K 0102:2019
(1)
追補1のまえがき
このJIS K 0102の追補1は,工業標準化法に基づき,日本工業標準調査会の審議を経て,経済産業大臣
がJIS K 0102:2016を改正した内容だけを示すものである。
JIS K 0102:2016は,この追補1の内容の改正がされ,JIS K 0102:2019となる。
K 0102:2019
K 0102:2019
(2)
目 次
ページ
追補1の序文 ······················································································································· 1
2. 共通事項 ························································································································ 1
7. 温度 ······························································································································ 1
12. pH ······························································································································ 2
13. 電気伝導率 ··················································································································· 2
28.1 フェノール類 ··············································································································· 2
31. 農薬 ···························································································································· 9
32. 溶存酸素 ······················································································································ 9
33. 残留塩素 ······················································································································ 9
34. ふっ素化合物 ··············································································································· 18
35. 塩化物イオン(Cl−) ····································································································· 25
38. シアン化合物 ··············································································································· 26
42. アンモニウムイオン(NH4+) ························································································· 36
43. 亜硝酸イオン(NO2−)及び硝酸イオン(NO3−) ································································ 47
44. 有機体窒素 ·················································································································· 48
45. 全窒素 ························································································································ 48
46. りん化合物及び全りん ··································································································· 57
47. ほう素(B) ················································································································ 68
48. ナトリウム(Na) ········································································································· 69
49. カリウム(K) ············································································································· 74
50. カルシウム(Ca) ········································································································· 76
51. マグネシウム(Mg) ····································································································· 82
52. 銅(Cu)····················································································································· 84
53. 亜鉛(Zn) ·················································································································· 93
54. 鉛(Pb) ····················································································································· 94
55. カドミウム(Cd)········································································································· 94
56. マンガン(Mn) ··········································································································· 95
57. 鉄(Fe) ····················································································································· 95
58. アルミニウム(Al) ······································································································ 95
59. ニッケル(Ni) ············································································································ 96
60. コバルト(Co) ············································································································ 96
61. ひ素(As) ·················································································································· 97
62. アンチモン(Sb) ········································································································· 97
63. すず(Sn) ·················································································································· 97
64. ビスマス(Bi)············································································································· 98
K 0102:2019
(3)
ページ
65. クロム(Cr) ··············································································································· 98
66. 水銀(Hg)················································································································· 102
67. セレン(Se) ·············································································································· 108
68. モリブデン(Mo) ······································································································· 108
69. タングステン(W) ····································································································· 109
70. バナジウム(V) ········································································································· 109
73. ウラン(U) ··············································································································· 109
74. ベリリウム(Be) ········································································································ 110
附属書1(参考)補足 ·········································································································· 110
附属書2(参考)JISと対応する国際規格との対比表 ································································· 117
日本工業規格 JIS
K 0102:2019
工場排水試験方法
(追補1)
Testing methods for industrial wastewater
(Amendment 1)
追補1の序文
この追補は,環境分析における近年の省力化,低コスト化及び低環境負荷のための新技術並びに各種の
測定機器を取り入れることによって,規格利用者の利便性の向上を図ることを目的としている。
導入した測定方法は,次のa)〜f)であり,これに伴い目次に示した項目を変更した。
a) フェノール類,ふっ素化合物,全シアン及びアンモニウムイオンへの少量の試料で蒸留操作を行う小
型蒸留操作の導入並びに全窒素及び全りんの加熱分解前処理操作の試料量及び試薬量の少量化
b) 誘導結合プラズマ発光分光分析法を用いたナトリウム及びカリウムの測定
c) ガスクロマトグラフィー質量分析法を用いたアルキル水銀の測定
d) 液体クロマトグラフィー誘導結合プラズマ質量分析法を用いた六価クロムの測定
e) 附属書のベリリウムの測定方法の本体への移行
f)
残留塩素測定における誤検出抑制手法の追加
JIS K 0102:2016を,次のように改正する。
2.(共通事項)o) 2)の“標準液は,各試験項目で調製方法を規定するもののほか,国家計量標準(計量法
第134条)に規定するトレーサビリティが確保されたもの又はそれを一定濃度に薄めたものを用いる(3)。”
を,“標準液は,各試験項目で調製方法を規定するもののほか,国家計量標準へのトレーサビリティが確保
されたもの又はそれを一定濃度に薄めたものを用いる(3)。”に置き換える。
2.(共通事項)o) 2)の参考を,次の文に置き換える。
参考 トレーサビリティが確保された標準液としては,計量標準供給制度(Japan Calibration Service
System)によるJCSSマークを付けたものがある。
2.(共通事項)o) 3)の“標準液の濃度は,1 mL中の質量(mg/mL,μg/mL又はng/mL)で表す。ただし,
残留塩素測定の塩素標準液,イオン電極法及びフレーム光度法に用いる標準液の濃度は,1 L中の質量
(mg/L)で表す。”を,“標準液の濃度は,1 mL又は1 L中の質量(mg/mL,μg/mL,ng/mL,g/L,mg/L
又はμg/L)で表す。”に置き換える。
7.(温度)の7.1(気温) a) 1)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
2
K 0102:2019
7.(温度)の7.2(水温) a) 1)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
12.(pH)の12.1(ガラス電極法) c) 2)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
12.(pH)の12.1(ガラス電極法)の注(3)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
13.(電気伝導率)のb) 2)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
28.(フェノール類)の28.1(フェノール類)の全文を,次に置き換える。
28.1 フェノール類 フェノール類の試験は,試料を28.1.1に示す前処理(蒸留法)後,28.1.2に示す4-
アミノアンチピリン吸光光度法又は28.1.3の4-アミノアンチピリン発色フローインジェクション分析法
(FIA法)を適用し,フェノール標準液を用いて定量した値で表す,又は流れの中で連続的に蒸留前処理
及び4-アミノアンチピリン発色吸光光度法を行う28.1.3の連続流れ分析法(CFA法)を適用する。
フェノール類は,フェノール分解菌によって分解されやすい。酸化性物質,還元性物質,アルカリなど
の影響も受けやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合は,3.3によって保存し,
できるだけ早く試験する。
この試験で,対象となるフェノール類は,ベンゼン及びその類似体のヒドロキシ誘導体で,規定の方法
によって4-アミノアンチピリンと反応して着色化合物を生成するものをいう。
なお,28.1.1及び28.1.2に示す方法は,1990年に第2版として発行されたISO 6439,流れ分析法は,1999
年に第1版として発行されたISO 14402との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6439:1990,Water quality−Determination of phenol index−4-Aminoantipyrine spectrometric
methods after distillation(MOD)
ISO 14402:1999,Water quality−Determination of phenol index by flow analysis (FIA and CFA)
(MOD)
この試験で求める“フェノール類”は,ISO 6439によって定義される“フェノール指標”に相当する。
28.1.1 前処理(蒸留法) りん酸酸性(pH約4)で,硫酸銅(II)の存在の下で加熱蒸留してフェノール
類を留出分離する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。ほうけい酸ガラス瓶に保存する。
2) りん酸(1+9) JIS K 9005に規定するりん酸を用いて調製する。
3) 硫酸銅(II)溶液 JIS K 8983に規定する硫酸銅(II)五水和物10 gを水に溶かして100 mLとする。
4) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
b) 装置 装置は,次による。
1) 蒸留装置 38.1.1.2 b) 1)による。
c) 蒸留操作 蒸留操作は,次による。ただし,試料に色又は濁りがなく,4-アミノアンチピリン吸光光
度法の妨害となる物質を含まない場合には,前処理(蒸留法)を省略できる。この場合は,3.3の試料
の保存処理は行わず,試料採取後,直ちに試験する。
3
K 0102:2019
なお,試料に一定量のフェノール標準液を添加して,備考2.又は備考3.の蒸留操作による回収率試
験を行い,回収率が80〜120 %であることを確認した場合は,備考2.又は備考3.によってもよい。備
考2.又は備考3.による留出液は,28.1.2.1,28.1.2.3,及び28.1.3のFIA法に適用する。
1) 試料250 mL又は試料中のフェノール濃度が50 mg/L以上の場合には,その適量をとり,水を加え
て250 mLにしたものを蒸留フラスコ500 mLにとり,メチルオレンジ溶液(1 g/L)5〜7滴を加え,
メチルオレンジが変色するまでりん酸(1+9)を加えてpHを約4にした後,硫酸銅(II)溶液2.5 mL
を加える。ただし,試料中のフェノール濃度が25 µg/L以下の場合は,試料500 mLを蒸留フラスコ
1 000 mLにとり,硫酸銅(II)溶液5 mLを加え,2)の受器の容量,3)の留出液量,4)の水の量及び
全留出液量を2倍とする。試料の保存にりん酸及び硫酸銅(II)五水和物の添加を行った場合は,
これらの添加は省略する。
2) 沸騰石(粒径2〜3 mm)を加えた後,蒸留フラスコを蒸留装置に取り付け,受器に容量250 mLの
メスシリンダー(有栓形)を用いて蒸留する。
3) メスシリンダー中の留出液が225 mLになったとき,一旦加熱を止める。
4) 蒸留フラスコ中の試料の沸騰がやんだ後,蒸留フラスコに水25 mLを加え,再び蒸留を続けて更に
25 mLを留出させ,全留出液量を250 mLとする。留出液が白濁している場合は,留出液に再びり
ん酸(1+9)を加えてpHを約4とし,硫酸銅(II)溶液2.5 mLを加え,蒸留操作を繰り返す。再
蒸留を行っても白濁が消えない場合には,備考1. 4)によって処理する。
備考 1. 試料の保存においてりん酸及び硫酸銅(II)五水和物を添加することによって,フェノール
類の生物化学的分解が抑制される。4-アミノアンチピリン吸光光度法の試験では,酸化性物
質,還元性物質,金属イオン,芳香族アミン類,油分,タール類などは妨害となる。大部分
は蒸留操作で取り除くことができるが,酸化性物質,還元性物質,硫黄化合物,油分及びタ
ール類が試料中に含まれる場合には,次のように処理する。
1) 酸化性物質 残留塩素のような酸化性物質が含まれている場合,又は試料に酸性でよう
化カリウムを加えるとよう素が遊離する場合は,試料採取直後に,JIS K 9502に規定す
るL(+)-アスコルビン酸の小過剰又はJIS K 8978に規定する硫酸鉄(II)七水和物の小過
剰量を加える。試料の保存には,これにりん酸を加えてpH約4とし,試料1 Lにつき
JIS K 8983に規定する硫酸銅(II)五水和物1 gを加える。
2) 還元性物質 還元性物質が存在する場合には,JIS K 8801に規定するヘキサシアノ鉄
(III)酸カリウムを過剰に加える。試料の保存には,JIS K 9005に規定するりん酸を加
えてpH約4とし,試料1 LにつきJIS K 8983に規定する硫酸銅(II)五水和物1 gを加
える。
3) 硫黄化合物 硫化水素及び亜硫酸イオンが含まれている場合には,試料採取直後にりん
酸を加えてpH約4とし,注意して試料に空気を吹き込むか,又はかき混ぜて,硫化水
素及び二酸化硫黄を追い出した後,硫酸銅(II)溶液を試料1 Lにつき10 mL加える。
又は,硫酸銅(II)溶液を過剰に加えて硫化銅(I)の沈殿とした後,りん酸を加えてpH
約4とする。
4) 油分及びタール類 油分及びタール類が含まれている場合には,次のいずれかの方法に
よるとよい。
4.1) 試料採取直後に硫酸銅(II)溶液を加えずに,水酸化ナトリウム溶液(100 g/L)[19. a)
2)による。]を加えてpH12〜12.5とし,分液漏斗に移してJIS K 8322に規定するクロ
4
K 0102:2019
ロホルムを加え,油分及びタール類を抽出して,クロロホルム層を捨てる。水層は,
沸騰水浴上で加熱して,残留するクロロホルムを除去する。試料の保存には,これに,
りん酸を加えてpH約4とし,試料1 Lにつき硫酸銅(II)五水和物1 gを加える。
4.2) 蒸留に当たって,試料250 mLをとり,メチルオレンジ溶液(1 g/L)数滴を加え,硫酸
(0.5 mol/L)(JIS K 8951に規定する硫酸を用いて調製する。)で酸性とする。分液漏
斗に移し,JIS K 8150に規定する塩化ナトリウム75 gを加える。クロロホルムを,最
初は20 mLで,以後12.5 mLずつで4回抽出分離する。クロロホルム層を集めて別の
分液漏斗に入れ,水酸化ナトリウム溶液(100 g/L)[19. a) 2)による。]を,最初は2.0 mL,
以後1.5 mLずつで2回逆抽出する。水層を集め,水浴上でクロロホルムがなくなるま
で加熱する。冷却し,水で250 mLとしてc)の蒸留操作を行う。
5) アミン類 特定の反応条件下では,ある種のアミン類はフェノールとして測定される。
この妨害は,pH0.5未満で蒸留することによって最小限に抑えられる。
備考 2. 蒸留フラスコに100 mL又は200 mLのものを用いてもよい。この場合の蒸留操作は,次によ
る。
なお,蒸留フラスコ200 mLを用いる場合は,試料量,硫酸銅(II)溶液の添加量,受器の
容量及び留出液量はそれぞれ2倍とする。
1) 蒸留フラスコ100 mLに試料50 mL及び水5 mLをとり,メチルオレンジ溶液(1 g/L)1,
2滴を加え,メチルオレンジが変色するまでりん酸(1+9)を加えてpHを約4にした後,
硫酸銅(II)溶液0.5 mLを加える。
2) 沸騰石(粒径2〜3 mm)を加えた後,蒸留フラスコを蒸留装置に取り付ける。受器に容
量50 mLのメスシリンダー(有栓形)などを用い,蒸留する。
3) 留出液が受器の容量(50 mL)になるまで蒸留を続ける。
3. 小型蒸留装置を用いる蒸留操作は,次による。小型蒸留装置の例を図28.1に示す。
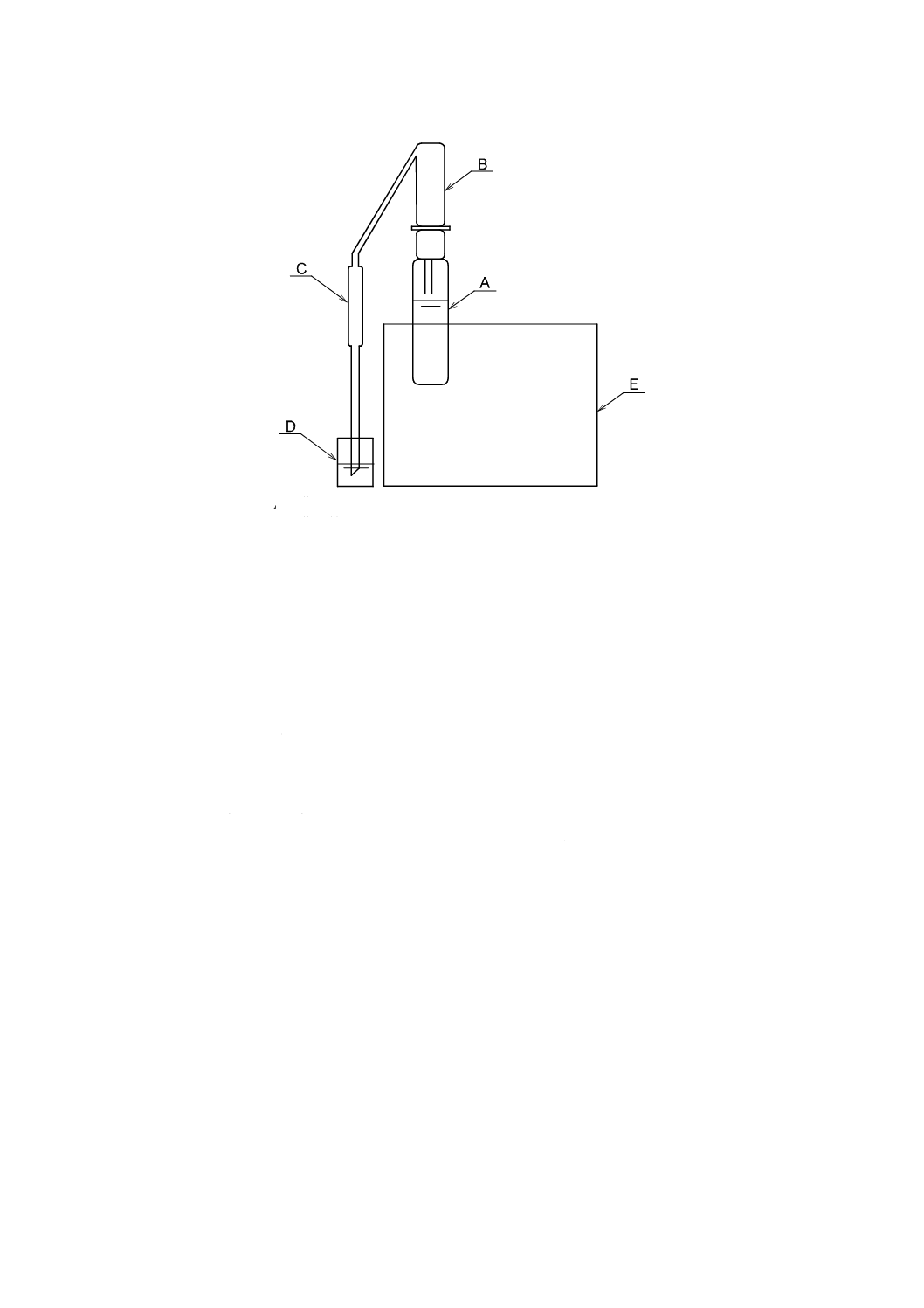
5
K 0102:2019
A: 蒸留容器(耐熱性のガラス容器で容量50〜80 mLのもの)
B: 蒸留管(気液分離が可能なもの)
C: 冷却器
D: 受器(容量50 mLの有栓形メスシリンダーなど)
E: 加熱器(150〜210 ℃の設定が可能なもの)
図28.1 小型蒸留装置の例
1) 試料50 mLを蒸留容器にとり,メチルオレンジ溶液(1 g/L)1,2滴を加え,メチルオレ
ンジが変色するまで,りん酸(1+9)を加えてpHを約4にした後,硫酸銅(II)溶液
0.5 mLを加える。
2) 沸騰石(粒径2〜3 mm)を加えた後,蒸留容器に蒸留管を取り付け,これを加熱器へセ
ットする。受器に容量50 mLのメスシリンダー(有栓形)などを用いて蒸留する。
3) 試料の体積の90 %が留出したら,蒸留容器を加熱器から外し,冷却器内を水で洗い,洗
液も受器に加えた後,更に水を50 mLの標線まで加える。
28.1.2 4-アミノアンチピリン吸光光度法 試料のpHを約10に調節し,これに4-アミノアンチピリン(4-
アミノ-1,2-ジヒドロ-1,5-ジメチル-2-フェニル-3H-ピラゾール-3-オン)溶液とヘキサシアノ鉄(III)酸カリ
ウム溶液とを加えて,生成する赤い色のアンチピリン色素の吸光度を波長510 nm付近で測定し,フェノ
ール標準液による検量線によってフェノール類を定量する(直接法)。発色の程度が弱い場合は,発色後の
溶液をクロロホルム又は安息香酸メチルで抽出する溶媒抽出法によって定量する(溶媒抽出法)。又は,試
料中のフェノール類を同様な原理で発色後,疎水性のカラムによる固相抽出法によって定量する(固相抽
出法)。
これらの方法では,フェノール(C6H5OH)のほかo-,m-位置に置換基があるフェノール誘導体及び多
環式化合物にヒドロキシル基が置換したものも4-アミノアンチピリンと反応してアンチピリン色素を生成
して定量される。p-位置に置換基があるフェノール誘導体は,4-アミノアンチピリンと反応しにくいため,
ほとんど発色しない。アンチピリン色素の発色の強さは,置換基の種類,位置,数などによって差がある。
定量範囲:直接法 C6H5OH 50〜500 μg,繰返し精度:3〜10 %
溶媒抽出法 C6H5OH 2.5〜50 μg,繰返し精度:3〜10 %
6
K 0102:2019
固相抽出法 C6H5OH 0.2〜15 μg,繰返し精度:3〜10 %
28.1.2.1 直接法 水溶液中で発色生成させた赤い色のアンチピリン色素の発色の強さを測定して定量す
る。
a) 試薬 試薬は,次による。
1) 水 28.1.1 a) 1)による。
2) 塩化アンモニウム-アンモニア緩衝液(pH10) JIS K 8116に規定する塩化アンモニウム67.5 gをJIS
K 8085に規定するアンモニア水570 mLに溶かし,水で1 Lとする。
3) ヘキサシアノ鉄(III)酸カリウム溶液 JIS K 8801に規定するヘキサシアノ鉄(III)酸カリウムの
大きな結晶9 gをとり,少量の水で表面を洗った後,水に溶かして100 mLとし,必要がある場合,
ろ過する。1週間ごとに調製するが,1週間以内でも色が暗い赤に変わったものは使用しない。
4) 4-アミノアンチピリン溶液(20 g/L) JIS K 8048に規定する4-アミノアンチピリン2.0 gを水に溶
かして100 mLとする。使用時に調製する。
5) フェノール標準液(C6H5OH 1 mg/mL) JIS K 8798に規定するフェノール1.00 gを水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加え,0〜10 ℃の暗所に保存する。
6) フェノール標準液(C6H5OH 10 μg/mL) フェノール標準液(C6H5OH 1 mg/mL)10 mLを全量フラ
スコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 28.1.1の前処理(蒸留法)を行った試料,又は前処理(蒸留法)を必要としない試料の適量(C6H5OH
として50〜500 μgを含む。)をメスシリンダー(有栓形)100 mLにとり,水を100 mLの標線まで
加える。ただし,備考2.及び備考3.の操作で,留出液の全量が50 mLの場合には,試料の適量(C6H5OH
として25〜250 μgを含む。)をメスシリンダー(有栓形)50 mLにとり,水を50 mLの標線まで加
える。メスシリンダー(有栓形)50 mLを用いた場合の操作では,2)及び3)の試薬の添加量を1/2
にし,4)の空試験を水50 mLで行う。
2) 次に,塩化アンモニウム-アンモニア緩衝液(pH10)3 mLを加えて振り混ぜ,pH10±0.2に調節す
る。
3) 4-アミノアンチピリン溶液(20 g/L)2 mLを加えて振り混ぜ,ヘキサシアノ鉄(III)酸カリウム溶
液2 mLを加えて十分に振り混ぜた後,約3分間放置する。
4) 別に,空試験として水100 mLについて2)及び3)の操作を行う。
5) 空試験の溶液を対照液として波長510 nm付近の吸光度を測定する。
6) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH mg/L)を算出
する。
d) 検量線 検量線の作成は,次による。
1) フェノール標準液(C6H5OH 10 μg/mL)5〜50 mLをメスシリンダー(有栓形)100 mLに段階的にと
り,水を100 mLの標線まで加える。c) 1)での発色前の試料量が50 mLの場合は,フェノール標準
液(C6H5OH 10 μg/mL)2.5〜25 mLをメスシリンダー(有栓形)50 mLに段階的にとり,水を50 mL
の標線まで加える。
2) c)の2)〜5)の操作を行ってフェノールの量と吸光度との関係線を作成する。
28.1.2.2 溶媒抽出法 水溶液中で発色生成させたアンチピリン色素の発色が弱い場合は,これをクロロホ
7
K 0102:2019
ルム又は安息香酸メチルに抽出して,有機層の吸光度を測定して定量する。ただし,備考2.及び備考3.に
よる留出液には適用しない。
a) 試薬 試薬は,次による。
1) 水 28.1.1 a) 1)による。
2) 塩化アンモニウム-アンモニア緩衝液(pH10) 28.1.2.1 a) 2)による。
3) ヘキサシアノ鉄(III)酸カリウム溶液 28.1.2.1 a) 3)による。
4) 4-アミノアンチピリン溶液(20 g/L) 28.1.2.1 a) 4)による。
5) フェノール標準液(C6H5OH 1 μg/mL) 28.1.2.1 a) 6)のフェノール標準液(C6H5OH 10 μg/mL)50 mL
を全量フラスコ500 mLにとり,水を標線まで加える。使用時に調製する。
6) クロロホルム JIS K 8322に規定するもの。
7) 安息香酸メチル
8) 硫酸ナトリウム JIS K 8987に規定するもの。
b) 器具及び装置 器具及び装置は,次による。
1) 光度計 分光光度計又は光電光度計
2) 分液漏斗 200 mL
c) 操作 操作は,次による。
1) 28.1.2.1 c) 3)で得た発色溶液の全量を分液漏斗200 mLに移し,クロロホルム又は安息香酸メチル10
mLを加えて1〜2分間激しく振り混ぜた後放置する。ただし,28.1.2.1 c) 1)で試料量50 mLで操作
したものには適用しない。
2) クロロホルム層又は安息香酸メチル層を分離し,乾いたろ紙でろ過するか,又はビーカーに移した
後,硫酸ナトリウム約1 gを加えて脱水する。
3) 別に,空試験として水100 mLについて28.1.2.1 c)の2)及び3)の操作を行った後,1)及び2)の操作を
行う。
4) 空試験のクロロホルム層又は安息香酸メチル層を対照液とし,2)のクロロホルム層の波長460 nm付
近の吸光度又は安息香酸メチル層の波長465 nm付近の吸光度を測定する。
5) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH mg/L)を算出
する。
d) 検量線 検量線の作成は,次による。
1) フェノール標準液(C6H5OH 1 μg/mL)2.5〜50 mLをメスシリンダー(有栓形)100 mLに段階的に
とり,水を100 mLの標線まで加える。
2) 28.1.2.1 c)の2)及び3),並びにc)の1)〜4)の操作を行い,フェノールの量と吸光度との関係線を作成
する。
備考 4. 28.1.1 c)で試料500 mLを用いた場合には,次の操作によって定量してもよい。
1) 前処理で得た留出液500 mLを分液漏斗1 000 mLにとり,塩化アンモニウム-アンモニア
緩衝液(pH10)10 mLを加えて振り混ぜ,pH 10±0.2とする。
2) 4-アミノアンチピリン溶液(20 g/L)3 mL,ヘキサシアノ鉄(III)酸カリウム溶液3 mL
を加え,十分に振り混ぜ,約3分間放置した後,クロロホルム20 mLを加えて約1分間
激しく振り混ぜる。
3) クロロホルム層を分離し,乾いたろ紙でろ過するか,ビーカーに移した後,硫酸ナトリ
ウム約1 gを加えて脱水する。
8
K 0102:2019
4) 別に,空試験として水500 mLについて,1)〜3)の操作を行う。
5) 空試験のクロロホルム層を対照液とし,3)のクロロホルム層の波長460 nm付近の吸光度
を測定する。
6) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH
mg/L)を算出する。検量線は,フェノール標準液(C6H5OH 1 μg/mL)2.5〜50 mLを分液
漏斗1 000 mLに段階的にとり,水を加えて500 mLとし,以下,試料の場合と同様に操
作して作成したものを用いる。
28.1.2.3 固相抽出法 水溶液中で発色生成させたアンチピリン色素を疎水性の固相カラムに捕集した後,
アセトニトリルで溶出させて,アセトニトリル層の吸光度を測定して定量する。
a) 試薬 試薬は,次による。
1) 水 28.1.1 a) 1)による。
2) 塩化アンモニウム-アンモニア緩衝液(pH10) 28.1.2.1 a) 2)による。
3) ヘキサシアノ鉄(III)酸カリウム溶液 28.1.2.1 a) 3)による。
4) 4-アミノアンチピリン溶液(20 g/L) 28.1.2.1 a) 4)による。
5) フェノール標準液(C6H5OH 1 μg/mL) 28.1.2.2 a) 5)による。
6) フェノール標準液(C6H5OH 0.1 μg/mL) 28.1.2.1 a) 6)のフェノール標準液(C6H5OH 10 μg/mL)5 mL
を全量フラスコ500 mLにとり,水を標線まで加える。使用時に調製する。
7) アセトニトリル JIS K 8032に規定するもの。
b) 器具及び装置 器具及び装置は,次による。
1) 光度計 分光光度計又は光電光度計
2) 固相カラム 市販のジビニルベンゼン共重合体充塡カラム又はODSカラム。
c) 操作 操作は,次による。
1) 28.1.1の前処理を行った試料,又は前処理を必要としない試料の適量(C6H5OHとして0.2〜15 μg
を含む。)をメスシリンダー(有栓形)50 mLにとり,水を50 mLの標線まで加える。
2) 塩化アンモニウム-アンモニア緩衝液(pH10)1.5 mLを加えて振り混ぜ,pH10±0.2に調節する。
3) 4-アミノアンチピリン溶液(20 g/L)1 mLを加えて振り混ぜ,ヘキサシアノ鉄(III)酸カリウム溶
液1 mLを加えて十分に振り混ぜた後,約10分間放置して,アンチピリン色素を生成させる。
4) 疎水性カラムにアセトニトリル10 mL,水10 mLを流して洗浄・コンディショニングを行った後,
3)の発色溶液を10 mL/minの流量で通液する。
5) 約5分間通気してカラムを乾燥する。
6) アセトニトリル5 mLをカラムに流し,アンチピリン色素を溶出させる。
7) 別に,空試験として水50 mLについて2)〜6)の操作を行う。
8) 空試験のアセトニトリル溶出液を対照液とし,6)のアセトニトリル溶出液の波長475 nm付近の吸光
度を測定する。
9) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH mg/L)を算出
する。
d) 検量線 検量線の作成は,次による。
1) フェノールが0.2〜15 μgとなるように,フェノール標準液(C6H5OH 0.1 μg/mL)又はフェノール標
準液(C6H5OH 1 μg/mL)をメスシリンダー(有線形)50 mLに段階的にとり,水を50 mLの標線ま
で加える。
9
K 0102:2019
2) c)の2)〜8)の操作を行ってフェノールの量と吸光度との関係線を作成する。
備考 5. フェノール標準液(C6H5OH 10 μg/mL)を用いて28.1.1 c),及び28.1.2.1 c),28.1.2.2 c)又は
28.1.2.3 c)のいずれかの操作を行い,フェノールの回収率が十分であることを確かめておくと
よい。
28.1.3 流れ分析法 試料中のフェノール類を,28.1.1及び28.1.2.1と同様な原理で蒸留,発色させるCFA
法又は28.1.1の前処理(蒸留法)後の留出液について28.1.2.1と同様な原理で発色させるFIA法によって
定量する。フェノールの種類によっては,生成したアンチピリン色素が水相で時間とともに退色する場合
がある。このため,JIS K 0170-5の試薬濃度,流量等の条件が28.1.2.1で行った場合と比較し,整合して
いることを確認して,その条件で定量する。
定量範囲:C6H5OH:0.01〜1 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-5による。ただし,JIS K 0170-5の6.3.2(4-アミノアンチピリン発色FIA
法)は28.1.1の前処理(蒸留法)後の留出液に適用し,6.3.4(くえん酸蒸留・4-アミノアンチピリン発色
CFA法)の方法は除く。
31.(農薬)の31.2.1 a)の“6) ヘキサシアノ鉄(III)酸カリウム溶液 28.1.2 a) 3)による。”を,“6) ヘ
キサシアノ鉄(III)酸カリウム溶液 28.1.2.1 a) 3)による。”に置き換える。
32.(溶存酸素)の32.3(隔膜電極法)b) 2)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換える。
32.(溶存酸素)の32.4(光学式センサ法)b) 3)(温度計)の“JIS B 7411-1”を,“JIS B 7414”に置き換
える。
33.(残留塩素)の全文を,次に置き換える。
33. 残留塩素 残留塩素とは,塩素剤が水に溶けて生成する次亜塩素酸及びこれがアンモニアと結合して
生じるクロロアミンをいい,前者を遊離残留塩素,後者を結合残留塩素,両者を合わせて残留塩素という。
残留塩素の定量には,濃度が低い場合にはo-トリジン比色法,ジエチル-p-フェニレンジアンモニウム
(DPD)比色法又はジエチル-p-フェニレンジアンモニウム(DPD)吸光光度法を適用し,濃度が比較的高
い場合には,よう素滴定法を適用する。
この試験は,試料採取後,直ちに行う。
なお,ジエチル-p-フェニレンジアンモニウム(DPD)比色法は,2017年に第2版として発行されたISO
7393-2,よう素滴定法は,1990年に第2版として発行されたISO 7393-3との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 7393-2:2017,Water quality−Determination of free chlorine and total chlorine−Part 2:
Colorimetric method using N,N-diethyl-1,4-phenylenediamine, for routine control purposes(MOD)
ISO 7393-3:1990,Water quality−Determination of free chlorine and total chlorine−Part 3: Iodometric
titration method for the determination of total chlorine(MOD)
33.1 o-トリジン比色法 試料に3,3'-ジメチルベンジジン(o-トリジン)溶液を加え,残留塩素との反応で
生じる黄色を,残留塩素標準比色液と比較して残留塩素を定量する方法である。亜ひ酸ナトリウム溶液で

10
K 0102:2019
処理し,残留塩素,遊離残留塩素及び結合残留塩素の三つに区別することができる。
定量範囲:Cl 0.01〜2.0 mg/L,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水(1)
2) o-トリジン溶液 二塩化3,3'-ジメチルベンジジニウム(o-トリジン二塩酸塩)0.14 gを水50 mLに
溶かし,塩酸(3+7)(JIS K 8180に規定する塩酸を用いて調製する。)50 mL中にかき混ぜながら
加える。着色瓶に入れて保存する。6か月以上経過したものは使用しない。
3) りん酸塩緩衝液(pH6.5) JIS K 9020に規定するりん酸水素二ナトリウムを110 ℃で約2時間加熱
し,デシケーター中で放冷した後,その22.86 gと,JIS K 9007に規定するりん酸二水素カリウム
46.14 gとを水に溶かして1 Lとする。沈殿が生じた場合には,ろ別する。この溶液200 mLをとり
水で1 Lとする。
4) クロム酸カリウム-二クロム酸カリウム溶液 JIS K 8312に規定するクロム酸カリウム3.63 gとJIS
K 8517に規定する二クロム酸カリウム1.21 gとをりん酸塩緩衝液(pH6.5)に溶かし,全量フラス
コ1 000 mLに移し入れ,りん酸塩緩衝液(pH6.5)を標線まで加える。
5) 残留塩素標準比色液 相当する残留塩素の濃度(Cl mg/L)に応じ,クロム酸カリウム-二クロム酸
カリウム溶液及びりん酸塩緩衝液(pH6.5)を表33.1に示す割合に比色管100 mLにとり,混ぜ合わ
せる。暗所に保存する。沈殿が生じた場合には使用しない。
6) 亜ひ酸ナトリウム溶液(5 g/L) メタ亜ひ酸ナトリウム0.5 gを水に溶かして100 mLとする。
注(1) この試験に用いる水は,残留塩素が存在しないこと及び塩素を消費しないことを,備考3.によ
って確かめておく。
b) 器具 器具は,次による。
1) 比色管 100 mL 底部から200±1.5 mmの高さに100 mLの標線を付けた平底のもの。
2) 比色管立 100 mL用 底部及び側面に乳白板を付けたもの。
表33.1 残留塩素標準比色液(液層200 mm用)
残留塩素
Cl mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH6.5
mL
残留塩素
Cl mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH6.5
mL
0.01
0.18
99.82
0.70
7.48
92.52
0.02
0.28
99.72
0.80
8.54
91.46
0.05
0.61
99.39
0.90
9.60
90.40
0.07
0.82
99.18
1.00
10.66
89.34
0.10
1.13
98.87
1.10
12.22
87.78
0.15
1.66
98.34
1.20
13.35
86.65
0.20
2.19
97.81
1.30
14.48
85.52
0.25
2.72
97.28
1.40
15.60
84.40
0.30
3.25
96.75
1.50
16.75
83.25
0.35
3.78
96.22
1.60
17.84
82.16
0.40
4.31
95.69
1.70
18.97
81.03
0.45
4.84
95.16
1.80
20.09
79.91
0.50
5.37
94.63
1.90
21.22
78.78
0.60
6.42
93.58
2.00
22.34
77.66
c) 操作 操作は,次による。
11
K 0102:2019
1) 比色管にo-トリジン溶液5 mLをとり,これに試料(2)の適量(残留塩素0.2 mg以下を含む。)を加え,
更に水を100 mLの標線まで加え,手早く栓をして振り混ぜる。
2) 5分間(3)暗所に放置する。
3) 上方から透視して残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当
する残留塩素の濃度a(Cl mg/L)を記録する。
4) 別の比色管にo-トリジン溶液5 mLをとり,これに1)の操作と同量の試料を加え,手早く栓をして
振り混ぜる。
5) 5秒間以内に亜ひ酸ナトリウム溶液(5 g/L)5 mLを加えて振り混ぜ,更に,水を100 mLの標線ま
で加えて振り混ぜる。
6) 残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する残留塩素の濃
度b(Cl mg/L)を記録する。
7) 空試験として比色管100 mLに亜ひ酸ナトリウム溶液(5 g/L)5 mLをとり,これに1)の操作と同量
の試料を加えて振り混ぜる。
8) o-トリジン溶液5 mLを加えて振り混ぜ,更に水を100 mLの標線まで加えて振り混ぜる。
9) 5秒間以内に残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する
残留塩素の濃度c1(Cl mg/L)を記録する。
10) さらに,5分間暗所に放置後,残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,
これに相当する残留塩素の濃度c2(Cl mg/L)を記録する。
11) 次の式によって残留塩素,遊離残留塩素及び結合残留塩素の濃度を算出する。
(
)
V
c
a
A
100
2×
−
=
(
)
V
c
b
B
100
1×
−
=
C=A−B
ここに,
A: 残留塩素の濃度(Cl mg/L)
a: 3)で求めた残留塩素の濃度(Cl mg/L)
c2: 10)で求めた残留塩素の濃度(Cl mg/L)
V: 試料量(mL)
B: 遊離残留塩素の濃度(Cl mg/L)
b: 6)で求めた残留塩素の濃度(Cl mg/L)
c1: 9)で求めた残留塩素の濃度(Cl mg/L)
C: 結合残留塩素の濃度(Cl mg/L)
注(2) 試料がアルカリ性の場合には,pH計を用い,塩酸(1+5)を加えてpHを約7にする。また,
発色時のpHは常に1.3以下とする。
(3) 残留塩素のうち結合残留塩素は,最高発色に達するのに0 ℃で6分間,20 ℃で3分間,25 ℃
で2分30秒間が必要である。
備考 1. 空試験を行わない場合には,鉄0.3 mg/L以上,マンガン10 μg/L以上又は亜硝酸イオン0.3
mg/L以上が含まれていると妨害する。鉄及びマンガンの妨害を防ぐには,試料100 mLにつ
き1,2-シクロヘキサンジアミン四酢酸溶液(10 g/L)(trans-1,2-シクロヘキサンジアミン四酢
酸一水和物1.05 gを水に溶かして100 mLとする。)3 mLを添加する。
2. 市販の残留塩素測定器を用いる場合には,あらかじめ残留塩素標準比色液と比較して,誤り
12
K 0102:2019
のないことを確認しておく。
備考 3. 試験に用いる水に残留塩素が存在しないこと及び塩素を消費しないことを確認する方法は,
次のいずれかによる。
1) 残留塩素が存在しないことの確認 水[a) 1)]約45 mLを比色管50 mLにとり,JIS K 8913
に規定するよう化カリウム約0.5 gを加えて振り混ぜ,約1分間後,りん酸塩緩衝液
(pH6.5)[33.2 a) 4)による。]2 mL,DPD希釈粉末[33.2 a) 3)による。]0.5 gを加えて振
り混ぜる。発色が認められないことを確認する。又は33.1 c)の1)及び2)の操作を行って
発色が認められないことを確認する。
2) 塩素を消費しないことの確認 水[a) 1)]約45 mLを比色管50 mLにとり,次亜塩素酸
ナトリウム溶液(有効塩素0.1 g/L)[42.2 a) 4)の次亜塩素酸ナトリウム溶液(有効塩素
10 g/L)を100倍に薄めて調製する。]1,2滴を加えて振り混ぜ,約2分間放置する。そ
の後,1)のりん酸塩緩衝液(pH6.5)以降の操作を行って発色が認められることを確認す
る。
33.2 ジエチル-p-フェニレンジアンモニウム(DPD)比色法 硫酸N,N-ジエチル-p-フェニレンジアンモニ
ウム(DPD)を比色管にとり,これに試料を加え,残留塩素との反応で生じる桃色から桃紅色を,残留塩
素標準比色液と比較して定量する。
定量範囲:Cl 0.05〜2 mg/L,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) よう化カリウム JIS K 8913に規定するもの。
3) DPD希釈粉末 硫酸N,N-ジエチル-p-フェニレンジアンモニウム(N,N-ジエチル-p-フェニレンジア
ミン硫酸塩)1.0 gをめのう乳鉢中で粉砕する。これにJIS K 8987に規定する硫酸ナトリウム24 g
を加えてよく混合し,着色ガラス瓶に入れ,湿気を避けて,0〜10 ℃の暗所に保存する。着色した
ものは使用しない。
4) りん酸塩緩衝液(pH6.5) りん酸二水素カリウム溶液(0.2 mol/L)(JIS K 9007に規定するりん酸二
水素カリウム27.2 gを水に溶かして1 Lとする。)100 mLをとり,水酸化ナトリウム溶液(0.2 mol/L)
(JIS K 8576に規定する水酸化ナトリウム8 gを水に溶かして1 Lとする。)をpH計を用いてpH6.5
になるまで加え,これにtrans-1,2-シクロヘキサンジアミン四酢酸一水和物0.13 gを加えて溶かす。
5) C. I. Acid Red 265溶液 C. I. Acid Red 265[1-(4-メチルベンゼンスルホンアミド)-7-(2-メチルフェニ
ルアゾ)-8-ヒドロキシ-3,6-ナフタレンジスルホン酸二ナトリウム]を105〜110 ℃で3〜4時間加熱
し,デシケーター中で放冷する。その0.329 gを1 mgの桁まではかりとり,少量の水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。この溶液50 mLを全量フラスコ500 mLに
とり,水を標線まで加える。0〜10 ℃の暗所に保存し,6か月間以上経過したものは使用しない。
6) DPD残留塩素標準比色液 C. I. Acid Red 265溶液を表33.2によって全量フラスコ50 mLにとり,水
を標線まで加える。これを比色管50 mLにそれぞれ移す。密栓して0〜10 ℃の暗所に保存する。6
か月間以上経過したものは使用しない。
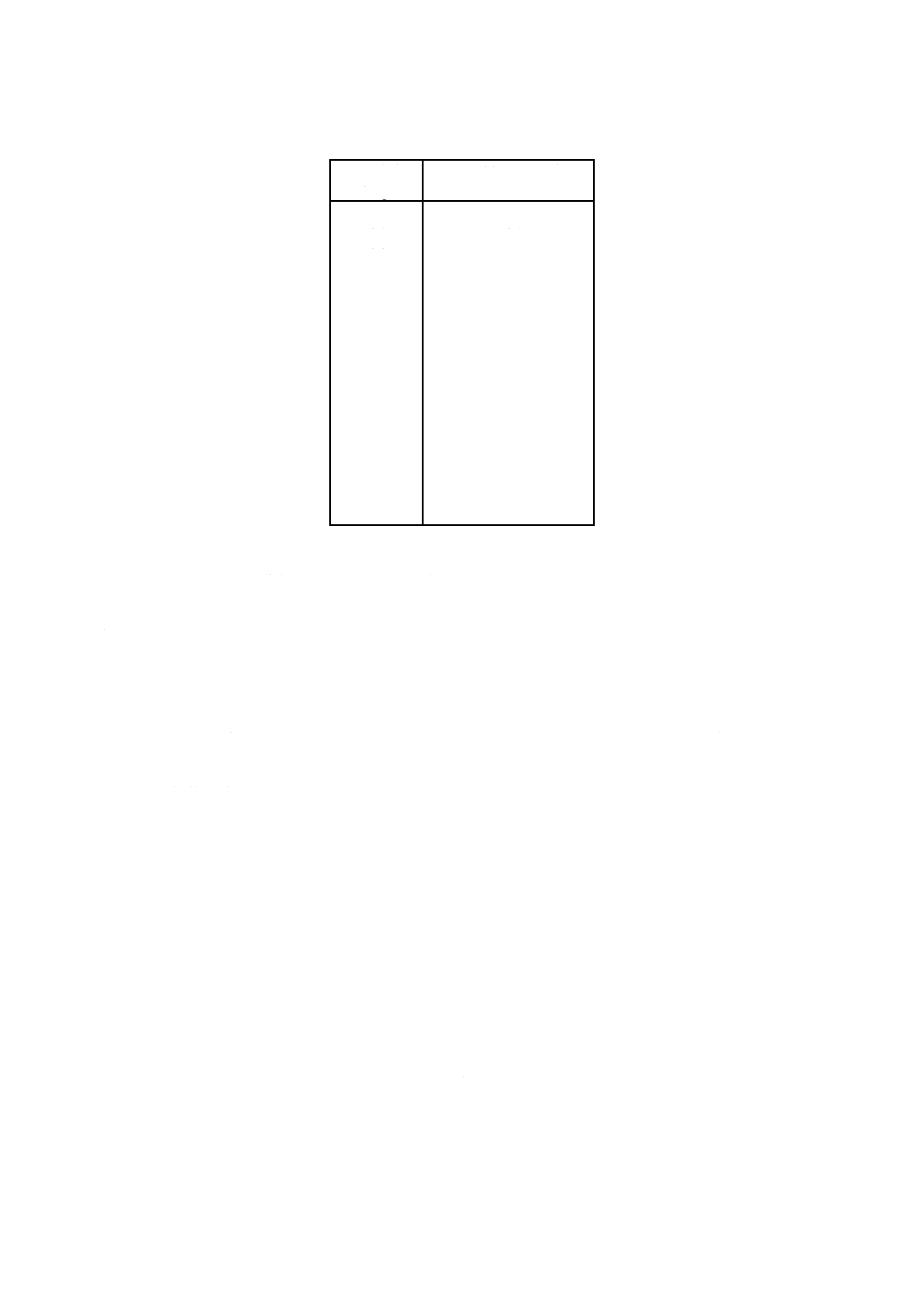
13
K 0102:2019
表33.2 DPD残留塩素標準比色液(50 mL中)
残留塩素
Cl mg/L
C. I. Acid Red 265溶液
mL
0.05
0.5
0.1
1.0
0.2
2.0
0.3
3.0
0.4
4.0
0.5
5.0
0.6
6.0
0.7
7.0
0.8
8.0
0.9
9.0
1.0
10.0
1.2
12.0
1.4
14.0
1.6
16.0
1.8
18.0
2.0
20.0
b) 器具 器具は,次による。
1) 比色管 50 mL 底部から150±1 mmの高さに50 mLの標線を付けた平底のもの。
2) 比色管立 50 mL用 底部及び側面に乳白板を付けたもの。
c) 操作 操作は,次による。
1) りん酸塩緩衝液(pH6.5)2.5 mLを比色管50 mLにとり,これにDPD希釈粉末0.5 gを加える。次
に,試料(4)の適量(残留塩素0.1 mg以下を含む。)を加え,更に水を標線まで加える。
2) 栓をしてよく振り混ぜ,1分間(5)以内にその発色を側面から透視して,DPD残留塩素標準比色液と
比較する。該当するDPD残留塩素標準比色液から,これに相当する残留塩素の濃度(Cl mg/L)を
求め,これを遊離残留塩素として,試料中の遊離残留塩素の濃度(Cl mg/L)を算出する。
3) 2)の操作が終了したら,よう化カリウム約0.5 gを加え,栓をして振り混ぜて溶かし,約2分間放置
後,その発色を2)と同様に残留塩素標準比色液と比較する。該当する残留塩素標準比色液からこれ
に相当する残留塩素の濃度(Cl mg/L)を求め,試料中の残留塩素の濃度を算出する。
4) 結合残留塩素の濃度(Cl mg/L)は,次の式によって算出する。
結合残留塩素の濃度(Cl mg/L)=残留塩素(Cl mg/L)−遊離残留塩素(Cl mg/L)
注(4) 試料の酸性又はアルカリ性が強い場合には,炭酸ナトリウム溶液(50 g/L)(JIS K 8625に規定
する炭酸ナトリウムを用いて調製する。)又は塩酸(1+11)[21. a) 2)による。]を用いてpHを
約6.5に調節する。
(5) 振り混ぜ時間を含める。DPD希釈粉末中の硫酸ナトリウムは完全には溶けなくてもよい。
備考 4. 市販の残留塩素測定器又はガラス色標準スケールを用いる場合には,あらかじめDPD残留塩
素標準比色液と比較して,誤りのないことを確認しておく。
5. 試料中のマンガン酸化物による妨害の補正方法は,次による。
1) 試料100 mLをとり,メタ亜ひ酸ナトリウム溶液(2 g/L)(メタ亜ひ酸ナトリウムを用い
て調製する。)又はチオアセトアミド(エタンチオアミド)溶液(2.5 g/L)1 mLを加え,
振り混ぜる。
14
K 0102:2019
2) この溶液を用いて,c)の1)及び2)の操作を行い,マンガン酸化物による発色を残留塩素
の濃度として求める。
3) 得られた値を用いて,遊離残留塩素の濃度及び残留塩素の濃度を補正する。
備考 6. 二酸化塩素(IV)は,残留塩素及び遊離残留塩素の値に含まれる。
7. DPDの酸化は,塩素化合物によるものだけではない。反応は,他の酸化剤によっても生じる。
これには臭素,よう素,ブロモアミン類,ヨードアミン類,オゾン,過酸化水素,クロム酸
塩,マンガン酸化物,亜硝酸塩,鉄(III)イオン及び銅イオンが挙げられる。ただし,この
方法では,銅イオン2 mg/L,鉄(II)イオン3 mg/L,アルミニウム4 mg/L及び亜硝酸イオン
4 mg/Lまではそれぞれ妨害しない。
8. 試料が海水の場合,一部の微細藻類等がDPDを発色させる場合がある。これを防止するには,
ガラス繊維又は有機高分子製のろ過材(孔径100 μm以下)を用い,自然ろ過又は加圧ろ過
によって試料をろ過し,そのろ液の適量をとり,c) 1)以降の操作を行う。ろ過操作前にはろ
過材及び器具を試料で共洗いし,接液面での残留塩素の損失を防ぐ。ろ過材及び器具はあら
かじめ,残留塩素の濃度を変化させないことを確認しておく。
9. c) 1)の操作後は日射によって発色が進むことがあるため,日射を当てないようにする。
33.3 よう素滴定法 残留塩素とよう化カリウムとが反応して遊離するよう素をチオ硫酸ナトリウム溶液
で滴定し,残留塩素を定量する。よう素を遊離させる酸化性物質が共存すると,残留塩素として定量され
る。
定量範囲:Cl 0.1 mg以上
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) よう化カリウム 33.2 a) 2)による。
3) 酢酸(1+1) JIS K 8355に規定する酢酸を用いて調製する。
4) でんぷん溶液(10 g/L) 19. a) 5)による。
5) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
b) 操作 操作は,次による。
1) 試料(2)の適量(Clとして0.1〜7 mgを含む。)を共栓三角フラスコ500 mLにとり,水を加えて約300
mLとし,よう化カリウム1 g及び酢酸(1+1)5 mLを加える。
2) 栓をして振り混ぜ,暗所に約5分間放置する。
3) 遊離したよう素を,10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,
指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴
定する。
4) 空試験として水100 mLをとり,1)〜3)の操作を行う。
5) 次の式によって試料中の残留塩素の濃度(Cl mg/L)を算出する。
(
)
5
354
.0
000
1
×
×
×
−
=
V
f
b
a
A
ここに,
A: 残留塩素の濃度(Cl mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 試料量(mL)
15
K 0102:2019
0.354 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する残留塩
素の質量(mg)
備考 10. 試料の着色又は濁りが著しく試験が困難な場合には,次の方法で残留塩素を分離し測定して
もよい。
1) 試料の適量(Clとして2 mg以上を含む。)を図33.1の蒸留フラスコ200 mLにとり,硫
酸(1+15)を加えてpHを0.9〜1.0に調節し,水で約80 mLとし,蒸留装置に接続する。
2) 10 mmol/Lチオ硫酸ナトリウム溶液を,ガス洗浄瓶(H)には20 mL,ガス洗浄瓶(I)
には5 mLを加え,それぞれ水を加えて50 mLとし,更に酢酸緩衝液(pH3.5)[JIS K 8371
に規定する酢酸ナトリウム三水和物240 gを水約300 mLに溶かし,酢酸460 mLを加え
た後,水で1 Lとする。]4 mLとよう化カリウム0.1 gとを加えて振り混ぜる。
3) 蒸留フラスコを40 ℃の恒温槽中(J)に入れ,緩やかに約40分間通気する。ガス洗浄
瓶中の溶液を三角フラスコ300 mLに移し,水でガス洗浄瓶の内部を洗い洗液を前の溶
液に合わせる。これに,よう素溶液(JIS K 8913に規定するよう化カリウム12 gを少量
の水に溶かし,JIS K 8920に規定するよう素4 gを加えて溶かし,水を加えて1 Lとす
る。)10 mLを加える。
4) 過剰のよう素を10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなった
ら指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が
消えるまで滴定する。
5) 空試験として10 mmol/Lチオ硫酸ナトリウム溶液25 mLを三角フラスコ300 mLにとり,
酢酸塩緩衝液(pH3.5)8 mL,よう化カリウム0.2 g及び水約120 mLを加えて振り混ぜ,
これによう素溶液10 mLを加え,試料と同様に10 mmol/Lチオ硫酸ナトリウム溶液で滴
定する。試料中の残留塩素の濃度(Cl mg/L)は,b) 5)の式によって算出する。
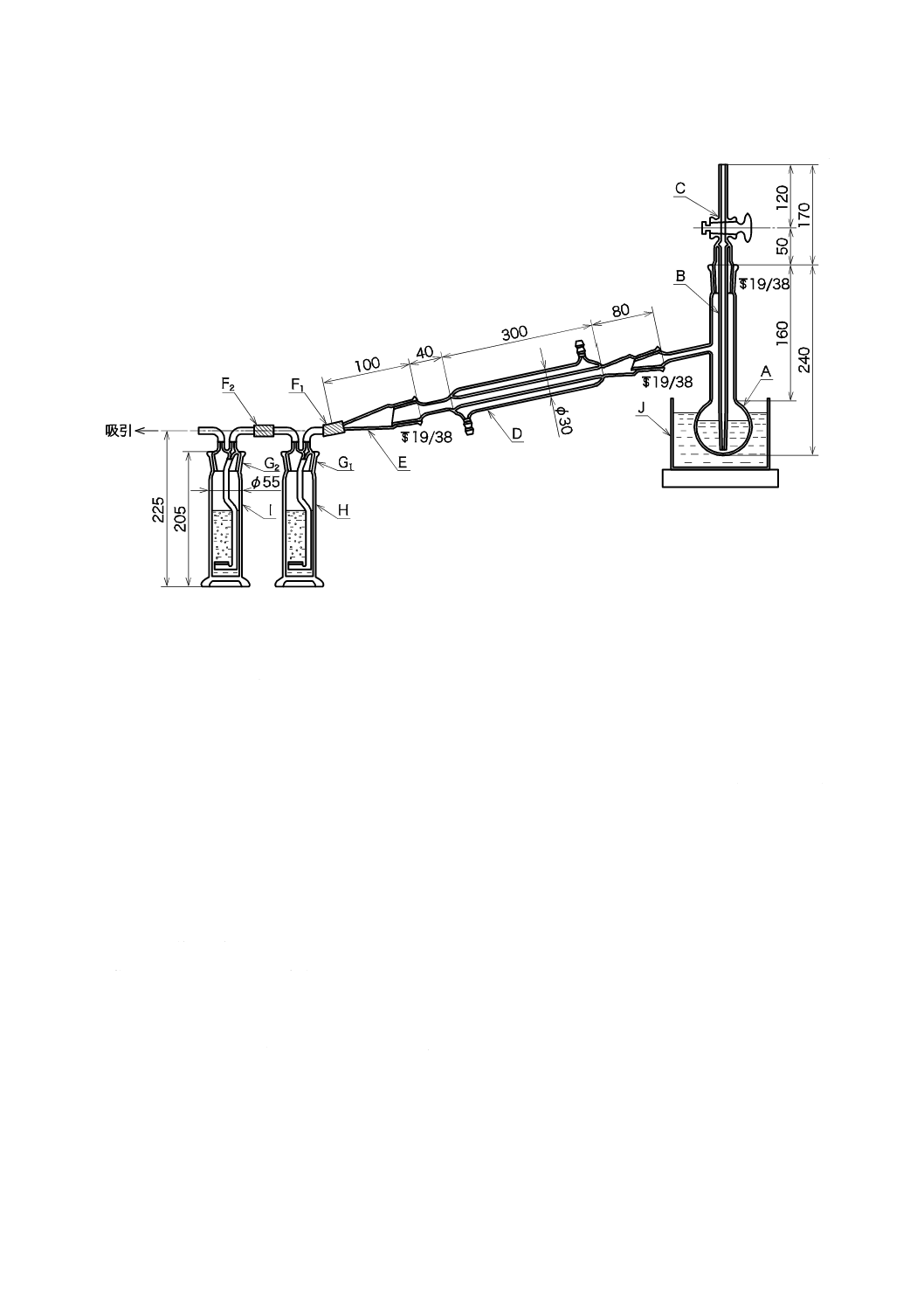
16
K 0102:2019
単位 mm
A:
B:
C:
D:
E:
蒸留フラスコ(共通すり合わせ枝付き)200 mL
中管(先端の内径約1 mm)
一方コック
共通すり合わせリービッヒ冷却器(200〜300 mm)
共通すり合わせアダプター
F1,F2:
G1,G2:
H,I:
J:
ゴム管
共通すり合わせ
共通すり合わせろ過板付きガス洗浄瓶
(250 mL)
恒温槽
図33.1 蒸留装置(共通すり合わせ)の例
33.4 ジエチル-p-フェニレンジアンモニウム(DPD)吸光光度法 試料に硫酸N,N-ジエチル-p-フェニレン
ジアンモニウム(DPD)を加え,残留塩素との反応で生じる桃色から桃紅色を,波長510 nm(又は555 nm)
付近の吸光度を測定して定量する。
定量範囲:Cl 2.5〜150 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) 希釈水 水1 Lに対し塩素水(濃度約50 mg/L)約3 mLを加え1時間放置した後,煮沸するか又は
紫外線を照射して残留塩素を除く。
3) よう化カリウム 33.2 a) 2)による。
4) DPD希釈粉末 33.2 a) 3)による。
5) りん酸塩緩衝液(pH6.5) 33.2 a) 4)による。
6) 0.1 mol/Lチオ硫酸ナトリウム溶液 19. a) 8)による。
7) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。ファクターは,19. a) 8)の0.1 mol/Lチオ硫酸
ナトリウム溶液のものを用いる。12時間以上経過したものは使用しない。
8) でんぷん溶液(10 g/L) 19. a) 5)による。
9) 塩素標準液 塩素水の調製,有効塩素濃度の測定及び塩素標準液の調製は,次による。
17
K 0102:2019
塩素標準液は,使用の都度その有効塩素濃度を測定する。
9.1) 塩素水の調製 次亜塩素酸ナトリウム溶液[有効塩素7〜12 %(質量百分率)]を水に溶かす。又
は,他の方法によって塩素水を調製してもよい。
9.2) 有効塩素濃度の測定
9.2.1) 9.1)で調製した塩素水100 mLを共栓三角フラスコ200 mLにとり,よう化カリウム1 g及び酢酸
(1+1)(JIS K 8355に規定する酢酸を用いて調製する。)5 mLを加える。
9.2.2) 栓をして振り混ぜ,暗所に5分間放置する。
9.2.3) 遊離したよう素を,0.1 mol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,
指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じた青い色が消えるまで滴定する。
9.2.4) 次の式によって,塩素水に含まれる有効塩素の濃度(Cl mg/L)を算出する。
545
.3
000
1
1
×
×
×
=
V
f
a
C
ここに,
C: 有効塩素の濃度(Cl mg/L)
a: 滴定に要した0.1 mol/Lチオ硫酸ナトリウム溶液量(mL)
f1: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
V: 9.2.1)で用いた塩素水(mL)
3.545: 0.1 mol/Lチオ硫酸ナトリウム溶液1 mLに相当する塩素の質
量(mg)
9.3) 塩素標準液(Cl 50 μg/mL) 塩素25 mgに相当するように9.2)で有効塩素の濃度を定量した塩素水
を全量フラスコ500 mLにとり,希釈水を標線まで加える。正確な濃度の標定は,次による。
この溶液100 mLをとり,9.2.1)〜9.2.3)の操作を行う。ただし,滴定には10 mmol/Lチオ硫酸ナ
トリウム溶液を用いる。次の式によって,塩素標準液(Cl 50 μg/mL)の正確な濃度を算出する。
5
354
.0
000
1
1
×
×
×
=
V
f
a
C
ここに,
C: 有効塩素の濃度(Cl mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f1: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 滴定に用いた塩素標準液(Cl 0.05 mg/mL)量(mL)
0.354 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する塩素の
質量(mg)
9.4) 塩素標準液(Cl 5 μg/mL) 塩素標準液(Cl 50 μg/mL)20 mLを全量フラスコ200 mLにとり,希
釈水を標線まで加える。この溶液は,使用時に調製する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(4)の適量について,33.2 c) 1)の操作を行う。
2) 栓をしてよく振り混ぜ,1分間(5)以内に,この溶液の一部を吸収セルにとり,水を対照液として波
長510 nm(又は555 nm)付近の吸光度を測定する。
3) 検量線から塩素の量(Cl μg)を求め,これを遊離残留塩素として試料中の遊離残留塩素の濃度(Cl
mg/L)を算出する。
4) 引き続き2)の残りの溶液によう化カリウム約0.5 gを加え,栓をして振り混ぜて溶かす。約2分間
放置した後,この溶液の一部を吸収セルにとり,水を対照液として波長510 nm(又は555 nm)付
18
K 0102:2019
近の吸光度を測定する。
5) 検量線から塩素の量(Cl μg)を求め,これを残留塩素として試料中の残留塩素の濃度(Cl mg/L)
を算出する。
6) 33.2 c) 4)によって結合残留塩素の濃度(Cl mg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) 塩素標準液(Cl 5 μg/mL)0.5〜30 mLについて,c)の1)及び2)の操作を行い,吸光度を測定し,塩
素(Cl)の量と吸光度との関係線を作成する。
備考 11. 備考8.による。
12. 備考9.による。
34.(ふっ素化合物)の全文を,次に置き換える。
34. ふっ素化合物 ふっ素化合物は,ふっ化物イオン,金属ふっ化物などの総称であり,ふっ化物イオン
として表す。ふっ化物イオンの定量には,ランタン-アリザリンコンプレキソン吸光光度法,イオン電極法,
イオンクロマトグラフ法又はランタン-アリザリンコンプレキソン発色流れ分析法を適用する。
なお,蒸留操作は,1992年に第1版として発行されたISO 10359-1,イオン電極法は,1994年に第1版
として発行されたISO 10359-2,イオンクロマトグラフ法は,2007年に第2版として発行されたISO 10304-1
との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 10359-1:1992,Water quality−Determination of fluoride−Part 1: Electrochemical probe method
for potable and lightly polluted water(MOD)
ISO 10359-2:1994,Water quality−Determination of fluoride−Part 2: Determination of inorganically
bound total fluoride after digestion and distillation(MOD)
ISO 10304-1:2007,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 1: Determination of bromide, chloride, fluoride, nitrate, nitrite, phosphate and sulfate
(MOD)
34.1 ランタン-アリザリンコンプレキソン吸光光度法 ふっ素化合物を蒸留分離し,ランタン(III)とア
リザリンコンプレキソンとの錯体を加え,これがふっ化物イオンと反応して生じる青い色の複合錯体の吸
光度を測定して,ふっ化物イオンを定量し,ふっ素化合物とする。
34.1.1 前処理(蒸留法) 水蒸気蒸留による前処理を行い,ふっ素化合物をふっ化物イオンとして分離す
る。
a) 試薬 試薬は,次による。
1) 過塩素酸 JIS K 8223に規定するものを,加熱して白煙を発生させた後,放冷したもの。
2) 硫酸 JIS K 8951に規定するものを,加熱して白煙を発生させた後,放冷したもの。
3) りん酸 JIS K 9005に規定するもの。
4) 水酸化ナトリウム溶液(100 g/L) 28.2.1 a) 3)による。
5) 二酸化けい素 JIS K 8885に規定する二酸化けい素(1)
6) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
注(1) 結晶質のもので粒径100〜150 μm程度のものを用いる。品質が分からない場合には,白金るつ
19
K 0102:2019
ぼ中で1 150 ℃以上で約1時間加熱し,デシケーター中で放冷したものを用いる。この場合,
ふっ化物イオン標準液(F− 2 μg/mL)50 mLをとり,c)の2)〜5)及び34.1.2 c)の1)〜5)を行って
回収率を確認する。
b) 装置 装置は,次による。
1) 蒸留装置 図34.1に例を示す。
c) 蒸留操作 蒸留操作は,次による。
なお,試料に一定量のふっ化物イオン標準液を添加して,備考1.の蒸留操作による回収率試験を行
い,回収率が80〜120 %であることを確認した場合は,備考1.によってもよい。
1) 試料の適量(F−として30 μg以上を含む。)を磁器蒸発皿,ビーカーなどにとり,フェノールフタレ
イン溶液(5 g/L)2,3滴を加え,水酸化ナトリウム溶液(100 g/L)を滴加して微アルカリ性とし
た後,加熱して約30 mLに濃縮する。
なお,溶存のふっ素化合物を試験するときは,3.2でろ過した試料を用いる。
2) 図34.1の蒸留フラスコ中に水約10 mLで洗い移す。次に,二酸化けい素約1 g,りん酸1 mL及び過
塩素酸40 mL又は硫酸30 mL及び沸騰石(粒径2〜3 mm)を加える。受器の全量フラスコ250 mL
には水20 mL(2)を加え,逆流止めの先端は水面下に保つ。
3) 蒸留フラスコを直接加熱し,蒸留フラスコ内の液温が約140 ℃に達してから,水蒸気を通す。
4) 蒸留温度を145±5 ℃,留出速度を3〜5 mL/minに調節し,受器の液量が約220 mLになるまで蒸留
を続ける。
5) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加え,更に水を標線まで加える。
注(2) 試料中にふっ化物イオン以外のハロゲン化物が多量に含まれる場合には,水酸化ナトリウム溶
液(40 g/L)[21. a) 3)による。]4〜5滴とフェノールフタレイン溶液(5 g/L)2〜3滴とを加え
ておく。受器中の溶液は,蒸留が終わるまで微紅色を保つように,必要に応じて水酸化ナトリ
ウム溶液(40 g/L)を滴加する。
なお,この場合は蒸留が終わった後,留出液に硫酸(1+35)[30.1.1 a) 2)による。]を微紅色
が消えるまで滴加し,その後,5)の操作を行う。
備考 1. 小型蒸留装置を用いる蒸留操作は,次による。小型蒸留装置の例を図34.2に示す。ただし,
この場合の留出液は,34.2(イオン電極法)には適用しない。
1) 試料の適量(F−として30 μg以上を含む。蒸留後に流れ分析法を適用する場合には10 μg
以上を含む。)を磁器蒸発皿,ビーカーなどにとり,フェノールフタレイン溶液(5 g/L)
2,3滴を加え,水酸化ナトリウム溶液(100 g/L)を滴加して微アルカリ性とした後,加
熱して約5〜10 mLに濃縮する。
2) 濃縮した試料を図34.2の蒸留容器中に少量の水で洗い流し,液量を10〜15 mLにする。
次に,二酸化けい素約0.25 g,りん酸0.5 mL及び過塩素酸12 mL,又はりん酸0.5 mL及
び硫酸8 mLを静かに加える。蒸留容器に蒸留管及び冷却器をセットする。過塩素酸を
加えた場合には,容量100 mLの目盛付共栓付受器に水20 mL,水酸化ナトリウム溶液及
びフェノールフタレイン溶液を数滴加え,冷却器の先端部を水面下に保つ。
なお,硫酸を加えた場合には,容量50 mLの目盛付共栓付受器を用い,加える水の量
を5 mLとする。また,受器中の溶液は,蒸留が終わるまで微紅色を保つように,必要
に応じて水酸化ナトリウム溶液を滴加する。
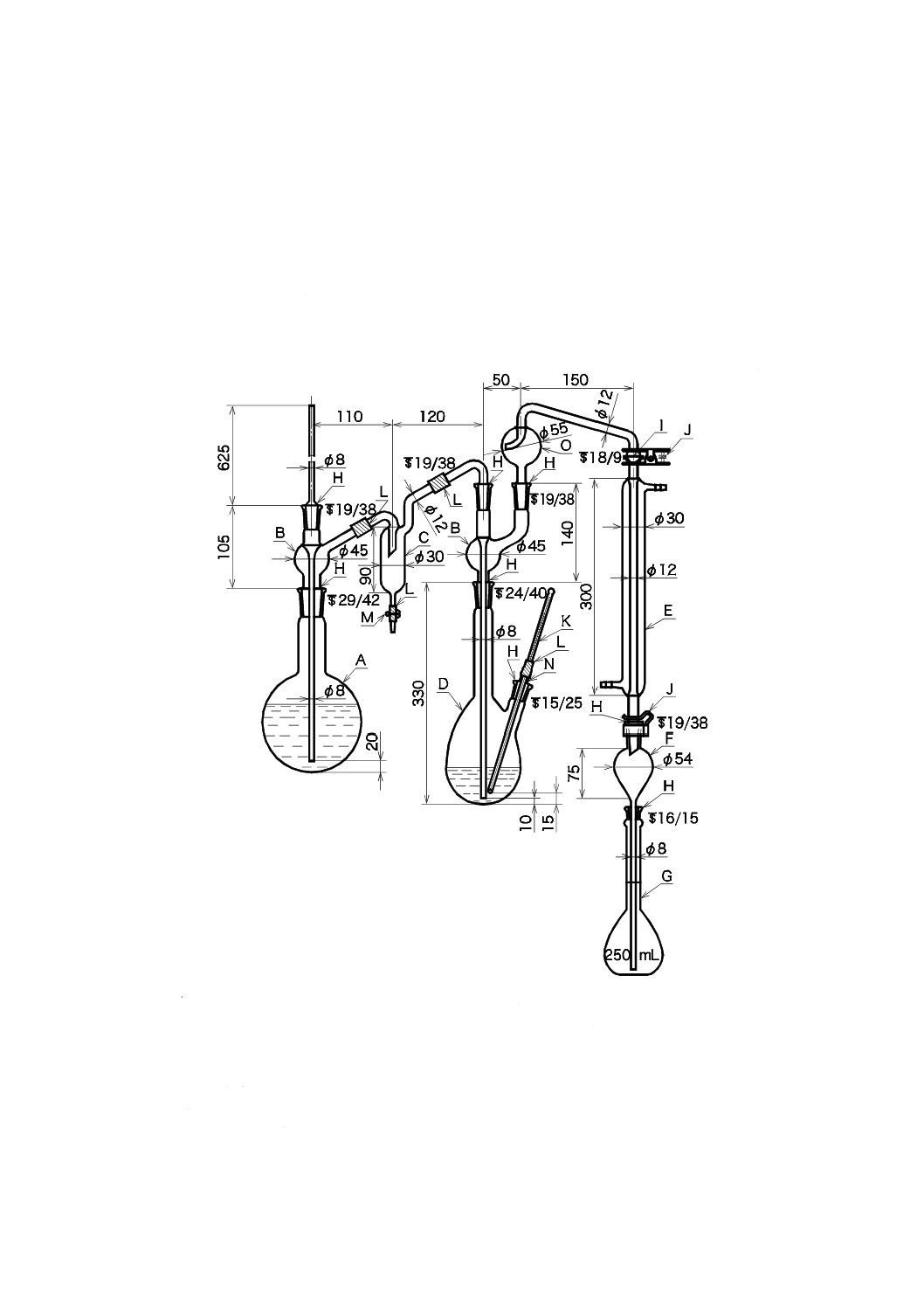
20
K 0102:2019
3) 蒸留容器と水蒸気発生用容器とをあらかじめ170〜210 ℃に加熱した加熱器(装置によ
って最適温度は異なる。)に設置し,蒸留容器内に水蒸気を安定的に供給するために,水
蒸気発生用容器に空気を0.1〜0.5 L/minで通す。
4) 留出速度1.0〜1.6 mL/minで,受器の液量が約70 mLになるまで蒸留を続ける。
なお,硫酸を加えた場合には,受器の液量が約45 mLになるまで蒸留を行う。
5) 蒸留後,冷却器などを取り外し,冷却器の内管を少量の水で洗い,洗液も受器に加え,
水を加えて全量を100 mLとする。
なお,硫酸を加えた場合には,全量を50 mLとする。
単位 mm
A:
B:
C:
D:
E:
F:
G:
H:
水蒸気発生フラスコ1 000 mL
連結導入管
トラップ
蒸留フラスコ500 mL
リービッヒ冷却器300 mm
逆流止め(約50 mL)
受器(全量フラスコ250 mL)
共通すり合わせ
I:
J:
K:
L:
M:
N:
O:
共通球面すり合わせ
押さえばね
温度計200 ℃
ゴム管
ピンチコック
温度計差し込み栓
トラップ球(ケルダール球)
図34.1 蒸留装置の例
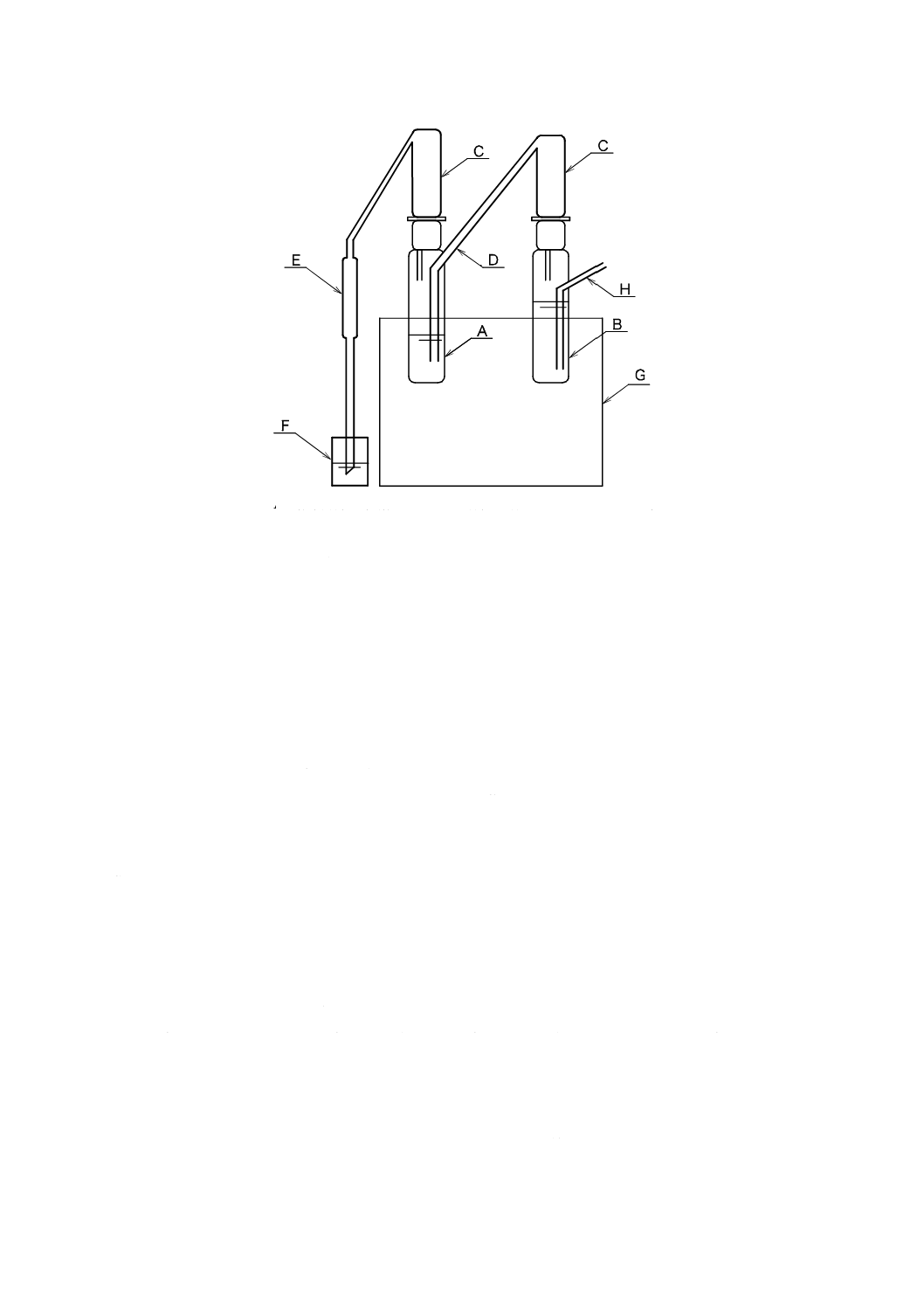
21
K 0102:2019
A:蒸留容器(耐熱性のガラス容器で容量50〜80 mLのもの)
B:水蒸気発生用容器(容量50〜80 mLのもの)
C:蒸留管(気液分離が可能なもの)
D:接続管(蒸留容器に水蒸気を供給できるもの)
E:冷却器
F:受器(有栓形メスシリンダー 50 mLなど)
G:加熱器(170〜210 ℃の設定が可能なもの)
H:空気導入口
図34.2 小型蒸留装置の例
34.1.2 ランタン-アリザリンコンプレキソン発色による定量法 34.1.1で得られた留出液中のふっ化物イ
オンをランタン-アリザリンコンプレキソン吸光光度法によって定量する。この方法は,陰イオンの妨害は
少ないが,陽イオンによる妨害を受けやすい。特に,アルミニウム,カドミウム,コバルト,鉄,ニッケ
ル,ベリリウム及び鉛などが妨害するので,あらかじめ蒸留してふっ化物イオンを分離する。
定量範囲:F− 4〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) ランタン-アリザリンコンプレキソン溶液(3) 調製は,次による。
− アリザリンコンプレキソン(1,2-ジヒドロキシアントラキノン-3-イルメチルアミン-N,N-二酢酸二
水和物)0.192 gを,アンモニア水(1+10)(JIS K 8085に規定するアンモニア水を用いて調製す
る。)4 mL及び酢酸アンモニウム溶液(200 g/L)(JIS K 8359に規定する酢酸アンモニウムを用い
て調製する。)4 mLに溶かす。
− これを酢酸ナトリウム溶液(JIS K 8371に規定する酢酸ナトリウム三水和物41 gを水400 mLに
溶かし,JIS K 8355に規定する酢酸24 mLを加えたもの。)中にかき混ぜながら加える。
− この溶液をかき混ぜながらJIS K 8034に規定するアセトン400 mLを徐々に加え,更にランタン
溶液[酸化ランタン(III)0.163 gを塩酸(1+5)(JIS K 8180に規定する塩酸を用いて調製する。)
10 mLに加え,加熱して溶かしたもの。]を加えてかき混ぜる。放冷後,酢酸又はJIS K 8085に
規定するアンモニア水でpH計を用いてpHを約4.7に調節した後,水を加えて1 Lとする。
2) ふっ化物イオン標準液(F− 100 mg/L) JIS K 8005に規定する容量分析用標準物質のふっ化ナトリ
22
K 0102:2019
ウムを白金皿にとり,500 ℃で約1時間加熱し,デシケーター中で放冷する。NaF 100 %に対して
その0.221 gをとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
ポリエチレン瓶に入れて保存する。
3) ふっ化物イオン標準液(F− 2 μg/mL) ふっ化物イオン標準液(F− 100 mg/L)10 mLを全量フラス
コ500 mLにとり,水を標線まで加える。標準液は,ポリエチレン瓶に貯蔵し,1か月間は使用でき
る。
注(3) 市販品を用いてもよい。市販のアルフッソンを用いる場合は,その2.5 gを水に溶かして50 mL
とする。使用時に調製する。
参考 アルフッソン(商品名)は,この規格の利用者の便宜を図って記載するもので,この製品を推
奨するものではない。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 定量操作 定量操作は,次による。
1) 34.1.1 c)の蒸留操作で得た留出液から30 mL以下の適量(F−として4〜50 μgを含む。)を全量フラ
スコ50 mLにとる。
2) ランタン-アリザリンコンプレキソン溶液(4) 20 mLを加え,更に水を標線まで加えて振り混ぜ,約1
時間放置する。
3) 別に,水30 mLを全量フラスコ50 mLにとり,2)の操作を行う。
4) 試料について2)で得た溶液の一部を吸収セルに移し,3)の溶液を対照液として波長620 nm付近の吸
光度を測定する。
5) 検量線からふっ化物イオンの量を求め,試料中のふっ化物イオンの濃度(F− mg/L)を算出する。
注(4) 注(3)で調製したアルフッソン溶液を用いる場合には,その5 mLとJIS K 8034に規定するアセ
トン10 mLとを1)の溶液に加えた後,水を標線まで加える。
d) 検量線 検量線の作成は,次による。
1) ふっ化物イオン標準液(F− 2 μg/mL)2〜25 mLを全量フラスコ50 mLに段階的にとる。
2) c)の2)〜4)の操作を行って吸光度を測定し,ふっ化物イオン(F−)の量と吸光度との関係線を作成
する。
34.2 イオン電極法 ふっ素化合物を前処理して蒸留分離し,緩衝液(イオン強度調節液)を加えてpHを
5.2±0.2に調節し,ふっ化物イオン電極を指示電極として電位を測定し,ふっ化物イオンを定量する。た
だし,備考1.による留出液には適用しない。
備考 2. 妨害物質を含まない清浄な試料中の溶存ふっ化物イオンを定量する場合は,蒸留に代え,試
料をろ過し,備考3.の操作で定量することができる。ただし,この方法では,溶存のふっ化
物イオン及び容易にふっ化物イオンとなる錯イオンが定量される。また,この前処理方法は,
汚濁のある排水には適用できない。
定量範囲:F− 0.1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 緩衝液(pH5.2) JIS K 8150に規定する塩化ナトリウム58 gとJIS K 8284に規定するくえん酸水
素二アンモニウム1 gとを水500 mLに加えて溶かし,JIS K 8355に規定する酢酸50 mLを加え,水
酸化ナトリウム溶液(200 g/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)を滴
加して,pH計を用いてpHを5.2に調節した後,水を加えて1 Lとする。
23
K 0102:2019
2) ふっ化物イオン標準液(F− 100 mg/L) 34.1.2 a) 2)による。標準液は,プラスチック製容器で貯蔵
し,1か月間は使用できる。
3) ふっ化物イオン標準液(F− 10 mg/L) ふっ化物イオン標準液(F− 100 mg/L)20 mLを全量フラス
コ200 mLにとり,水を標線まで加える。使用時に調製する。
4) ふっ化物イオン標準液(F− 1 mg/L) ふっ化物イオン標準液(F− 10 mg/L)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
5) ふっ化物イオン標準液(F− 0.1 mg/L) ふっ化物イオン標準液(F− 1 mg/L)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 0.1 mV又はそれ以下の電位差を読み取れるもの。高入力抵抗電位差計(例えば,デジタ
ル式pH-mV計,拡大スパン付きpH-mV計,イオン電極用電位差計など)
2) 指示電極 ふっ化物イオン電極,標準液を用いた起電力の応答は,25 ℃におけるふっ化物イオン濃
度の10倍濃度変化当たり55 mV以上のもの(5)。
3) 参照電極 銀-塩化銀電極を用いる(6)。
4) 測定容器 試料100 mLで扱えるもの。ポリプロピレン製で,恒温ジャケットが取り付けられてい
るもの。
5) 恒温槽 測定容器のジャケットに水温25±0.2 ℃の水を供給できるもの。
6) マグネチックスターラー 四ふっ化エチレン樹脂(PTFE)で被覆した回転子付きのものを用いる。
注(5) 使用時に電極をふっ化物イオン標準液(F− 0.1 mg/L)に浸し,指示値が安定してから使用する。
指示電極の感応膜にきずがつくと,検量線の勾配(電位勾配)が小さくなり,応答速度も遅く
なるので注意する。また,指示電極の感応膜が汚れると,応答速度が遅くなるので,エタノー
ル(95)を含ませた脱脂綿又は柔らかい紙で汚れを拭き取り,水で洗浄する。
(6) 参照電極は,抵抗の小さいものを選ぶ。一般に液間電位差の小さい単一液絡形のスリーブ形又
はセラミックス形を用いる。スリーブ形は,抵抗も小さく最適であるが,スリーブを締め過ぎ
ると抵抗が大きくなり,緩すぎると液の流出が多くなるため,適度の締付けが必要である。セ
ラミックス形は抵抗の大きい製品もあるため,イオン電極用を用いる。セラミックス形は乾燥
したり,汚れると抵抗が大きくなるため注意する。これらの電極は,内部液と同じ溶液中に浸
しておく。参照電極の内部液に塩化カリウム溶液(飽和)を使用する場合には,液温が低下す
ると塩化カリウムの結晶が析出し,固着して抵抗が大きくなることがあるため注意する。
c) 検量線 検量線の作成は,次による。
なお,測定する濃度によっては,次に記載する濃度以外の適切な濃度範囲のふっ化物イオン標準液
を用いる。
1) ふっ化物イオン標準液(F− 0.1 mg/L)100 mLを測定容器にとり,緩衝液(pH5.2)10 mLを加える
(7)。
2) 恒温槽から水を送り,この測定容器の溶液を25±0.5 ℃に保つ。
3) 指示電極と参照電極とを浸し固定した後,回転子を入れ,マグネチックスターラー(8)を用いて,泡
が電極に触れない程度に強くかき混ぜる(9)。
4) 液温を確認し,電位差計で電位を測定する(10)。
5) ふっ化物イオン標準液(F− 1 mg/L),ふっ化物イオン標準液(F− 10 mg/L)及びふっ化物イオン標
準液(F− 100 mg/L)のそれぞれ100 mLを測定容器にとり,それぞれに緩衝液(pH5.2)10 mLを
24
K 0102:2019
加える(7)。
6) 2)〜4)の操作を行って,それぞれのふっ化物イオン標準液の電位を測定する(11)(12)。
7) 横軸にふっ化物イオンの濃度の対数を,縦軸に電位をとり,ふっ化物イオンの濃度(mg/L)と電位
との関係線を作成する(11)。
注(7) 緩衝液(pH5.2)の添加によってpH5.2±0.2に調節し,イオン強度を一定にする。
(8) マグネチックスターラーを長時間使用すると,発熱して液温に変化を与えることがあるので,
液温の変化に注意する。
(9) かき混ぜ速度で電位差計の指示が不安定になる場合には,参照電極の抵抗が大きくなっている
ことが多い。
かき混ぜ速度は,約180〜200 min−1に調節するとよい。
(10) ふっ化物イオン電極の応答時間は,液温10〜30 ℃の場合には,ふっ化物イオンの濃度が0.1
mg/Lで約1分間,1 mg/L以上では約30秒間である。
セルの電位が,5分間で0.5 mV以上変わらなくなったら,マグネチックスターラーのスイッ
チを切る。少なくとも15秒間後に得られた値を記録する。
(11) ふっ化物イオン標準液(F− 1 mg/L)とふっ化物イオン標準液(F− 100 mg/L)との電位の差は,
110〜120 mV(25 ℃)の範囲に入り,ふっ化物イオンの濃度F− 0.1〜100 mg/Lの間の検量線は
直線になる。
(12) 次の測定を開始する前に,回転子,電極などを,次に測定する溶液ですすぐ。測定は,濃度の
薄いものから順に行う。高濃度の試料を測定した場合は,注(5)の操作を行った後,測定を続け
る。
d) 操作 操作は,次による。
1) 34.1.1 c)の蒸留操作で得た留出液から100 mLを測定容器にとり,緩衝液(pH5.2)10 mLを加える(7)。
2) c)の2)〜4)の操作を行って(12),検量線からふっ化物イオンの濃度を求め,試料中のふっ化物イオン
の濃度(F− mg/L)を算出する。
備考 3. 蒸留操作を行わず,ろ過による処理で測定する場合は,3.2に従って試料をろ過する。緩衝液
(pH5.2)(TISAB)25 mLを測定容器にとり,ろ過した試料25 mLを加える。次に,d) 2)の
操作を行う。検量線は,同じ操作で作成する。
緩衝液(pH5.2)(TISAB) JIS K 8150に規定する塩化ナトリウム58 g及びJIS K 8355に規
定する酢酸57 mLを,水500 mLを入れたビーカー1 000 mLに加える。溶けるまでかき混ぜ
る。水酸化ナトリウム溶液(5 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調
製する。)150 mLとtrans-1,2-シクロヘキサンジアミン四酢酸一水和物4 gとを加える。固形
物が全て溶けるまでかき混ぜ,pH計を用い,溶液を水酸化ナトリウム溶液(5 mol/L)でpH5.2
に調節する。
4. イオン濃度計の場合には,ふっ化物イオン標準液(F− 1 mg/L)と,ふっ化物イオン標準液
(F− 100 mg/L)とを用い,c)の2)及び3)の操作を行ってイオン濃度計の指示値を1 mg/L及
び100 mg/Lになるように調節する。さらに,ふっ化物イオン標準液(F− 0.1 mg/L)とふっ
化物イオン標準液(F− 10 mg/L)とを用いてイオン濃度計の指示値を確認する。
5. イオン電極法では,ふっ化物イオンだけが測定できるので,あらかじめふっ素化合物を蒸留
操作で全てふっ化物イオンにしてから測定する。
主な共存物質の許容限度を最大比率で次に示す。
25
K 0102:2019
HCO3−,Cl−,NO3−,I−,Br−,HPO42−:103
SO42−:104
水酸化物イオン,アルミニウムイオン及び鉄(III)イオンは,いずれも測定を妨害するが,
蒸留分離によって除去されるため影響はない。
備考 6. ふっ化物イオン電極による電位差滴定法 34.1.1 c)の蒸留操作で得た留出液から100 mLをビ
ーカーにとり,c)の2)〜4)の操作に準じて電位を測定しながら 〜 mol/Lの硝酸ランタ
ン(III)溶液で滴定して滴定曲線を作図し,滴定終点を求め,ふっ化物イオンの量を算出す
る。 mol/L硝酸ランタン(III)溶液1 mLは,F− 1.899 mgに相当する。
34.3 イオンクロマトグラフ法 試料をろ過した後,試料中のふっ化物イオンをイオンクロマトグラフ法
によって定量する。この方法を用いる場合には,試料採取後直ちに試験する。直ちに行えない場合には,0
〜10 ℃の暗所に保存し,できるだけ早く試験する。この方法は,清浄な試料に適用する。溶存のふっ化物
イオン及び容易にふっ化物イオンとなる錯イオンが定量される。
備考 7. 試料に妨害物質が含まれる場合は,34.1.1 c)の蒸留操作を行った後に適用する。また,ハロゲ
ン化物が多量に含まれる場合は,注(2)第3文(なお書きの部分)を除いた34.1.1 c)の蒸留操
作を行った後に適用し,留出液の液性の判定は,フェノールフタレイン溶液(5 g/L)の添加
によってではなく,pH試験紙によって行う。
蒸留操作を行った場合は,試料中のふっ素化合物が定量される。
試験操作などは,35.3による。
34.4 流れ分析法 試料中のふっ素化合物を,34.1.2と同様な原理で発色させる流れ分析法によって定量す
る。
定量範囲:F− 0.08〜10 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-6による。ただし,JIS K 0170-6の6.3.2(ランタン-アリザリンコンプレキ
ソン発色FIA法)による場合は,34.1.1 c)の蒸留操作を行った後に適用する。発色試薬にアルフッソンを
用いる場合には,蒸留終了後の留出液の中和に,塩酸(1+11)[21. a) 2)による。]を用いてもよい。
備考 8. 妨害物質,ハロゲン化物又はハロゲン化水素などが多量に含まれる試料に,JIS K 0170-6の
6.3.3(蒸留・ランタン-アリザリンコンプレキソン発色CFA法)を適用する場合は,試料に
一定量のふっ化物イオンを添加して試験操作を行ったときに得られる指示値の増加分と,同
量のふっ化物イオンを含む検量線用標準液について同様の操作を行ったときに得られる指示
値とを比較することによって回収率を求め,その値が80〜120 %の間にあることを確認し,
試料の分析値を回収率で補正する。回収率がこの範囲の外にある場合は,適用できない。
35.[塩化物イオン(Cl−)]の“備考4. 34.の備考3.による。”を削除する。
35.[塩化物イオン(Cl−)]の“注(5) 34.の注(13)による。”を,“注(5) 34.の注(5)による。”に置き換える。
35.[塩化物イオン(Cl−)]の“注(6) 34.の注(14)による。”を,“注(6) 34.の注(6)による。”に置き換える。
35.[塩化物イオン(Cl−)]の“注(9) 34.の注(15)による。”を,“注(9) 34.の注(8)による。”に置き換える。
35.[塩化物イオン(Cl−)]の“注(10) 34.の注(16)による。”を,“注(10) 34.の注(9)による。”に置き換える。
30
1
30
1
300
1
26
K 0102:2019
35.[塩化物イオン(Cl−)]の“注(11) 34.の注(17)による。”を,“注(11) 34.の注(10)による。”に置き換える。
35.[塩化物イオン(Cl−)]の“注(13) 34.の注(19)による。”を,“注(13) 34.の注(12)による。”に置き換え
る。
35.[塩化物イオン(Cl−)]の“備考5.”を,“備考4.”に置き換える。
35.[塩化物イオン(Cl−)]の“備考6.”を,“備考5.”に置き換える。
35.[塩化物イオン(Cl−)]の“備考7.”を,“備考6.”に置き換える。
35.[塩化物イオン(Cl−)]の35.3(イオンクロマトグラフ法)a) 2)(溶離液)の“備考8.”を,“備考7.”
に置き換える。
35.[塩化物イオン(Cl−)]の35.3(イオンクロマトグラフ法)a) 3)(再生液)の“備考8.”を,“備考7.”
に置き換える。
35.[塩化物イオン(Cl−)]の“注(21) 備考8.による。”を,“注(21) 備考7.による。”に置き換える。
35.[塩化物イオン(Cl−)]の“備考8.”を,“備考7.”に置き換える。
35.[塩化物イオン(Cl−)]の35.3(イオンクロマトグラフ法)c) 1)の“試料を3.2によってろ過する。”を,
“試料を孔径0.45 μm以下のフィルターによってろ過する。”に置き換える。
35.[塩化物イオン(Cl−)]の“備考9.”を,“備考8.”に置き換える。
35.[塩化物イオン(Cl−)]の“備考10.”を,“備考9.”に置き換える。
35.[塩化物イオン(Cl−)]の備考10.の“備考8.”を,“備考7.”に置き換える。
35.[塩化物イオン(Cl−)]の“備考11.”を,“備考10.”に置き換える。
38.(シアン化合物)の全文を,次に置き換える。
38. シアン化合物 シアン化合物は,水中のシアン化物イオン,シアノ錯体などを総称し,シアン化物イ
オンと全シアンとに区分する。
シアン化合物は,前処理でシアン化物イオンとし,定量には,ピリジン-ピラゾロン吸光光度法,4-ピリ
ジンカルボン酸-ピラゾロン吸光光度法,イオン電極法又は4-ピリジンカルボン酸-ピラゾロン発色流れ分
析法を適用する。
シアン化合物は変化しやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によ
って保存し,できるだけ早く試験する。
27
K 0102:2019
なお,流れ分析法は,2002年に第1版として発行されたISO 14403との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 14403:2002,Water quality−Determination of total cyanide and free cyanide by continuous flow
analysis(MOD)
38.1 前処理 試料を微酸性として通気又は加熱蒸留し,発生するシアン化水素を捕集する。
38.1.1 シアン化物 この前処理ではシアン化物イオン及び錯生成定数の小さい亜鉛,カドミウムなどのシ
アノ錯体からはほぼ完全に,また,ニッケル,銅などのシアノ錯体からは一部シアン化水素を発生する。
鉄(II)及び鉄(III)のシアノ錯体からは,シアン化水素は発生しない。
38.1.1.1 通気法(pH5.0で発生するシアン化水素) 試料のpHを5.0に調節し,恒温水槽で40 ℃に保持
しながら,約1.2 L/minで通気し,発生したシアン化水素を水酸化ナトリウム溶液に捕集する。
a) 試薬 試薬は,次による。
1) 酢酸(1+1) JIS K 8355に規定する酢酸を用いて調製する。
2) 酢酸(1+49) JIS K 8355に規定する酢酸を用いて調製する。
3) 水酸化ナトリウム溶液(200 g/L) JIS K 8576に規定する水酸化ナトリウム20 gを水に溶かして100
mLとする。
4) 水酸化ナトリウム溶液(20 g/L) 水酸化ナトリウム溶液(200 g/L)を水で10倍に薄める。
b) 装置 装置は,次による。
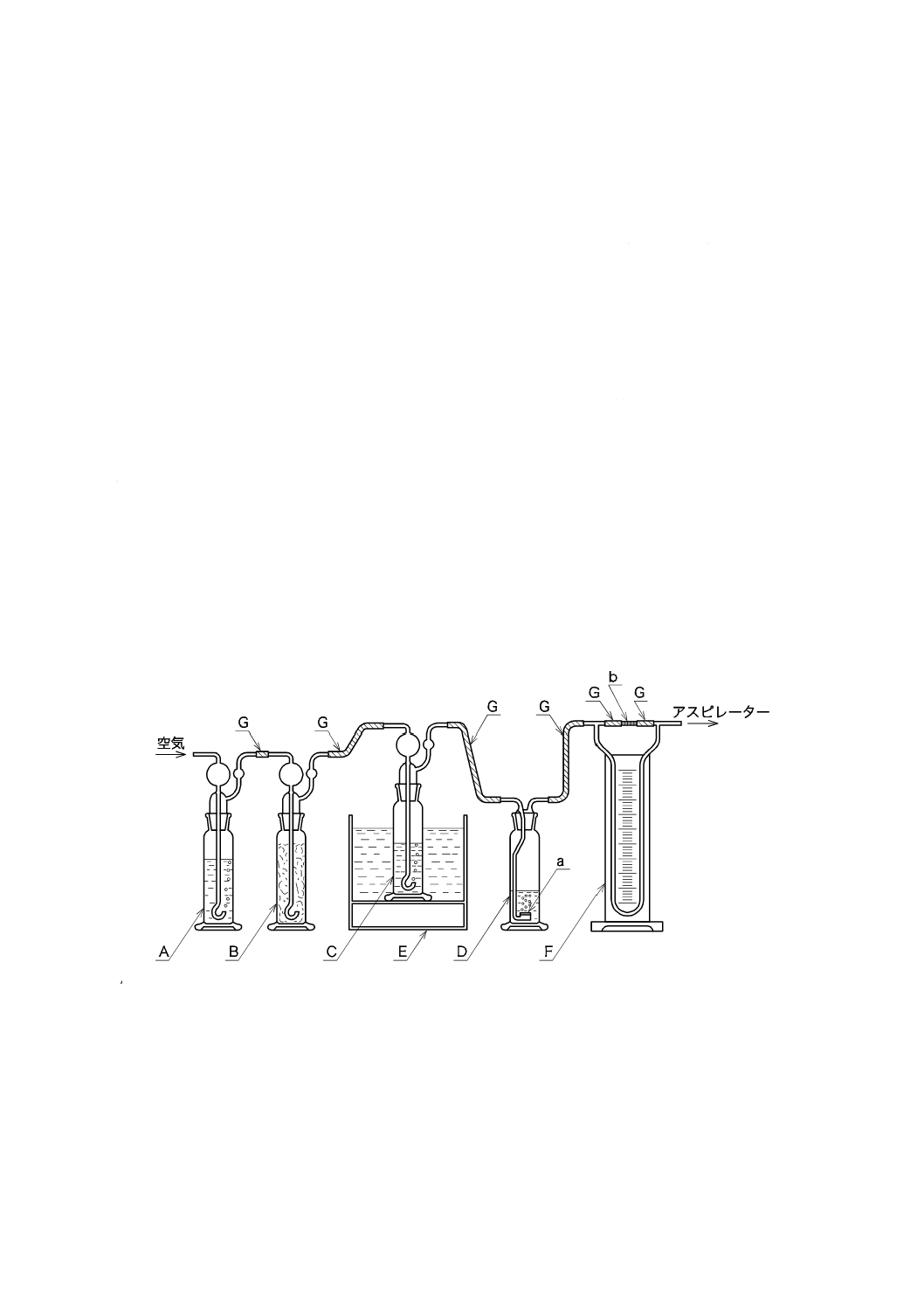
1) 通気装置 図38.1に例を示す。
A:
B:
C:
D:
ガス洗浄瓶250 mL
水酸化ナトリウム溶液(200 g/L)100 mLを入れる。
ガス洗浄瓶250 mL
ガラスウールを軽く詰めておく。
ガス洗浄瓶250 mL(試料用)
ろ過板付きガス洗浄瓶250 mL(シアン化水素吸収用)
E:
F:
G:
a:
b:
恒温水槽(40±2 ℃)
流量計
軟質塩化ビニル管又はシリコーンゴム管
ガラスろ過板G2
毛管
図38.1 通気装置の例
c) 通気操作 通気操作は,次による。
28
K 0102:2019
1) 通気装置を図38.1のように組み立て,ろ過板付きガス洗浄瓶(D)には,シアン化水素吸収用とし
て水40 mLと水酸化ナトリウム溶液(20 g/L)20 mLとを入れる。
2) 試料100 mL又は適量をビーカー300 mLにとり,pH計を用いてpH5.0±0.2になるまで酢酸(1+1)
及び酢酸(1+49)又は水酸化ナトリウム溶液(20 g/L)を滴加し,その量を求める。試料の適量は,
38.2〜38.4のそれぞれの方法に規定した定量範囲から求めた最適量で,100 mL以下とする。
3) ガス洗浄瓶(C)に2)と同量の試料を入れ,水を加えて100 mLにした後,2)の操作で求めた,酢酸
(1+1)及び酢酸(1+49)又は水酸化ナトリウム溶液(20 g/L)の量を加え,図38.1のように連結
する。試料中に,油脂類,残留塩素などの酸化性物質,又は硫化物などの還元性物質が含まれてい
る場合には,あらかじめ備考1.〜備考3.に示す方法によって除去する。
4) 恒温水槽を40±2 ℃に保持して,約1.2 L/minで1時間通気する。
5) 通気後ろ過板付きガス洗浄瓶(D)の中の水酸化ナトリウム溶液(吸収液)を全量フラスコ100 mL
に移し入れ,ろ過板付きガス洗浄瓶(D)を水で洗い,洗液も移し入れて水を標線まで加える。
備考 1. 試料中に多量の油脂類が含まれている場合には,あらかじめ酢酸又は水酸化ナトリウムを加
えてpHを6〜7に調節し,分液漏斗に移し入れる。試料の体積百分率約2 %量のヘキサンを
加えて,静かに振り混ぜ,放置して油脂類を分離した後,38.1.1.1の操作を行う。
2. 試料中に残留塩素などの酸化性物質が含まれている場合には,L(+)-アスコルビン酸溶液(100
g/L)[JIS K 9502に規定するL(+)-アスコルビン酸10 gを水に溶かして100 mLとする。]を
加えて還元する。
3. 硫化物が含まれている場合には,あらかじめ酢酸亜鉛溶液(100 g/L)[38.1.1.2 a) 4)による。]
2 mLを加える。酢酸亜鉛溶液(100 g/L)1 mLは,硫化物イオン約14 mgに相当する。
38.1.1.2 加熱蒸留法(pH5.5で酢酸亜鉛の存在下で発生するシアン化水素) 試料に酢酸亜鉛を加え,pH5.5
に調節して加熱蒸留し,発生するシアン化水素を水酸化ナトリウム溶液に捕集する。
a) 試薬 試薬は,次による。
1) 酢酸(1+1) 38.1.1.1 a) 1)による。
2) 酢酸(1+49) 38.1.1.1 a) 2)による。
3) 水酸化ナトリウム溶液(20 g/L) 38.1.1.1 a) 4)による。
4) 酢酸亜鉛溶液(100 g/L) JIS K 8356に規定する酢酸亜鉛二水和物12 gを水に溶かして100 mLと
する。
b) 装置 装置は,次による。
1) 蒸留装置 図38.2に例を示す。
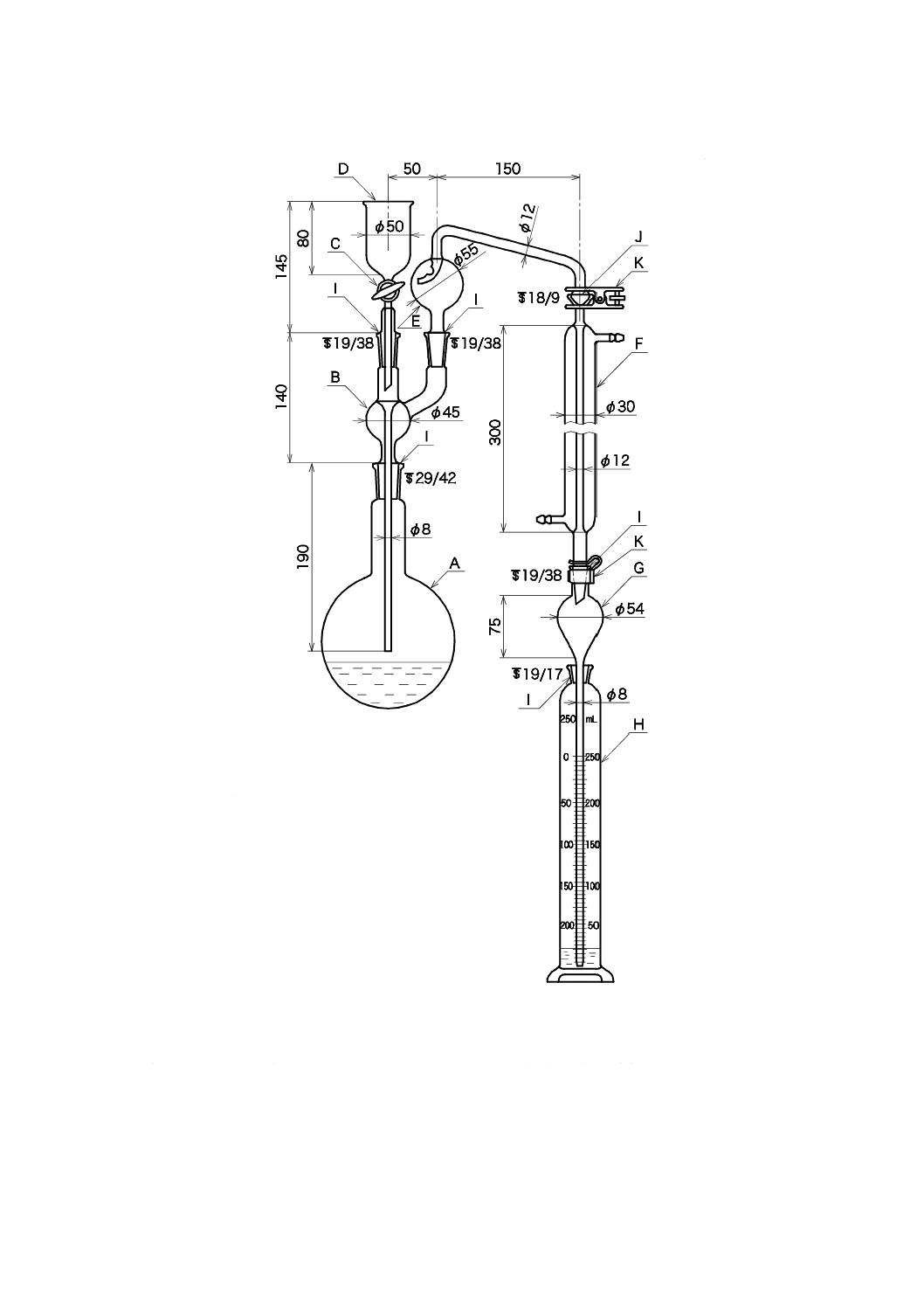
29
K 0102:2019
単位 mm
図38.2 蒸留装置の例
c) 蒸留操作 蒸留操作は,次による。
1) 試料500 mL又は適量をビーカー1 000 mLにとり,酢酸(1+1)を滴加し,pH計を用いてpH約7
とし,この中和に必要な添加量を求める。試料の適量は,38.2〜38.4のそれぞれの方法に規定した
定量範囲から求めた最適量で,500 mL以下とする。
2) これに酢酸亜鉛溶液(100 g/L)20 mLを加え,再び酢酸(1+49)を滴加し,pH計を用いてpH5.5
A: 蒸留フラスコ1 000 mL(又は500 mL)
B: 連結導入管
C: すり合わせコック
D: 注入漏斗
E: トラップ球(ケルダール球)
F: リービッヒ冷却器300 mm
G: 逆流止め(約50 mL)
H: 受器[メスシリンダー(有栓形)250 mL(又は100 mL)]
I: 共通すり合わせ
J: 共通球面すり合わせ
K: 押さえばね
30
K 0102:2019
に調節する。この酢酸(1+49)の添加量を求める。
3) 蒸留フラスコ1 000 mLに2)と同量の試料をとり,水で500 mLとした後,沸騰石(粒径2〜3 mm)
を入れる。
4) これに1)で求めた酢酸(1+1)を加え,蒸留フラスコを図38.2のように蒸留装置に接続する。
5) 蒸留装置の受器には,メスシリンダー(有栓形)250 mLを用い,これに水酸化ナトリウム溶液(20
g/L)20 mLを入れ,受器を図38.2のように接続する。
6) 次に,注入漏斗から,酢酸亜鉛溶液(100 g/L)20 mLを加え,更に2)で求めた酢酸(1+49)を加
える。
7) 蒸留フラスコを加熱し,留出速度を2〜3 mL/minに調節し,3 mL/min以上にしない。受器の液量が
約230 mLになるまで蒸留する。蒸留中は,逆流止めの先端を,常に受器の液面下約15 mmを保つ
ように,メスシリンダー(有栓形)250 mLの高さを調節する。
8) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加え,更に水を250 mLの標線まで加える。
備考 4. 試料中に多量の油脂類が含まれている場合には,備考1.と同じ操作を行う。
5. 試料中に残留塩素などの酸化性物質が含まれている場合には,備考2.と同じ操作を行う。
6. 試料中に硫化物などの還元性物質が含まれている場合には,c)の1)〜7)を行って得た,留出
液に対し,次のような酸化処理を行った後,再び蒸留操作を行って除去する。
− 蒸留操作を行った受器中の留出液と洗液とを再び蒸留フラスコに移し,指示薬としてフ
ェノールフタレイン溶液(5 g/L)[15.の備考2.による。]2,3滴を加え,酢酸(1+1)
で中和し,更に硝酸(50 mmol/L)(JIS K 8541に規定する硝酸3.8 mLを水に溶かして1
Lとする。)約30 mLを加える。
− 次に,過マンガン酸カリウム溶液(3 g/L)(JIS K 8247に規定する過マンガン酸カリウ
ムを用いて調製する。)を滴加し,過マンガン酸の微紅色になる点又は酸化マンガン(IV)
の褐色の濁りが生成した点から更に過剰に1 mLを加え,水を加えて約300 mLとする。
− 蒸留フラスコを図38.2のように蒸留装置に接続し,受器にはメスシリンダー(有栓形)
100 mLを用い,これに水酸化ナトリウム溶液(20 g/L)20 mLを入れ,受器を図38.2の
ように接続する。
− 蒸留フラスコを加熱し,留出速度を2〜3 mL/minに調節し,受器の液量が約90 mLにな
ったら蒸留を止める。冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内
外を少量の水で洗い,洗液も受器中に加えた後,水を100 mLの標線まで加える。
38.1.2 全シアン(pH2以下で発生するシアン化水素) 試料にりん酸を加えてpH2以下にし,エチレンジ
アミン四酢酸二水素二ナトリウムを加えて加熱蒸留し,発生したシアン化水素を水酸化ナトリウム溶液に
捕集する。
備考 7. 前処理によってシアン化物イオン及びほとんどのシアノ錯体中のシアンは,留出する。酸化
性物質が共存する状態で蒸留すると,チオシアン酸,2-プロペンニトリル(アクリロニトリ
ル)などが分解してシアン化水素が発生するので,あらかじめ酸化性物質を還元しておく。
a) 試薬 試薬は,次による。
1) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
2) 水酸化ナトリウム溶液(20 g/L) 38.1.1.1 a) 4)による。
3) 水酸化ナトリウム溶液(40 g/L)21. a) 3)による。
31
K 0102:2019
4) アミド硫酸アンモニウム溶液(100 g/L) JIS K 8588に規定するアミド硫酸アンモニウム10 gを水
に溶かして100 mLとする。
5) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物10 gを水
に溶かし,水酸化ナトリウム溶液(20 g/L)5〜7滴を加えて微アルカリ性とし,水を加えて100 mL
とする。
6) りん酸 JIS K 9005に規定するもの。
b) 装置 装置は,次による。
1) 蒸留装置 図38.2に例を示す。
c) 蒸留操作 蒸留操作は,次による。
なお,試料に一定量のシアン化物イオン標準液を添加して,備考11.の蒸留操作による回収率試験を
行い,回収率が80〜120 %であることを確認した場合は,備考11.によってもよい。
1) 試料50 mLを蒸留フラスコ500 mLにとり,水を加えて約250 mLとする。沸騰石(粒径2〜3 mm)
を加える。指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加える。
2) アルカリ性の場合には,溶液の紅色が消えるまで,りん酸を滴加し,溶液を弱酸性にする。
3) 次に,アミド硫酸アンモニウム溶液(100 g/L)1 mL (1)を加える。
4) 蒸留フラスコを図38.2のように接続し,受器にはメスシリンダー(有栓形)100 mLを用い,これ
に水酸化ナトリウム溶液(20 g/L)20 mLを入れ,図38.2のように接続する。
5) 注入漏斗から蒸留フラスコにりん酸10 mLを加え,次に,EDTA溶液10 mLを加え,少量の水で注
入漏斗を洗い,洗液を蒸留フラスコに加える。
6) 数分間放置した後,蒸留フラスコを加熱し,留出速度2〜3 mL/minで受器の液量が約90 mLになる
まで蒸留する。留出速度は3 mL/min以上にしない。蒸留中は,逆流止めの先端を,常に受器の液面
下約15 mmを保つように,メスシリンダー(有栓形)100 mLの高さを調節する。
7) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加えた後,更に水を100 mLの標線まで加える。
注(1) アミド硫酸アンモニウム溶液(100 g/L)は,試料中の亜硝酸イオンの妨害を除くために加える。
これを加えない場合には,亜硝酸イオンが存在すると,加熱蒸留時にEDTAと反応してシアン
化水素を生成する。アミド硫酸アンモニウム溶液(100 g/L)1 mLは,亜硝酸イオン約40 mgに
相当する。亜硝酸イオンが40 mg以上共存する場合には,その量に応じて添加量を増加する。
特殊な試料では,亜硝酸イオン以外にもEDTAとの反応によってシアン化水素を生成し,ア
ミド硫酸アンモニウム溶液(100 g/L)の添加によってもその妨害を除けないものもある。添加
したEDTAが関与すると考えられる場合は,EDTA溶液の添加を除いて1)〜7)の操作を行う。
なお,EDTA以外に類似の反応をする有機物もある。
備考 8. 油脂類の除去は,備考1.の操作を行う。
9. 試料中に残留塩素などの酸化性物質が含まれている場合には,備考2.の操作を行う。
10. 試料中に硫化物などの還元性物質が含まれている場合には,全シアンの蒸留操作を行って得
た留出液について備考6.の操作を行う。
11. 図28.1の小型蒸留装置を用いる蒸留操作は,次による。
1) 試料25 mLを蒸留容器にとり,水を加えて全量を約50 mLとする。沸騰石(粒径2〜3 mm)
を加える。指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加え,溶液の赤い色
が消えるまで,りん酸を滴加する。次に,アミド硫酸アンモニウム溶液(100 g/L)0.5 mL(*)
32
K 0102:2019
を加える。
2) 蒸留容器の上部からりん酸及びEDTA溶液2 mLずつを加え,直ちに蒸留容器に蒸留管,
冷却器をセットする。これを加熱器に取り付ける。受器にメスシリンダー(有栓形)50 mL
などを用い,これに水酸化ナトリウム溶液(40 g/L)5 mLを入れる。
3) 留出速度0.3〜0.7 mL/minで,受器の液量が約30 mLになるまで蒸留する。
なお,蒸留中は冷却器に接続された逆流止めの先端が受器中の溶液に浸かっているよ
うにする。
4) 冷却器を装置から外し,冷却器内を少量の水で洗い,洗液も受器に加えた後,更に水を
50 mLの標線まで加える。
注(*) 注(1)による。
38.2 ピリジン-ピラゾロン吸光光度法 38.1で前処理して得られたシアン化物イオン溶液の一部をとり,
酢酸で中和した後,クロラミンT溶液を加えて塩化シアンとし,これにピリジン-ピラゾロン溶液を加える。
このとき生じる青い色の吸光度を測定してシアン化物イオンを定量する。
定量範囲:CN− 0.5〜9 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 酢酸(1+8) JIS K 8355に規定する酢酸を用いて調製する。
2) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
3) りん酸塩緩衝液(pH6.8) JIS K 9007に規定するりん酸二水素カリウム17.0 gとJIS K 9020に規定
するりん酸水素二ナトリウム17.8 gとを水に溶かして500 mLとする。
4) クロラミンT溶液(10 g/L) JIS K 8318に規定するp-トルエンスルホンクロロアミドナトリウム三
水和物(クロラミンT)0.62 gを水に溶かして50 mLとする。使用時に調製する。
5) ピリジン-ピラゾロン溶液 JIS K 9548に規定する3-メチル-1-フェニル-5-ピラゾロン0.25 gをJIS K
8777に規定するピリジン20 mLに溶かし,これにJIS K 9545に規定するビス(3-メチル-1-フェニ
ル-5-ピラゾロン)20 mgを溶かし,更に水100 mLを加えて混ぜる。10 ℃以下であれば,1 週間は
使用できる。
6) 0.1 mol/L硝酸銀溶液 JIS K 8550に規定する硝酸銀17 gを水に溶かして1 Lとする。着色ガラス瓶
に保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを600 ℃で約1時間加熱し,デシケ
ーター中で放冷する。NaCl 100 %に対してその1.17 gを1 mgの桁まではかりとり,少量の水に溶
かして全量フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLをとり,水を加えて液量を約50 mLとし,デキストリン溶液[35.1 a) 4)による。]5 mL
及び指示薬としてジクロロフルオレセインナトリウム溶液(2 g/L)[35.1 a) 3)による。]3,4滴を
加え,0.1 mol/L硝酸銀溶液で滴定し,黄緑の蛍光が消え,僅かに赤くなるときを終点とする。次
の式によって0.1 mol/L硝酸銀溶液のファクター(f)を算出する。
844
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 塩化ナトリウムの量(g)
b: 塩化ナトリウムの純度(%)
x: 滴定に要した0.1 mol/L硝酸銀溶液(mL)
33
K 0102:2019
0.005 844: 0.1 mol/L硝酸銀溶液1 mLの塩化ナトリウム相当量(g)
7) シアン化物イオン標準液(CN− 1 mg/mL) JIS K 8443に規定するシアン化カリウム0.63 gを少量
の水に溶かし,水酸化ナトリウム溶液(20 g/L)2.5 mLを加え,水で250 mLとする。この溶液は,
使用時に調製し,その濃度は,次の方法で求める。
この溶液100 mLをとり,指示薬として5-(4-ジメチルアミノベンジリデン)ロダニンのアセト
ン溶液(0.2 g/L)[JIS K 8495に規定する5-(4-ジメチルアミノベンジリデン)ロダニン20 mgを
JIS K 8034に規定するアセトン100 mLに溶かす。]0.5 mLを加え,0.1 mol/L硝酸銀溶液で滴定し,
溶液の色が黄色から赤になったときを終点とする。次の式によってシアン化物イオン標準液の濃度
(CN− mg/mL)を算出する。
100
1
204
.5
×
×
×
=
f
a
C
ここに,
C: シアン化物イオン標準液(CN− mg/mL)
a: 滴定に要した0.1 mol/L硝酸銀溶液(mL)
f: 0.1 mol/L硝酸銀溶液のファクター
5.204: 0.1 mol/L硝酸銀溶液1 mLのシアン化物イオン相当量(mg)
8) シアン化物イオン標準液(CN− 1 μg/mL) シアン化物イオン標準液(CN− 1 mg/mL)10 mLを全
量フラスコ1 000 mLにとり,水酸化ナトリウム溶液(20 g/L)100 mLを加えた後,水を標線まで加
える。その10 mLを全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。この
溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/mL)の濃度から算出する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から10 mL(CN−として0.5〜9 μgを含む。)を全量
フラスコ50 mLにとる。
2) 指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加え,静かに振り混ぜながら溶液の紅色が
消えるまで酢酸(1+8)を滴加する。
3) りん酸塩緩衝液(pH6.8)10 mLを加え,pHを6.8とした後,密栓して静かに振り混ぜる。
4) これにクロラミンT溶液(10 g/L)0.25 mLを加え,直ちに密栓して静かに振り混ぜ,約5分間放置
する。
5) ピリジン-ピラゾロン溶液15 mLを加え,更に水を標線まで加え,密栓して静かに振り混ぜる。
6) 約25 ℃の水浴中に約30分間浸し,溶液の色がうすい紅から紫を経て安定な青になるまで発色させ
る。
7) 発色後1時間以内に溶液の一部を吸収セルに移し,波長620 nm付近の吸光度を測定する。
8) 空試験として水10 mLを全量フラスコ50 mLにとり,3)〜7)の操作を行って吸光度を測定し,試料
について得た吸光度を補正する。
9) 検量線からシアン化物イオンの量を求め,試料中のシアン化物イオンの濃度(CN− mg/L)を算出
する。
d) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 1 μg/mL)0.5〜9 mLを全量フラスコ50 mLに段階的にとり,水を
加えて約10 mLとする。
2) c)の2)〜8)の操作を行ってシアン化物イオン(CN−)の量と吸光度との関係線を作成する。
34
K 0102:2019
38.3 4-ピリジンカルボン酸-ピラゾロン吸光光度法 38.1の前処理で得られたシアン化物イオン溶液の一
部をとり,酢酸で中和した後,クロラミンT溶液を加えて塩化シアンとし,これに4-ピリジンカルボン酸
-ピラゾロン溶液を加え,生成する青い色の吸光度を測定してシアン化物イオンを定量する。
定量範囲:CN− 0.5〜9 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 酢酸(1+8) 38.2 a) 1)による。
2) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
3) りん酸塩緩衝液(pH7.2) JIS K 9020に規定するりん酸水素二ナトリウム17.8 gを水約300 mLに
溶かし,りん酸二水素カリウム溶液(200 g/L)(JIS K 9007に規定するりん酸二水素カリウムを用
いて調製する。)をpH7.2になるまで加え,水で500 mLとする。
4) クロラミンT溶液(10 g/L) 38.2 a) 4)による。
5) 4-ピリジンカルボン酸-ピラゾロン溶液 JIS K 9548に規定する3-メチル-1-フェニル-5-ピラゾロン
0.3 gを,JIS K 8500に規定するN, N-ジメチルホルムアミド20 mLに溶かす。別に,4-ピリジンカ
ルボン酸1.5 gを水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]約20 mLに溶かし,塩酸(1+
10)(JIS K 8180に規定する塩酸を用いて調製する。)を滴加してpHを約7とする。この溶液に代
え,4-ピリジンカルボン酸ナトリウム1.8 gを水約50 mLに溶かした溶液を用いることもできる。ピ
ラゾロン溶液と4-ピリジンカルボン酸溶液とを合わせ,水を加えて100 mLとする。この溶液は,
10 ℃以下の暗所に保存し,20日間以上経過したものは使用しない。
6) シアン化物イオン標準液(CN− 1 μg/mL) 38.2 a) 8)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から10 mL(CN−として0.5〜9 μgを含む。)を全量
フラスコ50 mLにとる。
2) 指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加え,静かに振り混ぜながら酢酸(1+8)
を滴加して中和した後,りん酸塩緩衝液(pH7.2)10 mLを加え,pHを7〜8にする。
3) クロラミンT溶液(10 g/L)0.5 mLを加え,約25 ℃の水浴中に約5分間放置する。
4) 4-ピリジンカルボン酸-ピラゾロン溶液10 mLを加え,更に水を標線まで加え,密栓して静かに振り
混ぜた後,約25 ℃の水浴中で約30分間放置する。
5) 発色後1時間以内に溶液の一部を吸収セルに移し,波長638 nm付近の吸光度を測定する。
6) 空試験として水10 mLを全量フラスコ50 mLにとり,りん酸塩緩衝液(pH7.2)10 mLを加えた後,
3)〜5)の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
7) 検量線からシアン化物イオンの量を求め,試料中のシアン化物イオンの濃度(CN− mg/L)を算出
する。
d) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 1 μg/mL)0.5〜9 mLを全量フラスコ50 mLに段階的にとり,水を
加えて約10 mLとする。
2) c)の2)〜6)の操作を行ってシアン化物イオン(CN−)の量と吸光度との関係線を作成する。
38.4 イオン電極法 38.1(ただし,備考10.は除く。)の前処理で得られたシアン化物イオン溶液(pH12
〜13)について,シアン化物イオン電極を指示電極として電位を測定し,シアン化物イオンを定量する。
35
K 0102:2019
定量範囲:CN− 0.1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 水酸化ナトリウム溶液(0.1 mol/L) JIS K 8576に規定する水酸化ナトリウム4 gを水に溶かして1
Lとする。
2) シアン化物イオン標準液(CN− 100 mg/L) 38.2 a) 7)のシアン化物イオン標準液(CN− 1 mg/mL)
20 mLを全量フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える。使用
時に調製する。この溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/mL)の濃度から算出する。
3) シアン化物イオン標準液(CN− 10 mg/L) シアン化物イオン標準液(CN− 100 mg/L)20 mLを全
量フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える。使用時に調製
する。この溶液の濃度は,シアン化物イオン標準液(CN− 100 mg/L)の濃度から算出する。
4) シアン化物イオン標準液(CN− 1 mg/L) シアン化物イオン標準液(CN− 10 mg/L)20 mLを全量
フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える。使用時に調製す
る。この溶液の濃度は,シアン化物イオン標準液(CN− 10 mg/L)の濃度から算出する。
5) シアン化物イオン標準液(CN− 0.1 mg/L) シアン化物イオン標準液(CN− 1 mg/L)20 mLを全量
フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える。使用時に調製す
る。この溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/L)の濃度から算出する。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 34.2 b) 1)による。
2) 指示電極 シアン化物イオン電極
3) 参照電極 34.2 b) 3)による。
4) 測定容器 35.2 b) 4)による。
5) 恒温槽 34.2 b) 5)による。
6) マグネチックスターラー 35.2 b) 6)による。
c) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 0.1 mg/L)100 mLを測定容器にとる。
2) 恒温槽から水を送り,測定容器の溶液を25±0.5 ℃にする。
3) 指示電極(2)(3)と参照電極(4)とを浸し,マグネチックスターラー(5)で泡が電極に触れない程度に強く
かき混ぜる(6)。
4) 液温を確認し,電位差計で電位を測定する(7)。
5) シアン化物イオン標準液(CN− 1 mg/L)100 mL,シアン化物イオン標準液(CN− 10 mg/L)100 mL
及びシアン化物イオン標準液(CN− 100 mg/L)100 mLをそれぞれ測定容器にとる。
6) 2)〜4)の操作を行って電位を測定する(8)(9)。
7) 横軸にシアン化物イオンの濃度の対数を,縦軸に電位をとり,シアン化物イオンの濃度(CN− mg/L)
と電位との関係線を作成する(8)。
注(2) 指示電極(シアン化物イオン電極)は,使用時にシアン化物イオン標準液(CN− 0.1 mg/L)に
浸し,指示値が安定してから使用する。シアン化物イオン電極の応答時間は,液温10〜30 ℃
の場合,シアン化物イオンの濃度が0.1 mg/Lで約1分間,1 mg/L以上であれば約30秒間であ
る。
(3) 34.の注(5)の第2文及び第3文による。
(4) 34.の注(6)による。
36
K 0102:2019
注(5) 34.の注(8)による。
(6) 34.の注(9)による。
(7) シアン化物イオン電極には,よう化銀を用いたものが多いので,直射日光を受けると電位が大
幅に変動して,正の誤差を与える。室内照明の影響は少ない。
(8) シアン化物イオン標準液(CN− 0.1 mg/L)とシアン化物イオン標準液(CN− 10 mg/L)との電
位の差は,110〜120 mV(25 ℃)の範囲に入り,シアン化物イオンの濃度0.1〜100 mg/Lの間
の検量線は直線になる。
(9) 34.の注(12)による。
d) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から100 mLを測定容器にとる。
2) c)の2)〜4)の操作を行って(9),検量線からシアン化物イオンの濃度を求め,試料中のシアン化物イ
オンの濃度(CN− mg/L)を算出する。
備考 12. イオン濃度計の場合には,シアン化物イオン標準液(CN− 0.1 mg/L)及びシアン化物イオン
標準液(CN− 10 mg/L)を用い,c)の2)〜4)の操作を行ってイオン濃度計の指示値をCN− 0.1
mg/L及びCN− 10 mg/Lになるように調節する。さらに,その他のシアン化物イオン標準液
(CN− 1 mg/L)及びシアン化物イオン標準液(CN− 100 mg/L)を用いて,イオン濃度計の
指示を確認する。
13. 硫化物イオン及びメルカプト酢酸(チオグリコール酸)の妨害は,前処理で除く。亜硫酸イ
オンは,シアン化物イオンの103倍以内であれば妨害しないので,備考6.の酸化処理を省略
することができる。ホルムアルデヒドは,負の誤差を与える。
主な共存物質の許容限度を最大比率で次に示す。
Cl−,F−,NO3−,CrO42−,K+,Na+: 10 4
Br−,SCN−,HCO3−,CO32−,SO32−,SO42−,PO43−: 10 3
S2O32−,Ag+: 10
I−: 0.1
14. イオン電極による電位差滴定法 38.1(ただし,備考10.は除く。)の前処理で得られたシア
ン化物イオン溶液のうち100 mLを測定容器にとり,指示電極(シアン化物イオン電極又は
銀イオン電極)を用い,c)の2)〜4)の操作に準じて電位を測定しながら1〜100 mmol/L硝酸
銀溶液で滴定し,滴定曲線を作図する。滴定曲線から滴定終点を求め,シアン化物イオンの
濃度を算出する。100 mmol/L硝酸銀溶液1 mLは,シアン化物イオン5.204 mgに相当する。
38.5 流れ分析法 試料中のシアン化物イオンを,38.3と同様な原理で発色させる流れ分析法によって定
量する。試料を,38.1.1.1,38.1.1.2又は38.1.2の操作で前処理した後に適用する。この場合は,試料中に
懸濁物が含まれても,蒸留前処理で除去可能である。
定量範囲:CN− 0.01〜1 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-9(シアン化物)による。ただし,7.3.5[蒸留(pH3.8)−4-ピリジンカル
ボン酸・ジメチルバルビツール酸発色CFA法]及び7.3.6[ガス拡散(pH3.8)−4-ピリジンカルボン酸・
ジメチルバルビツール酸発色CFA法]の方法は除く。
42.[アンモニウムイオン(NH4+)]の全文を,次に置き換える。
37
K 0102:2019
42. アンモニウムイオン(NH4+) アンモニウムイオンの定量には,インドフェノール青吸光光度法,中
和滴定法,イオン電極法,イオンクロマトグラフ法,インドフェノール青発色流れ分析法又はサリチル酸-
インドフェノール青吸光光度法を適用する。
アンモニウムイオンは変化しやすいから,試験は試料採取後,直ちに行う。直ちに行えない場合には,
3.3によって保存し,できるだけ早く試験する。
なお,蒸留法及び中和滴定法は,1984年に第1版として発行されたISO 5664,サリチル酸-インドフェ
ノール青吸光光度法は,1984年に第1版として発行されたISO 7150-1,イオン電極法は,1984年に第1
版として発行されたISO 6778,イオンクロマトグラフ法は,1998年に第1版として発行されたISO 14911,
流れ分析法は,2005年に第2版として発行されたISO 11732との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 5664:1984,Water quality−Determination of ammonium−Distillation and titration method(MOD)
ISO 7150-1:1984,Water quality−Determination of ammonium−Part 1: Manual spectrometric method
(MOD)
ISO 6778:1984,Water quality−Determination of ammonium−Potentiometric method(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+,Na+,NH4+,K+,Mn2+,Ca2+,
Mg2+,Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
ISO 11732:2005,Water quality−Determination of ammonium nitrogen−Method by flow analysis
(CFA and FIA) and spectrometric detection(MOD)
備考 1. インドフェノール青吸光光度法,中和滴定法,イオン電極法,インドフェノール青発色流れ
分析法及びサリチル酸-インドフェノール青吸光光度法は,試料を蒸留処理してアンモニウム
イオンを共存物から分離した後,適用する。イオン電極法及びインドフェノール青発色流れ
分析法では,妨害物質を含まない試料の場合は,蒸留処理を省略できる。また,イオンクロ
マトグラフ法は,妨害物質を含まない試料に適用し,3.3の試料の保存処理及び42.1の前処
理(蒸留法)は行わず,試料採取後,直ちに試験する。
42.1 前処理(蒸留法) 試料に酸化マグネシウムを加えて弱いアルカリ性とし,蒸留を行い,留出したア
ンモニアを硫酸(25 mmol/L)に吸収捕集する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) JIS K 8951に規定する硫酸約1.4 mLをあらかじめ水100 mLを入れたビーカー
に加えてよくかき混ぜ,水を加えて1 Lとする。
3) 硫酸(1+35) 30.1.1 a) 2)による。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 酸化マグネシウム JIS K 8432に規定する酸化マグネシウムを使用前に600 ℃で約30分間加熱し,
デシケーター中で放冷する。
6) ブロモチモールブルー溶液(1 g/L) 16.1の注(1)による。
b) 装置 装置は,次による。
1) 蒸留装置 図42.1に例を示す。ガラス器具類は,使用前に水でよく洗う。
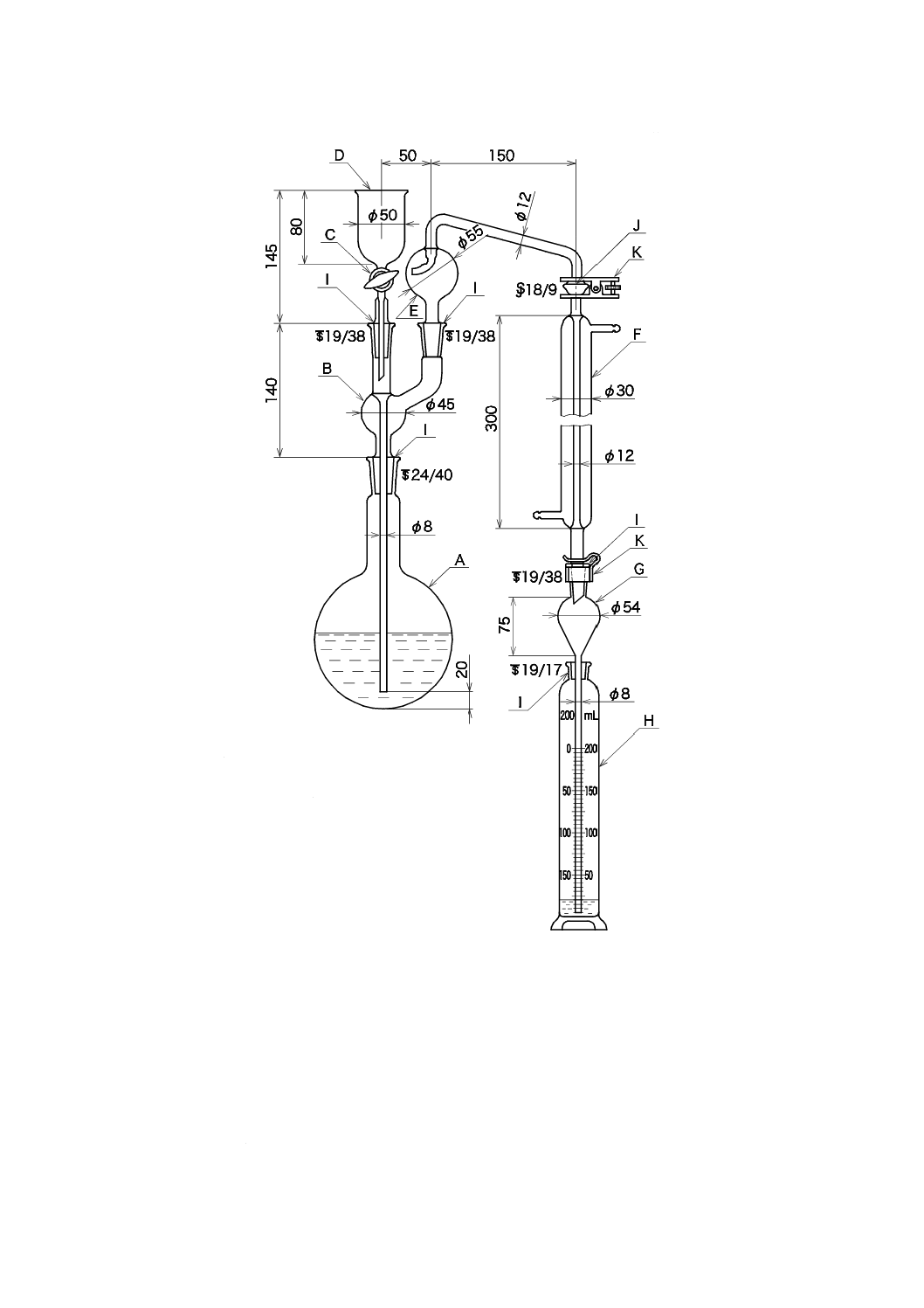
38
K 0102:2019
単位 mm
図42.1 蒸留装置の例
c) 蒸留操作 蒸留操作は,次による。
なお,試料に一定量のアンモニウムイオン標準液を添加して,備考2.又は備考3.の蒸留操作による
回収率試験を行い,回収率が80〜120 %であることを確認した場合は,備考2.又は備考3.によっても
よい。ただし,備考2.又は備考3.の蒸留操作は,42.3(中和滴定法)及び42.4(イオン電極法)には
適用しない。
1) 試料の適量をとり,中性でない場合には,ブロモチモールブルー溶液(1 g/L)5〜7滴を加え,水酸
化ナトリウム溶液(40 g/L)又は硫酸(1+35)でpHを6.0(黄色)〜7.4(青)に調節する。試料
A: 蒸留フラスコ500 mL
B: 連結導入管
C: すり合わせコック
D: 注入漏斗
E: トラップ球(ケルダール球)
F: リービッヒ冷却器300 mm
G: 逆流止め(約50 mL)
H: 受器[メスシリンダー(有栓形)200 mL]
I: 共通すり合わせ
J: 共通球面すり合わせ
K: 押さえばね
39
K 0102:2019
の採取量は定量方法によって異なり,NH4+として,インドフェノール青吸光光度法の場合には40 μg
以上,中和滴定法の場合には0.3〜40 mg,イオン電極法の場合には40 μg以上,流れ分析法の場合
には12 µg以上,サリチル酸-インドフェノール青吸光光度法では10 µg以上を含むようにとる。
なお,試料中に残留塩素が存在するときは,蒸留操作の前に,チオ硫酸ナトリウムの小結晶を加
えて除去する。
2) 蒸留フラスコに移し入れ,酸化マグネシウム0.25 g,沸騰石(粒径2〜3 mm)及び水を加えて液量
を約350 mLとする。
3) 蒸留装置を図42.1のように組み立て,受器のメスシリンダー(有栓形)200 mLに硫酸(25 mmol/L)
50 mLを入れる。
なお,留出液を中和滴定法に用いる場合には,受器には三角フラスコ500 mLを用い,これに硫
酸(25 mmol/L)50 mLを正しく加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合
溶液[15.の注(1)による。]5〜7滴を加える。
4) 蒸留フラスコを加熱し,留出速度5〜7 mL/minで蒸留を行う。冷却器の管の先端を,常に受器の液
面下約15 mmを保つようにする。
5) 受器の液量が約190 mLになるまで蒸留を続ける。
6) 冷却器及び逆流止めを外し,冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液は,受器の
メスシリンダー(有栓形)200 mLに入れ,水を200 mLの標線まで加える。留出液を中和滴定法に
用いる場合には,冷却器の内管及び逆流止めの内外の洗液は,三角フラスコ500 mLに合わせ,全
量を滴定に用いる。
備考 2. 小型の蒸留フラスコを用いる蒸留操作は,次による。
1) 試料の適量を蒸留フラスコ200 mLにとり,c) 1)と同様にpHを6.0〜7.4に調節する。酸
化マグネシウム0.11 g,沸騰石(粒径2〜3 mm)及び水を加えて液量を約150 mLとする。
試料の採取量としては,c) 1)に記載したNH4+量の1/2が含まれるようにする。
なお,試料中に残留塩素が存在するときは,蒸留操作の前に,チオ硫酸ナトリウムの
小結晶を加えて除去する。
2) 蒸留フラスコを蒸留装置に取り付け,受器のメスシリンダー(有栓形)100 mLに硫酸(25
mmol/L)25 mLを入れる。
3) 蒸留フラスコを加熱し,留出速度2〜3 mL/minで蒸留を行う。蒸留中は冷却器の管の先
端が受器の液面下にあるようにする。
4) 受器の液量が85〜95 mLになるまで蒸留を続ける。
5) 冷却器及び逆流止めを外し,冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液
は,受器のメスシリンダー(有栓形)100 mLに入れ,水を100 mLの標線まで加える。
3. 図28.1の小型蒸留装置を用いる蒸留操作は,次による。
1) 試料の適量を蒸留容器にとり,c) 1)と同様にpHを6.0〜7.4に調節する。酸化マグネシ
ウム0.03〜0.04 g,沸騰石(粒径2〜3 mm)及び水を加えて液量を約50 mLとする。試
料の採取量としては,c) 1)に記載したNH4+量の1/4が含まれるようにする。
なお,試料中に残留塩素が存在するときは,蒸留操作の前に,チオ硫酸ナトリウムの
小結晶を加えて除去する。
2) 受器に容量50 mLのメスシリンダー(有栓形)などを用い,これに硫酸(62.5 mmol/L)
(JIS K 8951に規定する硫酸約3.5 mLをあらかじめ水100 mLを入れたビーカーに加え
40
K 0102:2019
てよくかき混ぜ,水を加えて1 Lとする。)5 mLを入れる。
3) 蒸留容器に蒸留管及び冷却器を取り付け,これを加熱器にセットする。
4) 留出速度約0.5〜1 mL/minで蒸留を行う。
なお,蒸留中は冷却器の管の先端が受器の液面下にあるようにする。
5) 受器の液量が約45 mLになるまで蒸留を続ける。
6) 蒸留管及び冷却器を外し,それらの内管を少量の水で洗う。洗液も受器に加えた後,更
に水を50 mLの標線まで加える。
備考 4. 蒸留法として水蒸気蒸留法を用いてもよい。この場合は,図42.1の蒸留フラスコに水蒸気を
送るように装置を組み立て,蒸留フラスコを加熱する。沸騰し始めたら,水蒸気を蒸留フラ
スコに送り,留出速度3〜5 mL/minで蒸留し,受器の液量が約190 mLになるまで蒸留を続
ける。
5. 妨害物質 蒸留法においても,脂肪族アミン,芳香族アミン類なども留出するので,これら
の共存は妨害となる。
尿素,アセトアミド,ペプトン,アスパラギンなどの窒素を含む有機化合物は,蒸留する
とその一部が加水分解してアンモニアとなり正の誤差を生じる。その程度は,蒸留時のpH
が高くなるほど大きくなる。
42.2 インドフェノール青吸光光度法 アンモニウムイオンが次亜塩素酸イオンの共存のもとで,フェノ
ールと反応して,生じるインドフェノール青の吸光度を測定してアンモニウムイオンを定量する。
定量範囲:NH4+ 5〜100 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。使用時に調製する。
3) ナトリウムフェノキシド溶液 水酸化ナトリウム溶液(200 g/L)55 mLをビーカーにとり,冷水中
で冷却しながらJIS K 8798に規定するフェノール25 gを少量ずつ加えて溶かす。放冷後,JIS K 8034
に規定するアセトン6 mLを加え,水で200 mLとする。10 ℃以下の暗所に保存し,5日間以上経
過したものは使用しない。
4) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)の
有効塩素の濃度を36.の注(3)によって求め,有効塩素が約10 g/Lになるように水で薄める。使用時
に調製する。
5) アンモニウムイオン標準液(NH4+ 1 000 mg/L) 国家計量標準にトレーサブルなアンモニウムイオ
ン標準液(NH4+)1 000 mg/Lを使用するか,又は次による。JIS K 8116に規定する塩化アンモニウ
ムをデシケーター[JIS K 8228に規定する過塩素酸マグネシウム(乾燥用)を入れたもの。]中に
16時間以上放置し,その2.97 gをとり,水に溶かして全量フラスコ1 000 mLに移し入れ,水を標
線まで加える。
6) アンモニウムイオン標準液(NH4+ 10 μg/mL) アンモニウムイオン標準液(NH4+ 1 000 mg/L)10
mLを全量フラスコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
41
K 0102:2019
1) 42.1の留出液の適量(NH4+として5〜100 μgを含む。25 mL以下)を全量フラスコ50 mLにとり,
水を加えて約25 mLとする。
2) ナトリウムフェノキシド溶液10 mLを加えて振り混ぜる。
3) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L)5 mLを加え,水を標線まで加えた後,栓をして振り
混ぜる。
4) 液温を20〜25 ℃に保って,約30分間放置して発色させる。
5) 発色後約30分間以内に,この溶液の一部を吸収セルに移し,波長630 nm付近の吸光度を測定する。
6) 空試験として水25 mLをとり,2)〜5)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
7) 検量線からアンモニウムイオンの量を求め,試料中のアンモニウムイオンの濃度(NH4+ mg/L)を
算出する。
d) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 10 μg/mL)0.5〜10 mLを段階的に全量フラスコ50 mLにとり,
水を加えて約25 mLとする。
2) c)の2)〜6)の操作を行って吸光度を測定し,アンモニウムイオン(NH4+)の量と吸光度との関係線
を作成する。
備考 6. 微量のアンモニウムイオンを定量する場合には,c) 2)の操作でナトリウムフェノキシド溶液
10 mLに続き,ペンタシアノニトロシル鉄(III)酸ナトリウム溶液[JIS K 8722に規定する
ペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.15 gを水に溶かして100 mLとす
る。]1 mLを加えてもよい。
この場合の定量範囲は,NH4+として2.5〜50 μgとなる。検量線は,同一操作で作成する。
7. アンモニア体窒素で表示する場合は,次の換算式を用いる。
アンモニア体窒素(NH4+-N mg/L)=アンモニウムイオン(NH4+ mg/L)×0.776 6
また,アンモニアへの換算は,次の換算式を用いる。
アンモニア(NH3 mg/L)=アンモニウムイオン(NH4+ mg/L)×0.944 1
8. 脂肪族アミン類は妨害しないが,芳香族アミン類の一部は,次亜塩素酸塩によって酸化され
て着色物質を生じるので妨害する。p-アミノフェノールのような物質は,アルカリ性溶液中
でフェノールと反応してインドフェノール青を生じるので妨害する。p-ヒドロキノンは,妨
害しない。ヒドロキシルアミンも妨害するが,JIS K 8230に規定する過酸化水素の当量を加
えて酸化すれば,妨害を除くことができる。
42.3 中和滴定法 42.1に規定する前処理(蒸留法)(ただし,備考2.及び備考3.は除く。)を行って留出
したアンモニアを一定量の硫酸(25 mmol/L)中に吸収させた溶液について,50 mmol/L水酸化ナトリウム
溶液で,残った硫酸を滴定してアンモニウムイオンを定量する。
定量範囲:NH4+ 0.3〜40 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) メチルレッド-ブロモクレゾールグリーン混合溶液 15.の注(1)による。
4) 50 mmol/L水酸化ナトリウム溶液 水約30 mLをポリエチレン瓶にとり,冷却しながらJIS K 8576
に規定する水酸化ナトリウム約35 gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。その
42
K 0102:2019
上澄み液2.5 mLをポリエチレン製の気密容器1 Lにとり,2. n) 2)の二酸化炭素を含まない水を加え
て1 Lとし,混合した後,二酸化炭素を遮断して保存する。
4.1) 標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のアミド硫酸を,上口デシケーター中に圧力2 kPa
以下で約48時間放置して乾燥する。その約1 gを1 mgの桁まではかりとり,少量の水に溶かし
て全量フラスコ200 mLに移し入れ,水を標線まで加える。
− その20 mLを三角フラスコ300 mLにとり,指示薬としてブロモチモールブルー溶液(1 g/L)[16.
の注(1)による。]2,3滴を加え,この50 mmol/L水酸化ナトリウム溶液で滴定し,溶液の色が緑
になったときを終点とする。
− 次の式によって50 mmol/L水酸化ナトリウム溶液のファクター(f)を算出する。
855
004
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の量(g)
b: アミド硫酸の純度(%)
x: 滴定に要した50 mmol/L水酸化ナトリウム溶液(mL)
0.004 855: 50 mmol/L水酸化ナトリウム溶液1 mLのアミド硫酸相当
量(g)
b) 操作 操作は,次による。
1) 42.1 c)の前処理で得た留出液の全量を用い,50 mmol/L水酸化ナトリウム溶液で溶液の色が灰紫
(pH4.8)になるまで滴定する。
2) 別に,硫酸(25 mmol/L)50 mLを正しく三角フラスコ500 mLにとり,メチルレッド-ブロモクレゾ
ールグリーン混合溶液5〜7滴を加え,50 mmol/L水酸化ナトリウム溶液で溶液の色が灰紫(pH4.8)
になるまで滴定し,硫酸(25 mmol/L)50 mLに相当する50 mmol/L水酸化ナトリウム溶液のmL数
を求める。
3) 次の式によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を算出する。
902
.0
000
1
)
(
×
×
×
−
=
V
f
a
b
A
ここに,
A: アンモニウムイオン(NH4+ mg/L)
b: 硫酸(25 mmol/L)50 mLに相当する50 mmol/L水酸化ナトリ
ウム溶液(mL)
a: 滴定に要した50 mmol/L水酸化ナトリウム溶液(mL)
f: 50 mmol/L水酸化ナトリウム溶液のファクター
V: 試料(mL)
0.902: 50 mmol/L水酸化ナトリウム溶液1 mLのアンモニウムイオ
ン相当量(mg)
備考 9. 42.1 c) 3)の硫酸(25 mmol/L)の代わりに,ほう酸溶液(20 g/L)を用いてもよい。この場合
は,次のように操作する。
1) 三角フラスコ500 mLにほう酸溶液(20 g/L)(JIS K 8863に規定するほう酸を用いて調
製する。)50 mLを加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶
液5〜7滴を加え,42.1 c) 4)〜6)の操作を行う。
2) 次に,25 mmol/L硫酸(*)で溶液の色が灰紫(pH4.8)になるまで滴定する。
3) 別に,空試験としてほう酸溶液(20 g/L)50 mLを三角フラスコ500 mLにとり,水150 mL
を加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液5〜7滴を加え
43
K 0102:2019
る。次に,試料の場合と同様に滴定を行う。
4) 次の式によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を算出する。
902
.0
000
1
)
(
×
×
×
−
=
V
f
b
a
A
ここに,
A: アンモニウムイオン(NH4+ mg/L)
a: 滴定に要した25 mmol/L硫酸(mL)
b: 空試験に要した25 mmol/L硫酸(mL)
f: 25 mmol/L硫酸のファクター
V: 試料(mL)
0.902: 25 mmol/L硫酸1 mLのアンモニウムイオン相当量(mg)
注(*) 25 mmol/L硫酸の調製方法 42.1 a) 2)の硫酸(25 mmol/L)を標定して用いる。
標定
− JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムを600 ℃で約1時間
加熱した後,デシケーター中で放冷する。その0.53 gを1 mgの桁まではかりとり,
水に溶かして全量フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLをビーカーにとり,指示薬としてメチルレッド-ブロモクレゾールグリ
ーン混合溶液3〜5滴を加えた後,この硫酸(25 mmol/L)で滴定する。
− 溶液の色が灰紫になったら,煮沸して二酸化炭素を追い出し,放冷後,溶液の色
が灰紫になるまで滴定を続ける。
− 次の式によって25 mmol/L硫酸のファクター(f)を算出する。
650
002
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 炭酸ナトリウムの量(g)
b: 炭酸ナトリウムの純度(%)
x: 滴定に要した硫酸(25 mmol/L)(mL)
0.002 650: 25 mmol/L硫酸1 mLの炭酸ナトリウム相当量(g)
42.4 イオン電極法 42.1に規定する前処理(蒸留法)(ただし,備考2.及び備考3.は除く。)を行った試
料に水酸化ナトリウム溶液を加え,pHを11〜13に調節してアンモニウムイオンをアンモニアに変え,ア
ンモニア電極を指示電極として電位を測定し,アンモニウムイオンを定量する。電極を汚染したり,電位
応答を妨害するような共存物質が存在しなければ蒸留操作を省くことができる。
定量範囲:NH4+ 0.1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム溶液(100 g/L) 19. a) 2)による。
3) 塩化アンモニウム溶液(0.1 mol/L) JIS K 8116に規定する塩化アンモニウム5.4 gを水約800 mL
に溶かし,水で1 Lとする。
4) アルカリ性緩衝液 JIS K 8576に規定する水酸化ナトリウム40 g及びJIS K 8107に規定するエチレ
ンジアミン四酢酸二水素二ナトリウム二水和物(EDTA二ナトリウム塩)37 gを水約800 mLに溶か
し,水で1 Lとする。
なお,アンモニウムイオン0.5 mg/L以下の低濃度の定量では,この溶液を約20分間煮沸し,冷
却後水で1 Lとする。この溶液は,ポリエチレン瓶に貯蔵する。
5) アンモニウムイオン標準液(NH4+ 100 mg/L) 42.2 a) 5)のアンモニウムイオン標準液(NH4+ 1 000
44
K 0102:2019
mg/L)20 mLを全量フラスコ200 mLにとり,水を標線まで加える。
6) アンモニウムイオン標準液(NH4+ 10 mg/L) アンモニウムイオン標準液(NH4+ 100 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
7) アンモニウムイオン標準液(NH4+ 1 mg/L) アンモニウムイオン標準液(NH4+ 10 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
8) アンモニウムイオン標準液(NH4+ 0.1 mg/L) アンモニウムイオン標準液(NH4+ 1 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 34.2 b) 1)による。
2) 指示電極 アンモニア電極(隔膜形)
3) 参照電極 塩化カリウム溶液(飽和)を充塡した銀-塩化銀電極を用いる。
4) 測定容器 35.2 b) 4)による。
5) 恒温槽 34.2 b) 5)による。
6) マグネチックスターラー 35.2 b) 6)による。
c) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 0.1 mg/L)100 mLを測定容器にとり,水酸化ナトリウム溶液(100
g/L)1 mLを加えてpHを約12とする。このpHでは,アンモニウムイオンはアンモニアとなり,
揮散しやすいため,水酸化ナトリウム溶液の添加は電位測定の直前に加える。また,できるだけ口
の狭く,必要に応じて密閉できる容器を用いる。
2) 恒温槽から水を送り,測定容器の溶液を25±0.5 ℃にする。
3) 指示電極(1)と参照電極(2)とを浸し,マグネチックスターラー(3)で泡が電極に触れない程度に強くか
き混ぜる(4)。
4) 液温を確認し,電位差計で電位を測定する(5)。
5) NH4+濃度が1 mg/L,10 mg/L,100 mg/Lのアンモニウムイオン標準液のそれぞれについて1)〜4)の
操作を行う。電位の測定は,低濃度から順に行う。高濃度から低濃度へ移ると,応答速度は遅くな
るため,水で洗ってアンモニアを除き,次に,1)の水酸化ナトリウム溶液(100 g/L)1 mLを加えた
アンモニウムイオン標準液(NH4+ 0.1 mg/L)に浸し,指示値が安定してから測定する。また,水
酸化ナトリウムを加えた標準液は,5〜10分間経過するとアンモニアを消失するため,次の校正に
は新しい標準液を用いる。
6) 横軸にアンモニウムイオンの濃度(NH4+ mg/L)の対数,縦軸に電位(6)をとって,アンモニウムイ
オンの濃度と電位との関係線を作成する(7)。
注(1) 隔膜電極は,内部ガラス電極のガラス膜面に隔膜を強く押し付けると隔膜にきずがつくので注
意する。また,ガラス膜面と隔膜が離れ過ぎたり,電極の隔膜が汚れると,電位が不安定にな
り,応答時間が長くなる。
(2) 34.の注(6)による。
(3) かき混ぜが強すぎると,泡が膜を覆い誤差を生じるので注意する。
(4) 34.の注(9)による。
(5) アンモニア電極の応答時間は,液温10〜30 ℃の場合,アンモニウムイオン標準液(NH4+ 0.1
mg/L)では3〜5分間,アンモニウムイオン標準液(NH4+ 1 mg/L以上)では2〜3分間である。
(6) 電位は,ネルンスト式に従って,アンモニウムイオンの濃度が1桁の変化当たり約60 mV変化
45
K 0102:2019
する。
注(7) アンモニウムイオンの濃度NH4+ 0.1 mg/L付近からNH4+ 100 mg/Lまで検量線は直線になる。
備考 10. 試料中のアンモニウムイオンの濃度が想定できる場合は,その濃度範囲を含む標準液を調製
して検量線を作成してもよい。
d) 操作 操作は,次による。
1) 42.1の前処理を行った試料の適量(NH4+として0.02〜20 mgを含む。)をとり,水で約100 mLとし
た後,水酸化ナトリウム溶液(100 g/L)を加えてpHを約8.3に調節し,全量フラスコ200 mLに移
し入れ,水を標線まで加える。この溶液100 mLを三角フラスコ200 mLにとり,電位測定の直前に
水酸化ナトリウム溶液(100 g/L)1 mLを加える。
2) c)の2)〜4)の操作を行って,検量線からアンモニウムイオンの濃度(NH4+ mg/L)を求め,試料中
のアンモニウムイオンの濃度(NH4+ mg/L)を算出する。
備考 11. イオン濃度計の場合には,アンモニウムイオン標準液(NH4+ 0.1 mg/L)とアンモニウムイオ
ン標準液(NH4+ 10 mg/L)とを用い,c)の1)〜4)の操作を行ってイオン濃度計の指示値を
NH4+ 0.1 mg/L及びNH4+ 10 mg/Lになるように調節する。さらに,アンモニウムイオン標
準液(NH4+ 1 mg/L)とアンモニウムイオン標準液(NH4+ 100 mg/L)とを用いてイオン濃
度計の指示値を確認する。
12. 妨害物質がなく,蒸留処理をせず,ろ過による処理で測定する場合は,3.2に従って試料をろ
過する。ろ液の50 mLを測定容器にとり,アルカリ性緩衝液5 mLを加える。次いで,c)の
2)〜4)の操作を行う。検量線は,同じ操作で作成する。
13. アンモニア電極の保管方法 アンモニア電極を長い期間(例えば,一夜)保管するには,先
端を塩化アンモニウム溶液(0.1 mol/L)に浸しておく。使用前に,十分にすすぐ。
14. 妨害物質
1) 試料中のアンモニウムイオンの濃度が0.1 mg/Lの場合,ヒドラジニウムイオン(N2H5+)
は1 mg/L以下では影響しないが,10 mg/L及び100 mg/L共存すれば,それぞれ+35 %,
+100 %の誤差を生じる。
2) 試料中のアンモニウムイオンの濃度が1 mg/L以上の場合には,ヒドラジニウムイオン
100 mg/Lが共存しても影響しない。試料中のアンモニウムイオンの濃度が0.1 mg/Lの場
合,モルホリン(テトラヒドロ-1, 4-オキサジン,C4H8ONH)は10 mg/Lまで共存しても
影響しないが,100 mg/L共存すると,+100 %の誤差を生じる。
3) また,試料中のアンモニウムイオンの濃度が1 mg/L以上の場合には,モルホリンが100
mg/L共存しても影響しない。
4) アンモニア電極は,アンモニウムイオンの濃度が50 mg/Lを超える試料の測定に連続的
に使用すると,満足な応答をしない。このような水に対しては,測定試料をこの濃度未
満に薄めるのがよい。
5) アミン類は正の妨害を与える。表42.1に示す妨害物質としてヒドラジン,シクロヘキシ
ルアミン,モルホリン,オクタデシルアミン,メタノールアミンなどがある。
6) 界面活性剤及びある種の有機溶媒は,アンモニア電極の膜の寿命を短くする。このため
電極の手入れを必要とする頻度が増加する。この影響はかなり重大で,これらの妨害物
質の濃度が高い試料では,アンモニア電極の劣化を早めることになる。
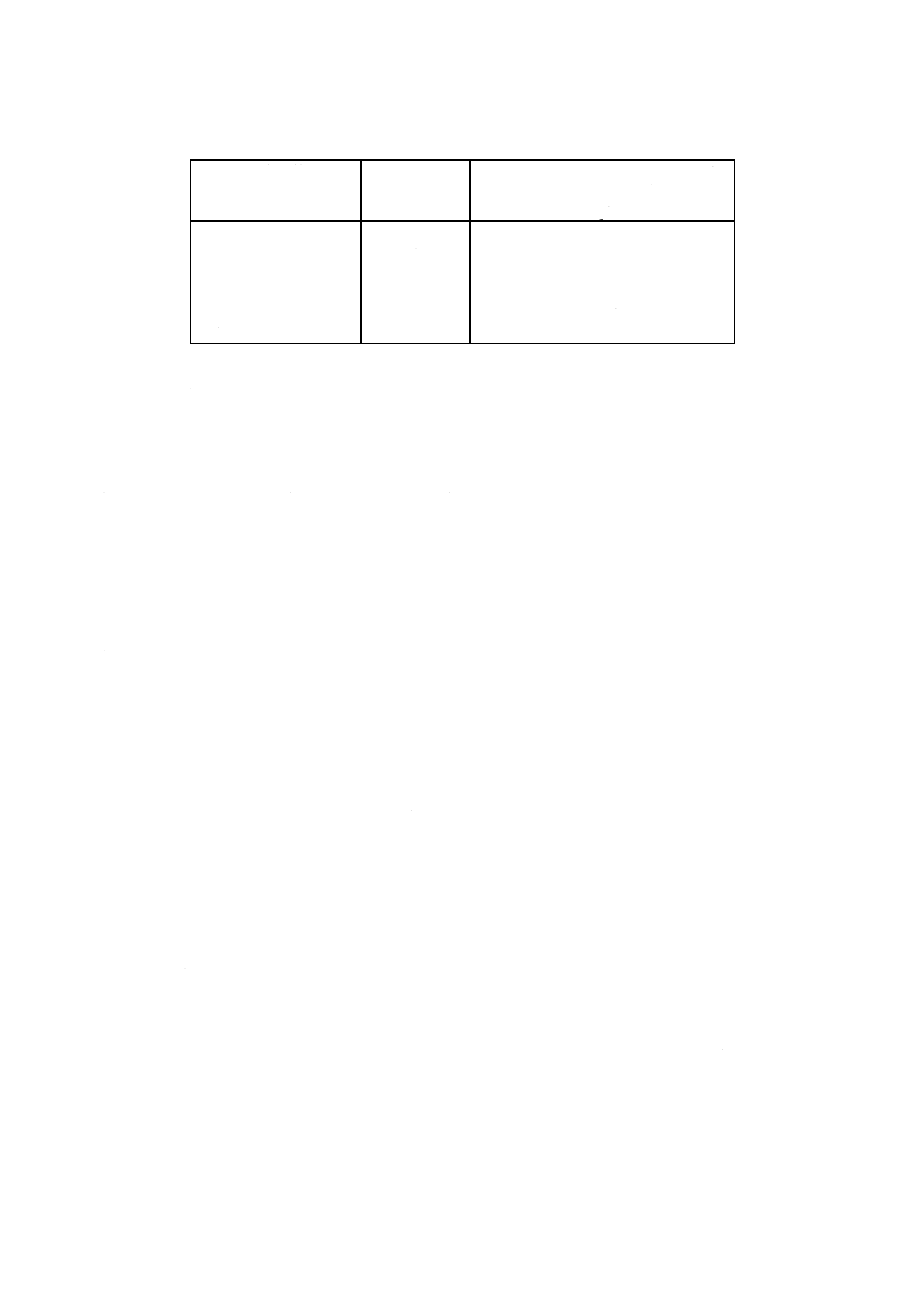
46
K 0102:2019
表42.1 妨害物質の例
妨害物質
妨害物質濃度
mg/L
アンモニウムイオンの濃度ρN=1 mg/L
における見掛けの増加
mg/L
ヒドラジン
4
0.06
シクロヘキシルアミン
1
0.03
モルホリン
10
0.03
オクタデシルアミン
0.4
0.14
メタノールアミン
3.4
0.15 *
尿素
11
0.01
注*
アンモニウムイオンの濃度ρN=0.5 mg/Lにおける見掛けの増加。
備考 15. アンモニウムイオン濃度をアンモニア体窒素で表示する場合は,備考7.による。
42.5 イオンクロマトグラフ法 試料中のアンモニウムイオンをイオンクロマトグラフ法によって定量す
る。この方法によって,表48.1に示す陽イオンが同時定量できる。この方法を適用する場合には,3.3の
試料の保存処理及び42.1の前処理(蒸留法)を行わず,試料採取後,直ちに試験する。直ちに行えない場
合は,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
試験操作などは,48.3による。
備考 16. 備考7.による。
42.6 流れ分析法 試料中のアンモニウムイオンを,42.2及び42.7と同様な原理で発色させる流れ分析法
によって定量する。試料を,42.1の前処理(蒸留法)の操作でアンモニウムイオンを共存物から分離した
後,適用する。ただし,妨害物質を含まない試料は,蒸留操作を省略してもよい。この場合は,懸濁物の
多い試料には適用できない。
試験操作などは,JIS K 0170-1の6.3.3(フェノールによるインドフェノール青発色FIA法),6.3.4(サ
リチル酸によるインドフェノール青発色CFA法)及び6.3.5(フェノールによるインドフェノール青発色
CFA法)による。
備考 17. アンモニウムイオン濃度をアンモニア体窒素で表示する場合は,備考7.による。
定量範囲:NH4+ 0.06〜13 mg/L(アンモニア体窒素として0.05〜10 mg/L),繰返し精度:10 %以下
試験操作などは,JIS K 0170-1による。ただし,JIS K 0170-1の6.3.2(ガス拡散・pH指示薬変色FIA法)
は除く。
42.7 サリチル酸-インドフェノール青吸光光度法 アンモニウムイオンがジクロロイソシアヌル酸の加
水分解で生じる次亜塩素酸イオンのもとで,サリチル酸と反応して生じるインドフェノール青の吸光度を
測定してアンモニウムイオンを定量する。
定量範囲:NH4+ 2〜40 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) サリチル酸ナトリウム溶液 JIS K 8397に規定するサリチル酸ナトリウム130 g及びJIS K 8288に
規定するくえん酸三ナトリウム二水和物130 gを水約900 mLに溶かし,これにJIS K 8722に規定
するペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.97 gを加え,水を加えて1 Lとする。
3) ジクロロイソシアヌル酸ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム32 gを水500
mLに溶かし,溶液を室温に冷却した後,ジクロロイソシアヌル酸ナトリウム二水和物2 gを加え,
水を加えて1 Lとする。
47
K 0102:2019
4) アンモニウムイオン標準液(NH4+ 100 μg/mL) 42.2 a) 5)のアンモニウムイオン標準液(NH4+ 1 000
mg/L)10 mLを全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
5) アンモニウムイオン標準液(NH4+ 10 μg/mL) 4)のアンモニウムイオン標準液(NH4+ 100 μg/mL)
10 mLを全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
6) アンモニウムイオン標準液(NH4+ 1 μg/mL) 5)のアンモニウムイオン標準液(NH4+ 10 μg/mL)
10 mLを全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 42.2 b) 1)による。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 42.1の留出液の適量(NH4+として2〜40 μgを含む。40 mL以下。)を全量フラスコ50 mLにとり,
水を加えて約40 mLにする。
2) サリチル酸ナトリウム溶液4 mLを加え,よく混ぜ合わせた後,ジクロロイソシアヌル酸ナトリウ
ム溶液4 mLを加え,よく混ぜ合わせる。
3) 水を標線まで加え,恒温槽中で25±1 ℃に保ち,30分間以上放置して発色させる。発色は,温度の
影響を大きく受けるので,発色時の液温は正しく保つ。
4) 発色後,恒温槽から取り出し,1時間以内に,この溶液の一部を吸収セルに移し,波長655 nm付近
で吸光度を測定する。
5) 空試験として,水40 mLについて2)〜4)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
6) 検量線からアンモニウムイオンの量を求め,試料中のアンモニウムイオンの濃度(NH4+ mg/L)を
算出する。
d) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 1 μg/mL)2〜40 mLを段階的に全量フラスコ50 mLにとり,水
を加えて40 mLとした後,c)の2)〜5)の操作を行って吸光度を測定し,アンモニウムイオン(NH4+)
の量と吸光度との関係線を作成する。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.1.1(ナフチルエチレンジアミン吸光光度法)
の“備考3. 亜硝酸イオンの濃度を亜硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考3.
亜硝酸イオンの濃度を亜硝酸体窒素で表示する場合は,35.の備考10. による。”に置き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.1.3(流れ分析法)の“備考4. 亜硝酸イオン
の濃度を亜硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考4. 亜硝酸イオンの濃度を亜
硝酸体窒素で表示する場合は,35.の備考10. による。”に置き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.2.1(還元蒸留-インドフェノール青吸光光度
法)の“備考6. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考6.
硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考10. による。”に置き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.2.2(還元蒸留-中和滴定法)の“備考7. 硝酸
48
K 0102:2019
イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考7. 硝酸イオンの濃度を
硝酸体窒素で表示する場合は,35.の備考10. による。”に置き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.2.3(銅・カドミウムカラム還元-ナフチルエ
チレンジアミン吸光光度法)の“備考8. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11.
による。”を,“備考8. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考10. による。”に置
き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.2.5(イオンクロマトグラフ法)の“備考11.
硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考11. 硝酸イオンの
濃度を硝酸体窒素で表示する場合は,35.の備考10. による。”に置き換える。
43.[亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)]の43.2.6(流れ分析法)の“備考12. 硝酸イオン
の濃度を硝酸体窒素で表示する場合は,35.の備考11. による。”を,“備考12. 硝酸イオンの濃度を硝酸
体窒素で表示する場合は,35.の備考10. による。”に置き換える。
44.(有機体窒素)の備考4.の第2文“その場合は42.の備考7.の操作を行う。”を,“その場合は42.の備考
9.の操作を行う。”に置き換える。
45.(全窒素)の全文を,次に置き換える。
45. 全窒素 亜硝酸イオンと硝酸イオンに相当する窒素と,アンモニウムイオンと有機体窒素に相当する
窒素とを求めて合計する総和法,全窒素化合物を硝酸イオンに変えた後の紫外線吸光光度法,硫酸ヒドラ
ジニウム還元法,銅・カドミウムカラム還元法,又は熱分解法,若しくは流れの中で全窒素化合物を硝酸
イオンに変えて紫外線吸光光度法,銅・カドミウムカラム還元法を用いた流れ分析法を適用する。
45.1 総和法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモニウムイオン及び一部の有機窒素化
合物の分解で生じたアンモニアを除いた後,デバルダ合金を加えて亜硝酸イオン及び硝酸イオンを還元し
てアンモニアとし,蒸留によって分離し,インドフェノール青吸光光度法又はサリチル酸-インドフェノー
ル青吸光光度法で窒素の量を定量する。別に,試料に硫酸銅(II)五水和物,硫酸カリウム及び硫酸を加
えて加熱分解して有機体窒素をアンモニウムイオンに変えた後,アルカリ性として蒸留し,試料中に含ま
れるアンモニウムイオンとともに蒸留分離し,インドフェノール青吸光光度法又はサリチル酸-インドフェ
ノール青吸光光度法によってその窒素の量を定量する。先に求めた亜硝酸イオン及び硝酸イオン相当の窒
素量を合わせて,全窒素の濃度を算出する。
定量範囲:N 8〜160 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) 硫酸(1+35) 30.1.1 a) 2)による。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 水酸化ナトリウム溶液(300 g/L) 43.2.1 a) 5)による。
6) デバルダ合金 43.2.1 a) 6)による。
49
K 0102:2019
7) 硫酸カリウム JIS K 8962に規定するもの。
8) 硫酸銅(II)五水和物 44.1 a) 8)による。
9) ナトリウムフェノキシド溶液 42.2 a) 3)による。
10) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L) 42.2 a) 4)による。
11) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
12) アンモニウムイオン標準液(NH4+ 1 000 mg/L) 42.2 a) 5)による。
13) アンモニウムイオン標準液(NH4+ 10 μg/mL) 42.2 a) 6)による。
14) アンモニウムイオン標準液(NH4+ 1 μg/mL) 42.7 a) 6)による。
15) サリチル酸ナトリウム溶液 42.7 a) 2)による。
16) ジクロロイソシアヌル酸ナトリウム溶液 42.7 a) 3)による。
17) 水酸化ナトリウム溶液(500 g/L) 44.1 a) 5)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) ケルダールフラスコ 200 mL。使用前に水でよく洗う。
3) 蒸留装置 42.1 b) 1)による。使用前に水でよく洗う。
4) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料50 mLをとり,中性でない場合には水酸化ナトリウム溶液(40 g/L)又は硫酸(1+35)でpH
約7に調節する。低濃度のものを定量する必要がある場合は,試料の量を増加する。
2) 43.2.1 c) の2)〜6)の操作を行って,試料中の亜硝酸イオンと硝酸イオンとを還元蒸留してアンモニ
アとして留出させる。
3) 得られた留出液200 mL中のアンモニウムイオンの定量を,42.2又は42.7で行う。全量フラスコ50
mLに留出液を,前者による場合は25 mL(1),後者による場合は40 mL(1)をとる。
4) 42.2 c)の2)〜5),又は42.7 c) の2)〜4)の操作を行って吸光度を測定する。
5) 空試験として水約50 mLをとり,水酸化ナトリウム溶液(300 g/L)10 mL及び沸騰石(粒径2〜3 mm)
を加えた後,43.2.1 c) の4)〜6)の操作を行い,得られた留出液について3)及び4)の操作を行って吸
光度を測定し,4)で得た吸光度を補正する。
6) 42.2 d)又は42.7 d)の検量線から3)で分取した留出液中のアンモニウムイオンの量(mg)を求める。
7) 別に,試料50 mLをケルダールフラスコ200 mLにとり,44.1 c) の3)〜6)の操作を行って,試料中
の有機体窒素を分解してアンモニアとし,試料中のアンモニアと一緒に留出させる。低濃度のもの
を定量する必要がある場合は,試料の量を増加する。
8) 得られた留出液200 mLについて,3)及び4)に従って操作する。
9) 空試験として水50 mLをとり,7)及び8)の操作を行って吸光度を測定し,8)で得た吸光度を補正す
る。
10) 42.2 d)又は42.7 d)の検量線から8)で分取した留出液中のアンモニウムイオンの量(mg)を求める。
11) 次の式によって試料中の全窒素の濃度(N mg/L)を算出する。
6
776
.0
25
200
000
1
6
776
.0
25
200
000
1
×
×
×
+
×
×
×
=
V
b
V
a
N
ここに,
N: 全窒素(N mg/L)
a: 6)の操作で得たアンモニウムイオン(mg)
50
K 0102:2019
b: 10)の操作で得たアンモニウムイオン(mg)
V: 蒸留に用いた試料(mL)
0.776 6: アンモニウムイオンを窒素の相当量に換算するときの係数
04
.
18
01
.
14
4)及び5)の操作で42.7 c)を用いた場合,又は8)及び9)の操作で42.7 c)を用いた場合は,c) 11)の式の
25の代わりに40を用いる。
注(1) アンモニウムイオン0.8 mg以上を含む留出液を42.2で定量する場合は,留出液の適量(NH4+
が0.4 mg未満になる量)を硫酸(25 mmol/L)25 mLが入った全量フラスコ100 mLにとり,水
を標線まで加えたものから25 mLを分取する。アンモニウムイオン0.2 mg以上を含む留出液を
42.7で定量する場合は,留出液の適量(NH4+が0.1 mg未満になる量)を同様に操作したものか
ら25 mLを分取する。これらの場合は,11)の式の1項及び2項に希釈率を乗じて補正を行って
全窒素濃度を算出する。
45.2 紫外線吸光光度法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃に加熱し
て窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液のpHを2〜3とした後,硝酸イ
オンによる波長220 nmの吸光度を測定して定量する。この方法は,試料中の有機物が分解されやすく,
少量である場合に適用する。また,試験に影響する臭化物イオンを10 mg/L以上,又はクロムを0.1 mg/L
以上を含む試料には適用できない。
定量範囲:N 5〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩酸(1+16) JIS K 8180に規定する塩酸を用いて調製する。
4) 塩酸(1+500) JIS K 8180に規定する塩酸を用いて調製する。
5) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 JIS K 8826に規定する水酸化ナトリウム(窒素
測定用)20 gを水500 mLに加えた後,JIS K 8253に規定するペルオキソ二硫酸カリウム(窒素り
ん測定用)15 gを溶かす。使用時に調製する。この溶液の窒素含有量は,0.4 mg/L以下とする。
6) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
7) 窒素標準液(N 0.1 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110 ℃で約2
時間加熱し,デシケーター中で放冷する。その0.722 gをとり,少量の水に溶かし,全量フラスコ1 000
mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
8) 窒素標準液(N 20 μg/mL) 窒素標準液(N 0.1 mg/mL)50 mLを全量フラスコ250 mLにとり,水を
標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 耐圧の四ふっ化エチレン樹脂瓶又は耐熱・耐圧のガラス瓶(容量約100 mL)で,高圧蒸気
滅菌器中(約120 ℃)で使用できるもの。また,ガラス製アンプル(容量約100 mL)で,高圧蒸
気滅菌器中(約120 ℃)で使用できるものを用いてもよい。
2) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので,約120 ℃に加熱できるもの。
3) 光度計 分光光度計
4) 吸収セル 石英ガラス製
51
K 0102:2019
c) 操作 操作は,次による。
1.1) 試料のpHが5〜9で,試料50 mLに含まれる全窒素が0.1 mg未満の場合は,分解瓶に試料50 mL
をとる。また,全窒素が0.1 mg以上含まれる場合は,試料の適量(Nとして0.2 mg未満を含む。)
を全量フラスコ100 mLにとり,水を標線まで加え,この溶液から50 mLをとる。
1.2) 試料のpHが5〜9の範囲にない場合で,試料50 mLに含まれる全窒素が0.1 mg未満の場合は,試
料50 mLを分解瓶にとり,塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)で中和し,中和に
要した両液の量(b mL)を記録する。
1.3) 試料のpHが5〜9の範囲にない場合で,試料50 mLに含まれる全窒素が0.1 mg以上の場合は,試
料の適量(Nとして0.2 mg未満を含む。)をビーカーなどにとり,塩酸(1+11)又は水酸化ナト
リウム溶液(40 g/L)で中和した後,全量フラスコ100 mLに移し入れ,水を標線まで加え,この
溶液から50 mLをとる。
2) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液10 mLを加え,直ちに密栓した後,混合する。
3) 高圧蒸気滅菌器に入れて加熱し,約120 ℃に達してから30分間加熱分解を行う。
4) 分解瓶を高圧蒸気滅菌器から取り出し,放冷する。
5.1) 上澄み液25 mLをビーカー50 mLに分取する。その際,水酸化物の沈殿を含まないように注意す
る。
5.2) 上澄み液の分取が困難なほど多量の水酸化物の沈殿が生じた場合は,孔径1 µm以下のガラス繊維
ろ紙を用いてろ過し,初めのろ液5〜10 mLを捨てた後のろ液25 mLを用いる。
6) 塩酸(1+16)5 mLを加えて溶液のpHを2〜3に調節する。
なお,5.2)で試料をろ過して分取した場合は,水酸化物の生成量に応じて濃度の低めた塩酸5 mL
を添加し,pHを2〜3に調節する。
7) 溶液の一部を吸収セルに移し,波長220 nmにおける吸光度を測定する。溶液中の全窒素の濃度が
0.4 mg/L未満の場合には,吸収セル50 mmを用いる。
8) 空試験として水50 mLを分解瓶にとり,2)〜7)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
9) 検量線から5)で分取した溶液中の全窒素の量を求め,次のいずれかの式によって試料中の全窒素の
濃度(N mg/L)を算出する。
v
a
N
b
a
N
100
50
000
1
25
60
50
000
1
25
60
×
×
×
=
×
+
×
=
ここに,
N: 全窒素(N mg/L)
a: 5.1)で分取した溶液25 mL中の全窒素(mg)
b: 1.2)で中和に要した塩酸及び水酸化ナトリウム溶液量(mL)
v: 1.1)及び1.3)で全量フラスコ100 mLに採取した試料量(mL)
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 20 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,それぞれに水を標線
まで加える。
2) その25 mLをそれぞれビーカー50 mLにとり,塩酸(1+500)5 mLを加えた後,一部を吸収セルに
移し,波長220 nmの吸光度を測定する。c) 7)で吸収セル50 mmを用いた場合には,窒素標準液(N
20 μg/mL)を5倍に薄めた窒素標準液(N 4 μg/mL)1〜10 mLをとり,1)及びここに規定する操作
を行い,吸光度の測定には,50 mmの吸収セルを用いる。
52
K 0102:2019
3) 別に,空試験として水25 mLをビーカー50 mLにとり,塩酸(1+500)5 mLを加えた後,波長220
nmの吸光度を求め,窒素標準液について得た吸光度を補正する。採取した溶液25 mL中の窒素(N)
の量と吸光度との関係線を作成する。
45.3 硫酸ヒドラジニウム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃
に加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅を
触媒として硫酸ヒドラジニウムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度
法によって定量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である
場合に適用する。
定量範囲:N 0.33〜3.3 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 45.2 a) 5)による。
3) 銅-亜鉛溶液 JIS K 8983に規定する硫酸銅(II)五水和物0.08 gとJIS K 8953に規定する硫酸亜鉛
七水和物1.76 gとを水に溶かして200 mLとし,その5 mLを水で薄めて250 mLとする。
4) 硫酸ヒドラジニウム溶液(7 g/L) JIS K 8992に規定する硫酸ヒドラジニウム3.5 gを水に溶かして
500 mLとする。
5) 硫酸ヒドラジニウム溶液(0.7 g/L) 硫酸ヒドラジニウム溶液(7 g/L)を水で10倍に薄める。使用
時に調製する。
6) 4-アミノベンゼンスルホンアミド溶液 43.1.1 a) 2)による。
7) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 43.1.1 a) 3)による。
8) 窒素標準液(N 20 μg/mL) 45.2 a) 8)による。
9) 窒素標準液(N 4 μg/mL) 窒素標準液(N 20 μg/mL)20 mLを全量フラスコ100 mLにとり,水を標
線まで加える。使用時に調製する。
10) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
11) 塩酸(1+11) 45.2 a) 2)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 45.2 b) 1)による。
2) 共栓試験管 材質及び形状が同じものを用いる。
3) 水浴 35±1 ℃に調節できるもの。
4) 高圧蒸気滅菌器 45.2 b) 2)による。
5) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 45.2 c)の1)〜4)の操作を行う。
2.1) 上澄み液10 mLを共栓試験管にとる。その際,水酸化物の沈殿を含まないように注意する。
なお,分解瓶にとった試料50 mL中の窒素が20 µg以上の場合には,上澄み液の適量c mL(N
として30 µg未満となる量)を全量フラスコ100 mLにとり,水酸化ナトリウム溶液(40 g/L)5 mL
を加えた後,水を標線まで加え,この溶液から10 mLをとる。上澄み液の適量c mLをとった場合
は,7)の式のaに100/cを乗じて全窒素の濃度を算出する。
2.2) 上澄み液の分取が困難なほど多量の水酸化物の沈殿が生じた場合は,孔径1 µm以下のガラス繊維
ろ紙を用いてろ過し,初めのろ液5〜10 mLを捨てた後のろ液10 mLを用いる。
53
K 0102:2019
3) 銅-亜鉛溶液1 mLを加えて振り混ぜた後,硫酸ヒドラジニウム溶液(0.7 g/L)1 mLを加えて振り混
ぜ,35±1 ℃の水浴中に浸す。
4) 2時間後,水浴から取り出し,室温まで冷却する。
5) 43.1.1 c)の2)及び3)の操作を行う。
6) 空試験として水50 mLを分解瓶にとり,1)〜5)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
7) 検量線から分解瓶にとった溶液50 mL中の全窒素の量を求め,次の式のいずれかによって試料中の
全窒素の濃度(N mg/L)を算出する。
v
a
N
b
a
N
a
N
100
50
000
1
50
50
50
000
1
50
000
1
×
×
=
+
×
×
=
×
=
ここに,
N: 全窒素(N mg/L)
a: 分解瓶にとった溶液50 mL中の全窒素(mg)
b: 1)の操作のうち,45.2のc) 1.2)で中和に要した塩酸及び水酸化
ナトリウム溶液量(mL)
v: 1)の操作のうち,45.2のc) 1.1)及びc) 1.3)で全量フラスコ100
mLに採取した試料量(mL)
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 4 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,それぞれに水を標線ま
で加える。
2) この溶液についてc)の1)〜6)の操作を行って分解瓶にとった溶液50 mL中の窒素(N)の量と吸光
度との関係線を作成する。
備考 1. 試料が海水などの場合は,含まれる無機物が硝酸イオンの還元率に影響するので,次の標準
添加法を行う。
試料40 mLを分解瓶にとり,水10 mLを加える。以下,45.2 c)の2)〜4)及びc)の2)〜6)の
操作を行って吸光度を測定し,下記の検量線から試料40 mL中の全窒素の量(mg)を求める。
別に,空試験として水50 mLを分解瓶にとり,45.2 c)の2)〜4)及びc)の2)〜6)の操作を行っ
て吸光度を測定し,d)の検量線から,相当する窒素の量(mg)を求める。次の式によって試
料中の全窒素の濃度(N mg/L)を算出する。
(
)
40
000
1
×
−
=
b
a
N
ここに,
N: 全窒素(N mg/L)
a: 試料40 mL中の全窒素(mg)
b: 空試験で得た窒素(mg)
検量線 窒素標準液(N 4 μg/mL)1〜8 mLを段階的に分解瓶にとり,それぞれに試料40 mL
を加えた後,水を加えて50 mLとし,45.2 c)の2)〜4)及びc)の2)〜6)の操作を行って吸光度
を測定し,その値から試料40 mLを用いて得た吸光度を差し引いて補正する。
なお,海水など多量のマグネシウムイオンが存在する試料の場合は,分解操作を行った溶
液のpHが低下して,マグネシウムの一部が上澄み液又はろ液に混入し,硝酸イオンの還元
率を低下させる。このため,分解後の溶液に水酸化ナトリウム溶液(40 g/L)を加えてpH12.6
〜12.8とした後の上澄み液を用いる。この操作を行った場合には,全窒素の算出式について
は,希釈による補正を行う。
試料40 mL中の窒素の量が10 μg以上の場合には,試料の適量(Nとして25 μg未満を含
54
K 0102:2019
む。)をとり,45.2 c)の1.1)及び1.3)に準じた操作を行い,これから40 mLを分解瓶にとる。
また,試料40 mL中の窒素の量が10 μg未満でpH5〜9の範囲にない場合には,45.2 c) 1.2)
に準じた中和操作を行い,これから40 mLを分解瓶にとる。これらの操作を行った場合には,
検量線の作成においても,この溶液を用い,全窒素の算出式については,それぞれ希釈に伴
う補正を行う。
45.4 銅・カドミウムカラム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃
に加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅・
カドミウムカラムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法によって定
量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である場合に適用す
る。
定量範囲:N 0.2〜2 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+11) 45.2 a) 2)による。
3) 塩化アンモニウム-アンモニア溶液 43.2.3 a) 3)による。
4) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 45.2 a) 5)による。
5) カラム活性化液 43.2.3 a) 4)による。
6) 銅・カドミウムカラム充塡剤 43.2.3 a) 5)による。
7) カラム充塡液 43.2.3 a) 6)による。
8) 4-アミノベンゼンスルホンアミド溶液 43.1.1 a) 2)による。
9) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 43.1.1 a) 3)による。
10) 窒素標準液(N 0.1 mg/mL) 45.2 a) 7)による。
11) 窒素標準液(N 2 μg/mL) 窒素標準液(N 0.1 mg/mL)10 mLを全量フラスコ500 mLにとり,水を
標線まで加える。使用時に調製する。
12) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 45.2 b) 1)による。
2) 高圧蒸気滅菌器 45.2 b) 2)による。
3) 銅・カドミウムカラム 43.2.3 b) 1)による。
4) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料50 mLを分解瓶にとる。試料のpHが5〜9の範囲にない場合には,分解瓶に塩酸(1+11)又
は水酸化ナトリウム溶液(40 g/L)を加えて中和する。試料50 mL中に含まれる全窒素が0.1 mg以
上の場合,試料の適量(Nとして0.2 mg未満を含む。)を全量フラスコ100 mLにとり,試料のpH
が5〜9の範囲にない場合には,塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)を用いて中和
した後,水を加えて100 mLとしたものから50 mLを分解瓶にとる。
2) 45.2 c)の2)〜4)の操作を行う。
3) 分解瓶に塩酸(1+11)10 mLを加えて振り混ぜた後,溶液を全量フラスコ100 mLに移す。
4) 分解瓶の内壁を少量の水で数回洗浄して洗液を3)の全量フラスコ100 mLに加える。
5) 塩化アンモニウム-アンモニア溶液10 mLを加え,水を標線まで加えて還元用溶液とする。1)で分解
55
K 0102:2019
瓶にとった溶液50 mL中の全窒素が20 μg以上の場合には,全量フラスコ200〜500 mLを用い,塩
化アンモニウム-アンモニア溶液を最終液量100 mL当たり10 mLとなるように添加した後,水を標
線まで加えたものを還元用溶液とする。この場合は,算出式に希釈率を乗じて補正する。
6) 43.2.3 c)の3)及び4)の操作を行う。
7) 空試験として水50 mLを分解瓶にとり,2)〜6)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
8) 検量線から還元用溶液中の全窒素の量を求め,次のいずれかの式によって試料中の全窒素の濃度(N
mg/L)を算出する。
v
a
N
a
N
100
50
000
1
50
000
1
×
×
=
×
=
ここに,
N: 全窒素(N mg/L)
a: 還元用溶液100 mL中の全窒素(mg)
v: 1)で全量フラスコ100 mLに採取した試料量(mL)
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 2 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,c)の5)及び6)の操作を
行って吸光度を測定する。
2) 別に,水約50 mLを全量フラスコ100 mLにとり,c)の5)及び6)の操作を行って吸光度を測定し,
窒素標準液(N 2 μg/mL)について得た吸光度を補正し,窒素(N)の量と吸光度との関係線を作成
する。
45.5 熱分解法 試料中の窒素化合物を熱分解してアンモニア又は窒素を生成させ,それらを定量する。
又は一酸化窒素に変えた後,化学発光法によって窒素を定量し,それぞれ全窒素を求める。
定量範囲:N 1〜200 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 全窒素標準液(N 0.2 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110 ℃で約2
時間加熱し,デシケーター中で放冷する。その1.444 gをとり,少量の水に溶かして全量フラスコ
1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
b) 器具及び装置 器具及び装置は,次による。
1) マイクロシリンジ 20〜150 μL
2) ホモジナイザー又はミキサー
3) 全窒素分析装置
c) 準備操作 準備操作は,次による。
1) 全窒素分析装置を作動できる状態にする。
2) 全窒素標準液(N 0.2 mg/mL)の一定量(例えば,20 μL)をマイクロシリンジで全窒素分析装置の
試料注入部から注入し,指示値(ピーク高さ)が最大目盛値の約80 %になるように装置の感度を調
節する。
3) 2)の操作を繰り返し,指示値が一定になることを確かめる。
4) 試料をよく振り混ぜて均一にした後,その一定量[例えば,2)と同量]をマイクロシリンジで試料
注入部から注入して指示値を読み取り,3)と比較して試料中の概略の全窒素の濃度を求める。全窒
素の濃度がN 200 mg/L以上の試料の場合には,水で薄めた試料を注入する。
56
K 0102:2019
d) 検量線 検量線の作成は,次による。
1) c) 4)で求めた試料中の概略の全窒素の濃度がほぼ中央になるように,全窒素標準液(N 0.2 mg/mL)
を全量フラスコ100 mLに段階的にとり,水を標線まで加えて各濃度の全窒素標準液を調製する。
2) 1)で調製した全窒素標準液の最高濃度のものの一定量[例えば,c) 2)と同量]をマイクロシリンジ
で試料注入部に注入し,指示値が最大目盛値の約80 %になるように全窒素分析装置の感度を調節す
る。
3) 順次,1)で調製した各濃度の全窒素標準液の一定量[2)で定めた量]をマイクロシリンジで試料注
入部から注入し,指示値を読み取る。
4) 空試験として3)と同量の水をマイクロシリンジでとり,3)と同様に操作して指示値を読み取り,3)
の指示値を補正して窒素(N)の濃度と指示値との関係線を作成する。
e) 操作 操作は,次による。
1) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
2) 試料の一定量[d) 2)で定めた量]をマイクロシリンジで装置の試料注入部に注入し,指示値を読み
取る。
3) 空試験として2)と同量の水をマイクロシリンジでとり,2)と同様に操作して指示値を読み取り,2)
の指示値を補正する。
4) あらかじめ作成した検量線から,注入試料中の全窒素の濃度を求め,試料中の全窒素の濃度(N
mg/L)を算出する。
備考 2. 全窒素分析装置には各種のものがある。アンモニアを生成させて定量する方式としては,試
料を水素気流中で熱分解し,全窒素を触媒によってアンモニアとした後,電量滴定法又は電
気伝導度測定法によるものがある。
窒素を生成させて定量する方式としては,試料をヘリウム気流中で熱分解し,触媒によっ
て全窒素を窒素とした後,熱伝導度測定法によるものがある。また,化学発光方式による方
式としては,試料を酸素気流中で熱分解して全窒素を一酸化窒素とし,更にこれをオゾンと
反応させ,二酸化窒素に酸化するとき生じる化学発光(波長650〜900 nm)を測定するもの
がある。
45.6 流れ分析法 試料中の窒素化合物の酸化分解,その結果生じる硝酸イオンの定量を45.2又は45.3と
同様な原理の流れ分析法によって行い,全窒素を定量する。
定量範囲:N:0.1〜2.0 mg/L(JIS K 0170-3の6.3.3及び6.3.5),1〜20 mg/L(JIS K 0170-3の6.3.2及
び6.3.4),繰返し精度:10 %以下
試験操作などは,JIS K 0170-3による。ただし,6.3.2(酸化分解・紫外検出FIA法)及び6.3.4(酸化分
解・紫外検出CFA法)は海水に適用できない。
備考 3. 試料中の窒素化合物を,45.2と同様な分解法である次の操作を行い,酸化分解して硝酸イオ
ンとし,JIS K 0170-2の箇条7(硝酸体窒素の測定)の試験操作によって定量してもよい。
分解法の操作は,次による。
1) 試料10 mL をマグネチックスターラーなどのかき混ぜ器でかき混ぜながら,耐圧・耐熱
性ねじ口試験管(耐圧・耐熱性のガラス製で容量15〜20 mLのもの)にとる。試料のpH
が5〜9の範囲にない場合には,分解瓶に塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)
を加えて中和し,中和に要した両液の量(mL)を記録し,濃度の計算時に補正する。
57
K 0102:2019
2) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液[45.2 a) 5)による。]2 mL を加え,密
栓して混合する。
3) 150〜180 ℃に加熱したブロックヒーターで約20分間加熱分解する。
4) 耐圧・耐熱性ねじ口試験管を取り出し,冷却後,pH8〜10になるように塩酸(1+13)(JIS
K 8180に規定する塩酸を用いて調製する)1 mLを加える。
なお,分解後の耐圧・耐熱性ねじ口試験管内の溶液に水酸化物の沈殿が生じている場
合は,上澄み液10 mLをとり,塩酸(1+13)に変えて塩酸(1+49)(JIS K 8180に規
定する塩酸を用いて調製する)1 mLを加える。濃度の計算時には加えた塩酸量の補正を
行う。
5) 空試験として水10 mLを耐圧・耐熱性ねじ口試験管にとり,2)〜4)の操作を行う。
46.(りん化合物及び全りん)の全文を,次に置き換える。
46. りん化合物及び全りん りん化合物は,りん酸,ポリりん酸,動物質及び植物質中のりんなど,水中
に存在するりん化合物のりんを意味し,りん酸イオン,加水分解性りん及び全りんに区分する。また,ろ
過した試料について試験することによって,それぞれを溶存及び懸濁のものに区別できる。りん化合物は
変化しやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,でき
るだけ早く試験する。
なお,モリブデン青吸光光度法,加水分解性りん及び全りんのペルオキソ二硫酸カリウム分解法は,2004
年に第2版として発行されたISO 6878,イオンクロマトグラフ法は,2007年に第2版として発行された
ISO 10304-1,流れ分析法は,2003年に第1版として発行されたISO 15681-1及びISO 15681-2との整合を
図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6878:2004,Water quality−Determination of phosphorus−Ammonium molybdate spectrometric
method(MOD)
ISO 10304-1:2007,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 1: Determination of bromide, chloride, fluoride, nitrate, nitrite, phosphate and sulfate
(MOD)
ISO 15681-1:2003,Water quality−Determination of orthophosphate and total phosphorus contents by
flow analysis (FIA and CFA)−Part 1: Method by flow injection analysis (FIA)(MOD)
ISO 15681-2:2003,Water quality−Determination of orthophosphate and total phosphorus contents by
flow analysis (FIA and CFA)−Part 2: Method by continuous flow analysis (CFA)(MOD)
46.1 りん酸イオン(PO43−) りん酸イオンの定量には,モリブデン青吸光光度法,イオンクロマトグラ
フ法又はモリブデン青発色による流れ分析法を適用する。
なお,溶存のりん酸イオンを定量する場合には,3.2によってろ過した試料を用いる。
46.1.1 モリブデン青吸光光度法 りん酸イオンが七モリブデン酸六アンモニウム及びタルトラトアンチ
モン(III)酸カリウムと反応して生成するヘテロポリ化合物をL(+)-アスコルビン酸で還元し,生成したモ
リブデン青の吸光度を測定してりん酸イオンを定量する。
定量範囲:PO43− 2.5〜75 μg,繰返し精度:2〜10 %
58
K 0102:2019
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) アスコルビン酸溶液(72 g/L) JIS K 9502に規定するL(+)-アスコルビン酸7.2 gを水に溶かして100
mLとする。0〜10 ℃の暗所に保存する。着色した溶液は使用しない。
3) モリブデン酸アンモニウム溶液 JIS K 8905に規定するモリブデン(VI)酸アンモニウム四水和物
6 gとJIS K 8533に規定するビス[(+)-タルトラト]二アンチモン(III)酸二カリウム三水和物0.24
gとを水約300 mLに溶かし,これに硫酸(2+1)(JIS K 8951に規定する硫酸を用いて調製する。)
120 mLを加え,次に,JIS K 8588に規定するアミド硫酸アンモニウム5 gを加えて溶かした後,水
を加えて500 mLとする。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 モリブデン酸アンモニウム溶液とアスコル
ビン酸溶液(72 g/L)とを体積比で5:1の割合になるように混合する。使用時に調製する。
5) りん酸イオン標準液(PO43− 0.1 mg/mL) JIS K 9007に規定するりん酸二水素カリウム(pH標準
液用)を105±2 ℃で約2時間加熱し,デシケーター中で放冷する。その0.143 3 gをとり,水に溶
かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
6) りん酸イオン標準液(PO43− 5 μg/mL) りん酸イオン標準液(PO43− 0.1 mg/mL)10 mLを全量フ
ラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
7) p-ニトロフェノール溶液(1 g/L) JIS K 8721に規定するp-ニトロフェノール0.1 gを水に溶かして
100 mLとする。
8) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1.1) 試料の適量(PO43−として2.5〜75 μgを含む。)をメスシリンダー(有栓形)25 mLにとる。
1.2) 試料が酸性の場合は,指示薬としてp-ニトロフェノール溶液(1 g/L)2,3滴を加え,水酸化ナト
リウム溶液(40 g/L)を用いて僅かに黄色になるまで中和する。ただし,このときアルミニウムな
どの水酸化物の沈殿が生じる場合には,沈殿が生じる直前でとどめる。中和後,水を25 mLの標
線まで加える。
2) モリブデン酸アンモニウム-アスコルビン酸混合溶液2 mLを加えて振り混ぜた後,20〜40 ℃で,約
15分間放置する。
3) 溶液の一部を吸収セルに移し,波長880 nm又は710 nm付近の吸光度を測定する(1)。
4) 空試験として水25 mLをとり,2)及び3)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
5) 検量線からりん酸イオンの量を求め,試料中のりん酸イオンの濃度(PO43− mg/L)を算出する。り
ん酸イオンの濃度をりん酸体りんで表示する場合は,35.の備考10.による。
注(1) 試料に濁り又は色がある場合は,1)と同量の試料をとり,モリブデン酸アンモニウム-アスコル
ビン酸混合溶液2 mLに代えてモリブデン酸アンモニウム溶液2 mLを用いて1)及び2)の操作を
行ってこの溶液を対照液として吸光度を測定するか,又はこの溶液の吸光度を測定して試料に
ついて得た吸光度を補正する。ただし,これらの場合は,試料に対しての4)による空試験の補
正は行わない。
なお,この操作による場合,濁りの著しい試料では誤差が大きくなる。
59
K 0102:2019
d) 検量線 検量線の作成は,次による。
1) りん酸イオン標準液(PO43− 5 μg/mL)0.5〜15 mLをメスシリンダー(有栓形)25 mLに段階的にと
り,水を25 mLの標線まで加える。
2) c)の2)〜4)の操作を行ってりん酸イオン(PO43−)の量と吸光度との関係線を作成する。温度は,試
料測定時と同じとなるようにする。
備考 1. 試料中にひ素(V)が含まれるときは,りん酸イオンと同様に発色するので,次の操作でそ
の妨害を除去する。この操作によってひ素(V)10 mg/Lの妨害が除去できる。
試料20 mLに硫酸(1 mol/L)(JIS K 8951に規定する硫酸5.6 mLを水約80 mL中に加え,
放冷後,水で100 mLとする。)1 mL,チオ硫酸ナトリウム溶液(7.65 g/L)(JIS K 8637に規
定するチオ硫酸ナトリウム五水和物1.2 gを水100 mLに溶かし,保存剤としてJIS K 8625に
規定する炭酸ナトリウム約50 mgを加える。)0.5 mLを加え,5〜10分間放置してひ素(V)
をひ素(III)とする。c) 1.2)の中和操作に従って中和し,水で25 mLとする。続いてc)の2)
〜5)の操作によってりん酸イオンを定量する。
2. 塩化物イオン及び硫酸イオンが多量(40 g/L程度)に共存しても妨害しない。しかし,多量
のアンモニウムイオン及びカリウムが共存すると,濁りが生じて妨害となる。
3. a) 3)のアミド硫酸アンモニウムを添加したモリブデン酸アンモニウム溶液を用いれば,亜硝
酸イオンによる発色妨害を防ぐことができる。a) 3)の溶液に含まれるアミド硫酸アンモニウ
ム量では,約7 mgまで亜硝酸イオンが共存しても発色は妨害されない。それ以上のときは,
別にアミド硫酸アンモニウム溶液を調製して追加する。
4. 鉄(III)約10 mg以上は,発色を妨害する。
5. りん酸塩が懸濁物として含まれるときは,試薬添加後,約15分間経過しても徐々に吸光度が
増加することがある。
6. りん酸イオンの濃度が低い試料の場合には,試料の量及びモリブデン酸アンモニウム-アスコ
ルビン酸混合溶液の量を増加してモリブデン青を発色させ,2,6-ジメチル-4-ヘプタノン[ジ
イソブチルケトン(DIBK)]で抽出して定量できる。操作は,46.3.1.3による。
7. 次のような操作で発色させ,定量することもできる。
試料の適量(PO43−として5〜150 μgを含む。)を全量フラスコ50 mLにとり,水を加えて
約40 mLとし,モリブデン酸アンモニウム-アスコルビン酸混合溶液3.5 mLを加え,水を標
線まで加えて振り混ぜた後,20〜40 ℃で約15分間放置して発色させ,波長880 nm又は710
nm付近の吸光度を測定する。水を用いた空試験を行って吸光度を補正する。
検量線は,りん酸イオン標準液(PO43− 5 μg/mL)1〜30 mLについて試料と同様に操作し
て作成する。
8. a) 3)のモリブデン酸アンモニウム溶液とa) 2)のアスコルビン酸溶液とを,混合せずに,別々
に添加することもできる。例えば,46.1.1の備考7.の操作で,モリブデン酸アンモニウム溶
液3 mL,続いてアスコルビン酸溶液0.5 mLを加え,水で50 mLとする。
46.1.2 欠番
46.1.3 イオンクロマトグラフ法 35.3による。
46.1.4 流れ分析法 試料中のりん酸イオンを,46.1.1と同様な原理で発色させる流れ分析法のJIS K
0170-4の箇条6(りん酸イオンの測定)によって定量する。
定量範囲:PO43− 0.03〜3 mg/L(Pとして0.01〜1 mg/L),繰返し精度:10 %以下
60
K 0102:2019
試験操作などは,JIS K 0170-4の箇条6のりん酸イオンの測定に関する規定による。りん酸イオンの濃
度をりん酸体りんで表示する場合は,35.の備考10.による。
46.2 加水分解性りん 試料を酸性として煮沸したとき,加水分解によってりん酸イオンとなるものをい
う。
試料に硫酸-硝酸の混酸を加え,煮沸してりん酸イオンとした後,46.1.1又は46.1.4によって定量し,こ
の値から加水分解前のりん酸イオンを差し引き,りん酸イオンに換算した値で表示する。
なお,溶存の加水分解性りんを定量する場合には,3.2によってろ過した試料を用いる。
定量範囲:PO43− 2.5〜75 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸-硝酸の混液 JIS K 8951に規定する硫酸300 mLを水約600 mL中に注意してかき混ぜながら
加えた後,放冷する。これにJIS K 8541に規定する硝酸4 mLと水とを加えて全量を1 Lとする。
3) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
4) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
5) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
6) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
7) p-ニトロフェノール溶液(1 g/L) 46.1.1 a) 7)による。
8) りん酸イオン標準液(PO43− 5 μg/mL) 46.1.1 a) 6)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(PO43−として1 mg以下を含む。)をビーカー200 mLにとる。試料が酸性の場合には,
46.1.1 c) 1.2)の中和操作に従って中和した後,水を加えて50〜100 mLとし,硫酸-硝酸の混酸1 mL
を加える。
2) 静かに煮沸する。液量が25 mL以下になったら水を加え,液量を25〜50 mLに保って約90分間煮
沸する。
3) 放冷後,ろ紙5種Bを用いてろ過し,温水で3,4回洗う。
4) ろ液と洗液とを合わせ,指示薬としてp-ニトロフェノール溶液(1 g/L)3〜5滴を加え,溶液が僅か
に黄色になるまで水酸化ナトリウム溶液(40 g/L)を滴加した後,全量フラスコ100 mLに移し入れ,
水を標線まで加える。
5) この溶液の適量をとり,46.1.1によってりん酸イオンの量を求め,試料中のりん酸イオンの濃度
(PO43− mg/L)に換算し,この値から別に46.1.1によって定量した試料中のりん酸イオンの濃度
(PO43− mg/L)を差し引いて加水分解性りんとし,りん酸イオンの濃度(PO43− mg/L)で表す。
46.3 全りん ペルオキソ二硫酸カリウム分解,硝酸-過塩素酸分解又は硝酸-硫酸分解によって試料中のり
ん化合物などを分解し,生成したりん酸イオンを46.3.1.2,46.3.1.3又は46.1.4によって定量し,これを全
りんとしてりんの濃度で表す。又は一連のペルオキソ二硫酸カリウム分解及びりん酸イオンのモリブデン
青吸光光度法による定量を46.3.4の流れ分析法によって行い,全りん濃度を求める。
46.3.1 ペルオキソ二硫酸カリウム分解法 試料にペルオキソ二硫酸カリウムを加え,高圧蒸気滅菌器中で
加熱して有機物などを分解し,この溶液についてりん酸イオンを定量して全りんの濃度を求める。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
61
K 0102:2019
46.3.1.1 分解法
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) ペルオキソ二硫酸カリウム溶液(40 g/L) JIS K 8253に規定するペルオキソ二硫酸カリウム(窒素・
りん測定用)4 gを水に溶かして100 mLとする。
3) 硫酸(1+35) 43.2.1 a) 3)による。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 亜硫酸水素ナトリウム溶液(50 g/L) JIS K 8059に規定する亜硫酸水素ナトリウム5 gを水に溶か
して100 mLとする。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 45.2 b) 1)による。
2) 高圧蒸気滅菌器 45.2 b) 2)による。
c) 操作 操作は,次による。
1.1) 試料のpHが5〜9で,試料50 mLに含まれる全りんが60 µg未満の場合は,分解瓶に試料50 mL
をとる。また,全りんが60 µg以上含まれる場合は,試料の適量(Pとして0.12 mg未満を含む。)
を全量フラスコ100 mLにとり,水を標線まで加え,この溶液から50 mLをとる。
1.2) 試料のpHが5〜9にない場合で,試料50 mLに含まれる全りんが60 µg未満の場合は,試料50 mL
を分解瓶にとり,硫酸(1+35)又は水酸化ナトリウム溶液(40 g/L)を用いて中和する。中和に
要した両液の合量(b mL)を記録し,全りんの濃度算出時[46.3.1.2 c) 4)]に補正する。
1.3) 試料のpHが5〜9の範囲にない場合で,試料50 mLに含まれる全りんが60 µg以上を含む場合は,
試料の適量(Pとして0.12 mg未満を含む。)をビーカなどにとり,硫酸(1+35)又は水酸化ナト
リウム溶液(40 g/L)を用いて中和した後,全量フラスコ100 mLに移し入れ,水を標線まで加え,
この溶液から50 mLをとる。
2) ペルオキソ二硫酸カリウム溶液(40 g/L)10 mLを加え,密栓して混合する。
3) 高圧蒸気滅菌器に入れて加熱し,約120 ℃に達してから30分間加熱分解する。
4.1) 分解瓶を取り出し,放冷する。
4.2) 塩化物イオンを多く含む試料の場合は,塩素が生成してモリブデン青の発色を妨害するおそれが
あるので,分解後の溶液に亜硫酸水素ナトリウム溶液(50 g/L)を1 mL加える。又は,塩素臭の
なくなるまで煮沸し,放冷後,水で60 mLとする。亜硫酸水素ナトリウムを添加した場合は,添
加量を全りん濃度の算出時に補正する。
4.3) 分解後の溶液にひ素(V)が含まれる場合には,備考1.に準じ,次の操作でひ素(III)に還元する。
分解後の溶液に硫酸(1 mol/L)3 mL(備考1.による。)及びチオ硫酸ナトリウム溶液(7.65 g/L)
(備考1.による。)1.5 mLを加え,5〜10分間放置してひ素(V)をひ素(III)とする。46.1.1 c) 1.2)
の中和操作に従って酸を中和する。その操作を行った場合は,用いた硫酸(1 mol/L),チオ硫酸ナ
トリウム溶液(7.65 g/L)及び水酸化ナトリウム溶液(40 g/L)の合量(mL)を記録し,全りん濃
度の算出時に補正する。
5) 空試験として水50 mLを分解瓶にとり,2)〜4)の操作を行う。
備考 9. c)の操作に代えて,耐圧・耐熱性ねじ口試験管とブロックヒーターとを用いる次の操作で分
解してもよい。
1.1) 試料のpHが5〜9で,試料10 mLに含まれる全りんが12 µg未満の場合は,耐圧・耐
62
K 0102:2019
熱性ねじ口試験管(45.6の備考3.による。)に試料10 mLをとる。また,全りんが12 µg
以上含まれる場合は,試料の適量(Pとして25 µg未満を含む。)を全量フラスコ20 mL
にとり,水を標線まで加え,この溶液から10 mLをとる。
1.2) 試料のpHが5〜9の範囲にない場合で,試料10 mLに含まれる全りんが12 µg未満の
場合は,試料10 mLを耐圧・耐熱性ねじ口試験管にとり,硫酸(1+35)又は水酸化ナ
トリウム溶液(40 g/L)を用いて中和する。この場合は,中和に要した両液の合量(b mL)
を記録する。
1.3) 試料のpHが5〜9の範囲にない場合で,試料10 mLに含まれる全りんが12 µg以上を
含む場合は,試料の適量(Pとして25 µg未満を含む。)をビーカーなどにとり,硫酸
(1+35)又は水酸化ナトリウム溶液(40 g/L)を用いて中和した後,全量フラスコ20
mLに移し入れ,水を標線まで加え,この溶液から10 mLをとる。
2) ペルオキソ二硫酸カリウム溶液(40 g/L)2 mLを加え,密栓して混合する。
3) 150〜180 ℃に加熱したブロックヒーターで約20分間加熱分解する。
4.1) 耐圧・耐熱性ねじ口試験管を取り出し,放冷する。
4.2) 塩化物イオンを多く含む試料の場合は,c) 4.2)に準じて,分解後の溶液に亜硫酸水素ナ
トリウム(50 g/L)を0.2 mL加える。この操作を行った場合は,この量を全りん濃度
の算出時に補正する。
4.3) 分解後の溶液にひ素(V)が含まれる場合には,c) 4.3)に準じて,分解後の溶液に硫酸
(1 mol/L)0.6 mL及びチオ硫酸ナトリウム溶液(7.65 g/L)0.3 mLを加え,5〜10 分
間放置してひ素(V)をひ素(III)とした後,水酸化ナトリウム溶液(40 g/L)で中和
し,この上澄み液10 mLをとり,備考10. 2)の操作を行う。
用いた硫酸(1 mol/L),チオ硫酸ナトリウム溶液(7.65 g/L)及び水酸化ナトリウム
溶液(40 g/L)の合量(mL)を記録し,全りん濃度の算出時に補正する。
5) 空試験として水10 mLを分解瓶にとり,2)〜4.1)の操作を行う。
46.3.1.2 定量法 46.3.1.1の操作で得られた分解液中のりん酸イオンをモリブデン青吸光光度法で定量し,
全りん濃度を求める。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
3) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。ただし,アミド硫酸アンモニウムは加えなく
てもよい。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
5) りん標準液(P 50 μg/mL) JIS K 9007に規定するりん酸二水素カリウム(pH標準液用)を105±2 ℃
で約2時間加熱し,デシケーター中で放冷する。その0.220 gをとり,少量の水に溶かして全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
6) りん標準液(P 5 μg/mL) りん標準液(P 50 μg/mL)10 mLを全量フラスコ100 mLにとり,水を標
線まで加える。使用時に調製する。
7) りん標準液(P 0.5 μg/mL) りん標準液(P 5 μg/mL)10 mLを全量フラスコ100 mLにとり,水を標
線まで加える。使用時に調製する。
63
K 0102:2019
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。備考9.を用いて分解を行った場合は,備考10.によって定量する。
1.1) 46.3.1.1 c)で分解した上澄み液25 mLをメスシリンダー(有栓形)25 mLに分取する。
1.2) 上澄み液に濁りが認められる場合には,ろ紙5種C又は孔径1 μm以下のガラス繊維ろ紙を用いて
ろ過し,初めのろ液5〜10 mLを捨てた後のろ液を用いる。
1.3) 分解後の溶液中に金属水酸化物の沈殿が認められる場合には,これらが溶ける点まで硫酸(1+35)
[及び必要に応じ水酸化ナトリウム溶液(40 g/L)]を用いて沈殿を溶解する。用いた両液の量(mL)
を記録し,全りん濃度の算出時に補正する。
なお,金属水酸化物の沈殿を溶かした後の溶液に濁りが認められる場合には,更にろ過の操作
を行う。
2) 46.1.1 c)の2)及び3)の操作を行って吸光度を測定する(2)。全りん濃度が0.1 mg/L未満の場合には,
吸光度の測定に光路長50 mmのセルを用いる。
3) 46.3.1.1 c) 5)の分解瓶の中から25 mLを分取し,2)の操作を行って吸光度を測定し,試料について得
た吸光度を補正する。
4) 検量線から1.1)で分取した溶液25 mL中のりんの量を求め,次の式のいずれかによって試料中の全
りんの濃度(P mg/L)を算出する。
v
a
P
b
a
P
100
50
000
1
25
60
50
000
1
25
60
×
×
×
=
×
+
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 1.1)で分取した溶液25 mL中の全りんの質量(mg)
b: 46.3.1.1 c) 1.2)で中和に要した硫酸及び水酸化ナトリウム溶液
の合量(mL)
v: 46.3.1.1 c)の1.1)又は1.3)で全量フラスコ100 mLに採取した試
料量(mL)
注(2) 分解後の溶液にひ素(V)が含まれる場合には,備考1.に準じ,次の操作でひ素(III)に還元
する。分解後の溶液に硫酸(1 mol/L)3 mL(備考1.による。)及びチオ硫酸ナトリウム溶液(7.65
g/L)(備考1.による。)1.5 mLを加え,5〜10分間放置してひ素(V)をひ素(III)とする。46.1.1
c) 1.2)の中和操作に従って酸を中和する。その操作を行った場合は,用いた硫酸(1 mol/L),チ
オ硫酸ナトリウム溶液(7.65 g/L)及び水酸化ナトリウム溶液(40 g/L)の合量(mL)を記録し,
全りん濃度の算出時に補正する。
d) 検量線 検量線の作成は,次による。
1) りんの採取量が1.25〜25 μgになるように,りん標準液(P 0.5 μg/mL)及びりん標準液(P 5 μg/mL)
の各適量をそれぞれメスシリンダー(有栓形)25 mLにとり,水を加えて25 mLとする。46.1.1 c) の
2)及び3)の操作を行って吸光度を測定する。
2) 別に,空試験として水25 mLをメスシリンダー(有栓形)25 mLにとり,46.1.1 c) の2)及び3)の操
作を行って吸光度を測定し,1)で得た吸光度を補正する。
3) 25 mL中のりん(P)の量と吸光度との関係線を作成する。
備考 10. 備考9.の分解操作を行った場合は,次の操作によって分解液中のりん酸イオンを定量し,試
料中の全りん濃度を求める。
1.1) 分解液の上澄み液10 mLをメスシリンダー(有栓形)10 mLに移し入れる。
64
K 0102:2019
1.2) 上澄み液に濁りが認められる場合には,遠心分離を行う。
1.3) 分解後の溶液中に金属水酸化物の沈殿が認められる場合には,46.3.1.2 c) 1.3)に準じて
これらが溶ける点まで硫酸(1+35)[及び必要に応じ水酸化ナトリウム溶液(40 g/L)]
を用いて沈殿を溶解する。用いた両液の量(mL)を記録し,全りん濃度の算出時に補
正する。金属水酸化物の沈殿を溶かした後の溶液に濁りが認められる場合には,更に
遠心分離を行う。
2) 46.1.1 c)の2)及び3)の操作を行って吸光度を測定する。ただし,モリブデン酸アンモニ
ウム-アスコルビン酸混合溶液の添加量は0.8 mLとする。
なお,試料中の全りん濃度が0.1 mg/L未満の場合には,吸光度の測定に光路長50 mm
のセルを用いる。
3) 備考9.の5)の分解後の耐圧・耐熱性ねじ口試験管から分解液10 mLをメスシリンダー(有
栓形)10 mLにとり,2)の操作を行って吸光度を測定し,試料について得た吸光度を補
正する。
4) 検量線から分取した上澄み液10 mL中のりんの量を求め,試料中の全りんの濃度(P
mg/L)を算出する。
v
a
P
b
a
P
20
10
000
1
10
12
10
000
1
10
)
12
(
×
×
×
=
×
+
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 備考10.の1.1)で分取した溶液10 mL中の全りんの質量(mg)
b: 備考9.の1.2)で中和に要した硫酸及び水酸化ナトリウム溶液
量(mL)
v: 備考9.の1.1)又は1.3)で採取した試料量(mL)
備考 11. りんの濃度が低く十分な定量精度が得にくい試料については,次の加熱濃縮操作を行うか,
又は46.3.1.3の溶媒抽出による定量を行う。
試料100〜250 mLをビーカー200〜500 mLにとり,硫酸(2+1)(JIS K 8951に規定する硫
酸を用いて調製する。)1,2滴を加えた後,加熱板上で加熱して液量が50 mL以下になるま
で濃縮する。この溶液を水酸化ナトリウム溶液(40 g/L)で中和した後,分解瓶(あらかじ
め50 mLの位置に印を付けたもの。)に移し,水を加えて50 mLとし,46.3.1.1 c) の2)〜5)
及び46.3.1.2 c)の操作を行う。
46.3.1.3 溶媒抽出法による定量法 46.3.1.1 c)の操作で得られた分解液を46.3.1.2 c)に準じて発色させてモ
リブデン青とし,2,6-ジメチル-4-ヘプタノン[ジイソブチルケトン(DIBK)]で抽出することによって定
量し,微量の全りん濃度を求める。
定量範囲:P 0.25〜6.25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
3) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
5) りん標準液(P 0.5 μg/mL) 46.3.1.2 a) 7)による。
6) 2,6-ジメチル-4-ヘプタノン[ジイソブチルケトン(DIBK)]
b) 器具及び装置 器具及び装置は,次による。
65
K 0102:2019
1) 光度計 分光光度計又は光電光度計
2) 分液漏斗 100 mL
c) 操作 操作は,次による。
1) 46.3.1.1 c)の1)〜4)の操作を行った後,分解瓶中の溶液を分液漏斗100 mLに移し,分解瓶は水10 mL
で洗浄し洗液を分液漏斗に合わせる。
2) モリブデン酸アンモニウム-アスコルビン酸混合溶液5.5 mLを加えて20〜40 ℃で約15分間放置す
る。
3) 分液漏斗に2,6-ジメチル-4-ヘプタノン5 mLを加えて約5分間振り混ぜる。
4) 静置後,水層を捨て,2,6-ジメチル-4-ヘプタノン層(水滴などによる濁りがあれば,乾燥したろ紙
で手早くろ過する。)の一部を吸収セルに移し,波長640 nm付近の吸光度を測定する。
5) 46.3.1.1 c) 5)の操作を行った後,分解瓶中の溶液を分液漏斗100 mLに移し,分解瓶は水10 mLで洗
浄し,洗液を分液漏斗に合わせる。2)〜4) の操作を行って吸光度を測定し,試料について得た吸光
度を補正する。検量線から試料中の全りんの量を求め,次の式によって試料中の全りんの濃度(P
mg/L)を算出する。
V
a
P
000
1
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 測定した全りんの質量(mg)
V: 試料量(mL)
d) 検量線 検量線の作成は,次による。
1) りん標準液(P 0.5 μg/mL)の0.5〜12.5 mLを段階的に分液漏斗(あらかじめ,70 mLの位置に印を
付けたもの)100 mLにとり,水を加えて70 mLとした後,c)の2)〜4)の操作を行って吸光度を測定
する。
2) 空試験として水70 mLを分液漏斗100 mLにとり,同様の操作を行って吸光度を測定し,各りん標
準液について得た吸光度を補正し,採取したりん(P)の量と吸光度との関係線を作成する。
46.3.2 硝酸-過塩素酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸及び過塩素酸を加え,再び加熱して
有機物などを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は,多
量の有機物を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
46.3.2.1 分解法
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) 硝酸 JIS K 8541に規定するもの。
3) 過塩素酸 JIS K 8223に規定するもの。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。
6) p-ニトロフェノール溶液(1 g/L) 46.1.1 a) 7)による。
b) 操作 操作は,次による。
1) 試料50 mLをビーカーにとる。試料中の全りんの濃度が低い場合には,50 mL以上の適量をとる。
多量の塩化物イオンを含む試料で全りんの濃度が高い場合には,50 mL未満の適量をとる。
2) 硝酸を加えて弱酸性とし,加熱板上で静かに加熱して15〜20 mLに濃縮する。
66
K 0102:2019
3) これに硝酸2〜5 mLを加えて再び加熱し,約10 mLになるまで濃縮した後,更に硝酸2 mLを加え
て加熱し,約10 mLになるまで濃縮し,放冷する。
4) 過塩素酸5 mLを少量ずつ加える。
なお,試料に多量の塩化物イオンが含まれる場合には,塩化物イオンの当量よりも多い量を更に
加える。加熱板上で再度加熱し,過塩素酸の白煙が発生し始めたらビーカーを時計皿で覆い,過塩
素酸がビーカーの内壁を還流する状態に保つ(3)。この操作によっても有機物が分解されず,溶液に
色が残った場合には,硝酸2 mLを加えて加熱する操作を繰り返す。
5) 放冷後,水約30 mLを加える。必要に応じ加熱して可溶性塩を溶かす。加熱しても不溶解物が残っ
た場合には,ろ紙5種C又は孔径1 μm以下のガラス繊維ろ紙を用いて溶液をろ過し,次に,ろ紙
を少量の水で洗浄し,ろ液と洗液とを合わせる。
6) この溶液に指示薬としてp-ニトロフェノール溶液(1 g/L)3〜5滴を加え,初めに水酸化ナトリウム
溶液(200 g/L)を,次に,水酸化ナトリウム溶液(40 g/L)を加えて溶液が僅かに黄色になるまで
中和する。中和するときに金属水酸化物の沈殿が認められる場合には,水酸化ナトリウム溶液(40
g/L)の添加は,沈殿の生じる直前でとどめる。必要に応じ硫酸(1+35)[30.1.1 a) 2)による。]を
用いて調節する。
7) 溶液を全量フラスコ50 mLに移し入れ,水を標線まで加える。
8) 空試験として1)で採取した試料と同量の水をビーカーにとり,2)〜7)の操作を行う。
注(3) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこと
に注意する。
− 酸化されやすい有機物は,過塩素酸を加える前に,2)及び3)の操作によって十分に分解し
ておく。
− 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
− 必ず過塩素酸と硝酸とを共存させた状態で加熱分解を行う。
− 濃縮液を乾固させない。
46.3.2.2 定量法 46.3.2.1の操作で得られた分解液中のりん酸イオンをモリブデン青吸光光度法で定量し,
全りん濃度を求める。
なお,りん酸イオンの定量を46.1.4で行ってもよい。また,りん酸イオン濃度が低い場合は46.3.1.3に
準じて操作を行い,全りん濃度を求める。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
3) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
5) りん標準液(P 5 μg/mL) 46.3.1.2 a) 6)による。
6) りん標準液(P 0.5 μg/mL) 46.3.1.2 a) 7)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 46.3.2.1 b) 7)の溶液25 mL又は全りんが25 μg以上になる場合は,この溶液の適量(りん含有量25 μg
67
K 0102:2019
未満となる量)をメスシリンダー(有栓形)25 mLにとる。適量(b mL)をとった場合は,水を加
えて25 mLとする。
2) 46.1.1 c)の2)及び3)の操作を行って吸光度を測定する。
3) 空試験として46.3.2.1 b) 8)の溶液25 mLをメスシリンダー(有栓形)25 mLにとり,1)及び2)を行
い,試料について得た吸光度を補正する。
4) 検量線から1)で分取した溶液中のりんの量を求め,次の式によって試料中の全りんの濃度(P mg/L)
を算出する。
v
b
a
P
000
1
50×
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 1)で分取した25 mL又はb mL中の全りんの質量(mg)
b: 1)で分取した溶液量(mL)
v: 試料量(mL)
d) 検量線 46.3.1.2 d)の検量線と同じ操作によって作成する。
46.3.3 硝酸-硫酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸及び硫酸を加え,更に加熱して有機物な
どを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は,多量の有機
物を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
46.3.3.1 分解法
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) 硝酸 46.3.2.1 a) 2)による。
3) 硫酸(1+1) 5.4 a) 2)による。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。
6) p-ニトロフェノール溶液(1 g/L) 46.1.1 a) 7)による。
b) 操作 操作は,次による。
1) 46.3.2.1 b)の1)及び2)の操作を行う。
2) 1)の操作を行った後の溶液に硫酸(1+1)2 mL及び硝酸2〜5 mLを加える。
なお,試料に多量の塩化物イオンが含まれる場合には,塩化物イオンの当量よりも多い量の硫酸
(1+1)を更に加える。加熱して硫酸の白煙が発生するまで濃縮し,ビーカーを時計皿で覆い,更
に加熱して硫酸の白煙を短時間強く発生させた後,放冷する。
3) この溶液に硝酸5 mLを加えて再び加熱し,硫酸の白煙が発生するまで加熱する。この操作によっ
ても有機物が分解されず,溶液に色が残った場合には,硝酸2 mLを加えて加熱する操作を繰り返
す。
4) 放冷後,水約30 mLを加え,約10分間静かに煮沸する。不溶解物が残った場合には,ろ紙5種C
又は孔径1 μm以下のガラス繊維ろ紙を用いて溶液をろ過し,次に,ろ紙を少量の水で洗浄し,ろ
液と洗液とを合わせる。
5) 46.3.2.1 b)の6)及び7)の操作を行う。
6) 空試験として1)で採取した試料と同量の水をビーカーにとり,2)〜5)の操作を行う。
46.3.3.2 定量法 46.3.3.1の操作で得られた分解液中のりん酸イオンをモリブデン青吸光光度法で定量し,
68
K 0102:2019
全りん濃度を求める。
なお,りん酸イオンの定量を46.1.4で行ってもよい。また,りん酸イオン濃度が低い場合は46.3.1.3に
準じて操作を行い,全りん濃度を求める。
定量範囲: P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 46.3.1.1 a) 1)による。
2) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
3) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
5) p-ニトロフェノール溶液(1 g/L) 46.1.1 a) 7)による。
6) りん標準液(P 5 μg/mL) 46.3.1.2 a) 6)による。
7) りん標準液(P 0.5 μg/mL) 46.3.1.2 a) 7)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 46.3.3.1 b) 5)の溶液25 mL又は全りんが25 μg以上になる場合には,この溶液の適量(りん含有量
25 μg未満となる量)をメスシリンダー(有栓形)25 mLにとる。適量(b mL)をとった場合は,
水を加えて25 mLとする。
2) 46.1.1 c)の 2)及び3)の操作を行って吸光度を測定する。
3) 空試験として46.3.3.1 b) 6)の溶液25 mLをメスシリンダー(有栓形)25 mLにとり,1)を行って吸
光度を測定し,試料について得た吸光度を補正する。
4) 検量線からりんの量を求め,46.3.2.2 c) 4)の式によって試料中の全りんの濃度(P mg/L)を算出する。
d) 検量線 46.3.1.2 d)の検量線と同じ操作によって作成する。
46.3.4 流れ分析法 試料中のりん化合物などを,46.3.1と同様な原理で加水分解又は酸化分解してりん酸
イオンとした後,りん酸イオンをモリブデン青吸光光度法によって定量する一連の操作を流れ分析法の
JIS K 0170-4の箇条7(全りんの測定)によって行い,全りん濃度を求める。
定量範囲:P 0.01〜10 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-4の箇条7(全りんの測定)による。ただし,JIS K 0170-4の7.3.2(UV照
射酸化分解・モリブデン青発色FIA法)及び7.3.4(UV照射酸化分解・モリブデン青発色CFA法)は除く。
47.[ほう素(B)]の注(9)を,“注(9) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を
用いて測定してもよい。また,感度,精度及びスペクトル干渉が許容できるものであれば,他の波長を用
いてもよい。”に置き換える。
47.[ほう素(B)]の備考5.を,次に置き換える。
備考 5. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることが
できる。操作は,次による。
1) 試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50 μg/mL)10 mLを加
えた後,水を標線まで加える。
2) この溶液についてc) 1)の操作を行ってほう素[249.773 (I) nm]及びイットリウム[464.370
69
K 0102:2019
(I) nm]の発光強度を測定し,ほう素の発光強度とイットリウムの発光強度との比を求め
る(*)。
3) 空試験として,試料に代えて水を用い,1)及び2)の操作を行い,ほう素の発光強度とイ
ットリウムの発光強度との比を求め,2)で得た発光強度比を補正する。
4) 検量線から,ほう素の量を求め,試料中のほう素の濃度(B μg/L)を算出する。
5) 検量線 全量フラスコ100 mL数個に,ほう素標準液(B 20 μg/mL)0.1〜40 mLを段階
的にとり,イットリウム溶液(Y 50 μg/mL)10 mLを加え,水を標線まで加える。この
溶液について2)の操作を行ってほう素及びイットリウムの発光強度を測定し,ほう素の
発光強度とイットリウムの発光強度との比を求める。別に,空試験として,ほう素標準
液に代えて水を用い,同じ操作を行って,同様にほう素とイットリウムとの発光強度の
比を求め,ほう素標準液でのほう素とイットリウムとの発光強度比を補正し,ほう素の
量と,ほう素とイットリウムとの発光強度比との関係線を作成する。
6) イットリウム溶液(Y 50 μg/mL)の調製 酸化イットリウム(III)0.318 gをとり,JIS K
9901に規定する高純度試薬−硝酸5 mLを加え,加熱して溶かし,煮沸して窒素酸化物
を追い出し,放冷後,全量フラスコ250 mLに移し,水を標線まで加える。この溶液10 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
注(*) 表52.1の注参照。Iは中性線を示す。
47.[ほう素(B)]の備考6.を,次に置き換える。
備考 6. ほう素を含む溶液を発光部に導入した場合には,メモリー効果が他の元素の場合より大きい
ため,次の溶液を噴霧する前に,酸溶液又は水を十分な時間噴霧して前の試料の影響を除去
する。また,水で十分洗浄した後でも,酸溶液を導入するとほう素のメモリーが現れる場合
があることに留意し,次の測定値にメモリーの影響が出ないことを確認する。
48.[ナトリウム(Na)]の全文を,次に置き換える。
48. ナトリウム(Na) ナトリウムの定量には,フレーム光度法,フレーム原子吸光法,イオンクロマト
グラフ法又はICP発光分光分析法を適用する。
なお,フレーム光度法は,1993年に第1版として発行されたISO 9964-3,フレーム原子吸光法は,1993
年に第1版として発行されたISO 9964-1,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885との整合を図ったもので
ある。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9964-1:1993,Water quality−Determination of sodium and potassium−Part 1: Determination of
sodium by atomic absorption spectrometry(MOD)
ISO 9964-3:1993,Water quality−Determination of sodium and potassium−Part 3: Determination of
sodium and potassium by flame emission spectrometry(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+,
Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
70
K 0102:2019
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
48.1 フレーム光度法 試料をアセチレン-空気フレーム,水素-酸素フレームなどの中に噴霧し,このとき
生じる波長589.0 nmの輝線の強さを測定してナトリウムを定量する。
定量範囲:Na 30〜300 μg/L,0.3〜3 mg/L,3〜30 mg/L,繰返し精度:3〜10 %(装置及び測定条件に
よって異なる。)
a) 試薬 試薬は,次による。
1) ナトリウム標準液(Na 1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウム
を600 ℃で約1時間加熱し,デシケーター中で放冷する。NaCl 100 %に対してその2.542 gをはか
りとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。ポリエチレ
ン瓶に保存する。
2) ナトリウム標準液(Na 3〜30 mg/L) ナトリウム標準液(Na 1 000 mg/L)を段階的にとり,これを
水で薄めてNa 3〜30 mg/Lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,Na 30〜300 μg/L又はNa 0.3〜3 mg/Lの標準液を調製する。
b) 装置 装置は,次による。
1) フレーム光度計
c) 操作 操作は,次による。
1) ナトリウム標準液(Na 30 mg/L)(2)をフレーム光度計のフレーム中に噴霧し,波長589.0 nmの指示
値が100を示すように調節する。
2) 水を噴霧して指示値がゼロを示すように調節する。
3) ナトリウム標準液(Na 3〜30 mg/L)(1)を順次噴霧し,ナトリウム(Na)の濃度と指示値との関係線
を作成し,検量線とする。
4) 試料(3) (4)(ナトリウムの濃度が30 mg/L以上の場合は薄める。)を噴霧して指示値を読み取り,検
量線から試料中のナトリウムの濃度(Na mg/L)を求める。
注(2) 低い濃度の測定用には,Na 3 mg/L又はNa 0.3 mg/Lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むナトリウム標準液(Na 3〜30 mg/L)を調製し,検量線を作成す
る。
備考 1. カリウム及びカルシウムが共存すると正の誤差を生じる。
このような試料には,塩化セシウム溶液(25 g/L)[塩化セシウム25 gをJIS K 8180に規
定する塩酸50 mL及び水450 mLに溶かし,水を加えて1 Lとする。この溶液1 Lは,セシウ
ム(Cs)を約20 gを含む。]を試料40 mLに対して5 mLを加えることで,カリウム,カル
シウムなどの影響を抑制できる。この操作を行った場合は,検量線作成時の操作も塩化セシ
ウム溶液(25 g/L)を試料と同様に加えて行う。また,リチウム,バリウム,遊離酸,りん
酸塩,ほう酸塩,しゅう酸塩,シリカ,グルコース,ゼラチンなどが共存すると負の誤差を
生じる。マグネシウム及び硫酸イオンはほとんど干渉しない。
多量のけい酸塩が共存する場合には,試料の適量を石英ガラスビーカー又は白金皿にとり,
塩酸(1+1)[24.2 a) 2)による。]を加えて酸性とした後,蒸発乾固する。放冷後,塩酸(1
+1)5滴及び少量の水を加え,加熱して溶かし,ろ紙5種Bでろ過し,ろ液を水で一定量と
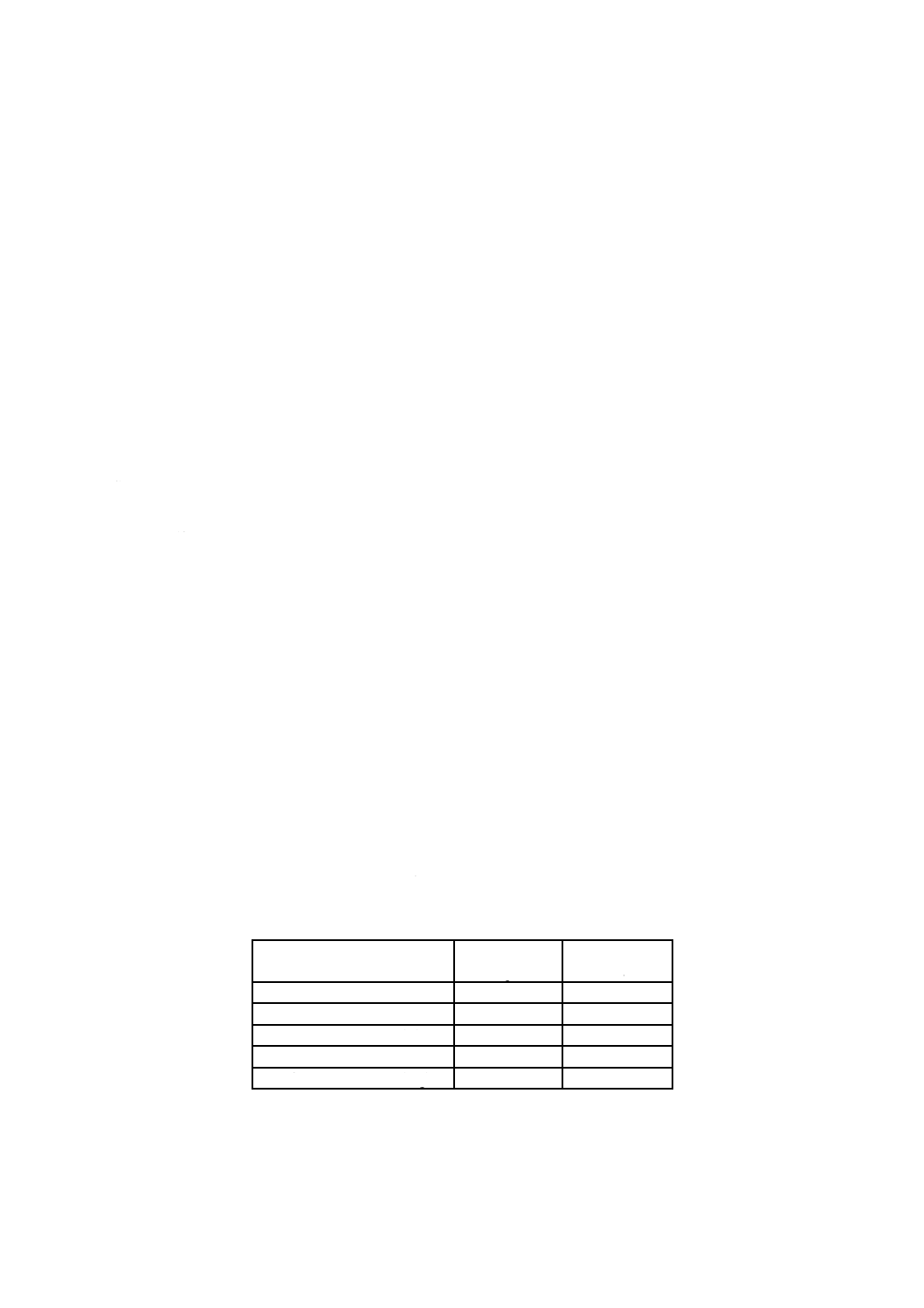
71
K 0102:2019
する。
48.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,ナトリウムによる原子吸光を波
長589.0 nmで測定してナトリウムを定量する。
定量範囲:Na 0.05〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) ナトリウム標準液(Na 100 mg/L) 48.1 a) 1)のナトリウム標準液(Na 1 000 mg/L)10 mLを全量フ
ラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
2) ナトリウム標準液(Na 10 mg/L) ナトリウム標準液(Na 100 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 測定対象元素用の光源を備え,かつ,バックグラウンド補正が可能な
もの。
c) 操作 操作は,次による。
1) 試料(3)をフレーム中に導入し,波長589.0 nmの指示値(5)を読み取る。
2) 空試験として,水について,1)の操作を行って試料について得た指示値を補正する。
3) 検量線からナトリウムの量を求め,試料中のナトリウムの濃度(Na mg/L)を算出する。
注(5) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) ナトリウム標準液(Na 10 mg/L)0.5〜40 mLを全量フラスコ100 mLに段階的にとり,水を標線ま
で加える。この溶液についてc) 1)の操作を行う。
2) 別に,空試験として水についてc) 1)の操作を行って標準液について得た指示値を補正し,ナトリウ
ム(Na)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 2. 塩化セシウム溶液(25 g/L)を加えることで,カリウム,カルシウムなどの影響を抑制でき
る。添加量などは,備考1.による。
48.3 イオンクロマトグラフ法 試料中の陽イオンをイオンクロマトグラフ法によって定量する。検出器
には電気伝導率検出器を用いる。この方法によって,表48.1に示す陽イオンが同時定量できる。アンモニ
ウムイオンを同時定量する場合は,3.3の保存処理を行わず,試料採取後,直ちに行う。直ちに行えない場
合は,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
それぞれの陽イオンの定量範囲,繰返し精度などの例を,表48.1に示す。
表48.1 各陽イオンの定量範囲などの例*
対象陽イオン
定量範囲
mg/L
繰返し精度
%
アンモニウムイオン (NH4+)
0.1〜30
2〜10
ナトリウム
(Na)
0.1〜30
2〜10
カリウム
(K)
0.1〜30
2〜10
カルシウム
(Ca)
0.2〜50
5〜10
マグネシウム
(Mg)
0.2〜50
5〜10
注*
定量範囲は,検出器,試料注入量,カラムのイオン交換容
量などによって変わる。
72
K 0102:2019
a) 試薬 試薬は,次による。これらは,ポリエチレン瓶に保存する。
1) 水 JIS K 0557に規定するA2又はA3の水
2) 溶離液 溶離液は,装置の種類及び分離カラムに充塡した陽イオン交換体の種類によって異なるの
で,あらかじめ,備考3.によって,アンモニウムイオン,ナトリウム,カリウム,カルシウム及び
マグネシウムの分離を確認する。
3) 再生液 再生液は,サプレッサーを用いる場合に使用するが,装置の種類及びサプレッサーの種類
によって異なる。あらかじめ分離カラムと組み合わせて,備考3.の操作を行って再生液の性能を確
認する。
4) ナトリウム標準液(Na 1 000 mg/L) 48.1 a) 1)による。
5) アンモニウムイオン標準液(NH4+ 1 000 mg/L) 42.2 a) 5) による。
6) カリウム標準液(K 1 000 mg/L) JIS K 8121に規定する塩化カリウムを500 ℃で約4時間加熱し,
デシケーター中で放冷する。その1.907 gをはかりとり,少量の水に溶かして全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。
7) カルシウム標準液(Ca 1 000 mg/L) JIS K 8617に規定する炭酸カルシウムを105±2 ℃で約2時間
加熱し,デシケーター中で放冷する。その2.498 gをはかりとり,水約50 mLに分散させ,これに
塩酸(1+1)[24.2 a) 2)による。]40 mLを加えて溶かす。沸騰しない程度に数分間加熱して二酸化
炭素を除く。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
8) マグネシウム標準液(Mg 1 000 mg/L) JIS K 8432に規定する酸化マグネシウムを約800 ℃で約2
時間加熱し,デシケーター中で放冷する。その1.658 gをはかりとり,塩酸(1+1)[24.2 a) 2)によ
る。]40 mLに溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
9) 陽イオン混合標準液[(NH4+ 100 mg,Na 100 mg,K 100 mg,Ca 200 mg,Mg 200 mg)/L](6) ア
ンモニウムイオン標準液(NH4+ 1 000 mg/L)10 mL,ナトリウム標準液(Na 1 000 mg/L)10 mL,
カリウム標準液(K 1 000 mg/L)10 mL,カルシウム標準液(Ca 1 000 mg/L)20 mL及びマグネシウ
ム標準液(Mg 1 000 mg/L)20 mLをそれぞれ全量フラスコ100 mLにとり,水を標線まで加える。
使用時に調製する。
10) 陽イオン混合標準液[(NH4+ 10 mg,Na 10 mg,K 10 mg,Ca 20 mg,Mg 20 mg)/L](6) アンモ
ニウムイオン標準液(NH4+ 1 000 mg/L)5 mL,ナトリウム標準液(Na 1 000 mg/L)5 mL,カリウ
ム標準液(K 1 000 mg/L)5 mL,カルシウム標準液(Ca 1 000 mg/L)10 mL及びマグネシウム標準
液(Mg 1 000 mg/L)10 mLをそれぞれ全量フラスコ500 mLにとり,水を標線まで加える。又は,
陽イオン混合標準液[(NH4+ 100 mg,Na 100 mg,K 100 mg,Ca 200 mg,Mg 200 mg)/L]10 mL
を全量フラスコ100 mLにとり,水を標線まで加える。いずれも使用時に調製する。
注(6) 陽イオンをそれぞれ単独に測定する場合,又はいずれかの同時測定の場合は,この操作に準じ
て必要な混合標準液を調製して用いてもよい。
b) 器具及び装置 器具及び装置は,次による。
1) イオンクロマトグラフ イオンクロマトグラフは,分離カラムとサプレッサー(7)とを組み合わせた
方式のもの,分離カラム単独の方式のものいずれかで,次に掲げる条件を満たすもので,アンモニ
ウムイオン,ナトリウム,カリウム,カルシウム及びマグネシウムが分離定量できるもの。
1.1) 分離カラム ステンレス鋼製又は合成樹脂製(8)のものに,陽イオン交換体を充塡したもの(9)。
1.2) 検出器 電気伝導率検出器
1.3) データ処理部 JIS K 0127の5.7(データ処理部)による。
73
K 0102:2019
2) マイクロシリンジ 10〜200 μLの適切なもの。又は自動注入装置
注(7) 溶離液中の陰イオンを水酸化物イオンに変換するためのもので,溶離液中の陰イオンの濃度に
対して十分なイオン交換容量をもった陰イオン交換膜(膜形,電気透析形がある。)又はこれと
同等な性能をもった陰イオン交換体を充塡したもの。
(8) 例えば,四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などがある。
(9) 備考3.による。
c) 準備操作 準備操作は,次による。
1) 試料を孔径0.45 μm以下のフィルターによってろ過する。
2) 試料の電気伝導率が10 mS/m(100 μS/cm)(25 ℃)以上の場合には,電気伝導率が10 mS/m以下に
なるように,水で一定の割合に薄める。
d) 操作 操作は,次による。
1) イオンクロマトグラフを作動できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。再生液を必要とするサプレッサー装置では,再生液を一定の流量で流して
おく。
2) 陽イオン混合標準液[(NH4+ 10 mg,Na 10 mg,K 10 mg,Ca 20 mg,Mg 20 mg)/L](例えば,20
〜200 μLの一定量)をマイクロシリンジ(10)を用いて,イオンクロマトグラフに注入してクロマトグ
ラムを記録し,各陽イオンの保持時間に相当するピークの位置を確認しておく。
3) c)の準備操作を行った試料の一定量(例えば,20〜200 μLの一定量)をマイクロシリンジ(10)を用い
て,イオンクロマトグラフに注入し,クロマトグラムを記録する。
4) クロマトグラム上の各陽イオンに相当するピークについて,指示値(11)を読み取る。
5) 試料を薄めた場合には,空試験として試料と同量の水について,1)〜4)の操作を行って試料につい
て得た結果を補正する。
6) 検量線から各陽イオンの濃度を求め,試料中の各陽イオンの濃度(mg/L)を算出する(12)。
注(10) 検量線作成時と同じものを用いる。
(11) ピーク高さ又はピーク面積。
(12) 注(6)によった場合は,各陽イオンのそれぞれの量を求め,濃度を算出する。
e) 検量線 検量線の作成は,次による。
1) 陽イオン混合標準液[(NH4+ 100 mg,Na 100 mg,K 100 mg,Ca 200 mg,Mg 200 mg)/L]0.1〜30
mLを段階的に全量フラスコ100 mLにとり,水を標線まで加え,d)の1)〜4)の操作を行って各陽イ
オンに相当する指示値を読み取る。
2) 別に,空試験として,水についてd)の1)〜4)の操作を行って各陽イオンに相当する指示値を補正し
た後,各陽イオンの量と指示値との関係線を作成する(13)。検量線の作成は,試料測定時に行う。
検量線は,必ずしも直線関係を示さない。
注(13) 注(6)によった場合は,その陽イオンについて作成する。
備考 3. 溶離液を一定の流量(例えば,1〜2 mL/min)で流し,陽イオン混合標準液[(NH4+ 10 mg,
Na 10 mg,K 10 mg,Ca 20 mg,Mg 20 mg)/L]の一定量をイオンクロマトグラフに注入し,
クロマトグラムを求め,各陽イオンの分離度(R)が1.3以上に分離できるものを用いる。分
離度(R)は,35.の備考7.の式によって求める。また,定期的に分離カラムの性能を確認す
るとよい。
4. 分離カラムは,使用を続けると性能が低下するので,定期的に備考3.の操作で確認する。性
74
K 0102:2019
能が低下している場合には,溶離液の20〜200倍の濃度のものを調製し,分離カラムに注入
し,洗浄した後,性能を確認する。性能が回復しない場合は,新品と取り替える。
試料中の懸濁物,有機物などによっても汚染されて性能が徐々に低下する。懸濁物を含む
試料は,c)の準備操作で除去した後,試験する。また,有機物(たんぱく質,油脂,界面活
性剤など)を含む試料は限外ろ過膜でろ過し,できるだけ有機物を除去した後,試験する。
試料中に分離カラムの充塡剤と親和力の強い陽イオン(例えば,カルシウム,マグネシウ
ムなど)が存在すると,これらが充塡剤に吸着され,分離性能が徐々に低下するので,定期
的に溶離液の20〜200倍の濃度のものを調製し,試料と同様に分離カラムに注入し洗浄する。
その他,酸化性物質又は還元性物質が共存すると,分離カラムの分離性能が低下する。こ
のような場合には,試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐこと
ができる。
備考 5. カルボン酸形の陽イオン交換カラムと,溶離液として,硝酸溶液,メタンスルホン酸溶液,
[2,6-ピリジンジカルボン酸-L(+)-酒石酸]溶液などを用いると,1価陽イオンのほかにカル
シウム,マグネシウムなど2価の陽イオンの溶離及び同時定量が可能になる。
6. 妨害物質
− アミノ酸及び脂肪族アミンのような有機化合物は,無機陽イオンの定量を妨害する可能
性がある。
− 2,6-ピリジンジカルボン酸(PDA)のような強い錯形成剤が溶離液に含まれてなく,サ
プレッサーを用いない場合は,亜鉛,ニッケル及びカドミウムのような陽イオンによる
妨害があり得る。
− マンガンのような他の陽イオンによる妨害の程度は,用いる分離カラムの選択性に依存
する。
− アンモニウムイオン及びナトリウムの定量において,それらの濃度に大きな違いがある
場合,相互の影響があり得る。ナトリウムの濃度が1 mg/Lのときアンモニウムイオン及
びカリウムは,いずれも100 mg/L以下であれば妨害しない。
− 試料中の固形物及び鉱物油・洗剤・フミン酸の有機物は,分離カラムの寿命を短くする
ので除去する。
48.4 ICP発光分光分析法 50.3による。
49.[カリウム(K)]の全文を,次に置き換える。
49. カリウム(K) カリウムの定量には,フレーム光度法,フレーム原子吸光法,イオンクロマトグラフ
法又はICP発光分光分析法を適用する。
なお,フレーム光度法は,1993年に第1版として発行されたISO 9964-3,フレーム原子吸光法は,1993
年に第1版として発行されたISO 9964-1,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885との整合を図ったもので
ある。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9964-1:1993,Water quality−Determination of sodium and potassium−Part 1: Determination of
75
K 0102:2019
sodium by atomic absorption spectrometry(MOD)
ISO 9964-3:1993,Water quality−Determination of sodium and potassium−Part 3: Determination of
sodium and potassium by flame emission spectrometry(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+,
Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
49.1 フレーム光度法 試料をアセチレン-空気フレーム,水素-酸素フレームなどの中に噴霧し,このとき
生じる波長766.5 nm又は769.9 nmの輝線の強さを測定してカリウムを定量する。
定量範囲:K 40〜400 μg/L,0.4〜4 mg/L,4〜40 mg/L,繰返し精度:3〜10 %(装置及び測定条件によ
って異なる。)
a) 試薬 試薬は,次による。
1) カリウム標準液(K 1 000 mg/L) 48.3 a) 6)による。
2) カリウム標準液(K 4〜40 mg/L) カリウム標準液(K 1 000 mg/L)を段階的にとり,これを水で薄
めてK 4〜40 mg/Lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,K 0.4〜4 mg/L又はK 40〜400 μg/Lの標準液を調製する。
b) 装置 装置は,次による。
1) フレーム光度計
c) 操作 操作は,次による。
1) カリウム標準液(K 40 mg/L)(2)をフレーム光度計のフレーム中に噴霧し,波長766.5 nm又は769.9
nmの指示値が100を示すように調節する。
2) 水を噴霧して指示値がゼロを示すように調節する。
3) カリウム標準液(K 4〜40 mg/L)(1)を順次噴霧し,カリウム(K)の濃度と指示値との関係線を作
成し,検量線とする。
4) 試料(3) (4)(カリウム濃度が40 mg/L以上の場合は薄める。)を噴霧して指示値を読み取り,検量線
から試料中のカリウムの濃度(K mg/L)を求める。
注(2) 低い濃度の測定用には,K 4 mg/L又はK 0.4 mg/Lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むカリウム標準液(K 4〜40 mg/L)を調製し,検量線を作成する。
備考 1. ナトリウムが共存すると正の誤差を生じる。その他の共存物質による影響は,48.の備考1.
による。
49.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,カリウムによる原子吸光を波長
766.5 nmで測定してカリウムを定量する。
定量範囲:K 0.05〜5 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) カリウム標準液(K 100 mg/L) 48.3 a) 6)のカリウム標準液(K 1 000 mg/L)10 mLを全量フラスコ
100 mLにとり,水を標線まで加える。使用時に調製する。
2) カリウム標準液(K 10 mg/L) カリウム標準液(K 100 mg/L)20 mLを全量フラスコ200 mLにとり,
水を標線まで加える。使用時に調製する。
76
K 0102:2019
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 48.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(3)をフレーム中に導入し,波長766.5 nmの指示値(5)を読み取る。
2) 空試験として水について,1)の操作を行って試料について得た指示値を補正する。
3) 検量線からカリウムの量を求め,試料中のカリウムの濃度(K mg/L)を算出する。
注(5) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) カリウム標準液(K 10 mg/L)0.5〜50 mLを全量フラスコ100 mLに段階的にとり,水を標線まで加
える。この溶液についてc) 1)の操作を行う。
2) 別に,空試験として,水についてc) 1)の操作を行って標準液について得た指示値を補正し,カリウ
ム(K)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 2. 塩化セシウム溶液(25 g/L)を加えることで,カルシウムなどの影響を抑制できる。添加量
などは,48.の備考1.による。
49.3 イオンクロマトグラフ法 48.3による。
備考 3. 48.の備考6.による。カリウムの濃度が1 mg/Lのとき,アンモニウムイオン及びナトリウム
はいずれも100 mg/L以下であれば妨害しない。また,アミン類のうちメタンアミン(モノメ
チルアミン)が共存するとカリウムのピークと重なり妨害する。
49.4 ICP発光分光分析法 50.3による。
50.[カルシウム(Ca)]の全文を,次に置き換える。
50. カルシウム(Ca) カルシウムの定量には,キレート滴定法,フレーム原子吸光法,イオンクロマト
グラフ法又はICP発光分光分析法を適用する。
なお,キレート滴定法は,1984年に第1版として発行されたISO 6058,フレーム原子吸光法は,1986
年に第1版として発行されたISO 7980,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885との整合を図ったもので
ある。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6058:1984,Water quality−Determination of calcium content−EDTA titrimetric method(MOD)
ISO 7980:1986,Water quality−Determination of calcium and magnesium−Atomic absorption
spectrometric method(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+,
Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
50.1 キレート滴定法 試料をpH12以上とし,指示薬としてHSNN{2-ヒドロキシ-1-[(2'-ヒドロキシ-4'-
スルホ-1'-ナフタレニル)アゾ]-3-ナフタレンカルボン酸(IUPACによる名称)}を加え,エチレンジアミ
ン四酢酸二水素二ナトリウム溶液で滴定してカルシウムを定量する。
77
K 0102:2019
定量範囲:Ca 0.2〜5 mg
a) 試薬 試薬は,次による。
1) 水酸化カリウム溶液 JIS K 8574に規定する水酸化カリウム250 gを水に溶かして500 mLとする。
ポリエチレン瓶に保存する。
2) シアン化カリウム溶液(100 g/L) JIS K 8443に規定するシアン化カリウム10 gを水に溶かして100
mLとする。ポリエチレン瓶に保存する。
3) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
4) HSNN溶液(1) JIS K 8776に規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-
ナフトエ酸0.5 gをJIS K 8891に規定するメタノール100 mLに溶かし,JIS K 8201に規定する塩化
ヒドロキシルアンモニウム0.5 gを加える。着色ガラス瓶に保存する。
5) 10 mmol/L EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和
物3.8 gをとり,水1 Lに溶かした後,ポリエチレン瓶に保存する。
標定 10 mmol/L 亜鉛溶液25 mLをビーカー200 mLにとり,水75 mLを加え,水酸化ナトリウム
溶液(100 g/L)[19. a) 2)による。]でpH6〜8に調節する。塩化アンモニウム-アンモニア緩衝液
(pH10)[28.1.2.1 a) 2)による。]2 mLと指示薬としてEBT溶液(5 g/L)[51.1 a) 4)による。]2,3
滴又はEBT粉末指示薬(JIS K 8736に規定するエリオクロムブラックT 0.10 gとJIS K 8150に規定
する塩化ナトリウム10 gをすり潰し混合する。)約0.05 gとを加え,調製した10 mmol/L EDTA溶液
で滴定する。終点は,溶液の色が赤から青に変わる点とする。
ファクターは,次の式によって算出する。
V
f
f
)
25
(1×
=
ここに,
f: 10 mmol/L EDTA溶液のファクター
f1: 10 mmol/L 亜鉛溶液のファクター
V: 滴定に要した10 mmol/L EDTA溶液量(mL)
6) 10 mmol/L亜鉛溶液 JIS K 8001に準じ,次の方法で調製する。
− JIS K 8005に規定する容量分析用標準物質の亜鉛約0.5 gを塩酸(1+3)(JIS K 8180に規定する
塩酸を用いて調製する。)で洗い,水洗いし,更にJIS K 8101に規定するエタノール(99.5)及び
JIS K 8103に規定するジエチルエーテルで順次洗った後,直ちに上口デシケーターに入れ,2.0 kPa
以下で数分間保った後,減圧下で約12時間保つ。
− その0.33 gを0.1 mgの桁まではかりとり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調
製する。)20 mL中に加え,加熱して溶かし,煮沸して窒素酸化物を追い出し,放冷後,全量フラ
スコ500 mLに移し入れ,水を標線まで加える。
この10 mmol/L 亜鉛溶液のファクターは,次の式による。
×
=
100
0
327
.0
1
A
a
f
ここに,
f1: 10 mmol/L 亜鉛溶液のファクター
a: はかりとった亜鉛の質量(g)
A: 亜鉛の純度(質量分率%)
0.327 0: 10 mmol/L亜鉛溶液500 mL中の亜鉛の相当量(g)
注(1) HSNN溶液に代え,次の方法で調製したHSNN粉末指示薬を用いてもよい。
− JIS K 8776に規定するHSNN 0.2 gとJIS K 8962に規定する硫酸カリウム10 gとをよくす
78
K 0102:2019
り潰し混合する。
− JIS K 8776に規定するHSNN 0.2 gとJIS K 8150に規定する塩化ナトリウム100 gとをよく
すり潰し混合する。
参考 HSNNは,カルコンカルボン酸という名称でも知られている。
b) 操作 操作は,次による。
1) 試料(2)の適量(Caとして5 mg以下を含む。)をビーカーにとり,水を加えて約50 mLとする。
2) 水酸化カリウム溶液4 mLを加え,よくかき混ぜた後,約5分間放置する(3)。
3) シアン化カリウム溶液(100 g/L)0.5 mL(4)及び塩化ヒドロキシルアンモニウム溶液(100 g/L)0.5 mL
を加えてかき混ぜる。
4) 指示薬としてHSNN溶液(5) 5,6滴を加え(6),10 mmol/L EDTA溶液で,溶液の色が赤紫から青にな
るまで滴定する。
5) 次の式によって試料中のカルシウムの濃度(Ca mg/L)を算出する。
8
400
.0
000
1
×
×
×
=
V
f
a
C
ここに,
C: カルシウムの濃度(Ca mg/L)
a: 滴定に要した10 mmol/L EDTA溶液量(mL)
V: 試料量(mL)
f: 10 mmol/L EDTA溶液のファクター
0.400 8: 10 mmol/L EDTA溶液1 mLに相当するカルシウムの質量
(mg)
注(2) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。また,この試験に影響
を与える有機物及び着色物質を含む場合には,5.の操作を行った後,中和する。ただし,5.4の
方法は適用しない。
(3) 放置したときに生じる沈殿の量が多いと,終点が不明瞭になる。このような場合には,1回目
の滴定で,概略の滴定量を求めておき,別のビーカーに同量の試料をとり,1回目の滴定に要
した10 mmol/L EDTA溶液の量よりも約1 mL少ない量の10 mmol/L EDTA溶液を加え,水酸化
カリウム溶液4 mLを加えて,よく振り混ぜた後,約5分間放置する。次に,シアン化カリウム
溶液(100 g/L)0.5 mL(4)と塩化ヒドロキシルアンモニウム溶液(100 g/L)0.5 mLとを加えて振
り混ぜる。これに指示薬としてHSNN溶液5,6滴を加え(5) (6),再び10 mmol/L EDTA溶液で,
溶液の色が青になるまで滴定する。
(4) 亜鉛,銅などシアン化カリウムによってマスキングする金属類が共存しない場合は,添加しな
くてよい。
(5) HSNN溶液の代わりに注(1)のHSNN粉末指示薬の適量を用いてもよい。ただし,粉末指示薬は
HSNNが溶けるのに時間を要するので注意する。
(6) この指示薬は,添加後放置すると酸化され,終点の変色が不明瞭になる。
備考 1. シアン化カリウムは,有毒である。取扱い及び廃棄には,必要な予防措置をとる。シアン化
カリウムを含む溶液は,酸性にしてはならない。
2. アルミニウム,バリウム,鉛,鉄,コバルト,銅,マンガン,すず及び亜鉛の金属イオンは,
カルシウムと同じように滴定される。また,終点の色の変化を不明瞭にする。
オルトりん酸イオンの濃度が1 mg/L以上の場合は,滴定時のpH条件ではカルシウムと沈
殿する。滴定に時間がかかった場合,又はカルシウムの含有量が高い場合(100 mg/L又は2.5
79
K 0102:2019
mmol/Lより高い)には,炭酸カルシウムの沈殿が生じる。
鉄30 mg/L以下の妨害は,シアン化カリウム250 mg又はJIS K 8663に規定する2,2′,2''-ニ
トリロトリエタノール(トリエタノールアミン)5〜7 mLを滴定直前に添加することによっ
て,マスキングできる。シアン化物イオンは,亜鉛,銅及びコバルトの妨害も最小にする。
また,トリエタノールアミンはアルミニウムの妨害を小さくする。シアン化カリウムを加え
る前に溶液がアルカリ性であることを確認する。
50.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,カルシウムによる原子吸光を波
長422.7 nmで測定してカルシウムを定量する。
定量範囲:Ca 0.2〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) ランタン(III)溶液(La 50 g/L) 酸化ランタン(III)29 gを少量ずつ塩酸(1+1)500 mLに加え
て溶かす。
3) カルシウム標準液(Ca 20 mg/L) 48.3 a) 7)のカルシウム標準液(Ca 1 000 mg/L)10 mLを全量フラ
スコ500 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 48.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(7)の適量(Caとして20〜400 μgを含む。)を全量フラスコ100 mLにとり,塩酸(1+1)2 mL
を加えた後,水を標線まで加える。
2) この溶液10 mLを乾いたビーカーにとり,ランタン(III)溶液(La 50 g/L)1 mLを加える。
3) 2)の溶液をフレーム中に導入し,波長422.7 nmの指示値(8)を読み取る。
4) 空試験として試料と同量の水について,1)〜3)の操作を行って試料について得た指示値を補正する。
5) 検量線からカルシウムの量を求め,試料中のカルシウムの濃度(Ca mg/L)を算出する。
注(7) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(8) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) カルシウム標準液(Ca 20 mg/L)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料と同じ酸
の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。この溶液についてc)の2)及び
3)の操作を行う。
2) 別に,空試験として水について試料と同じ酸の濃度になるように塩酸(1+1)を加えた後,c)の2)
及び3)の操作を行って標準液について得た指示値を補正し,カルシウム(Ca)の量と指示値との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考 3. りん酸イオン,硫酸イオン,アルミニウムなどは妨害するが,ランタン(III)溶液(La 50 g/L)
を加えることによって妨害を抑制することができる。
4. アセチレン-一酸化二窒素フレームを用いるときは,ランタン(III)溶液(La 50 g/L)に代え
て塩化セシウム溶液(25 g/L)を用いる。この溶液の調製は,次による。
塩化セシウム溶液(25 g/L) 塩化セシウム25 g(Csとして約20 g)を塩酸(0.1 mol/L)(JIS
K 8180に規定する塩酸を用いて調製する。)1 Lに溶かす。
5. 多量のマグネシウム(1 000 mg/L以上)の共存は,負の誤差を与える。
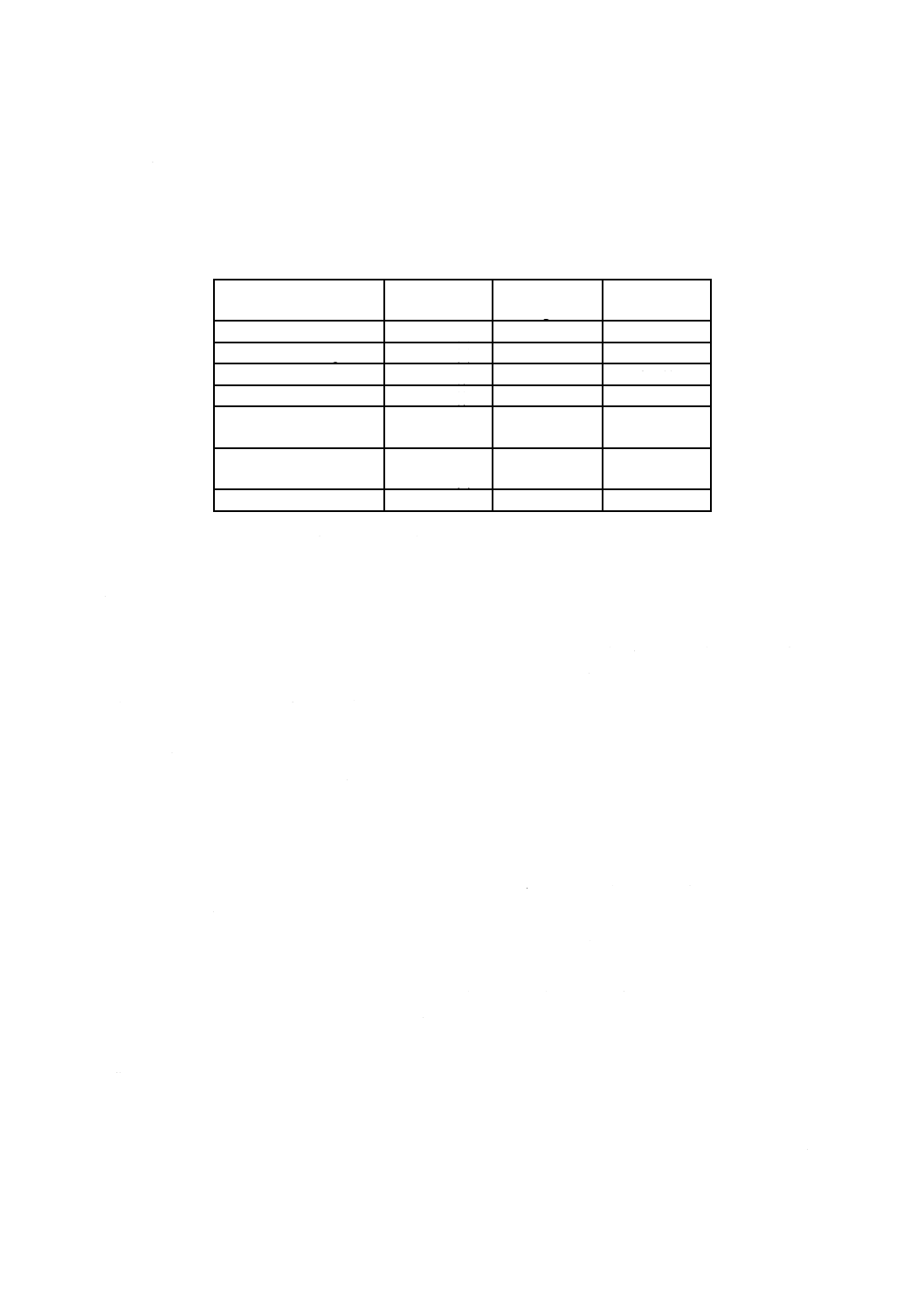
80
K 0102:2019
50.3 ICP発光分光分析法 試料を試料導入部を通して誘導結合プラズマ中に噴霧し,カルシウムによる
発光を波長393.367 nmで測定してカルシウムを定量する。この方法によって,表50.1に示す元素が同時
定量できる。
それぞれの元素の測定波長,定量範囲,繰返し精度などの例を,表50.1に示す。
表50.1 測定波長,定量範囲及び繰返し精度の例*
対象元素
測定波長**
nm
定量範囲
mg/L
繰返し精度
%
カルシウム(Ca)
393.367 (II)
0.01〜10
2〜10
マグネシウム(Mg)
279.553 (II)
0.005〜5
2〜10
ナトリウム(Na)
589.592 (I)
0.5〜50
2〜10
カリウム(K)
766.491 (I)
0.1〜10
2〜10
イットリウム(Y)***
371.029 (II)
464.370 (I)
−
−
インジウム(In)***
325.609 (I)
230.606 (II)
−
−
イッテルビウム(Yb)***
369.420 (II)
−
−
注*
装置及び測定条件によって異なる。
** Iは中性線,IIはイオン線。
*** 内標準元素
a) 試薬 試薬は,次による。
1) 塩酸(1+1) 50.2 a) 1)による。
2) カルシウム標準液(Ca 100 mg/L) 48.3 a) 7)のカルシウム標準液(Ca 1 000 mg/L)10 mLを全量フ
ラスコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
3) カルシウム標準液(Ca 1 mg/L) カルシウム標準液(Ca 100 mg/L)10 mLを全量フラスコ1 000 mL
にとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
4) マグネシウム標準液(Mg 100 mg/L) 48.3 a) 8)のマグネシウム標準液(Mg 1 000 mg/L)10 mLを全
量フラスコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
5) マグネシウム標準液(Mg 1 mg/L) マグネシウム標準液(Mg 100 mg/L)10 mLを全量フラスコ1 000
mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
6) ナトリウム標準液(Na 1 000 mg/L) 48.1 a) 1) による。
7) ナトリウム標準液(Na 100 mg/L) ナトリウム標準液(Na 1 000 mg/L)10 mLを全量フラスコ100 mL
にとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
8) カリウム標準液(K 100 mg/L) 48.3 a) 6)のカリウム標準液(K 1 000 mg/L)10 mLを全量フラスコ
100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
9) カリウム標準液(K 10 mg/L) カリウム標準液(K 100 mg/L)10 mLを全量フラスコ100 mLにとり,
塩酸(1+1)5 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(7)の適量(Caとして1〜1 000 μg,Mgとして0.5〜500 μg,Naとして50〜5 000 μg,Kとして
10〜1 000 μgを含む。)を全量フラスコ100 mLにとり,塩酸(1+1)2 mLを加えた後,水を標線ま
81
K 0102:2019
で加える。
2) 1)の溶液を試料導入部を通して発光部に導入し,各測定対象元素の発光強度を測定する(9) (10)。
3) 空試験として1)で用いた試料と同量の水をとり,1)及び2)の操作を行って各測定対象元素に相当す
る発光強度を測定し,試料について得た発光強度を補正する。
4) 検量線から各測定対象元素の量を求め,試料中の各測定対象元素の濃度(mg/L)を算出する(11)。
注(9) 47.の注(8)による。
(10) 47.の注(9)による。
(11) 測定対象の元素の中から,必要な元素だけの濃度を算出する。
d) 検量線 検量線の作成は,次による。
1) a)の測定対象元素の各標準液のそれぞれ一定量をとり,希釈して一定量として混合標準液を調製す
る。この混合標準液は,測定対象の元素を含み(12),測定する濃度範囲よりも高い濃度(13)とする。
2) 全量フラスコ100 mLに,測定濃度範囲を含むように,この混合標準液を段階的にとり,試料と同
じ酸の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。
3) 2)で調製した溶液についてc) 2)の操作を行う。
4) 別に,空試験として水について試料と同じ酸の濃度になるように塩酸(1+1)を加えた後,c) 2)の
操作を行って,標準液について得た発光強度を補正する。
5) 各測定対象元素について,それぞれの元素の量と発光強度との関係線を作成する。
注(12) 測定対象の単独又は限られた複数の元素だけでもよい。
(13) 作成する検量線の最高の濃度の5〜10倍程度。
備考 6. イオン化エネルギーの低いナトリウム及びカリウムは,共存元素によるイオン化干渉を受け
やすい。特に,測定対象元素がナトリウムではカリウムによる干渉が大きく,測定対象元素
がカリウムではナトリウムによる干渉が大きい。ICPからの光の観測方式には,横方向観測
方式と軸方向観測方式があり,軸方向観測方式は高感度であるがイオン化干渉を受けやすい
ため,共存元素によるイオン化干渉が懸念される試料には次の方法によって干渉を抑制する。
塩化セシウム溶液(25 g/L)[塩化セシウム25 gをJIS K 8180に規定する塩酸50 mL及び
水450 mLに溶かし,水を加えて1 Lとする。この溶液1 Lは,セシウム(Cs)を約20 gを
含む。]を試料に適量(共存元素の約5倍のモル濃度となるように)加えることでイオン化干
渉を抑制できる。
この操作を行った場合は,検量線作成時の操作も塩化セシウム溶液(25 g/L)を試料と同
様に加えて行う。
7. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置を用いるときは,内標準法に
よることができる。内標準元素としては,ICP中で分析元素と類似の発光挙動を示す元素が
適切であり,したがって内標準線及び分析線は中性線同士又はイオン線同士で,その励起エ
ネルギー差が小さく,かつ,分析線に対し分光干渉を生じない発光線を選択する。また,元
の試料中に含まれる濃度が,添加する濃度に比べ,無視できる濃度であることを確認してお
く。内標準元素としてはイットリウム(Y),インジウム(In),イッテルビウム(Yb)など
が用いられるが,上記の条件を満足すれば他の元素を用いてもよい。内標準物質としてイッ
トリウムを用いる場合の操作は,次による。
1) 試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50 μg/mL)[47.の備考
5. 6)による。]10 mL及び塩酸(1+1)2 mLを加えた後,水を標線まで加える。
82
K 0102:2019
2) この溶液について,c) 2)の操作を行って各測定対象元素及びイットリウムの発光強度を
測定し,各測定対象元素の発光強度とイットリウムの発光強度との比を求める。
3) 空試験として,試料に代えて同量の水を用い,1)及び2)の操作を行い,各測定対象元素
の発光強度とイットリウムの発光強度との比を求め,2)で得た発光強度の比を補正する。
4) 検量線から,各測定対象元素の量を求め,試料中の各測定対象元素の濃度(mg/L)を算
出する。
5) 検量線 全量フラスコ100 mL数個にd) 2)に従って混合標準液を段階的にとり,イット
リウム溶液(Y 50 μg/mL)10 mL及び1)の試料と同じ酸の濃度となるように塩酸(1+1)
を加えた後,水を標線まで加える。この溶液について,2)の操作を行い,各測定対象元
素及びイットリウムの発光強度を測定し,各測定対象元素の発光強度とイットリウムの
発光強度との比を求める。別に,空試験として,混合標準液に代えて水を用い,同じ操
作を行って,同様に発光強度の比を求め,混合標準液での各測定対象元素の発光強度比
を補正し,各測定対象元素の量と,それぞれの発光強度とイットリウムの発光強度との
比との関係線を作成する。
50.4 イオンクロマトグラフ法 48.3による。
51.[マグネシウム(Mg)]の全文を,次に置き換える。
51. マグネシウム(Mg) マグネシウムの定量には,キレート滴定法,フレーム原子吸光法,イオンクロ
マトグラフ法又はICP発光分光分析法を適用する。
なお,キレート滴定法は,1984年に第1版として発行されたISO 6059,フレーム原子吸光法は,1986
年に第1版として発行されたISO 7980,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885との整合を図ったもので
ある。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6059:1984,Water quality−Determination of the sum of calcium and magnesium−EDTA
titrimetric method(MOD)
ISO 7980:1986,Water quality−Determination of calcium and magnesium−Atomic absorption
spectrometric method(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+,Na+,NH4+,K+,Mn2+,Ca2+,
Mg2+,Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
51.1 キレート滴定法 試料に緩衝液を加えてpHを約10に調節し,指示薬としてエリオクロムブラック
T[3-ヒドロキシ-4-[(1-ヒドロキシ-2-ナフタレニル)アゾ]-7-ニトロ-1-ナフタレンスルホン酸ナトリウム
(IUPACの名称による。)]を加え,エチレンジアミン四酢酸二水素二ナトリウム溶液で滴定し,カルシウ
ムとマグネシウムとの合量に対する滴定量を求め,カルシウムに対する滴定量を差し引き,マグネシウム
を定量する。
定量範囲:MgとCaとの合量がCaとして0.15〜5 mg
83
K 0102:2019
a) 試薬 試薬は,次による。
1) シアン化カリウム溶液(100 g/L) 50.1 a) 2)による。
2) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
3) 塩化アンモニウム-アンモニア緩衝液(pH10) 28.1.2.1 a) 2)による。
4) EBT溶液(5 g/L) JIS K 8736に規定するエリオクロムブラックT[1-(1-ヒドロキシ-2-ナフチル
アゾ)-6-ニトロ-2-ナフトール-4-スルホン酸ナトリウム]0.5 gをJIS K 8891に規定するメタノール
100 mLに溶かし,JIS K 8201に規定する塩化ヒドロキシルアンモニウム0.5 gを加える(1)。着色ガ
ラス瓶に入れて保存する。
5) 10 mmol/L EDTA溶液 50.1 a) 5)による。この溶液1 mLは,Mg 0.243 1 mgに相当する。
注(1) 指示薬にメタニル(metanil)塩(メタニルイエロー,4-アニリドアゾベンゼンスルホン酸ナト
リウム塩),{[3-[4-(フェニルアミノ)フェニル]アゾ]ベンゼンスルホン酸ナトリウム}0.17
gを加えると終点の検出が容易になる。この場合には,赤から灰青(pale grey)又は緑に変わる。
b) 操作 操作は,次による。
1) 試料(2)の適量(MgとCaとの合量がCaとして5 mg以下を含む。)をビーカーにとり,水を加えて
約50 mLとする。
2) シアン化カリウム溶液(100 g/L)0.5 mL(3),塩化ヒドロキシルアンモニウム溶液(100 g/L)5〜7
滴及び塩化アンモニウム-アンモニア緩衝液(pH10)1 mLを加える。
3) 指示薬としてEBT溶液(5 g/L)2,3滴(4)を加える。
4) 10 mmol/L EDTA溶液で,溶液の赤みが消えて青になるまで滴定(5)する。
5) 別に,同量の試料をとり,50.1 b)の1)〜4)の操作を行ってカルシウムの量に相当する10 mmol/L
EDTA溶液の滴定量(mL)を求める。
6) 次の式によって試料中のマグネシウムの濃度(Mg mg/L)を算出する。
1
243
.0
000
1
Ca
×
×
×
−
=
f
V
b
V
a
M
ここに,
M: マグネシウムの濃度(Mg mg/L)
a: 滴定に要した10 mmol/L EDTA溶液量(mL)
b: 50.1 b)で滴定に要した10 mmol/L EDTA溶液量(mL)
V: 試料量(mL)
f: 10 mmol/L EDTA溶液のファクター
VCa: 50.1 b)での試料量(mL)
0.243 1: 10 mmol/L EDTA溶液1 mLに相当するマグネシウムの質量
(mg)
注(2) 50.の注(2)による。
(3) 試料に亜鉛,銅,コバルトなどが共存しない場合は,添加しなくてもよい。
(4) EBT溶液(5 g/L)に代えて,備考1.に示す指示薬を用いてもよい。
(5) エリオクロムブラックTの変色は遅いので,変色点近くではよくかき混ぜながら徐々に滴定す
る。
備考 1. EBT溶液(5 g/L)以外の指示薬の調製方法は,次による。
− JIS K 8736に規定するエリオクロムブラックT 0.5 gをJIS K 8663に規定する2,2′,2″-ニ
トリロトリエタノール(トリエタノールアミン)100 mLに溶かす。この溶液の粘性を減
らすため,25 mLまではトリエタノールアミンの代わりにJIS K 8102に規定するエタノ
84
K 0102:2019
ール(95)を加えてもよい。
備考 2. 50.の備考2.による。
51.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,マグネシウムによる原子吸光を
波長285.2 nmで測定してマグネシウムを定量する。
定量範囲:Mg 0.02〜0.4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸(1+1) 50.2 a) 1)による。
2) ランタン(III)溶液(La 50 g/L) 50.2 a) 2)による。
3) マグネシウム標準液(Mg 500 mg/L) 48.3 a) 8)のマグネシウム標準液(Mg 1 000 mg/L)50 mLを全
量フラスコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
4) マグネシウム標準液(Mg 2 mg/L) マグネシウム標準液(Mg 500 mg/L)10 mLを全量フラスコ100
mLにとり,水を標線まで加える。この溶液10 mLを全量フラスコ250 mLにとり,塩酸(1+1)5 mL
を加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 48.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(6)の適量(Mgとして2〜40 μgを含む。)を全量フラスコ100 mLにとり,塩酸(1+1)2 mLを
加えた後,水を標線まで加える。
2) この溶液10 mLを乾いたビーカーにとり,ランタン(III)溶液(La 50 g/L)1 mLを加える。
3) 2)の溶液をフレーム中に導入し,波長285.2 nmの指示値(7)を読み取る。
4) 空試験として試料と同量の水について,1)〜3)の操作を行って試料について得た指示値を補正する。
5) 検量線からマグネシウムの量を求め,試料中のマグネシウムの濃度(Mg mg/L)を算出する。
注(6) 50.の注(7)による。
(7) 50.の注(8)による。
d) 検量線 検量線の作成は,次による。
1) マグネシウム標準液(Mg 2 mg/L)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料と同じ
酸の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。この溶液についてc)の2)及
び3)の操作を行う。
2) 別に,空試験として水について試料と同じ酸の濃度になるように塩酸(1+1)を加えた後,c)の2)
及び3)の操作を行って標準液について得た指示値を補正し,マグネシウム(Mg)の量と指示値との
関係線を作成する。検量線の作成は,試料測定時に行う。
備考 3. アルミニウムは少量(2 mg/L)でも妨害するが,ランタン(III)溶液(La 50 g/L)を添加す
ることによって妨害を抑制することができる。
4. 50.の備考4.による。
51.3 ICP発光分光分析法 50.3による。
51.4 イオンクロマトグラフ法 48.3による。
52.[銅(Cu)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光
分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,“なお,
フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,2007年に
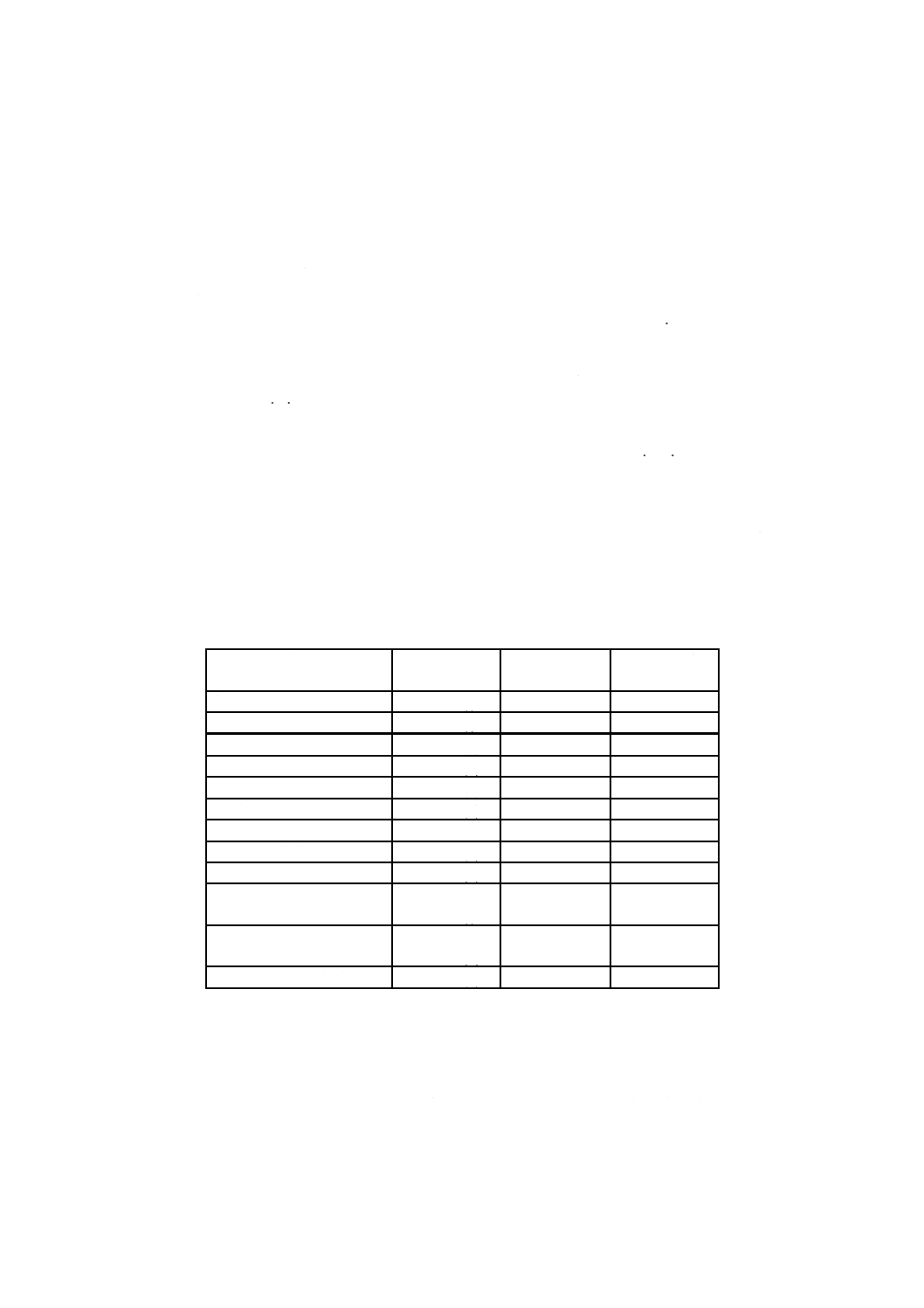
85
K 0102:2019
第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2
との整合を図ったものである。”に置き換える。
52.[銅(Cu)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
52.[銅(Cu)]の52.4(ICP発光分光分析法)の全文を,次に置き換える。
52.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銅
による発光を波長324.754 nmで測定して銅を定量する。この方法によって表52.1に示す元素が同時定量
できる。それぞれの元素ごとの測定波長,定量範囲及び繰返し精度の例を,表52.1に示す。
表52.1 測定波長,定量範囲及び繰返し精度の例*
対象元素
測定波長**
nm
定量範囲
mg/L
繰返し精度
%
銅(Cu)
324.754 (I)
0.02〜5
2〜10
亜鉛(Zn)
213.856 (I)
0.01〜5
2〜10
鉛(Pb)
220.351 (II)
0.05〜5
2〜10
カドミウム(Cd)
214.438 (II)
0.01〜5
2〜10
マンガン(Mn)
257.610 (II)
0.01〜5
2〜10
鉄(Fe)
238.204 (II)
0.01〜5
2〜10
ニッケル(Ni)
221.647 (II)
0.04〜5
2〜10
コバルト(Co)
228.616 (II)
0.03〜5
2〜10
ベリリウム(Be)
313.042 (II)
0.005〜5
2〜10
イットリウム(Y)***
371.029 (II)
464.370 (I)
−
−
インジウム(In)***
325.609 (I)
230.606 (II)
−
−
イッテルビウム(Yb)***
369.420 (II)
−
−
注*
装置及び測定条件によって異なる。
** Iは中性線,IIはイオン線。
*** 内標準元素
a) 試薬 試薬は,次による。
1) 銅標準液(Cu 1 mg/mL) JIS K 8005に規定する容量分析用標準物質の銅を塩酸(1+3)(JIS K 8180
に規定する塩酸を用いて調製する。)で洗い,水洗いし,JIS K 8101に規定するエタノール(99.5)
で洗い,次に,JIS K 8103に規定するジエチルエーテルで洗った後,直ちに上口デシケーター中に
86
K 0102:2019
入れ,2.0 kPa以下で数分間保った後,減圧下で約12時間保つ。Cu 100 %に対してその1.00 gをは
かりとり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調製する。)30 mL中に加え,煮沸し
て溶かし,窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加え
る。
2) 銅標準液(Cu 10 μg/mL) 銅標準液(Cu 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸(1
+1)10 mLを加えた後,水を標線まで加える。
3) 亜鉛標準液(Zn 1 mg/mL) JIS K 8005に規定する容量分析用標準物質の亜鉛を塩酸(1+3)で洗
い,水洗いし,JIS K 8101に規定するエタノール(99.5)で洗い,次に,JIS K 8103に規定するジ
エチルエーテルで洗った後,直ちに上口デシケーター中に入れ,圧力2 kPa以下で数分間保った後,
減圧下で約12時間保つ。Zn 100 %に対してその1.00 gをはかりとり,硝酸(1+1)30 mLに溶かし,
煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
4) 亜鉛標準液(Zn 10 μg/mL) 亜鉛標準液(Zn 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸
(1+1)10 mLを加えた後,水を標線まで加える。
5) 鉛標準液(Pb 1 mg/mL) JIS K 8701に規定する鉛(99.9 %以上)1.00 gをはかりとり,硝酸(1+1)
30 mLに溶かし,加熱して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水
を標線まで加える。又はJIS K 8563に規定する硝酸鉛(II)1.60 gをはかりとり,硝酸(1+1)20 mL
及び適量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
6) 鉛標準液(Pb 10 μg/mL) 鉛標準液(Pb 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸(1
+1)10 mLを加え,水を標線まで加える。
7) カドミウム標準液(Cd 0.1 mg/mL) カドミウム(99.9 %以上)0.100 gをはかりとり,硝酸(1+1)
20 mLに溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水
を標線まで加える。
8) カドミウム標準液(Cd 10 μg/mL) カドミウム標準液(Cd 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
9) マンガン標準液(Mn 1 mg/mL) JIS K 8247に規定する過マンガン酸カリウム2.88 gをはかりとり,
水150 mLに硝酸(1+1)10 mLを加えた溶液に溶かす。過酸化水素水(1+9)(JIS K 8230に規定
する過酸化水素を用いて調製する。)を滴加し,かき混ぜて脱色した後,煮沸して過剰の過酸化水素
を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はマンガン(99.9 %
以上)1.00 gをはかりとり,硝酸(1+3)20 mLに溶かし,煮沸して窒素酸化物を追い出す。放冷
後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
10) マンガン標準液(Mn 10 μg/mL) マンガン標準液(Mn 1 mg/mL)5 mLを全量フラスコ500 mLに
とり,硝酸(1+1)10 mLを加え,水を標線まで加える。
11) 鉄標準液(Fe 1 mg/mL) 鉄(99.5 %以上)1.00 gをはかりとり,塩酸(1+1)30 mL中に入れ,加
熱して溶かし,放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はJIS K 8979
に規定する硫酸アンモニウム鉄(II)六水和物[ビス(硫酸)鉄(II)アンモニウム六水和物]7.02
gをとり,塩酸(1+1)20 mL及び適量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標
線まで加える。
12) 鉄標準液(Fe 0.1 mg/mL) 鉄標準液(Fe 1 mg/mL)10 mLを全量フラスコ100 mLにとり,塩酸(1
+1)2 mLを加えた後,水を標線まで加える。
13) 鉄標準液(Fe 10 μg/mL) 鉄標準液(Fe 0.1 mg/mL)10 mLを全量フラスコ100 mLにとり,塩酸(1
87
K 0102:2019
+1)2 mLを加えた後,水を標線まで加える。
14) ニッケル標準液(Ni 0.1 mg/mL) JIS K 9062に規定するニッケル(99.9 %以上)0.100 gをはかりと
り,硝酸(1+1)20 mLに溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。
15) ニッケル標準液(Ni 10 μg/mL) ニッケル標準液(Ni 0.1 mg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
16) コバルト標準液(Co 0.1 mg/mL) コバルト(99.5 %以上)0.100 gをはかりとり,硝酸(1+1)20 mL
に溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線
まで加える。
17) コバルト標準液(Co 10 μg/mL) コバルト標準液(Co 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
18) ベリリウム標準液(Be 0.1 mg/mL) ベリリウム(99.5 %以上)0.100 gをはかりとり,塩酸(1+1)
20 mLに溶かす。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
19) ベリリウム標準液(Be 10 μg/mL) ベリリウム標準液(Be 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
備考 9. 試料の噴霧に超音波ネブライザー又はこれと同等の性能をもったものを用いてもよい。この
場合は,定量下限値を1桁程度下げることができる。ただし,メモリー効果に注意し,十分
に洗浄を行う。
c) 準備操作 準備操作は,次による。
1) 試料を5.5によって処理する。
備考 10. 試料の銅又は測定対象元素の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イ
オンなどの共存物質の濃度が高く,測定を妨害する場合の準備操作は,次の1)〜3)の操作に
よるか,又は備考6.による。次の準備操作は,亜鉛,鉛,カドミウム,マンガン,鉄,ニッ
ケル,コバルト,モリブデン及びバナジウムの定量にも使用できる。
1) 試料500 mL(又は100〜500 mLの一定量)をビーカーにとり,JIS K 8180に規定する塩
酸5 mLを加え,約5分間煮沸する。
2) 放冷後,酢酸-酢酸ナトリウム緩衝液(pH5)(JIS K 8371に規定する酢酸ナトリウム三水
和物19.2 gとJIS K 8355に規定する酢酸3.4 mLとを水に溶かして1 Lとする。)(*12) 10
mLを加え,アンモニア水(1+1)(JIS K 8085に規定するアンモニア水を用いて調製す
る。)又は硝酸(1+10)(JIS K 8541に規定する硝酸を用いて調製する。)でpHを5.2に
調整する。
注(*12) この操作に用いる酢酸-酢酸ナトリウム緩衝液(pH5)は,使用前に1-ピロリジ
ンカルボジチオ酸アンモニウム溶液,ヘキサメチレンアンモニウム-ヘキサメチ
レンカルバモジチオ酸(ヘキサメチレンアンモニウム-ヘキサメチレンジチオカ
ルバミド酸)のメタノール溶液及びJIS K 8271に規定するキシレンを加えて振
り混ぜ,精製する。
3) この溶液を分液漏斗1 000 mL(又は200〜500 mL)に移し,1-ピロリジンカルボジチオ
酸アンモニウム溶液(20 g/L)2 mL及びヘキサメチレンアンモニウム-ヘキサメチレンカ
88
K 0102:2019
ルバモジチオ酸(HMA-HMDC)のメタノール溶液(20 g/L)2 mLを加えて混合した後,
JIS K 8271に規定するキシレンの一定量(5〜20 mL)を加えて約5分間激しく振り混ぜ
て静置する。水層を捨てキシレン層を共栓試験管に入れる。定量操作は,備考12.による。
d) 操作 操作は,次による。
1) c) 1)の準備操作を行った試料を試料導入部を通して発光部に導入し(11) (12),各測定対象元素の波長
の発光強度を測定する(13) (14)。
2) 空試験としてc) 1)の準備操作で用いた試料と同量の水をとり,c) 1)及び1)の操作を行って各測定対
象元素に相当する発光強度を測定し,試料について得た発光強度を補正する。
3) 検量線から各測定対象元素の量を求め,試料中の各元素の濃度(mg/L)を算出する(15)。
注(11) 試料の測定を始める前に,硝酸(1+20)(JIS K 8541に規定する硝酸を用いて調製する。)を噴
霧して測定系を洗い流す。また,各試料測定の間にも洗い流しを行う。
(12) 備考11.による。
(13) 47.の注(8)による。
(14) 47.の注(9)による。
(15) 50.の注(11)による。
e) 検量線 検量線の作成は,次による。
1) a)の測定対象元素の各標準液のそれぞれ一定量をとり,希釈して一定量として混合標準液を調製す
る。この混合標準液は,測定対象の元素を含み(16),測定する濃度範囲よりも高い濃度(17)とする。
2) 全量フラスコ100 mLに,測定濃度範囲を含むように,混合標準液を段階的にとり,c) 1)の試料と同
じ酸の濃度となるように硝酸(1+1)(18)を加えた後,水を標線まで加える。
3) 2)で調製した各溶液についてd) 1)の操作を行う。
4) 空試験として,全量フラスコ100 mLにc) 1)の試料と同じ酸の濃度となるように硝酸(1+1)を加
え,水を標線まで加えた溶液について,d) 1)の操作を行い,3)で得た発光強度を補正する。
5) 各測定対象元素について,それぞれの元素の量と発光強度との関係線を作成する。
注(16) 測定対象の単独又は限られた複数の元素だけでもよい。
(17) 作成する検量線の最高濃度の5〜10倍程度。
(18) 又は塩酸(1+1)。ただし,指定されている場合は,それに従う。
備考 11. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることが
できる。内標準元素としては,ICP中で分析元素と類似の発光挙動を示す元素が適切であり,
したがって内標準線及び分析線は中性線同士又はイオン線同士で,その励起エネルギー差が
小さく,かつ,分析線に対し分光干渉を生じない発光線を選択する。また,元の試料中に含
まれる濃度が,添加する濃度に比べ,無視できる濃度であることを確認しておく。内標準元
素としてはイットリウム(Y),インジウム(In),イッテルビウム(Yb)などが用いられる
が,上記の条件を満足すれば他の元素を用いてもよい。内標準物質としてイットリウムを用
いる場合の操作は,次による。
1) c) 1)で処理した試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50
μg/mL)[47.の備考5. 6)による。]10 mL及びc) 1)の試料と同じ酸の濃度となるように硝
酸(1+1)(*)を加えた後,水を標線まで加える。
2) この溶液についてd) 1)の操作を行って各測定対象元素の発光強度及びイットリウムの
発光強度を測定し,各測定対象元素の発光強度とイットリウムの発光強度との比を求め
89
K 0102:2019
る。
3) 空試験として,試料に代えて同量の水を用い,c) 1),1)及び2)の操作を行い,各測定対
象元素の発光強度とイットリウムの発光強度との比を求め,2)で得た発光強度比を補正
する。
4) 検量線から,各測定対象元素の量を求め,試料中の各測定対象元素の濃度(mg/L)を算
出する。
5) 検量線 全量フラスコ100 mL数個にe) 2)に従って混合標準液を段階的にとり,イット
リウム溶液(Y 50 μg/mL)10 mL及び1)の試料と同じ酸の濃度となるように硝酸(1+1) (*)
を加えた後,水を標線まで加える。この溶液について2)の操作を行い,各測定対象元素
及びイットリウムの発光強度を測定し,各測定対象元素の発光強度とイットリウムの発
光強度との比を求める。別に,空試験として,混合標準液に代えて水を用い,同じ操作
を行って,同様に発光強度の比を求め,混合標準液での各測定対象元素の発光強度比を
補正し,各測定対象元素の量と,それぞれの発光強度とイットリウムの発光強度との比
との関係線を作成する。
注(*) 注(18)による。
備考 12. 備考10.の1)〜3)によって準備操作を行った場合は,キシレン層をそのまま噴霧して測定対象
の各元素の発光強度を測定して定量する。その場合の検量線の作成は,次による。
1) e) 1)に準じ,測定対象元素を含む混合標準液を調製する。ただし,各測定対象元素はe) 1)
の混合標準液よりも低濃度(0.1〜1 μg/mL)とする。
2) 調製した混合標準液を段階的にとり,水で500 mLとし,備考10.の1)〜3)の操作及び各
測定対象元素の発光強度の測定操作を行う。
3) 空試験として,水500 mLを用いて,2)の操作を行い,2)で得た各測定対象元素の発光強
度を補正し,各測定対象元素の量とその発光強度との関係線を作成する。
52.[銅(Cu)]の52.5(ICP質量分析法)の全文を,次に置き換える。
52.5 ICP質量分析法 試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中
に噴霧し,銅及び内標準元素のそれぞれの質量/電荷数における指示値(19)を測定し,銅の指示値と内標準
元素の指示値との比を求めて銅を定量する。この方法によって,表52.2に示す元素が同時定量できる。そ
れぞれの元素ごとの定量範囲,繰返し精度などの例を,表52.2に示す。
注(19) イオンカウント値又はその比例値。
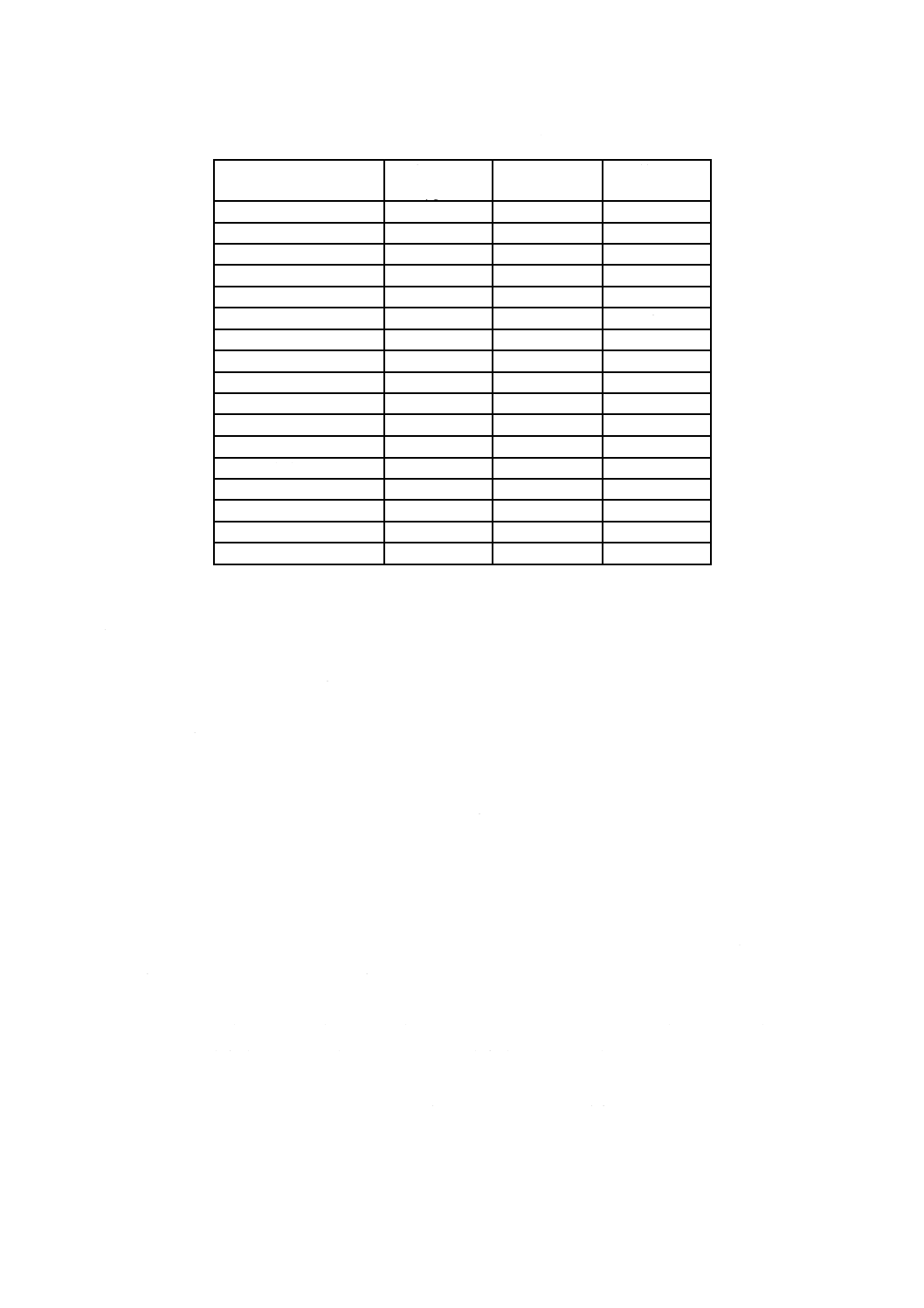
90
K 0102:2019
表52.2 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
銅(Cu)
0.5〜500
2〜10
63,65
亜鉛(Zn)
0.5〜500
2〜10
66,68,64
鉛(Pb)
0.3〜500
2〜10
208,206,207
カドミウム(Cd)
0.3〜500
2〜10
111,114
マンガン(Mn)
0.5〜500
2〜10
55
アルミニウム(Al)
0.5〜500
2〜10
27
ニッケル(Ni)
0.5〜500
2〜10
60,58
コバルト(Co)
0.5〜500
2〜10
59
ベリリウム(Be)
0.5〜500
2〜10
9
ひ素(As)
0.5〜500
2〜10
75
ビスマス(Bi)
0.3〜500
2〜10
209
クロム(Cr)
0.5〜500
2〜10
53,52,50
セレン(Se)
0.5〜500
2〜10
82,77,78
バナジウム(V)
0.5〜500
2〜10
51
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 内標準液(1 μg/mL) 内標準元素としてイットリウム,インジウム又はビスマスを用いる。内標準
液の調製には,次の3.1)〜3.3)に規定する溶液のうち内標準とする元素の溶液2 mLを全量フラスコ
100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する(20) (21)。
3.1) イットリウム溶液(Y 50 μg/mL) 47.の備考5. 6)による。
3.2) インジウム溶液(In 50 μg/mL) 47.4 a) 2.2)による。
3.3) ビスマス溶液(Bi 50 μg/mL) 酸化ビスマス(III)0.279 gをとり,硝酸(1+1)10 mLを加え,
加熱して溶かし,放冷後,全量フラスコ250 mLに移し入れ,水を標線まで加える。この溶液25 mL
を全量フラスコ500 mLにとり,硝酸(1+1)10 mLを加え,水を標線まで加える。
4) 混合標準液[(Cu 10 μg,Zn 10 μg,Pb 10 μg,Cd 10 μg,Mn 10 μg,Al 10 μg,Ni 10 μg,Co 10 μg,
Be 10 μg,As 10 μg,Bi 10 μg,Cr 10 μg,Se 10 μg,V 10 μg)/mL](16) (21) 52.4 a)の1) 銅標準液(Cu
1 mg/mL),3) 亜鉛標準液(Zn 1 mg/mL),5) 鉛標準液(Pb 1 mg/mL),9) マンガン標準液(Mn 1
mg/mL)のそれぞれ5 mL,58.2 a) 2) アルミニウム標準液(Al 0.5 mg/mL)10 mL及び67.1 a) 11) セ
レン標準液(Se 0.2 mg/mL)25 mL,並びに52.4 a)の7) カドミウム標準液(Cd 0.1 mg/mL),14) ニ
ッケル標準液(Ni 0.1 mg/mL),16) コバルト標準液(Co 0.1 mg/mL),18) ベリリウム標準液(Be 0.1
mg/mL),58.4 a)の3) クロム標準液(Cr 0.1 mg/mL),7) バナジウム標準液(V 0.1 mg/mL),61.1 a)
12) ひ素標準液(As 0.1 mg/mL),64.1 a) 5) ビスマス標準液(Bi 0.1 mg/mL)のそれぞれ50 mLを全
量フラスコ500 mLにとり,硝酸(1+1)10 mLを加えた後,水を標線まで加える(22)。
5) 混合標準液[(Cu 0.5 μg,Zn 0.5 μg,Pb 0.5 μg,Cd 0.5 μg,Mn 0.5 μg,Al 0.5 μg,Ni 0.5 μg,Co 0.5
91
K 0102:2019
μg,Be 0.5 μg,As 0.5 μg,Bi 0.5 μg,Cr 0.5 μg,Se 0.5 μg,V 0.5 μg)/mL](16) (21) 4)の混合標準液
5 mLを全量フラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調
製する。
注(20) 3種類の内標準元素は,単独又は混合して用いてもよい。ICP質量分析法では,主成分(マトリ
ックス)による非スペクトル干渉の大きさは質量数に依存するため,測定対象元素と比較的質
量数の近いものを内標準元素とするとよい。ここに挙げた3種類以外にも,元の試料に無視で
きる量より少ない量しか含まれていないことが確認できれば,内標準元素として用いてもよい。
ビスマスが測定対象元素であるときは,内標準元素としてビスマスを用いることはできない。
ビスマスの代わりにタリウムを用いることがある。
(21) 定期的に濃度の安定性を,新たに調製した標準液と比較して確認する。特に,濃度の低い標準
液は濃度が低下しやすいため注意する。
(22) 標準液は,混合したときに沈殿を生じないものを用いる。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 13. イオン源として,高周波プラズマと同等の性能をもつものを用いてもよい。
14. サンプリングコーン及びスキマーコーンの材質からの汚染が認められないことを確認する。
c) 準備操作 準備操作は,次による(23)。
1) 試料を5.5によって処理する。ただし,クロムを定量する場合は,前処理に5.3は用いない。
2) 1)で処理した試料の適量(測定対象元素として0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,
内標準液(1 μg/mL)1 mLを加え,硝酸の最終濃度が0.1〜0.5 mol/Lとなるように硝酸(1+1)を
加えた後,水を標線まで加える。
注(23) 分析者からの汚染がないように注意する。JIS T 9107に規定する単回使用手術用ゴム手袋(打
粉のないもの)などを用いるとよい。
備考 15. 銅又は測定対象元素の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンな
どの共存物質の濃度が高く,測定を妨害する場合の準備操作は,備考6.による[ただし,3.6)
は除く。]。得られた液は全量フラスコ20 mLに移し入れ,内標準液(1 μg/mL)0.2 mLを加
え,標線まで水を加える。
d) 操作 操作は,次による(24)。
1) ICP質量分析装置を作動できる状態にし,c) 2)の溶液を試料導入部を通してイオン化部に導入して
測定対象元素及び内標準元素(イットリウム,インジウム又はビスマス)のそれぞれの質量/電荷数
(25)における指示値を読み取り,測定対象元素の指示値と内標準元素の指示値との比をそれぞれ求め
る。
2) 空試験として,c) 1)での試料と同量の水をとり,試料と同様にc)及びd) 1)の操作を行って測定対象
元素の指示値と内標準元素の指示値との比を求め,試料について得た測定対象元素と内標準元素と
の指示値の比を補正する。
3) 検量線から測定対象元素の量を求め,試料中の測定対象元素の濃度(μg/L)を算出する。
注(24) 妨害物質の存在が不明の場合には,定量に先立ってICP質量分析計による定性分析を行うこと
によって,測定対象元素及び内標準元素の測定質量数に対する妨害(スペクトル干渉及び非ス
ペクトル干渉)の有無と程度を推定することができる。スペクトル干渉が認められる場合には,
測定質量数の変更,試料の希釈又は前処理を行って妨害の軽減を図る。スペクトル干渉のため,
92
K 0102:2019
上記のイットリウム,インジウム又はビスマスを内標準元素として使用できない場合もあるが,
その場合には,他の内標準元素を用いる。スペクトル干渉の例を,表52.3に示す。非スペクト
ル干渉(マトリックス干渉ともいい,検量線の傾きに影響する。)は,一般にこの方法で採用し
ている内標準法によって補正できるが,妨害物質の濃度が高い場合には,補正が不十分となる
ことがある。このような場合には,試料の希釈又は前処理を行った後,内標準法を適用して妨
害の軽減を図る。非スペクトル干渉の程度は,標準液を添加して回収率を求めることによって
推定することができる。すなわち,試料(元の試料又は希釈・前処理後の試料)中の測定対象
元素の濃度が10 ng/mL分だけ(ただし,試料中の測定元素の濃度が高い場合には,増加分が精
度よく測定できるように,試料中と同程度の濃度だけ)増加するように,測定対象元素の標準
液(0.5 μg/mL)の適量を試料に添加後,d)に準じた操作を行って測定対象元素の濃度を求め,
その回収率を求める。回収率が90〜110 %の範囲にあれば,非スペクトル干渉は,ほぼ無視し
得るものと考えられる。
注(25) 質量数を設定するには,表52.2及び表52.3を参考にするとよい。複数の安定同位体がある場合,
複数の同位体の質量/電荷数を用いて測定を行うことによって,スペクトル干渉による妨害を推
定することができる。
e) 検量線 検量線の作成は,次による。
1) a) 4)の混合標準液(各元素濃度10 μg/mL)又はa) 5)の混合標準液(各元素濃度0.5 μg/mL)いずれ
か(26)の混合標準液0.1〜5 mLを,全量フラスコ100 mLに段階的にとり,内標準液(1 μg/mL)1 mL
を加え,c) 2)の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を標線まで加える。
使用時に調製する。
2) この溶液についてd) 1)の操作を行う。
3) 別に,空試験として全量フラスコ100 mLに内標準液(1 μg/mL)1 mLを加え,c) 2)の試料と同じ酸
の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,d) 1)の操作を行って標準液につ
いて得た指示値の比をそれぞれ補正し,測定対象元素の濃度に対する,測定元素の指示値と内標準
元素の指示値との比の関係線をそれぞれ作成する。検量線の作成は,試料測定時に行う。
注(26) a) 4)の混合標準液の調製に用いた各元素の標準液のうち,測定対象元素の各標準液のそれぞれ
一定量をとり,希釈して一定量として混合標準液を調製して用いてもよい。この混合標準液は,
測定対象の元素を含み,測定する濃度範囲よりも高い濃度とする。
備考 16. 注(24)の操作で,主成分の元素又は有機物の含有量が少なく,非スペクトル干渉が無視できる
試料の場合は,内標準元素の添加を省略し,検量線法によって定量してもよい。
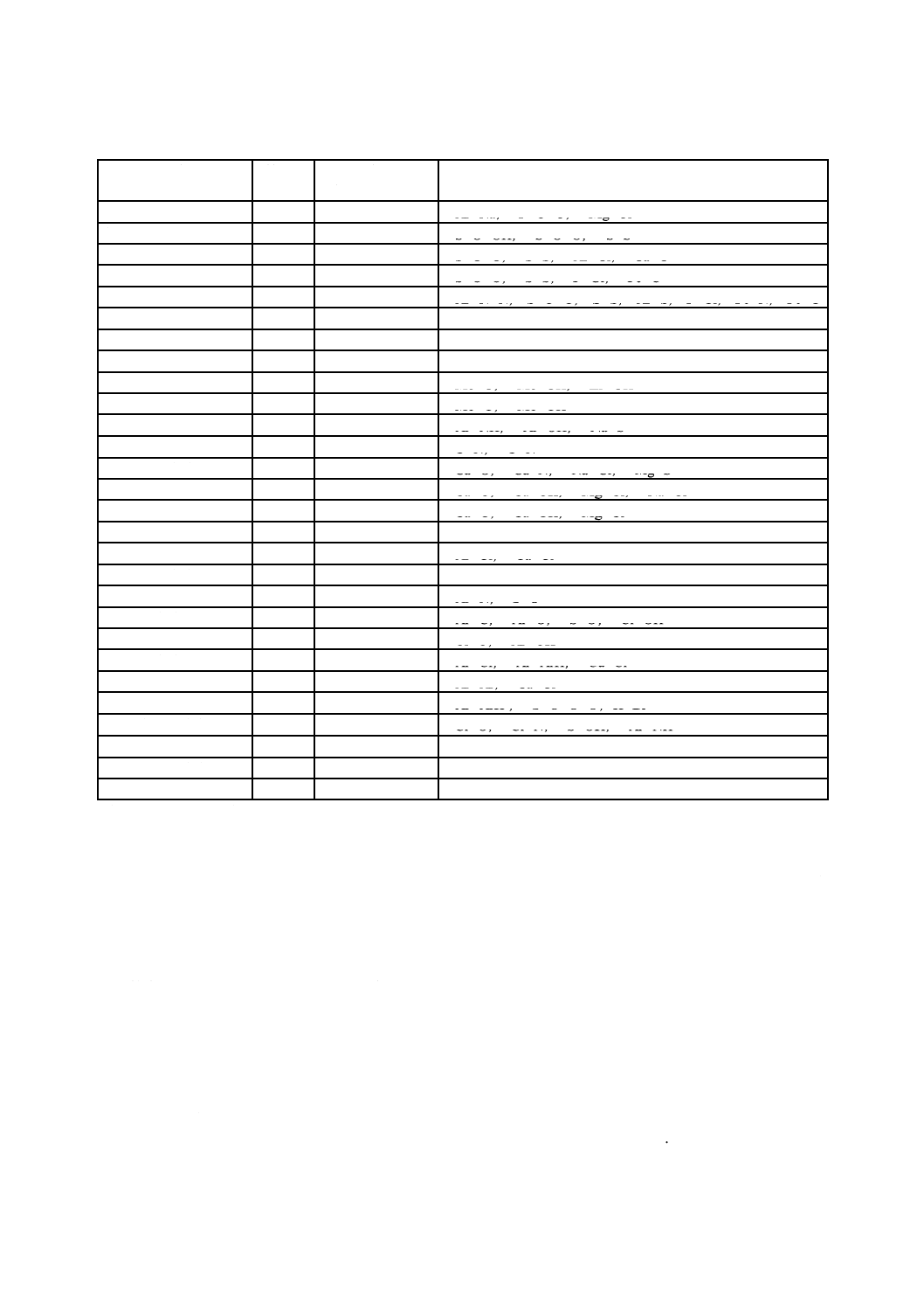
93
K 0102:2019
表52.3 スペクトル干渉の例*
元素
質量数
同重体及び
2価イオンの干渉
多原子イオンの干渉
銅(Cu)
63
40Ar23Na,31P16O16O,26Mg37Cl
銅(Cu)
65
32S16O16OH,33S16O16O,32S33S
亜鉛(Zn)
64
Ni+
32S16O16O,32S32S,27Al37Cl,48Ca16O
亜鉛(Zn)
66
Ba++
34S16O16O,32S34S,31P35Cl,54Fe12C
亜鉛(Zn)
68
Ba++,Ce++
40Ar14N14N,36S16O16O,32S36S,36Ar32S,31P37Cl,54Fe14N,56Fe12C
鉛(Pb)
206
鉛(Pb)
207
鉛(Pb)
208
カドミウム(Cd)
111
95Mo16O,94Mo16OH,94Zr16OH
カドミウム(Cd)
114
Sn+
98Mo16O,97Mo16OH
マンガン(Mn)
55
40Ar14NH,38Ar16OH,23Na32S
アルミニウム(Al)
27
12C15N,13C14N
ニッケル(Ni)
58
Fe+
42Ca16O,44Ca14N,23Na35Cl,24Mg34S
ニッケル(Ni)
60
44Ca16O,43Ca16OH,25Mg35Cl,23Na37Cl
コバルト(Co)
59
43Ca16O,42Ca16OH,24Mg35Cl
ベリリウム(Be)
9
ひ素(As)
75
Nd++,Sm++
40Ar35Cl,40Ca35Cl
ビスマス(Bi)
209
クロム(Cr)
50
36Ar14N,34S16O
クロム(Cr)
52
40Ar12C,36Ar16O,36S16O,35Cl16OH
クロム(Cr)
53
37Cl16O,36Ar16OH
セレン(Se)
77
40Ar37Cl,36Ar40ArH,40Ca37Cl
セレン(Se)
78
Kr+
38Ar40Ar,43Ca35Cl
セレン(Se)
82
Kr+
40Ar40ArH2,34S16O16O16O,H81Br
バナジウム(V)
51
35Cl16O,37Cl14N,34S16OH,36Ar14NH
イットリウム(Y)**
89
インジウム(In)**
115
Sn+
ビスマス(Bi)**
209
注*
装置及び測定条件によって異なる。
** 内標準元素
53.[亜鉛(Zn)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発
光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,“なお,
フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,2007年に
第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2
との整合を図ったものである。”に置き換える。
53.[亜鉛(Zn)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
94
K 0102:2019
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
54.[鉛(Pb)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光
分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,“なお,
フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,2007年に
第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2
との整合を図ったものである。”に置き換える。
54.[鉛(Pb)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
55.[カドミウム(Cd)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288
及び1994年に第1版として発行されたISO 5961,電気加熱原子吸光法は,1994年に第1版として発行さ
れたISO 5961,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったも
のである。”を,“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288及び1994年
に第2版として発行されたISO 5961,電気加熱原子吸光法は,1994年に第2版として発行されたISO 5961,
ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2
版として発行されたISO 17294-2との整合を図ったものである。”に置き換える。
55.[カドミウム(Cd)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 5961:1994,Water quality−Determination of cadmium by atomic absorption spectrometry(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
95
K 0102:2019
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
56.[マンガン(Mn)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885と
の整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
56.[マンガン(Mn)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
57.[鉄(Fe)]の“なお,フェナントロリン吸光光度法は,1988年の第2版として発行されたISO 6332,
ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,
“なお,フェナントロリン吸光光度法は,1988年に第2版として発行されたISO 6332,ICP発光分光分析
法は,2007年に第2版として発行されたISO 11885との整合を図ったものである。”に置き換える。
57.[鉄(Fe)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6332:1988,Water quality−Determination of iron−Spectrometric method using 1,10-
phenanthroline(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
58.[アルミニウム(Al)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
58.[アルミニウム(Al)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
96
K 0102:2019
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
58.[アルミニウム(Al)]の58.4(ICP発光分光分析法)e)(検量線)の後に,“備考9. 52.の備考11.に
よる。”を追加する。
59.[ニッケル(Ni)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP
発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,“な
お,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,2007
年に第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2
との整合を図ったものである。”に置き換える。
59.[ニッケル(Ni)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
60.[コバルト(Co)]の“なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP
発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,“な
お,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,2007
年に第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2
との整合を図ったものである。”に置き換える。
60.[コバルト(Co)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
97
K 0102:2019
61.[ひ素(As)]の“なお,水素化物発生原子吸光法は,1996年に第1版として発行されたISO 11969,
ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。”を,
“なお,水素化物発生原子吸光法は,1996年に第1版として発行されたISO 11969,ICP発光分光分析法
は,2007年に第2版として発行されたISO 11885,ICP質量分析法は,2016年に第2版として発行された
ISO 17294-2との整合を図ったものである。”に置き換える。
61.[ひ素(As)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11969:1996,Water quality−Determination of arsenic−Atomic absorption spectrometric method
(hydride technique)(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
62.[アンチモン(Sb)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
62.[アンチモン(Sb)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
63.[すず(Sn)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整
合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,
ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”に置き
換える。
63.[すず(Sn)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
98
K 0102:2019
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
64.[ビスマス(Bi)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885と
の整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
64.[ビスマス(Bi)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
65.[クロム(Cr)]の65.1(全クロム)の“なお,フレーム原子吸光法及び電気加熱原子吸光法は,1990
年に第1版として発行されたISO 9174,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,フレーム原子吸光法及び電気加熱原子吸光法は,1990年に第
1版として発行されたISO 9174,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,
ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”に置き
換える。
65.[クロム(Cr)]の65.1(全クロム)の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9174:1990,Water quality−Determination of total chromium−Atomic absorption spectrometric
methods(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
65.[クロム(Cr)]の65.2{クロム(VI)[Cr(VI)]}の“クロム(VI)の定量には,ジフェニルカルバジ
ド吸光光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法,ICP質量分析法又はジ
フェニルカルバジド発色による流れ分析法を適用する。なお,ジフェニルカルバジド吸光光度法は,1994
99
K 0102:2019
年に第1版として発行されたISO 11083,ICP発光分光分析法は,1996年に第1版として発行されたISO
11885,ジフェニルカルバジド発色流れ分析法は2006年に第1版として発行されたISO 23913との整合を
図ったものである。”を“クロム(VI)の定量には,ジフェニルカルバジド吸光光度法,フレーム原子吸
光法,電気加熱原子吸光法,ICP発光分光分析法,ICP質量分析法,ジフェニルカルバジド発色による流
れ分析法又は液体クロマトグラフィー誘導結合プラズマ質量分析法を適用する。なお,ジフェニルカルバ
ジド吸光光度法は,1994年に第1版として発行されたISO 11083,ICP発光分光分析法は,2007年に第2
版として発行されたISO 11885,ジフェニルカルバジド発色流れ分析法は,2006年に第1版として発行さ
れたISO 23913,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったも
のである。”に置き換える。
65.[クロム(Cr)]の65.2{クロム(VI)[Cr(VI)]}の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11083:1994,Water quality−Determination of chromium (VI)−Spectrometric method using 1,5-
diphenylcarbazide(MOD)
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
ISO 23913:2006,Water quality−Determination of chromium (VI)−Method using flow analysis (FIA
and CFA) and spectrometric detection(MOD)
65.[クロム(Cr)]の65.2.5(ICP質量分析法)の“試料を前処理した後,内標準物質を加え,試料導入部
を通して誘導結合プラズマ中に噴霧し,クロム及び内標準物質のそれぞれの質量/荷電数におけるイオンの
電流を測定し,クロムのイオンの電流と内標準物質のイオンの電流との比を求めてクロム(VI)を定量す
る。”を,“試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,
クロム及び内標準物質のそれぞれの質量/荷電数における指示値(11)を測定し,クロムの指示値と内標準物
質の指示値との比を求めてクロム(VI)を定量する。”に置き換える。
65.[クロム(Cr)]の65.2.5(ICP質量分析法)の“それぞれの元素ごとの定量範囲,繰返し精度などの例
を,表52.2に示す。”の後に,“注(11) イオンカウント値又はその比例値。”を追加する。
65.[クロム(Cr)]の65.2.6(流れ分析法)の後に65.2.7(液体クロマトグラフィー誘導結合プラズマ質量
分析法)を設け,次の文を追加する。
65.2.7 液体クロマトグラフィー誘導結合プラズマ質量分析法 試料中のクロム(VI)を液体クロマトグラ
フの分離カラムによって分離して,誘導結合プラズマ中に噴霧し,クロムの質量/荷電数における指示値(11)
を測定してクロム(VI)を定量する。
定量範囲:Cr(VI)5 μg/L〜5 mg/L,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
100
K 0102:2019
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 溶離液 溶離液は,装置の種類及び分離カラムに充塡した充塡剤の種類によって異なるので,あら
かじめ,備考15.によってクロム(VI)及びクロム(III)の分離の確認を行う。
4) クロム(VI)標準液[Cr(VI)0.1 mg/mL] 58.4 a) 3)による。
5) クロム(VI)標準液[Cr(VI)5 μg/mL] a) 4)のクロム標準液[Cr(VI)0.1 mg/mL]50 mLを全
量フラスコ1 000 mLにとり,水を標線まで加える。
6) クロム(III)標準液[Cr(III)0.1 mg/mL] 硝酸クロム九水和物を0.770 gとり,少量の水に溶か
して全量フラスコ1 000 mLに移し入れ,硝酸(1+1)50 mLを加えた後,水を標線まで加える。
7) クロム(III)標準液[Cr(III)5 μg/mL] a) 6)のクロム(III)標準液[Cr(III)0.1 mg/mL]50 mL
を全量フラスコ1 000 mLにとり,水を標線まで加える。
8) 2,6-ピリジンジカルボン酸
9) りん酸水素二ナトリウム JIS K 9020に規定するもの。
10) 酢酸アンモニウム JIS K 8359に規定するもの。
11) 2,6-ピリジンジカルボン酸溶液(20 mmol/L) 2,6-ピリジンジカルボン酸3.3 g,りん酸水素二ナト
リウム2.8 g及び酢酸アンモニウム38.5 gを水900 mLに加えて溶かし,水酸化ナトリウム溶液でpH
6.8に調節して,水を加えて1 000 mLにする。
b) 器具及び装置 器具及び装置は,次による。
1) 液体クロマトグラフICP質量分析装置 液体クロマトグラフICP質量分析装置は,液体クロマトグ
ラフとICP質量分析装置とを組み合わせたもの。液体クロマトグラフの分離カラムによって,クロ
ム(VI)と2,6-ピリジンジカルボン酸によって錯形成化したクロム(III)[2,6-ピリジンジカルボン
酸クロム(III)錯体]とを分離し,カラムからの溶出液をICP質量分析装置に導入してクロム(VI)
を分離定量できるもの。また,液体クロマトグラフの溶離液及び接液部からのクロムの汚染がクロ
ム(VI)の定量に影響を及ぼさないもの。
1.1) 分離カラム クロム(VI)及び2,6-ピリジンジカルボン酸クロム(III)錯体を分離できるもの(12)。
1.2) ICP質量分析装置 52.5 b) 1)による。
2) マイクロシリンジ 10〜200 μLの容量で,接液部がポリプロピレン,チタンなどの材質で,クロム
の溶出が測定に影響を及ぼさないもの。又は自動注入装置。
3) 蓋付き試験管 耐熱・耐圧のねじ蓋付き試験管で,接液部がポリプロピレンなどの材質で,クロム
の溶出が測定に影響を及ぼさないもの。
4) ブロックヒーター 80±3 ℃を維持できるもので,ヒーターの壁が蓋付き試験管に密着するもの。
内容物を加熱するのに十分な深さがあるものが望ましい。
注(12) 例えば,ポリエーテルエーテルケトン製のカラムに,陰イオン交換樹脂を充塡したもの,又は
陽イオン交換樹脂及び陰イオン交換樹脂を混合充塡したもの。
備考 15. 分離カラムの性能として,クロム(VI)及び2,6-ピリジンジカルボン酸クロム(III)の分離
度(R)が1.3以上に分離できるものを用いる。分離度(R)は,35.の備考7.の式によって求
める。分離カラムの性能は,定期的に確認するとよい。
c) 準備操作 準備操作は,次による。
1) 試料(13)を10 mLとる。試料に懸濁物を含む場合には,ろ紙5種C又は孔径0.45 μmのろ過材(14)で
ろ過し,初めのろ液約50 mLを捨て,その後のろ液を用いる。
101
K 0102:2019
2) 1)の操作を行った試料に2,6-ピリジンジカルボン酸溶液(20 mmol/L)を10 mL加え,水を加えて100
mLにする。1)の操作を行った試料が酸性の場合には,水酸化ナトリウム溶液(40 g/L)[21. a) 3)に
よる。]で,また,アルカリ性の場合は,硝酸(1+10)(JIS K 8541に規定する硝酸を用いて調製す
る。)を加えて中和した後に,2,6-ピリジンジカルボン酸溶液(20 mmol/L)を10 mL加え,更に水
を加えて100 mLにする。
3) 蓋付き試験管に2)の試料適量をとって蓋をし,ブロックヒーターで,80 ℃,30分間加熱し放冷す
る(15)。
注(13) 試料中のクロム(VI)濃度が定量範囲を超える場合は,水で一定の割合に薄める。
(14) シリンジフィルターでろ過を行うときは,初めのろ液約5 mLを捨て,その後のろ液を用いる。
(15) 放冷後の溶液中に懸濁物を含む場合には,孔径0.45 μmのろ過材でろ過し,初めのろ液約5 mL
を捨て,その後のろ液を用いる。
d) 操作 操作は,次による。
1) 液体クロマトグラフICP質量分析装置を作動できる状態にし,分離カラムに溶離液を一定の流量(例
えば,0.2〜2 mL/min)で流しておく。
2) c)の準備操作を行った試料の一定量(例えば,10〜200 μLの一定量)をマイクロシリンジ(16)を用い
て,液体クロマトグラフICP質量分析装置に注入してクロマトグラムを記録する。
3) クロマトグラム上のクロム(VI)に相当するピークについて,指示値(17)を読み取る。
4) 空試験として,試料と同量の水について,c)及び2)〜3)の操作を行って,試料について得た結果を
補正する。
5) 検量線からクロム(VI)の濃度を求め,試料中のクロム(VI)の濃度(μg/L)を算出する。
注(16) 検量線作成時と同じものを用いる。
(17) ピーク高さ又はピーク面積。
e) 検量線 検量線の作成は,次による。
1) クロム(VI)標準液[Cr(VI)5 μg/mL]0.1〜100 mLを全量フラスコ100 mLに段階的にとり,ク
ロム(VI)標準液100 mLとった場合以外は水を標線まで加える。この溶液についてc)並びにd)の
2)及び3)の操作を行う。
2) 別に空試験として水についてc)並びにd)の2)及び3)の操作を行い クロム(VI)に相当する指示値
を補正した後,クロム(VI)の量と指示値との関係線を作成する。検量線の作成は測定時に行う。
備考 16. クロム(VI)標準液[Cr(VI)5 μg/mL]及びクロム(III)標準液[Cr(III)5 μg/mL]につ
いてc)の準備操作及びd)の操作を行い,クロム(VI)及び2,6-ピリジンジカルボン酸クロム
(III)錯体の保持時間に相当するピークの位置と各価数のクロムの分離度(R)が1.3以上で
あることをあらかじめ確認する。また,c)の操作由来のクロムの価数変化によって,クロム
(VI)の定量に影響を及ぼさないことも確認する。
17. 試料中に鉛,バリウム及び銀イオン(塩類)が存在し,難溶性クロム酸塩を生じる場合は,
定量が困難である。また,試料中に酸化性又は還元性の物質が存在し,クロムの原子価の変
化による測定妨害が生じる場合は,備考9.の除去方法を用いる。
なお,このクロムの原子価の変化は,クロム(VI)及びクロム(III)のクロムの添加回収
試験をそれぞれ行い,クロム(VI)の回収率及びクロム(III)からのクロム(VI)への価数
変化率を調べて確認するとよい。
102
K 0102:2019
66.[水銀(Hg)]の66.2[アルキル水銀(II)化合物]の全文を,次に置き換える。
66.2 アルキル水銀(II)化合物 アルキル水銀(II)化合物の定量は,アルキル水銀(II)化合物のうち,
エチル水銀(II)化合物及びメチル水銀(II)化合物を対象とし,水銀の量で表示する。定量にはガスクロ
マトグラフ法又はガスクロマトグラフィー質量分析法を適用する。
66.2.1 ガスクロマトグラフ法 アルキル水銀(II)化合物をトルエンで抽出し,L-2-アミノ-3-メルカプト
プロピオン酸(L-システイン)によって選択的に逆抽出した後,トルエンで再び抽出し,ガスクロマトグ
ラフ法を用いて定量する。
定量範囲:Hg 0.5 μg/L以上(試料換算)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) アンモニア水 JIS K 8085に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
4) アンモニア水(1+1) 3)のアンモニア水を用いて調製する。
5) 塩化ナトリウム溶液(200 g/L) JIS K 8150に規定する塩化ナトリウム200 gを水に溶かして1 Lと
する。予期保持時間付近にピークを生じないもの。
6) トルエン JIS K 8680に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
7) L-システイン-酢酸ナトリウム混合溶液 JIS K 8470に規定するL-システイン塩酸塩一水和物1 g,
JIS K 8371に規定する酢酸ナトリウム三水和物0.8 g及びJIS K 8987に規定する硫酸ナトリウム12.8
gを水に溶かして100 mLとする。予期保持時間付近にピークを生じないもの。
8) 塩化エチル水銀標準液(Hg 10 g/L)又は塩化メチル水銀標準液(Hg 10 g/L) クロロエチル水銀(II)
[塩化エチル水銀(II)]0.132 g又はクロロメチル水銀(II)[塩化メチル水銀(II)]0.125 gを少量
のトルエンに溶かし,全量フラスコ10 mLに移し入れ,トルエンを標線まで加える。
9) 塩化エチル水銀標準液(Hg 100 mg/L)又は塩化メチル水銀標準液(Hg 100 mg/L) 塩化エチル水
銀標準液(Hg 10 g/L)又は塩化メチル水銀標準液(Hg 10 g/L)1 mLを全量フラスコ100 mLにとり,
トルエンを標線まで加える。
10) 塩化エチル水銀標準液(Hg 1 mg/L)又は塩化メチル水銀標準液(Hg 1 mg/L) 塩化エチル水銀標
準液(Hg 100 mg/L)又は塩化メチル水銀標準液(Hg 100 mg/L)1 mLを全量フラスコ100 mLにと
り,トルエンを標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 50 mL及び500 mL。コックにワセリンなどの滑材を塗布しない。
2) 共栓試験管 5〜10 mL
3) マイクロシリンジ 1〜10 μL
4) ガスクロマトグラフ 次に挙げる条件を満たすもの。
4.1) 分離カラム ガラス製管(内径3 mm,長さ400〜1 500 mm)に4.2)のカラム充塡剤を詰めたもの
(19)。
4.2) カラム充塡剤 酸洗浄した後,シラン処理(20)を行った粒径180〜250 μmの耐火れんが(21)にエステ
ル系固定相液体5〜25 %を含浸させたもの。
4.3) 検出器 電子捕獲検出器又はこれと同等の性能をもつもの。
4.4) キャリヤーガス JIS K 1107に規定する窒素2級 流量30〜80 mL/min
4.5) 試料気化室湿度 140〜240 ℃
103
K 0102:2019
4.6) カラム槽温度 130〜180 ℃
4.7) 検出器槽温度 140〜200 ℃
4.8) 装置の感度 4.1)〜4.7)の条件下で塩化エチル水銀(又は塩化メチル水銀)を水銀(Hg)として40
pgを注入したときのS/N比が3以上とする。
注(19) キャピラリーカラムを用いてもよい。ただし,低濃度においてもアルキル水銀のハロゲン化物
の鋭いピークが得られるもの。キャリヤーガスはヘリウムを用い,流量は1〜3 mL/min程度と
する。
(20) ジクロロジメチルシランのトルエン溶液(体積百分率 1 %)中に担体を浸し,水浴上で約1時
間保った後,乾燥する。この処理をした担体が市販されている。また,あらかじめ担体に臭化
カリウム又は塩化ナトリウム(予期保持時間付近にピークを生じないもの。)5〜10 %を含浸さ
せた後,液層を被覆したものを用いると鋭いピークが得られる。
(21) 31.の注(4)による。
参考 カラム充塡剤には市販品がある。
c) 操作 操作は,次による。
1) 試料200 mLを分液漏斗500 mLにとり,中性でない場合は,アンモニア水(1+1)又は塩酸(1+1)
で中和した後,塩酸を加えて約2 mol/Lとする(22)。
2) トルエン50 mLを加え,約2分間激しく振り混ぜて放置した後,水層を別の分液漏斗500 mLに移
し,トルエン層は保存する。
3) 水層にトルエン50 mLを加えて約2分間激しく振り混ぜ,放置した後,水層を捨てる。
4) トルエン層を合わせ塩化ナトリウム溶液(200 g/L)20 mLを加え,約1分間激しく振り混ぜ,トル
エン層を洗浄し(23),放置後,水層を捨てる。
5) トルエン層にL-システイン-酢酸ナトリウム混合溶液8 mLを加え,約2分間激しく振り混ぜ,放置
後,水層を分液漏斗50 mLに移す。
6) 塩酸2 mLとトルエン5 mLとを加えて約2分間激しく振り混ぜ,放置後,水層を捨て,トルエン層
を共栓試験管に移す(24)。
7) マイクロシリンジを用い,トルエン層の一定量をガスクロマトグラフに注入し,ガスクロマトグラ
ムを記録する。
8) 塩化エチル水銀(II)又は塩化メチル水銀(II)の保持時間(25)に相当する位置のピークについて,
指示値(26)を読み取る(27)。
9) 先に測定に使用した共栓試験管内のトルエン層の残部1 mLを別の共栓試験管にとり,L-システイ
ン-酢酸ナトリウム混合溶液1 mLを加えて約2分間激しく振り混ぜ,放置する。
10) 上部のトルエン層から,先にガスクロマトグラフに注入したトルエン層と同量をマイクロシリンジ
を用いてガスクロマトグラフに注入する。この結果,先に得られたピークが消滅した場合には,先
のピークはエチル水銀(II)化合物又はメチル水銀(II)化合物によるものと判断する。
11) 空試験として水200 mLをとり,1)〜10)の操作を行って指示値を読み取り,試料について得た指示
値を補正する。
12) 検量線から水銀の量を求め,試料中のアルキル水銀(II)化合物の濃度を水銀の濃度(Hg μg/L)と
して算出する。
注(22) 試料中に硫化物イオン及びチオシアン酸イオンが含まれているときは,約2 mol/Lの塩酸酸性
とした試料にJIS K 8138に規定する塩化銅(I)の粉末100 mgを加えて十分にかき混ぜ,しば
104
K 0102:2019
らく放置する。沈殿をろ別し,ろ紙を塩酸(1+5)[a) 1)の塩酸を用いて調製する。]で2,3回
洗浄する。
注(23) 多量の無機水銀が存在する場合は,電子捕獲検出器を用いたとき,メチル水銀の位置に無機水
銀によるピークを生じることがあるので,洗浄を繰り返す。また,トルエン層に塩酸が残留す
るとL-システインによるアルキル水銀の逆抽出が不完全になるので,洗液が中性になるまで洗
浄を繰り返す。
(24) 水分が存在すると,ガスクロマトグラフに注入したとき異常ピークが生じることがあるので,
JIS K 8987に規定する硫酸ナトリウムなどを用いて脱水する。
(25) 操作において塩酸を使用するため,エチル水銀(II)化合物又はメチル水銀(II)化合物は,そ
れぞれ塩化エチル水銀(II)又は塩化メチル水銀(II)として挙動する。
(26) ピーク高さ又はピーク面積。
(27) 測定時に標準液の一定量を注入して,検出器の感度の経時変化を補正する。また,ガスクロマ
トグラフへの試料の注入量と得られる指示値との関係が直線になる範囲をあらかじめ求めてお
き,測定される指示値がこの範囲内となるように試料の注入量を調節する。
d) 検量線 検量線の作成は,次による。
1) 分液漏斗50 mLにL-システイン-酢酸ナトリウム混合溶液8 mLをとり,これに塩化エチル水銀標準
液(Hg 1 mg/L)又は塩化メチル水銀標準液(Hg 1 mg/L)を検出器の感度に応じて段階的に加え,
c)の5)〜8)及び11)の操作を行い,塩化エチル水銀(II)又は塩化メチル水銀(II)に相当する水銀
(Hg)の量と指示値との関係線を作成する。
備考 4. 試料中にアルキル水銀(II)化合物のトルエン抽出を妨害する成分が含まれている場合には,
試料に一定量の塩化エチル水銀又は塩化メチル水銀標準液を加えた後,c)の操作を行ってそ
の回収率を求め,定量値を補正する。
66.2.2 ガスクロマトグラフィー質量分析法 アルキル水銀(II)化合物をテトラフェニルほう酸ナトリウ
ムによって誘導体化(フェニル化)後,トルエンで抽出し,ガスクロマトグラフィー質量分析法を用いて
定量する。
試料は,ふっ素系樹脂製容器又はほうけい酸ガラス製容器に入れ,塩酸を加えてpHを約1.4として密栓
し,冷暗所に保存してできるだけ早く試験する。
定量範囲:Hg 0.2〜10 μg/L(試料換算)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。ただし,ガスクロマトグラフ質量分析計でアルキル水銀が検出
されないもの。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) 酢酸 JIS K 8355に規定するもの。ただし,ガスクロマトグラフ質量分析計でアルキル水銀が検出
されないもの。
4) 水酸化ナトリウム溶液(3 mol/L) JIS K 8576に規定する水酸化ナトリウム12 gを水に溶かして100
mLとする。ただし,ガスクロマトグラフ質量分析計でアルキル水銀が検出されないもの。
5) 酢酸緩衝液 全量フラスコ1 Lに水500 mLをとり,酢酸11.5 mL及び水酸化ナトリウム溶液(3
mol/L)42.5 mLを加えた後,水を標線まで加える。
6) テトラフェニルほう酸ナトリウム溶液(20 g/L) ガスクロマトグラフ分析用又は同等純度のテトラ
フェニルほう酸ナトリウム2 gを水に溶かして100 mLとする。使用時に調製する。
105
K 0102:2019
7) トルエン JIS K 8680に規定するもの。ただし,ガスクロマトグラフ質量分析計でアルキル水銀が
検出されないもの。
8) メタノール JIS K 8891に規定するもの。
9) 塩酸-酢酸希釈水 全量フラスコ1 Lに水500 mLをとり,塩酸2 mL及び酢酸5 mLを加えた後,水
を標線まで加える。
10) 硫酸ナトリウム JIS K 8987に規定するもの。
11) 塩化エチル水銀標準液(Hg 1 000 mg/L)又は塩化メチル水銀標準液(Hg 1 000 mg/L) 塩化エチル
水銀0.132 g又は塩化メチル水銀0.125 gを全量フラスコ100 mLにとり,メタノール又は塩酸-酢酸
希釈水を加え,緩やかに振り混ぜて溶解し,メタノール又は塩酸-酢酸希釈水を標線まで加える。
12) 塩化エチル水銀標準液(Hg 10 mg/L)又は塩化メチル水銀標準液(Hg 10 mg/L) 塩化エチル水銀
標準液(Hg 1 000 mg/L)又は塩化メチル水銀標準液(Hg 1 000 mg/L)1 mLを全量フラスコ100 mL
にとり,メタノール又は塩酸-酢酸希釈水を標線まで加える。
13) 塩化エチル水銀標準液(Hg 1 mg/L)又は塩化メチル水銀標準液(Hg 1 mg/L) 塩化エチル水銀標
準液(Hg 10 mg/L)又は塩化メチル水銀標準液(Hg 10 mg/L)1 mLを全量フラスコ10 mLにとり,
メタノール又は塩酸-酢酸希釈水を標線まで加える。
14) 2,4,6-トリクロロアニソール-d3内標準液(40 mg/L) 2,4,6-トリクロロアニソール-d3 10 mgを少量
のメタノールに溶かして,全量フラスコ10 mLに移し入れ,メタノールを標線まで加える。この溶
液をメタノール又はトルエンで25倍に希釈する。
15) 装置性能確認用フェニルアルキル水銀混合標準液 b) 1)の共栓平底フラスコ150 mLに水100 mLを
入れた後,マイクロシリンジを用いて塩化エチル水銀標準液(Hg 10 mg/L)又は塩化メチル水銀標
準液(Hg 10 mg/L)100 μLを壁面に接触させることなく加える(28)。酢酸緩衝液5 mLを加え,水酸
化ナトリウム溶液(3 mol/L)又は塩酸(1+1)でpHを5.0±0.1とした後(29),操作d)の2)〜5)を行
ってフェニル化されたアルキル水銀(II)化合物のトルエン溶液を調製する。この溶液を水銀濃度
として2〜200 μg/Lとなるようにトルエンで希釈する。この溶液は,ガスクロマトグラフ質量分析
計の性能(感度,検量線の直線性,分析対象成分の分離度など)を確認するために使用する。
16) PEG300溶液(100 g/L) ポリエチレングリコール(PEG)300 1 gを共栓試験管にとり,トルエン
を加えて10 mLとする。
注(28) フェニル誘導体化及びトルエン抽出操作において溶液と接触しない高さの内壁に付着した成分
は,誘導体化・抽出されない。これを防止するため,塩化エチル水銀標準液又は塩化メチル水
銀標準液は共栓平底フラスコ内壁に接触させずに直接水中に加える。
(29) pH測定に伴う汚染を避けるため,pH電極は溶液に浸さない。溶液の少量をパスツールピペッ
トで共栓試験管に分取しpHを測定する,又は極少量(例えば,0.1 mL)をパスツールピペット
でpHセンサー感応部に移しpHを測定する。
b) 器具及び装置 器具及び装置は,次による。
1) 共栓平底フラスコ 容量150〜200 mL,底部は回転子が安定に回転する形状であり,静置後のトル
エン層を効率よく回収するために,上部は内径が約1.5〜3.0 cmの細口であるもの。例えば,共栓三
角フラスコ,共栓短形メスフラスコ。
2) 共栓試験管 5〜10 mL
3) マイクロシリンジ 1〜100 μL
4) パスツールピペット ほうけい酸ガラス製

106
K 0102:2019
5) マグネチックスターラー 四ふっ化エチレン樹脂(PTFE)で被覆した回転子付きのものを用いる。
6) ガスクロマトグラフ質量分析計 次に挙げる条件を満たすもの。
6.1) キャピラリーカラム 内径0.2〜0.32 mm,長さ約25〜60 mの石英ガラス製,硬質ガラス製又は内
面を不活性処理したステンレス鋼製のキャピラリー管の内壁にジメチルポリシロキサン又はフェ
ニルメチルポリシロキサンを0.1〜3 μmの厚さで被覆したもの,又はこれと同等の分離性能をもつ
もので,アルキル水銀(II)化合物及び無機水銀(II)化合物のフェニル化体の分離が十分なもの。
6.2) キャリヤーガス ヘリウム[純度99.999 9 %(体積百分率)以上]。線速度は,20〜50 cm/sの範囲
に調節して用いる。
6.3) カラム槽温度 35〜300 ℃で0.5 ℃以内の温度調節の精度があり,昇温が可能なもの。
6.4) 試料注入方式 スプリットレス注入が可能なもの。
6.5) 試料注入口温度 280 ℃での設定が可能なもの。
6.6) インターフェース部温度 280 ℃での設定が可能なもの。
6.7) イオン化方式 電子イオン化(EI)
6.8) 電子加速電圧 70 Vでの設定が可能なもの。
6.9) イオン源温度 150〜280 ℃で機器の最適条件に設定する。
6.10) 検出方式 選択イオン検出(SIM)又は全イオン検出(TIM)が行え,所定の定量範囲に感度が調
節できるもの。
c) 準備操作 準備操作は,次による。
1) 装置性能確認用フェニルアルキル水銀混合標準液(30)及び2,4,6-トリクロロアニソール-d3内標準液
を用いて,ガスクロマトグラフ質量分析計の分析感度,検量線の直線性,分析対象成分の保持時間
などを確認する。
2) フェニルエチル水銀,フェニルメチル水銀,2,4,6-トリクロロアニソール-d3のマススペクトルから
定量イオン及び確認イオンを設定する。定量イオン及び確認イオンは,イオン強度の大きいもの,
実試料で妨害を受けないものから選定する。定量イオン及び確認イオンの例を表66.1に示す。
表66.1 アルキル水銀(II)化合物及び無機水銀(II)のフェニル化体,
内標準物質の定量イオン及び確認イオンの一例
水銀化合物
フェニル化後の
水銀化合物
フェニル化後の
分子式
定量イオン
m/z
確認イオン
m/z
メチル水銀(II)化合物
フェニルメチル水銀
CH3HgC6H5
200,202,217,
279,292,294
292,294
エチル水銀(II)化合物
フェニルエチル水銀
C2H5HgC6H5
200,202,231,
279,306,308
306,308
無機水銀(II)化合物
(参考)
ジフェニル水銀
C6H5HgC6H5
200,202,279,
354,356
354,356
2,4,6-トリクロロアニソール-d3
(内標準)
−
C7H2D3Cl3O
213,215
213,215
注(30) 標準液1 mLに対しPEG300溶液(100 g/L)を2 μLの割合で加えておく。PEG300はガスクロ
マトグラフの注入口及びキャピラリーカラムの活性点を不活化し,フェニル化アルキル水銀濃
度が低い領域における検量線の直線性及びピーク形状の改善に必要である。
d) 操作 操作は,次による。
107
K 0102:2019
1) 試料100 mLを共栓平底フラスコ150 mL(31)にとり,中性でない場合は,水酸化ナトリウム溶液(3
mol/L)又は塩酸(1+1)で中和した後,酢酸緩衝液5 mL及び2,4,6-トリクロロアニソール-d3内標
準液5 μLを加え(32),更に水酸化ナトリウム溶液(3 mol/L)又は塩酸(1+1)でpHを5.0±0.1と
する(29)。
2) テトラフェニルほう酸ナトリウム溶液(20 g/L)1 mLを加えて緩やかに振り混ぜた後,トルエン5 mL
を加える。
3) 回転子を入れて共栓をし,マグネチックスターラーで約60分間激しくかき混ぜた後,約10分間放
置する。
4) 水を共栓平底フラスコの内壁を伝わせながら緩やかに加え,トルエン層を細口部にまで上昇させた
後,パスツールピペットでトルエン層を共栓試験管に移す。
5) トルエン層に硫酸ナトリウム2 gを加えて振り混ぜ,水分を除く。
6) トルエン層から約1 mLをガスクロマトグラフ質量分析計用のバイアルに移し,PEG300溶液(100
g/L)2 μLを加えた後(30),マイクロシリンジを用いてトルエン層の一定量をガスクロマトグラフ質
量分析計に注入する。
7) フェニルエチル水銀,フェニルメチル水銀,2,4,6-トリクロロアニソール-d3の定量イオンの指示値
と確認イオンの指示値との比が,検量線作成時の指示値の比の±20 %以内であれば,試料中に存在
するとみなして,その定量イオンのクロマトグラムを記録する。
8) フェニルエチル水銀,フェニルメチル水銀,2,4,6-トリクロロアニソール-d3の保持時間(33)に相当す
る位置のピークについて,指示値(26)を読み取り,フェニルエチル水銀又はフェニルメチル水銀の指
示値と2,4,6-トリクロロアニソール-d3の指示値との比を求める。
9) 空試験として水100 mLをとり,1)〜8)の操作を行って指示値を読み取り,試料について得たフェニ
ルエチル水銀又はフェニルメチル水銀の指示値と2,4,6-トリクロロアニソール-d3の指示値との比を
補正する。
10) 検量線から水銀の量を求め,試料中のアルキル水銀(II)化合物の濃度を水銀の濃度(Hg μg/L)と
して算出する。
注(31) 中和操作等により液量が増加する場合には,適宜,他の容量のものを用いる。
(32) 2,4,6-トリクロロアニソール-d3内標準液は,塩化エチル水銀標準液,塩化メチル水銀標準液と
同様に,共栓平底フラスコ内壁に接触させずに直接溶液に加える。トルエン抽出操作において
溶液と接触しない高さの内壁に付着した成分は抽出されないためである。
(33) 操作においてフェニル化されるため,エチル水銀(II)化合物又はメチル水銀(II)化合物は,
それぞれフェニルエチル水銀又はフェニルメチル水銀として挙動する。また,無機水銀(II)化
合物は,ジフェニル水銀として挙動する。各々の保持時間が検量線作成時の保持時間に対して
±6秒以内であること確認する。
e) 検量線 検量線の作成は,次による。
1) 共栓平底フラスコ150 mLに水100 mLをとり,これに塩化エチル水銀標準液(Hg 1 mg/L)又は塩
化メチル水銀標準液(Hg 1 mg/L)20〜100 μL,塩化エチル水銀標準液(Hg 10 mg/L)又は塩化メチ
ル水銀標準液(Hg 10 mg/L)20〜100 μLを水銀濃度として0.2〜10 μg/Lとなるよう段階的に加える
(28)。これに酢酸緩衝液5 mL及び2,4,6-トリクロロアニソール-d3内標準液5 μLを加え(32),水酸化
ナトリウム溶液(3 mol/L)又は塩酸(1+1)でpHを5.0±0.1とした後(29),d)の2)〜8)の操作を行
う。
108
K 0102:2019
2) 別に,空試験として水100 mLをとり,これに酢酸緩衝液5 mL及び2,4,6-トリクロロアニソール-d3
内標準液5 μLを加え(32),水酸化ナトリウム溶液(3 mol/L)又は塩酸(1+1)でpHを5.0±0.1と
した後(29),d)の2)〜8)の操作を行い,標準液について得た指示値の比を補正し,塩化エチル水銀(II)
又は塩化メチル水銀(II)に相当する水銀(Hg)の量と指示値の比との関係線を作成する。検量線
の作成は,試料測定時に行う。
備考 5. 試料中にアルキル水銀(II)化合物の誘導体化又はトルエン抽出を妨害する溶存成分が含ま
れている場合には,別にとった試料に一定量の塩化エチル水銀又は塩化メチル水銀標準液を
加えた後,d)の操作を行ってその回収率を求め,定量値を補正する。
6. 試料中に懸濁物が含まれている場合には,別に取った試料に一定量の塩化エチル水銀又は塩
化メチル水銀標準液を加え,約1時間放置して吸着平衡に近づけた後,d)の操作を行ってそ
の回収率を求め,定量値を補正する。
7. 試料中に多量の細胞が含まれている場合には,ホモジナイザー等で細胞を破砕した後,d)の
操作を行う。また,細胞を破砕した後の試料を別にとり,一定量の塩化エチル水銀又は塩化
メチル水銀標準液を加え,約1時間放置して吸着平衡に近づけた後,d)の操作を行ってその
回収率を求め,定量値を補正する。
67.[セレン(Se)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との
整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,
ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”に置き
換える。
67.[セレン(Se)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
68.[モリブデン(Mo)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
68.[モリブデン(Mo)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
109
K 0102:2019
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
69.[タングステン(W)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
69.[タングステン(W)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
70.[バナジウム(V)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885
との整合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO
11885,ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”
に置き換える。
70.[バナジウム(V)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
73.[ウラン(U)]の“なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整
合を図ったものである。”を,“なお,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,
ICP質量分析法は,2016年に第2版として発行されたISO 17294-2との整合を図ったものである。”に置き
換える。
73.[ウラン(U)]の備考を,次に置き換える。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
110
K 0102:2019
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
73.[ウラン(U)]の後に74.[ベリリウム(Be)]を設け,次の文を追加する。
74. ベリリウム(Be) ベリリウムの定量には,ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,2007年に第2版として発行されたISO 11885,ICP質量分析法は,2016
年に第2版として発行されたISO 17294-2との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)(MOD)
ISO 17294-2:2016,Water quality−Application of inductively coupled plasma mass spectrometry
(ICP-MS)−Part 2: Determination of selected elements including uranium isotopes(MOD)
74.1 ICP発光分光分析法 52.4による。
74.2 ICP質量分析法 52.5による。
付表1(引用規格)の“JIS B 7411-1 一般用ガラス製温度計−第1部:一般計量器”を,“JIS B 7414 ガ
ラス製温度計”に置き換える。
付表1(引用規格)の“JIS K 0117 赤外分光分析方法通則”を,“JIS K 0117 赤外分光分析通則”に置
き換える。
付表1(引用規格)の“JIS K 8495 p-ジメチルアミノベンジリデンロダニン(試薬)”を,“JIS K 8495 5-
(4-ジメチルアミノベンジリデン)ロダニン(試薬)”に置き換える。
付表1(引用規格)の“JIS K 8905 七モリブデン酸六アンモニウム四水和物(試薬)”を,“JIS K 8905 モ
リブデン(VI)酸アンモニウム四水和物(試薬)”に置き換える。
付表1(引用規格)の“JIS T 9107 使い捨て手術用ゴム手袋”を,“JIS T 9107 単回使用手術用ゴム手袋”
に置き換える。
附属書1(補足)のX.(サリチル酸塩を用いるインドフェノール青吸光光度法)を削除する。
附属書1(補足)のXI.(イオン電極法によるアンモニウムイオン定量のための標準添加法)の全文を,次
に置き換える。
XI. イオン電極法によるアンモニウムイオン定量のための標準添加法 本体42.4のイオン電極法による
111
K 0102:2019
アンモニウムイオンの定量法において,ある種の排水試料の試験は,試料中の妨害物質の影響によって複
雑となる。その影響を最小にするため,また,疑わしい結果を確かめるために,次に示す標準添加法を用
いることができる。ただし,本体42.の備考14.で記載した妨害物質を含む試料の例のように,このような
影響が著しい場合は,試料の蒸留処理を行うとよい。
なお,この方法は,1984年に第1版として発行されたISO 6778を参考として記載した。
備考 ISO 6778:1984,Water quality−Determination of ammonium−Potentiometric method
− 本体42.4の備考12.に従って操作する。ただし,電位測定後,プローブは測定試料中に浸したままに
しておく。
− 測定試料の予想するアンモニウムイオンの濃度が50〜100 %増加する程度の量のアンモニウムイオン
標準液を,測定試料に加える。新しい電位及び添加したアンモニウムイオン標準液の体積を記録する。
次の試料を分析する前に,アンモニア電極を水で十分にすすぐ(1) (2) (3)。
注(1) 試料中のアンモニウムイオンの濃度が予測できないならば,添加するアンモニウムイオン標準
液の体積は,電位変化が少なくとも20 mVを示すのに十分な量とする。
(2) アンモニウムイオン標準液の添加量は,測定試料の希釈を最小にするためできるだけ少なくす
ることが望ましい。
(3) この操作は,プローブの応答が直線となる範囲においてだけ適用する。
− 測定結果の表現 アンモニウムイオンの濃度,ρN(mg/L)は,次の式によって示す。
1
)1
(
)
(
log
2
1
N2
N
−
+
−
=
k
S
E
E
anti
kρ
ρ
ここに, ρN2: 添加に使用したアンモニウムイオン標準液のアンモニウムイ
オンの濃度(NH4+ mg/L)
E1: プローブの最初の電位(mV)
E2: プローブの最終の電位(mV)
S: アンモニウムイオン濃度10倍の変化当たりのmVで表した検
量線の傾き(k)
0
1
V
V
k=
ここに,
V0: 測定試料の量(mL)
V1: 添加に用いた標準液の量(mL)
附属書1(補足)のXII.[ナトリウム(Na)のイオン電極法]の全文を,次に置き換える。
XII. ナトリウム(Na)のイオン電極法 試料のpHを10.2〜10.6に調節し,ナトリウムイオン電極を指
示電極として電位を測定し,ナトリウムを定量する。
定量範囲:Na 1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) トリス緩衝液 JIS K 9704に規定する2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール[トリス
(ヒドロキシメチル)アミノメタン]60 gをとり,水に溶かして1 Lとする。
2) ナトリウム標準液(Na 100 mg/L) 本体48.1 a) 1)のナトリウム標準液(Na 1 000 mg/L)20 mLを全
量フラスコ200 mLにとり,水を標線まで加える。
3) ナトリウム標準液(Na 10 mg/L) ナトリウム標準液(Na 100 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。
112
K 0102:2019
4) ナトリウム標準液(Na 1 mg/L) ナトリウム標準液(Na 10 mg/L)20 mLを全量フラスコ200 mLに
とり,水を標線まで加える。
b) 装置 装置は,次による。
1) 電位差計 本体34.2 b) 1)による。
2) 指示電極 ナトリウム電極[ガラス膜(1)電極]
3) 参照電極 本体34.2 b) 3)による。ただし,外筒液には硝酸アンモニウム溶液(100 g/L)(JIS K 8545
に規定する硝酸アンモニウムを用いて調製する。)又は硝酸カリウム溶液(100 g/L)(JIS K 8548に
規定する硝酸カリウムを用いて調製する。)を用いる。
4) 測定容器(セル) 本体34.2 b) 4)による。
5) 恒温槽 本体34.2 b) 5)による。
6) マグネチックスターラー 本体34.2 b) 6)による。
注(1) 液体膜のナトリウム電極を用いる場合は,備考3.による。
c) 検量線 検量線の作成は,次による。
1) ナトリウム標準液(Na 1 mg/L)100 mLを測定容器にとり,トリス緩衝液10 mL (2)を加える。
2) この測定容器(セル)のまま水浴を用いて,液温を一定温度(例えば,25±0.5 ℃)にする。
3) 水浴で液温が一定に保たれている測定容器(セル)に,指示電極(3) (4)と参照電極(5)とを浸し,固定
した後,回転子を入れ,マグネチックスターラー(6)を用いて,泡が電極に触れない程度に強くかき
混ぜながら(7),電位差計で電位を測定する(8)。
4) ナトリウム標準液(Na 10 mg/L)及びナトリウム標準液(Na 100 mg/L)100 mLを測定容器にとり,
トリス緩衝液10 mL (2)をそれぞれ加える。2)及び3)の操作を行ってナトリウム標準液(Na 10〜100
mg/L)の電位を測定する(8)。
5) 縦軸にナトリウムの濃度の対数を,横軸に電位(mV)をとって,ナトリウムの濃度(Na mg/L)と
電位との関係線を作成する(9) (10)。
注(2) トリス緩衝液は,測定時のpHを10.2〜10.6に調節し,イオン強度を一定にするためのもので
ある。
(3) ナトリウムイオン電極は,使用時にナトリウム標準液(Na l mg/L)に浸し,指針が安定してか
ら電位を測定する。
(4) 本体34.の注(5)による。
(5) 本体34.の注(6)による。
(6) 本体34.の注(8)による。
(7) 本体34.の注(9)による。
(8) ナトリウムイオン電極の応答時間は,液温10〜30 ℃の場合は2〜3分間である。
(9) ナトリウム標準液Na 1 mg/LとNa 100 mg/Lとの電位の差は,100〜120 mV(25 ℃)の範囲に
入り,ナトリウムの濃度1〜100 mg/Lの間の検量線は直線になる。
(10) 本体34.の注(12)による。
d) 操作 操作は,次による。
1) 試料100 mL(11)を測定容器(セル)にとり,トリス緩衝液10 mL(2)を加える。
2) c)の2)及び3)の操作を行って(10),検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度
(Na mg/L)を算出する。
3) 空試験として,試料の代わりに水を用いてc)の1)及び3)の操作を行って,試料について得たナトリ
113
K 0102:2019
ウムの濃度を補正する。
注(11) 試料が酸性又は強アルカリ性で,トリス緩衝液を加えてもpHが10.2〜10.6に入らない場合に
は,あらかじめ試料を全量フラスコ200 mLに入れ,これをアンモニア水(1+1)[本体52.1 a)
1)による。]又は塩酸(1+2)[本体58.1 a) 1)による。]で,pHを約10に調節し,水を標線まで
加える。これから100 mLをとり,1)以降の操作を行って測定したナトリウムの濃度(Na mg/L)
を希釈割合に応じて補正し,試料中のナトリウムの濃度(Na mg/L)を求める。
備考 1. イオン濃度計の場合には,ナトリウム標準液(Na 1 mg/L)とナトリウム標準液(Na 100 mg/L)
とを用いて,c)の1)〜3)の操作を行ってイオン濃度計の指針をそれぞれNa 1 mg/LとNa 100
mg/Lとになるように調節する。さらに,ナトリウム標準液(Na 10 mg/L)を用いてイオン濃
度計の指示値を確認する。
2. ナトリウムイオン電極(ガラス膜)における主な共存物質の許容限度を,最大比率で次に示
す。
Li+:4.5×10,K+:4×102,NH4+:1.3×103,Ag+:0.2
3. 液体膜のナトリウムイオン電極を用いる場合は,参照電極の外筒液に酢酸リチウム溶液(1
mol/L)又は酢酸リチウム緩衝液(酢酸リチウム二水和物102 gを水約500 mLに溶かし,こ
れにJIS K 8355に規定する酢酸2 mL及び水を加えて全量1 Lとする。pH6〜7になる。)を
用いる。検量線は,ナトリウム標準液100 mL当たり酢酸リチウム緩衝液10 mLを加え,pH
を6〜7に調節し,c) 2)以降の操作を行って作成する。測定は,検量線の作成と同じ条件にな
るように操作する。
この場合の共存物質の許容限度を,最大比率で次に示す。
K+:1.25×102Li+:6.25×103,NH4+:1.4×103,Ca2+:1.5×104
また,界面活性剤が共存するとドリフトを生じるので注意する。
附属書1(補足)のXIV.“銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP発光分光分析法”の題
名を,“銀(Ag)及びバリウム(Ba)のICP発光分光分析法”に変更し,全文を,次に置き換える。
XIV. 銀(Ag)及びバリウム(Ba)のICP発光分光分析法 試料を前処理した後,試料導入部を通して
誘導結合プラズマ中に噴霧し,これらの金属元素による発光を所定の波長で測定して定量する。
なお,この方法は,2007年に第2版として発行されたISO 11885を参考として記載した。
備考 ISO 11885:2007,Water quality−Determination of selected elements by inductively coupled plasma
optical emission spectrometry (ICP-OES)
1. 銀(Ag) 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銀による発光を
波長328.068 nm(又は338.289 nm)で測定して定量する。
定量範囲:Ag 20〜5 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 銀標準液(Ag 0.1 mg/mL) JIS K 8550に規定する硝酸銀0.157 5 gを,全量フラスコ1 000 mLにと
り,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調製する。)20 mLを加えた後,水を標線ま
で加える(1)。暗所で保存する。
注(1) 原子吸光用銀標準液(Ag 1 000 mg/L)を用いてもよい。
2) 銀標準液(Ag 10 μg/mL) 銀標準液(Ag 0.1 mg/mL)10 mLを全量フラスコ100 mLにとり,硝酸(1
114
K 0102:2019
+1)(JIS K 8541に規定する硝酸を用いて調製する。)2 mLを加えた後,水を標線まで加える。使
用時に調製する。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を本体5.によって処理する。
d) 操作 操作は,次による。
1) 本体52.4 d) 1)による。ただし,波長328.068 (I) nm[又は338.289 (I) nm]の発光強度を測定する(2) (3)
(4) (5)。
2) 本体52.4 d)の2)及び3)の操作を行い,試料中の銀の濃度(Ag μg/L)を算出する。
注(2) 本体52.の備考11.による。
(3) 本体47.の注(8)による。
(4) 本体47.の注(9)による。
(5) 表52.1の注参照。Iは中性線を示す。
e) 検量線 検量線の作成は,次による。
1) 銀標準液(Ag 10 μg/mL) 0.2〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き本体52.4 e)
1)〜3)の操作を行い,銀(Ag)の量と発光強度との関係線を作成する。検量線の作成は,試料測定
時に行う。
2. バリウム(Ba) 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,バリウム
による発光を波長233.527 nm(又は455.403,493.409 nm)で測定して定量する。
定量範囲:Ba 10〜5 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) バリウム標準液(Ba 0.1 mg/mL) JIS K 8565に規定する硝酸バリウム0.190 3 gを,全量フラスコ
1 000 mLにとり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調製する。)20 mLを加えた後,
水を標線まで加える(1)。
注(1) 原子吸光用バリウム標準液(Ba 1 000 mg/L)を用いてもよい。
2) バリウム標準液(Ba 10 μg/mL) バリウム標準液(Ba 0.1 mg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調製する。)2 mLを加えた後,水を標線ま
で加える。使用時に調製する。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を本体5.によって処理する。
d) 操作 操作は,次による。
1) 本体52.4 d)の1)による。ただし,波長233.527 (II) nm[又は455.403 (II),493.409 (II) nm]の発光強
度を測定する(2) (3) (4) (5)。
2) 本体52.4 d)の2)及び3)の操作を行い,試料中のバリウムの濃度(Ba μg/L)を算出する。
注(2) 本体52.の備考11.による。
(3) 本体47.の注(8)による。
(4) 本体47.の注(9)による。
115
K 0102:2019
注(5) 表52.1の注参照。IIはイオン線を示す。
e) 検量線 検量線の作成は,次による。
1) バリウム標準液(Ba 10 μg/mL) 0.1〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き本
体52.4 e)の1)〜3)の操作を行い,バリウム(Ba)の量と発光強度との関係線を作成する。検量線の
作成は,試料測定時に行う。
附属書1(補足)のXV.“水銀(Hg),銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP質量分析法”
の題名を,“水銀(Hg),銀(Ag)及びバリウム(Ba)のICP質量分析法”に変更し,全文を,次に置き
換える。
XV.
水銀(Hg),銀(Ag)及びバリウム(Ba)のICP質量分析法 試料を前処理した後,内標準元素を
加え,試料導入部を通して高周波プラズマ中に噴霧し,測定対象元素及び内標準元素のそれぞれの質量/
電荷数における指示値(1)を測定し,測定対象元素の指示値と内標準元素の指示値との比を求めて測定対象
元素を定量する。この方法によって附属書1表2に示す元素が同時定量できる。それぞれの元素ごとの定
量範囲,繰返し精度などの例を,附属書1表2に示す。
注(1) 本体52.の注(19)による。
附属書1表2 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
水銀(Hg)
0.5〜500
2〜10
202,200
銀(Ag)
0.5〜500
2〜10
107,109
バリウム(Ba)
0.5〜500
2〜10
137,138
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 水 本体52.3 a) 1)による。
2) 硝酸(1+1) 本体52.3 a) 2)による。
3) 内標準液(1 μg/mL) 本体52.5 a) 3)による(2)。
4) 水銀標準液(Hg 10 μg/mL) 本体66.1.1 a) 9)による(3)。
5) 水銀標準液(Hg 0.5 μg/mL) 水銀標準液(Hg 10 μg/mL)5 mLを全量フラスコ100 mLにとり,硝
酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
6) 銀標準液(Ag 10 μg/mL) 附属書1 XIV.の1. a) 2)による(3)。
7) 銀標準液(Ag 0.5 μg/mL) 銀標準液(Ag 10 μg/mL)5 mLを全量フラスコ100 mLにとり,硝酸(1
+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
8) バリウム標準液(Ba 10 μg/mL) 附属書1 XIV.の2. a) 2)による(3)。
9) バリウム標準液(Ba 0.5 μg/mL) バリウム標準液(Ba 10 μg/mL)5 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
10) 混合標準液[(Hg 10 μg,Ag 10 μg,Ba 10 μg)/mL](3) (4) 各元素の標準液(0.1 mg/mL)(5) 50 mL

116
K 0102:2019
[水銀の場合は,本体66.1.1 a) 8)水銀標準液(Hg 0.5 mg/mL)10 mL]を全量フラスコ500 mLにと
り,硝酸(1+1)10 mLを加えた後,水を標線まで加える。使用時に調製する。
11) 混合標準液[(Hg 0.5 μg,Ag 0.5 μg,Ba 0.5 μg)/mL](3) (4) 10)の混合標準液5 mLを全量フラス
コ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する。
注(2) 本体52.の注(20)による。
(3) 本体52.の注(21)による。
(4) 本体52.の注(16)による。
(5) 本体52.の注(22)による。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 1. 本体52.の備考13.による。
2. 本体52.の備考14.による。
c) 準備操作 準備操作は,本体52.5 c)による。
d) 操作 操作は,本体52.5 d)による(6)。
注(6) 本体52.の注(24)による。ただし,スペクトルの干渉の例は,附属書1表3による。
e) 検量線 検量線の作成は,本体52.5 e)による。
備考 3. 本体52.の備考16.による。
附属書1表3 スペクトル干渉の例*
元素
質量数
同重体及び2価
イオンの干渉
多原子イオンの
干渉
水銀(Hg)
200
184W16O
水銀(Hg)
202
銀(Ag)
107
91Zr16O
銀(Ag)
109
93Nb16O,92Zr16OH
バリウム(Ba)
137
バリウム(Ba)
138
La+,Ce+
イットリウム(Y)**
89
インジウム(In)**
115
Sn+
ビスマス(Bi)**
209
注*
装置及び測定条件によって異なる。
** 内標準元素
4. 水銀及び銀はメモリー効果が大きいため,これらの元素を含む試料及び標準液を導入した後
は,十分な洗浄を行って,次の測定値にメモリーの影響が出ないことを確認する。
附属書1(補足)のXVI.“薄層クロマトグラフ分離−原子吸光法”の“a) 11) 塩化エチル水銀標準液(Hg
1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL)”を,“a) 11) 塩化エチル水銀標準液(Hg 1 mg/L)又
は塩化メチル水銀標準液(Hg 1 mg/L)”に置き換える。
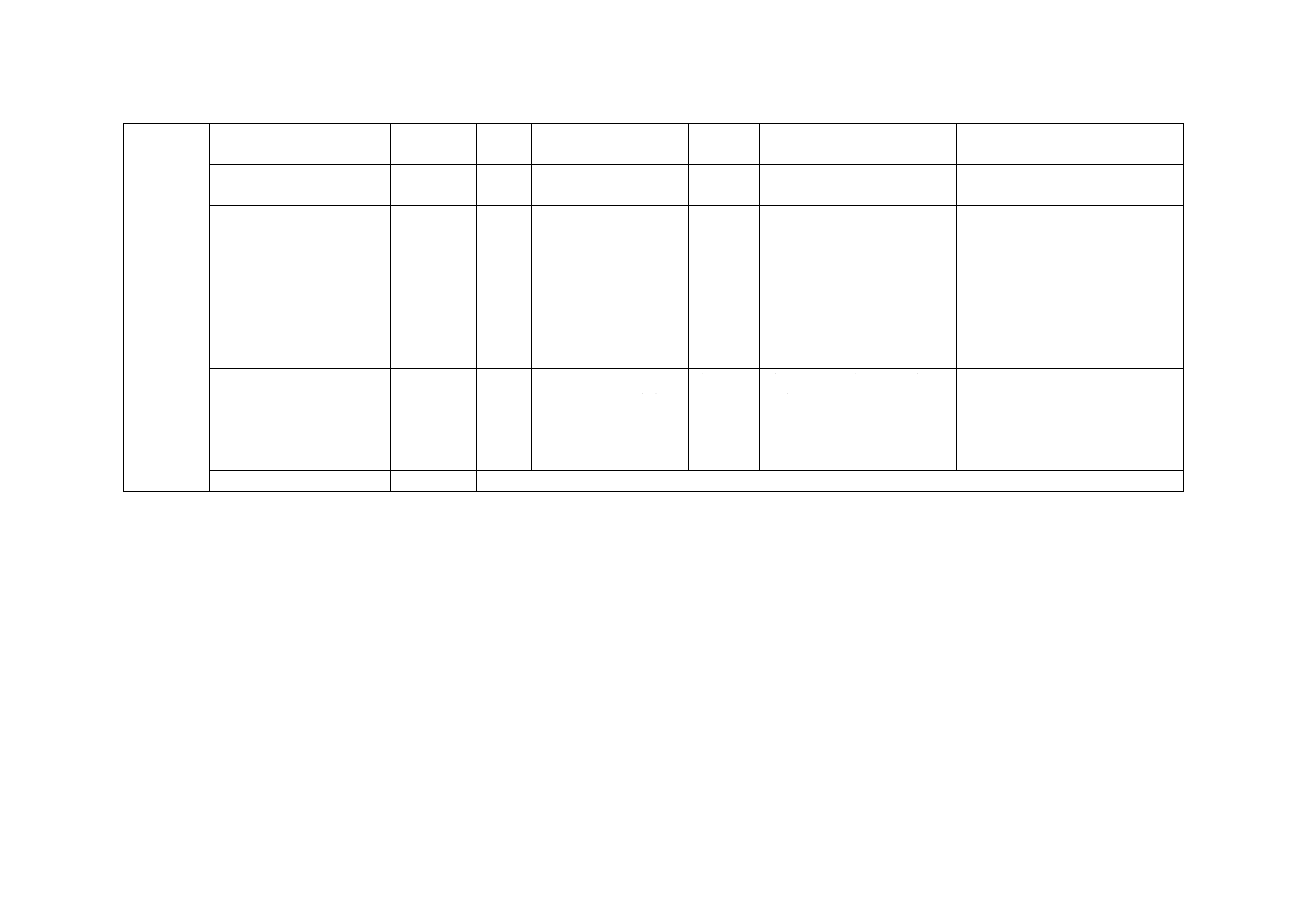
117
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の42.[アンモニウムイオン(NH4+)]に係る欄を,次に置き換える。
42. アンモ
ニウムイオ
ン(NH4+)
42.1 前処理(蒸留法)
ISO 5664
2〜9
蒸留後の中和滴定法
における蒸留操作
変更
11.2と同じ。
11.2と同じ。
42.2 インドフェノール青
吸光光度法
ISO 7150-1
NH4+の定量範囲は最
大1 mg
追加
JISのNH4+定量範囲は5〜
100 μg
強制法規で引用されている項目。
42.3 中和滴定法
ISO 5664
2〜9
蒸留後の中和滴定法
における中和滴定操
作
変更
JISは,塩酸標準液(滴定溶
液)を硫酸滴定溶液に変更。
塩酸標準液(滴定溶液)の濃度が
薄く,低濃度域での終点が明確に
判断できるか疑問であるため。
ISO規格見直し時に,修正提案を
検討する。
42.4 イオン電極法
ISO 6778
2〜10
電気プローブ法(イオ
ン電極法)
変更
JISは,ISO規格の測定時の
アルカリ性緩衝液の添加を
削除。
ISO規格見直し時に,修正提案を
検討する。
42.5 イオンクロマトグラ
フ法
ISO 14911
3〜13
イオンクロマトグラ
フ法を用いた溶存ア
ンモニウムイオンな
どの定量
変更
適用範囲から飲料水を除く。
対象イオンからリチウムイ
オン,マンガンイオン,スト
ロンチウムイオン及びバリ
ウムイオンを除いた。
11.2と同じ。
42.6流れ分析法
ISO 11732
JIS K 0170-1:2019附属書JA(参考)JISと対応国際規格との対比表を参照
1
3
K
0
1
0
2
:
2
0
1
9
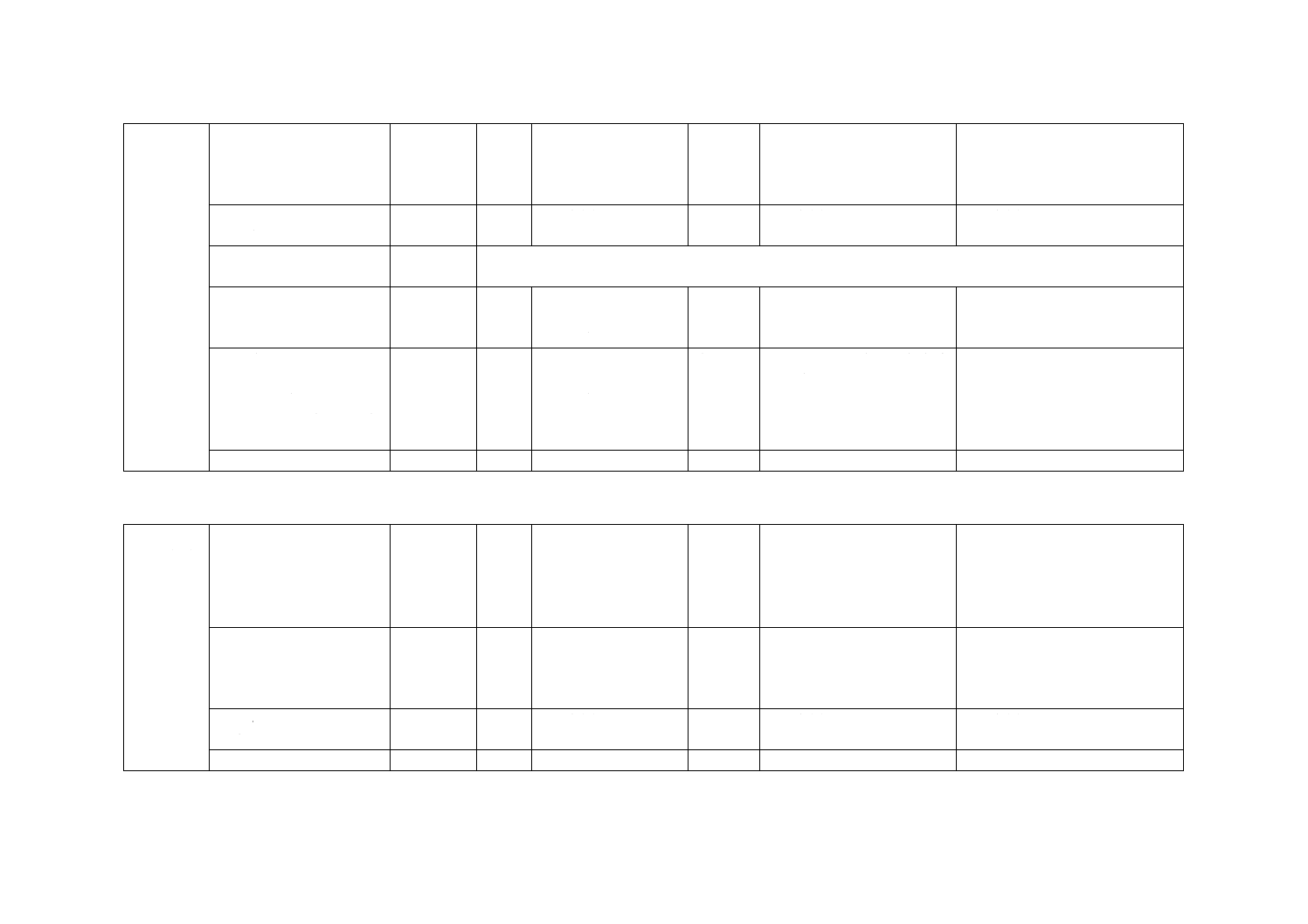
118
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の46.(りん化合物及び全りん)に係る欄を,次に置き換える。
46. りん化
合物及び
全りん
46.1 りん酸イオン
(PO43−)
46.1.1 モリブデン青吸光
光度法
ISO 6878
2〜8
りんの定量−モリブ
デン酸アンモニウム
吸光光度法
変更
11.2と同じ。
11.2と同じ。
46.1.3 イオンクロマトグ
ラフ法
ISO 10304-1
35.3と同じ。
変更
35.3と同じ。
35.3と同じ。
46.1.4 流れ分析法
ISO 15681-1
ISO 15681-2
JIS K 0170-4:2019附属書JA(参考)JISと対応国際規格との対比表を参照
46.2 加水分解性りん
ISO 6878
2〜8
りんの定量−モリブ
デン酸アンモニウム
吸光光度法
変更
JISは,ISO規格の分解条件
を削除。
ISO規格見直し時に,修正提案を
検討する。
46.3 全りん
46.3.1 ペルオキソ二硫酸
カリウム分解法
46.3.2 硝酸-過塩素酸分解
法
ISO 6878
2〜8
りんの定量−モリブ
デン酸アンモニウム
吸光光度法
変更
JISは,ISO規格の分解条件
を削除。
ISO規格見直し時に,修正提案を
検討する。
46.3.3 硝酸-硫酸分解法
−
−
−
追加
−
対応国際規格がない。
附属書2(JISと対応する国際規格との対比表)の(I)欄の48.[ナトリウム(Na)]に係る欄を,次に置き換える。
48. ナトリ
ウム(Na)
48.1 フレーム光度法
ISO 9964-3
3〜9
ナトリウム及びカリ
ウムの定量−第3部:
フレーム発光法によ
るナトリウム及びカ
リウムの定量
変更
11.2と同じ。
11.2と同じ。
48.2 フレーム原子吸光法
ISO 9964-1
3〜9
ナトリウム及びカリ
ウムの定量−第1部:
原子吸光法によるナ
トリウムの定量
変更
11.2と同じ。
11.2と同じ。
48.3 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
42.5と同じ。
42.5と同じ。
48.4 ICP発光分光分析法
ISO 11885
4〜12
50.3と同じ。
追加
50.3と同じ。
50.3と同じ。
1
3
K
0
1
0
2
:
2
0
1
9
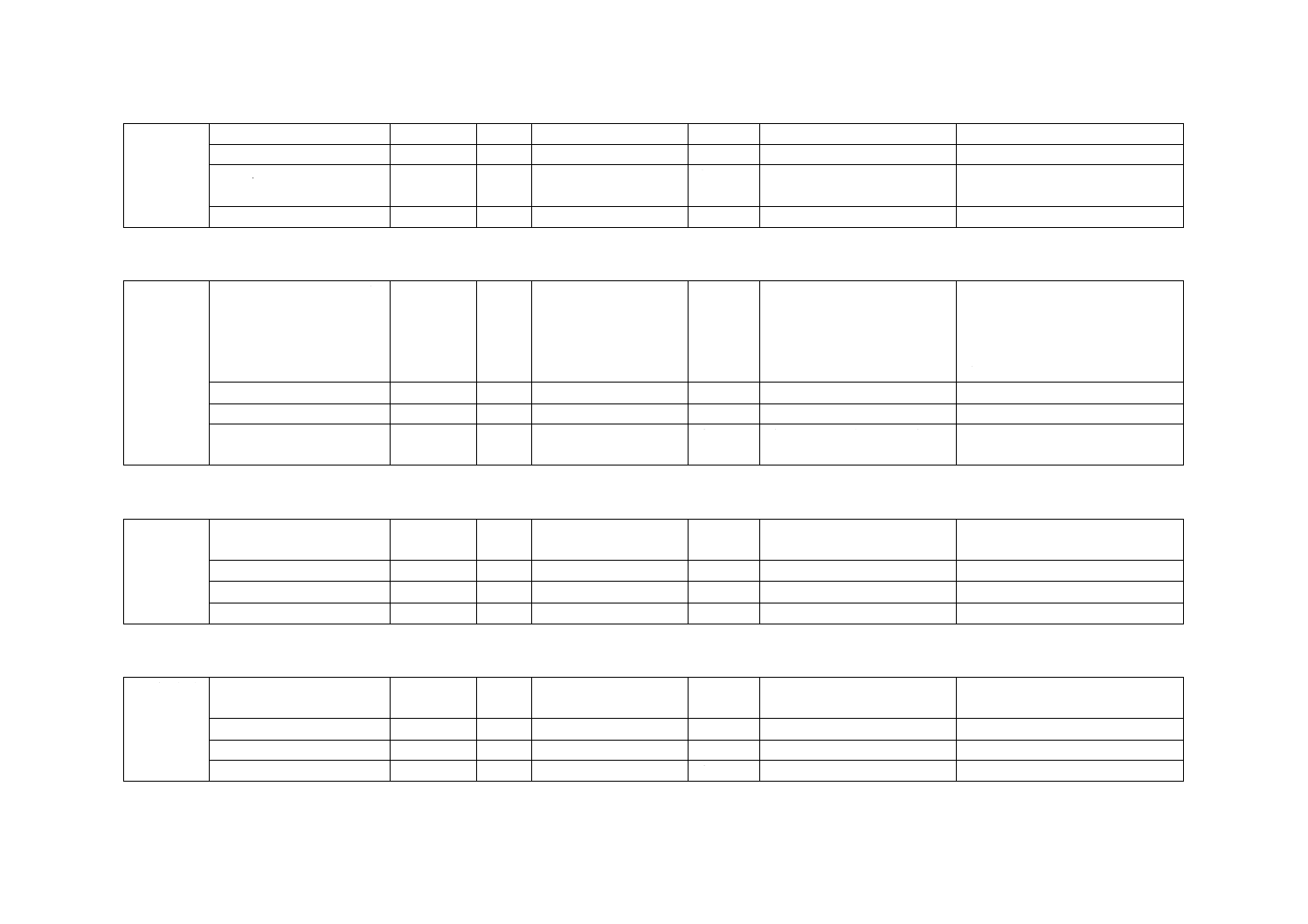
119
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の49.[カリウム(K)]に係る欄を,次に置き換える。
49. カリウ
ム(K)
49.1 フレーム光度法
ISO 9964-3
3〜9
48.1と同じ。
変更
11.2と同じ。
11.2と同じ。
49.2 フレーム原子吸光法
ISO 9964-1
3〜9
48.2と同じ。
変更
11.2と同じ。
11.2と同じ。
49.3 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
42.5と同じ。
42.5と同じ。
49.4 ICP発光分光分析法
ISO 11885
3〜12
50.3と同じ。
追加
50.3と同じ。
50.3と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の52.[銅(Cu)]に係る欄を,次に置き換える。
52. 銅(Cu) 52.2 フレーム原子吸光法
ISO 8288
A〜C
法
コバルト,ニッケル,
銅,亜鉛,カドミウム
及び鉛の定量−原子
吸光法
変更
JISは,ISO規格の混合標準
液の調製を削除。
原子吸光法は,個別に金属元素を
測定するものであり,混合標準液
を調製する必要性はない。
ISO規格見直し時に,修正提案を
検討する。
52.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
52.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
52.5 ICP質量分析法
ISO 17294-2 3〜12
ICP質量分析法を用い
た63元素の定量
追加
適用範囲から飲料水を除く。 強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の53.[亜鉛(Zn)]に係る欄を,次に置き換える。
53. 亜鉛
(Zn)
53.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
追加
52.2と同じ。
52.2と同じ。
53.2 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
53.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
53.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の54.[鉛(Pb)]に係る欄を,次に置き換える。
54. 鉛(Pb) 54.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
54.2 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
54.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
54.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
1
3
K
0
1
0
2
:
2
0
1
9
120
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の55.[カドミウム(Cd)]に係る欄を,次に置き換える。
55. カドミ
ウム(Cd)
55.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
ISO 5961
第2章 原子吸光法によるカ
ドミウムの定量
変更
JISは,ISO規格の試料の前
処理法を変更。
ISO規格見直し時に,修正提案を
検討する。
55.2 電気加熱原子吸光法
ISO 5961
第3章 原子吸光法によるカ
ドミウムの定量
変更
発熱体を用いた直接濃縮法
を削除。
ISO規格見直し時に,修正提案を
検討する。
55.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
55.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の56.[マンガン(Mn)]に係る欄を,次に置き換える。
56. マンガ
ン(Mn)
56.1 過よう素酸吸光光度
法
−
−
−
追加
−
対応国際規格がない。
56.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
56.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
56.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
56.5 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の58.[アルミニウム(Al)]に係る欄を,次に置き換える。
58. アルミ
ニ
ウ
ム
(Al)
58.1 キノリノール吸光光
度法
−
−
−
追加
−
対応国際規格がない。
58.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
58.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
58.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
58.5 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の59.[ニッケル(Ni)]に係る欄を,次に置き換える。
59. ニッケ
ル(Ni)
59.1 ジメチルグリオキシ
ム吸光光度法
−
−
−
追加
−
対応国際規格がない。
59.2 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
59.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
59.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
1
3
K
0
1
0
2
:
2
0
1
9
121
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の60.[コバルト(Co)]に係る欄を,次に置き換える。
60. コバル
ト(Co)
60.1 ニトロソR塩吸光光
度法
−
−
−
追加
−
対応国際規格がない。
60.2 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
60.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
60.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の61.[ひ素(As)]に係る欄を,次に置き換える。
61. ひ素
(As)
61.1 ジエチルジチオカル
バミド酸銀吸光光度法
−
−
−
追加
−
対応国際規格がない。
61.2 水素化物発生原子吸
光法
ISO 11969
3〜12
ひ素の水素化物発生
による原子吸光法
変更
11.2と同じ。
11.2と同じ。
61.3 水素化物発生ICP発
光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
61.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の62.[アンチモン(Sb)]に係る欄を,次に置き換える。
62. アンチ
モン(Sb)
62.1 ローダミンB吸光光
度法
−
−
−
追加
−
対応国際規格がない。
62.2 水素化物発生原子吸
光法
−
−
−
追加
−
対応国際規格がない。
62.3 水素化物発生ICP発
光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
62.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の63.[すず(Sn)]に係る欄を,次に置き換える。
63. すず
(Sn)
63.1 フェニルフルオロン
吸光光度法
−
−
−
追加
−
対応国際規格がない。
63.2 ケルセチン吸光光度
法
−
−
−
追加
−
対応国際規格がない。
63.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
63.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
1
3
K
0
1
0
2
:
2
0
1
9
122
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の64.[ビスマス(Bi)]に係る欄を,次に置き換える。
64. ビスマ
ス(Bi)
64.1 よう化物抽出吸光光
度法
−
−
−
追加
−
対応国際規格がない。
64.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
64.3 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の65.[クロム(Cr)]に係る欄を,次に置き換える。
65. クロム
(Cr)
65.1 全クロム
65.1.1 ジフェニルカルバ
ジド吸光光度法
−
−
−
追加
−
対応国際規格がない。
65.1.2 フレーム原子吸光
法
ISO 9174
3
全クロムの定量−原
子吸光法
変更
JISは,ISO規格の溶存クロ
ム及び塩化ランタンを不採
用。
当該試験方法は,強制法規に引用
されている。
ISO規格見直し時に,修正提案を
検討する。
65.1.3 電気加熱原子吸光
法
ISO 9174
4
全クロムの定量−電
子加熱原子吸光法
変更
JISは,ISO規格の標準添加
法を削除。
ISO規格見直し時に,修正提案を
検討する。
65.1.4 ICP発光分光分析法 ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
65.1.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
65.2 クロム(VI)[Cr
(VI)]
65.2.1 ジフェニルカルバ
ジド吸光光度法
ISO 11083
2〜9
クロム(VI)の定量−
1,5-ジフェニルカルバ
ジド吸光光度法
変更
JISは,ISO規格の試料の前
処理(凝集処理)を削除。
当該試験方法は,強制法規に引用
されている。
ISO規格見直し時に,修正提案を
検討する。
65.2.2 フレーム原子吸光
法
−
−
−
追加
−
対応国際規格がない。
65.2.3 電気加熱原子吸光
法
−
−
−
追加
−
対応国際規格がない。
65.2.4 ICP発光分光分析法 ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
65.2.5 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
65.2.6流れ分析法
ISO 23913
JIS K 0170-7:2019附属書JC(参考)JISと対応国際規格との対比表を参照
1
3
K
0
1
0
2
:
2
0
1
9
123
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の67.[セレン(Se)]に係る欄を,次に置き換える。
67. セレン
(Se)
67.1 3,3′-ジアミノベンジ
ジン吸光光度法
67.2 水素化合物発生原子
吸光法
−
−
−
追加
−
対応国際規格がない。
67.3 水素化合物発生ICP
発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
67.4 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
強制法規で引用されている項目。
附属書2(JISと対応する国際規格との対比表)の(I)欄の68.[モリブデン(Mo)]に係る欄を,次に置き換える。
68. モリブ
デン(Mo)
68.1 チオシアン酸吸光光
度法
−
−
−
追加
−
対応国際規格がない。
68.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
68.3 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の69.[タングステン(W)]に係る欄を,次に置き換える。
69. タング
ステン(W)
69.1 チオシアン酸吸光光
度法
−
−
−
追加
−
対応国際規格がない。
69.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
69.3 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の70.[バナジウム(V)]に係る欄を,次に置き換える。
70. バナジ
ウム(V)
70.1 N-ベンゾイル-N-フェ
ニルヒドロキシルアミン
吸光光度法
−
−
−
追加
−
対応国際規格がない。
70.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
70.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
70.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
70.5 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
1
3
K
0
1
0
2
:
2
0
1
9
124
K 0102:2019
附属書2(JISと対応する国際規格との対比表)の(I)欄の73.[ウラン(U)]に係る欄を,次に置き換える。
73. ウラン
(U)
73.1 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
73.2 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の(I)欄の73.[ウラン(U)]に係る欄の下に,次の欄を追加する。
74. ベリリ
ウム(Be)
74.1 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
74.2 ICP質量分析法
ISO 17294-2 3〜12
52.5と同じ。
追加
52.5と同じ。
11.2と同じ。
附属書2(JISと対応する国際規格との対比表)の附属書2付表1(対応国際規格)の“ISO 7027:1990”の下に,“ISO 7150-1:1984,Water quality−
Determination of ammonium−Part 1: Manual spectrometric method”を追加する。
附属書2(JISと対応する国際規格との対比表)の附属書2付表1(対応国際規格)の“ISO 10304-1:1992”の下に,“ISO 10304-1:2007,Water quality
−Determination of dissolved anions by liquid chromatography of ions−Part 1: Determination of bromide, chloride, fluoride, nitrate, nitrite, phosphate and sulfate”
を追加する。
附属書2(JISと対応する国際規格との対比表)の附属書2付表1(対応国際規格)の“ISO 11885:1996”の下に,“ISO 11885:2007,Water quality−
Determination of selected elements by inductively coupled plasma optical emission spectrometry (ICP-OES)”を追加する。
附属書2(JISと対応する国際規格との対比表)の附属書2付表1(対応国際規格)の“ISO 17289:2014”の下に,“ISO 17294-2:2016,Water quality−
Application of inductively coupled plasma mass spectrometry (ICP-MS)−Part 2: Determination of selected elements including uranium isotopes”を追加する。
1
3
K
0
1
0
2
:
2
0
1
9