K 0123:2018
(1)
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 1
4 概要······························································································································· 5
5 装置······························································································································· 5
5.1 装置の構成 ··················································································································· 5
5.2 ガスクロマトグラフ ······································································································· 5
5.3 インターフェース(GC/MS接続部) ················································································· 7
5.4 質量分析計 ··················································································································· 8
5.5 システム制御・データ処理部 ·························································································· 17
5.6 附属装置 ····················································································································· 17
6 安全······························································································································ 19
7 装置の設置 ····················································································································· 20
8 装置の運転 ····················································································································· 21
8.1 運転の手順 ·················································································································· 21
8.2 ガスクロマトグラフの準備 ····························································································· 21
8.3 質量分析計の準備 ········································································································· 21
8.4 始動 ··························································································································· 21
8.5 調整 ··························································································································· 21
8.6 測定条件の設定 ············································································································ 22
8.7 試料の導入 ·················································································································· 22
8.8 測定(質量スペクトルの採取) ······················································································· 22
8.9 データ処理 ·················································································································· 25
8.10 誘導体化 ···················································································································· 25
9 試料の調製及び測定 ········································································································· 26
9.1 試料の調製 ·················································································································· 26
9.2 測定 ··························································································································· 26
10 定性分析 ······················································································································ 27
11 定量分析 ······················································································································ 29
11.1 試料の前処理 ·············································································································· 29
11.2 内標準法 ···················································································································· 29
11.3 絶対検量線法 ·············································································································· 30
11.4 標準添加法 ················································································································· 30
11.5 検量線データベース法 ·································································································· 30
11.6 検量線又は関係線の作成方法 ························································································· 30
K 0123:2018 目次
(2)
ページ
11.7 定量操作 ···················································································································· 33
12 データの質の管理 ·········································································································· 35
12.1 一般事項 ···················································································································· 35
12.2 計量計測トレーサビリティの確保 ··················································································· 35
12.3 分析値の信頼性の確保 ·································································································· 36
12.4 データの質の管理のための測定 ······················································································ 36
12.5 分析方法の妥当性確認の実施 ························································································· 36
12.6 検出下限の求め方 ········································································································ 37
12.7 空試験値の測定 ··········································································································· 38
12.8 定期的な装置性能の点検 ······························································································· 38
12.9 クロマトグラムのピーク形状及び分離の確認 ···································································· 39
12.10 質量スペクトルの質の確認 ·························································································· 39
12.11 作業手順書の作成 ······································································································· 40
12.12 分析値の不確かさの求め方 ·························································································· 40
13 分析報告書 ··················································································································· 40
14 個別規格に記載すべき事項 ······························································································ 41
附属書A(参考)化学イオン化 ······························································································ 42
附属書B(参考)分解能パラメーターの定義及び評価方法 ··························································· 44
附属書C(参考)分析値の不確かさの見積り手順 ······································································ 45
附属書D(参考)四重極形 ···································································································· 53
附属書E(規定)サイクルタイム,サンプリング時間及びドゥエルタイム ······································ 54
K 0123:2018
(3)
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人日本
分析機器工業会(JAIMA)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業
規格を改正すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業
規格である。
これによって,JIS K 0123:2006は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
K 0123:2018
ガスクロマトグラフィー質量分析通則
General rules for gas chromatography / mass spectrometry
1
適用範囲
この規格は,ガスクロマトグラフ質量分析計を用いて,常温で気体又は十分な蒸気圧をもつ安定な有機
化合物,有機金属化合物,無機化合物などの定性分析及び定量分析を行う場合の通則について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0114 ガスクロマトグラフィー通則
JIS K 0133 高周波プラズマ質量分析通則
JIS K 0211 分析化学用語(基礎部門)
JIS K 0214 分析化学用語(クロマトグラフィー部門)
JIS K 0215 分析化学用語(分析機器部門)
JIS R 3505 ガラス製体積計
JIS Z 8000-1 量及び単位−第1部:一般
JIS Z 8000-9 量及び単位−第9部:物理化学及び分子物理学
ISO 17034,General requirements for the competence of reference material producers
ISO Guide 34:2009,General requirements for the competence of reference material producers
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS K 0050,JIS K 0114,JIS K 0133,JIS K 0211,JIS K 0214,
JIS K 0215,JIS Z 8000-1及びJIS Z 8000-9によるほか,次による。
なお,括弧内の対応英語は参考のために示す。
3.1
アナライザー(analyzer)
質量分離部の総称。分析管ともいう。
3.2
イオン化室(ionization chamber)
イオン源内で,試料分子が電子又は反応イオンと相互作用を起こし,イオンが生成する場所。
2
K 0123:2018
3.3
イオン源(ion source)
質量分析計を構成する一部分で,試料成分のイオン化及び生成したイオンのアナライザーへの移送を行
う箇所。イオン化室,フィラメント,イオンの加速,収束などを行う電極群などからなる。
3.4
m/z(mass-to-charge number ratio)
イオンの質量mとその電荷数zとの比。質量スペクトルの横軸に用いられる。質量電荷数比ともいう。
3.5
サンプリング時間(sampling time)
装置が検出対象とするイオンを取り込む時間。モニタリング方法によって異なる(附属書E参照)。
3.6
試薬ガス(reagent gas)
化学イオン化法において試料分子をイオン化するために用いるガス。通常,高純度のメタン,2-メチル
プロパン(イソブタン),アンモニアなどが用いられる。
3.7
衝突誘起解離(collision induced dissociation:CID)
運動エネルギーをもったイオンが衝突ガスと衝突し,衝突エネルギーの一部が内部エネルギーに変換さ
れ励起されることでイオンの解離が起こる現象。衝突活性化解離(CAD)ともいう。
3.8
精密質量(exact mass or accurate mass)
イオン又は分子の質量を10−3 uまで計算又は測定した値。それぞれ,計算精密質量(exact mass)及び
測定精密質量(accurate mass)ということもある。
注記 uは統一原子質量単位を表し,静止して基底状態の質量数12の炭素(12C)原子の質量の12分
の1の質量と定義される。
3.9
全イオン電流クロマトグラム(total ion current chromatogram:TICC)
ある範囲のm/zのイオン電流の総和を連続的に検出・記録した図。
3.10
選択イオンモニタリング(selected ion monitoring:SIM)
あらかじめ選択した特定のm/zのイオンを連続的に検出する方法。
3.11
走査(scan)
ある範囲のm/zのイオンを検出するために,磁場及び電場強度又は四重極ロッドへの電圧を変化させる
こと。
注記 附属書E参照。
3.12
相対感度(relative response)
基準にする成分(内標準物質)の単位量当たりのピーク面積(又はピーク高さ)に対する分析種の単位
量当たりのピーク面積(又はピーク高さ)との比。相対感度の逆数を,相対応答係数(RRF:relative response
factor)という。
3
K 0123:2018
3.13
タンデム質量分析(tandem mass spectrometry:MS/MS)
同種又は異種の質量分析計を直列に接続し,1台目のイオン化室で生成したイオン種のうち一つをプリ
カーサーイオン(前駆イオンともいう)として選択し,2台目の質量分析計でそのプリカーサーイオンの
分解から生じるプロダクトイオンを検出する方法。
3.14
デコンボリューション(deconvolution)
クロマトグラム上で完全に分離できなかった重複ピークから,それを構成する個々の成分ピーク及び/
又は質量スペクトルを得る操作。
3.15
ドゥエルタイム(dwell time)
SIM又はSRMにおいて選択したイオンの1測定当たりの取込み時間。
注記 附属書E参照。
3.16
ハートカット[heart-cut (s)]
カラムから溶出する特定の画分を流路切換えで次のカラムに導入する,又は系外に排出するカラムスイ
ッチング手法。
注記 最初に溶出する画分を系外に排出する場合を,特にプレカットという。
3.17
バックフラッシュ(back flash)
キャリヤーガスを逆方向に流してカラム内に保持された成分を一括してカラムから溶出させる手法。
注記 分析時間の短縮,分離カラムの保護(不要成分の排出)などのため,また,狭いバンドで溶出
させ次のカラムに導入する,又は一つのピークとして検出器に導入し定量する目的で使用する。
3.18
半定量分析(semi quantitative analysis)
量的概念を加味して行う定性分析。
3.19
反応イオン(reaction ion or reagent ion)
試薬ガスを起源とする,分析種のイオン化を促進するイオン種。
3.20
標準作業手順書(standard operating procedure:SOP)
試験,検査などの実施方法及び手順について詳細に規定した文書。
3.21
フラグメンテーション(fragmentation)
イオンを構成する一つ以上の結合が開裂することによって,そのイオンより小さい質量のイオンを生じ
る反応。
3.22
フラグメントイオン(fragment ion)
フラグメンテーションで生じたイオン。
4
K 0123:2018
3.23
プリカーサーイオン(precursor ion)
あるイオンから別のイオンが生じるときのイオン。前駆イオンともいう。
3.24
プロダクトイオン(product ion)
特定のイオンから生成した全てのイオン。生成イオンともいう。
3.25
プロファイルデータ(profile data)
質量分析計で測定されたm/zと信号強度からなる生データであり,質量スペクトルに加工する前のデー
タである。生データの分解能は装置によって異なる。
3.26
分解能(resolution)
質量分析計においてm/zの異なる質量(M)のピークを区別できる尺度となる数値。質量スペクトルの
任意の質量ピークm/z=M及びm/z=M+ΔMの2本のピークは区別できるが,m/zの差がΔMより小さく2
本のピークは区別できないとき,R=M/ΔMをこの装置の分解能とする。検出器が識別できる質量の差を表
現するもので質量分解能ともいう。
注記 分解能の計算は,ピークの半値幅(FWHM:full width at half maximum)から求めるものと隣接
したピーク間の重なりの度合い(10 %谷など)から求めるものがある。
3.27
保持指標(retention index)
ガスクロマトグラフィーにおいて,成分を同定するための指標の一つで,次の式によって定義される数
値。
z
z
z
i
X
X
X
X
z
I
log
log
log
log
100
100
1−
−
+
=
+
ここに,
Xi: 対象成分の空間補正保持時間(s)
Xz: Xiより空間補正保持時間が小さく,かつ,最もXiに近い
直鎖アルカンの空間補正保持時間(s)
z: 直鎖アルカンの炭素数
Xz+1: z+1の炭素数の直鎖アルカンの空間補正保持時間(s)
なお,直線昇温分析の場合,上式のlogXはXとなる。Kovats Index(KI)と呼ぶこともある。
3.28
マスクロマトグラム(mass chromatogram)
一定の時間間隔で質量スペクトルを測定し,コンピュータに記憶させた後,特定のm/zのイオン強度を
取り出して表示した図。抽出イオンクロマトグラム(extracted ion chromatogram:EIC)ともいう。
3.29
モジュレータ(modulator)
包括的二次元GCにおいて,前段のカラムから溶出する全成分を一定時間間隔で捕集と脱離とを繰り返
し,順次,後段のカラムに導入する装置。
3.30
ライブラリー検索(library search)
5
K 0123:2018
GC/MSなどで測定した特定成分の質量スペクトルを,データ処理部に内蔵の又は外部の質量スペクトル
ライブラリーと照合し,一致度などから同定又は分子構造推定を行う手法。
4
概要
ガスクロマトグラフィー質量分析法(GC/MS法)は,混合物試料の分離分析に優れているガスクロマト
グラフ(GC)と,試料成分の構造解析及びごく微量分析に優れている質量分析計(MS)とを直結した装
置であるガスクロマトグラフ質量分析計(GC/MS装置)を用いて,それぞれの特徴を生かして試料に関す
る物質情報を高感度に得るための分析方法である。
気体又は液体の混合物試料をGC/MS装置に導入すると,分析種はガスクロマトグラフで分離され,連
続的に質量分析計のイオン源に導かれてイオン化される。生じた正又は負のイオンは,アナライザー(質
量分離部)に入り,m/zに応じて分離される。分離されたイオンは,順次,検出部でその量に対応する電
気信号に変換され,各種クロマトグラム及び質量スペクトルとして記録される。分析種ピークの保持時間
(ここでは空間補正保持時間)及び質量スペクトルから定性分析を行い,ピーク面積(又はピーク高さ)
から定量分析を行う。
5
装置
5.1
装置の構成
装置は,ガスクロマトグラフ,インターフェース(GC/MS接続部),質量分析計及びシステム制御・デ
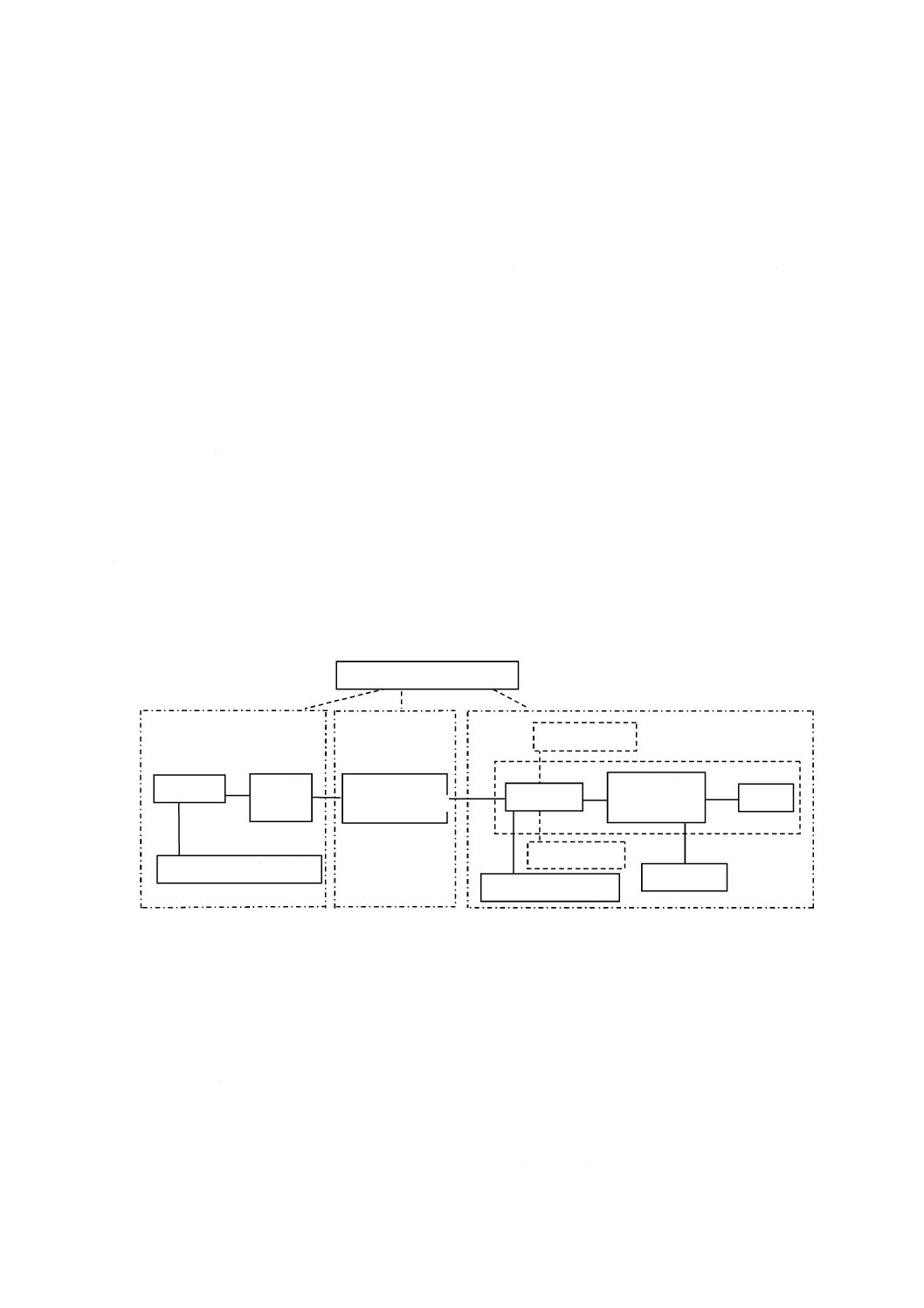
ータ処理部からなる。装置の構成の例を,図1に示す。
図1−装置の構成図(例)
5.2
ガスクロマトグラフ
ガスクロマトグラフの構成は,次による。
a) キャリヤーガス流量制御部 キャリヤーガス流量を制御するための部分である。基本的には圧力調節
弁,流量調節弁,圧力計などで構成されるが,最近の装置の多くは各部の小形化,電子化がなされ,
圧力センサー,流量センサー,調節弁等を用いてキャリヤーガスの流量,圧力,線速度などを電子制
御できるようになっている。また,キャリヤーガスの消費量を削減するための機能を備えているもの
アナライザー
(質量分離部)
ガスクロマトグラフ
質量分析計
直接試料導入部
システム制御・データ処理部
キャリヤーガス流量制御部
試料導入部
カラム槽
カラム
(分離部)
インターフェース
(GC/MS接続部)
試薬ガス導入部
校正用標準試料導入部
真空排気部
検出部
イオン化部
6
K 0123:2018
が多い。
b) 試料導入部 試料をキャリヤーガス流路中に導入する部分である。液体又は気体試料をシリンジで導
入するもの及び気体又は液体試料を計量管などで採取し,弁操作で導入するものがある。また,キャ
ピラリーカラム用の試料導入部には,分割導入方式と非分割導入方式とがある。分割導入方式には,
注入気化した試料の一部だけをカラムに導入するスプリット注入法がある。非分割導入方式には,開
閉可能な排気口を利用して溶媒処理を工夫し,そのピークの大きなテーリングを生じることなく試料
をほぼ全量導入するスプリットレス注入法,大口径キャピラリーカラムを用いて気化した試料の全量
を導入する直接注入法,試料導入部の温度を溶媒の沸点温度以下に設定し,注入口を通してカラムに
直接試料を導入するコールドオンカラム注入法,試料導入部の温度を溶媒の沸点温度以下に設定し,
試料導入後,低温で溶媒を排出した後,急速加熱して試料をカラムに導入する温度プログラム気化注
入法などがある。
また,各種前処理導入装置を直結して,試料の導入を行い,適用範囲を広げる。
c) カラム カラムは,キャピラリーカラムと充塡カラムとの2種類に大別できる。目的に応じて長さ,
内径,固定相の種類などを選ぶ。キャピラリーカラムは,材質が金属,ガラス,石英ガラス,合成樹
脂などで,一般に内径が0.1 mm〜1.2 mm,長さが5 m〜100 m及び固定相の膜厚が0.1 µm〜10 µmの
ものが使われる。吸着形キャピラリーカラムは,通常は吸着剤が数μm〜数10 μmの厚さでカラム内
壁に塗布され,各種のバインダーを用いて固定化されている。分配形キャピラリーカラムは,固定相
液体をカラム内壁に塗布したものである。固定相液体の移動を防ぐため,化学結合・架橋を施したも
のがほとんどである。充塡カラムは,材質が金属,ガラス,合成樹脂などでできた内径が2 mm〜6 mm,
長さが0.5 m〜5 mの管に充塡剤を充塡したものである。充塡剤には,固定相液体を担体に担持させた
もの,吸着剤その他が使われる。担体及び吸着剤の粒度は150 µm〜180 µm,180 µm〜250 µmなど各
種ある。
d) カラム槽 カラム槽は,内部を設定された温度に保持し,かつ,温度分布を一定に保つことが可能な
加熱機構をもつ。また,適切な分離が行え,分離にかかる時間を調節可能な昇温分析を行う場合,初
期温度,継続時間(ホールド時間),昇温速度,最終温度及び継続時間(ホールド時間)の設定が可能
でなければならない。
e) 高速GC 高速ガスクロマトグラフィー(高速GC)は通常,内径の小さい0.1 mm程度のカラムを用
い,高速昇温することによって,分離を損なうことなく測定時間を通常の数分〜数10分の一に短縮す
る手法である。内径の小さく膜厚が薄いカラムを用いるため試料負荷容量は小さいが,測定時間の短
縮が可能なためGC/MSへの適用も多い。高速GC及びGC×GC[f) 2) 参照]に対応したカラムは,
その上に直接ヒーターと温度センサーを巻きつけた形でカラム槽とは独立に高速昇温ができる仕様に
なっている場合が多い。
なお,通常のカラム槽でも可能な高速の昇温を行う場合も高速GCと呼ばれている。
f)
二次元GC 特性の異なる2本のカラムと検出器(質量分析計含む)との組合せによって一つのカラ
ムでは分離が困難な試料の定性分析及び定量分析,並びに複雑な組成の試料の定性分析及び定量分析
を可能にするものとして次の二次元ガスクロマトグラフィー(二次元GC)がある。
1) GC-GC 二次元GCの一種で最初のカラムで分離した特定成分(分画)をハートカットし,選択的
に二番目のカラムに導入し,再度分離を行う。通常,一つのカラム槽内で流路切替え装置(部品)
を用いて測定するが,独立した二つのカラム槽を使用する場合もある。
2) GC×GC 包括的二次元ガスクロマトグラフィーと呼ばれ,最初のカラムで分離した成分(分画)
7
K 0123:2018
を全領域にわたって連続的に捕集と脱離とを繰り返し,極性の異なる2番目のカラムで高速分離を
行う。捕集及び脱離はモジュレータと呼ばれる装置を用いる。また,通常のカラム槽とは別に直接
加熱が可能なカラム[e) 参照]を取り付け,個々のカラム温度を独立に制御して測定する場合があ
る。
5.3
インターフェース(GC/MS接続部)
インターフェースは,GCとMSとの接続部をいう。大気圧となっているガスクロマトグラフのカラム
出口と高真空になっている質量分析計のイオン源とを接続する部分で,カラムの種類及び真空ポンプの排
気速度によって,次のいずれかを用いる。
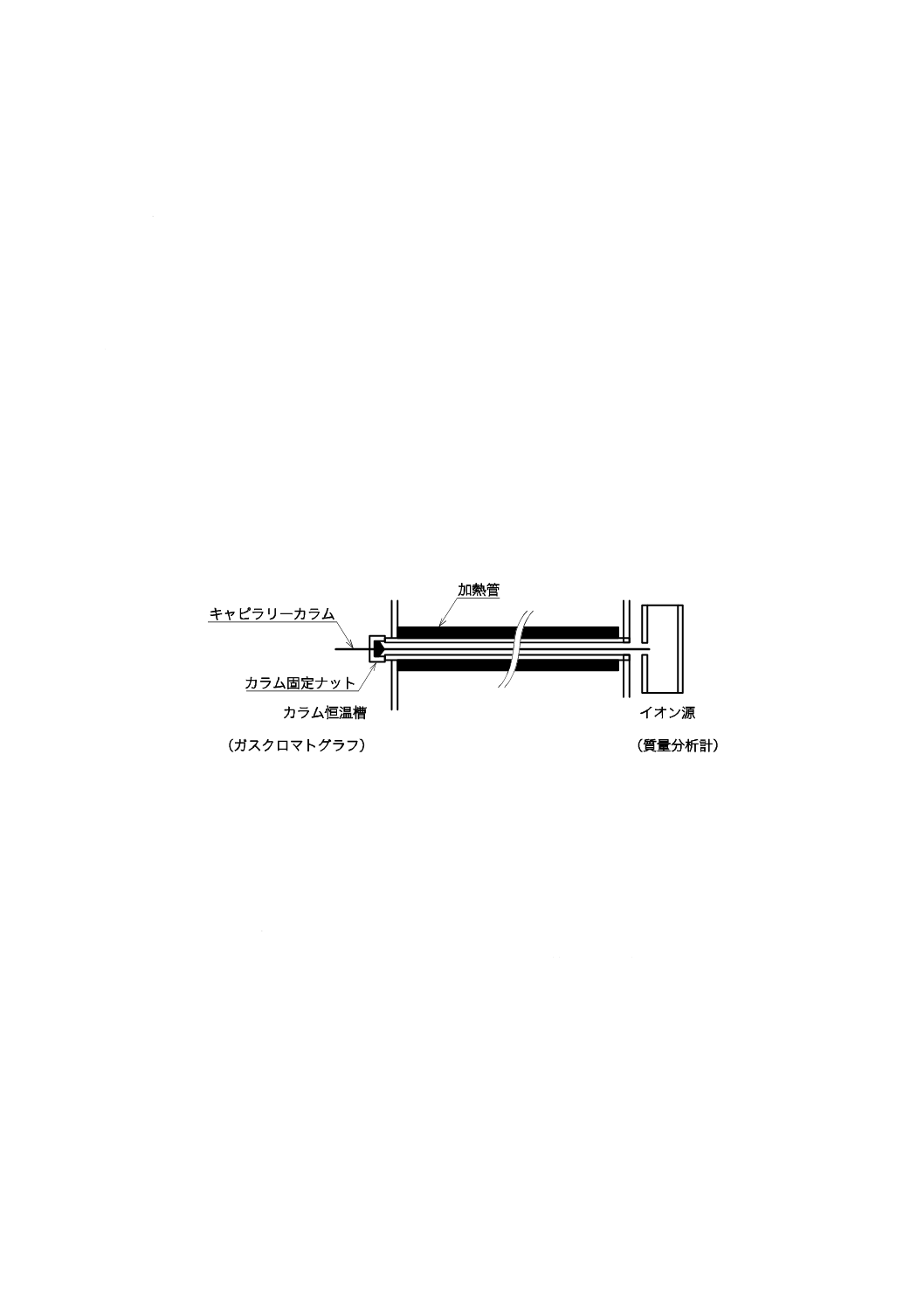
a) キャピラリーカラム 内径が約0.3 mm以下のキャピラリーカラムの場合は,その出口側の一端が質
量分析計のイオン源に直接又はごく近くに位置するように接続する。また,内径が約0.3 mm以上の
大口径キャピラリーカラムの場合は,直接イオン源に接続する場合もあるが,キャピラリーカラムの
前段又は後段に抵抗管を接続して使用することも多い。キャピラリーカラム又は抵抗管を介しての接
続時にはこのインターフェースの箇所を特にトランスファーラインと呼ぶ。また,b) に示す接続方法
を用いる場合もある。GC/MS接続部は,小形の恒温槽又は加熱管によって,カラム槽と同じかそれ以
上の温度に保つことができる加熱・温度制御機構及び温度測定機構をもつ。キャピラリーカラムの接
続部の例を図2に示す。
図2−キャピラリーカラムの接続部(例)
b) 充塡カラム及び大口径キャピラリーカラム 充塡カラム及び大口径キャピラリーカラムのキャリヤ
ーガス流量は5 mL/min〜20 mL/minであり,a) で示した方式では真空維持が困難,測定感度低下,流
量制御精度の低下などが起こる。そのため,キャリヤーガスの分離除去及び分析種の濃縮のため,通
常ジェットセパレーターを用いる。このセパレーターの材質はガラス製が一般的で,排気系をもつ。
セパレーターの構造の例を,図3に示す。この原理は,キャリヤーガスであるヘリウムと分析種との
拡散係数の差を利用するもので,図3のA部のようにガラス管の先端を細くすることによって,質量
の小さいヘリウムのほとんどを油回転ポンプで吸引する。一方,質量が相対的に大きい分析種をさほ
ど損失することなく質量分析計に導く。
また,カラムから流出するキャリヤーガス及び試料成分を口径の小さいキャピラリーを介してスプ
リットさせて,一部を質量分析計に導入する方法もある。
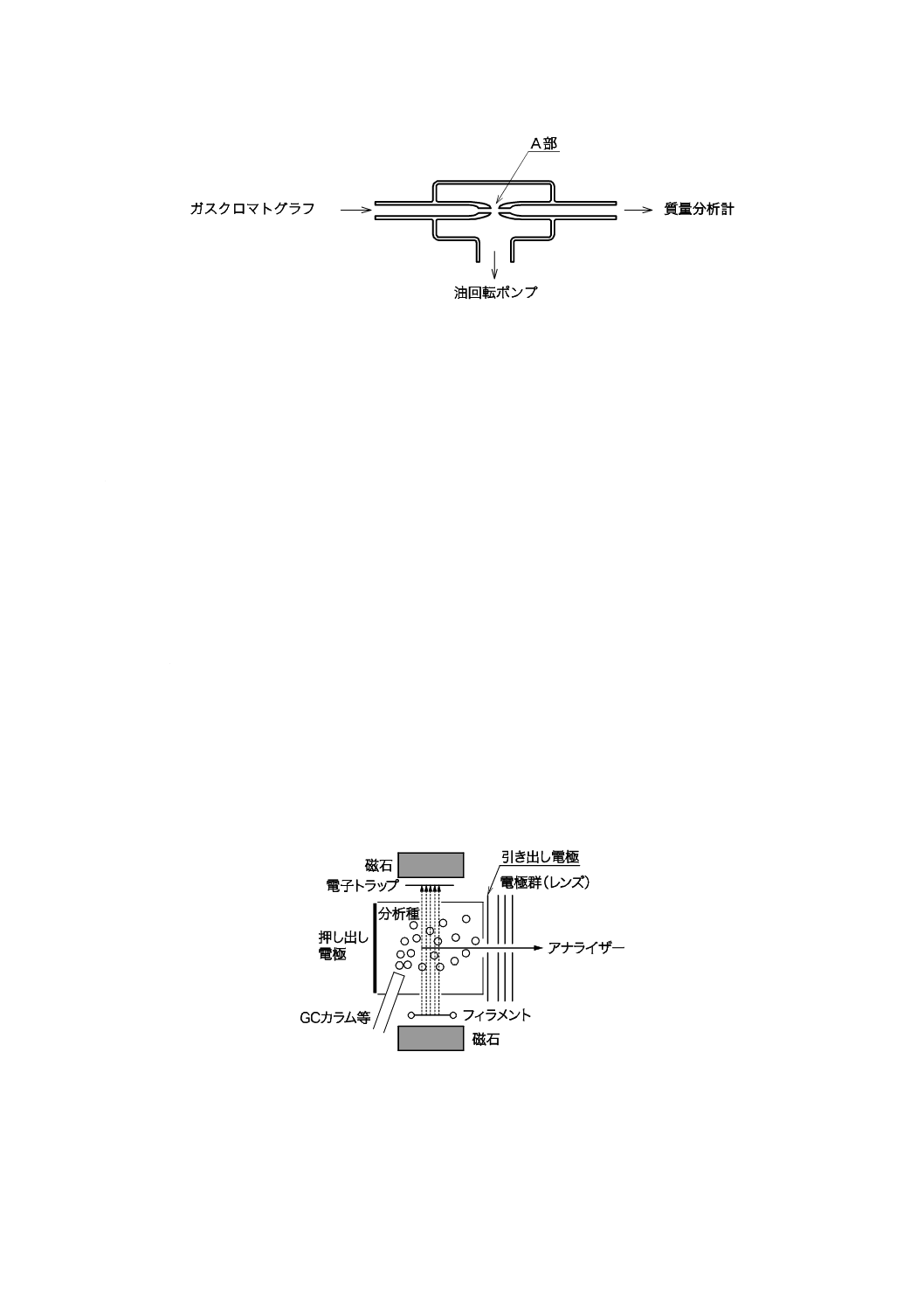
8
K 0123:2018
図3−セパレーター(例)
5.4
質量分析計
質量分析計は,ガスクロマトグラフによって分離された分析種をイオン化し,m/zに応じて分離した後,
これを検出する部分で,次のイオン化部,アナライザー(質量分離部),検出部,真空排気部及び校正用標
準試料導入部からなる。
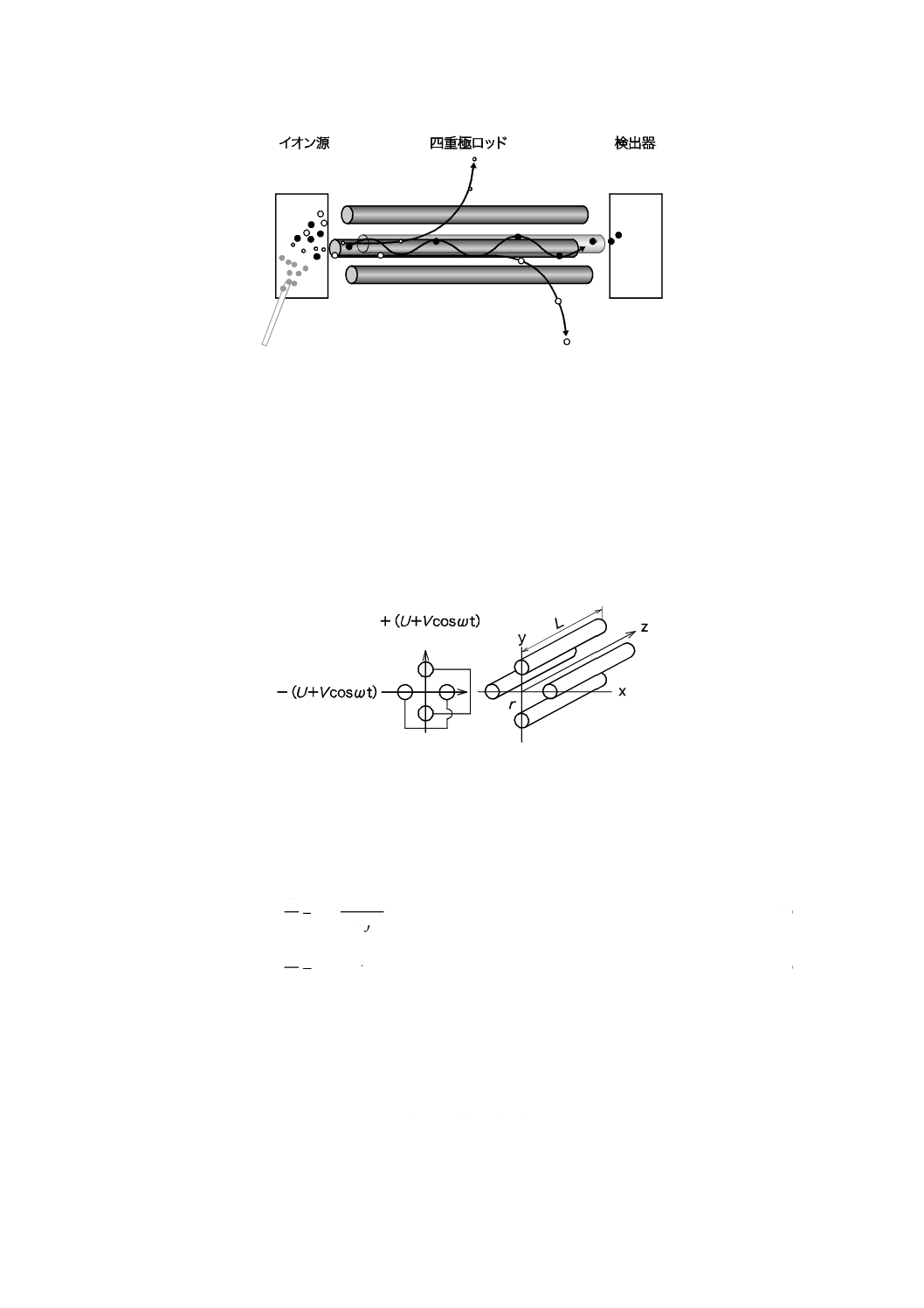
a) イオン化部 イオン化部は,ガスクロマトグラフのカラムから溶出した分析種をイオン化し,アナラ
イザーに導く部分である。イオン化法には,電子イオン化(EI)法,化学イオン化(CI)法その他の
方法がある。化学イオン化法には正イオン化学イオン化法(PICI)と負イオン化学イオン化法(NICI)
がある。最も一般的なEI法のイオン源は,イオンを生成するイオン化室,生成したイオンを効率よく
アナライザーに導くための押し出し電極又は引き出し電極,及びイオン化室直後に配置された電極群
(レンズ)から成る。イオン源にはイオン化室内に電子を供給するフィラメント(陰極),電子トラッ
プ(陽極)が附属し,更にフィラメントと電子トラップの外側に磁石を装着し,電子をら(螺)旋状
の軌道で運動させることでイオン源内での広がりを防ぎ,分析種のイオン化の効率を高める。イオン
の押し出し電極又は引き出し電極によってイオン化室から射出したイオンは,イオン化室直後に配置
された電極群(レンズ)によってイオンの加速,イオンビームの収束などの作用を受けながら,アナ
ライザーに導かれる。また,イオン源はイオン化室内壁への分析種及び共存成分の吸着を軽減させる
ため,水分などの残留を抑制するためにヒーターを備え,目的に応じた温度に設定可能である。CIイ
オン源には,イオン化時に,試薬ガスを導入する。イオン化部(EIイオン源)の例を図4に示す。
図4−イオン化部(EIイオン源)(例)
電子イオン化法,正イオン化学イオン化法,負イオン化学イオン化法その他のイオン化法は,次に
よる。
9
K 0123:2018
1) 電子イオン化(EI)法 真空下でフィラメントから放出された数10 eV以上のエネルギーをもつ電
子をイオン化室内の気体状の分析種に照射し,その運動エネルギーの一部を電子エネルギーの形で
付与してイオン化する方法。最初に分子イオン [M]+・を生じるが,一般に過剰な内部エネルギーに
よって分子構造に依存したフラグメンテーションが起こり,フラグメントイオンを生じる。分子構
造によっては,分子イオンが残らない場合もある。GC/MSで最も利用されている。
2) 正イオン化学イオン化(PICI)法 イオン化室に高純度メタンなどの試薬ガスを100 Pa程度の圧力
となるように導入し,電子などを照射して試薬ガス由来の反応イオン(例えば,メタンの場合,
CH5+,C2H5+など)を生成させる。次に,これらの反応イオンと分析種との間のイオン−分子反応
によって分析種をイオン化する。構造的に安定な偶数電子イオンであるプロトン付加分子 [M+H]+
又は脱ヒドリド分子 [M−H]+を生じやすい。また,反応熱の一部が過剰な内部エネルギーとして蓄
えられるが,反応エネルギーの小さいソフトなイオン化のため,EI法と比較して,フラグメンテー
ションは少ない。PICI法によって分子の質量の推定が容易な場合が多い。
試薬ガスの高純度メタンは,純度99.99 %以上を推奨する。
なお,メタン以外の試薬ガスとしてはイソブタン,アンモニアなどが用いられ,純度99.9 %以上
を推奨する。
注記 附属書A参照。
3) 負イオン化学イオン化(NICI)法 イオン化室をメタンなどの試薬ガスで満たすことはPICI法と
同じであるが,イオン化の機構は,反応イオン形と電子捕獲形との2種類に大別される。前者は,
試薬ガスから生じた反応イオン(OH−,CH3O−,Cl−など)又は系内に存在する水から生じた反応
イオン(OH−など)と分析種との間のイオン−分子反応によってイオン化し,[M−H]−などのイオ
ンを生じさせる。後者は,イオン化室内で運動エネルギーを失った熱電子が分析種と共鳴捕獲反応
を起こしてイオン化するもので,電子親和性の高い化合物に対して分子イオン [M]−・などを生成す
る。
注記 附属書A参照。
4) その他のイオン化法 GC/MS装置で用いられるその他のイオン化法としては,フィールドイオン化
(FI)法,光イオン化(PI)法,レーザーイオン化(LI)法,大気圧化学イオン化(APCI)法,誘
導結合プラズマによるイオン化(ICP)法などがある。ICP法は本来,無機元素の分析に用いられる
が,GC/MS装置では有機金属化合物などの元素分析的測定などに用いられる。GC/MSとしてAPCI
法を用いる場合,フラグメントイオンの生成が少ないため,構造情報を得るためにはMS/MSを行
う必要がある。
b) アナライザー イオン化部で生じた分析種由来のイオンをそのm/zに従って分離する部分で,分離方
式の違いによって次の四重極形(Q),イオントラップ形(IT),磁場形,飛行時間形(TOF),三連四
重極形,フーリエ変換形(FT),ハイブリッド形などがある。
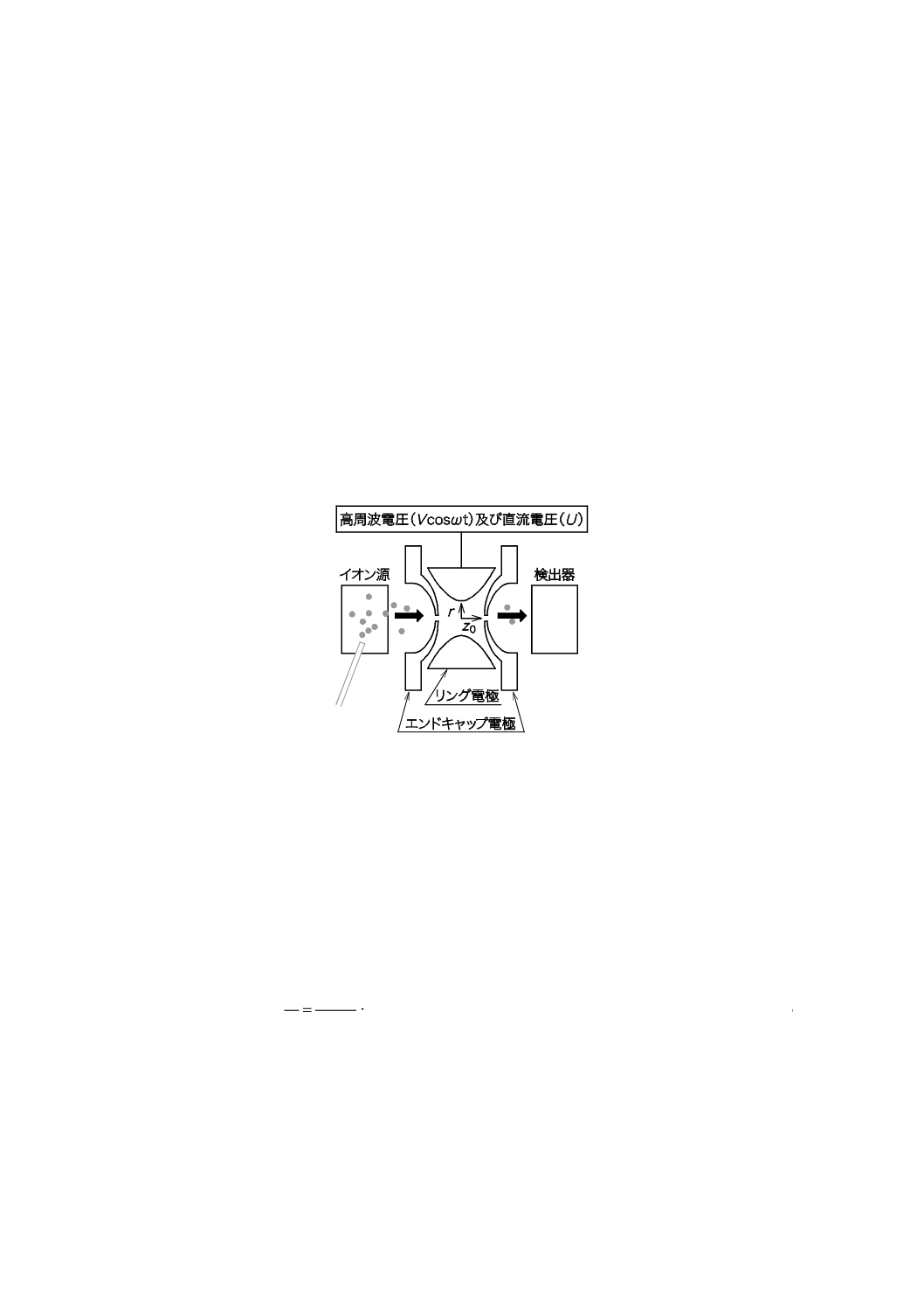
1) 四重極形 四重極形質量分析計の概略図の例を図5に示す。
10
K 0123:2018
図5−四重極形質量分析計の概略図(例)
金属製又は金属を蒸着したガラス製の4本の円柱若しくは双曲面をもつロッドをお互いに平行に
イオン源の後方に設置する。この四重極ロッドの模式図の例を図6に示す。対向するロッドには同
じ電圧[高周波電圧(Vcosωt)に直流電圧(U)を重ね合わせたもの]を,隣り合うロッドには正
負逆電位をかけ,高速で切り替える。イオン源で発生したイオンを中心軸に沿って入射させると,
イオンは,x軸又はy軸方向に振動しながらz軸方向に進む。このとき,ある特定のm/z範囲のイ
オンだけ振幅が大きくならずに通過することができ,検出器に到達する。
図6−四重極ロッドの模式図(例)
四重極ロッドを通過後,検出器に到達するイオンは,式(1)による。
ロッドまでの半径及び周波数を一定にすると,式(1)は式(2)となる。検出器に到達するイオンは,
ロッド電位に比例する。このロッド電位を走査することで,質量スペクトルが得られる。
2
2f
r
V
K
z
m
×
=
········································································· (1)
V
K
z
m
×′
=
··············································································· (2)
ここに,
m: イオンの質量(kg)
z: 電荷数
K: 定数
K': 定数
V: ロッド電圧(V)
r: ロッドまでの半径(m)
f: 周波数(Hz)
注記 附属書D参照。
11
K 0123:2018
2) イオントラップ形 イオントラップ形質量分析計は,イオン化部で生成されたイオンを,高周波を
加えた電場によってアナライザー内に閉じ込めた後,高周波電圧を連続的に変化させることによっ
てm/zの大きさの順にイオンを排出して質量分離する装置である。
イオントラップ形質量分析計の模式図の例を図7に示す。アナライザーは,双曲面をもったz軸
対称の3個の回転体の電極から構成される。左右2個の電極をエンドキャップ電極,中央の1個の
電極をリング電極と呼ぶ。
リング電極に1 MHz程度の周波数で,小さな振幅Vの高周波電圧を印加しておくことによって,
イオンの安定な振動の条件が作られ,ある値以上のm/zをもったイオンが振動空間の中心部に集ま
って閉じ込められる。次に,この高周波電圧の振幅を徐々に増大させると,閉じ込められたイオン
はm/zの小さい順にエンドキャップ電極にある孔を通過して排出され,検出器に到達して質量スペ
クトルが測定される。
イオントラップ形では,同一のアナライザー内で時間を分けて2回以上のMS/MSが可能なため
MSnとも表記される。
図7−イオントラップ形質量分析計の模式図(例)
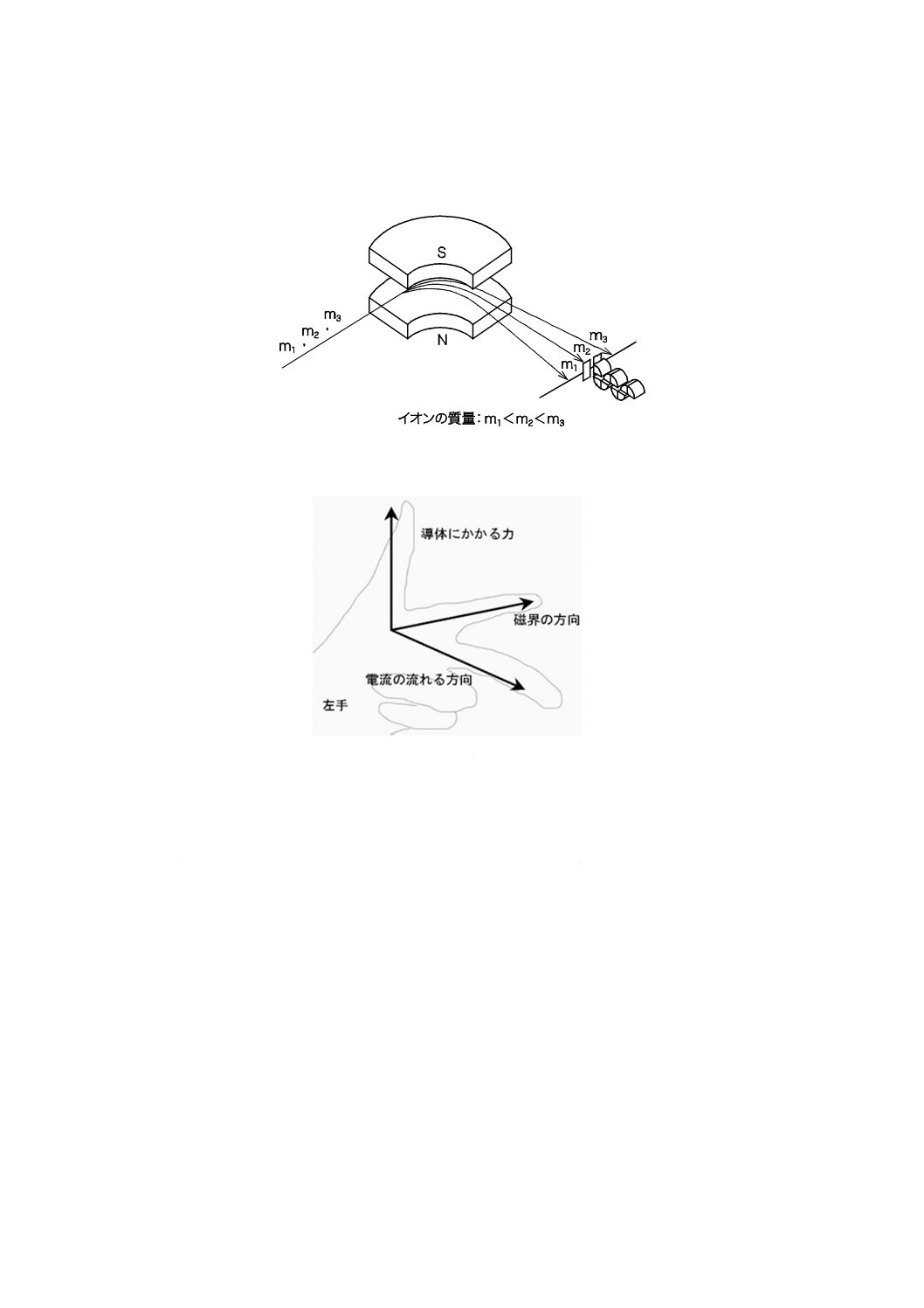
3) 磁場形 磁場形質量分析計は,通常,加速電圧一定で磁場強度(磁束密度)を変化させることによ
って広いm/z範囲にあるイオンを分離,検出する装置である。磁場セクターにおけるイオンの分離
のイメージを図8に示す。
一様の磁場の中を移動する電荷をもった粒子(イオン)は,図9に示すフレミングの左手の法則
に従い,イオンの進行方向と磁場の向きとの両方に対して垂直方向に力(ローレンツ力)がかかる
ため,その平面内で偏向される。したがって,磁場内の軌道半径がrに固定されている装置では,
式(3)を満足するm/zのイオンだけが通過することができる。
a
2
2
2V
B
er
z
m=
··············································································· (3)
ここに,
m: イオンの質量(kg)
Va: 加速電圧(V)
z: 電荷数
e: 電気素量(C)
r: 軌道半径(m)
B: 磁場強度(磁束密度)(T)
12
K 0123:2018
注記 磁場形質量分析計は,測定の目的に応じて,磁場強度一定で加速電圧(及び電場電圧)を
変化させて質量分離する方法を用いることがある。
図8−磁場セクターにおけるイオンの分離
図9−フレミングの左手の法則
ここで,一般に多くの磁場形質量分析計は,磁場セクターのほかに電場セクターをもつ二重収束
形となっている。磁場セクターと電場セクターとを組み合わせることによって,イオンの方向収束
とエネルギー収束との二つの収束作用をもつこととなり,高感度及び高質量分解能を実現している。
二重収束形質量分析計には,磁場セクターと電場セクターとの順序の違いによって,正配置形(電
場−磁場)と逆配置形(磁場−電場)とがある。その構成図の例を図10に示す。
13
K 0123:2018
図10−正配置形及び逆配置形の構成図(例)
可変スリットをもつ磁場形質量分析計では,スリット幅を調節することによって,分解能を調節
することが可能である。磁場形質量分析計は,SIMによる定量,質量スペクトルによる定性などの
日常的な質量分析のほか,高分解能SIM測定などが可能である。また,安定同位体比測定に特化し
た磁場形質量分析計もある。
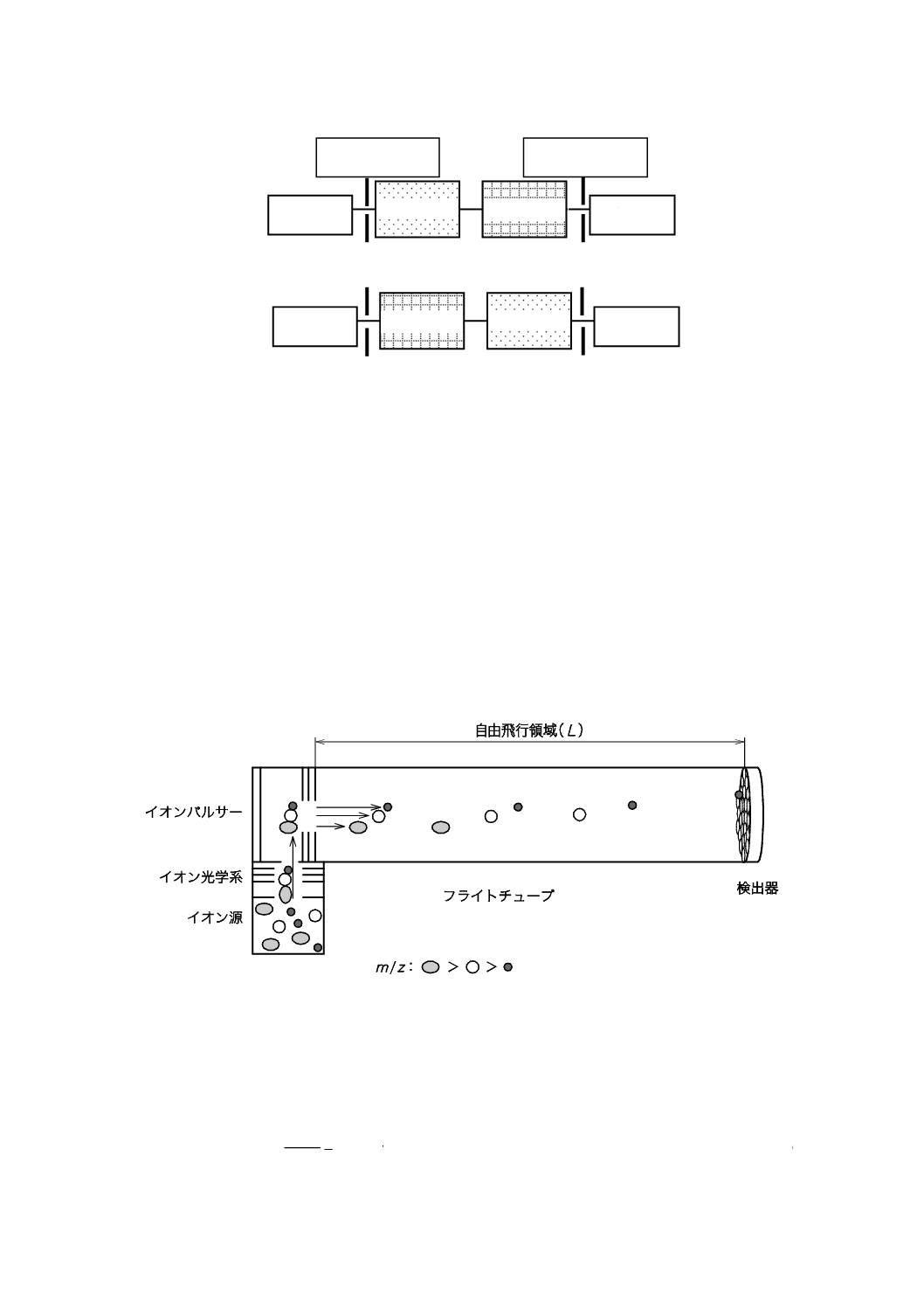
4) 飛行時間形 飛行時間形質量分析計(TOF-MS)は,イオン源で発生した分析種由来のイオンがパ
ルス電圧によって加速され,検出器まで到達する時間によって質量スペクトルを得る装置である。
イオン化室で生成する全てのイオンは,加速された後,検出器までフライトチューブ(無電界空
間)内をm/zに対応する速度で飛行し検出器に到達する。この飛行時間形質量分析計の模式図を図
11に示す。
図11−リニア形飛行時間形質量分析計の模式図(例)
このときの飛行速度,加速電圧,質量及び電荷数の間には,式(4)のような関係が成り立つ。した
がって,飛行速度は式(5)で,飛行時間は式(6)で求められる。式(6)を書き換えるとm/zを与える式(7)
となる。
V
e
z
v
m
=
2
2
··········································································· (4)
a) 正配置形
イオン源
電場セクター
磁場セクター
検出器
可変スリット
可変スリット
イオン源
検出器
b) 逆配置形
電場セクター
磁場セクター
14
K 0123:2018
m
V
e
z
v=
2
··········································································· (5)
V
e
z
m
L
v
L
t
×
=
=
2
·································································· (6)
2
2
2
L
eVt
z
m=
··············································································· (7)
ここに,
V: 加速電圧(V)
v: 飛行速度(m/s)
m: イオンの質量(kg)
z: 電荷数
e: 電気素量(C)
t: 飛行時間(s)
L: 飛行距離(m)
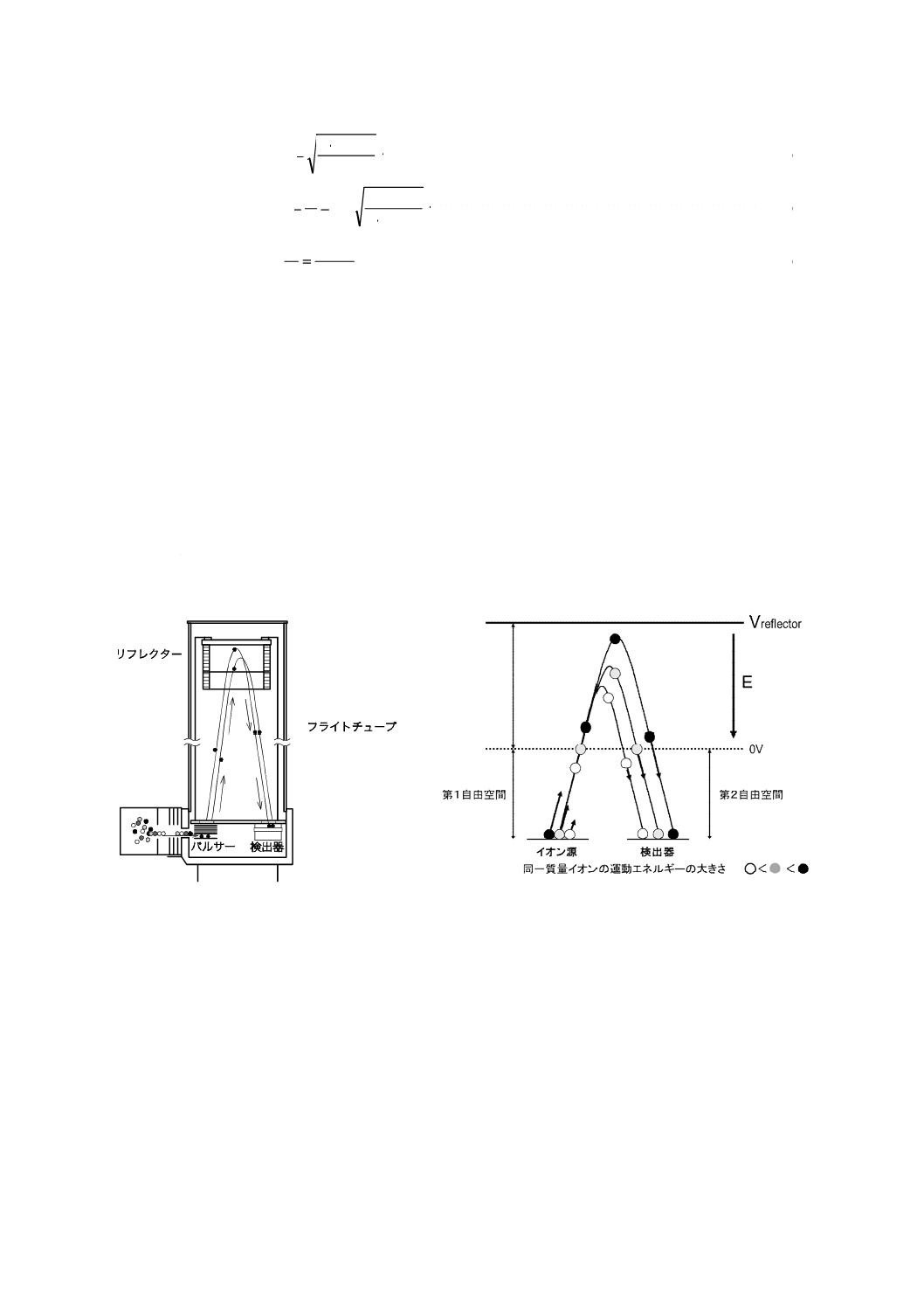
一般のGC-TOFMSは,イオンが直進し,検出器に到達するリニア形,リフレクターをもつリフレ
クトロン形及び同一軌道上を周回させる多重周回形がある。その模式図及び原理図の例を図12に示
す。リニア形は分解能が低いため,GC/MSとしての使用はほとんどない。リフレクトロン形では,
リフレクターによって同じ質量をもつが運動エネルギーが異なるイオンを同時に検出器に到達させ
るエネルギー収束作用をもつため分解能を向上できる。
図12−リフレクトロン形質量分析計の模式図(左)及び原理図(右)(例)
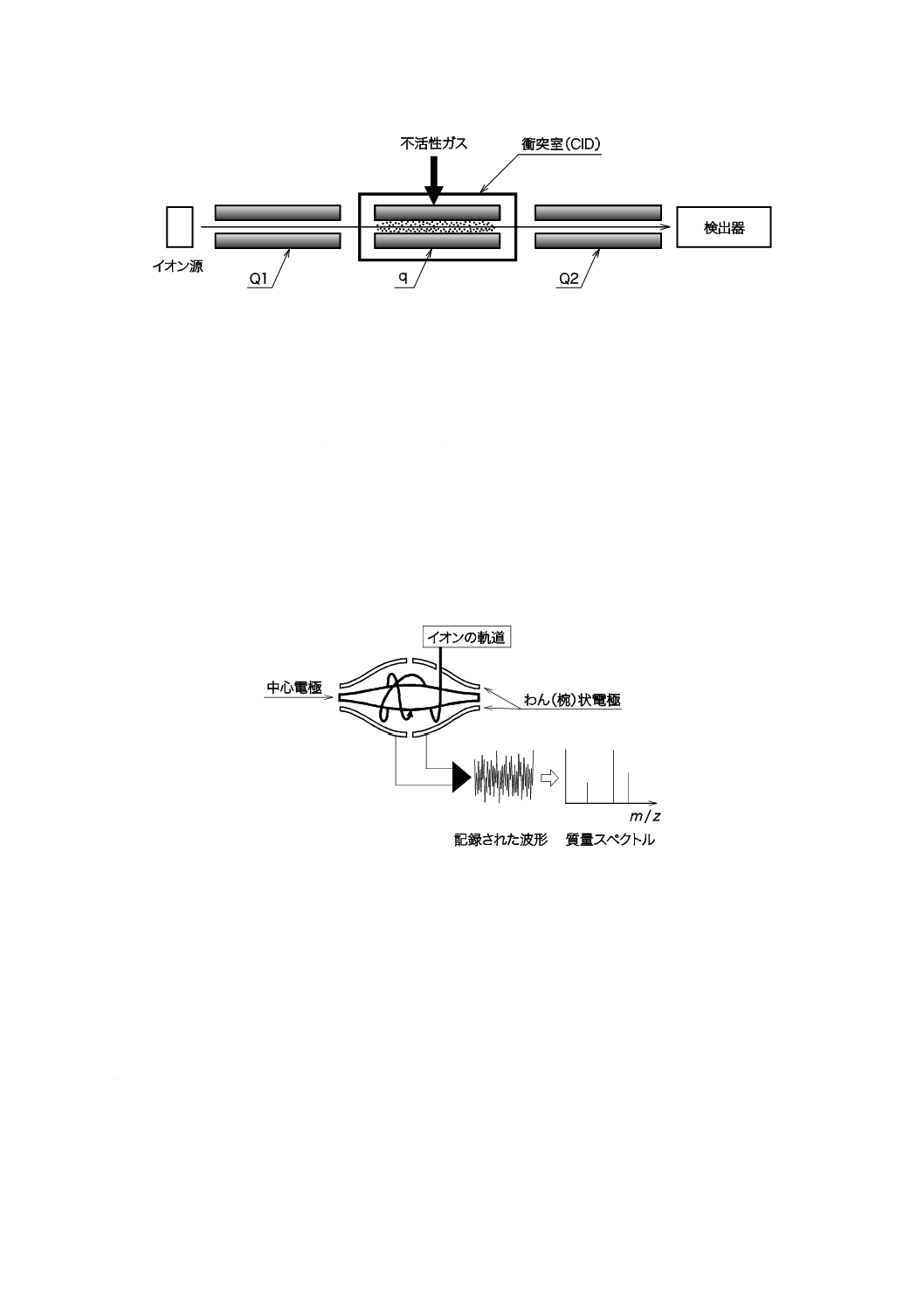
5) 三連四重極形 三連四重極(QqQ)形は空間的に四重極を直列又は曲線上に配した質量分析計で
MS/MSが可能である。三連四重極形の例を図13に示す。三連四重極では第1のアナライザー(Q1)
で特定のプリカーサーイオンを分離選択し,そのイオンを中央のqでアルゴンなどの不活性ガスと
衝突させてフラグメンテーション[衝突誘起解離(CID)]を起こさせる。CIDによって生成された
プロダクトイオンは第2のアナライザーで(Q2)で更に分離選択され検出器で検出される。Q1と
Q2の動作モードとしてはイオンの走査,特定のイオンの選択,全イオンの通過のいずれかが可能で,
その組合せによって幾つかの分析モードを実現できる。また,qでは,プリカーサーイオンの衝突
エネルギーを変えることでフラグメンテーションの程度を変えることもできる。
リフレクター
15
K 0123:2018
図13−三連四重極形質量分析計の模式図(例)
6) フーリエ変換形 フーリエ変換形には,イオンサイクロトロン共鳴(ICR)及びキングドントラッ
プがある。ICRは,一様の高磁場中で回転運動するイオンに高周波電場を印加すると,その周波数
に一致する回転周波数をもつ特定のm/zのイオンだけが共鳴的にエネルギーを吸収し,軌道半径を
増大させて検出電極に誘導電流を生じる現象を利用する。キングドントラップ形は,紡すい(錘)
形曲面をもつ中心電極とそれを覆うように配置したわん(椀)状電極から成り,中心電極とわん(椀)
状電極との間に高電圧が印加されている。この電極間の電場内に一定のエネルギーをもつイオンを
入射したとき,イオンが中心電極の周りを回転及び振動運動し,わん(椀)状電極に誘導電流を生
じる。これらの記録された波形をフーリエ変換し,質量スペクトルを得る。その模式図の例を図14
に示す。
図14−キングドントラップ形の模式図(例)
7) ハイブリッド形 ハイブリッド形質量分析計は,MS/MSを行うために異なるタイプのアナライザー
を結合した装置である。GC/MS/MSとして使用される装置には,四重極形及び飛行時間形若しくは
四重極形及びフーリエ変換形(FT)を組み合わせたQqTOF形,QqFT形などがある。
c) 検出部 検出部は,次の二次電子増倍管,マイクロチャンネルプレート及びファラデーカップに大別
する。また,イオンを検出器に直接入射せず,事前に電子に変換する変換ダイノードと組み合わせて,
検出部を構成することがある。
1) 二次電子増倍管 二次電子増倍管は,検出器に入射したイオン又は電子が,ガラス又はセラミック
製の曲がったパイプ(チャンネル形),若しくは連続して設置したダイノード(ディスクリート形)
への衝突を繰り返す間に電子数を増倍させ,応答量を増加させる。検出器入口にシンチレータを配
した光電子増倍管は,変換ダイノードによって発生した電子をシンチレータに照射し,発生した光
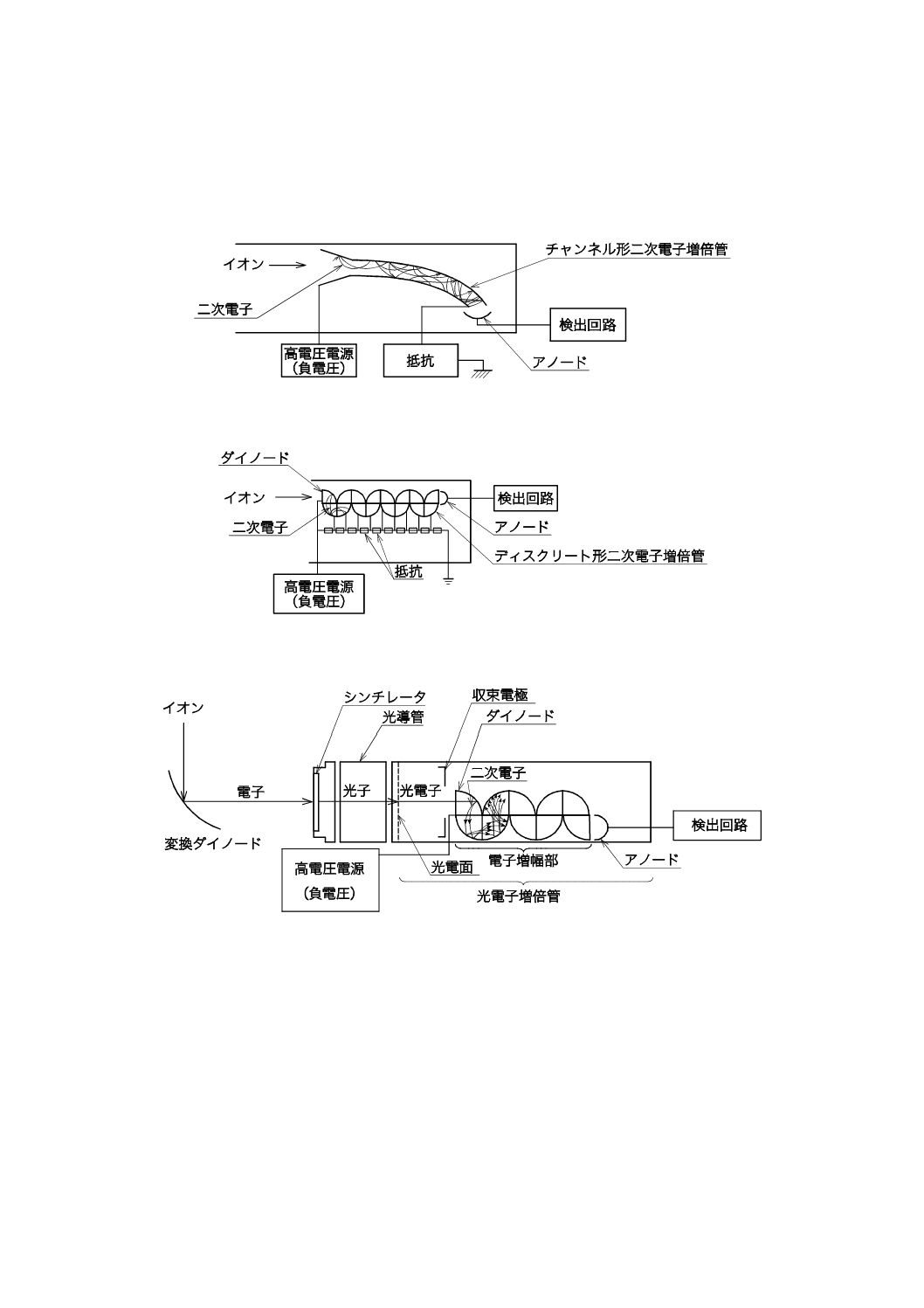
16
K 0123:2018
子を光電管に通した後,光電子に変換,その後はディスクリート形と同じく二次電子を増倍させる
ことができる。二次電子増倍管では,106倍〜108倍に電流値は増幅される(増幅率)。上記3例を図
15〜図17に示す。
図15−チャンネル形二次電子増倍管(例)
図16−ディスクリート形二次電子増倍管(例)
図17−光電子増倍管(例)
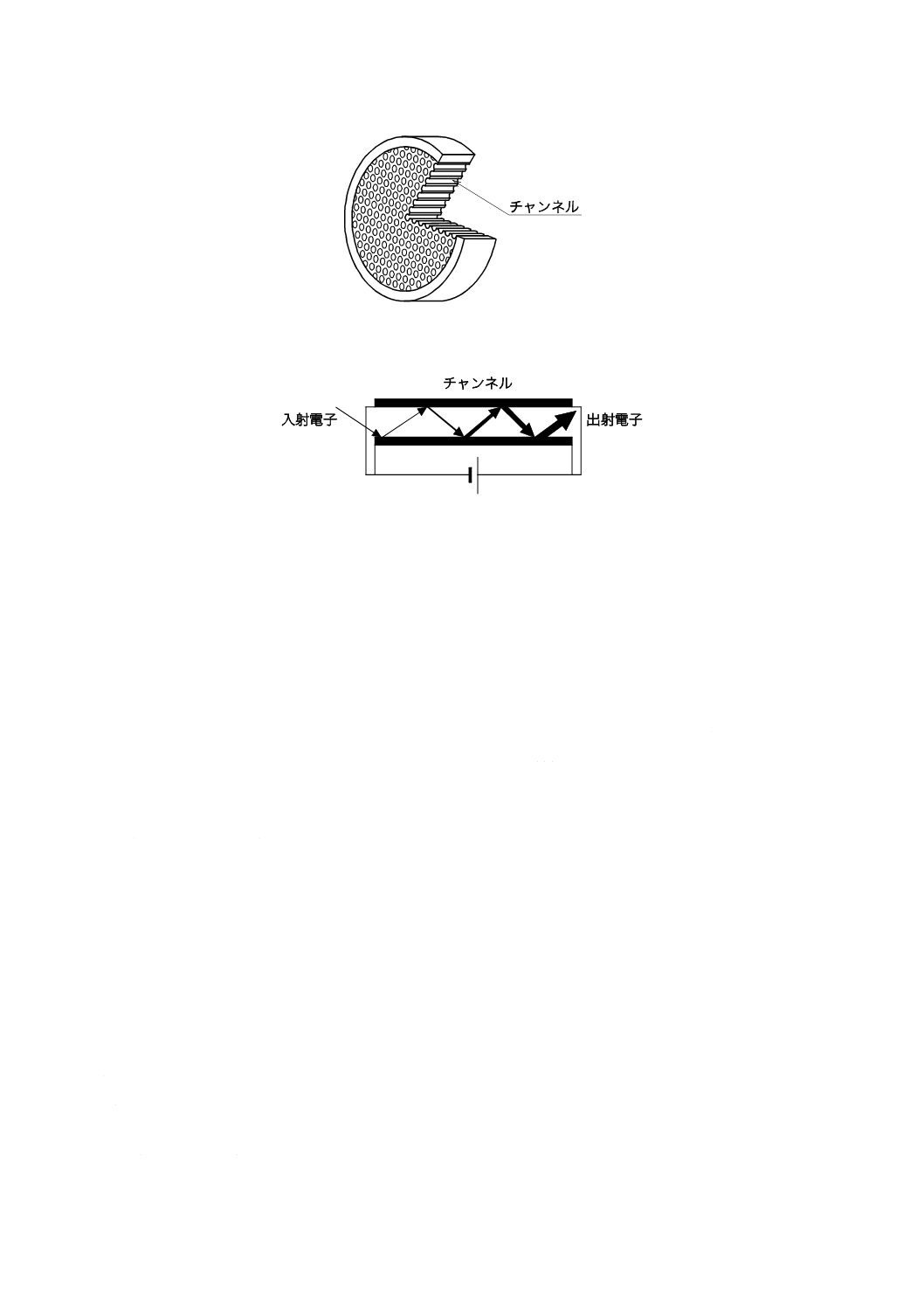
2) マイクロチャンネルプレート マイクロチャンネルプレート(microchannel plate:MCP)はチャン
ネルと呼ばれる細孔が蜂の巣状に配置されている金属被覆された薄いガラス板である。その例を図
18に示す。チャンネル入口付近にイオンが入射すると,チャンネル内壁で衝突を繰り返し,電子数
を増加させる。MCP1枚の増幅率は103程度であるため,複数枚を組み合わせる,又はMCPと二次
電子増倍管検出器とを組み合わせることで増幅率を向上させる。その概略図の例を図19に示す。主
に飛行時間形質量分析計の検出器として用いられている。
17
K 0123:2018
図18−マイクロチャンネルプレート(MCP)(例)
注記 矢印の太きさは,発生二次電子量を示す。
図19−チャンネル内での電子の増幅の概略図(例)
3) ファラデーカップ カップ状の導電体で捕捉したイオン(荷電粒子)によって発生した電子電流を
計測することによって,捕捉したイオンの数を決定する。電子増倍管のような間接的に感度を増幅
する機構はもたないが,捕捉した荷電粒子と計測する電流の値は直接的に比例するため,精度は高
いといわれている。同位体質量分析計に使用される場合が多い。
d) 真空排気部 真空排気部は,イオン化部及びアナライザー及び検出部を真空に保つためのもので,前
段に油回転ポンプ,後段にターボ分子ポンプ又は油拡散ポンプが用いられることが多い。イオン化部
は,通常,EI法の場合0.01 Pa以下,CI法の場合10 Pa〜100 Pa程度,アナライザーは1×10−5Pa〜
1×10−3Pa以下に保たれる。イオン化部とアナライザーの間に隔壁を設け,それぞれ異なる排気シス
テムをもつ装置がある。また,これらの排気システムは附属の校正用標準ガス導入部,CI用試薬ガス
導入部,直接試料導入部などの排気にも用いられる。
e) 校正用標準試料導入部 m/z,感度,分解能を調整するための標準試料をイオン化部に導入するための
装置で,圧力(流量)制御機構,開閉弁,排気機構などからなる。
5.5
システム制御・データ処理部
システム制御部は,装置各部の制御,質量スペクトル,クロマトグラムの採取,記録,処理,表示など
を主に行う部分で,中央演算ユニット(CPU),記録媒体などから構成され,オペレーティングシステム及
び各種ソフトウェアが組み込まれたコンピュータ本体,キーボード,ディスプレイなどからなる。通信機
能をもち,ローカルエリアネットワーク(LAN)などを通して装置の遠隔制御,測定データへの接続が可
能な装置もある。また,GC/MS装置本体に接続した各種前処理,試料導入装置を同時に制御できるものも
多い。
5.6
附属装置
附属装置は,次による。
a) ガスクロマトグラフへの試料導入附属装置 ガスクロマトグラフへの試料導入附属装置は,次によ
18
K 0123:2018
る。
1) 液体試料導入装置 液体試料を自動でガスクロマトグラフに導入するために使用する。試料を数
mL程度の試料容器に入れ,試料容器に応じた栓で封じる。試料容器及び栓は使用する装置の仕様
に適合する容量・寸法のものから,溶媒の種類・試料の吸着しやすさ・試料の分解しやすさなどを
考慮して目的に合うものを選択する。試料導入にはマイクロシリンジを用い,一定量の試料を試料
容器の栓を通して採取し,これをガスクロマトグラフの試料導入部へ導入する。マイクロシリンジ
は,試料採取の前後に洗浄溶媒で洗浄する。
なお,待機時間が長くなる場合には,ガスクロマトグラフからの熱による試料の気化・変性など
に注意する。
注記 試料を冷却できる装置もある。
2) 気体試料導入装置 流路切替弁に附属した0.1 mL〜数mL程度の計量管に気体試料を採取し,流路
を切り替えることでガスクロマトグラフに導入する。分析種が凝縮するおそれがある場合,必要に
応じて温度調節のできる流路切替弁を用いる。
3) ヘッドスペースサンプリング装置 液体又は固体中の揮発性成分の分析に用いられる。液体又は固
体を一定量密栓できるバイアルに入れ,一定温度,一定時間加温・保温し,気相部分(以下,ヘッ
ドスペースという。)の一定量を採取し,カラムに導入する装置。液体試料では,主成分の溶媒の沸
点より20 ℃以下の一定温度が望ましい。
注記1 繰返し精度を上げるには,試料容器を加温する温度の正確さ,相比の平衡・均一性,試
料容器に使用する栓の試料接触面の成分吸着特性の影響などを考慮するとよい。
注記2 ヘッドスペースの一部を濃縮管へ移動させた後,ガスクロマトグラフに導入する方式も
ある。
4) 熱脱着装置 捕集管に充塡した吸着剤に捕集した気体試料中の低濃度揮発性成分の分析などに利用
するもので,ヘリウム,窒素などの不活性ガスを通じながら,分析種を捕集した捕集管を加熱して
熱脱着させ,カラムに導入する。固体,液体試料などを直接加熱し,試料中の分析種を熱抽出する
分析にも用いられる。
注記 捕集した分析種をカラムに導入し,捕集管から脱離した分析種を低温に冷やされた固体吸
着剤に再捕集し,この固体吸着剤を不活性ガス流の下で急速に加熱して脱着してカラムに
導入する方式もある。この再捕集操作では,分析種の破過を常に考慮するとともに,適切
な固体吸着剤を選択することが重要である。
5) パージ・トラップ装置 液体試料中の低濃度揮発性成分の分析に用いられる。数mL〜数10 mL程
度の液体試料を適切な容量の容器に入れ,ヘリウム,窒素などの不活性ガスを試料に導き,大部分
の揮発性成分を気相に抽出し(パージという。),その揮発性成分を冷却又は吸着捕集又は両方を用
いて捕集する(トラップという。)。捕集した揮発性成分を,不活性ガス流の下で加熱によって再び
気化させ,ガスクロマトグラフに導入する。倍率の高い濃縮を行うために,装置又は気相抽出に用
いる不活性ガス中の不純物は,できる限り除去しておく。
6) ガス自動濃縮導入装置 気体試料中の低濃度揮発性成分の分析に用いられる。一般に内面を不活性
化処理したステンレス製容器(キャニスターと呼ばれる。容量は0.2 L〜10 L程度)又はガラス製の
真空瓶をあらかじめ減圧又は真空にし,試料を吸引採取する。採取した気体試料は,濃縮部に一定
量捕集・濃縮し1),必要に応じて水分及び二酸化炭素を除去した後,加熱して捕集・濃縮した成分
をカラムに導入する。揮発性の高い成分のピーク幅を狭めるためには,カラム前段部に移送し,冷
19
K 0123:2018
却・再濃縮させる。その後,急速加熱を行い濃縮成分をGCカラムに送り込み測定を行う。
注1) 捕集量は通常,0.1 L〜1.0 L程度。そのため,同じ試料から複数回の測定が可能。
7) 熱分解装置 試料を高温度まで急速加熱し,熱分解によって生成する揮発性成分をガスクロマトグ
ラフに導入する。
なお,試料の熱分解温度以下に温度設定することによって,固体及び液体試料中の分析種を熱抽
出する分析にも用いられる。
8) 多機能サンプラー 一つのサンプラーで複数の試料導入方法等を兼用する場合に利用される。液体
試料注入装置(オートインジェクター),ヘッドスペースサンプリング(シリンジ形)装置,固相マ
イクロ抽出による注入装置,熱脱着装置等が組み合わさっている。また,試料の希釈,誘導体化,
内標準物質の添加等の機能が組み込まれた装置もある。
b) その他のガスクロマトグラフの附属装置 その他のガスクロマトグラフの附属装置は,次による。
1) カラム槽冷却装置 低沸点成分の分析などにおいて,分析種のカラムへの保持が十分でない場合に
用いられる。カラム槽を室温付近又はそれ以下に調節する装置で,冷媒として液体二酸化炭素又は
液体窒素を電磁弁操作でカラム槽内に吹き込むことで冷却する。
冷却を効率よく行うために,冷媒容器とガスクロマトグラフとの距離を最短にする。
2) 流路切替装置 沸点,極性などの異なる多成分からなる試料に対して,複数の分離カラムを組み合
わせ,試料中の一部の成分だけを分析したり,分析時間を短縮したり,不適切な成分のカラム流入
を防いだり,バックフラッシュする目的に使用される。流路を切り替える手段として,切替弁を利
用する方法,圧力差を利用する方法などがある。
c) 質量分析計の附属装置 質量分析計の附属装置は,次による。
1) 試薬ガス導入部 化学イオン化に用いる試薬ガスを供給する部分で,圧力(流量)制御機構,開閉
弁及び排気機構からなる。
2) 直接試料導入部 ガスクロマトグラフに通さずに試料をイオン源に直接導入するための部分であ
る。プローブ,加熱機構,開閉弁及び排気機構からなり,試料をプローブ先端に固定し,開閉弁及
び排気機構を操作してプローブを質量分析計のイオン化部まで挿入し,試料を加熱しイオン源に導
入する。高沸点化合物及び熱に不安定な化合物を分析するのに適している。特に,単一成分からな
る試料の質量スペクトルを容易に得ることができる。
6
安全
安全のために,十分な配慮が必要で,次のことを考慮するとよい。
a) 試料及び分析に用いる薬品の取扱いは,爆発性,引火性,毒性又は有害性に十分注意して行う。それ
らの廃棄については,安全化及び無害化に配慮し,環境汚染防止の諸規定に従う。作業及び作業環境
全般に関しては“労働安全衛生法”,危険物の取扱いについては“消防法”・“危険物の規制に関する政
令”,毒物及び劇物の取扱いについては“毒物及び劇物取締法”などの諸規定に従う。
b) 装置及び周辺機器は,接地点に接地する。
c) キャピラリーカラム接続時に,カラム先端が目に入らないように,防護めがねなどを着用することが
望ましい。
d) マイクロシリンジを測定者が洗浄又は試料溶液採取などで直接取り扱うときは,指などへの針刺し又
は洗浄有機溶媒の吸込みなどに注意する。
e) 高圧容器詰めのガスを使用するときは,“高圧ガス保安法”の諸規定に従う。装置の運転に先立ち,ガ
20
K 0123:2018
ス配管の接続部その他流路系からのガス漏れがないかどうかを十分に検査する。特に,可燃性ガス(水
素など)を使用する場合は,厳重に検査する。運転中にガス漏れが生じた場合は,直ちに運転を停止
し,漏えい箇所を修復する。また,ガスの入った高圧容器は,火気のない製造業者が指定する温度の
場所に転倒しないように固定して設置する。
なお,高圧ガス保安法に規定する圧力以下の容器を装置の近くで用いる場合にも,容器が転倒しな
いように,架台,壁,実験台などに固定しなければならない。
f)
油回転ポンプ,GCのベントなどから排出される有害成分は,吸着剤などでトラップすることが望ま
しい。また,油回転ポンプのオイルは,オイル中に含まれる排出成分を考慮し,適切に処理する。
g) 電源が入っている状態で,装置又は周辺機器の内部に直接触れると感電するおそれがあるので,装置
の点検及び修理は,通常電源を切って取扱説明書に従って行う。また,電源が切れていても高温にな
っている部位があるので十分に注意し,冷却後に作業を行う。
警告1 装置にリチウム電池が内蔵されている場合は,取扱いを誤ると破裂する危険があるため注
意する。
h) 装置が他の機器,ペースメーカーなどに電磁波障害を与えたり,ほかの機器から電磁波妨害を受けた
りする場合があるので注意する。
i)
生体試料(血清,血しょう,組織,尿など)は,ウイルス,細菌などの感染のおそれがあるため,素
手で取り扱ってはならない。
なお,生体試料は,必要に応じて所定の場所で取り扱う。また,使用したシリンジなどは適切な方
法で廃棄する。
警告2 生体試料の取扱いについては,皮膚への直接接触,ピペット操作による誤飲,針刺しなど
に注意し,保護具(保護めがね,ゴム手袋,マスクなど)を着用しなければならない。
7
装置の設置
装置は,次のa)〜j) の条件を備えた部屋に設置することが望ましい。また,必要な場合はk) の確認を
行う。
a) 装置に定められた仕様範囲の温度及び湿度内であり,急激な変化を生じない。
b) 実験台及び床は水平であり,装置の総質量に十分耐えられる。
c) 振動が少なく,直射日光及び風が直接当たらない。
d) 使用する有機溶媒及び有害物質を安全に排出処理でき,換気がよく,また,ほこりが少ない。油回転
ポンプ及びスプリットベントからの排気ガス並びにガスクロマトグラフから発生する熱を排出するた
めの室内換気設備又は強制排気設備を必要に応じて設置する。
e) 装置の放熱及び保守を考慮した設置スペースを確保する。
f)
供給電源は,装置の仕様に指定された電圧,電気容量及び周波数で使用する。
g) 大形変圧器,高周波加熱炉,発信機などから電磁誘導を受けない。
h) 指定された接地抵抗を満足する接地端子をもつ。
i)
油拡散ポンプなどに冷却水を使用する場合は,指定された水圧,水量及び水温で,ごみ及び異物を含
まず,スケールが生成しない水を供給する。
j)
高圧ガスの配管が可能で,その接続部などに漏れがない。
k) 可燃性ガス(水素など)又は腐食性ガスを利用する場合,装置がその使用を許容していることを確認
する。
21
K 0123:2018
8
装置の運転
8.1
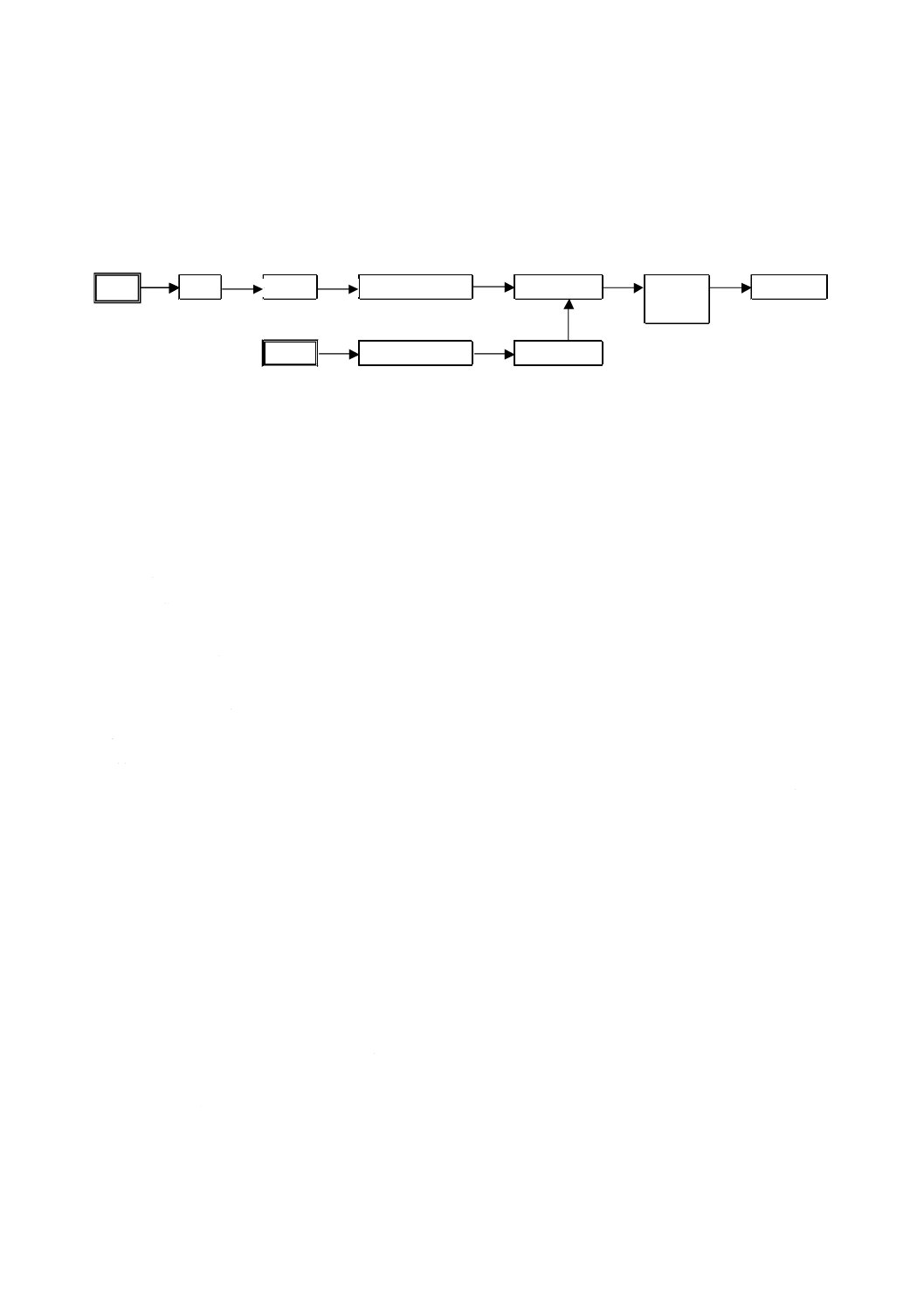
運転の手順
装置の運転に当たっては,取扱説明書に従って始動した後,調整を行う。装置の始動から測定,データ
管理までの手順の例を図20に示す。
図20−装置の始動から測定,データ管理までの手順(例)
8.2
ガスクロマトグラフの準備
ガスクロマトグラフの準備は,次による。
a) カラム 分析目的に適したカラムを用いる。あらかじめカラムの性能を確認し,必要に応じてカラム
交換,コンディショニング2) などを行う。注入口側及び質量分析計側にカラムを取り付ける直前に,
カラム両端部を必要に応じて切り取る。カラムナットから出すカラムの長さは,取扱説明書に従って
調節し取り付ける。その場合,注入口に分析目的に適したライナー及びセプタムを取り付ける。
注2) カラムをコンディショニングする場合には,カラム槽はカラムの最高使用温度より10 ℃〜
20 ℃程度低い温度にするとよい。
b) キャリヤーガス 一般に,キャリヤーガスとして高純度ヘリウムを用いるが,高純度水素,高純度窒
素を使うこともできる。分析に用いるカラム内径,長さなどに応じて,十分な感度が得られるように
最適な流量,線速度又は圧力を設定する。また,ガス漏れのないことを確認する。
8.3
質量分析計の準備
質量分析計の準備は,次による。
a) 校正用標準試料 分析目的に適した校正用標準試料を用いる。一例として,ペルフルオロトリブチル
アミン(PFTBA),ペルフルオロケロセン(PFK)などを用いる。操作は,取扱説明書に従う。
b) 試薬ガス 化学イオン化に用いる試薬ガスには,一般的に純度の高いメタン,2-メチルプロパン(イ
ソブタン),アンモニアなどを用いる。分析対象成分に応じて,十分な感度が得られるように最適な試
薬ガスを選択し流量を設定する。
8.4
始動
装置の運転に当たっては,取扱説明書に従って始動する。まず,ガスクロマトグラフを始動させる。分
析試料の性質,注入方法などに応じて,分析目的に適した注入口温度,カラム槽温度,昇温条件などを設
定する。質量分析計は前段の粗引き真空ポンプを稼動させ,ある程度の真空度になってから,後段のター
ボ分子ポンプ又は油拡散ポンプを稼動させる。イオン化部,アナライザー,インターフェースの温度など
を設定する。イオン化部及びアナライザーが所定の真空度に達するまで待機する。イオン化部の変更若し
くは取付け,カラムの交換などを行った後に真空系を立ち上げたときは,装置の取扱説明書に従って漏れ
がないことを確認する。
8.5
調整
装置の据付け時,一時停止後の始動時のほか,特に必要な場合に調整を行う3)。装置が作動している状
装置
始動
調整
測定条件の設定
測定
定性
データ管理
定量
試料
前処理
試料導入
装置
22
K 0123:2018
態で必要な項目の条件設定を行った後,主として次の項目について実施する。この操作では,装置の取扱
説明書に従う。
注3) 最近の装置では,システム制御部のオートチューニングによって,m/z校正,強度比,分解能,
感度などを自動的に調整することができる。
a) m/z m/z目盛校正用標準試料を用いて校正する。校正目的に適した標準試料を選択する。校正方法は,
取扱説明書に従う。
b) 強度比 目的に適した標準試料を用いてスペクトル強度比(パターン)を校正する。校正方法は,取
扱説明書に従う。
c) 分解能 分析目的に応じて必要な分解能が得られるように,取扱説明書に従って調節する。
d) 感度 必要に応じて各装置で指定された標準試料などを用いて所定の感度が得られるようにレンズ,
検出器などを調節する。
8.6
測定条件の設定
測定条件の設定は,分析目的・内容によって異なる。次に示す項目などがあり,必要に応じて手動又は
システム制御部によって設定する。装置によっては設定項目が異なるので,各装置の取扱説明書を参照す
る。長時間使用しなかった装置は,必要に応じた調整を行い,性能を確認する。
a) ガスクロマトグラフ ガスクロマトグラフ部の主な設定条件は,次による。
1) 注入口の種類及びライナーの種類,温度条件
2) キャリヤーガスの種類及び流量,線速度又は圧力
3) 注入方法(スプリット注入法,スプリットレス注入法,直接注入法,温度プログラミング気化注入
法など)
4) カラムの種類(カラムの長さ,内径,膜厚,固定相の種類),カラムの昇温条件(初期温度から昇温
速度,最終温度,コンディショニングの温度など)
b) インターフェース インターフェース部では主にインターフェース温度を設定する。
c) 質量分析計 質量分析計部の主な設定条件は,次による。
1) イオン化方法の選択[電子イオン化(EI)法,正イオン化学イオン化(PICI)法,負イオン化学イ
オン化(NICI)法など]
2) イオン化電流,電子加速電圧,イオン源温度
3) 分解能
4) サンプリング時間,走査速度,測定質量範囲,選択イオンモニタリング(SIM:selected ion monitoring)
のm/z,ドゥエルタイム
5) 検出器の印加電圧,検出イオンの種別(正イオン又は負イオン)
6) タンデム質量分析(MS/MS)におけるプリカーサーイオン,プロダクトイオン,衝突誘起解離電圧,
ドゥエルタイム,衝突ガスの種類,圧力など
8.7
試料の導入
試料の導入量に応じて適切な容量の計量器(マイクロシリンジ又はガスタイトシリンジ)を用いて注入
口から試料を導入する。又は5.6 a) に規定するガスクロマトグラフへの試料導入附属装置を,適切な条件
に設定して試料を導入する。
8.8
測定(質量スペクトルの採取)
ガスクロマトグラフで分離された分析種は,イオン化部でイオン化されアナライザーで分離された後,
検出されて質量スペクトルが取得される。データの採取・記録方法には,次に示す各種の手法がある。
23
K 0123:2018
なお,測定終了後は,設定時のガス流量及び温度のままの状態で装置を待機させ,測定を長時間行わな
い場合,質量分析計及びガスクロマトグラフの温度を常温にしてもよい。また,装置を停止させる場合は,
各装置の取扱説明書に従う。そのとき,キャリヤーガスは必要最小限の流量で流し,冷却水循環装置など
は停止してもよい。
a) 全イオンモニタリング(TIM:total ion monitoring) あらかじめ設定したm/z範囲と時間間隔に基づい
て,そのm/z範囲に検出されるイオンのm/z及び強度をその時間間隔ごとに質量スペクトルとして記
録して保存する。全イオン電流クロマトグラム(TICC)は,各走査で記録されたイオン強度(電流値)
を積算したガスクロマトグラフィーの保持時間に対してプロットしたクロマトグラムとして得られる。
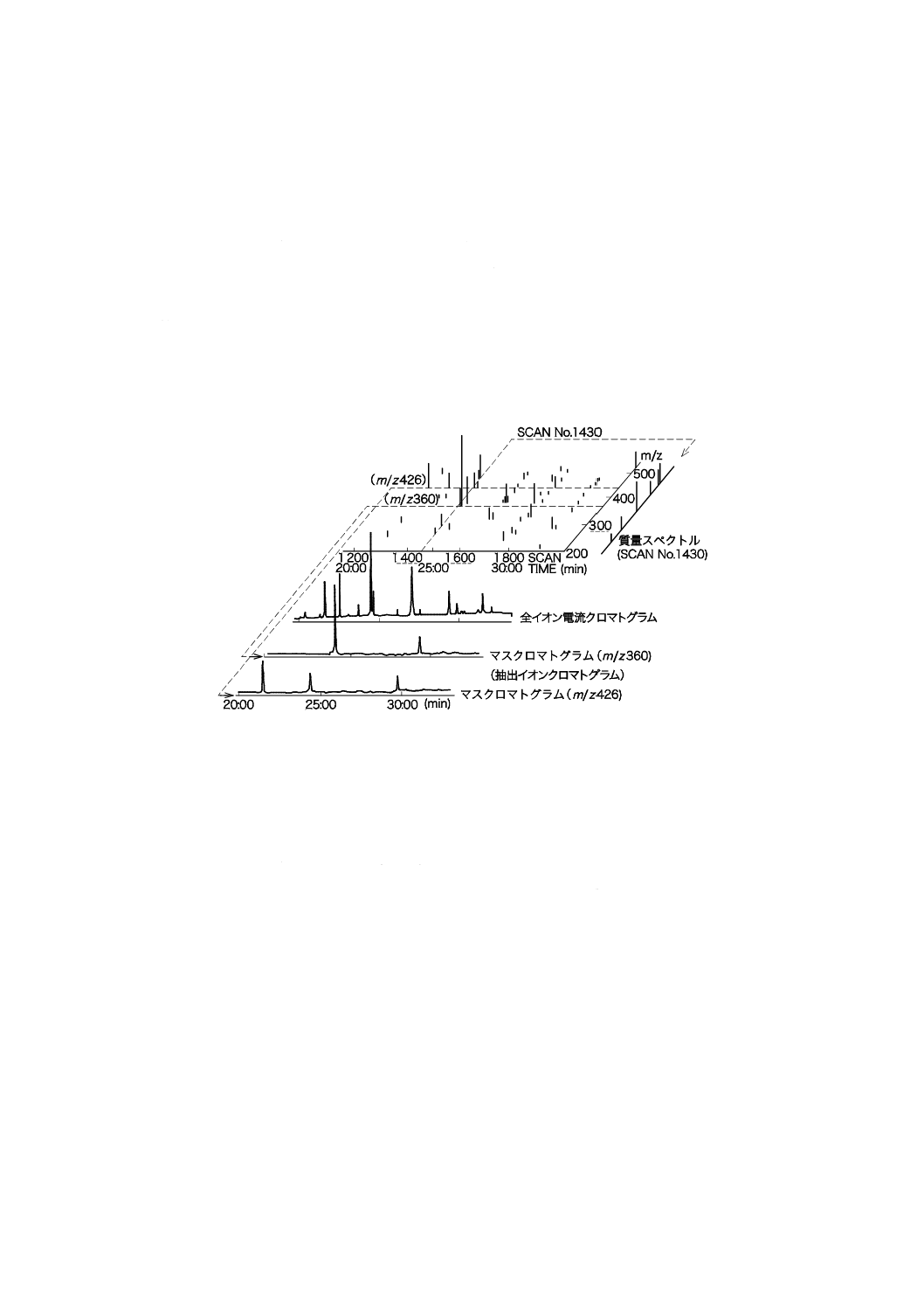
走査ごとにそれぞれ特定のm/zを抽出し,そのイオン強度を保持時間に対してプロットしたものがマ
スクロマトグラムである。質量スペクトルとマスクロマトグラムとの関係の例を,図21に示す。
図21−質量スペクトルとマスクロマトグラムとの関係(例)
b) 選択イオンモニタリング(SIM:selected ion monitoring) 分析種に応じて,あらかじめ決めた特定の
m/zのイオンを検出する手法。選択イオンモニタリングは特定分析種の高感度定量分析に利用される。
選択したイオンだけを測定するのでSN比が向上する。
c) 選択反応モニタリング(SRM:selected reaction monitoring)又は多重反応モニタリング法(MRM:multiple
reaction monitoring) 特定のプリカーサーイオンを第一のアナライザー(MS1)で選択し,そのイオン
を衝突誘起解離(CID)させて生じたプロダクトイオンの幾つかを選んで第二のアナライザー(MS2)
で分離及び検出する手法。その模式図の例を図22に示す。一般に,選択イオンモニタリング(SIM)
と比べて選択性が向上する。
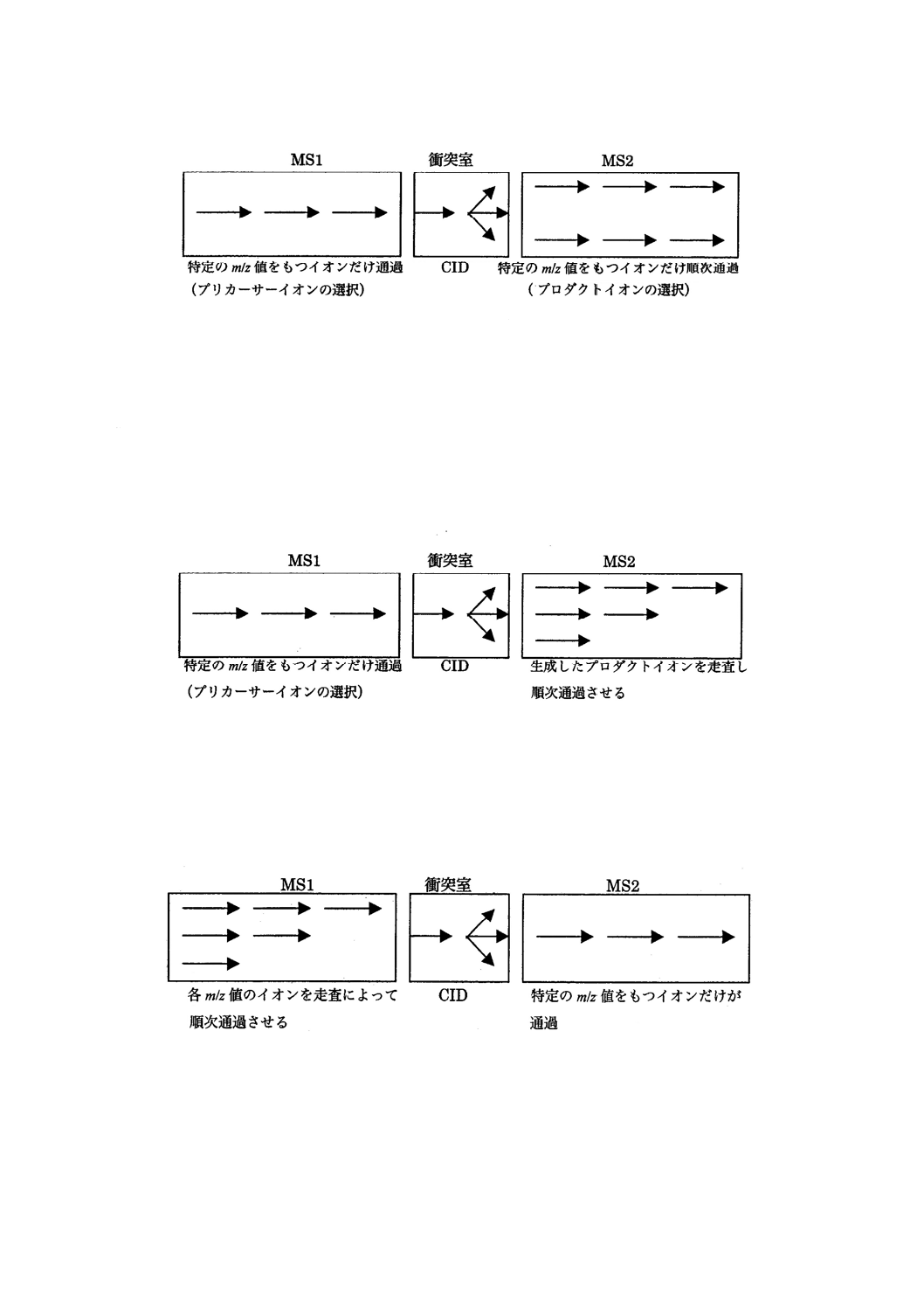
24
K 0123:2018
図22−選択反応モニタリングの模式図(例)
d) その他のMS/MSの測定モード 三連四重極形(QqQ)に代表されるMS/MS装置の操作方法は3種あ
り,その模式図は,次のとおりである。
1) プロダクトイオン走査 分析種から生じた特定のプリカーサーイオンのCIDから生じるプロダクト
イオンを検出する走査法。IT形の場合は,プロダクトイオンの走査だけとなる。その模式図の例を
図23に示す。
図23−プロダクトイオン走査の模式図(例)
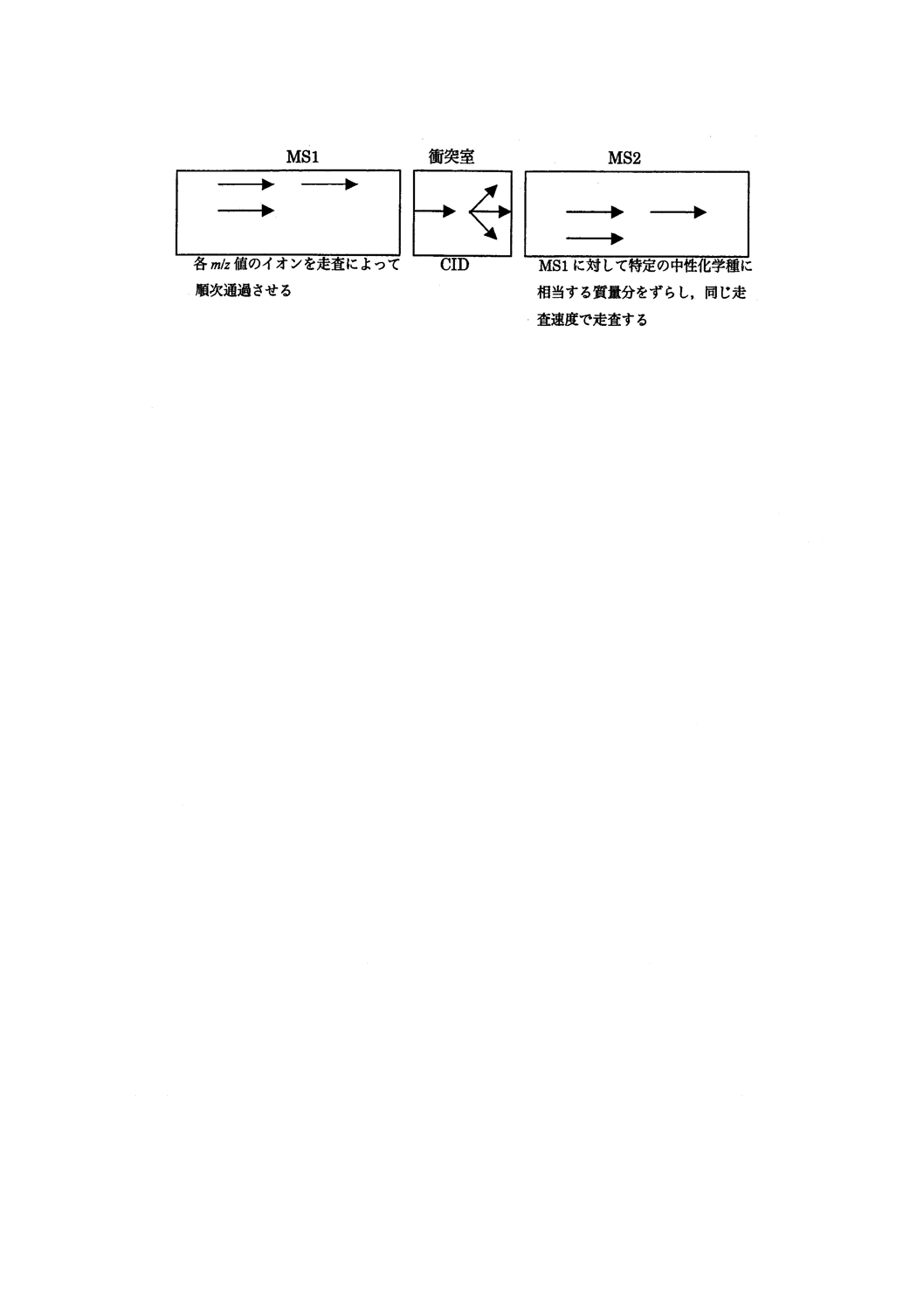
2) プリカーサーイオン走査 CIDによって生じる特定のフラグメントイオンを生じる全てのプリカー
サーイオンを検出する走査方法。その模式図の例を図24に示す。
図24−プリカーサーイオン走査の模式図(例)
3) コンスタントニュートラルロス走査 CIDにより,特定m/zの減少が起こる全てのプリカーサーイ
オンを検出する走査方法である。特定の部分構造をもつ分析種を特異的に検出するために用いられ
る。その模式図の例を図25に示す。
25
K 0123:2018
図25−コンスタントニュートラルロス走査の模式図
8.9
データ処理
取得されたデータは,試料ごとのクロマトグラム及びスペクトルとしてシステム制御・データ処理部の
データ処理システムに保存される。全イオンモニタリングによって測定されたイオンは,データ処理の方
法によって質量スペクトルとして表したり,マスクロマトグラムとして表したりすることができる。質量
スペクトルは,必要に応じて,バックグランドの差し引きを行い,スペクトルとして表示する。デコンボ
リューション機能をもつシステムでは,成分ごとの質量スペクトルの抽出を行うことによって[箇条10 e)
参照],ターゲット成分の同定,未知成分の探索が可能で,試料に含まれる成分の判断が容易になる。全イ
オン電流クロマトグラム及びマスクロマトグラムを利用して定量を行う場合,一般的には標準物質を用い
て得られるクロマトグラム上のピーク面積を測定し,検量線を作成する。また,高分解能条件下で求めら
れた精密質量から,未知化合物の組成式を推定することができる。
8.10
誘導体化
8.10.1
一般
試料をガスクロマトグラフで分離・分析しやすい形態又は検出しやすい形態への変換のための前処理法
として誘導体化がある。
8.10.2
効果
効果は,次による。
a) 分離の改善 誘導体化することで,揮発性又は安定性を増すことによって分離が容易になり,使用可
能なカラムの選択肢が増える。また,光学異性体をジアステレオマーに導くことで,光学活性カラム
を使わずに分離可能にできるなどの効果がある。
b) 定量性の向上 極性の低下などによってカラムなどへの不可逆的な吸着を軽減できるため,定量性が
向上する。また,より微量な成分の検出を可能にする。
c) 検出感度の向上 検出しやすい化学形にすることで,選択性を向上し高感度検出を可能にする。NICI
において,ペンタフルオロベンジルブロミド,ペンタフルオロベンゾイルクロリド,ペンタフルオロ
ベンジルヒドロキシルアミンなどを用いると,電子親和性の向上によってEIと比較して数十倍以上の
高感度化が可能になる場合がある。
d) 化学構造に関する知見 保持時間変化から官能基の数などを推定でき,また,分離挙動を比較し,特
定の成分を見分けることができる。重水素標識化したトリメチルシリル化剤を用いた誘導体化によっ
て,生成イオンのm/zをシフトさせ構造解析を容易にする利点がある。
8.10.3
誘導体化の種類
26
K 0123:2018
誘導体化の種類は,次による。
a) 誘導体化後にガスクロマトグラフに中に注入する方法
1) エステル化 カルボキシル基をもつ化合物である脂肪酸及び有機酸の誘導体化に用いられる。酸触
媒とメタノールの混合溶液,ジアゾメタンなどのメチル化剤が広く用いられる。
2) シリル化 水酸基,カルボキシル基,アミノ基,メルカプト基などをもつ化合物の誘導体化に広く
用いられるが,最も多いのはトリメチルシリル(TMS)化である。
3) アシル化 水酸基をもつアルコール,アミノ基をもつアミンなどの化合物の誘導体化に主に用いら
れる。アセチル化,トリフルオロアセチル(TFA)化,ペンタフルオロプロピオニル化などがよく
行われるが,TFA化が多用される。
4) その他 シッフ塩基生成,環状誘導体化,光学異性体のジアステレオマー化,アルデヒドの分析に
“2,4-ジニトロフェニルヒドラジン”を用いた“2,4-ジニトロフェニルヒドラゾン”(2,4-DNPH)化,
ペンタフルオロベンジルブロミドによる誘導体化などがある。
b) オンカラム誘導体化 誘導体化試薬と混合した溶液を加熱した注入口に注入して反応させ,そのまま
測定する。代表的な誘導体化試薬としては,アルキルアンモニウムヒドロキシドなどがあり,エステ
ル化剤の一種である。これらは,トリグリセリドとエステル交換反応によって構成脂肪酸のメチルエ
ステルを生成する。
9
試料の調製及び測定
9.1
試料の調製
試料導入法に合わせた試料調製を行う。必要に応じて,試料の溶解,精製,濃縮,希釈,誘導体化など
を行う。定量分析の場合には,検量線作成のための標準試料の調製も行う。
9.2
測定
測定は,次による。
a) 8.1〜8.5によって,目的に適したイオン化のモード,カラムなどを選択して測定準備を行った後,8.6
及び8.7によって測定条件を設定する。
注記 カラムの種類,長さ,温度条件の選定,走査速度などの設定は,カラムの固定相液体から生
じるバックグラウンドが試料の質量スペクトルに影響を与えないようにする。全イオンモニ
タリング(TIM)では,分析種1ピーク当たり,少なくとも10回程度の質量スペクトルの採
取を行うことが望ましい。また,選択イオンモニタリング(SIM)若しくは選択反応モニタ
リング(SRM)による定量分析又はデコンボリューション機能を用いる場合は,分析種1ピ
ーク当たり12回以上のデータ採取を行うことが望ましい。
b) 8.8及び8.9によって,必要な測定及びデータ処理を行い,次の測定データのうち,必要なものを整え
る。
1) 質量スペクトル バックグラウンドを差し引き,GCピークの頂点付近における数回の走査又はス
ペクトル採取時の平均スペクトルを求めておく。
なお,質量スペクトルに対応した表(質量スペクトル上の各m/zとそのイオン強度をまとめた表)
をもって代用してもよい。
2) 全イオン電流クロマトグラム(TICC)
3) マスクロマトグラム(抽出イオンクロマトグラム)
4) 選択イオンモニタリング(SIM)のクロマトグラム(低分解能)
27
K 0123:2018
5) 選択イオンモニタリング(SIM)のクロマトグラム(高分解能)
6) 選択反応モニタリング(SRM)のクロマトグラム
7) 特定イオンの精密質量
8) MS/MSの各種スペクトル[衝突誘起解離(CID)スペクトル含む]
10
定性分析
定性又は同定は,次のいずれかの方法又はそれらの組合せによって得られた情報を総合して行う。
注記 質量分析法だけでは,異性体などの区別が付けにくい場合がある。赤外吸収スペクトル,核磁
気共鳴スペクトルなど,他の分析手段によるデータも有用な情報を与える。
a) 質量スペクトルの解析による方法 質量スペクトルの解析による方法は,次による。
1) 相対分子質量 分子イオン,プロトン付加分子,脱ヒドリド分子,付加イオン(CI法の場合)など
を利用して相対分子質量を推定する4)。
注4) EI法では,分子イオンが出現しない場合も多いCI法ではプロトン付加分子等が生じやすい
ので相対分子質量の推定に役立つことが多いが,その生成は試薬ガスの種類などに依存し
て変化するため,注意しなければならない。
2) 特定元素の種類及びその原子数 炭素,けい素,硫黄,塩素,臭素など同位体が存在する特定元素
について,それらの同位体イオンを確認し,その同位体存在比(相対強度比)から原子数を推定す
る5)。
注5) 存在が予想される元素の安定同位体を含む標識化合物の一定量を試料に混合し,その質量
スペクトルにおける特定イオンの同位体存在比の変化から,特定元素の存在を確認する方
法も用いられる。
3) 官能基の種類,数及び部分構造 開裂の経験則に基づくフラグメントイオンなどから官能基の種類,
数及び部分構造を推定する6)。最近はm/zとその元素組成推定,部分構造推定及び開裂の予測によ
ってスペクトルの解析を手助けするソフトウェアもある。また,特定プリカーサーイオンのプロダ
クトイオンスペクトルなどを利用し,分子の部分構造推定を行うことができる。特に精密質量を測
定した質量スペクトルの場合,蓄積されたデータベース及び専用のソフトウェアを用いて,実際の
プロダクトイオンスペクトルと照合しながら,フラグメントイオンの構造(元素組成含む)を推定
することが可能である。
注6) シリル化剤,アシル化剤,エステル化剤などの誘導体化剤を用いて,試料を誘導体化し,
質量スペクトル中の特性イオンについて求めた元の化合物及び誘導体間,又は誘導体化剤
中のアルキル基等を変えて測定したときの各誘導体間の質量シフトから,官能基の種類,
置換位置及びその数などを推定する方法も用いられる。
4) 分子式及びイオンの組成式 分子イオン又はフラグメントイオンについて高分解能測定が可能な場
合は,精密質量を求め,データシステムによって分子式又はイオンの組成式を推定することができ
る7)。
注7) 四重極形質量分析計による低分解能測定でも質量スペクトルをプロファイルデータの形で
取り込み,精密質量での質量スペクトルに変換可能とするソフトウェアもある。
b) ライブラリー検索による方法 データシステムによって,未知化合物の質量スペクトルとデータファ
イルされている既知の標準スペクトルとを比較し,類似する質量スペクトルをもつ化合物名を順序付
きで選び出し,同定を行う8)。ライブラリーにはMS/MSのスペクトルが登録されたものがあり,質量
28
K 0123:2018
スペクトル上の特定イオンのMS/MSスペクトルを同定に用いることもできる。また,一部の化合物
群については精密質量で測定された質量スペクトルのライブラリーがあり,より確実な同定が可能と
なる。
この手法の変形として横軸を時間に,縦軸をライブラリー上の特定の質量スペクトルとTICC上の
各質量スペクトルとの一致度に設定し,クロマトグラムの形で特定成分がいつ,検出されている可能
性が高いかを確認する方法もある。
注8) この方法では,該当する化合物がファイルされていない場合でも,結果が表示されるので,
分析者は検査結果を十分確認した方がよい。
なお,未知化合物のCIスペクトルから相対分子質量が推定できる場合には,これと検索結
果とを照合するとよい。
c) 特定イオンの検出による方法 試料中に共存成分が多く,目的の測定成分(分析種)の質量スペクト
ルの抽出が困難な場合は目的成分に特徴的な特定のイオンを選び,そのm/zを用いてマスクロマトグ
ラム(抽出イオンクロマトグラム)を描かせれば目的成分の検出が可能である。また,試料中の目的
成分の濃度が低く,質量スペクトルの測定が困難な場合には,存在する可能性がある複数のイオンを
選択イオンモニタリング又は選択反応モニタリングによって検出し,そのイオンの相対強度又は同位
体存在比を求めて同定を行う。また,この方法は既知の成分(一連の同族体の場合もある)の有無を
調べる場合にも用いられ,特徴的なイオン1個〜数個を選んで同様にその有無を検出する。いずれの
場合も同定にはイオンの相対強度又は同位体存在比が,推定された化合物の値と一致することを確認
する。
d) 保持時間による方法 この方法は,a)〜c) の方法と併用する。同一条件下で測定した未知化合物と推
定化合物とのクロマトグラム[マスクロマトグラム,選択イオンモニタリング(SIM)のクロマトグ
ラム又は選択反応モニタリング(SRM)のクロマトグラム]の保持時間との一致を確認する9)。また,
保持指標が登録されたデータベース10) との比較によってもよい。
なお,必要に応じ分離特性の異なる複数のカラムを用いて測定を行い,未知化合物及び推定化合物
の保持時間が両者で一致していることを確認する。
注9) 内標準物質を用いれば,保持時間の信頼性は非常によくなる。
10) 11.5参照。
e) デコンボリューションによる方法 NIST(National Institute of Standards and Technology)で開発した手
法に端を発すもので,全イオンモニタリングにおいて,TICC上でピークの重なりがある場合,測定
質量領域全体で各イオンの時間的な強度変化をモニターし,幾つかの特定のm/zのイオン強度変化の
パターン(各イオン強度の割合)が一致する場合は,同一の成分由来とする。これらを複数の変化の
パターンを再構築することによって,TICC上では重なり合っているピークから個々の成分ピークと
してクロマトグラム上に描かせることを可能にする11)。質量スペクトルも個々の成分のものを得るこ
とが可能になる。原理上,保持時間が僅かでも異なれば,個々の成分ピークに分けることが可能にな
り,ライブラリー検索も用いれば,従来は困難であった複雑な組成をもつ試料の分析種ごとの定性が
可能になる。
なお,最近は目的成分を定めず,測定された全成分を対象とする分析もある(ノンターゲット分析)。
後者は,一般的にケモメトリックスの範ちゅうに入り多変量解析を用いるが,専用の解析ソフトウェ
アもある。このソフトウェアを用いれば,非常に多くの成分が観測され,全く同じように見えるTICC
でも,特定の微量成分の有無,量の差異などを見分けることが可能である。
29
K 0123:2018
注11) 例えば,3成分が重なっている場合は,三つの成分に分解されたピークが重なり合った形で
表示される。
11
定量分析
11.1
試料の前処理
定量に適する試料とするために,分析種の抽出(溶解)・妨害成分の除去・分離・濃縮,誘導体化などの
前処理を行う。分析種がごく微量の場合には,前処理操作における損失及びコンタミネーションに特に注
意し,また,光,熱,酸化などの影響を避けなければならない。主な前処理操作は,次による。
a) 試料からの分析種の抽出 溶液試料では,溶媒抽出,固相抽出,ヘッドスペース法などを用い,固体
試料では微粉とした後,適切な溶媒を用いてソックスレー抽出,超音波抽出などの方法を用いる。超
臨界流体抽出及び直接熱抽出を行うこともある12)。
注12) ガスクロマトグラフへの導入まで,自動的に連続して行う場合もある。
b) 妨害成分の除去(クリーンアップ) 溶媒抽出,酸又は塩基による抽出,シリカゲル,アルミナ(中
性,酸性又は塩基性),合成けい酸マグネシウム,シリカ系又はポリマー系吸着剤などによる固相抽出
(カラム分離),液体クロマトグラフィー(サイズ排除クロマトグラフィーなど)による分画などの方
法を用いる。
c) 分析種の濃縮 溶液試料では,エバポレーター,クデルナダニッシュ形(KD)濃縮器などによる常圧
又は減圧下の蒸発濃縮,気体試料では吸着剤による常温,低温濃縮などの方法を用いる。
d) 誘導体化 分析種の揮発性・熱安定性の向上,高感度化のために,エステル化,シリル化,アシル化
などを行う。また,電子親和性を高めて負イオンによる検出感度を向上させるために,含ふっ素誘導
体化などを行うこともある(8.10参照)。
11.2
内標準法
前処理における回収率の変動,試料導入量の変動,分析種の試料導入部での吸着など,測定時における
各種の変動の影響を避け,正確な定量値を得るためには,内標準法を用いるとよい。特に,分析種がごく
微量の場合には変動が大きくなるので,この方法によることが望ましい。
内標準物質には,試料の前処理及び測定時における挙動が分析種と類似している物質を選択する。特に,
ガスクロマトグラフィー質量分析では,分析種と同じか又はその異性体で,それらの同位体標識化合物(D
体,13C体など)を用いることが望ましい13)。内標準物質を添加する時期は,試料採取から測定の直前ま
で,目的及び状態によって異なるが14),同位体標識化合物の場合には,できるだけ早い段階で添加すると,
定量値の正確さも向上する。
注13) ごく微量の多数の同族体又は異性体を同時に定量する場合(例えば,ダイオキシン類など)に
は,複数の同位体標識化合物を内標準物質として使用するとよい。また,物性の異なる非常に
多数の分析種(例えば,数百種類の農薬など)を一斉に定量する場合には,複数の分析種(例
えば,数十種類程度の農薬など)の同位体標識化合物を選択し,ガスクロマトグラフィー質量
分析における挙動ができるだけ類似している同位体標識化合物を各分析種の内標準物質として
使用するとよい。
なお,ある分析種を含む試料に,その同位体標識化合物を添加して分析を行う場合,特に,
同位体希釈法ということがある。
14) 内標準物質には,抽出からクリーンアップまでの前処理操作の全体の結果を確認し,分析種を
定量するための基準として添加するクリーンアップスパイク用内標準物質,試料採取から抽出
30
K 0123:2018
前までの操作の結果を確認するために添加するサンプリングスパイク用内標準物質,ガスクロ
マトグラフ質量分析計への試料の注入量の変動など測定時の影響を取り除くために添加するシ
リンジスパイク用内標準物質などがある。
11.3
絶対検量線法
精度のよい結果が得られる場合又は適切な内標準物質がないときに用いる。
11.4
標準添加法
適切な内標準物質がなく,試料に由来する共存物質などによる影響で,単位濃度当たりのピーク面積(又
はピーク高さ)に相違が見られるときに用いる。
11.5
検量線データベース法
分析種が非常に多い場合及び標準物質がないときに用いる。正確な定量値を得る必要がある場合には,
内標準法,絶対検量線法又は標準添加法を用いる。所定の条件であらかじめ標準試料を測定し,得られた
保持指標(retention index:RI),質量スペクトル及び内標準法による検量線などをデータベース化してお
き,それらのデータベースを用いて標準試料を用いることなくスクリーニング(又は半定量分析)を行う
方法である。ここでいうスクリーニングは,基準値を基に許容差をもって設定された濃度範囲に対して,
その範囲以上,又は以下であるかを判定する場合などが相当する。
作成したデータベースは求められる精度が担保できるならば,データベース作成に使用した装置以外の
装置にも適用することができる。保持指標用の標準試料の混合試料(例えば,直鎖アルカン)を測定し,
それらの保持時間及びデータベースに登録された保持指標からその装置での分析種の保持時間を推測する。
推測された保持時間と登録されている質量スペクトル情報(例えば,定量イオンに対する確認イオンの強
度比など)から分析種のピークを同定する。同定されたピークの面積(又はピーク高さ)と未知試料にあ
らかじめ添加された内標準物質のピーク面積(又はピーク高さ)との比を求め,検量線データベースから
濃度値を算出する。内標準物質として同位体標識化合物を用いると定量精度が向上する。
11.6
検量線又は関係線の作成方法
検量線又は関係線は,次の方法によって作成する。以下では,ピーク面積を用いた作成方法を示すが,
ピーク面積を用いた場合と同等以上の精度が得られるなら,ピーク高さを代わりに用いてよい。
11.6.1
検量線又は検量線作成用試料の調製
検量線又は関係線作成用の試料は,次のように調製する15)。検量線又は関係線作成に用いる試薬などは,
計量計測トレーサビリティの確保された標準物質を使用することが望ましい。計量計測トレーサビリティ
を確保するには12.2を参照する。
注15) 検量線作成用試料の濃度範囲は,直線性が得られる範囲内で必要以上に広くせず,かつ,上限
は実際試料中の予想濃度の数十倍程度までとすることが望ましい。また,下限は装置及びその
整備状態,測定条件,検出に利用するイオンの種類,バックグラウンドなどによって著しく変
わることがあるが,およそpg前後で,fgレベルまで可能な場合もある。一般に,定量下限は,
検出下限の数倍程度と考えてよい。
a) 内標準法及び絶対検量線法
1) 液体又は固体成分の場合 分析種の高純度試薬などの一定量を正確にはかりとり,適切な溶媒に溶
解した後,一定濃度に調製する。その一部を段階的に採取し,溶媒で希釈して数個の検量線作成用
試料を調製した後,内標準法の場合は内標準物質を一定量加え,密栓して保存する。
なお,濃度変化がないように,保存方法及び期間に注意が必要である。
2) 気体成分の場合 シリコーンゴム栓などの付いた内容量既知のガラス製捕集瓶数本を,あらかじめ
31
K 0123:2018
十分に洗浄し,乾燥する。これに分析種を含まない精製した不活性ガスなどを満たす。シリコーン
ゴム栓を通して,気体試料用シリンジで分析種の高純度ガス又は濃度既知のガスを段階的にとり,
注入する。内標準法の場合は,内標準物質を一定量注入する。分析種がガラス内壁に吸着するおそ
れがある場合には,分析種が吸着しない素材の捕集バックなどを用いて同様の操作をする。いずれ
の場合も数段階の希釈を行ってよく,また,調製にパーミエーション管及びガス自動濃縮導入装置
を用いてもよい。低濃度に希釈した検量線作成用試料は,調製後,速やかに測定を行う16)。
注16) 使用時,容器を加熱し,内壁へ吸着した分析種を脱離させる場合もあるが,再現性に十分
注意するのが望ましい。
b) 標準添加法 試料溶液から一定量を複数採取する。採取した試料溶液のそれぞれに既知量の分析種を
濃度が段階的に異なるように添加し,均一に混合する。そのとき,1個は分析種を添加しない。これ
らの溶液をそのまま,又はそれぞれ一定量に希釈して関係線作成用試料とする。
なお,添加量は,分析種を添加した試料の分析種のピーク面積が分析種を添加しない試料の分析種
のピーク面積の3倍程度以内になるように調製する。
11.6.2
検量線又は関係線の作成方法
検量線又は関係線は,次のいずれかの方法によって作成する。
a) 内標準法 箇条8によって,装置を試料の場合と同一条件17) に設定しておき,あらかじめ,分析種の
純物質(X)の既知量(Mx)に内標準物質(S)の既知量(Ms)を加えた一連の検量線作成用試料の一
定量を,順次装置に導入する。選択イオンモニタリング(SIM),選択反応モニタリング(SRM)又は
全イオンモニタリング(TIM)によって分析種及び内標準物質に特徴的なイオンを検出し,得られた
それぞれのクロマトグラム上のピーク面積を測定する。
注17) 11.7.2参照。
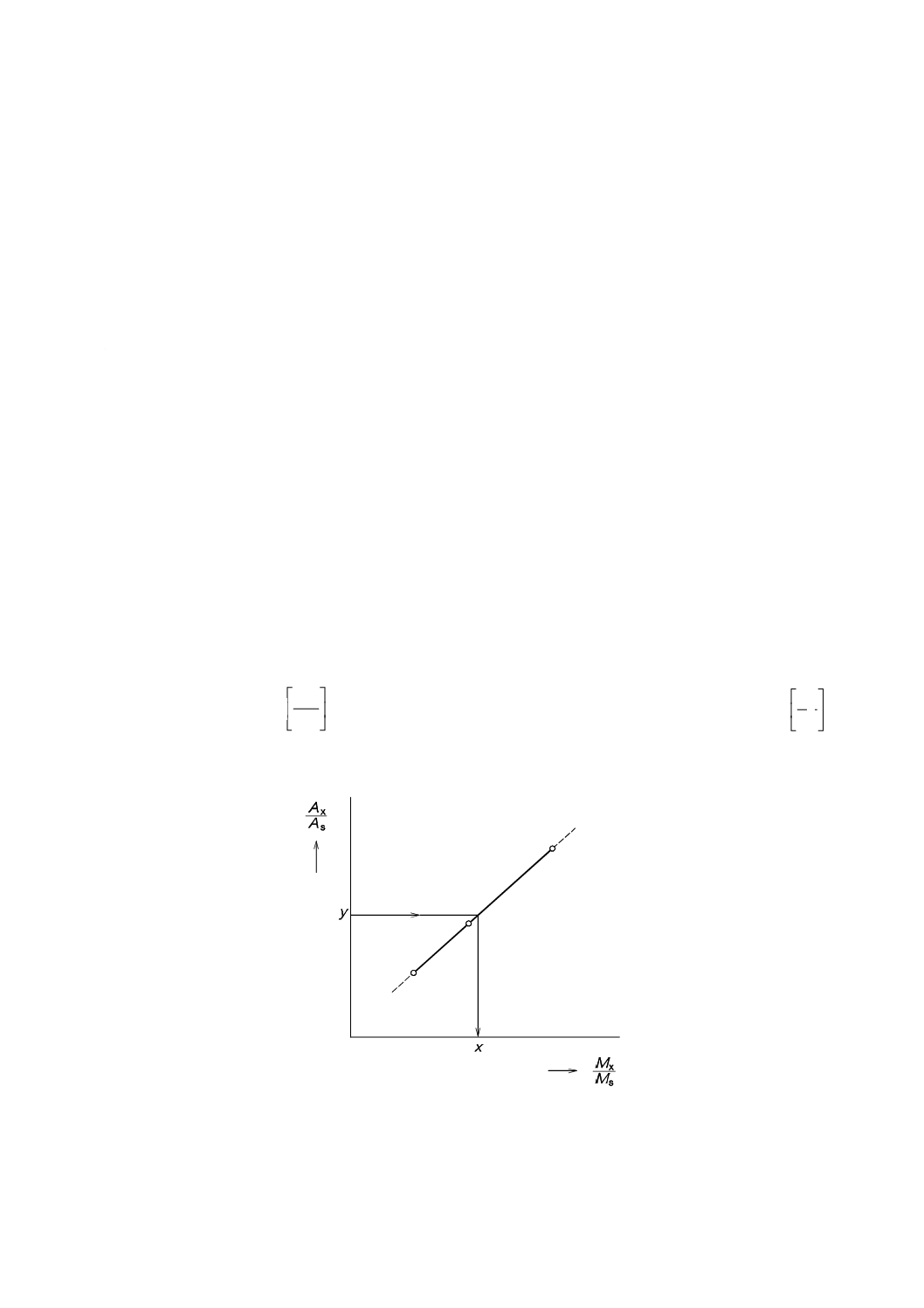
横軸にMxとMsとの
s
x
M
M
比をとり,縦軸にXのピーク面積(Ax)とSのピーク面積(As)との比
s
x
A
A
をとって,関係線を作成する。その関係式の例を図26に示す。
図26−内標準法による関係線(例)
試料の既知量(p)に対して内標準物質の既知量(q)を関係線の範囲内に入るように適切に加えて
32
K 0123:2018
均一に混合し,内標準物質のピークが関係線作成の場合とほぼ同じ大きさになるように導入量を加減
し,同一条件の下でクロマトグラムを記録する。
クロマトグラムから分析種のピーク面積(A'x)と内標準物質のピーク面積(A's)との比y=
′′sxAA
を
求め,次に関係線から分析種量(M'x)と内標準物質量(M's)との比x=
′
′
s
x
M
M
を求めて,次の式によ
って含有率C(%)を算出する。
100
×
×
=
p
q
x
C
ここに,
C: 分析種の含有率(%)
x: 図26の関係線から求めた分析種量と内標準物質量との
比
p: 試料の既知量
q: 内標準物質の既知量
ただし,p,qは同じ単位を用いる。
b) 絶対検量線法 一連の検量線作成用試料18) の一定量を,a) と同様に測定し,クロマトグラムを記録
して分析種のピーク面積を求める。次に成分量を横軸に,ピーク面積を縦軸にとって検量線を作成す
る。このときの既知量は測定点を代表するものを選ぶ。検量線の例を図27に示す。
同一条件の下で試料を導入し,クロマトグラムを記録し,ピーク面積から検量線によって被検各成
分の量を求め,試料中の含有量を算出する。この方法では,全測定操作を厳密に一定条件にして行わ
なければならない。
試料注入量のばらつきを押さえるために,自動の試料導入装置を使用することが望ましい。
注18) 定量は,一般的には数点を取り,検量線を作成して行われる。簡便法としてあらかじめ,原
点を通る直線性が確かめられている場合に既知量の導入を1点だけとし,単位ピーク面積当
たりの成分量を算出し,定量値を求める方法もある。
図27−検量線(例)
c) 標準添加法 11.6.1 b) によって調製した関係線作成用試料の一定量をa) と同様に測定し,クロマト
グラムを記録して分析種のピーク面積を求める。
なお,分析種を添加する溶液を1段階だけとする場合は,添加量とピーク面積との間に直線関係が
33
K 0123:2018
成立することを事前に確認する作業を行わなければならない。それぞれの試料への分析種添加量を横
軸,ピーク面積を縦軸として,関係線を作成する。関係線の例を図28に示す。分析種を添加しない試
料溶液から得られたピーク面積に相当する分析種の濃度は,関係線と横軸との交点から求める。この
方法は,関係線が直線の場合にだけ適用できる。分析種を添加した試料のピーク面積が分析種を添加
しなかった試料のピーク面積の3倍程度以内となるよう,分析種の添加量を加減する。
図28−標準添加法による関係線(例)
図28において,横軸との切片を−∆w,原試料量をWとして,次の式によって分析種の含有率Cを
算出する。
100
×
∆
=Ww
C
ここに,
C: 分析種の含有率(%)
∆w: 関係線から求めた横軸との切片の絶対値
W: 原試料量
ただし,∆w,Wは同じ単位を用いる。
また,分析種の添加による原試料の体積変化が無視できない場合は,その補正を行う。
11.7
定量操作
11.7.1
ピーク同定
標準試料を測定して得られたデータを基に,未知試料中の分析種の保持時間又は保持指標,質量スペク
トル情報(例えば,定量イオンと確認イオンの強度比)などの一致から同定する。
なお,定量イオンに対する確認イオンの強度比の許容幅は,標準試料の20 %程度とする。
11.7.2
測定条件の選定
定量のための測定条件は,次のとおり選定する。
a) イオンの検出及びイオン化の方式 目的に応じ,測定の感度,選択性,精度などを考慮して,イオン
化の方式,正・負イオンの別,及び検出の方式を選択する。
b) カラム 分析種の望ましい分離が行われるように選定する19)。この場合,カラム温度は,最高使用温
度を超えないように,また,バックグラウンドが大きくならないようにする。
注19) カラムの選定は,選択イオンモニタリング(SIM)などの選択性とも関連する。選択性が優
れていれば,カラムによる分析種の完全分離を必要としない場合もある。また,このことは
34
K 0123:2018
内標準物質についても同様である。
c) 選択イオン 分析種を検出するイオンは,その成分が特徴的でイオン強度が大きく,共存成分,カラ
ム,質量分析計に起因するバックグラウンドなどの影響がないものを選択する。内標準物質について
も同様にして選択する。この場合,1成分について二つ以上のイオンで選択イオンモニタリング(SIM)
又は選択反応モニタリング(SRM)などを行い,得られたイオンの強度比を確認しながら定量するこ
とが望ましい。
d) サンプリング時間 全イオンモニタリング(TIM),選択イオンモニタリング(SIM)又は選択反応モ
ニタリング(SRM)において得られる各種クロマトグラムでは分析種1ピーク当たり12点以上の測
定点が得られるようにサンプリング時間を設定する。TIMにおける定量のために用いるクロマトグラ
ム(マスクロマトグラム)では測定質量範囲に合わせ走査速度などを調整し,m/zごとのモニターに
費やす時間の積算値(附属書E参照)が大きくなりすぎないようにする。また,あまりサンプリング
時間を短くしすぎても測定精度が落ちるので注意が必要である。
なお,通常はTICCを用いての定量は行わないが,明らかにピークが単離している場合は定量に用
いることも可能である。
SIM及びSRMにおいては選択イオンのドゥエルタイムは10 ms〜100 ms程度が望ましいが,同時に
モニターするイオンの数及びクロマトグラム(SIM及びSRMクロマトグラム)上の測定点の数に合
わせて適宜調整を行うようにする。モニターするイオンの数が多くドゥエルタイムが短すぎる又はサ
ンプリング時間が長くなりすぎると測定精度が落ちるので注意が必要である。
なお,モニターするイオンの数が多く,感度不足等が懸念される場合は特定時間間隔ごとに適切な
数のモニターイオンのm/zと各ドゥエルタイムとを設定して組分け(グルーピング)を行い,測定感
度及び精度を確保するようにする。
e) 分解能 通常の測定では低分解能(磁場形では1 000程度)でよいが,分析種がごく微量の場合で,
特に妨害となる共存成分がクリーンアップ操作で十分に除けない場合,又はバックグラウンドの影響
を除けない場合には,できるだけ高分解能(約5 000〜10 000以上)で測定することが望ましい20)。
注20) 分解能を高くしていくとバックグラウンド及び不明ピーク(ケミカルノイズ)が徐々に小さ
くなりSN比は向上するが,分解能を高くし過ぎると絶対感度が低下するので,あらかじめ
最適な分解能を経験的に求めておくとよい。また,分解能を高くしても共存成分,ケミカル
ノイズなどの影響が除けない場合には,GC/MS/MSを用いて選択反応モニタリング(SRM)
によって測定すると除ける場合がある。
f)
試料導入部(インジェクター) 溶媒と分析種との沸点差などを考慮し,適切な試料導入部を選択す
る。
11.7.3
測定
実際試料の測定は,次による。
なお,ごく微量測定を行う場合には,例えば,濃度の高い検量線又は関係線作成用試料を装置から遠ざ
け,シリンジも高濃度試料を扱ったものは使わないなど,コンタミネーションには特に注意して操作しな
ければならない。また,流路系及び注入口セプタムからのコンタミネーション並びにカラム接続部の僅か
な漏れによる感度の低下にも注意しなければならない。
a) 11.1によって試料に応じた適切な前処理を行った後,定容し,密栓のできる容器に保存する。
b) 検量線又は関係線作成と同一条件で,一定量を装置に導入する。
c) 選択イオンモニタリング(SIM),選択反応モニタリング(SRM)によって得られた分析種(内標準法
35
K 0123:2018
の場合には,分析種及び内標準物質)のクロマトグラム上,又は全イオンモニタリング(TIM)によ
って得られた分析種(内標準法の場合には,分析種及び内標準物質)のマスクロマトグラム上のピー
クを検出する。あらかじめ標準試料を分析し得られたピーク同定の条件[保持時間(又は保持指標),
質量スペクトル情報(例えば,定量イオンと確認イオンとの強度比)]などを基に検出されたピークか
ら定量する分析種(内標準法の場合には,分析種及び内標準物質)のピークを同定する。それらのピ
ーク面積(内標準法の場合には,ピーク面積の比)を求め,検量線又はその回帰式から定量する21)。
標準添加法の場合は,11.6.2 c) による。
なお,全イオンモニタリングによって得られたマスクロマトグラム上に導入した試料の全成分のピ
ークが現れ,これら全成分の相対感度が求められ,それぞれの相対感度が測定濃度域にわたって一定
とみなされる場合,式(8)によって含有率Ci(%)を求めることができる。また,相対応答係数(補正
係数ともいう。)を利用する場合は,式(9)を利用する。
100
1
×
=∑
=
n
i
i
i
i
i
i
f
A
f
A
C
········································································ (8)
100
1
×
=∑
=
n
i
i
i
i
i
i
r
A
r
A
C
····································································· (9)
ここに,
Ci: i成分の含有率(%)
Ai: i成分の面積
fi: i成分の相対感度
ri: i成分の相対応答係数
n: 全ピーク数
注21) 必要に応じて,同様に操作して得られた空試験値を差し引く。
d) 必要に応じて,分離特性の異なる2種類のカラムによって測定を行い,定量値などを確認するとよい。
また,同一の試料について2回以上又は別に処理した試料について測定を繰り返し,平均値を算出す
る。
12
データの質の管理
12.1
一般事項
分析値の信頼性を確保し,データの質を管理するために必要な事項を次に示す。分析値が真値を含み普
遍性をもつようにするには,計量計測トレーサビリティを確保し,測定の手順を明確にして不確かさの要
因を示し,不確かさを見積もって分析値の信頼性を確保することが必要である。分析機関内での分析値の
同等性を確保するには,適切な内部精度管理を実施するとよい。分析機関間の分析値の同等性を確保する
には,適切な外部精度管理を行うとよい。一般に,データの質の管理を行うには,データの質の管理のた
めの測定,分析方法の妥当性確認の実施,検出限界の確認,空試験値の測定,定期的な装置性能の点検,
クロマトグラムのピーク形状及び分離の確認,質量スペクトルの質の確認,作業手順書の作成などが必要
である。これらの評価結果は,測定記録,クロマトグラムと共に保管する。
12.2
計量計測トレーサビリティの確保
計量計測トレーサビリティを確保するには,検量線作成時に用いる標準物質及び調製に用いる器具(分
銅,全量ピペット,全量フラスコ,ピペット,天びんなど)の計量計測トレーサビリティを示さなければ
36
K 0123:2018
ならない。トレーサビリティソースが明確で,かつ,計量計測トレーサビリティが確保されている標準物
質及び器具は,計量法トレーサビリティ制度(JCSS)によって供給されており,不確かさが明記された校
正証明書が付いている。JCSS以外の標準物質は,ISO Guide 34:2009(ISO規格制定に伴い,2018年11月
以降はISO 17034)の認定を取得した標準物質生産者から認証標準物質(CRM)が供給されており,特性
値(不確かさ付き)が明記されている。検量線作成用標準液の調製に用いるJCSS以外の器具には,次の
ものがある。JIS R 3505に沿って製造されたガラス製体積計は許容誤差が表記されている。その他,調製
に用いる体積計は,JIS K 0050の附属書H(体積計の校正方法)に従って校正を行う。質量比混合法での
調製に用いる天びんは,JCSS校正証明書付の分銅を用いて定期校正を行う。
国際単位系(SI)にトレーサブルな最上位の標準物質又はトレーサビリティソースとなる標準物質は,
国家計量機関[国立研究開発法人産業技術総合研究所計量標準総合センター(NMIJ),NISTなど]から
CRMとして供給されている。JCSSで供給されている標準物質は全てSIにトレーサブルである。計量証明
ではSIにトレーサブルな標準物質を用いることが望ましいが,供給されていない標準物質については標準
物質生産者が供給するCRMを用いるとよい。しかし,全ての市販のCRMがSIにトレーサブルではない
ので,使用時には確認が必要である。
計量計測トレーサビリティが確保されている標準物質を用いて,検量線を作成して濃度未知の物質を定
量する場合,標準物質の不確かさ,検量線作成時の調製及び測定の不確かさ並びに未知試料測定時の測定
不確かさから分析値の測定不確かさを求める分析値の不確かさの表記は拡張不確かさUを用いることが適
切である。その際,包含係数kの値を明記する。化学分析においてはUの信頼の水準が約95 %に相当する
k=2を用いることが一般的である。
SIに計量計測トレーサビリティが取れない試薬を用いた分析値の不確かさの求め方は12.12による。
12.3
分析値の信頼性の確保
要求に応じて分析値の信頼性を確保するためには,測定の手順を明確にして不確かさの要因を示し不確
かさを見積もることが必要である。その上で,品質管理用標準物質を用いて実施される内部精度管理,共
同分析,技能試験などの外部精度管理を適切に実施して,その結果を記録し,一定の精度を維持すること
が望ましい。
12.4
データの質の管理のための測定
質量分析計は,測定ごとに感度と質量スペクトルとが変化するおそれがあるので,定量操作を行う場合
は,測定試料に内標準物質を添加し,測定ごとに感度変化の確認とスペクトルの変化とを確認するとよい。
データの質の管理のために,必要に応じて既知濃度の標準物質又は検量線用標準物質を別途用意し,その
平均濃度値,平均保持時間,理論段数,分離度などを測定し,分離及び安定性を確認し記録する。また,
クロマトグラムのピーク形状も確認し記録する。測定対象物質の一部をこの目的に用いることができる。
12.5
分析方法の妥当性確認の実施
新たに開発した分析方法(分析条件を変更した場合を含む。)を試験に用いる場合には,その分析方法が
試験の目的に合致していることを立証するために,分析方法の妥当性を確認する。分析方法の妥当性確認
では,附属書Bの例にある選択性及び特異性,範囲,直線性,検出下限,定量下限,堅ろう性(頑健性),
精度(併行精度,室内再現精度,室間再現精度),真度などの分析能パラメーターについて評価を行うとよ
い。公定法を採用する場合でも,その試験室で要求される項目について,分析方法の妥当性確認を実施す
ることが望ましい。
なお,評価結果は文書に記録し,保管する。
37
K 0123:2018
12.6
検出下限の求め方
検出下限は,統計的手法によって求める方法,合意された方法などもあるが,代表的な方法として次の
二つの方法を示す。
なお,検出下限を記載する場合には,用いた方法を明記する。
a) シグナル対ノイズ比に基づく方法 既知の低濃度の分析対象物を含む試料の一定量をガスクロマトグ
ラフに注入し,得られたシグナルS及びベースラインノイズNを同条件で測定する22), 23)。シグナル対
ノイズ比が2又は3の場合の目的成分量を検出下限とする24), 25)。シグナル対ノイズ比が2のピークの
例を図29に示す。
図29−シグナル対ノイズ比が2のピーク(例)
検出下限は次で示される。
検出下限D=2N / S又は3N / S
注22) この規格とJIS K 0124及びJIS K 0136とでは,Nの採り方が異なる。
23) 日本薬局方によるガスクロマトグラフィー質量分析では,Nの採り方が異なる液体クロマト
グラフィーの用語の定義を準用している。
24) IUPAC(国際純正・応用化学連合)が発行している手引書[The Compendium on Analytical
Nomenclature(The Orange Book)]では,シグナル対ノイズ比が2の場合を目的成分量の検出
下限としている。
25) ドリフト,ふらつき,汚染などの影響で,経験的には2〜3にするのが妥当であると判断され
ている。
b) 定量値の標準偏差及び検量線の傾きに基づく方法 検出下限Dは,次の式から求めることができる。
検出下限D=t·σ / a
ここに,
t: 表1参照
σ: シグナルの標準偏差
a: 検量線の傾き(横軸:成分量 縦軸:シグナル)
検出下限付近の濃度の分析対象物を含む標準物質を用いて検量線を作成する。得られた検量線から
傾きaを求める。回帰直線の残差の標準偏差又は回帰直線から推定した成分量ゼロにおけるシグナル
の標準偏差,ブランク試料の応答量の標準偏差などをσとして利用できる。
38
K 0123:2018
このσに,表1の測定回数に対応したt値を乗じ,傾きaで除した値を検出下限とする。検出下限
をより精密に推定するためには,測定の繰返し回数を20回以上とすることが望ましい。この場合には
t値として3.3を用いる。
表1−測定回数n及びt値
測定回数n
3
4
5
6
7
8
9
10
11
12
13
14
15
t値
5.84
4.71
4.26
4.03
3.89
3.79
3.72
3.67
3.62
3.59
3.56
3.54
3.52
測定回数n
16
17
18
19
−
∞
t値
3.51
3.49
3.48
3.47
−
3.29
20回以上測定することは,実際の測定ではあまり現実的ではない。ただし,測定回数が少ない場合
には,t値を乗じることによってより安全な値を得ることができる。また,定量下限については,適
用する分析方法の要求する精度によるが,おおむね検出下限の10倍が妥当である。個別の測定方法に
適用するには,規定する測定方法が定量したい濃度と精度の目標を定め,これを十分満足する定量下
限を得るためにはどの程度の濃度の装置性能試験用標準物質を用いればよいかについて明確に記載す
る必要がある。
注記 一例として,JIS K 0311:2008“排ガス中のダイオキシン類の測定方法”がある。
12.7
空試験値の測定
ガスクロマトグラフィー質量分析では,昇温操作を含む高感度分析時には,特に試料気化室,カラムの
汚れ,注入口ゴム栓からの溶出成分などに起因するピーク(ゴーストピーク)の出現によって精確なデー
タが得られない場合がある。このため,同条件で試料を導入せず空昇温をし,ブランクを測定しゴースト
ピークの有無を確認する。ゴーストピークが出る場合は,注入口ライナーの交換,セプタムのコンディシ
ョニング,カラムのコンディショニングなどの適切な処置を施す。また,試料気化室内又はカラムに残留
した試料成分,カラム固定相の分解などで,検出器のベースラインの変化又は試料成分の分解・吸着など
が生じる可能性がある。これらの確認のためにも,試料を注入せずに又は溶媒だけを注入し,ベースライ
ンの変化又はゴーストピークの有無を確認し記録することは重要である26)。
注26) 分析種を含まないことが明らかな試料を,分析種を含む試料と並行して処理した測定用試料溶
液を測定すると,試料由来のきょう(夾)雑物の影響及び全分析操作にわたる影響を明らかに
できる。一方,純溶媒を測定すれば,溶媒の空試験及び試料導入時の汚染,相互汚染などの装
置からの影響を分離して明らかにできる。
12.8
定期的な装置性能の点検
装置構成部の点検は,各装置の取扱説明書に従い,定められた頻度で実施する。装置性能の点検のため,
定期的に濃度既知の標準物質又は検量線用標準物質を用意し,規定の感度,規定の分離及び保持時間,良
好なクロマトグラムなどが得られることを確認する。装置性能の点検は,使用開始時の点検(初期性能の
確認),一連の分析操作時に適切な間隔で実施する点検及び一連の分析操作終了時に行う確認に分けられる。
いずれも初期性能を維持していることが望ましいが,感度変化の大きい質量分析計ではある程度の許容範
囲を設けておくことが現実的である。また,測定時にも性能評価の基準となる物質と質量スペクトルの質
とについての基準を設けておくことで適切な管理が行える。測定ごとに内標準物質を添加している場合は,
内標準物質を用いてもよい。装置性能の点検記録は操作条件と共に文書にして保管する。効果的な点検は,
点検計画書を作成し,日常点検簿(ユーティリティを含む。),及び定期点検簿を付けておくことで異常の
39
K 0123:2018
未然防止及び早期発見が可能となる。
12.9
クロマトグラムのピーク形状及び分離の確認
クロマトグラムのピーク形状には正常な対象ピークのほかに,リーディングピーク及びテーリングピー
クがある。カラム中で分析種の相平衡関係が成り立ち,固定相中濃度と移動相中濃度に直線関係があれば,
対称なピークとなる。直線関係にないと正常な対称ピークのようでも,ピーク頂点から垂線を下ろしたと
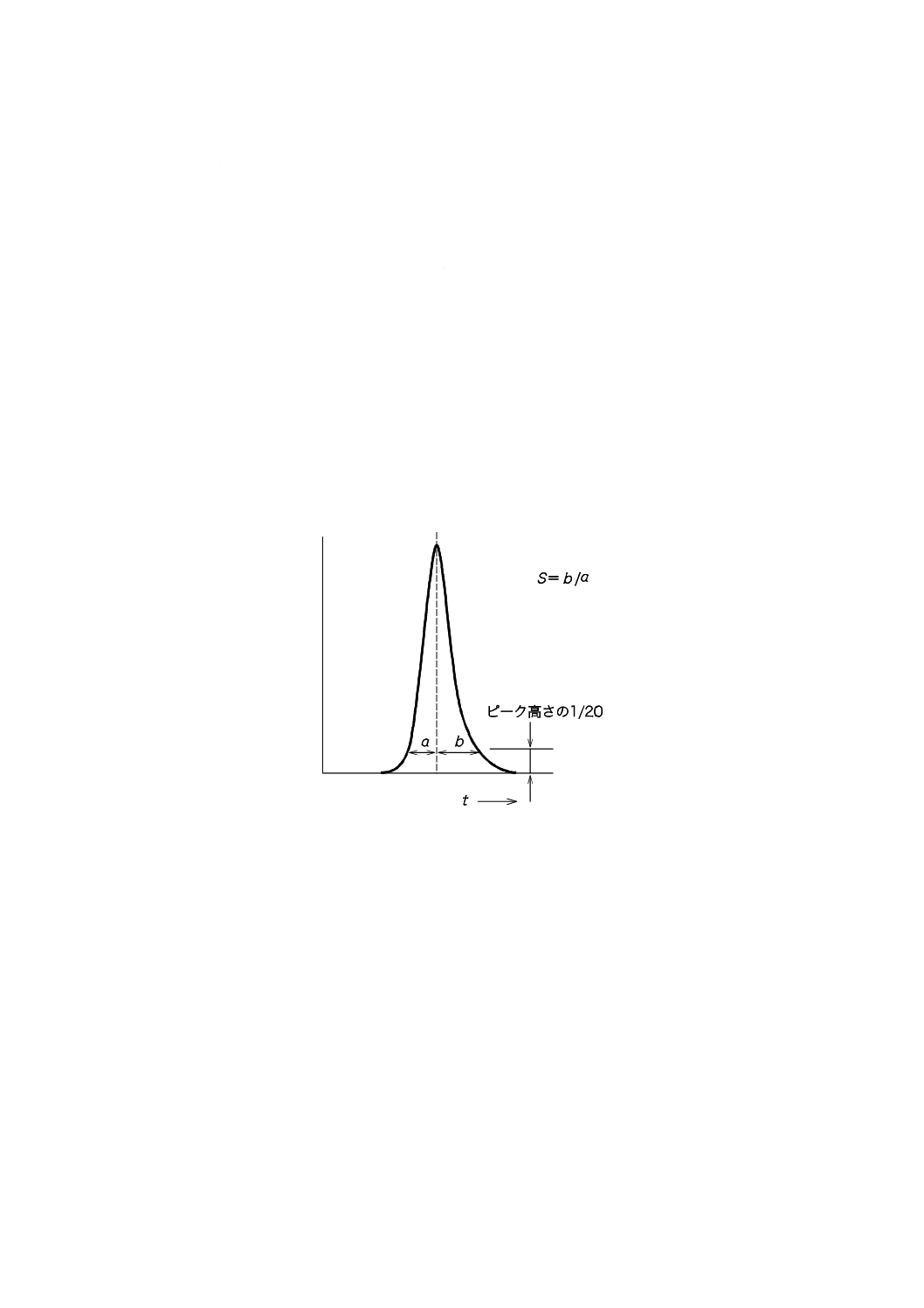
きに前後の対称性が異なる。これをピークの非対象性といい,アシンメトリー係数(S)で表す。Sはピー
ク頂点からベースラインに垂線を下ろし,ベースラインから1/20の所(0.05 h)で垂線前後のピーク幅を
測定してb/aで表す。その例を図30に示す。S>1:テーリングピーク,S<1:リーディングピークと呼ば
れる。テーリングピークでは,この数値がカラムの劣化,試料による汚染などで変化するので記録してお
くことが望ましい。リーディングピークは定量に差し支えない。テーリングピークは一般的に濃度が低く
なると徐々に検量線の直線部分から外れ,ある程度のところで応答しなくなるので定量下限を求めるとき
には正しく評価しなければならない。tは試料注入からの経過時間である。
定量分析の場合,クロマトグラムのピーク形状と合わせてピーク面積処理,ベースラインの引き方,不
分離ピークの場合のピーク分割方法などを目視で確認することが必要である。
図30−アシンメトリー係数
12.10 質量スペクトルの質の確認
検出器から得られる質量スペクトルの質の管理は,定量分析及び定性分析の場合において留意しなけれ
ばならない。実際の測定では微量成分の高感度測定を目的とした場合には,定量イオンを定めて選択イオ
ンモニタリングを行う。この場合に,成分ピークを確認するための確認イオンも同時に定めて測定する。
定量イオンと確認イオンとの比率が一定(許容範囲)であれば定量しても問題はないが,検出下限に近づ
いてくると定量イオンは確認できるが,確認イオンが確認できなくなってくる。このような場合に,定量
イオンのSN比から定量下限を定めると共存成分の影響を評価できなくなる。質量スペクトルの質の管理
基準を設けて,定量イオンと確認イオンとの比率が許容範囲を超えた場合の数値の処理基準を定めておく
必要がある。また,定量イオンと確認イオンとの比を管理することで,検出器の状態及び共存する成分の
影響についても評価を行うことができるので,質量スペクトルの質の管理は必要である。定性を行う場合
は,標準物質による質量スペクトルとの比較を行うとよい。
40
K 0123:2018
12.11 作業手順書の作成
分析操作の手順,機器の操作方法及び点検方法を記載した作業手順書(標準作業手順書)を作成し,こ
れを遵守する。
12.12 分析値の不確かさの求め方
通常の分析では計量計測トレーサビリティを確保し,不確かさを付与して分析値の信頼性を表記するこ
とはかなり困難である。さらに,標準物質が入手できない場合には試薬を用いて検量線を作成しなければ
ならない。試薬には純度と不確かさが付与されていないので使用時に見積もらなければならないが,手順
は明確に示されていない。附属書Cに例として試薬の純度評価から検量線の作成,分析値とその不確かさ
を求める方法を示す。
JCSSで供給されている標準物質又はCRMが入手できる場合は,附属書Cを参考にしてその特性値を用
いれば計量計測トレーサビリティを確保して分析値の信頼性を示すことができる。
13
分析報告書
分析報告書には,次のうち必要な項目を記載する。
なお,分析結果の単位は,JIS Z 8000-1及びJIS Z 8000-9の規定に従って表示する。
a) 一般的な事項 次に示す。
1) 分析年月日
2) 分析者名
3) 分析装置の製造会社名及び形式名
4) 分析種及び対象成分
5) 試料採取場所及び採集方法
6) 定性及び定量結果
b) ガスクロマトグラフの操作条件 次に示す。
1) キャリヤーガスの種類及び流量,線速度又は圧力
2) カラムの種類
2.1) 分配形キャピラリーカラムの場合 次に示す。
− 固定相液体名及び固定相液体の膜厚
− カラム用キャピラリーの材質,内径及び長さ
− 試料の量及び試料導入方法
2.2) 充塡カラムの場合 次に示す。
− カラム充塡剤の種類及び粒径範囲(分配形充塡剤の場合は,担体名,処理方法,固定相液体
名及び担体上への塗布量)
− カラム用管の材質,内径及び長さ
− 試料の量及び試料導入方法
2.3) 吸着形キャピラリーカラムの場合 次に示す。
− 多孔質層の名称及びその膜厚
− カラム用キャピラリーの材質,内径及び長さ
− 試料の量及び試料導入方法
3) 試料気化室温度
4) カラム槽温度(昇温法を用いる場合は,初期温度,昇温速度及び最終温度)
41
K 0123:2018
c) 質量分析計操作条件 次に示す。
1) イオン化法の種類,正・負イオンの別,及びイオン化電圧
2) 試薬ガスの種類及び流量又は圧力(化学イオン化の場合)
3) データの種類(質量スペクトル,全イオン電流クロマトグラム,選択イオンクロマトグラム,マス
クロマトグラム,などの別)
4) インターフェース(GC/MS接続部)の温度
5) 校正用標準試料の種類及び使用条件
d) 定量法 次に示す。
1) ピーク面積又は高さの測定方法
2) 定量方法の種別及び測定回数
3) 検出の方式及び選択イオン
4) 被検成分及び内標準物質名
14
個別規格に記載すべき事項
ガスクロマトグラフィー質量分析法(GC/MS)を用いる個別規格では,少なくとも,次の事項を記載し
なければならない。
a) 適用範囲
b) 試料の調製方法
c) 検量線及び関係線作成用試料の調製方法
d) 測定条件
e) 結果の整理及び表示方法
f)
データの質の管理
42
K 0123:2018
附属書A
(参考)
化学イオン化
A.1 正イオン化学イオン化(PICI)法
PICI法では,まず,イオン化室に満たされた試薬ガスに電子線を照射させるなどして生じた一次イオン
(例えば,メタンの場合,CH4+,CH3+など)と試薬ガス自身の分子との反応によって,二次又は三次イ
オンである反応イオン(例えば,メタンの場合,CH5+,C2H5+,C3H5+など)を生成させる。これらの反
応イオンと試料分子との間のイオン−分子反応によって種々のイオンが生成する。メタン試薬ガスの場合
の主なイオン化反応は,次のとおりである(Mは試料分子)。
M+CH5+ → (M+H)++CH4,M+C2H5+ → (M+H)++C2H4
(プロトン移動反応)
M+CH5+ → (M−H)++CH4+H2,M+C2H5+ → (M−H)++C2H6
(ヒドリド引き抜き反応)
M+C2H5+ → (M+C2H5)+,M+C3H5+ → (M+C3H5)+
(反応イオンの付加反応)
その他,電荷交換反応による分子イオンの生成反応なども起こる。生成したイオンは,イオン化反応の
エンタルピー変化を内部エネルギーとして蓄えるため,結合エネルギーが小さい箇所をもつイオンはフラ
グメンテーションを起こすが,蓄えられるエネルギーは,一般にEIにおけるそれよりも小さいため,その
度合いは少ない。イオン化反応の起こりやすさは,主反応であるプロトン移動反応の場合,試料分子と反
応イオンとの共役塩基であるメタンとエチレンとのプロトン親和力の差による。すなわち,それらのプロ
トン親和力の差が大きいほどイオン化は起こりやすいが,メタン及びエチレンのプロトン親和力は通常の
有機化合物よりも小さいため,効率的に有機化合物をイオン化できる。そのため感度の要求されるGC/MS
での測定には,メタンを試薬ガスとして用いることが多い。
なお,プロトン化分子,(M+H)+(MH+と表記する場合も多い),付加イオン,(M+C2H5)+,(M+C3H5)+
の質量数はそれぞれ (M+1),(M+29),(M+41) なので,これらの質量数を示すイオンをスペクトルから
読み取ることによって,相対分子質量が容易に推測できる場合が多い。また,試薬ガスに2-メチルプロパ
ン(イソブタン)とアンモニアとを用いた場合,生成する主な反応イオンは,それぞれt−(CH3)3 C+,
NH4+であるが,それらによるイオン化反応は試料分子とのプロトン親和力の関係から,メタンの場合と比
較すると効率が悪い。ただ,よりソフトにイオン化が行われることから,過剰な内部エネルギーの蓄積が
少ないため,フラグメンテーションは起こりにくく,(M+H)+及び付加イオンが質量スペクトル上に生成
しやすい。
A.2 負イオン化学イオン化(NICI)法
NICI法は,反応イオン形と電子捕獲形とに大別される。前者はPICI法と同じく,反応イオンと試料分
子との間のイオン−分子反応によってイオン化が行われるが,反応イオンとしてはプロトン親和力が大き
く試料分子から,プロトンを引き抜いて (M−H)−などのイオンを生じさせやすいOH−,CH3O−などが用
いられることが多い。OH−を用いた場合の反応式は,次のとおりである。
M+OH− → (M−H)−+H2O
OH−はN2O/He/CH4混合系,CH3O−はCH3ONO/CH4混合系などから効率よく生成されるが,これらの系
43
K 0123:2018
は酸化性ガスを含んでいることが多く,装置の運用上は望ましくないので汎用性は低い。OH−は効率的で
はないもののメタンなどの試薬ガス存在下で,系内に残存する水からも生成する。系内には水が必ず残留
するため,メタン試薬ガスでもOH−由来のプロトン引き抜き反応が起こり,生成量は少ないものの (M−
H)−が生成する場合が多い。このイオン化反応は,PICIと相補的でPICIで (M+H)+,NICIで (M−H)−が
生成すれば相対分子質量推定などが容易になる。また,Cl−などのハロゲン化物イオンを反応イオンに用
いる場合もある。Cl−はクロロホルムなどのハロゲン化アルキルをメタンなどに少量添加すれば効率よく
生成し,試料分子に付加して付加イオン (M+Cl)−を生成しやすい。
電子捕獲形のNICI法は,フィラメントなどから放出された電子と試薬ガスから放出された電子とが主
役となる。すなわち,次の式のようにメタンなどの試薬ガスを満たしたイオン化室に電子線を当てると電
子の一部は,試薬ガスの一次イオン化に使われ,試薬ガス分子から電子を放出させる。
CH4+e− → CH4++2e−
これらの電子及びイオン化に使用されなかったフィラメントからの電子は,試薬ガス分子と衝突を繰り
返して運動エネルギーを失い,運動エネルギーがゼロ又はほとんどない熱電子を生じる。この熱電子を,
電子親和性の高い試料分子が捕獲してイオン化する。主なイオン化過程には,分子イオン,M−を生じる非
解離共鳴捕獲反応と最初から,フラグメントイオンを生じる解離共鳴捕獲反応とがある。
M+e− → M−(非解離共鳴捕獲反応)
M+e− → (M−A)−+A(解離共鳴捕獲反応)(Aは解離によって生じるラジカルなど)
電子捕獲形NICI法の特徴は,その反応速度の速さにあり,試料分子の電子親和性が高い(例えば,電
子親和力が大きい)とそのイオン化反応は通常のPICIにおけるイオン化反応よりもかなり速く,結果とし
て極めて効率の良いイオン化が行われる。そのためGC/MS法は,ごく微量の成分の検出に適しており,
適用例も多い。また,極性化合物を誘導体化してGC/MS装置で測定を行う場合,電子捕獲形のNICI法で
の測定に適した含ふっ素誘導体化剤による誘導体化もよく行われる。

44
K 0123:2018
附属書B
(参考)
分解能パラメーターの定義及び評価方法
分析能パラメーターの定義及び評価方法の例を表B.1に示す(JIS K 0136の解説表2)。個別分析方法に
おける評価方法の適用例としては,JIS K 0124が参考になる。
表B.1−分析能パラメーターの定義及び評価方法(例)
分析能パラメーター
定義
評価方法の一例
選択性/特異性
試料に混在する他の成分の影
響を受けないで,特異的に分析
種を測定する能力。
分析種のクロマトグラムのピークが単一成分であること
を,そのピークの質量スペクトルによって確認する。
不純物が入手可能な場合,不純物を試料に添加して分析し
て,目的対象成分のピークとの分離度を評価する。
範囲
分析方法が適切な精度,真度及
び直線性を与える試料中の分
析対象成分の上限及び下限の
濃度(量)の間隔。
範囲の上限値,下限値及び中央付近の値の試料について,
精度,真度及び直線性を評価する。
直線性
(一定の範囲内で)試料中の分
析種の濃度(量)と直線関係に
ある測定値を与える分析方法
の能力。
分析種の濃度を関数としてシグナルをプロットしたもの
を視覚的に観察し,直線関係が認められた場合,最小二乗
法によって回帰分析し,その直線の傾き,y切片,相関係
数及び残差平方和を求める。濃度点数は5点以上とする。
検出下限
試料中に存在する分析種の検
出可能な最低の濃度(量)。
本体12.6を参照。
定量下限
適切な精度と真度を伴って定
量できる,試料中に存在する分
析種の最低の濃度(量)。
クロマトグラムのシグナルSとノイズNとの比S/Nが10
となるときの分析種の濃度(量)を求める。
堅ろう性(頑健性)
分析方法の条件を小さい範囲
で故意に変動させたときに,測
定値が影響を受けにくい能力。
測定値に影響を及ぼす操作上及び環境上の条件を適切な
範囲で変化させ,測定値の安定性を評価する。
精
度
併行精度
(繰返し性)
同一施設内において,試験日,
試験実施者,器具,機器などを
変えないで,短時間の間に同一
条件下で測定する場合の精度。
規定する範囲を含む濃度について,分析方法の全操作を少
なくとも9回繰返しの測定(例えば,3濃度において3回
繰返しの測定)又は試験濃度の100 %に相当する濃度で分
析方法の全操作を少なくとも6回繰返しの測定を行い,測
定値の分散,標準偏差又は相対標準偏差,分散の90 %信
頼区間及びこれに対応する標準偏差の区間を評価する。
室内再現精度
同一施設内において,試験日,
試験実施者,器具,機器などを
変えて測定する場合の精度。
分析方法の目的に応じて試験日,試験実験者,器具,機器
などを変え,同一試料を測定し,測定値の分散,標準偏差
又は相対標準偏差,分散の90 %信頼区間及びこれに対応
する標準偏差の区間を評価する。
室間再現精度
異なった施設間で測定する場
合の精度。
異なる施設で同一試料を測定し,測定値の分散,標準偏差
又は相対標準偏差,分散の90 %信頼区間及びこれに対応
する標準偏差の区間を評価する。
真度
真値として認証又は合意され
た値と実測値との間の一致の
程度。
室内再現精度又は室間再現精度を求めた場合の測定値の
総平均と,標準品の認証値又は合意値との差を求める。
45
K 0123:2018
附属書C
(参考)
分析値の不確かさの見積り手順
C.1 概要
計量計測トレーサビリティを確保し,不確かさを付与して分析値の信頼性を表記することは通常の分析
ではかなり困難である。ここでの記載が唯一のものではないが,標準液調製時に使用する溶質(原料)の
純度評価から検量線の作成,分析値とその不確かさを求める方法を参考として次に示す。
分析値の不確かさの見積り手順は,次のとおりである。
a) 標準液の調製,その濃度値及びその不確かさの見積り(C.2)
b) 検量線の作成及びその検量線由来の不確かさの見積り(C.3)
c) 未知試料の測定,分析値及びその不確かさの見積り(C.4)
なお,ここで示す方法は,限られた情報から不確かさを推定することによる不確かさの過小評価のリス
クと,簡易化された方法を用いることによる不確かさの過大評価のリスクが混在していることに留意しな
ければならない。また,過小評価及び過大評価,それぞれの程度についての情報がないので,それぞれを
相殺して不確かさを求めることはできない。
例に沿って実際の数値を使い分析値と不確かさを見積もる例を示す。
C.2 標準液の調製,その濃度値及びその不確かさの見積り手順
C.2.1 一般
検量線を作成するために用いる標準液には,認証標準物質(CRM)のように不確かさが付与されている
ものを使用することが望ましい。しかし,そのような標準液が入手できない場合には,使用者が標準液を
調製しその濃度値及びその不確かさを見積もる。その方法について,次のC.2.2〜C.2.4に示す。
C.2.2 標準液調製時に使用する溶質(原料)の純度及びその不確かさの見積り手順の例
標準液調製時に使用する原料の純度に関する情報について,使用者に与えられているものの種類は,大
きく分けて次の三つである。
a) 純度及びその不確かさの表記があるもの(例 認証標準物質)
b) “純度:xx.x %以上”などといった表記のあるもの(例 製造業者保証値記載の試薬)
c) 純度又は不純物に関する情報がないもの
それぞれについて,原料の純度及びその不確かさの見積り方法を次に示す。
a) 純度及びその不確かさの表記があるもの 認証標準物質のように純度値及びその不確かさが表記され
ているものについては,その値を使用する。ただし,付与されている値の単位が異なっている場合が
あるので注意する。単位が異なっている場合には,使用者が自身の責任において単位変換を行う必要
がある。必要に応じて,c) に従って,使用者が原料の純度値及びその不確かさを見積もってもよい。
b) “純度:xx.x %以上”などといった表記のあるもの 市販の試薬のように,製造業者が保証する純度
値などの表記があるものについては,次に示すようにその表記から得られる情報を用いて,原料の純
46
K 0123:2018
度及びその不確かさを見積もる。
1) 表記されている純度値を,その原料の純度の中心値,すなわち,純度値として使用する。
例C-1 “純度98.0 %以上”と表記されていた場合は,98.0 %を純度値として使用する。
2) 純度値の推定純度の情報から見積もられる不確かさ,及び不純物の情報から見積もられる不確かさ
を次によってそれぞれ推定し,これらの不確かさを合成することによって原料の純度の不確かさを
見積もる。
2.1) 純度の情報から見積もられる不確かさ 表記されている純度xを原料の純度の中心値とし,最大
値を1(100 %)とする。原料の純度の中心値と最小値の範囲は,最大値と中心値までの範囲と同
じであるとする。すなわち,
原料の純度の最大値:1(100 %)
原料の純度の中心値:x
原料の純度の最小値:x−(1−x)=2x−1
である。この中での分布がく(矩)形分布であるとみなして,純度の情報から見積もられる不確
かさu(xp) を見積もる。すなわち,
()
(
)
3
1
3
2
2
2
3
2
1
2
1
x
x
x
x
u
p
−
=
−
=
−
−
=
················································ (C.1)
である。
2.2) 不純物の情報から見積もられる不確かさ 表記されている純度x及びその純度の評価方法を確認
する。合わせて表記されている不純物iの濃度xi及びその評価方法を確認する。原料の純度の評
価方法を用いて不純物iの評価を行うことができない場合には,原料の純度の評価において不純物
iの評価が行われていないものとみなし,不純物iが含まれていることによる不確かさuiを見積も
る。原料に表記されている不純物iの濃度を不純物iの濃度の中心値とし,最小値を0とする。不
純物iの濃度の中心値と最大値との範囲は,最小値と中心値までの範囲と同じであるとする。すな
わち,
不純物iの濃度の最小値:0
不純物iの濃度の中心値:xi
不純物iの濃度の最大値:2xi
である。この中での分布がく(矩)形分布であるとみなして,不純物iの濃度の標準不確かさu(xi)
を見積もる。すなわち,
()
3
3
2
2
i
i
i
x
x
x
u
=
=
···································································· (C.2)
である。
全ての不純物iの濃度の不確かさを見積もり,それらを合成することによって,不純物の情報か
ら見積もられる不確かさu(xip) を見積もる。すなわち,
()
()
∑
=
i
ip
x
u
x
u
2
·································································· (C.3)
である。
47
K 0123:2018
なお,不純物の情報が表記されていない試薬の場合には,不純物の情報から見積もられる不確
かさu(xip) を0とする。
2.3) 原料の純度の不確かさの見積り 純度の情報から見積もられる不確かさu(xp) と不純物の情報か
ら見積もられる不確かさu(xip) を合成することによって,原料の純度の不確かさu(x) を見積もる。
すなわち,
()
()
()
ip
p
x
u
x
u
x
u
2
2
+
=
····························································· (C.4)
である。
なお,この純度の不確かさの見積り方法には,不確かさをより小さく見積もってしまうリスク
(例えば,原料純度安定性の不確かさが含まれないなど。)がある。そのため,原料の純度の不確
かさu(x) が1 %未満であった場合には,特別な理由がない限り,不確かさの値を1 %に切り上げ
たほうがよい。次にその例を示す。
例C-2 試薬に表記されている純度が99.0 %であり不純物の情報が記載されていない場合,純
度の情報から見積もられる不確かさは式(C.1)から,
()
2
10
58
.0
3
990
.0
1
3
1
−
×
=
−
=
−
=
x
x
u
p
である。不純物の情報が表記されていない試薬であるので,不純物の情報から見積も
られる不確かさを0とする。
()0
=
ip
x
u
この試薬の純度の標準不確かさu(x) は,見積もられたそれぞれの不確かさを合成す
ることによって求めることができる。すなわち,式(C.4)から,
()
()
()
(
)
%
58
.0
10
58
.0
0
10
58
.0
2
2
2
2
2
2
=
×
=
+
×
=
+
=
−
−
ip
p
x
u
x
u
x
u
である。得られた値は1 %未満であるため,1 %に切り上げる。
()
%
1
01
.0
=
=
∴x
u
例C-3 表C.1に示すような純度及び不純物の情報をもつ試薬Aの場合
表C.1−試薬Aの純度及びそれに含まれる不純物の情報
単位 %
成分
濃度
純度(GC-FID)
>97.0
水
< 1.0
酸(HClとして)
< 0.5
不純物B(GC-FID)
< 2.0
物質Aに含まれる不純物は水,酸及び不純物Bである。不純物Bの濃度の評価方法
は,物質Aの純度の評価方法と同じである。それゆえ,物質Aの純度の評価及び不純
物Bの濃度の評価は同時に行われているものとみなすことができる。水及び酸の濃度
はGC-FIDを用いて評価することができないので,これらの不純物に由来する不確か
さを見積もる必要がある。純度の情報から見積もられる不確かさは式(C.1)から,

48
K 0123:2018
()
2
10
73
.1
3
970
.0
1
3
1
−
×
=
−
=
−
=
x
x
u
p
である。一方,不純物の情報から見積もられる不確かさは式(C.2)及び式(C.3)から,
(
)
3
10
77
.5
3
010
.0
3
−
×
=
=
=
water
water
x
x
u
(
)
3
10
89
.2
3
005
.0
3
−
×
=
=
=
acid
acid
x
x
u
()
()
(
)(
)
3
5
2
3
2
3
2
10
45
.6
10
16
.4
10
89
.2
10
77
.5
−
−
−
−
×
=
×
=
×
+
×
=
=∑
i
ip
x
u
x
u
と見積もることができる。この試薬の純度の標準不確かさu(x) は,純度の情報から見
積もられる不確かさと不純物の情報から見積もられる不確かさとを合成することによ
って求めることができる。すなわち,式(C.4)から,
()
()
()
(
)(
)
%
85
.1
10
85
.1
10
41
.3
10
45
.6
10
73
.1
2
4
2
3
2
2
2
2
=
×
=
×
=
×
+
×
=
+
=
−
−
−
−
ip
p
x
u
x
u
x
u
である。
c) 純度又は不純物に関する情報がないもの 純度が確保された標準液が入手できない場合,不確かさの
見積りは,表C.2に示す評価方法を複数組み合わせるか,又は一つの方法を選択して行う。
表C.2−純度の評価方法
差数法
面積百分率法
修正面積百分率法
示差走査熱量計(DSC)等による凝固点降下法
定量NMRによる方法
純度を評価した場合には,例C-4のように採用した評価方法の選択理由又は基準を示す。
なお,b) 2.3) で述べたように,不確かさをより小さく見積もってしまうリスクがある。したがって,
適切な不確かさを含むように留意する。
例C-4 物質AはBという性質をもっており,不純物としてCがV %程度含まれると考えられ
る。また,使用する濃度はW %で目標とする不確かさはX %である。したがって,評価
方法として,D(表C.2の方法)を採用した。物質Aの純度はY %,その標準不確かさ
はZ %であった。
C.2.3 調製時に使用する溶媒について
標準液調製のために使用する溶媒は,分析種を不純物として含まないものを使用する。分析種が不純物
として含まれている溶媒を用いる場合,その不純物の濃度を推定し,その推定濃度に1 000を乗じた濃度
値以上の標準液調製に使用したほうがよい。
例C-5 濃度1 µg/Lのベンゼン標準液を調製する場合には,用いる溶媒に含まれるベンゼンの濃度は
1 ng/L未満のものが適している。
C.2.4 検量線を作成するための標準液の調製方法と不確かさの見積り手順
49
K 0123:2018
不確かさが付与された標準物質,又は,C.2.1で見積もった原料を用いて検量線作成用原液を調製し,こ
れを段階希釈して検量線を作成する方法での不確かさの見積り方法について次に示す。
a) 質量比混合法による標準液の調製 密栓可能な容器を密栓して,その質量(m0)をひょう量する。次
に,原料i(又は高濃度の標準液)を手早く入れて密栓し,その質量(mi)をひょう量する。最後に,
溶媒を手早く加えて密栓し,その質量(msolvent)をひょう量する。この溶液の濃度は,次の式(C.5)か
ら求める。
total
i
raw
i
i
m
C
m
C
,
×
∆
=
····································································· (C.5)
ここに,
Ci: 調製した標準液の濃度(kg/kg)
Δmi: 入れられた原料iの質量(kg)。(mi−m0)から求める
Craw,i: 質量分率で表された原料iの濃度(純度)
mtotal: 容器に入れた原料と溶媒の総質量(kg)。(msolvent−m0)
から求める
b) 原料の質量をひょう量し全量フラスコを用いた標準液の調製 全量フラスコにその容量の約80 %の
量の溶媒を入れて密栓し,その質量(m0)をひょう量する。次に,原料iを手早く加えて密栓し,そ
の質量(mi)をひょう量する。最後に,溶媒を標線まで加える。この溶液の濃度は,次の式(C.6)から
求める。
V
C
m
C
i
raw
i
i
,
×
∆
=
····································································· (C.6)
ここに,
Ci: 調製した標準液の濃度(kg/L)
Δmi: 入れられた原料iの質量(kg)。(mi−m0)から求める
Craw,i: 質量分率で表された原料iの濃度(純度)
V: 全量フラスコの容量(L)
c) 全量ピペットと全量フラスコを用いた標準液の希釈(調製) 全量フラスコにその容量の約80 %の量
の溶媒を入れる。次に,全量ピペットを用いて高濃度の標準液iをとり,全量フラスコに加える。最
後に,溶媒を標線まで加える。この溶液の濃度は,次の式(C.7)から求める。
V
C
V
C
i
raw
i
pipette
i
,
,×
=
·································································· (C.7)
ここに,
Ci: 調製した標準液の濃度(kg/L)
Vpipette,i: 標準液iをとった全量ピペットの容量(L)
Craw,i: 標準液iの濃度(kg/L)
V: 全量フラスコの容量(L)
C.3 検量線の作成及びその検量線由来の不確かさの見積りの例
質量分析計(MS)は,存在イオン種の物質量に比例した応答を示す。MSを使用する場合には,n個の
(異なる濃度の)標準液を用いた機器の校正,すなわち,検量線を作成しこれを用いて定量を行う。検量
線を作成する場合,特別な理由がない限り,用いる標準液の数nは3以上でなければならない。標準液の
数nが1又は2の場合には,次の方法で作成された検量線由来の不確かさを見積もることはできない。ま
た,検量線の作成に必要な測定回数は特別な理由がない限り,1濃度水準当たり最低3回,総測定回数は
20回以上であることが望ましい。
MSを用いて作成された検量線は一般的に直線である。MSでは絶対検量線法,標準添加法,内標準法の
三つの方法が用いられるが,これ以降の説明では絶対検量線法を用いて定量するときの検量線作成時の不
50
K 0123:2018
確かさを見積もる例を示す。標準添加法及び内標準法へ適用する場合は,表C.3を参考に読替えを行わな
ければならない。
i番目の標準液の濃度に関するパラメーターをxi,i番目の標準液に対するMSの応答に関するパラメー
ターをyiとすると,作成された検量線の式は式(C.8)で示される。
bx
a
y
+
=
············································································· (C.8)
ここに,
a: 作成された検量線のy切片
b: 検量線の傾き
表C.3−検量線法を用いる代表的な方法と各方法におけるパラメーターの意味
パラメーターxi
パラメーターyi
絶対検量線法
標準液の濃度
MSの応答値
標準添加法
試料に添加された標準液の量
MSの応答値
内標準法
内標準物質1単位濃度当たりの
被検成分標準液の濃度
被検成分と内標準物質の応答比
標準液を測定し,得られた結果を用いて濃度と応答値との真の関係式を推定する,すなわち,このaと
bとの誤差が最小になる組合せを推定する。この組合せを推定するために,最小二乗法が使用される。今
まで述べてきたように,標準液の濃度及びMSの応答値はそれぞれ不確かさをもっている。この場合の完
全な最小二乗法の取扱いは,非常に複雑な表現が必要となる。それゆえ,特別な理由がない限りは,不確
かさが含まれない条件,すなわち,一般的によく知られている最小二乗法を用いて,検量線の式を推定す
る。
最小二乗法を用いてa及びbを求めると,
∑
∑
∑
∑
∑
∑
=
=
=
=
=
=
−
−
=
n
i
n
i
i
i
n
i
i
i
n
i
i
n
i
i
n
i
i
x
x
n
y
x
x
y
x
a
1
2
1
2
1
1
1
1
2
··················································· (C.9)
∑
∑
∑
∑
∑
=
=
=
=
=
−
−
=
n
i
n
i
i
i
n
i
i
n
i
i
n
i
i
i
x
x
n
y
x
y
x
n
b
1
2
1
2
1
1
1
························································· (C.10)
となる。a及びbそれぞれの標準誤差をsn(a),及びsn(b) とすると,
()
(
)
−
−
−
−
−
−
=
∑
∑
∑
∑
∑
∑
∑
∑
∑
∑
=
=
=
=
=
=
=
=
=
=
2
1
1
2
1
2
2
1
1
2
2
1
1
1
2
1
1
2
2
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
i
n
i
i
n
i
i
n
x
x
n
n
n
x
x
x
n
y
x
y
x
n
y
y
n
a
s
········· (C.11)
51
K 0123:2018
()
(
)
−
−
−
−
−
−
=
∑
∑
∑
∑
∑
∑
∑
∑
∑
=
=
=
=
=
=
=
=
=
2
1
1
2
2
1
1
2
2
1
1
1
2
1
1
2
2
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
n
i
i
i
n
i
i
n
i
i
n
x
x
n
n
x
x
n
y
x
y
x
n
y
y
n
b
s
···················· (C.12)
となる。
切片aの標準誤差に由来する標準不確かさu(a) は,く(矩)形分布を仮定し切片aの標準誤差を3で
除することによって推定することができる。すなわち,
()
()
3
a
s
a
u
n
=
·········································································· (C.13)
である。傾きbの標準誤差に由来する標準不確かさu(b) も,く(矩)形分布を仮定し傾きbの標準誤差を
3で除することによって推定することができる。すなわち,
()
()
3
b
s
b
u
n
=
·········································································· (C.14)
である。
C.4 未知試料の測定,分析値及びその不確かさの見積り例
未知試料の測定を行い,C.3で作成した検量線から未知試料の濃度値とその不確かさを見積もる。これ
にC.2で見積もった標準液の不確かさとC.3で求めた標準液測定時の不確かさを合成して,分析値の不確
かさを見積もる方法を示す。
未知試料に対するMSの応答y' が得られたとき,検量線式(C.8)を用いて未知試料の濃度x' を推定すると,
b
a
y
x
−′
=
′
··········································································· (C.15)
となる。このときのx' の標準不確かさu(x') を見積もるためには,式(C.16)を用いる。
()
()
()
()
()
()
()
b
u
b
a
y
a
u
b
y
u
b
b
u
b
x
a
u
a
x
y
u
y
x
x
u
2
2
2
2
2
2
2
2
2
2
2
2
2
1
1
−
′
−
+
−
+
′
=
∂′
∂
+
∂′
∂
+
′
′
∂
′
∂
=
′
··················· (C.16)
また,x' の相対標準不確かさurel(x') を見積もるためには,式(C.17)を用いる。
()
()
x
x
u
x
urel
′
=
′
······································································· (C.17)
一般的によく知られている最小二乗法では,検量線の作成のために使用された標準液の濃度の不確かさ
と標準液を測定したときの不確かさ(検量線作成時のMSの応答値の不確かさ)を,ゼロと仮定している。
そのため,この検量線を用いて得られた定量値の不確かさには,標準液の濃度の不確かさとその標準液を
測定したときの不確かさが含まれていない。簡易的に標準液の濃度の不確かさを推定する場合には,次の
方法が用いられる。検量線の作成に使用された標準液全てについて,濃度値Cn及びその標準不確かさu(Cn)
を用いて濃度値の相対標準不確かさurel(Cn) を求める。次に,得られた各urel(Cn) の中で最も大きな値を,
52
K 0123:2018
検量線の作成のために使用された標準液の濃度の相対標準不確かさurel(C) であるとみなす。検量線の作成
では標準液ごとに繰返し測定を行ってMSの応答値を得ているので,それぞれについて応答値の平均値yn
及びその標準不確かさu(yn) を用いて応答値の相対標準不確かさurel(yn) を求める。得られた各urel(yn) の中
で最も大きな値を,標準液を測定したときの相対標準不確かさurel(y) であるとみなす。
検量線を用いて未知試料の濃度x' を推定したときの濃度値の相対標準不確かさurelは,式(C.17)で求めた
x' の相対標準不確かさurel(x') 及び検量線の作成のために使用された標準液の濃度の相対標準不確かさ
urel(C) 並びに標準液を測定したときの相対標準不確かさ(検量線作成時の分析機器応答値の相対標準不確
かさ)urel(y) を合成したものであるので,
()
()
()
y
u
C
u
x
u
u
rel
rel
rel
rel
2
2
2
+
+
′
=
··············································· (C.18)
となる。これを絶対値に変換することによって,未知試料の濃度の合成標準不確かさuを求めることがで
きる。
rel
u
x
u
′
=
············································································ (C.19)
参考文献
・ 実験精度と誤差−測定の確からしさとはなにか N. C. バーフォード著 酒井英行訳 丸善(1997)
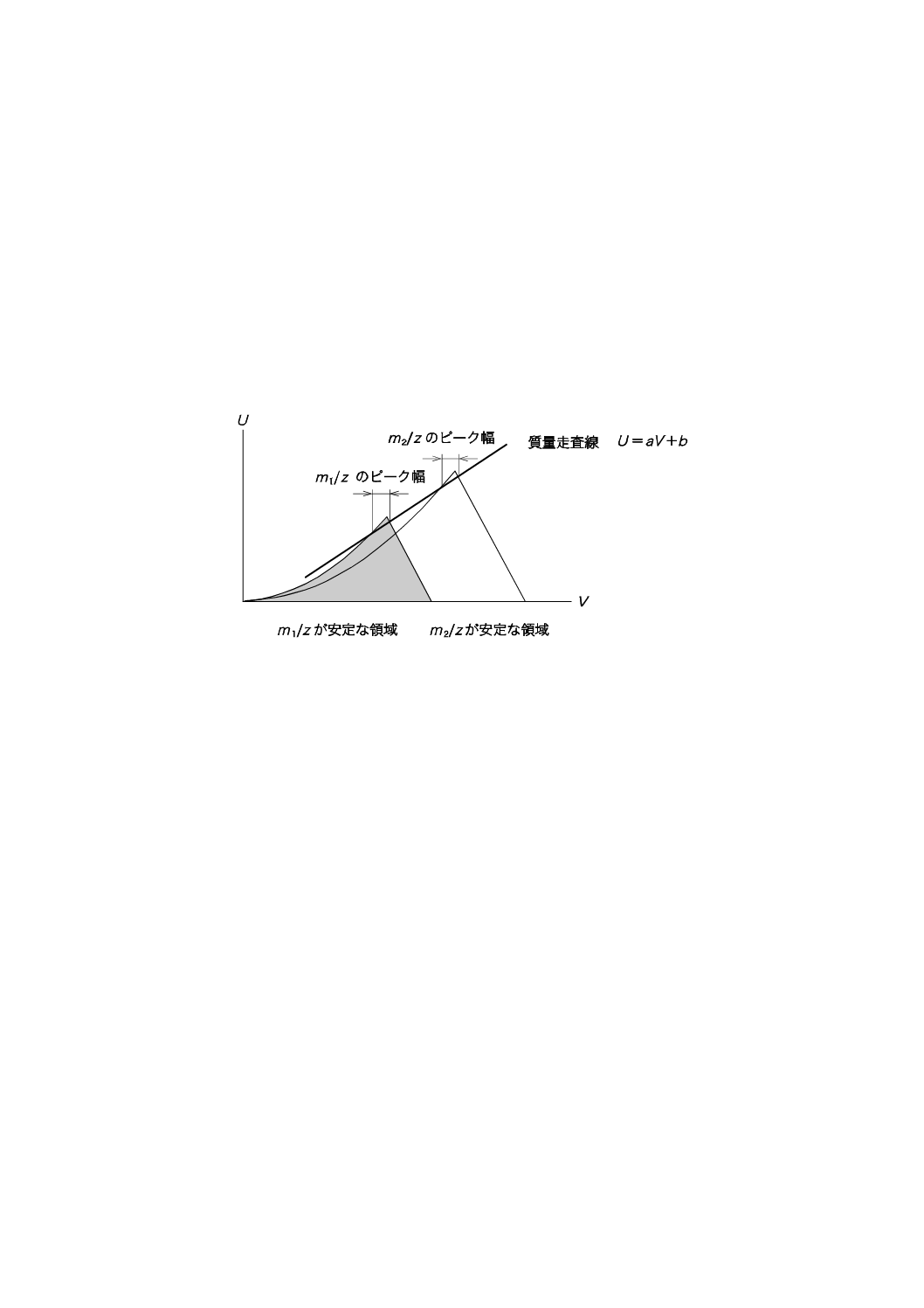
53
K 0123:2018
附属書D
(参考)
四重極形
四重極形質量分析計の分解能(質量スペクトルのピーク幅)四重極形質量分析計では,図D.1に示すよ
うな安定領域のイオンが検出器に導かれる。ここで質量走査線:U=aV+bの上部がイオンとして検出さ
れる。しかも安定領域と質量走査線との接線がピークの幅となり,分解能を示す。a,bの値を変更するこ
とで分解能は変化する。a,bの値によっては低質量域と高質量域との分解能を同一にすることが可能であ
る。
図D.1−安定領域の模式図
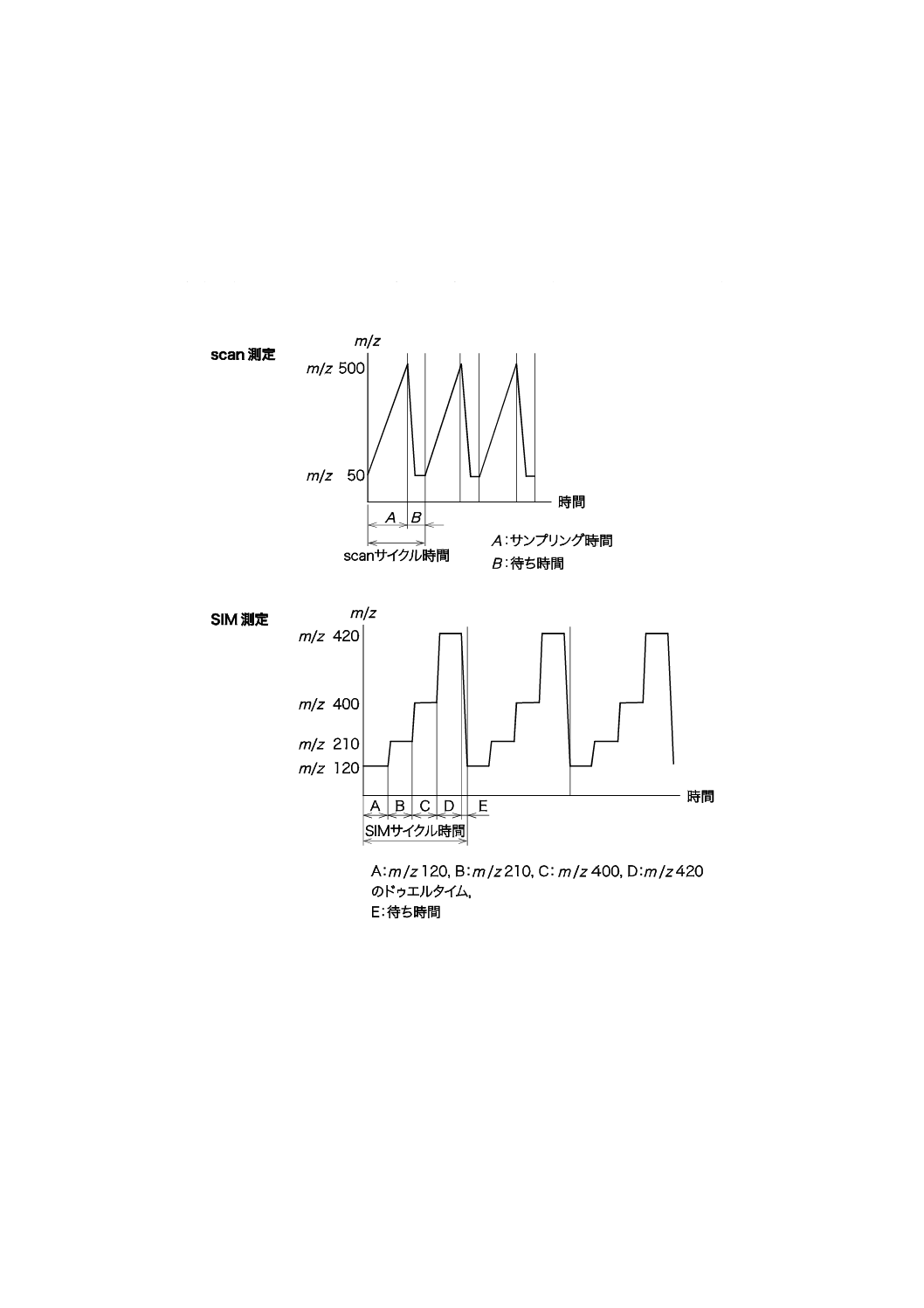
54
K 0123:2018
附属書E
(規定)
サイクルタイム,サンプリング時間及びドゥエルタイム
四重極形又は磁場形のGC/MSによる測定には,全イオンモニタリング(TIM)による方法と選択イオン
モニタリング(SIM)法とがある。この測定法の比較を,図E.1(上段:TIM,下段:SIM)に示す。
図E.1−測定方法の概念図[上段:TIM(四重極形のscanによる測定の場合),下段:SIM]
TIM法による測定の場合,scanサイクル時間は走査に要するサンプリング時間(A)と待ち時間(B)と
の合計となる。SIM法の場合,ドゥエルタイムは選択イオンの1測定当たりの取込み時間(A,B,C,D)
であり,SIMサイクル時間はそれらの合計と待ち時間(E)との合算となる。
注記 ドゥエルタイムは各イオンへの変更時間を除いた時間とすることもある。
55
K 0123:2018
参考文献 JIS K 0088 排ガス中のベンゼン分析方法
JIS K 0124 高速液体クロマトグラフィー通則
JIS K 0125 用水・排水中の揮発性有機化合物試験方法
JIS K 0128 用水・排水中の農薬試験方法
JIS K 0136 高速液体クロマトグラフィー質量分析通則
JIS K 0311 排ガス中のダイオキシン類の測定方法
JIS K 0312 工業用水・工場排水中のダイオキシン類の測定方法
JIS K 0450-10-10 工業用水・工場排水中のビスフェノールA試験方法
JIS K 0450-20-10 工業用水・工場排水中のアルキルフェノール類試験方法
JIS K 0450-30-10 工業用水・工場排水中のフタル酸エステル類試験方法
JIS K 0450-40-10 用水・排水中のアジピン酸ビス(2-エチルヘキシル)試験方法
JIS K 0450-50-10 用水・排水中のベンゾフェノン試験方法
JIS S 3200-7 水道用器具−浸出性能試験方法