる。逆に,分析種が
非イオン化するpHの緩衝液を使用し,素通り
で単離する方法もある。樹脂製のイオン交換体
を用いる系では,イオン交換のほかに,疎水性
相互作用,水素結合,サイズ排除などの相互作
用が付随することがあるため,溶離液に有機溶
媒を添加することによって,分離が調節可能に
なる場合がある。
イオン排除クロマトグラ
フィー
カルボン酸など弱酸分離の溶離液には水
又は1〜10 mmol/Lの酸水溶液(pH 2〜3),
弱塩基性化合物分離の溶離液には主に水
を使用する。
一般に,分析種がイオン化している状態では早
く溶出し,非イオン化状態では遅く溶出する。
通常溶離液のpHには分析種の間のイオン化率
の差が最も大きくなるpHを選択する。

25
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表4−分離モードに基づく選択の例(続き)
分離モード
溶離液
備考
サ
イ
ズ
排
除
ク
ロ
マ
ト
グ
ラ
フ
ィ
ー
非水系サイズ排除
クロマトグラフィ
ー
サイズ排除クロマトグラフィーの溶離液
には分析種及びカラム充塡剤の両方に高
い親和性をもち,吸着,分配などサイズ
排除クロマトグラフィー以外の分離モー
ドが働かない溶媒を使用する。非水系サ
イズ排除クロマトグラフィーでは,通常
カラム充塡剤としてポリスチレンゲルが
使用されるため,ポリスチレンゲルと溶
解パラメーターが近いTHFa)を多く使用
する。また,極性物質を分析種とする場
合はDMFb)などの極性溶媒を使用しても
よい。
サイズ排除クロマトグラフィーを100 ℃以上
の高温で行う必要がある場合はTCBc)を使用
してもよいが,常温において固体である溶媒を
使用する場合は,分析後のカラム保存に当たっ
てほかの溶媒に置換しなければならない。溶媒
置換においてはカラム使用耐圧を超えない流
速で行わなければならない。不必要な溶媒交換
はカラム寿命を短くするため行わないことが
望ましい。
水系サイズ排除ク
ロマトグラフィー
カラム充塡剤と分析種との静電的相互作
用を抑制するために塩類を添加し,イオ
ン強度を上げた溶離液を使用することが
多い。たんぱく質などpHによって安定
性又は立体配置が変化する試料の場合,
適切な緩衝液を使用してもよい。
分析対象成分がカラム充塡剤と疎水性相互作
用を起こす可能性がある場合,メタノールなど
の極性有機溶媒をカラム使用可能範囲内で溶
離液に添加してもよい。
アフィニティークロマト
グラフィー
使用する相互作用及びリガンドの種類,
分析種に適したpH,イオン強度の緩衝液
を使用する。たんぱく質などのアフィニ
ティークロマトグラフィーでは,トリス-
塩酸緩衝液,りん酸緩衝液,酢酸緩衝液
などを使用することが多い。
分析種がリガンドと結合しやすい溶離液で平
衡化したカラムに試料を導入後,イオン強度,
pHを変化させる非特異的な溶離方法,遊離の
大過剰リガンド,その類似物質の溶液を使用す
る特異的溶離方法などを使用する。
注a) テトラヒドロフラン
b) N, N-ジメチルホルムアミド
c) 1, 2, 4-トリクロロベンゼン
8.2.1.3
検出器に基づく選択
検出器に基づく溶離液の選択例を表5に示す。
表5−検出器に基づく溶離液の選択の例
検出器
溶離液の選択
紫外可視吸光光度検
出器
通常は検出波長において透明,又は無視できる程度の吸収しか示さないものを使用する。紫
外吸収の少ない高純度な溶媒,水及び試薬を用いて調製する。一般に,溶媒は短波長におい
て吸収が大きくなり,緩衝液においても紫外部での吸収が強いものがあるため注意を要する。
また,グラジエント溶離を行う場合は,溶離液組成の変化によって検出波長における吸収強
度に変化が起こり,ベースラインの変動を生じるおそれがあるため,用いる全ての溶媒が検
出波長において十分に透明であることが望ましい。
蛍光検出器
励起波長及び蛍光波長のいずれにも強い吸収をもたないものとする。発蛍光性不純物の少な
い高純度な溶媒,水及び試薬を用いて調製する。
電気化学検出器
電気化学的に不活性な高純度な溶媒,水及び試薬を用いて調製する。
示差屈折率検出器
屈折率が分析種と異なるものとする。濃度差が生じないように十分にかくはんすること。
質量分析計
揮発性であることが望ましい。不純物の少ない高純度な溶媒,水及び試薬を用いて調製する。
化学発光検出器
検出波長において吸収がないものとする。発光性不純物の少ない高純度な溶媒,水及び試薬
を用いて調製する。
26
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.2.2
溶離液の調製
8.2.2.1
混合溶離液の調製法
分離に最適な溶離液は,サイズ排除クロマトグラフィーを除けば単一の溶媒が選ばれることは少なく,
通常はよく混和する2種以上の溶媒を用いて調製する。混合溶離液調製に当たっては次の点に留意する。
a) 互いに完全に混和する溶媒を使用する。
b) 均一な組成を得るため,十分にかくはんする。
c) 混合溶離液の組成は室温における混合前の体積比で表す。混合溶離液の体積が混合前の各溶媒の体積
の和になるとは限らない。そのため各溶媒の体積を個別にはかりとることが望ましい。
d) 混合時に液温変化が生じる場合,室温になるまでしばらく放置する。
e) 有機溶媒と緩衝液又はイオン対試薬などを混合したときに沈殿が生じる可能性が高い組合せの場合
は,混合後に孔径0.5 µm以下のメンブランフィルターなどで沈殿をろ過する。
f)
溶離液によっては保存状態によって組成変化,化学変化,吸湿などが生じるおそれがある。
8.2.2.2
溶離液の脱水及び溶媒の水飽和
分析種が水に対し不安定な場合,又はシリカゲルのように水に対する親和性が大きな物質を固定相に用
いる場合,溶離液中の水分には注意を要し,必要に応じて溶離液を使用前に脱水する。また,吸着クロマ
トグラフィーのように水分含量が分離選択性に大きな影響をもつ場合,所定の水飽和度になるよう調製し
た有機溶媒を使用し溶離液を調製してもよい。
a) 溶離液の脱水 溶離液に使用している溶媒に適した乾燥剤を使用して脱水することが望ましい。モレ
キュラーシーブ3A及び4Aはアルコール,アセトニトリルなどを含む多くの溶媒の脱水に適用しても
よい。
b) 溶媒の水飽和 有機溶媒及び飽和量より少し過剰量の水を,使用温度より5〜10 ℃高い温度で十分に
かくはんした後,使用温度に変えてかくはんし,静置後に有機層を分取し,100 %水飽和溶媒とし,
完全に脱水した溶媒と必要な比率で混合し所定飽和度の水飽和溶媒を調製する。
8.2.2.3
溶離液のpH調整法
逆相分配クロマトグラフィーなどにおいて便宜上,酸又はアルカリを少量添加して溶離液のpHを調整
することがあるが,溶離液は,空気中の二酸化炭素の溶解によってpH変化を受けることがあるため,pH
が分離に大きな影響を与える場合は使用pH範囲において十分な緩衝能をもつ緩衝液を使用することが望
ましい。有機溶媒と緩衝液との混合溶離液の場合,混合比によってその水素イオン濃度は変化するが,通
常,混合前の緩衝液のpHをもって溶離液のpHとする。
8.2.2.4
溶離液の脱気
減圧脱気,超音波脱気,加圧脱気などがあり,必要によって使い分け又は補完して用いるとよい。
8.3
分析種による検出器の選定
高速液体クロマトグラフィーの検出器はカラムを通過した移動相中の分析種を検出し,その定性・定量
を行うものである。様々な検出器があり,その中から実際の測定試料に合わせて最適なものを選択する。
代表的な液体クロマトグラフの検出器,及び検出可能な測定対象を表6に示す。中には誘導体化が必要な
化合物も記載されている。誘導体化については8.1.4及び8.4を参照。
27
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表6−代表的な検出器及び測定対象
検出器の種類
分析種
紫外可視吸光光度検出器
有機酸,エステル,アルデヒドなどのカルボニル化合物,アルカロイド,N-アセチル
化糖,アミノ酸,ペプチド,たんぱく,糖たんぱく,核酸,ジカルボン酸,ステロイ
ド化合物,カテコールアミン,アニリン類,ビタミン類,グルクロン酸類,キサンチ
ン類,芳香族化合物全般など基本的に不飽和結合をもつ化合物。それ以外の化合物で
も,誘導体化によって紫外部又は可視部に吸収をもたせることで検出可能。汎用性は
高い。
蛍光検出器
リボフラビン類,(カテコールアミン,胆汁酸,糖類,アミノ酸,ポリアミン,アルキ
ルアミン,アルキルすず,イミダゾール化合物,カルバメート)など。( )内は,誘
導体化して蛍光検出される化合物。
示差屈折率検出器
糖類,グリセロール,ヒドロキシ脂肪酸,有機酸,アミノ糖,たんぱく,アルコール,
トリグリセリド,キレート化合物,ポリエチレンオキサイド,ポリエチレングリコー
ルなどのポリマー,りん酸化糖,グルクロン酸類,レシチン,ジオール類,ポリシロ
キサン,シクロデキストリンなど,基本的に化合物の種類に制限されない。誘導体化
及び他の光学的検出が困難な化合物でも適用可能。溶離液として,光学的検出器によ
って検出される化合物を使用する場合。
電気化学検出器
コリン,アセチルコリン,マクロライド系抗生物質,ひ酸,尿酸,アスコルビン酸,
過酸化水素,カテコールアミン,糖,糖アルコール,アルコールなど電極酸化又は還
元を受けやすい化合物。
電気伝導度検出器
アルキルアミン,陰イオン全般,陽イオン全般,有機酸など。
質量分析計
エレクトロスプレー,大気圧化学イオン化,高速原子衝撃法,パーティクルビームな
ど数種類のイオン化法があり,使い分けることでほとんどの種類の化合物を検出可能。
最も汎用性の高いエレクトロスプレーでは,アミノ基,水酸基などのプロトン授与性
又はスルホ基,カルボキシ基,酸性水酸基などのプロトン供与性の官能基をもつ化合
物全般,多環芳香族化合物,フラーレン,金属錯体など。
化学発光検出器
基本的に酸化還元を受ける際に光を発する化合物,酸化還元反応などの反応エネルギ
ーによって励起されて発光する化合物。代表例は,ニトロアレーン。アミノ酸,ステ
ロイド化合物,多環芳香族化合物,チオール化合物,カテコールアミン,アスコルビ
ン酸などは,いずれも誘導体化が必要。酸化剤の例として,過しゅう酸,過酸化水素
など。
8.4
ポストカラム誘導体化法の選択
分析種をカラムで分離後,溶出液に誘導化試薬,pH調整液などを混合させ,又はあらかじめ誘導体化試
薬を移動相に添加しておき,加熱,光照射,固定化酵素カラムなどのリアクターを通過させ,誘導体化反
応を行った後,検出する。ポストカラム誘導体化は,元の分析対象成分そのままの構造では検出できない,
検出感度が低い,きょう(夾)雑成分の妨害が大きいなどの理由で,選択的で高感度な検出を行えない場
合に用いる。参考として主な誘導体化反応用試薬及びその反応を表7に示す。
表7−主なポストカラム誘導体化反応用試薬及びその反応
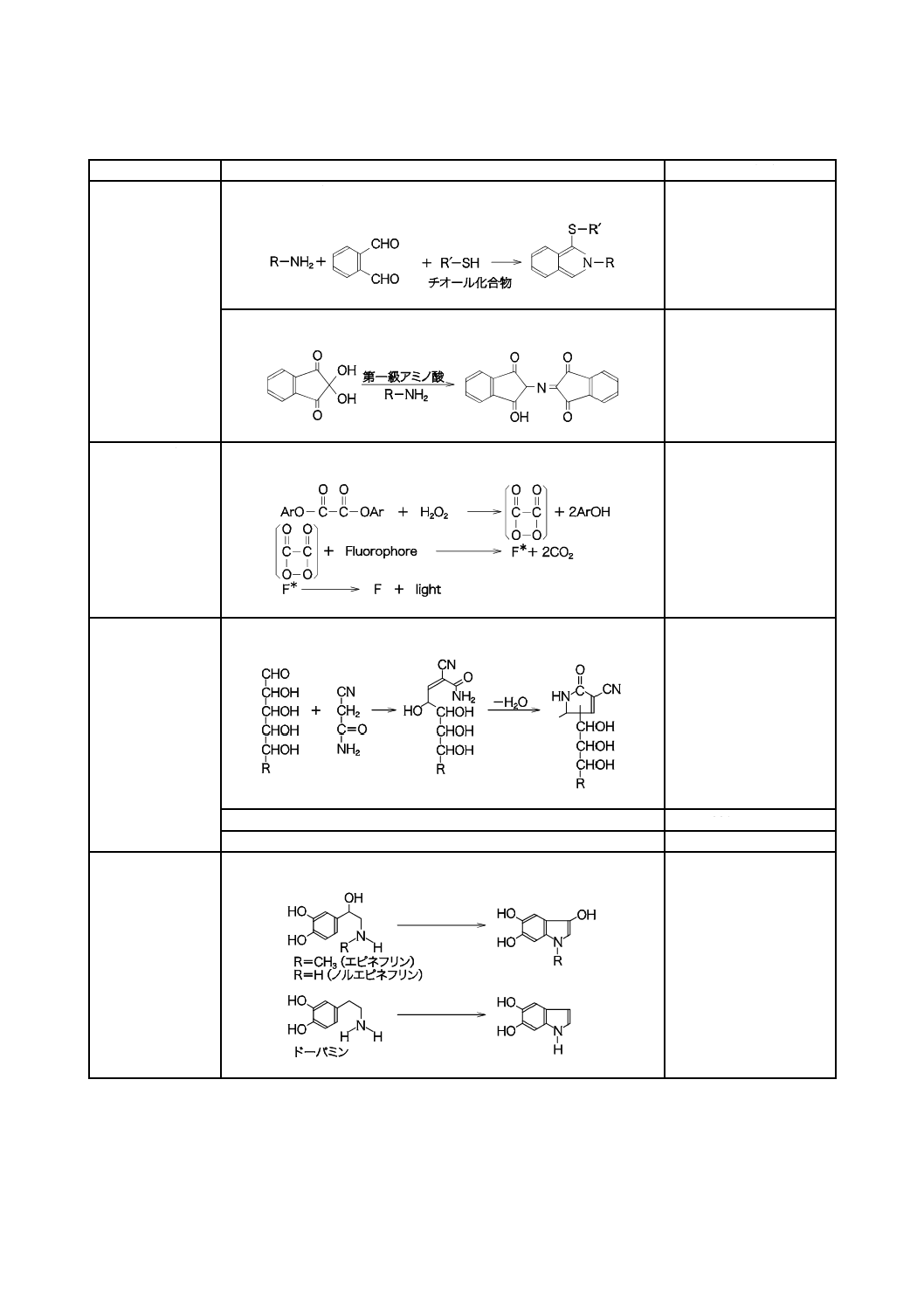
測定対象
試薬及び反応式
検出条件
アミノ酸(アミン) フルオレサミン
FL:Ex 385 nm,Em 485 nm
28
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7−主なポストカラム誘導体化反応用試薬及びその反応(続き)
測定対象
試薬及び反応式
検出条件
アミノ酸(アミン)
(続き)
o-フタルアルデヒド
FL:Ex 345 nm,Em 450 nm
ニンヒドリン
VIS:570 nm,440 nm
Dns-アミノ酸
TCPO/H2O2
CL
糖類
2-シアノアセトアミド
FL:Ex 331 nm,Em 383 nm
UV:280 nm
アルギニン
FL:Ex 320 nm,Em 430 nm
ベンズアミジン
FL:Ex 287 nm,Em 470 nm
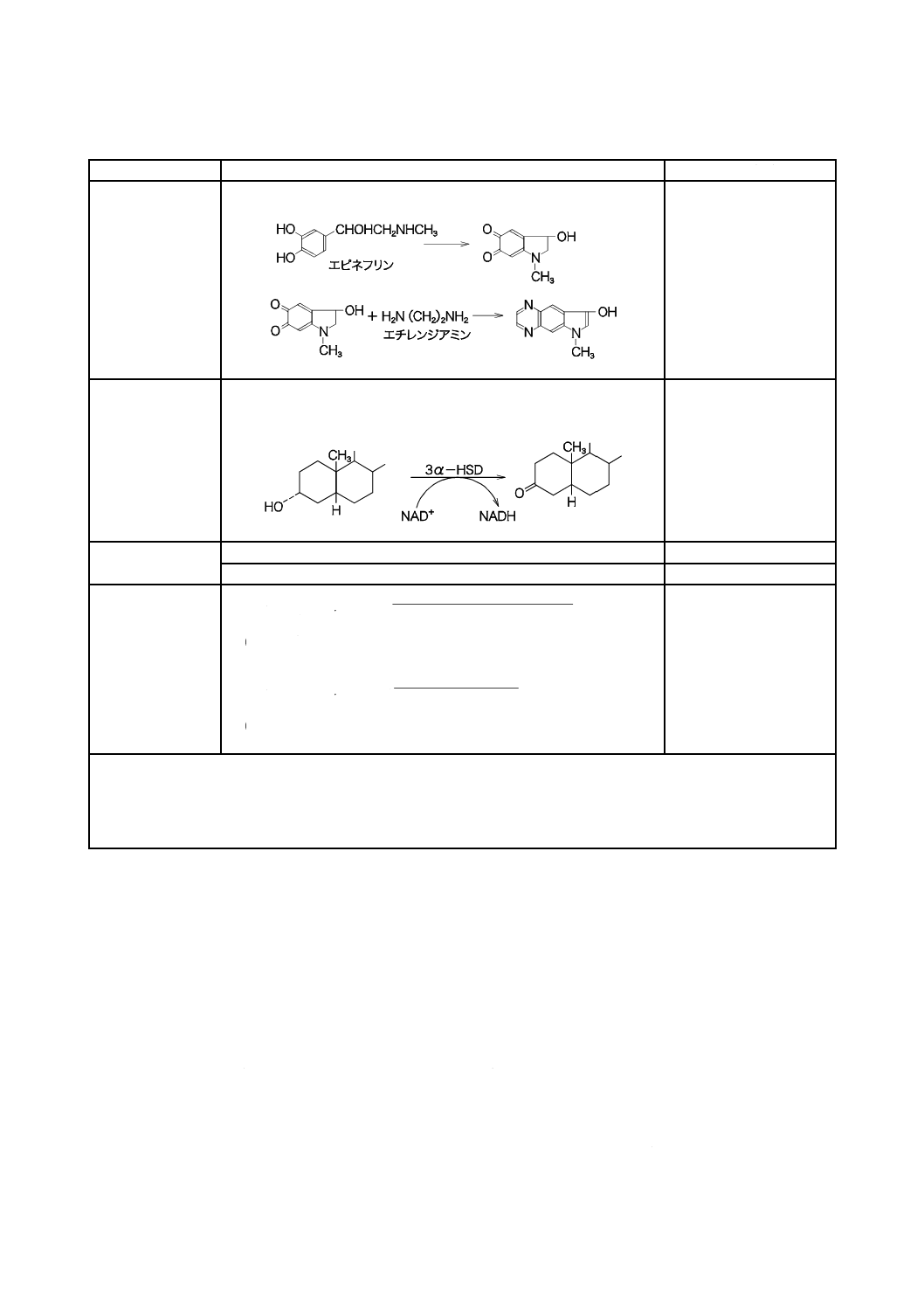
カテコールアミン
酸化剤(トリヒドロキシインドール法)
FL:Ex 410 nm,Em 520 nm
FL:Ex 330 nm,Em 380 nm
29
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7−主なポストカラム誘導体化反応用試薬及びその反応(続き)
測定対象
試薬及び反応式
検出条件
カテコールアミン
エチレンジアミン
FL:Ex 400 nm,Em 510 nm
胆汁酸
ニコチンアミドアデニンジヌクレオチド/3α-HSD
(固定化カラム)
FL:Ex 345 nm,Em 470 nm
有機酸
ブロモクレゾールパープル
VIS:430 nm
ブロモチモールブルー
VIS:445 nm
アセチルコリン,
コリン
(
)
→
+
ラーゼ
アセチルコリンエステ
アセチルコリン
O
H
OCOCH
CH
CH
N
CH
2
3
2
2
3
3
(
)
コリン
+
COOH
CH
OH
CH
CH
N
CH
3
2
2
3
3
+
(
)
→
+
コリンオキシダーゼ
コリン
+
O
H
2O
OH
CH
CH
N
CH
2
2
2
2
3
3
(
)
2
2
2
3
3
O
H
COOH
CH
N
CH
+
ベタイン
+
ECD:0.45 V vs Ag/AgCl,
電極:白金
略号 1) 試薬 TCPO:ビス(2, 4, 6-トリクロロフェニル)オキザレート,3α-HSD:3α-ヒドロキシステロイドデヒ
ドロゲナーゼ
2) 検出 FL:蛍光検出器,VIS:可視吸収検出器,UV:紫外吸収検出器,CL:化学発光検出器,ECD:電気
化学検出器,Ex:励起波長,Em:検出波長
8.5
溶離液の選択
8.5.1
イソクラティック溶離
単一組成の溶離液を用いて,分析種をカラムから溶出させる。イソクラティック溶離法は,液体クロマ
トグラフを用いるルーチン分析に広く用いられている。保持時間の再現性がよいという利点がある一方で,
極性化合物と非極性化合物とを分離するには長時間を要する欠点がある。また,非極性成分が充塡剤に吸
着して溶出されない可能性もある。
溶出時間を短くするためには,溶離液の流量を増加させる方法もあるが,粒子径の小さい充塡剤を詰め
たカラムを用いる場合には,カラム圧が上昇するので注意する必要がある。
8.5.2
グラジエント溶離
時間の経過とともに溶離液の組成を変化させて分析種をカラムから溶出させる。イソクラティック溶離
と比較すると溶出時間を短縮でき,ピーク形状が鋭くなる。溶離液の組成を直線的,段階的又は指数関数
30
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
的に変える方法などがある。ポンプから吐出した溶出力の異なる移動相を,カラム背圧による圧力がかか
る系内で混合して行う,高圧グラジエント分離方式及び,送液ポンプで加圧する前段において,様々な方
法によって移動相を構成する複数の液の混合比を連続又は段階的に変える方法である低圧グラジエント分
離方式がある。
グラジエント溶離法は,溶出しないままでカラム中に残っていると思われる分析種を速やかに溶出させ
たり,分析時間が短くなるなどの利点がある反面,カラム内をはじめの状態に戻すのに時間を要したり,
保持時間の再現性,定量性が悪くなるなどの欠点もある。
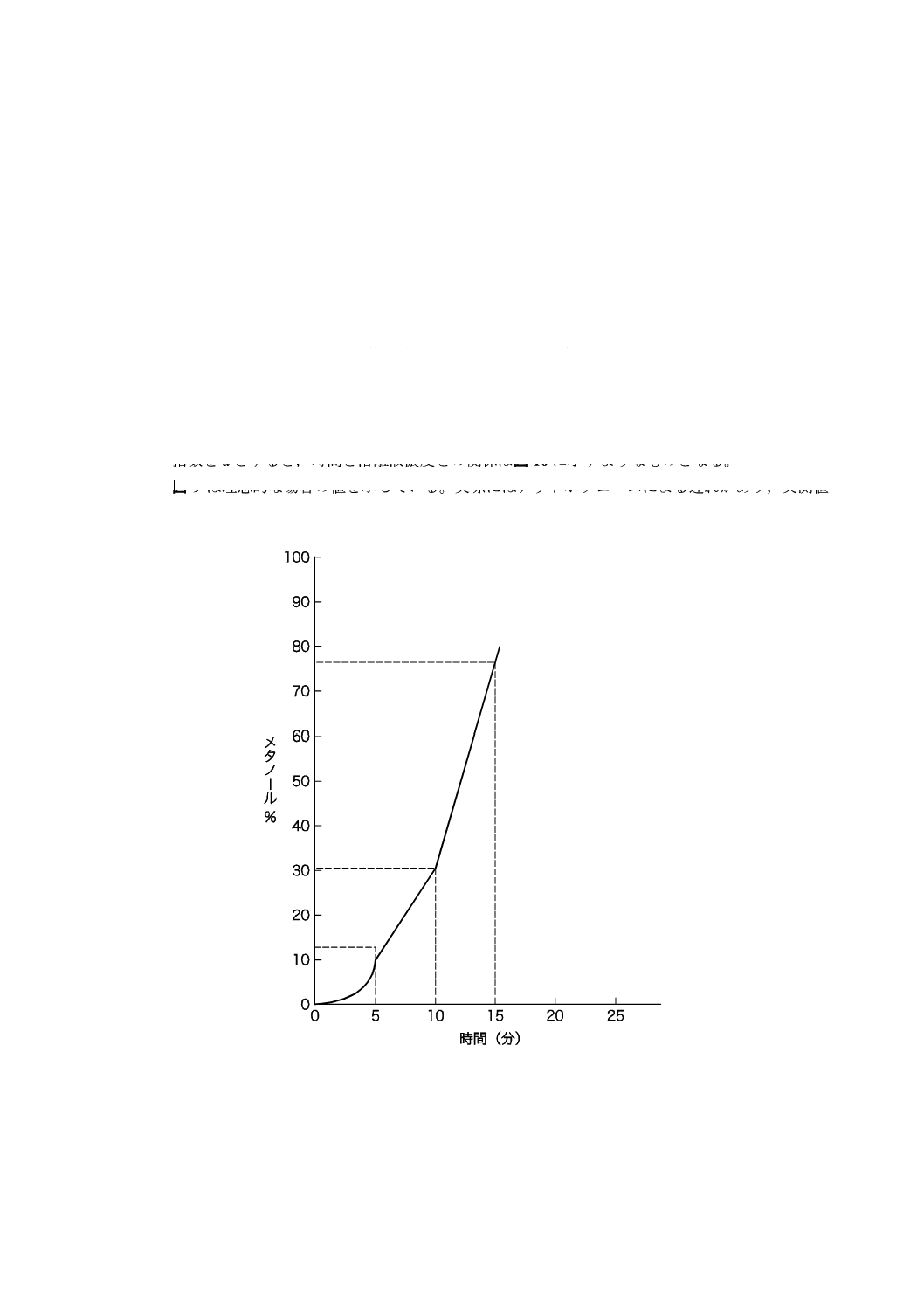
水/メタノール2液の混合比8)を変えて行うとき,試料導入時水100 %,その後5分間で指数関数的9)
(a=5)にメタノールを10 %に,その後5分間で30 %に,更に5分間で80 %に直線的に変えた場合の時
間と溶媒中のメタノール濃度の関係は図9 10)のようになる。グラジエント溶離法においては,時間に対す
る溶離液組成の高い再現性が得られる装置であることが必要である。
注8) 溶離液の組成は,特記しない限り体積比(v/v)で表す。
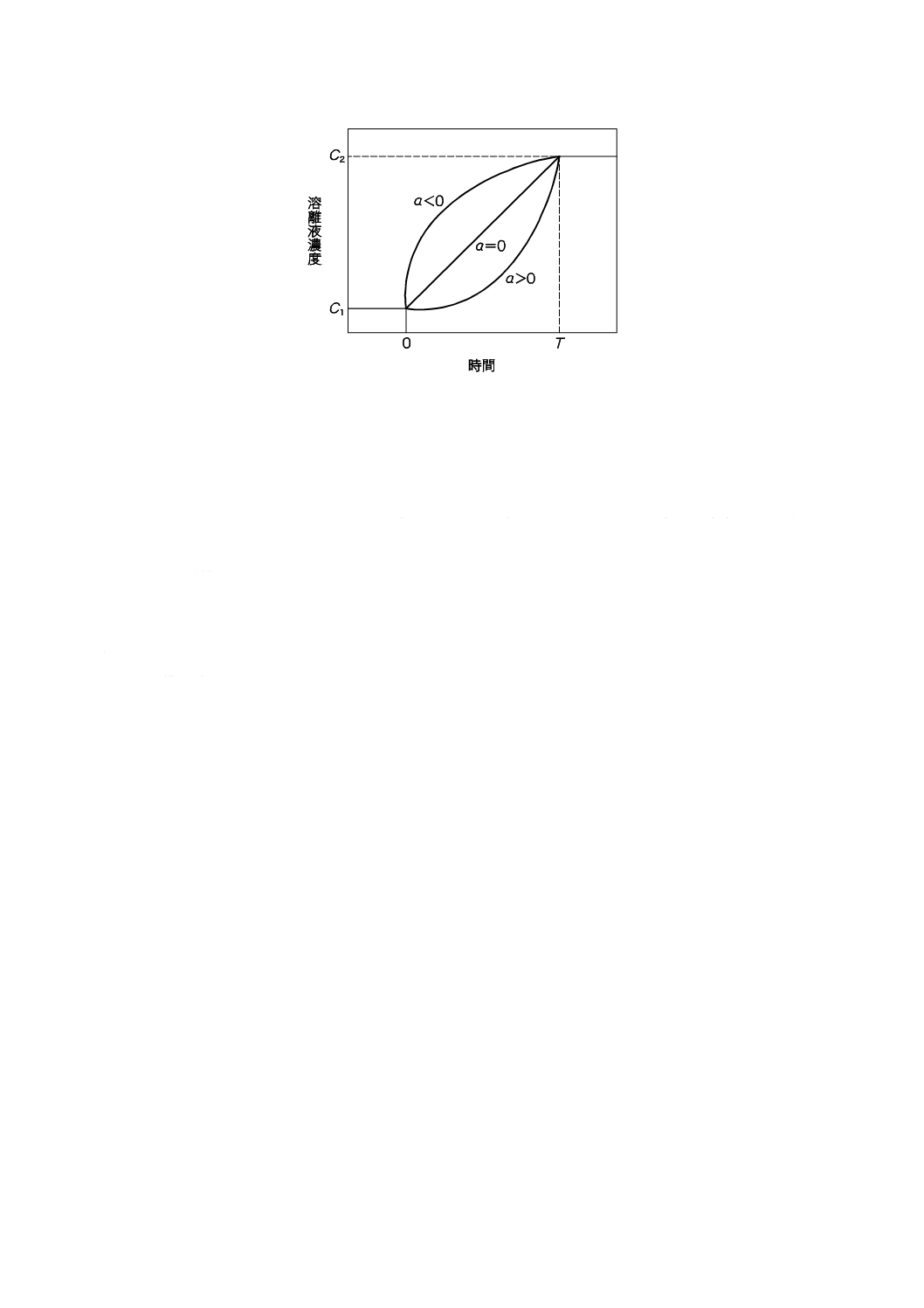
9) 指数をaとすると,時間と溶離液濃度との関係は図10に示すようなものとなる。
10) 図9は理想的な場合の値を示している。実際にはデッドボリュームによる遅れがあり,実測値
はこの図とは異なったものとなる。
図9−グラジエント溶離法
31
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図10−指数関数的曲線
8.6
測定操作
8.6.1
分析条件の設定
分析条件の設定は,次による。
a) 移動相の種類及び流量(グラジエント溶離を用いる場合は,移動相の初期組成,組成変化率,最終組
成などのプログラム)
b) カラムの種類
c) カラム温度
d) 検出器の感度
e) データ処理部[データ処理装置11)]の条件
注11) データ処理装置には,“インテグレーター”と呼ばれる専用の装置又はパーソナルコンピュー
ターを用いたデータ処理装置が用いられる。クロマトグラムの印刷,データの印字など紙へ
の記録については,通常前者はプリント機能を含んでおり,後者はプリントにプリンターを
用いる。
8.6.2
ベースラインの安定性の確認
8.6.1で設定された条件で作動したとき,ベースラインの変動が測定に支障がないことを確認する。
8.6.3
試料の導入
試料導入量に応じて適切な容量の計量器具を使用し,試料導入装置から速やかに導入する。自動試料導
入装置を使用する場合は,適切な条件を設定する。
8.6.4
クロマトグラムの記録
クロマトグラムの記録は,専用又はパーソナルコンピューターを用いたデータ処理装置で行う。データ
処理装置は,クロマトグラム,保持時間,ピーク面積値,定量値などを表示できるものとする。正確なデ
ータを得るために,データの取込みに関するパラメーターの数値を状況に応じ設定する。
データの取込み及びピークの計算処理に関するパラメーターには,サンプリング周期12),時定数13),ピ
ーク検出パラメーター14)などがある。また,クロマトグラムを描画,印刷するためのパラメーターには,
データ収集時間,クロマトグラム表示時間,縦軸の単位,スケールなどがある。データ処理部にこれらの
パラメーター15)を所定の数値に設定する。クロマトグラムは,印刷物として描画し,又は情報として保存
する。
32
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注12) 1ピーク当たり20〜30点のデータ数となるようピーク形状に合わせて設定するのが望ましい。
13) 時定数が短い場合は,記録が信号の変化に追従するのに有利な一方,ノイズ程度の僅かな信号
の変化も反映しやすい。逆に,時定数が長い場合は,記録が信号の変化に追従しにくくなり,
小さな又は短時間のピークを見逃す可能性がある。時定数を検出器側で設定する場合もある。
時定数の呼び方及び表記は,メーカ又は使用する装置の種類によって,時定数はレスポンス,
タイムコンスタント,レスポンスタイムとの呼び方及び表記となる場合がある。
14) 分析条件に合わせて不要なピーク検出,不適切なベースライン設定などを避け,ピークの正確
な判定を行うためのパラメーターであり,ノイズ,感度,スロープなどの項目がある。
15) パラメーターは,メーカによって,また,使用するデータ処理装置の種類によって異なる名称
が使われる場合がある。
8.6.5
クロマトグラムの整理
クロマトグラムの整理に当たり,次の事項を記録する。単位は一例として示す。
a) 測定日及び測定者
b) 高速液体クロマトグラフの製造業者名又はその形式記号
c) 試料名
d) 測定用試料溶液の調製方法及び導入量(µL又はmL)
e) カラム充塡剤の種類又は充塡カラムの形式名
f)
クロマトグラフィー管の材質,内径(mm),及び長さ(mm)
g) カラム温度(℃)
h) 溶離液(移動相)の種類
i)
溶離液(移動相)の流速又は流量(mL/min)及びカラム入口の圧力(Pa)
j)
グラジエント溶離の条件
k) 検出器の種類及び設定条件
l)
化学反応を利用した場合は,反応液及び反応試薬の種類,流速又は流量(mL/min),移動相と反応液
の混合比並びに反応条件
m) データ処理部の種類及び記録条件
n) その他必要事項
9
定性分析
定性分析は,同一条件下の測定で得られたクロマトグラムを用いての希釈標準液又は混合希釈標準液と
試料溶液中の未知成分の保持値16)の比較,試料に希釈標準液を添加しても試料中の分析種のピーク形状が
崩れないことを確認するなどして行う。併せて,未知成分ピークの単一性を確認するためには,固定相の
変更,移動相を変えるなど分離条件を変えて測定する。オンライン及びオフラインでの操作を含む次の定
性手法によって確かめることもできる。
a) 質量分析計の利用
b) フォトダイオードアレイ検出器の利用
c) 原理の異なる複数の検出器(赤外分析,発光分析,核磁気共鳴など)の利用
d) 化学反応,酵素反応などの利用
注16) 保持値は,保持時間,保持容量, 空間補正保持容量, 比保持容量, 保持比, 保持係数などの総
称。

33
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10 定量分析
10.1 定量法
定量は,記録部によって得られたクロマトグラムからピーク面積17)を求め,絶対検量線法, 内標準法,
標準添加法によって行う18),19)。
注17) ピーク面積の代わりにピーク高さを用いてもよい。
18) ピーク面積を求めるに当たっては,ピークの形状に合わせ,適切なベースラインを引かなけれ
ばならない。
19) 適切な内標準物質が得られる場合は,内標準法による。この場合,導入誤差の補正,前処理誤
差の補正を行うことができる。また,標準添加法は,試料マトリックスの影響が補正できる特
長がある。
10.2 ピーク高さの測定
ピークの頂点の信号値から,ピーク頂点の保持時間と同一の保持時間におけるベースラインの信号値を
差し引いたもの,又はピークの頂点から時間軸に下ろした垂線がベースラインと交わる点と頂点との距離
をピーク高さとする。
10.3 ピーク面積の測定
ピークの始まりから終わりにわたってピークの信号値とベースラインの信号値の差とを積算したもの,
又はピーク高さの中点(h/2)におけるピーク幅(半値幅w0.5h)にピーク高さ(h)を乗じたものをピーク
面積(A)とする。ただし,後者の場合,著しいリーディング又はテーリングが認められるピークには適
用しない。
なお,データ処理装置を用いる場合は,表示された記録値,又は指示値による。データ処理装置にはピ
ークの形状に合わせ,適切な設定条件を選ばなければならない。
半値幅法によるピーク面積の測定を図11に示す。
図11−半値幅法によるピーク面積測定
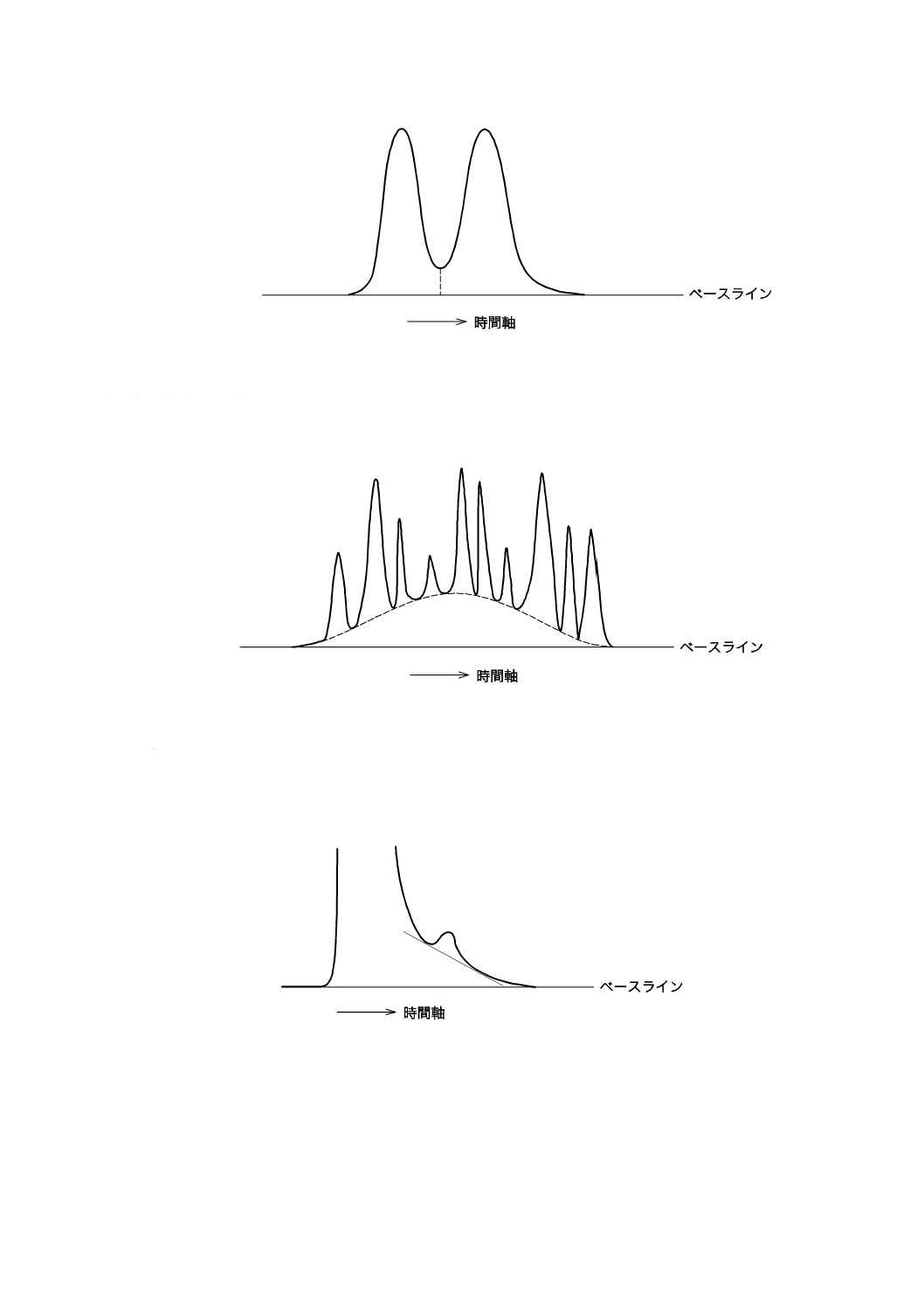
重複ピークの面積分割方法には,次の方法がある。
a) 垂線法 図12のように,二つのピークの大きさがほぼ等しい場合,ピークの谷から時間軸に下ろした
垂線によってベースライン上のピークを二つに分割し,それぞれの面積を求める。
34
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図12−垂線法によるピーク面積の分割
b) 谷−谷(valley to valley)法 図13のように,バックグラウンドの上に重複したピークに対して適用
する。隣接する谷と谷とを結ぶ線分及びクロマトグラムによって囲まれた面積を求める。
図13−谷−谷法によるピーク面積の分割
c) 接線法 図14のように,大きなピークのテーリングに重なった小さなピークの場合,ピークの谷と大
きなピークの裾とを結ぶ接線上の部分をピーク面積とする。接線の代わりに,指数関数曲線によるピ
ーク分割もできる。
図14−接線法によるピーク面積の分割
10.4 絶対検量線法
分析対象成分の標準液を3段階以上の濃度20)に調製し,各希釈標準液の一定量を導入しクロマトグラム
を記録してピーク面積を測定する。次に導入された希釈標準液中の分析対象成分の量を横軸に,ピーク面
積を縦軸にして図15に示すような検量線を作成する。
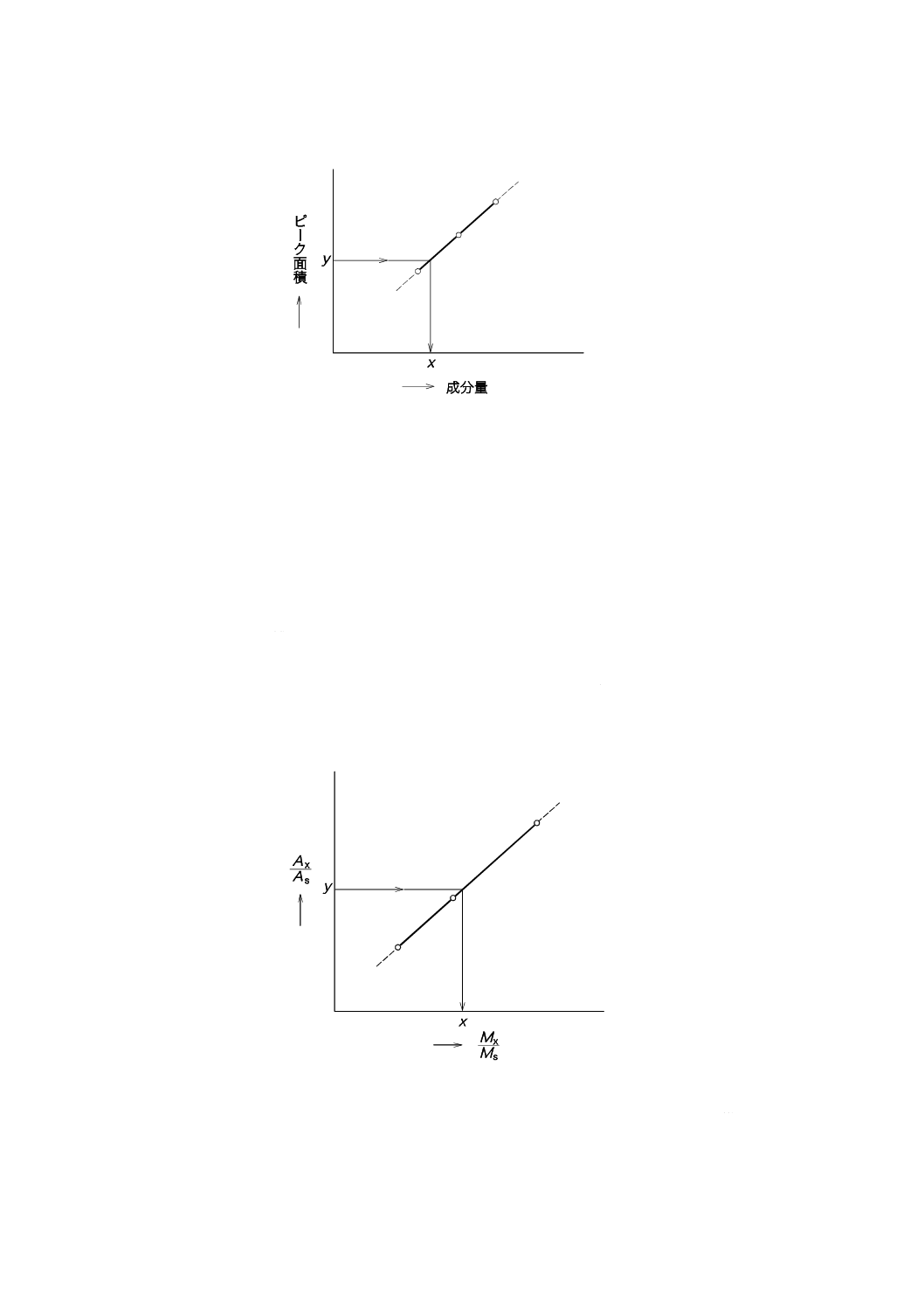
35
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なお,検量線は測定点を代表するように引く。
図15−絶対検量線法による検量線
同一条件の下で試料を導入し,クロマトグラムを記録し,ピーク面積(y)から検量線によって分析対象
成分の量(x)を求め,試料中の濃度を算出する。この方法は,全測定操作を厳密に一定条件にして行わな
ければならない。この定量方法を外標準法ともいう。
注20) 検量線が原点を通る直線であることがあらかじめ確かめられている場合は,分析対象成分の濃
度を一点だけとしてこれを導入した場合のAx/Asを測定し,これに基づいて検量線を求めてもよ
い。
10.5 内標準法
一定濃度の内標準物質21)を含む,3段階以上の濃度の分析対象成分希釈標準液を調製する。各希釈標準
液を一定量導入し,クロマトグラムを記録してピーク面積を測定する。 次いで,導入された分析対象成分
の量(Mx)と内標準物質の量(Ms)との比(Mx/Ms)を横軸に,分析対象成分のピーク面積(Ax)と内標
準物質のピーク面積(As)との比(Ax/As)を縦軸にして図16に示すような検量線を作成する。
なお,検量線は,測定点を代表するように引く。
図16−内標準法による検量線
次に,試料溶液に,希釈標準液中とほぼ同じ濃度になるように内標準物質を既知量添加22)した測定用試
料溶液を調製する。このとき,分析対象成分のピーク面積(Ax′)と内標準物質のピーク面積(As′)との比
(Ax′/As′)が検量線の範囲内になるようにする。この測定用試料溶液を希釈標準液と同一条件の下で導入

36
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
してクロマトグラムを記録する。クロマトグラムから分析対象成分のピーク面積(Ax′)と内標準物質のピ
ーク面積(As′)との比(Ax′/As′)を算出し,検量線から分析対象成分量と内標準物質の量の比を求め,導
入された内標準物質の量から分析対象成分の量を算出する。これから試料中の分析対象成分の濃度を求め
る。
注21) 内標準物質には,分析対象成分及び化学的性状が類似した安定な物質で,そのピークが分析対
象成分ピークの位置になるべく近く,試料中の成分ピークとも完全に分離するものを選択する。
22) 試料溶液に内標準物質を添加したとき,分析対象成分及び内標準物質の濃度に変化を生じさせ
る沈殿などの化学変化があってはならない。
10.6 標準添加法
試料溶液から一定量の溶液を3個以上採取する。これらの採取溶液のそれぞれに分析対象成分の標準液
を濃度が異なるように段階的に加える。この分析対象成分を加えた試料溶液及び分析対象成分を加えてい
ない試料溶液をそのまま又はそれぞれ一定量に希釈して測定用試料溶液とする。測定用試料溶液を一定量
導入してクロマトグラムを記録し,分析対象成分のピーク面積を測定する。添加による分析対象成分の濃
度を横軸23)に,ピーク面積を縦軸にとり関係線を作成する。標準液を添加しない試料溶液から得られたピ
ーク面積(図17に示すy)に相当する分析対象成分濃度は,関係線と横軸との交点(図17に示すx)の絶
対値から求める24)。
注23) 分析対象成分濃度の増加量と標準液の添加量又は一定量の添加回数との間に比例関係が成立す
る場合は,横軸に添加量又は添加回数をとることができる。
24) この方法は,関係線が直線の場合にだけ適用する。
図17−標準添加法による関係線
10.7 定量値の表し方
成分名及び定量値を併記する。定量値は,g/L,mg/L,μg/L,mol/Lなどで表す。
11 分子量分布の測定
分子量分布の測定は,試料及び分子量標準物質25)が同一条件で測定されたサイズ排除クロマトグラムの
保持容量又は保持時間と質量分率26)を用いて行う。試料のクロマトグラムにおける任意の溶出時間に対応
する分子量は,標準物質を用いて作成した校正曲線によって求める。相対分子量として正確な分子量を測
定するために標準物質を測定したとき,できるだけ対称的なピークが得られ,かつ十分な理論段数をもつ
カラムを使用するものとする。
37
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注25) 標準物質には,Mp(ピーク頂点の分子量)に応じて次のような狭い分子量分布(多分散度d=
Mw/Mn)をもつ物質を用いることが望ましい。
Mp<2×103 Mw/Mn<1.20
2×103≦Mp<1×106 Mw/Mn<1.10
1×106≦Mp Mw/Mn<1.20
ここに,
Mn: 数平均分子量
Mw: 質量平均分子量
Mp: ピーク頂点の分子量を示し,上記標準物質でカタログ値
など正しい値が入手できない場合は,近似式
Mp=(Mn×Mw)1/2を用いることができる。
26) 質量分率は,ピーク高さを分子量及びその分子のモル吸光係数又は屈折率濃度変化の分子量依
存性を勘案・補正して求めるべきであるが,化学構造が同じで重合度だけ異なる試料に関して
はその影響を無視し,ピーク高さで代用することができる。
11.1 校正曲線を用いて平均分子量を測定する方法
校正曲線作成と同一条件で測定されたクロマトグラムの保持容量又は保持時間を,分子量1桁当たり20
等分以上に区分し,各分割点の高さとの関係から求める。
11.1.1 校正曲線の作成
分子量既知の単分散試料を高速液体クロマトグラフに導入し,クロマトグラムのピークから得られる保
持容量又は保持時間と分子量との関係線27)(校正曲線)を作成する。
注27) 校正曲線は,標準物質のlogM及び標準物質のピーク頂点の溶出容量Tを基に多項式によって近
似する。次の一次又は三次の多項式が近似式として広く用いられる。
logM=A0+A1T
logM=A0+A1T+A2T2+A3T3
ここに,
M: 分子量
A0,A1,A2,A3: 係数
T: 溶出容量
11.1.2 保持容量の分子量への換算
あらかじめ作成された校正曲線によって行う。このとき得られる分子量は,標準物質(例えば,ポリス
チレン)に換算された分子量である。
11.1.3 数平均分子量,質量平均分子量,z平均分子量,多分散度d値の算出
クロマトグラムの横軸(保持容量)を50等分以上に区分し,各点から縦軸に平行に線を引き,基線から
クロマトグラムの交点までの長さ(高さ)を求める。各点における分子量(Mi)及びその高さ(Hi)から,
次の式によって算出する。
∑
∑
=
=
i
i
i
i
i
i
n
M
H
H
N
N
M
M
∑∑
=
i
2
i
i
3
i
z
N
M
N
M
M
∑
∑
∑∑
=
=
i
i
i
i
i
i
2
i
w
H
M
H
N
M
N
M
M
38
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
n
w
M
M
d=
ここに,
Mz: z平均分子量
Mi: 各溶出時間における分子量
Ni: 各溶出時間における分子数
Hi: 各溶出時間における高さ(信号強度)
d: 多分散度
11.1.4 分子量分布曲線の作成
分子量を横軸に,縦軸に質量分率をとり,関係線(分子量分布曲線)を作成する。
11.2 パターンによる分子量分布の測定
同一条件,同族物質で測定されたクロマトグラムの頂点,適切に区分された分子量間の面積,又は11.1.1
及び11.1.2の操作によって得られた各点における高さ(Hi)の分率を縦軸とした分子量分布の面積など,
パターンの比較を行うことによって,試料の分子量分布を推定する。
12 分取液体クロマトグラフィー
クロマトグラムの特定のピーク部分に相当する溶出液を採取することによって,分離された分析種の分
取・精製を行うことができる。この手法を分取液体クロマトグラフィーと呼ぶ。
12.1 分取の準備
分取の準備は,次の条件を満たすことが望ましい。
12.1.1 分取用試料溶液
分取用試料溶液は,次による。
a) 試料溶媒は,試料を溶解できる溶媒であって,分析種を変質させないものを使用する。また,負荷試
料は溶離液と同じか,又は溶離液よりも溶出力の弱い溶媒に溶解させる。
b) 試料溶液中の微粒子及び不溶物は,あらかじめメンブランフィルターなどで除去する。
c) 負荷試料は,分析種を溶媒抽出などで,あらかじめ粗分画しておく。
12.1.2 溶離液(移動相)
溶離液(移動相)は,次による。
a) 溶離液は,充塡剤,試料に影響を与えず回収が容易であることが必要である。回収濃縮には沸点の低
いものを使用した方が有利である。不揮発性の塩などは,脱塩操作が必要となるが,検出及び試料の
変質に問題がなければ,ぎ酸,アンモニウム,ピリジンなどの揮発性の塩も使用できる。
不純物の混入を防ぐため高純度の溶媒を使用する。しかしながら,通常添加されている安定剤が除
去されている溶媒は,場合によっては,装置を腐食したり,爆発する危険性もあるので注意を要する。
クロロホルムには,通常安定剤としてエタノールなどが入っているが,回収した溶媒を蒸留してそ
のまま使用すると装置を腐食する原因となる場合がある。
テトラヒドロフランは,分解して過酸化物が生成しやすいので通常酸化防止剤が入っている。酸化
防止剤を含まないものは取扱いに注意を要する。また,大量の溶媒を使用する場合には,特に引火性,
毒性に注意して使用する。
b) 粘度の低い溶媒を選択した方が,使用圧力が下がり装置を安定に運転しやすい。
c) 溶離液(移動相)中の微粒子及びごみは,あらかじめメンブランフィルターなどで除去する。
d) 試料の検出に適したものを使用する。
39
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.1.3 分取装置
分取装置は,次による。
a) 送液ポンプ,配管,カラム管など接液部の材質は,試料及び溶離液に侵されることがなく,また,試
料及び溶離液を汚染,変質及び吸着することのないものを使用する。
b) 使用圧力に耐える,十分な強度をもつ。
c) 使用するカラムの大きさ,流速に応じて,デッドボリュームが小さい。
d) 負荷試料量に応じて,検出器の感度を最適化する。
12.1.4 分取カラム
分取カラムは,次による。
a) 充塡剤は,操作条件下で物理的,化学的に安定なもので,不可逆的吸着,酸化などによる目的成分の
変質が少ないものを選択する。分取の目的によっては,装置及びカラムをアルカリなどで洗浄するこ
とが要求される。その場合には洗浄によって充塡剤の性能が劣化しないものを選択する。
b) カラムの効率は負荷する試料容量,及び試料濃度が増すと低下する。あらかじめ分析用カラムなどに
よる小さなスケールで,実試料を用いて試料容量及び試料濃度と分離の関係を求めておき,目的成分
の必要量からカラムの大きさを選択するとよい。また,試料の負荷量は,他の条件が同じならばカラ
ムの断面積と長さに比例するが,スケールアップに当たっては,充塡剤の粒子径,カラムの温度,流
速等と分離の関係を確認しておき,最適の条件を選ぶことが望ましい。カラムは大きすぎると回収率
低下の原因となることもある。
c) あらかじめ決められた条件で,分取カラムの性能確認を行う。
12.2 分取法
分取法は,次による。
a) カラムから溶出した液を分取分画する方法には,時間によって分取する方法,液滴数で分取する方法,
検出器からの信号でコントローラーと組み合わせることによってピークで分取する方法,更に,これ
らの方法を複数組み合わせた方法などがある。分取液体クロマトグラフの検出器の下流側配管,フラ
クションコレクターなど(バルブ類も含め)は,目的のスケールに合ったデッドボリュームの小さい
もので,分離した成分が混合しない方法を選択する。
b) 分離が不十分な場合には,目的成分のピークをリサイクルして分離を上げる方法もある。1本のカラ
ムで,検出器からの必要画分について,リサイクルバルブを用いて,フラクションコレクターから,
ポンプへ戻す流路へ切り替え,ポンプの吸引側からリサイクルする方法,又は切り替えバルブを用い
て2本のカラムを接続し,1本目のカラムで粗分離した目的の画分についてバルブを切り替え,2本目
のカラムに送って,更に分離する方法などがある。いずれの場合もカラムの大きさに応じて,ポンプ
内,配管などの装置のデッドボリュームを小さくして分離を低下させないようにすることが必要であ
る。
12.2.1 分取の操作(微量成分分取)
分取の操作(微量成分分取)は,次による。
a) 必要とする分取量に応じて,装置及びカラムを選択する。特に微量成分の分取の場合には,デッドボ
リュームが小さい装置,カラム,配管などを選択して,分析種の損失を少なくするために非特異的吸
着の少ないカラム,配管の材質及び分取容器を使用することが望ましい。また,事前に同じ種類の充
塡剤で得られた分析スケールのデータを準備することによって,分取量に応じた適切なカラム,条件
など(カラムの内径と長さ,試料負荷量,溶離液流量,分画位置)を推定する情報となる。
40
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 分取装置のラインを適切な溶媒で洗浄して,分取用溶離液に置換する。
c) カラムを接続して送液し,ベースラインの安定度を確認する。必要ならばカラムから溶出した液を分
析して分析種の分取に問題がないことを確認する。
d) フラクションコレクターが接続されている場合には,検出器から分取容器までの配管の容量と移動相
流量の関係を計算し,分析種が,検出されてから,分取容器に到達する時間を考慮し,分取時間を設
定する。
e) 試料導入装置などを使用してカラムに試料を負荷する。
f)
分取する分析種を必要に応じて分画する。
12.2.2 分取画分の分析
分取画分の純度分析及び定量は,分析種に適した方法で行う。
12.3 操作
箇条8に準じて測定を行う。
13 データの質の保証
13.1 分析法バリデーション
データの質を保証するために,新たに開発した分析法(分析条件の変更を含む)については,分析法バ
リデーション(妥当性確認)を実施する28)。公定法(個別JISなど)を採用する場合でも,その試験室で
要求される項目についての分析法バリデーションを行うことが望ましい。その試験室で開発した方法(イ
ンハウスメソッド)による場合は,その方法を適用する前に必ず分析法バリデーションを実施し,少なく
ともその分析法が適用される間は,その分析条件を維持する。
注28) 実際に分析を開始する際には,カラム,溶離液などを選定し,その最適化を図る。すなわち,
分析法を開発又は改良するのである。この場合には,開発した方法が分析の目的にかなう方法
であることを,確認することをここでは要求している。分析法バリデーションの項目には,精
確さ,精度,真度又は正確さ(回収率),直線性,検出下限,定量下限,適用範囲,堅ろう性試
験などがある。
13.2 データの質の管理
データの質は,それぞれの分析条件における定量値の“不確かさ”をもって表示する。この規格では,
包含係数k=2とする拡張不確かさUを用いることとする。
また,品質管理用の試料29)を準備し,これを定期的に分析することによって,データの質を監視する。
ルーチン分析では,試料20個に1個の品質管理用の試料及び1個のブランク試料を含め,並行分析を実施
してデータの質の管理を行うのが望ましい。
注29) 品質管理用の試料には,通常インハウス標準物質(試験室で値付けした標準物質),購入した標
準物質,認証標準物質のいずれかを用いるが,均質で安定な試料であれば,大量に残っている
分析試料を用いてもよい。
13.3 検出下限の求め方
検出下限を報告する場合は,統計的手法で誤りの確率を説明できる検出下限の求め方を定め,それを記
述する。
検出下限は,ブランク試料及びそれに一定量スパイクした試料を各々少なくとも20回以上測定して,正
味の(ブランク値を差し引いた)測定値の標準偏差σの3.3倍の値を濃度(量)換算した値を検出下限と
する。測定回数を,例えば,5回などと減らした場合は,母標準偏差を推定して算出することになるが,n
41
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
回測定の標準偏差の値に3.3の代わりに,表8のt値(n=5では4.26)を乗じることによって求められる。
すなわち,検出下限は“95 %の確率[誤りの確率(危険率)5 %]でブランク信号と区別できる信号を与
える分析種の濃度(量)”とする。また,シグナルSとノイズNとの比,S/Nの値が3のときの目的成分
量又は濃度を検出下限としてもよい30)。この方法で検出下限を述べる場合は,必ずS/Nの値と,記録計の
応答時間又はデータ処理装置のパラメーター(積分時間など)を併記する。これは,単一の分析種を極め
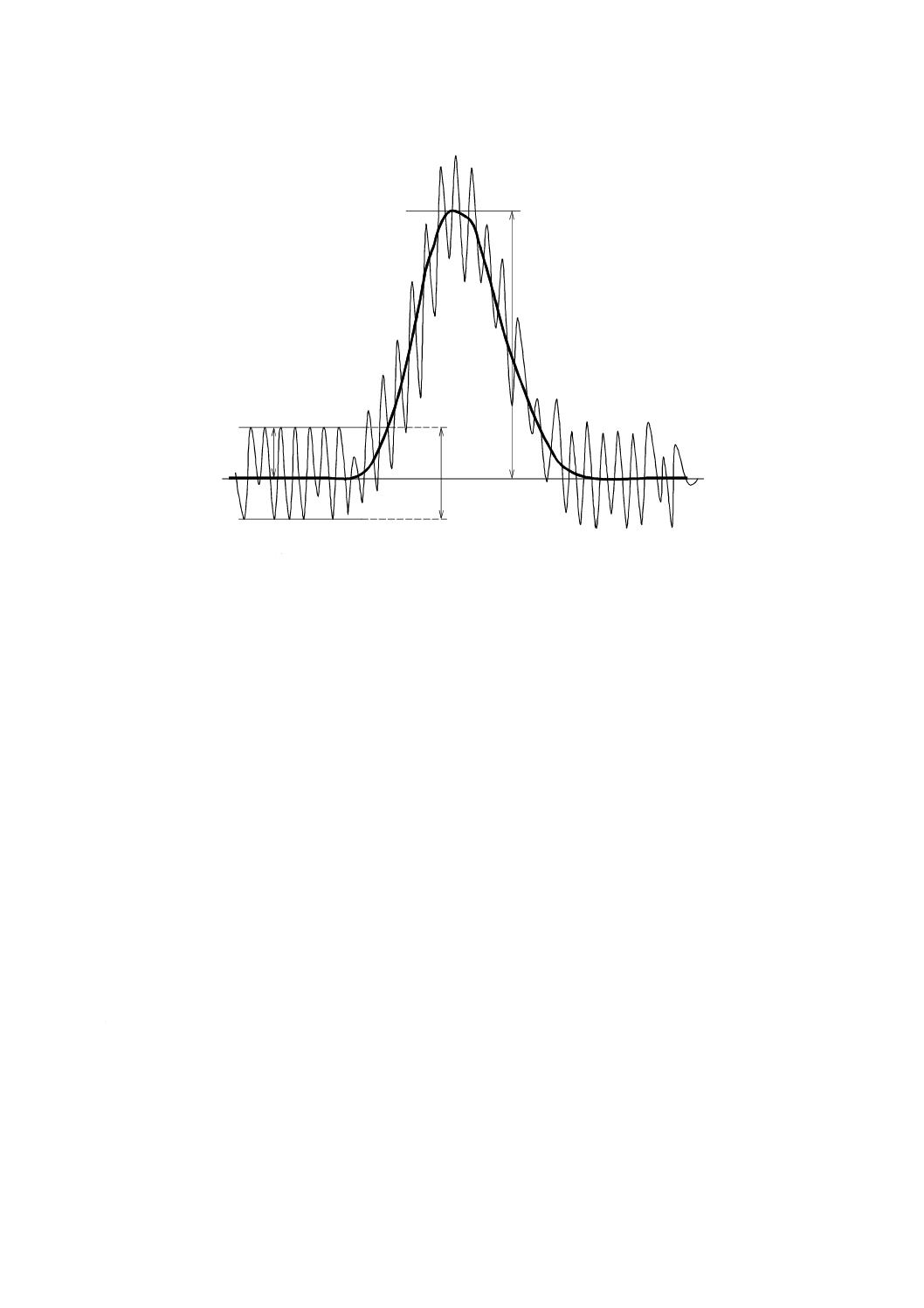
て低濃度に含む試料のクロマトグラムを図18に示すように,ノイズレベルが十分に測定できるように高
感度に記録して描き(図18の細い実線),次のようにして求める。
シグナル:検出器出力の平均値を線で結びノイズを含まないクロマトグラム(図18の太い実線)を得て,
ベースラインからピークの頂点までのピーク高さhをシグナルSとする。
S=h ······················································································· (1)
ノイズ:ピークの前後におけるベースラインの,ピーク半値幅の20倍の間における出力信号の最大値と
最小値の差の振れ幅の1/2をノイズNとする31)。
2
h
N
N
=
··················································································· (2)
注記1 検出下限は,ISO 11843-1:1997“Capability of detection”第1部(用語及び定義)では“minimum
detectable value of the net state variable”“検出可能な最小正味状態変数値”と呼び,JIS Z
8462-1:2001としてJIS化され,“検出下限”という用語は使用しない方向にある。
注記2 検出下限の定義には,次に示すように数種類ある。
a) 確率的手法 ブランク試料及びそれに一定量スパイクした試料を繰り返し測定し,その
ときの測定値の標準偏差から算出する。繰返し回数が多い場合は表8から3.29倍するこ
とになるが,繰返し回数が少ない場合にはその回数に応じた倍率tを乗じる。計算上は
3.29であるが,ICH:International Conference on Harmonisation of Technical Requirements for
Registration of Pharmaceuticals for Human Use[米国,EC,日本の3地域間で新医薬品の製
造(輸入)承認申請に際して要求される分析方法の評価方式について調和を図る国際会
議]では3.3を採用しており,日本薬局方もこの数値を採用している。また,IUPAC:
International Union of Pure and Applied Chemistry(国際純正及び応用化学連合)では3を採
用しているが,今なお検討を進めている。
この規格では,高速液体クロマトグラフィーの使用比率の高い日本薬局方の数値を採
用した。
b) S/Nを用いる手法 クロマトグラフィーにおいては,従来から,シグナルSとノイズN
との比から検出下限を算出する方法が用いられている。
この規格では,従来からの,高速液体クロマトグラフィーにおける検出下限の求め方
を採用し,クロマトグラムベースラインのノイズレベルNをpeak to peakの幅の1/2とと
らえ,これがおおむね3σであるとみなす場合を取り上げる。すなわち,クロマトグラム
のベースラインのノイズからブランク信号の母標準偏差σを推定するのである。また,
“検出可能な最小の正味の濃度又は量”に対する出力信号の平均値もクロマトグラムか
ら推定し,その分布も標準偏差をσとする正規分布と仮定する。
ここで,S/N=2を検出下限と定義すると,これは,おおむね危険率0.1 %となり,検
出下限は“確率99.9 %でブランク信号と区別できる信号を与える目的成分の濃度”を表
すことになる。しかし,ここでノイズレベルの求め方によっては大きな落とし穴がある。

42
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
例えば,信号系の電気回路にダンパーを入れることによってもノイズレベルは著しく下
げることができる。また,試料を1回だけ測定し,しかも短時間のノイズレベルを評価
しているので,必ずしも上記は適用されるとは言い難い。しかし,従来から広く用いら
れている方法であり,従来の値との比較が必要になることもあるので,この方法を,こ
の規格の方法として,採用することを認めることとした。
なお,この方法はICH及び日本薬局方に採用されている。また,S/Nを用いる方法が
IUPAC,ASTM:American Society for Testing and Materials(米国材料試験協会)などにも
記載されているが,Nの採り方がこの規格と異なっているので注意を要する。
注30) S/N=3を検出下限として定めるのは,S/N=2では理論的には誤りの確率が0.1 %であるとはい
え,ドリフト,ふらつき,汚染などの影響で,経験的には2〜3にするのが妥当であると判断さ
れているからである。
31) JIS K 0114とは定義が異なるので注意する。
定量下限を報告する場合は,定量下限の定義を定め,それに従って定量下限の値を報告する。
定量下限の求め方には,検出下限の3〜5倍の値を採る方法,S/N=10のときの分析種量又は濃度とする
方法,分析種量を下げていったときに,定量値の精度が低下し,変動係数が10 %以上になる値を定量下限
とする方法などがある。
表8−測定回数nを減らしたときの倍率t
測定回数n
倍率t
3
5.84
4
4.71
5
4.26
6
4.03
7
3.89
8
3.79
9
3.72
10
3.67
11
3.62
12
3.59
13
3.56
14
3.54
15
3.52
∞
3.29
43
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
細い実線:ノイズがあるクロマトグラム
太い実線:ノイズがない場合のクロマトグラム
図18−分析種が検出下限付近の濃度の高感度記録クロマトグラム概念図
13.4 ブランクの測定
ブランク試料[例えば,分析種を含まないことが明らかな試料を分析種と並行して処理した測定用試料
溶液,又は純溶媒32)]を用いてブランクのクロマトグラムを得る。
注32) 分析種を含まないことが明らかな試料を分析種と並行して処理した測定用試料溶液を測定する
と,試料マトリックス及び全分析操作にわたる影響を明らかにできる。純溶媒をブランクとし
て測定すれば,溶媒ブランク,試料導入時の汚染,クロスコンタミネーションなど,装置から
の影響を分離して求められる。
13.5 定期的な装置性能の点検
分析機器製造業者の取扱説明書に従い,各装置構成部の点検を定められた頻度で実施する。点検の記録
は保管する。
14 装置の設置
設置場所は,次の条件を備えた室内が望ましい。
a) 結露せず,温度及び湿度が装置に定められた仕様の範囲内にあり,急激な変化を生じないところ。
b) 振動がなく,直射日光が当たらないところ。
c) 腐食性ガス及びほこりが少なく,換気のよいところ。
d) 強い磁気及び電場の影響がないところ。
e) 供給電源は装置の仕様に指定された電圧,電気容量及び周波数のもとで,電圧変動は±10 %以下でか
つ周波数の変動がないところ。
f)
接地抵抗100 Ω以下の接地点があるところ。
44
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 実験台は,装置の総質量に十分耐えられる。
h) 装置の放熱及びメンテナンスを考慮した設置スペースを確保する。
i)
有機溶媒等による汚染を防ぐための室内換気設備又は強制排気設備を必要に応じて設ける。
15 安全
安全のために,次の事項に十分注意しなければならない。
a) 試料及び分析に使用する薬品の取扱いは,爆発性,引火性,毒性又は有害性に十分注意して行う。そ
れらの廃棄については,安全化,無害化などの配慮を行い環境汚染防止の諸規定に従った方法で行う。
危険物の取扱いについては,“消防法”及び“危険物の規制に関する政令”など,毒物,劇物の取扱い
については,“毒物及び劇物取締法”の諸規定に従う33)。
注33) 毒性,有害性のある薬品の取扱いには,必要に応じて保護具(保護めがね,ゴム手袋,防毒
マスクなど)を着用する。
b) 装置を接地点に接地する。
c) 高圧容器詰のガスを利用するときは,“高圧ガス保安法”の諸規定に従う。
なお,高圧ガス保安法に規定する圧力以下の容器を装置の近くで用いるときにも,容器が転倒しな
いように,架台,壁,実験台などに固定しなければならない。
d) 装置の運転に先立ち,配管の接続部,流路などからの液漏れ及びガス漏れがないことを十分に点検す
る。
e) 装置内部に直接触れると感電のおそれがあるので,装置の点検,修理は,通常電源を切って行う34)。
注34) 装置にリチウム電池が内蔵されている場合は,取扱い方を誤ると破裂する危険があるため注
意する。
f)
検出器に用いられる光源ランプの保守及び交換に際し,検出器によっては点灯時,紫外線が放射され
ている場合があるので必ず眼の保護具を用いる。また,発熱していることもあるので,直接触れると
きは冷えていることを確認する。
g) 装置が他の機器に電磁波障害を与えたり,他の機器から電磁波妨害を受ける場合があるので注意する。
h) 生体試料[血清,血しょう(漿),組織,尿など]を素手で取り扱うと,感染のおそれがあるため,慎
重に取り扱う必要がある。試料の皮膚への直接接触,ピペッティングによる誤飲,針刺しなどに注意
し,保護具(保護めがね,ゴム手袋,マスクなど)を着用する。
i)
有機溶媒による発火を防ぐため,廃液チューブ及び廃液容器の静電気対策を行う。
16 個別規格に記載すべき事項
高速液体クロマトグラフィーを分析方法に用いる場合は,少なくとも次の事項を規定しなければならな
い。
なお,データの質の管理は,必要に応じて分析法バリデーション,システム適合性試験などで規定する。
a) 分析成分及びその濃度範囲
b) 試料の採取方法,前処理方法及び保存方法
c) 分析条件
1) カラム充塡剤の種類及び粒子径
2) カラム管の材質,内径及び長さ
3) カラム温度(必要な場合)
45
K 0124:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 移動相の種類,流量,組成及び溶離条件
5) 試料導入方法並びに試料導入量及びその希釈倍率
6) ポストカラム誘導体化反応を用いる場合は,反応液の組成,流量及び反応条件
7) 検出器の種類及び所要感度35)
注35) 特定成分の一定量を導入したときクロマトグラムのピークのシグナルとノイズとの比
(S/N)又はシグナルとブランクとの比(S/B)で規定し,その試験方法も明記する。
8) データ処理部(データ処理装置,記録計)
9) 測定時間範囲(データ取込み時間)
d) 成分の確認方法 代表的なクロマトグラム例を示す。
なお,クロマトグラムには分析条件,ピークの成分名,保持時間及び検出器の出力信号のスケール
を付記しなければならない。
e) 定量方法
1) ピーク面積又は高さの測定方法
2) 定量方法の種別及び分析回数
3) 定量分析に用いた物質[分析種の純物質,内標準物質(種類,純度)又は分析種純物質の混合物の
場合には,組成,濃度範囲及び調製方法]
f)
分析結果の表示及び濃度の単位
参考文献 [1] C. T. Viswanathan , Surendra Bansal , Brian Booth , Anthony J. DeStefano , Mark J. Rose , Jeffrey
Sailstad , Vinod P. Shah , Jerome P. Skelly , Patrick G. Swann , and Russell Weiner ,
Workshop/Conference
Report−Quantitative
Bioanalytical
Methods
Validation
and
Implementation: Best Practices for Chromatographic and Ligand Binding Assays ,The AAPS
Journal 2007; 9 (1) Article 4
[2] Chambers, E., Wagrowski-Diehl, D. M., Lu, Z., Mazzeo, J. R. , J. Chromatogr. B 852 (2007) 22-34
[3] JIS K 0114 ガスクロマトグラフ分析通則
[4] JIS Z 8462-1 測定方法の検出能力−第1部:用語及び定義
注記 対応国際規格:ISO 11843-1,Capability of detection−Part 1: Terms and definitions
(IDT)