K 0101 : 1998
(1)
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS K 0101 : 1991は改正され,この規格に置き換えられる。
JIS K 0101には,次に示す附属書がある。
附属書(参考) 補足
K 0101 : 1998
(1)
目次
ページ
1. 適用範囲 ························································································································ 1
2. 共通事項 ························································································································ 1
3. 試料 ······························································································································ 3
3.1 試料の採取,試料容器,採水器及び採取操作 ······································································· 3
3.2 試料の取扱い ················································································································ 3
3.3 試料の保存処理 ············································································································· 3
4. 試料の前処理 ·················································································································· 5
4.1 塩酸又は硝酸酸性で煮沸·································································································· 5
4.2 塩酸又は硝酸による分解·································································································· 5
4.3 硝酸と過塩素酸とによる分解 ···························································································· 5
4.4 硝酸と硫酸とによる分解·································································································· 6
4.5 フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する場合
の前処理 ····························································································································· 7
5. 結果の表示 ····················································································································· 7
6. 温度 ······························································································································ 7
6.1 気温 ···························································································································· 7
6.2 水温 ···························································································································· 8
7. 外観 ······························································································································ 9
8. 臭気及び臭気強度 (TON) ·································································································· 9
8.1 臭気 ···························································································································· 9
8.2 臭気強度 (TON) ············································································································ 10
9. 濁度 ····························································································································· 11
9.1 視覚濁度 ····················································································································· 11
9.2 透過光濁度 ·················································································································· 12
9.3 散乱光濁度 ·················································································································· 13
9.4 積分球濁度 ·················································································································· 15
10. 色 ······························································································································ 16
10.1 白金・コバルトによる色度 ···························································································· 16
10.2 刺激値Y及び色度座標x,yによる表示 ··········································································· 17
11. pH ······························································································································ 23
11.1 ガラス電極法 ·············································································································· 23
12. 電気伝導率 ·················································································································· 27
13. 酸消費量 ····················································································································· 30
13.1 酸消費量 (pH4.8) ········································································································· 30
13.2 酸消費量 (pH 8.3) ········································································································ 31
K 0101 : 1998 目次
(2)
ページ
14. アルカリ消費量 ············································································································ 32
14.1 アルカリ消費量 (pH 8.3) ······························································································· 32
14.2 アルカリ消費量 (pH4.8) ································································································ 34
14.3 アルカリ消費量(遊離酸) ···························································································· 34
15. 硬度 ··························································································································· 35
15.1 全硬度 ······················································································································· 35
15.1.1 キレート滴定法 ········································································································· 35
15.1.2 フレーム原子吸光法 ··································································································· 36
15.1.3 ICP発光分光分析法 ··································································································· 36
15.2 カルシウム硬度 ··········································································································· 36
15.2.1 キレート滴定法 ········································································································· 36
15.2.2 フレーム原子吸光法 ··································································································· 36
15.2.3 ICP発光分光分析法 ··································································································· 37
15.3 マグネシウム硬度 ········································································································ 37
15.3.1 キレート滴定法 ········································································································· 37
15.3.2 フレーム原子吸光法 ··································································································· 37
15.3.3 ICP発光分光分析法 ··································································································· 37
16. 懸濁物質及び蒸発残留物································································································· 37
16.1 懸濁物質 ···················································································································· 37
16.2 全蒸発残留物 ·············································································································· 39
16.3 溶解性蒸発残留物 ········································································································ 39
16.4 強熱残留物 ················································································································· 40
16.4.1 懸濁物質の強熱残留物 ································································································ 40
16.4.2 全蒸発残留物の強熱残留物 ·························································································· 40
16.4.3 溶解性蒸発残留物の強熱残留物 ···················································································· 40
16.5 強熱減量 ···················································································································· 40
17. 100℃における過マンガン酸カリウムによる酸素消費量 (CODMn)··········································· 41
18. 二クロム酸カリウムによる酸素消費量 (CODCr) ·································································· 43
19. 生物化学的酸素消費量 (BOD) ·························································································· 45
20. 有機体炭素 (TOC) ········································································································· 51
20.1 燃焼酸化-赤外線式TOC分析法 ······················································································· 51
20.2 燃焼酸化-赤外線式TOC自動計測法 ················································································· 53
21. 全酸素消費量 (TOD) ······································································································ 54
22. フェノール類及びp-クレゾール類 ···················································································· 56
22.1 フェノール類 ·············································································································· 56
22.1.1 前処理 ····················································································································· 56
22.1.2 4-アミノアンチピリン吸光光度法 ················································································· 57
22.2 p-クレゾール類 ··········································································································· 60
22.2.1 p-ヒドラジノベンゼンスルホン酸吸光光度法··································································· 60
K 0101 : 1998 目次
(3)
ページ
23. 界面活性剤 ·················································································································· 62
23.1 陰イオン界面活性剤 ····································································································· 62
23.1.1 メチレンブルー吸光光度法 ·························································································· 62
23.1.2 エチルバイオレット吸光光度法 ···················································································· 66
23.1.3 溶媒抽出-フレーム原子吸光法 ······················································································ 68
23.2 非イオン界面活性剤 ····································································································· 69
23.2.1 テトラチオシアナトコバルト (II) 酸吸光光度法 ······························································ 69
24. 溶存酸素 ····················································································································· 72
24.1 ウインクラー法 ··········································································································· 72
24.2 ウインクラー-アジ化ナトリウム変法 ··············································································· 75
24.3 ミラー変法 ················································································································· 77
24.4 隔膜電極法 ················································································································· 78
25. 全炭酸 ························································································································ 81
25.1 塩化ストロンチウム-塩酸滴定法 ····················································································· 81
25.2 赤外線分析法 ·············································································································· 84
26. ヘキサン抽出物質 ········································································································· 85
26.1 試料採取 ···················································································································· 85
26.2 抽出法 ······················································································································· 87
27. 欠番 ··························································································································· 89
28. 残留塩素 ····················································································································· 89
28.1 o-トリジン比色法 ········································································································ 89
28.2 ジエチル-p-フェニレンジアミン (DPD) 比色法 ·································································· 91
28.3 よう素滴定法 ·············································································································· 93
28.4 DPD-硫酸アンモニウム鉄 (II) 滴定法 ·············································································· 94
29. 塩素要求量 ·················································································································· 98
30. 水酸化物イオン (OH-) ··································································································· 100
31. ふっ素化合物 ·············································································································· 100
31.1 ランタン-アリザリンコンプレキソン吸光光度法 ······························································· 100
31.2 イオン電極法 ············································································································· 104
32. 塩化物イオン (Cl-) ······································································································· 106
32.1 チオシアン酸水銀 (II) 吸光光度法 ················································································· 106
32.2 硝酸水銀 (II) 滴定法 ··································································································· 107
32.3 硝酸銀滴定法 ············································································································· 108
32.4 イオン電極法 ············································································································· 109
32.5 イオンクロマトグラフ法 ······························································································ 111
33. よう化物イオン (I-) ······································································································ 113
33.1 よう素抽出吸光光度法 ································································································· 114
33.2 よう素滴定法 ············································································································· 115
34. 臭化物イオン (Br-) ······································································································· 116
K 0101 : 1998 目次
(4)
ページ
34.1 よう素滴定法 ············································································································· 116
34.2 イオンクロマトグラフ法 ······························································································ 118
35. シアン化合物 ·············································································································· 118
35.1 前処理 ······················································································································ 119
35.1.1 シアン化物 ·············································································································· 119
35.1.1.1 通気法(pH 5.0で発生するシアン化水素) ·································································· 119
35.1.1.2 加熱蒸留法(pH5.5で酢酸亜鉛の存在下で発生するシアン化水素) ·································· 121
35.1.2 全シアン(pH 2以下で発生するシアン化水素) ····························································· 123
35.2 4-ピリジンカルボン酸-ピラゾロン吸光光度法 ··································································· 124
35.3 イオン電極法 ············································································································· 126
36. アンモニウムイオン (NH4+) ··························································································· 128
36.1 前処理 ······················································································································ 128
36.1.1 凝集沈殿法 ·············································································································· 128
36.1.2 蒸留法 ···················································································································· 129
36.2 インドフェノール青吸光光度法 ····················································································· 131
36.3 中和滴定法 ················································································································ 132
36.4 イオン電極法 ············································································································· 133
36.5 イオンクロマトグラフ法 ······························································································ 135
37. 亜硝酸イオン (NO2-) 及び硝酸イオン (NO3-) ····································································· 138
37.1 亜硝酸イオン (NO2-)···································································································· 138
37.1.1 ナフチルエチレンジアミン吸光光度法 ·········································································· 138
37.1.2 イオンクロマトグラフ法 ···························································································· 139
37.2 硝酸イオン (NO3-)······································································································· 140
37.2.1 還元蒸留-インドフェノール青吸光光度法 ······································································ 140
37.2.2 還元蒸留-中和滴定法 ································································································· 142
37.2.3 銅・カドミウムカラム還元−ナフチルエチレンジアミン吸光光度法 ··································· 143
37.2.4 ブルシン吸光光度法 ·································································································· 146
37.2.5 イオンクロマトグラフ法 ···························································································· 147
38. 有機体窒素 ················································································································· 148
38.1 前処理(ケルダール法) ······························································································ 148
38.2 インドフェノール青吸光光度法 ····················································································· 149
38.3 中和滴定法 ················································································································ 150
39. 全窒素 ······················································································································· 151
39.1 総和法 ······················································································································ 151
39.2 紫外線吸光光度法 ······································································································· 152
39.3 硫酸ヒドラジニウム還元法 ··························································································· 154
39.4 銅・カドミウムカラム還元法 ························································································ 156
39.5 熱分解法 ··················································································································· 158
40. 硫化物イオン (S2-) ········································································································ 159
K 0101 : 1998 目次
(5)
ページ
40.1 メチレンブルー吸光光度法 ··························································································· 159
40.2 よう素滴定法 ············································································································· 161
41. 亜硫酸イオン (SO32-)····································································································· 164
41.1 よう素滴定法 ············································································································· 164
42. 硫酸イオン (SO42-)········································································································ 166
42.1 クロム酸バリウム−ジフェニルカルバジド吸光光度法 ······················································· 166
42.2 クロム酸バリウム吸光光度法 ························································································ 168
42.3 重量法 ······················································································································ 169
42.4 イオンクロマトグラフ法 ······························································································ 170
43. りん化合物及び全りん··································································································· 171
43.1 りん酸イオン (PO43-) ··································································································· 171
43.1.1 モリブデン青(アスコルビン酸還元)吸光光度法 ··························································· 171
43.1.2 モリブデン青[塩化すず (II) 還元]吸光光度法 ····························································· 173
43.2 加水分解性りん ·········································································································· 175
43.3 全りん ······················································································································ 176
43.3.1 ペルオキソ二硫酸カリウム分解法 ················································································ 176
43.3.2 硝酸−過塩素酸分解法 ······························································································· 179
43.3.3 硝酸-硫酸分解法 ······································································································· 180
44. シリカ (SiO2) ·············································································································· 181
44.1 イオン状シリカ ·········································································································· 181
44.1.1 モリブデン黄吸光光度法 ···························································································· 181
44.1.2 モリブデン青吸光光度法 ···························································································· 182
44.1.3 モリブデン青抽出吸光光度法 ······················································································ 183
44.2 溶存及びコロイド状シリカ ··························································································· 184
44.3 全シリカ ··················································································································· 185
44.3.1 炭酸ナトリウムによる融解 ························································································· 185
44.3.2 重量法 ···················································································································· 186
45. ほう素 (B) ·················································································································· 187
45.1 メチレンブルー吸光光度法 ··························································································· 187
45.2 アゾメチンH吸光光度法 ····························································································· 188
45.3 ICP発光分光分析法 ····································································································· 189
46. ひ素 (As)···················································································································· 190
46.1 ジエチルジチオカルバミド酸銀吸光光度法 ······································································ 190
46.2 水素化物発生原子吸光法 ······························································································ 194
46.3 水素化物発生ICP発光分光分析法 ·················································································· 197
47. ナトリウム (Na) ·········································································································· 198
47.1 フレーム光度法 ·········································································································· 198
47.2 フレーム原子吸光法 ···································································································· 199
47.3 イオン電極法 ············································································································· 199
K 0101 : 1998 目次
(6)
ページ
47.4 イオンクロマトグラフ法 ······························································································ 201
48. カリウム (K) ··············································································································· 202
48.1 フレーム光度法 ·········································································································· 202
48.2 フレーム原子吸光法 ···································································································· 203
48.3 イオンクロマトグラフ法 ······························································································ 203
49. カルシウム (Ca) ··········································································································· 204
49.1 キレート滴定法 ·········································································································· 204
49.2 フレーム原子吸光法 ···································································································· 205
49.3 ICP発光分光分析法 ····································································································· 206
50. マグネシウム (Mg) ······································································································· 207
50.1 キレート滴定法 ·········································································································· 207
50.2 フレーム原子吸光法 ···································································································· 208
50.3 ICP発光分光分析法 ····································································································· 209
51. 銅 (Cu) ······················································································································ 210
51.1 ジエチルジチオカルバミド酸吸光光度法 ········································································· 210
51.2 フレーム原子吸光法 ···································································································· 212
51.3 電気加熱原子吸光法 ···································································································· 213
51.4 ICP発光分光分析法 ····································································································· 214
51.5 ICP質量分析法··········································································································· 216
52. 亜鉛 (Zn)···················································································································· 218
52.1 フレーム原子吸光法 ···································································································· 218
52.2 電気加熱原子吸光法 ···································································································· 219
52.3 ICP発光分光分析法 ····································································································· 220
52.4 ICP質量分析法··········································································································· 221
53. カドミウム (Cd) ·········································································································· 222
53.1 フレーム原子吸光法 ···································································································· 222
53.2 電気加熱原子吸光法 ···································································································· 224
53.3 ICP発光分光分析法 ····································································································· 225
53.4 ICP質量分析法··········································································································· 226
54. ニッケル (Ni) ·············································································································· 227
54.1 ジメチルグリオキシム吸光光度法 ·················································································· 227
54.2 フレーム原子吸光法 ···································································································· 229
54.3 ICP発光分光分析法 ····································································································· 230
55. すず (Sn) ···················································································································· 231
55.1 フェニルフルオロン吸光光度法 ····················································································· 231
55.2 ケルセチン吸光光度法 ································································································· 233
55.3 ICP発光分光分析法 ····································································································· 234
56. 鉛 (Pb) ······················································································································· 235
56.1 フレーム原子吸光法 ···································································································· 235
K 0101 : 1998 目次
(7)
ページ
56.2 電気加熱原子吸光法 ···································································································· 236
56.3 ICP発光分光分析法 ····································································································· 237
56.4 ICP質量分析法··········································································································· 238
57. 水銀 (Hg) ··················································································································· 239
57.1 還元気化原子吸光法 ···································································································· 239
57.2 加熱気化原子吸光法 ···································································································· 243
58. マンガン (Mn) ············································································································· 245
58.1 過よう素酸吸光光度法 ································································································· 245
58.2 フレーム原子吸光法 ···································································································· 246
58.3 電気加熱原子吸光法 ···································································································· 247
58.4 ICP発光分光分析法 ····································································································· 248
58.5 ICP質量分析法··········································································································· 249
59. アルミニウム (Al) ········································································································ 251
59.1 キノリノール吸光光度法 ······························································································ 251
59.2 フレーム原子吸光法 ···································································································· 253
59.3 電気加熱原子吸光法 ···································································································· 254
59.4 ICP発光分光分析法 ····································································································· 255
60. 鉄 (Fe) ······················································································································· 256
60.1 フェナントロリン吸光光度法 ························································································ 256
60.2 フレーム原子吸光法 ···································································································· 258
60.3 電気加熱原子吸光法 ···································································································· 259
60.4 ICP発光分光分析法 ····································································································· 260
61. クロム (Cr) ················································································································· 261
61.1 全クロム ··················································································································· 261
61.1.1 ジフェニルカルバジド吸光光度法 ················································································ 262
61.1.2 フレーム原子吸光法 ·································································································· 264
61.1.3 電気加熱原子吸光法 ·································································································· 265
61.1.4 ICP発光分光分析法 ·································································································· 266
61.1.5 ICP質量分析法 ········································································································ 267
61.2 クロム (VI) [Cr (VI)] ···································································································· 268
61.2.1 ジフェニルカルバジド吸光光度法 ················································································ 268
61.2.2 フレーム原子吸光法 ·································································································· 269
61.2.3 電気加熱原子吸光法 ·································································································· 269
61.2.4 ICP発光分光分析法 ·································································································· 270
61.2.5 ICP質量分析法 ········································································································ 270
62. バナジウム (V) ············································································································ 271
62.1 N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法 ··················································· 271
62.2 フレーム原子吸光法 ···································································································· 272
62.3 電気加熱原子吸光法 ···································································································· 273
K 0101 : 1998 目次
(8)
ページ
62.4 ICP発光分光分析法 ····································································································· 274
63. 細菌試験 ···················································································································· 275
63.1 試料の採取及び細菌の捕集 ··························································································· 275
63.2 一般細菌 ··················································································································· 276
63.3 従属栄養細菌 ············································································································· 278
63.4 大腸菌群 ··················································································································· 280
63.5 ふん便性大腸菌群 ······································································································· 282
64. 生物試験 ···················································································································· 283
64.1 生物試験 ··················································································································· 283
64.2 細菌類 ······················································································································ 285
64.3 藻類 ························································································································· 289
64.4 動物 ························································································································· 289
附属書(参考) 補足 ········································································································· 291
I. 透視度 ························································································································· 291
II. アルカリ性過マンガン酸カリウムによる酸素消費量 (CODOH) ················································ 292
III. 陽イオン界面活性剤 ····································································································· 294
IV. 油類··························································································································· 296
V. 炭化水素及び動植物油脂類 ······························································································ 297
VI. よう化物イオンのイオン電極法 ······················································································ 299
VII. 臭化物イオンのイオン電極法 ························································································ 301
VIII. 硝酸イオンのイオン電極法 ·························································································· 302
IX. 硫化物イオンのイオン電極法 ························································································· 304
X. 硫酸イオンの硫酸バリウム比濁法 ···················································································· 306
付表1 引用規格 ················································································································ 307
日本工業規格 JIS
K 0101 : 1998
工業用水試験方法
Testing methods for industrial water
1. 適用範囲 この規格は,工業用水の試験方法について規定する。
備考 この規格の引用規格を付表1に示す。
2. 共通事項 共通事項は,次のとおりとする。
(1) 通則 化学分析に共通する一般事項は,JIS K 0050による。
(2) 定義 この規格で用いる主な用語の定義は,JIS K 0102,JIS K 0211又はJIS K 0215による。
また,誘導結合プラズマ質量分析法は,以下,ICP質量分析法という。
(3) ガスクロマトグラフ法 ガスクロマトグラフ法に共通する一般事項は,JIS K 0114による。
(4) 吸光光度法 吸光光度法に共通する一般事項は,JIS K 0115による。
(5) 誘導結合プラズマ発光分光分析法 誘導結合プラズマ発光分光分析法(以下,ICP発光分光分析法と
いう。)に共通する一般事項は,JIS K 0116による。
(6) 赤外分光法 赤外分光法に共通する一般事項は,JIS K 0117による。
(7) 原子吸光法 原子吸光法には,フレーム原子吸光法,電気加熱方式原子吸光法(以下,電気加熱原子
吸光法という。)及びその他の原子吸光法がある。これらに共通する一般事項は,JIS K 0121による。
(8) イオン電極法 イオン電極法に共通する一般事項は,JIS K 0122による。
(9) イオンクロマトグラフ法 イオンクロマトグラフ法に共通する一般事項は,JIS K 0127による。
(10) 定量範囲 それぞれの試験方法に示してある定量範囲は,最終溶液中の質量(mg,μg又はng)で示
す。ただし,原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオンクロマト
グラフ法,イオン電極法,有機体炭素 (TOC) ,全酸素消費量 (TOD) ,溶存酸素及び残留塩素の試験
方法においては,最終溶液中の濃度(mg/l又はμg/l)で示す。
(11) 繰返し分析精度 繰返し分析精度は,標準液についてそれぞれの試験方法の定量範囲内で,繰り返し
試験によって求めた変動係数 (%) で示す(1)。
注(1) 変動係数(%)=
100
×
x
σ
ここに,
σ: 標準偏差
x: 平均値
(12) 水 この規格で用いる水は,JIS K 0557に規定するA1〜A4の水とするが,項目中で規定されている
場合には,それに従う。
(a) 溶存酸素を含まない水 使用時にJIS K 0557に規定するA2〜A3の水をフラスコに入れ,約5分間
煮沸して溶存酸素を除去した後,図2.1のようにアルカリ性ピロガロール溶液(2)を入れたガス洗浄
瓶を連結して,空気中の酸素と遮断して放冷する。又は煮沸する代わりにJIS K 1107に規定する高
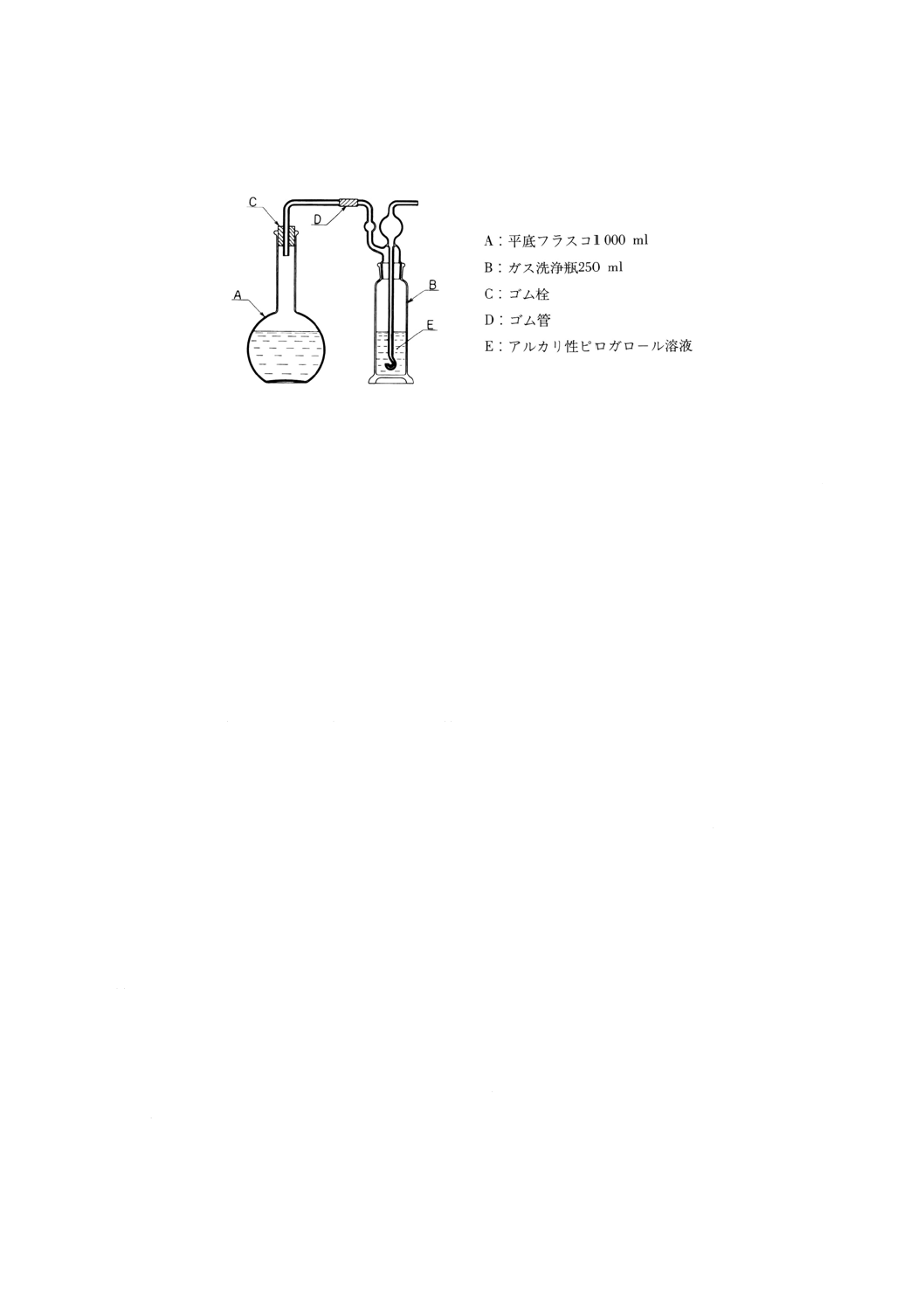
2
K 0101 : 1998
純度窒素2級を約15分間通気して溶存酸素を除去してもよい。
図2.1 溶存酸素を含まない水の冷却,保存の一例
(b) 炭酸を含まない水 JIS K 0557に規定するA2〜A3の水をフラスコに入れ,約5分間沸騰させて溶
存気体及び炭酸を除去した後,図2.1と同様の装置を用い,ガス洗浄瓶に水酸化カリウム溶液
(250g/l) を入れ,空気中の二酸化炭素を遮断して放冷する。
注(2) JIS K 8780に規定するピロガロール(1, 2, 3‐ベンゼントリオール)6gを水50mlに溶かし,着
色瓶に保存する。別に,JIS K 8574に規定する水酸化カリウム30gを水50mlに溶かす。使用時
に両液を混合する。この溶液1mlは,酸素約12ml(約17mg)を吸収する。
(13) 試薬
(a) 試薬は,品目指定されている場合には,JISマーク表示品の最上級品質のものを用い,JISマーク表
示品がない場合には,試験に支障のないものを用いる(3)。
滴定液類の標定には,JIS K 8005に規定する容量分析用標準物質を用いる。
(b) 試薬類の溶液の濃度は,特に断らない限り質量濃度はg/l又はmg/l,モル濃度はmol/l又はmmol/l
で示す。
なお,化合物については無水物としての質量を用いる。
標準液の濃度は,イオン電極法及びフレーム光度法以外は,1ml中の質量(mg/ml又はμg/ml)で
表す。
(c) 試薬類の溶液名称の後に括弧で示されている濃度は,標準液以外は概略の濃度であることを意味す
る。例えば,水酸化ナトリウム溶液 (0.1mol/l) は約0.1mol/lの水酸化ナトリウム溶液であることを
示す。
また,溶液名の前に示される濃度は,正確な濃度を意味する。ただし,一般には,端数のない数
値で示し,別にファクターを求めておく。
(d) 試薬類の調製に用いる水は,(12)の水とするが,それぞれの項目中で規定されている場合には,そ
れに従う。
(e) 標準液を薄めて低濃度の標準液を調製する場合には,特に断らない限り10ml以上の全量ピペット
でとる。
(f) 試薬類の名称は,特に断らない限り国際純正及び応用化学連合 (IUPAC) の無機化学命名法及び有
機化学命名法によった社団法人日本化学会が定めた化合物命名法及びJIS試薬の名称に整合させる。
(g) 試薬類及び廃液などの取扱いについては,関係法令規則などに従い十分に注意する。
注(3) 電気加熱原子吸光法,ICP質量分析法など,ごく微量の試験には,特に高純度の試薬を用いる。
(14) 器具類 この規格で用いるガラス器具類,磁器るつぼ,磁器蒸発皿及びろ紙は,次のとおりとする。
3
K 0101 : 1998
(a) ガラス器具は,特に断らない限りJIS R 3503,及びJIS R 3505に規定するものを使用する。ただし,
特殊な器具を必要とする場合には,それぞれの項目に,その一例を図示又は説明する。
また,加熱操作を伴う場合には,JIS R 3503に規定するほうけい酸ガラス-1を用いる。
デシケーターに用いる乾燥剤は,特に断らない限りシリカゲル(4)とする。
(b) 磁器るつぼ及び磁器蒸発皿は,JIS R 1301及びJIS R 1302に規定するものを使用する。
(c) ろ紙は,JIS P 3801に規定する定量分析用を使用する。ただし,ろ紙の種類は,それぞれの項目で
規定する。
注(4) JIS Z 0701に規定する包装用シリカゲル乾燥剤A形1種を用いる。
備考 シリカ,ほう素,ナトリウム,カリウム,ひ素,亜鉛などを試験する場合には,ほうけい酸ガ
ラスからのこれらの成分の溶出に十分に注意する。
(15) 吸光度の測定(吸光光度法) 吸収セルについて特に記載がない場合には,光路長が10mmのものを
用いる。
(16) 検量線[吸光光度法,原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオン
クロマトグラフ法,イオン電極法,有機体炭素 (TOC) ,全酸素消費量 (TOD) ] 検量線の作成に
当たっては,試験方法に示される定量範囲内を4〜6段階に分け,これに一致するように標準液をとる。
検量線は定量範囲内について作成する。
原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオンクロマトグラフ法,
イオン電極法,有機体炭素 (TOC) 及び全酸素消費量 (TOD) の試験においては,試験に際して新たに
作成した検量線を用い,同一項目を多数の試料について連続して試験する場合には,試験の途中にお
いて,適宜,標準液を用いて指示値の確認を行う。
吸光光度法においては,あらかじめ作成した検量線を用いることができる。
(17) 注,備考,図,表及び式 注,備考,図,表及び式は,各項目ごとに一連番号を付ける。
3. 試料
3.1
試料の採取,試料容器,採水器及び採取操作 試料とは試験を行うために採取した水をいう。試料
の採取,試料容器,採水器及び採取操作は,JIS K 0094に従う。
3.2
試料の取扱い 試験は,特に断らない限り試料中に含まれる全量について行う。このため,試料に
懸濁物がある場合には,十分に振り混ぜて均一にした後,試料を採取して試験に用いる。ただし,陰イオ
ンの試験では,特に断らない限りろ過した試料を用いる。全量を求める場合には,それぞれの項目で規定
する。
その他,溶存状態のものだけを試験する場合には,試料採取後,直ちにろ紙5種C(1)でろ過し,初めの
ろ液約50mlを捨て,その後のろ液を試料とする。
注(1) ろ紙6種又は孔径1μm以下のろ過材を用いてもよい。
3.3
試料の保存処理 試験は,特に断らない限り試料採取後直ちに行う。直ちに試験ができずに保存す
る場合は,JIS K 0094の7.(試料の保存処理)に従って,次のように行い,なるべく早く試験する。冷所
に保存する場合には,凍結させないようにする。
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 塩酸(ひ素分析用) JIS K 8180に規定するもの。
(c) 硝酸 JIS K 8541に規定するもの。
4
K 0101 : 1998
(d) 硫酸 JIS K 8951に規定するもの。
(e) りん酸 JIS K 9005に規定するもの。
(f) L (+) ‐アスコルビン酸 JIS K 9502に規定するもの。
(g) 水酸化ナトリウム溶液 (200g/l) JIS K 8576に規定する水酸化ナトリウム20gを水に溶かして
100mlとする。
(h) 塩基性炭酸亜鉛懸濁液 JIS K 8953に規定する硫酸亜鉛七水和物20gを水に溶かし,これと等体積
の炭酸ナトリウム溶液 (100g/l) と混合する。使用時に調製する。
(i) 硫酸銅 (II) 五水和物 JIS K 8983に規定するもの。
(j) クロロホルム JIS K 8322に規定するもの。
(2) 保存処理 保存処理は,次のとおり行う。
(a) 100℃における過マンガン酸カリウムによる酸素消費量 (CODMn) ,二クロム酸カリウムによる酸素
消費量 (CODCr) ,生物化学的酸素消費量 (BOD) ,有機体炭素 (TOC) ,全酸素消費量 (TOD) 及
び界面活性剤の試験に用いる試料は,0〜10℃の暗所に保存する。
(b) アンモニウムイオン,有機体窒素及び全窒素の試験に用いる試料は,塩酸又は硫酸を加え,pHを2
〜3に調節し,0〜10℃の暗所に保存する。短い日数であれば,保存処理を行わずそのままの状態で
0〜10℃の暗所に保存してもよい。
(c) 亜硝酸イオン及び硝酸イオンの試験に用いる試料は,試料1lにつきクロロホルム約5mlを加えて0
〜10℃の暗所に保存する。短い日数であれば,保存処理を行わずそのままの状態で0〜10℃の暗所
に保存してもよい。
(d) よう化物イオン,臭化物イオンの試験に用いる試料は,水酸化ナトリウム溶液 (200g/l) を加えてpH
を約10にして保存する(試料1lにつき水酸化ナトリウム2〜4粒を加えてもよい。)。
(e) シアン化合物及び硫化物イオンの試験に用いる試料は,水酸化ナトリウム溶液 (200g/l) を加えて
pHを約12にして保存する(試料1lにつき水酸化ナトリウム4〜6粒を加えてもよい。)。シアン化
合物の試験に用いる試料で,残留塩素など酸化性物質が共存する場合は,L (+) -アスコルビン酸を
加えて還元した後,pHを約12とする。
また,硫化物イオンの場合には,試料を溶存酸素測定瓶に採取し,塩基性炭酸亜鉛懸濁液を試料
100mlにつき約2mlを加え,硫化亜鉛として固定し保存してもよい。
(f) フェノール類の試験に用いる試料は,りん酸を加えてpHを約4にし,試料1l当たり硫酸銅 (II) 五
水和物1gを加えて振り混ぜ,0〜10℃の暗所に保存する。
(g) りん化合物及び全りんの試験に用いる試料は,そのままの状態で試料1lにつきクロロホルム約5ml
を加えて0〜10℃の暗所に保存する。この場合,短い日数であれば,保存処理を行わずにそのまま
の状態で0〜10℃の暗所に保存してもよい。ただし,溶存りん化合物の試験に用いる試料は,3.2に
よってろ過した後,試料1lにつきクロロホルム約5mlを加えて0〜10℃の暗所に保存する。この場
合,短い日数であれば,保存処理を行わずそのままの状態で0〜10℃の暗所に保存してもよい。
全りんの試験に用いる試料は,硫酸又は硝酸を加えてpHを約2にして保存してもよい。
(h) 銅,亜鉛,鉛,カドミウム,マンガン,鉄,アルミニウム,ニッケル,コバルト,ひ素,すず,全
クロム,水銀,バナジウムなどの金属元素の試験に用いる試料は,硝酸を加えてpHを約1にして
保存する。
ひ素の試験に用いる試料で,有機物や多量の硝酸塩,亜硝酸塩を含まず,試験に際して硫酸と硝
酸又は硝酸と過マンガン酸カリウムによる処理を行わない場合には,塩酸(ひ素分析用)を加えて
5
K 0101 : 1998
pHを約1にして保存する。
クロム (VI) の試験に用いる試料は,そのままの状態で0〜10℃の暗所に保存する。
溶存状態の金属元素の試験に用いる試料は,3.2によって試料をろ過した後,硝酸を加えてpHを
約1にして保存する。
4. 試料の前処理 試料の前処理操作は,各試験項目で規定するが,金属元素の試験における前処理操作
は,金属元素の種類に関係なく共通するものがほとんどであるため,一括して以下に規定する。ただし,
金属元素のうちナトリウム,カリウム,カルシウム,マグネシウム,ひ素,クロム (VI) ,水銀などの試
験の前処理は,それぞれの試験項目において規定する。
金属元素試験での前処理は,主として共存する有機物,懸濁物及び金属錯体の分解を目的としている。
前処理には,試料に各種の酸を加えて加熱する方法を用いるが,試料の状態や試験の種類によって適当
な方法を選択する。
4.1
塩酸又は硝酸酸性で煮沸 この方法は,有機物や懸濁物が極めて少ない試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 硝酸 JIS K 8541に規定するもの。
(2) 操作 操作は,次のとおり行う。
(a) 試料(1)100mlにつき塩酸5ml又は硝酸5mlを加える。
(b) 加熱して約10分間静かに沸騰させる。
(c) 放冷後,必要に応じて水で一定量にする。
注(1) 溶存状態の金属元素を試験する場合には,3.2によってろ過した試料を用いる。
4.2
塩酸又は硝酸による分解 この方法は,有機物が少なく,懸濁物として水酸化物,酸化物,硫化物,
りん酸塩などを含む試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 硝酸 JIS K 8541に規定するもの。
(2) 操作 操作は,次のとおり行う。
(a) 試料(2)をよく振り混ぜた後,直ちにその適量をビーカーにとり,試料100mlにつき塩酸5ml又は硝
酸5mlを加える。
(b) 加熱して液量が約15mlになるまで濃縮する。
(c) 不溶解物が残った場合には,ろ紙5種Bでろ過した後,水でよく洗浄する。
(d) 放冷後,ろ液と洗液を適当な容量の全量フラスコに移し入れ,水を標線まで加える。
注(2) 溶存状態の金属元素を試験する場合には,3.2に従ってろ過した試料を用い,4.1の方法を用いる。
備考 塩酸と硝酸の混酸による分解が有利な試料の場合には,(b)までの操作を行った後,室温まで放
冷する。(a)で塩酸を使用したときは硝酸5mlを,硝酸を使用したときは塩酸5mlを加え,時計
皿で覆い再び加熱し,激しい反応が終わったら時計皿を取り除き,更に加熱して窒素酸化物を
追い出し,約5mlになるまで濃縮する。この操作で酸が不足している場合は,適量の塩酸及び
硝酸を加えて同じ操作で加熱し溶解する。不溶解物が残った場合は,温水15mlを加え,(c)及
び(d)の操作を行う。
4.3
硝酸と過塩素酸とによる分解 この方法は,酸化されにくい有機物を含む試料に適用する。
6
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 過塩素酸 JIS K 8223に規定するもの。
(b) 硝酸 JIS K 8541に規定するもの。
(2) 操作 操作は,次のとおり行う。
(a) 試料(2)をよく振り混ぜ,直ちにその適量をビーカー又は磁器蒸発皿にとる。
(b) 硝酸5〜10mlを加え,加熱板上で静かに加熱して約10ml(3)になるまで濃縮し,放冷する。
(c) 硝酸5mlを加え,過塩素酸(4)10mlを少量ずつ加え,加熱を続け,過塩素酸の白煙が発生し始めたら,
時計皿で容器を覆い,過塩素酸が器壁を流下する状態に保って有機物を分解する。
(d) 有機物が分解しないで残ったときは,更に,硝酸5mlを加えて(c)の操作を繰り返し,有機物を分解
する。
(e) 放冷後,水を加えて液量を約50mlに薄め,不溶解物が残った場合には,ろ紙5種Bを用いてろ過
し,水で洗い,ろ液と洗液を適当な容量の全量フラスコに移し入れ,水を標線まで加える。
注(3) ケルダールフラスコに移して分解してもよい。
(4) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこと
に注意する。
i)
酸化されやすい有機物は,過塩素酸を加える前に,(b)の硝酸を加え,加熱する操作によって
十分に分解しておく。
ii) 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
iii) 必ず過塩素酸と硝酸を共存させた状態で加熱分解を行う。
iv) 濃縮液を乾固させない。
4.4
硝酸と硫酸とによる分解 この方法は,多種類の試料に適用(5)することができる。
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 JIS K 8541に規定するもの。
(b) 硫酸 (1+1) 水1容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加える。
(2) 操作 操作は,次のとおり行う。
(a) 試料(2)をよく振り混ぜ,直ちにその適量をビーカー又は磁器蒸発皿にとり,硝酸5〜10mlを加える。
(b) 加熱して,液量が約10ml(3)になったら,再び硝酸5mlと硫酸 (1+1) 10mlとを加え,硫酸の白煙が
発生し,有機物が分解するまで加熱する。
(c) 有機物の分解が困難なときは,更に,硝酸10mlを加えて(b)の操作を繰り返し,有機物を分解する。
(d) 放冷後,水で液量を約50mlに薄める。不溶解物(6)が残った場合には,ろ紙5種Bを用いてろ過し,
水で洗い,ろ液と洗液を適当な容量の全量フラスコに移し入れ,水を標線まで加える。
注(5) 水溶液をそのまま噴霧するフレーム原子吸光法を適用する場合には,好ましくない。
(6) 鉛が含まれていて沈殿を生じる場合には,4.3又は次の操作を行う。
(b)の操作を行って溶液をほとんど蒸発乾固し,水約30mlと塩酸15mlとを加えて加熱して溶
かす。不溶解物がある場合には,ろ紙5種Bを用いてろ過した後,温塩酸 (1+10) で洗浄する。
放冷後,ろ液と洗液を適当な容量の全量フラスコに移し入れ,水を標線まで加える。
7
K 0101 : 1998
4.5
フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する場
合の前処理 試料に含まれている有機物及び懸濁物の量,その存在状態及び適用しようとする原子吸光法,
ICP発光分光分析法又はICP質量分析法などの方法を十分に考慮して4.1〜4.4に示した方法のうち最適な
ものを選択して前処理する(7)(8)。
調製した試料をそのまま噴霧するフレーム原子吸光法又はICP発光分光分析法を適用する場合には,特
に断らない限り試料は塩酸又は硝酸酸性(9),電気加熱原子吸光法又はICP質量分析法を適用する場合には,
硝酸酸性とし,適当な濃度(10)に調節する。
注(7) フレーム原子吸光法又はICP発光分光分析法に先立って溶媒抽出法を適用する場合の前処理は,
原則として各項目のとおりとし,妨害する可能性のある有機物その他の妨害物質を十分に分解
する。
試料をそのまま噴霧するフレーム原子吸光法又はICP発光分光分析法を行う場合には,次の
ように前処理を行ってもよい。
有機物及び懸濁物が極めて少ない試料の場合は,4.1の操作を行う。有機物及び懸濁物を含む
試料の一般的な前処理法としては,4.3又は4.4を適用する。この場合,白煙を十分に発生させ
て大部分の硫酸及び過塩素酸を除去しておく。
ICP質量分析法の場合は,酸の種類と濃度によって空試験値が無視できないことがあるので,
測定する元素についてあらかじめ酸の種類と濃度の影響について調べておく。
いずれの前処理方法を適用するかは,試料に一定量の目的成分を添加して回収試験を行い,
その結果に基づいて判断するとよい。
(8) 2.の注(3)による。高純度の試薬には,JIS K 9901に規定する高純度試薬‐硝酸,JIS K 9902に
規定する高純度試薬‐塩酸,JIS K 9904に規定する高純度試薬-過塩素酸,JIS K 9905に規定す
る高純度試薬‐硫酸などがある。
(9) ICP発光分光分析法の場合,硫酸酸性では試料導入量が少なく感度が悪くなることがあるから,
4.4の適用は止むを得ない場合だけとする。
(10) フレーム原子吸光法及び電気加熱原子吸光法の場合には,0.1〜1mol/l,ICP発光分光分析法にお
いては,すず (Sn) を対象としない場合には,0.1〜0.5mol/lとする。すず (Sn) を対象とする場
合には,1〜1.5mol/lとする。また,ICP質量分析法の場合には,0.1〜0.5mol/lとする。ただし,
いずれの場合も検量線作成時の場合とほぼ同じ濃度とする。
5. 結果の表示 試験方法が二つ以上ある場合には,試験方法を付記する。
6. 温度 気温と水温に分け,試料採取時に測定する。
6.1
気温 気温は,次によって測定する。
(1) 器具 器具は,次のとおりとする。
(a) 温度計 JIS B 7411に規定する一般用ガラス製棒状温度計の50度温度計。
(2) 操作 操作は,次のとおり行う。
(a) 温度計を採水現場において直射日光及び周囲の強い熱放射を避けた風通しのよい場所で,地上1.2
〜1.5mの位置に保ち,感温液の止まるときの目盛を読み取る。
備考1. 測定期間内の最高と最低の温度の測定には,最高最低温度計(図6.1参照)を使用する。
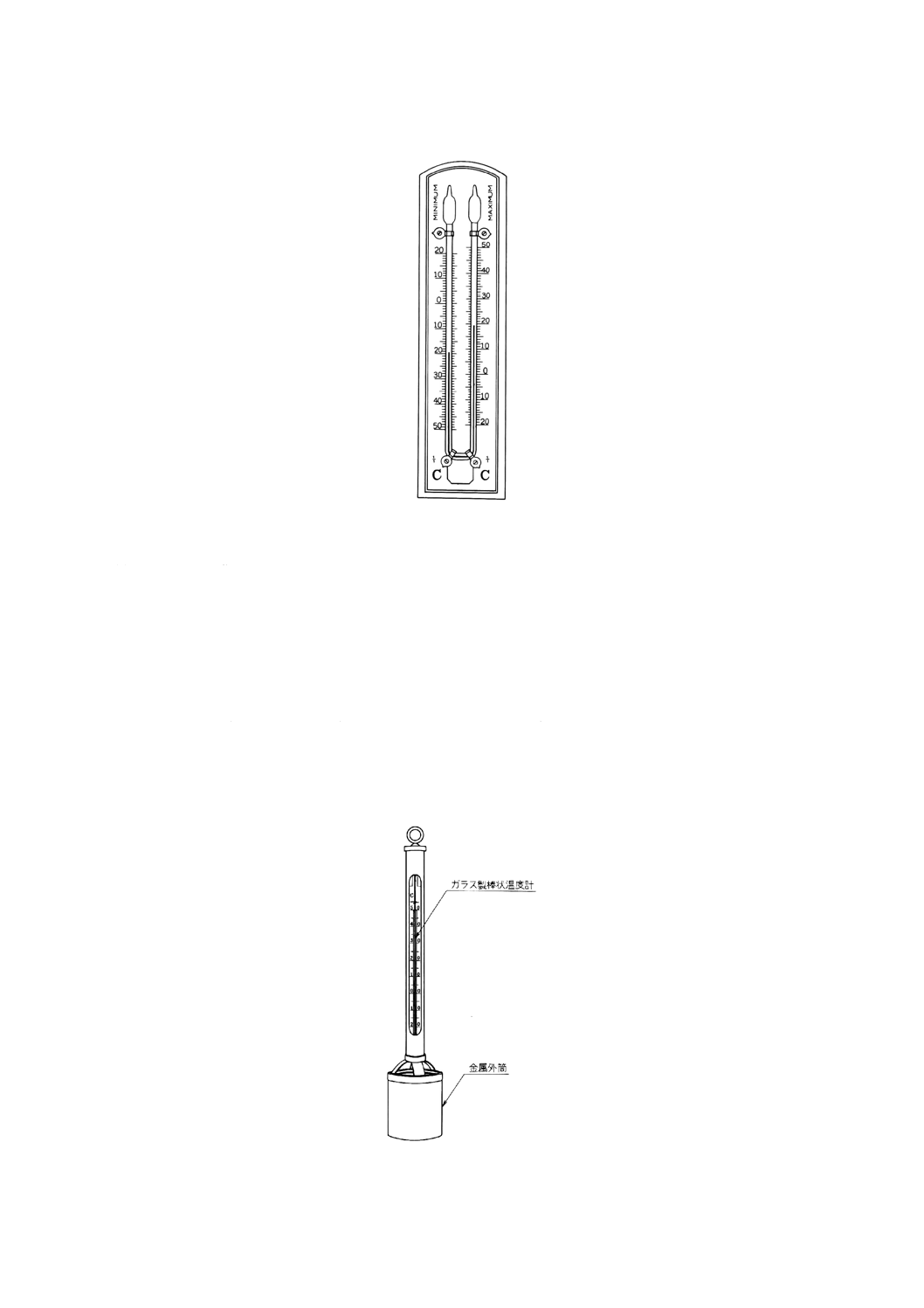
8
K 0101 : 1998
図6.1 最高最低温度計の一例
6.2
水温 水温は,次によって測定する。
(1) 器具 器具は,次のとおりとする。
(a) 温度計 JIS B 7411に規定する一般用ガラス製棒状温度計の50度温度計又は100度温度計
(2) 操作 操作は,次のとおり行う。
(a) 温度計を現場の水に直接差し入れるか,採取直後の試料(1)の中に差し入れて感温液の止まる目盛付
近まで浸没した状態に保ち,感温液の止まるときの目盛を読み取る。
注(1) 容器及び外気の温度の影響を避けるため,多量の試料を採取する。
備考2. ペッテンコーヘル水温計(図6.2参照)を用いる場合には,金属筒内に試料を3回入れ替えた
後,試料を満たし,感温液の止まるときの目盛を読み取る。
3. サーミスタ温度計及び金属抵抗温度計は,温度検出部を測定する水中に保ち,指示部の指針
が一定したときの目盛を読み取る。
図6.2 ペッテンコーヘル水温計

9
K 0101 : 1998
7. 外観 外観は,採取直後の試料について観察する。
(1) 器具 器具は,次のとおりとする。
(a) ビーカー 300〜500ml(無色のもの)
(2) 操作 操作は,次のとおり行う。
(a) 採水直後の試料をビーカーにとり,次の事項について肉眼で観察する。
i)
試料全体の色の種類と程度
ii)
上澄み液の色の種類と程度
iii)
浮上物質,懸濁物などの色の種類と量の程度
iv)
油類,タール類などの状態と程度
v)
その他,試料の泡立ち及び臭気など特異な状態
8. 臭気及び臭気強度 (TON) 臭気の試験は,臭気と臭気強度 (TON) (1)とに区分する。
水の臭気は,細菌,藻類,微生物などの繁殖及び死滅,都市下水,畜舎排水,工場排水の混入,貯水槽
及び配管系統の内面処理物質の溶出,塩素処理による残留塩素などの影響による。
臭気の試験は,きゅう覚によるので,個人差が大きく,さらに,温度及び湿度,測定者の食事及び喫煙
などにも影響される。
注(1) TONは,Threshold odor numberの略称で,臭気いき(閾)値の希釈倍数,すなわち,明らかに
臭気を感じるときの希釈の倍数値である。
8.1
臭気 試料を約40℃に温め,臭気の種類とその程度を試験する。
(1) 器具 器具は,次のとおりとする。
(a) 共栓三角フラスコ 300ml
(2) 操作 操作は,次のとおり行う。
(a) 試料200mlを共栓三角フラスコにとり,軽く栓をして約40℃に温める。
(b) 共栓三角フラスコを揺り動かしながら栓をとり,直ちに臭気の有無及び臭気の種類とその程度を試
験する。
(c) 臭気の表示は表8.1に倣い,試料の臭気の種類と程度を,概略の理解ができるように表示する。
表8.1 臭気の分類と種類の一例
臭気の大分類
臭気の種類
(1) 芳香性臭気
メロン臭,すみれ臭,にんにく臭,きゅうり臭,芳香臭,薬味臭など
(2) 植物性臭気
藻臭,青草臭,木材臭,海藻臭など
(3) 土臭,かび臭
土臭,沼沢臭,かび臭など
(4) 魚貝臭
魚臭,肝油臭,はまぐり臭など
(5) 薬品性臭気
フェノール臭,タール臭,油臭,油脂臭,パラフィン臭,塩素臭,硫化
水素臭,クロロフェノール臭,薬局臭,薬品臭など
(6) 金属性臭気
かなけ臭,金属臭など
(7) 腐敗性臭気
ちゅうかい(厨芥)臭,下水臭,豚小屋臭,腐敗臭など
(8) 不快臭
魚臭,豚小屋臭,腐敗臭などが強烈になった不快なにおい
備考1. きゅう覚の個人差を少なくするため,同一試料を数人で試験するとよい。
2. 試料採取時に試料を温めずにその臭気を試験し,記録しておくとよい(これを冷
時臭という。)。
10
K 0101 : 1998
8.2
臭気強度 (TON) 臭気の強さを表すもので,約40℃に保った水に試料を加え,明らかに臭気を感
じるときの希釈の倍数値[臭気いき(閾)値の希釈倍数]で表す。きゅう覚の個人差を少なくするため,
同一試料について少なくとも5人,できれば10人程度で試験する。
(1) 試薬 試薬は,次のものを用いる。
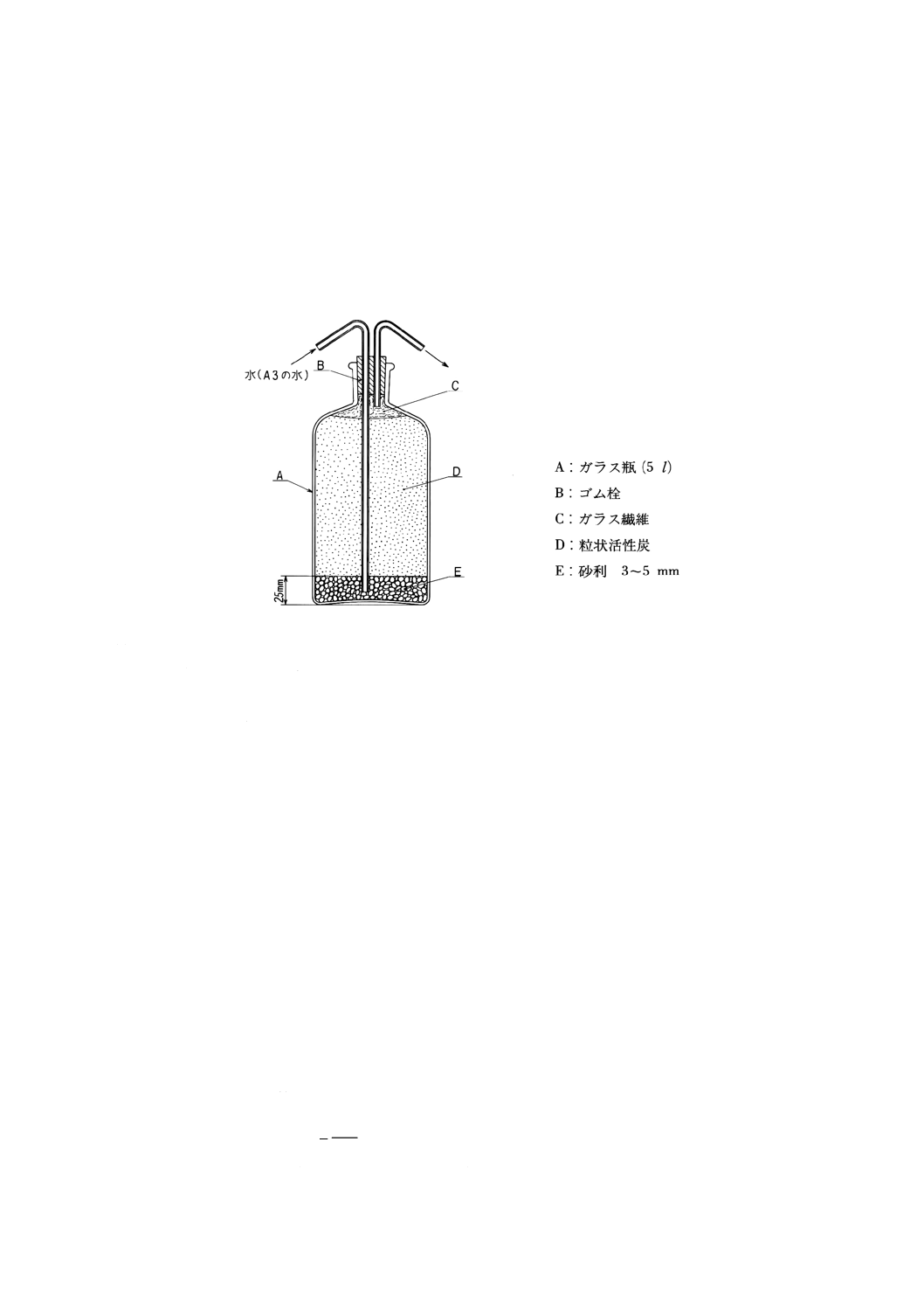
(a) 臭気のない水 図8.1に示すような装置にJIS K 0557に規定するA3の水を5l(l‐活性炭・h)で通
す。
図8.1 臭気のない水の作り方の一例
(2) 器具 器具は,次のとおりとする。
(a) 共栓三角フラスコ 300ml
(b) 足長ビュレット 50ml
(c) 水浴 温度調節器の付いたもの。
(3) 予備試験 予備試験は,次のとおり行う。
(a) 試料200,40,10,4mlをそれぞれ共栓三角フラスコにとり,臭気のない水を加えて200mlとし,
予備試験の試料とする。
(b) 別に,対照水として臭気のない水200mlを共栓三角フラスコにとる。
(c) 予備試験の試料と対照水とを水浴上で40〜50℃に温めた後,対照水を振り混ぜ,開栓と同時に発生
するにおいをかぐ。
(d) 次に,試料の量の少ないほうから同様に操作して予備試験の試料のにおいを対照水と比較し,にお
いが感じられる最少の試料の量 (ml) を求める。
(4) 操作 操作は,次のとおり行う。
(a) (3)(d)で求めた試料の量から表8.2によって試験に用いる試料の量を縦系列に示すml数として求め
る。
(b) (a)で求めた試料の各量をそれぞれ別の共栓三角フラスコにとり,臭気のない水を加えて200mlとし,
これを本試験の試料とする。
(c) 次に,(3)(b)〜(d)と同様に操作してにおいを感知できる最少の試料のml数を求め,次の式によって
臭気強度 (TON) を算出する。
V
TON
200
=
ここに, TON: 臭気強度 (TON)

11
K 0101 : 1998
V: 希釈に用いた試料 (ml)
表8.2 試験に用いる試料の量
単位ml
予備試験の試料の量
200
40
10
4
試験に用いる試料の量
200
40
10
4.0
100
28.5
8.0
2.9
67
20
6.7
2.0
50
13.3
5.0
1.3
40
10
4.0
1.0
備考3. 試料の臭気が強すぎるときは,試料を臭気のない水で10倍に薄めてから予備試験及び試験
の操作を行う。
4. 試験に用いる共栓三角フラスコは,あらかじめ,臭気のない水でよく洗浄しておく。
5. 試験は,環境に左右されることが多いから,においのない静かな室内で行う。
6. 試験直前の喫煙,喫茶,食事などは避け,さらに手及び指に石けん,ローション,香水な
どのかおりがないようにする。
7. 試験を続けて行うと,4〜5回ぐらいできゅう覚が鈍るから,15〜30分間程度休憩する。
8. 臭気度 (pO) を求める場合には,次の式による。
TON
TON
pO
log
32
.3
log
2
log
1
×
=
×
=
9. 濁度 水の濁りの程度を表すもので,視覚濁度,透過光濁度,散乱光濁度及び積分球濁度に区分し表
示する。カオリン標準液と比較して測定する場合には,“度(カオリン)”を単位とし,ホルマジン標準液
と比較して測定する場合には,“度(ホルマジン)”を単位として表す。
濁度は変化しやすいので,試料採取後直ちに試験することが望ましい。
9.1
視覚濁度 試料の濁りを肉眼によってカオリン標準液と比較して求める。
測定範囲:1〜10度(カオリン)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水を孔径約0.1μmのろ過材を用いてろ過し,初めの約200mlを捨
てた後のろ液。
(b) 精製カオリン はくとう土(カオリン)約10gをビーカー500mlにとり,これに水300mlとJIS K 8785
に規定する二りん酸ナトリウム十水和物0.2gとを加え,マグネチックスターラーで約3分間激しく
かき混ぜる。これをメスシリンダー(有栓形)1 000mlに移し入れ,水を1 000mlの標線まで加え,
栓をして約1分間激しく振り混ぜる。室温で約1時間静置した後,サイホンを用いて上部から250ml
の液を捨て,次の500mlまでの液を採取する。
採取した液を約3 000min-1(遠心分離器の回転部分の半径によって回転数を加減する。)で約20
分間遠心分離するか,又は孔径1μm以下のろ過材によってろ過する。ろ別したカオリンを105〜
110℃で約3時間加熱し,デシケーター中で放冷した後,広口瓶に保存する。
(c) カオリン標準液[1 000度(カオリン)] 精製カオリン1.00gをとり適量の水に分散させた後,全
量フラスコ1 000mlに移し入れ,水約800mlとJIS K 8872に規定するホルムアルデヒド液約10ml
を加えた後,水を標線まで加える。
(d) カオリン標準液[100度(カオリン)] カオリン標準液[1 000度(カオリン)]をよく振り混ぜた
後,直ちにその100mlを全量フラスコ1 000mlにとり,水を標線まで加える。
12
K 0101 : 1998
(2) 器具 器具は,次のとおりとする。
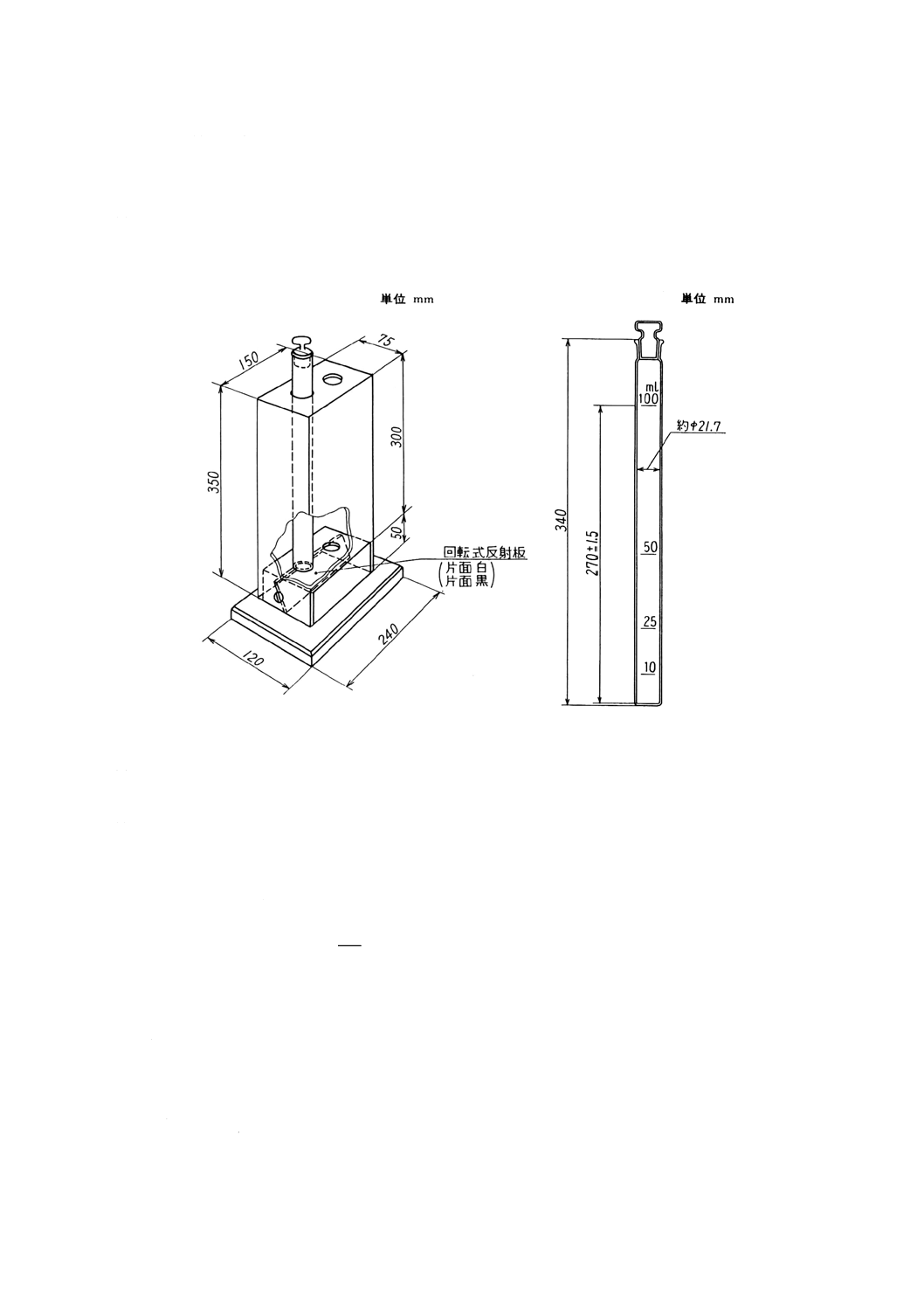
(a) 暗箱 図9.1に示すような暗箱を使用すると肉眼で濁りを比較するのに便利である。電灯を暗箱の
下窓に近付けて測定すると見やすくなる。
(b) 比色管 図9.2に示すように,底部から270±1.5mmの高さに100mlの標線の付いた共栓平底の比
色管で,ガラスに色のないもの。標線の高さのそろったもの (±1.5mm) を用いる。
図9.1 暗箱の一例
図9.2 比色管
(3) 操作 操作は,次のとおり行う。
(a) 試料をよく振り混ぜた後,これから適量(1)を比色管にとり,水を100mlの標線まで加える。
(b) 別に,カオリン標準液[100度(カオリン)]1〜10mlを比色管に段階的にとり,水を100mlの標線
まで加え,比濁用カオリン標準液[1度〜10度(カオリン)]を調製する。
(c) 試料を入れた比色管及び比濁用カオリン標準液を入れた比色管をよく振り混ぜた後,直ちに暗箱に
入れ,上方から透視して濁りを比較し,試料に該当する比濁用カオリン標準液を選び出す。
(d) 該当する比濁用カオリン標準液の濁度[度(カオリン)]から,次の式によって試料の視覚濁度[度
(カオリン)]を算出する。
V
T
T
s
100
×
=
ここに,
T: 視覚濁度[度(カオリン)]
Ts: 該当する比濁用カオリン標準液の濁度[度(カオリン)]
V: 試料 (ml)
注(1) 試料の濁度が10度(カオリン)以下の場合は,そのまま100mlをとる。
9.2
透過光濁度 試料を通過した波長660nm付近の透過光の強度を測定し,カオリン標準液又はホルマ
ジン標準液を用いて作成した検量線から求める。
測定範囲 : 吸収セル50mmのとき5〜50度(カオリン)又は4〜80度(ホルマジン),吸収セル
10mmのとき25〜250度(カオリン)又は20〜400度(ホルマジン)
13
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 水 9.1(1)(a)による。
(b) カオリン標準液[100度(カオリン)] 9.1(1)(d)による。
(c) ホルマジン標準液[400度(ホルマジン)] JIS K 8992に規定する硫酸ヒドラジニウム1.00gをと
り,適量の水に溶かして,全量フラスコ100mlに移し入れ,水を標線まで加える。
別に,JIS K 8847に規定するヘキサメチレンテトラミン10.0gをとり,適量の水に溶かして,全
量フラスコ100mlに移し入れ,水を標線まで加える。
この両溶液それぞれ10mlを全量フラスコ200mlにとり,よく振り混ぜる。液温25±3℃で約24
時間放置した後,水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(3.1) カオリン標準液を用いる場合
(a) 試料をよく振り混ぜた後,吸収セル50mm(2)にとり,波長660nm(3)付近における透過光の強度を見
掛けの吸光度で測定する。
(b) カオリン標準液を用いて作成した検量線から試料の透過光濁度[度(カオリン)]を求める。
検量線 カオリン標準液[100度(カオリン)]5〜50mlを全量フラスコ100mlに段階的にとり,
水を標線まで加えて検量線用カオリン標準液[5〜50度(カオリン)]を調製する(4)。以下,(a)の
操作を行って検量線用カオリン標準液の透過光濁度[度(カオリン)]と吸光度との関係線を作成
する。
注(2) 試料の透過光濁度が25〜250[度(カオリン)]の場合は,吸収セル10mmを用いる。
(3) 試料に色がある場合(特に,波長660nm付近に吸収があるとき)は,試料を孔径1μm以下のろ
過材を用いてろ過したろ液又は遠心分離[約3 000min-1(遠心分離器の回転部分の半径によって
回転数を加減する。)で約20分間]した上澄み液を対照液にして,透過光の強度を吸光度で測
定する。
(4) 吸収セル10mmを用いる場合には,9.1(1)(c)のカオリン標準液[1 000度(カオリン)]2.5〜25ml
を段階的にとり,検量線用カオリン標準液[25〜250度(カオリン)]を調製する。
(3.2) ホルマジン標準液を用いる場合
(a) 試料をよく振り混ぜた後,(3.1)(a)の操作(5)を行う。
(b) ホルマジン標準液を用いて作成した検量線から試料の透過光濁度[度(ホルマジン)]を求める。
検量線 ホルマジン標準液[400度(ホルマジン)]1〜20mlを全量フラスコ100mlに段階的にと
り,水を標線まで加えて検量線用ホルマジン標準液[4〜80度(ホルマジン)]を調製する(6)。以
下,(a)の操作を行って検量線用ホルマジン標準液の透過光濁度[度(ホルマジン)]と吸光度との
関係線を作成する。
注(5) 試料の透過光濁度が20〜400度(ホルマジン)の場合は,吸収セル10mmを用いる。
(6) 吸収セル10mmを用いる場合には,9.2(1)(c)のホルマジン標準液[400度(ホルマジン)]5〜100ml
を段階的にとり,検量線用ホルマジン標準液[20〜400度(ホルマジン)]を調製する。
9.3
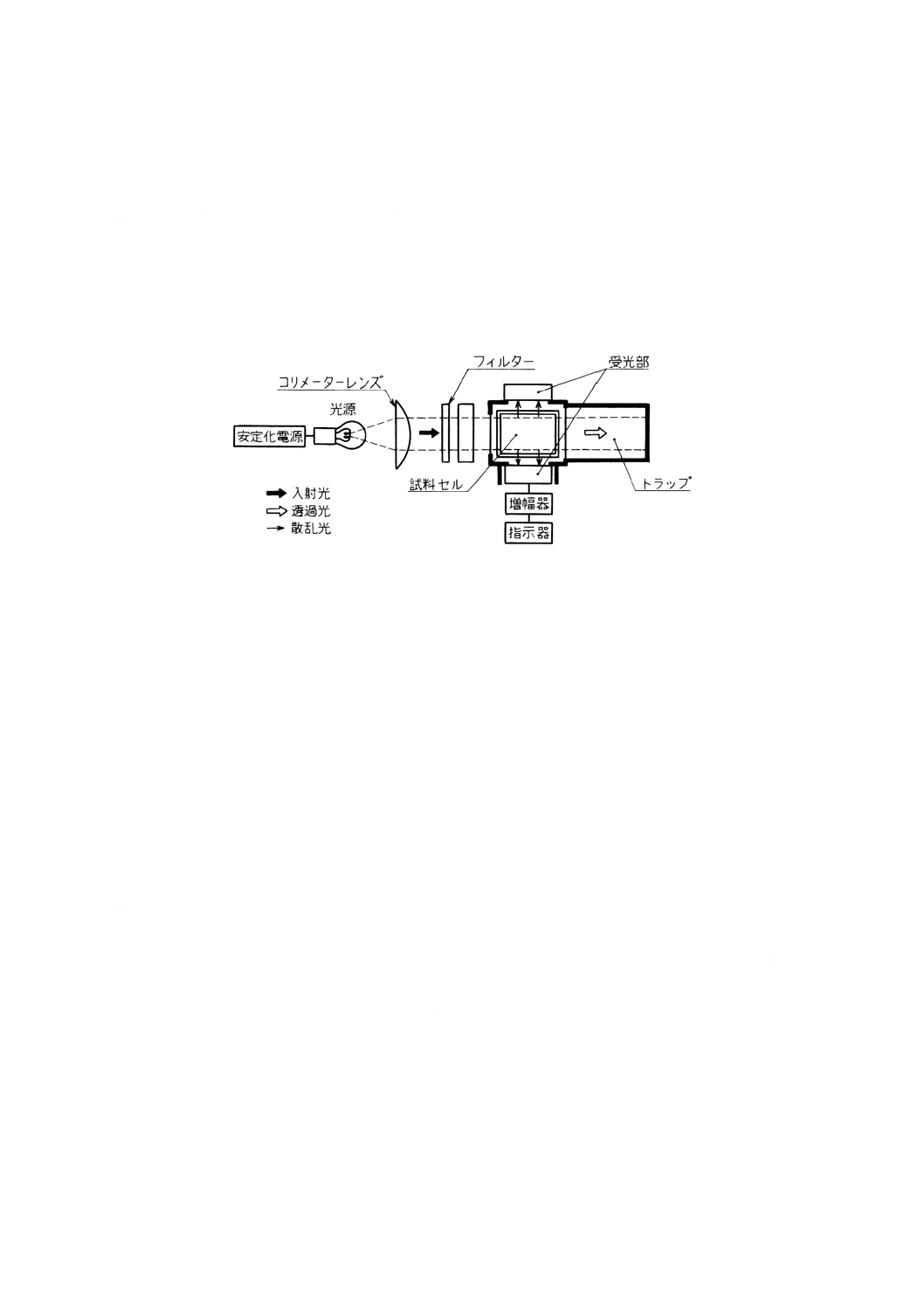
散乱光濁度 試料中の粒子によって散乱した光の強度を波長660nm付近で測定し,カオリン標準液
又はホルマジン標準液を用いて作成した検量線から求める。
測定範囲:1〜5度(カオリン)又は0.4〜5度(ホルマジン)(装置によって異なる。)
14
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 水 9.1(1)(a)による。
(b) カオリン標準液[100度(カオリン)] 9.1(1)(d)による。
(c) ホルマジン標準液[40度(ホルマジン)] 9.2(1)(c)のホルマジン標準液[400度(ホルマジン)]
10mlを全量フラスコ100mlにとり,水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 散乱光濁度計 図9.3に散乱光濁度計の一例を示す。
図9.3 散乱光濁度計の構成例
(3) 操作 操作は,次のとおり行う。
(3.1) カオリン標準液を用いる場合
(a) 水を吸収セルにとり,散乱光濁度計の指示値(7)を0に調節し,次に,検量線用カオリン標準液[5
度(カオリン)]を用いて散乱光濁度計の指示値(7)を100%に調節する。
(b) 試料をよく振り混ぜた後,吸収セルにとり,波長660nm付近における指示値(散乱光の強度)を測
定する。
(c) カオリン標準液を用いて作成した検量線から試料の散乱光濁度[度(カオリン)]を求める。
検量線 カオリン標準液[100度(カオリン)]1〜5mlを全量フラスコ100mlに段階的にとり,水
を標線まで加えて検量線用カオリン標準液[1〜5度(カオリン)]を調製する。以下,(a)及び(b)
の操作を行って検量線用カオリン標準液の散乱光濁度[1〜5度(カオリン)]と指示値(散乱光の
強度)との関係線を作成する。
注(7) 散乱光の強度は,入射光に対し90°又は270°の位置における測定が一般に行われている。
(3.2) ホルマジン標準液を用いる場合
(a) 水を吸収セルにとり,散乱光濁度計の指示値(7)を0に調節し,次に,検量線用ホルマジン標準液[5
度(ホルマジン)]を用いて散乱光濁度計の指示値(7)を100%に調節する。
(b) 試料をよく振り混ぜた後,吸収セルにとり,波長660nm付近における指示値(散乱光の強度)を測
定する。
(c) ホルマジン標準液を用いて作成した検量線から試料の散乱光濁度[度(ホルマジン)]を求める。
検量線 ホルマジン標準液[40度(ホルマジン)]1〜12.5mlを全量フラスコ100mlに段階的にと
り,水を標線まで加えて検量線用ホルマジン標準液[1〜5度(ホルマジン)]を調製する。以下,
(a)及び(b)の操作を行って検量線用ホルマジン標準液の散乱光濁度[1〜5度(ホルマジン)]と指
示値(散乱光の強度)との関係線を作成する。
15
K 0101 : 1998
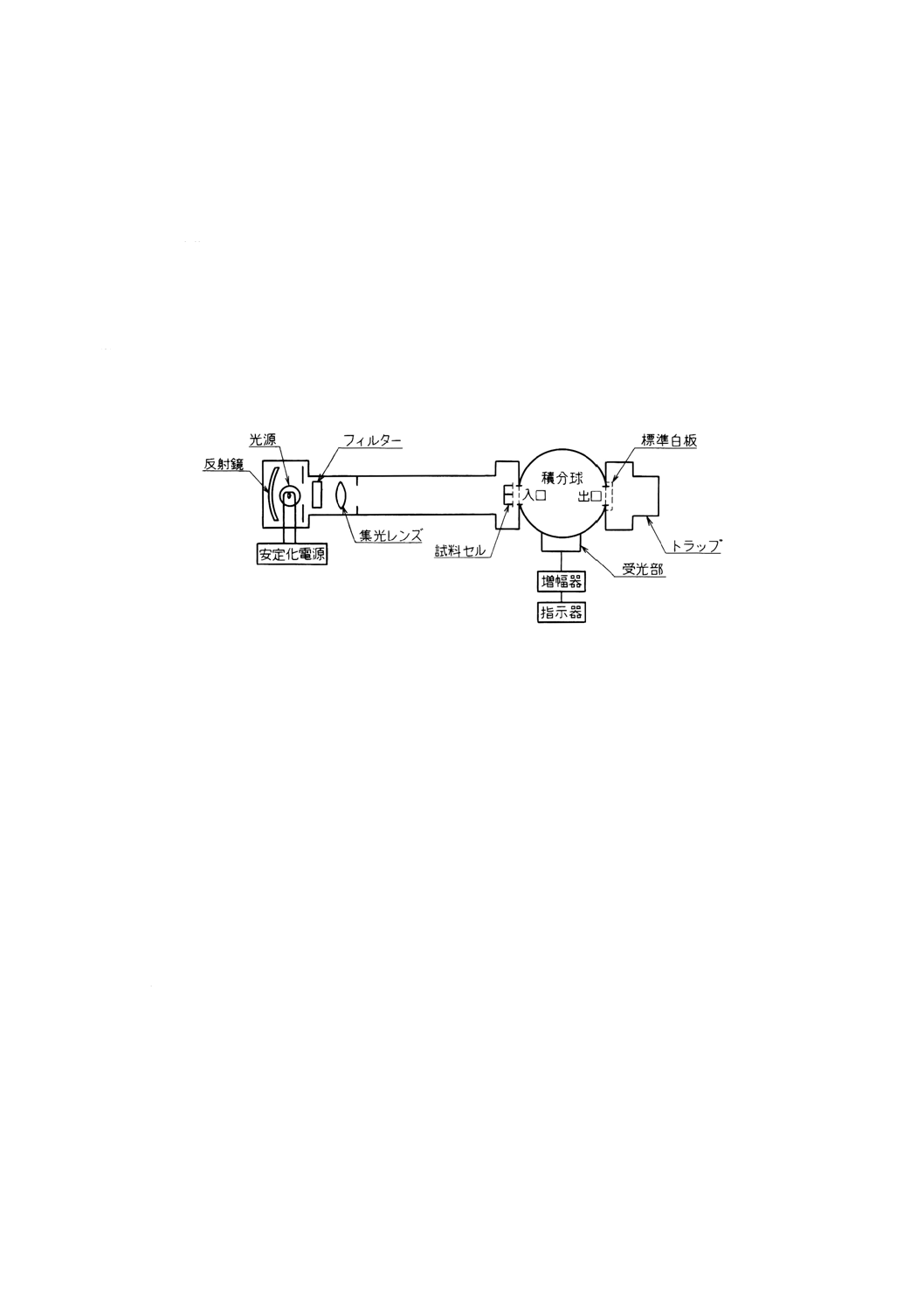
9.4
積分球濁度 水中の粒子による散乱光の強度と透過光の強度との比を求め,カオリン標準液又はホ
ルマジン標準液を用いて作成した検量線から求める。
測定範囲 :吸収セル50mmのとき0.2〜5度(カオリン)又は0.2〜5度(ホルマジン),吸収セル
10mmのとき5〜100度(カオリン)又は5〜100度(ホルマジン)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 9.1(1)(a)による。
(b) カオリン標準液[100度(カオリン)] 9.1(1)(d)による。
(c) ホルマジン標準液[40度(ホルマジン)] 9.3(1)(c)による。
(2) 装置 装置は,次のとおりとする。
(a) 積分球濁度計 図9.4に積分球濁度計の一例を示す。
図9.4 積分球濁度計の構成例
(3) 操作 操作は,次のとおり行う。
(3.1) カオリン標準液を用いる場合
(a) 水を入れた吸収セル50mmとトラップを光路に入れて指示値を0に調節する。次に,トラップに標
準白板を挿入し,指示値が100になるように調節する。
(b) 次に,水を入れた吸収セル50mmに代えて,よく振り混ぜた試料を入れた吸収セル50mmとトラッ
プ(標準白板は入れない。)を光路に入れて散乱光の強度Tdを測定する。
(c) 続いてトラップに標準白板を挿入して,試料の全透過光の強度Ttを測定する。
(d) Td/Tt×100の値を算出し,カオリン標準液を用いて作成した検量線から,試料の積分球濁度[度(カ
オリン)]を求める。
検量線 カオリン標準液[100度(カオリン)]0.2〜5mlを全量フラスコ100mlに段階的にとり,
水を標線まで加えて検量線用カオリン標準液[0.2〜5度(カオリン)]を調製する(8)。以下,(a)〜
(d)の操作を行って検量線用カオリン標準液の積分球濁度[度(カオリン)]とTd/Tt×100の値との
関係線を作成する。
注(8) 吸収セル10mmを用いて測定するときは,検量線用カオリン標準液[5〜100度(カオリン)]を
調製する。
備考1. Td/Tt×100の計算を行わない方法 (a)の操作を行った後,水を入れた吸収セルに代えて試料
を入れたセルを入れ,このときの指示値が100になるように調節する。続いて標準白板を外し
て指示値を読む。検量線はカオリン標準液(又はホルマジン標準液)について同様に操作し,
横軸に積分球濁度[度(カオリン)]〔又は[度(ホルマジン)]〕,縦軸に指示値(Td/Tt×100
に相当する。)をとって作成し,この検量線から,試料の積分球濁度を求める。
16
K 0101 : 1998
(3.2) ホルマジン標準液を用いる場合
(a) (3.1)(a)〜(c)の操作を行う。
(b) Td/Ttの値を算出し,ホルマジン標準液を用いて作成した検量線から試料の積分球濁度[度(ホルマ
ジン)]を求める。
検量線 ホルマジン標準液[40度(ホルマジン)]0.5〜12.5mlを全量フラスコ100mlに段階的に
とり,水を標線まで加えて検量線用ホルマジン標準液[0.2〜5度(ホルマジン)]を調製する(9)。
以下,(a)の操作を行って検量線用標準液の積分球濁度[度(ホルマジン)]とTd/Tt×100の値との
関係線を作成する。
注(9) 吸収セル10mmを用いて測定するときは,検量線用ホルマジン標準液[5〜100度(ホルマジン)]
を調製する。
10. 色 色の表示には,白金・コバルトによる色度又は刺激値Y及び色度座標x,yによる方法を用いる。
白金・コバルトによる色度は試料が淡黄色から黄褐色系統の色の場合にだけ適用する。
10.1 白金・コバルトによる色度 白金・コバルトによる色度とは,水に溶存又はコロイド状で存在する
物質による淡黄色から黄褐色の程度を示すもので,水1l中に白金・コバルト色度標準液1ml(白金1mg及
びコバルト0.5mg)を加えたときの色を白金・コバルト色度1度とする。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水を孔径約0.1μmのろ過材を用いてろ過し,初めの約200mlを捨
てた後のろ液。
(b) 白金・コバルト色度標準液(1 000度) JIS K 8163に規定するヘキサクロロ白金 (IV) 酸カリウム
(1)2.49gとJIS K 8129に規定する塩化コバルト (II) 六水和物2.00gとをとり,JIS K 8180に規定す
る塩酸200ml中に加え,水を加えて溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加え
る。着色瓶に保存する。
注(1) JIS K 8163に規定するヘキサクロロ白金 (IV) 酸カリウムの代わりに白金を用いる場合には,白
金1.00gを王水(塩酸3+硝酸1)の適量に溶かし,塩酸を過剰に加えて沸騰水浴上で蒸発乾固す
る。この操作を2,3回繰り返して硝酸を除去した後,JIS K 8129に規定する塩化コバルト (II) 六
水和物2.00g及びJIS K 8180に規定する塩酸200mlとともに溶かして全量フラスコ1 000mlに移
し入れ,水を標線まで加える。
(2) 器具 器具は,次のとおりとする。
(a) 暗箱 9.1(2)(a)による。
(b) 比色管 100ml 9.1(2)(b)による。
(3) 操作 操作は,次のとおり行う。
(a) 試料は,ろ紙5種C又は孔径1μm以下のろ過材でろ過するか,又は約3 000min-1(遠心分離器の回
転部分の半径によって回転数を加減する。)で20分間遠心分離して濁りを除去する。
(b) この試料の適量を比色管100mlにとり,水を100mlの標線まで加える。
(c) 白金・コバルト色度標準液(1 000度)0.1〜2.0mlを比色管100mlに段階的にとり,水を100mlの標
線まで加え,栓をしてよく振り混ぜて1〜20度の白金・コバルト色度標準液列を調製する。
(d) 試料及び色度標準液列を白紙上に置くか,又は暗箱に入れて上方から透視し,試料の色を白金・コ
バルト色度標準液列と比較し,該当する白金・コバルト色度標準液を求める。
(e) 次の式によって試料の白金・コバルト色度を算出する。
17
K 0101 : 1998
V
C
C
s
100
×
=
ここに,
C: 白金・コバルト色度
Cs: 該当する白金・コバルト色度標準液(度)
V: 試料 (ml)
10.2 刺激値Y及び色度座標x,yによる表示 水を対照液として波長400〜700nmの所定の波長における
試料についての透過パーセントを測定して,三刺激値(2)X,Y,Zを算出し,次に,色度座標x,yを算出し,
試料の色を刺激値Y及び色度座標x,yによって表示する。
注(2) 三刺激値とは,色に対する視神経の感じ方が3種類あると考え,その3種類の視神経に対する刺
激の混合の割合によって色に対する感じ方が変わるとし,それぞれの刺激を与える波長を種類
別にX,Y,Zの系列にまとめ,透過パーセントを集計して求めた値である。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 10.1(1)(a)による。
(2) 装置 装置は,次のとおりとする。
(a) 遠心分離器
(b) 光度計 分光光度計 吸収セル100mmを使用できるもので,波長間隔10nm以下で可視部全域が測
定できるものか,又はこれと同等の性能をもつ色度計。
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ紙5種C又は孔径1μm以下のろ過材でろ過するか,又は約3 000min-1(遠心分離器の回転
部分の半径によって回転数を加減する。)で約20分間遠心分離して濁りを除去する。
(b) この一部を吸収セル100mm(3)に移し,水を対照液として表10.1(4)の各波長における透過パーセント
を測定する。
注(3) 色が濃く,吸収セル100mmで測定できない場合には,適当な光路長の吸収セルを用いて測定す
る。この場合には,所定波長における吸光度を測定し,この値から吸収セル100mmの吸光度に
換算し,この吸光度から透過パーセントを求めて計算する。
(4) 表10.1は,JIS Z 8719に規定する条件等色指数‐照明光条件等色度の評価方法の付表5-1-1(波
長間隔20nm)に従ったものである。
表10.1 波長間隔20nmで三刺激値X,Y,Zを計算するための重価係数fX,fY,fZ
波長
λ (nm)
標準の光 C
fX
fY
fZ
400
0.019
−0.003
0.062
420
2.993
0.087
14.387
440
7.634
0.510
38.438
460
6.642
1.382
38.130
480
2.360
3.206
19.545
500
0.068
6.907
5.746
520
1.196
12.876
1.444
540
5.590
18.261
0.356
560
11.751
19.592
0.073
580
16.795
15.991
0.026
600
17.897
10.694
0.013
620
14.021
6.261
0.002

18
K 0101 : 1998
波長
λ (nm)
標準の光 C
fX
fY
fZ
640
7.453
2.901
0.000
660
2.731
1.003
0.000
680
0.756
0.273
0.000
700
0.166
0.059
0.000
合計
98.072
100.000
118.225
色度座標
x=0.3101
y=0.31622
z=0.3737
(c) 三刺激値及び色度座標x,yの求め方 表10.1のX,Y,Zの所定波長における透過パーセントをそ
れぞれ合計して,次の式によって三刺激値X,Y,Z及び色度座標x,yを算出する。
)
(
)
(
1700
400
λ
τ
λ
∑
•
=
X
f
K
X
)
(
)
(
1700
400
λ
τ
λ
∑
•
=
Yf
K
Y
)
(
)
(
1700
400
λ
τ
λ
∑
•
=
Zf
K
Z
Z
Y
X
X
x
+
+
=
Z
Y
X
Y
y
+
+
=
ここに,
X: 刺激値X
Y: 刺激値Y
Z: 刺激値Z
K: 100.000
fX(λ): 波長λでのfX
fY(λ): 波長λでのfY
fZ(λ): 波長λでのfZ
τ(λ): 波長λでの透過パーセント
(d) 刺激値Yの求め方 刺激値Yの値をそのまま刺激値とする。
備考1. 主波長及び補色主波長の求め方 色度(図10.1)の中の点は無色の色度座標でx=0.310 1,y
=0.316 2。
色度座標が直線RC,直線VC及びスペクトル軌跡によって囲まれた面積内の点S1で表さ
れる色の場合,直線CS1の延長とスペクトル軌跡との交点S1'に対応する波長を図10.3から求
める。この波長をその色の主波長といい,記号λdで表す。
また,色度座標が三角形CRV内の点S2で表される色の場合には,直線CS2の延長とスペ
クトル軌跡との交点S2''に対応する波長を図10.3から求める。この波長をその色の補色主波
長といい,記号λeで表す。
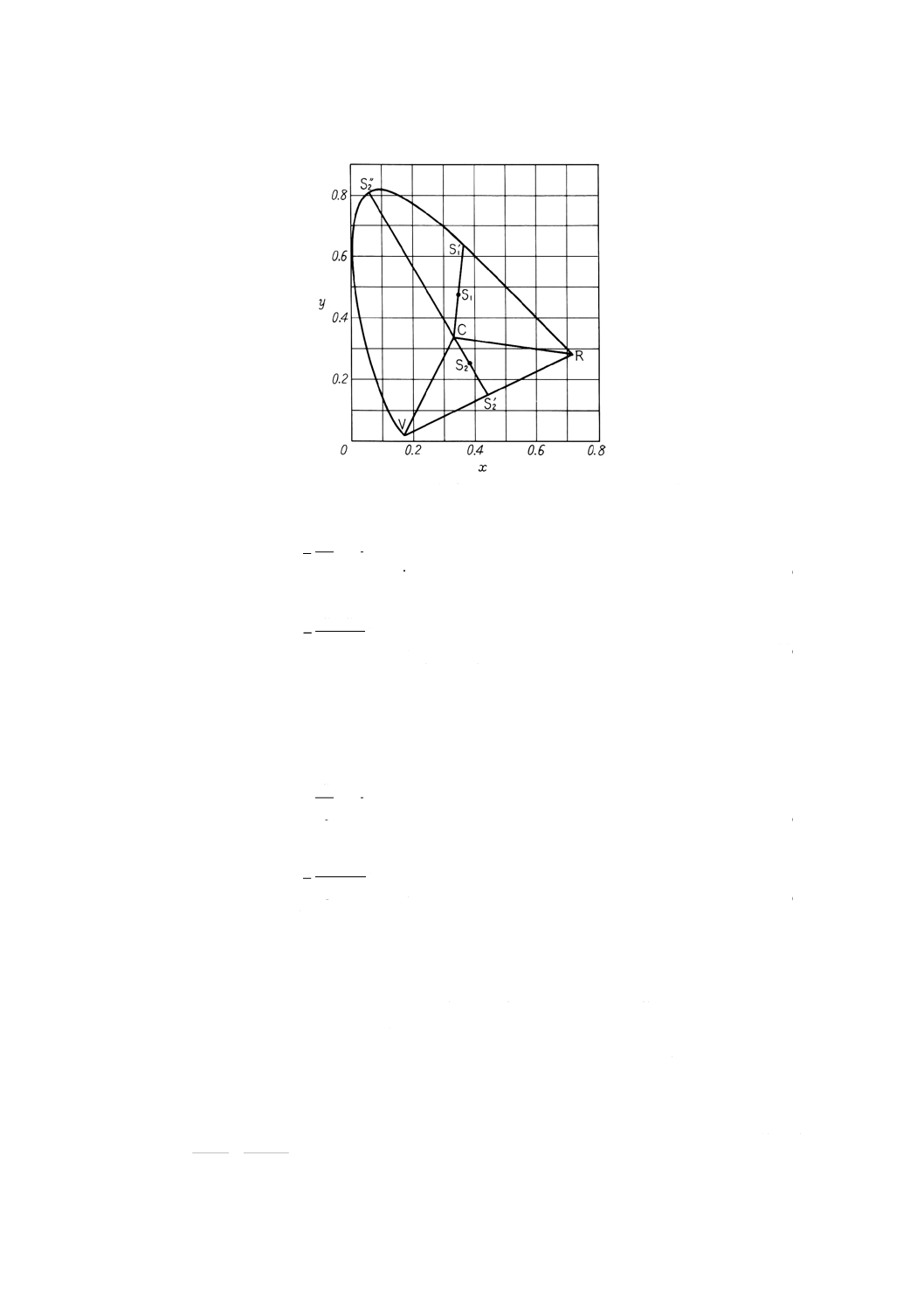
19
K 0101 : 1998
図10.1 色度図
備考2. 刺激純度の求め方 図10.1において色度座標が点S1によって表される色の場合には,刺激純
度Peは次の式(*)によって求め,その値に%を付ける。
100
×
−
−
=
c
c
e
x
x
x
x
P
λ
······································································· (1)
又は
100
×
−
−
=
c
c
e
y
y
y
y
P
λ
······································································ (2)
ここに,
x, y: 点S1の座標
xc, yc: 点Cの座標
xλ, yλ: 点S1'の座標
また,色度座標が点S2で表される色の場合には,刺激純度Peは次の式(*)によって求め,
その値に%を付ける。
100
×
−
−
=
c
p
c
e
x
x
x
x
P
······································································ (3)
又は
100
×
−
−
=
c
p
c
e
y
y
y
y
P
······································································ (4)
ここに,
x, y: 点S2の座標
xc, yc: 点Cの座標
xp, yp: 点S2'(直線CS2の延長と純紫軌跡との交点)の座標
注(*) Peを計算するには,分母又は分子の絶対値の大きい式で求める。
刺激純度及び主波長の求め方の一例 試料を測定して得られた三刺激値Xが40.14,Yが76.50,
Zが34.25であったとすれば,色度座標はxが0.266,yが0.507で刺激値Yは76.50である。こ
の色度座標を図10.2上の点 (S1) として求め,次に無色の点 (x=0.310 1, y=0.316 2) Cとして
求める。CとS1とを直線で結び,更に延長してスペクトル軌跡と交わる点をS1'として求める。
S1'はスペクトル軌跡上の534nmの位置にある。すなわち主波長λdは534nmである。
また,図10.2からS1ʼの座標xλが0.200,yλが0.785であるので,式(2)によって刺激純度Pe
は
100
3162
.0
785
.0
3162
.0
507
.0
×
−
−
=40.7%となる。
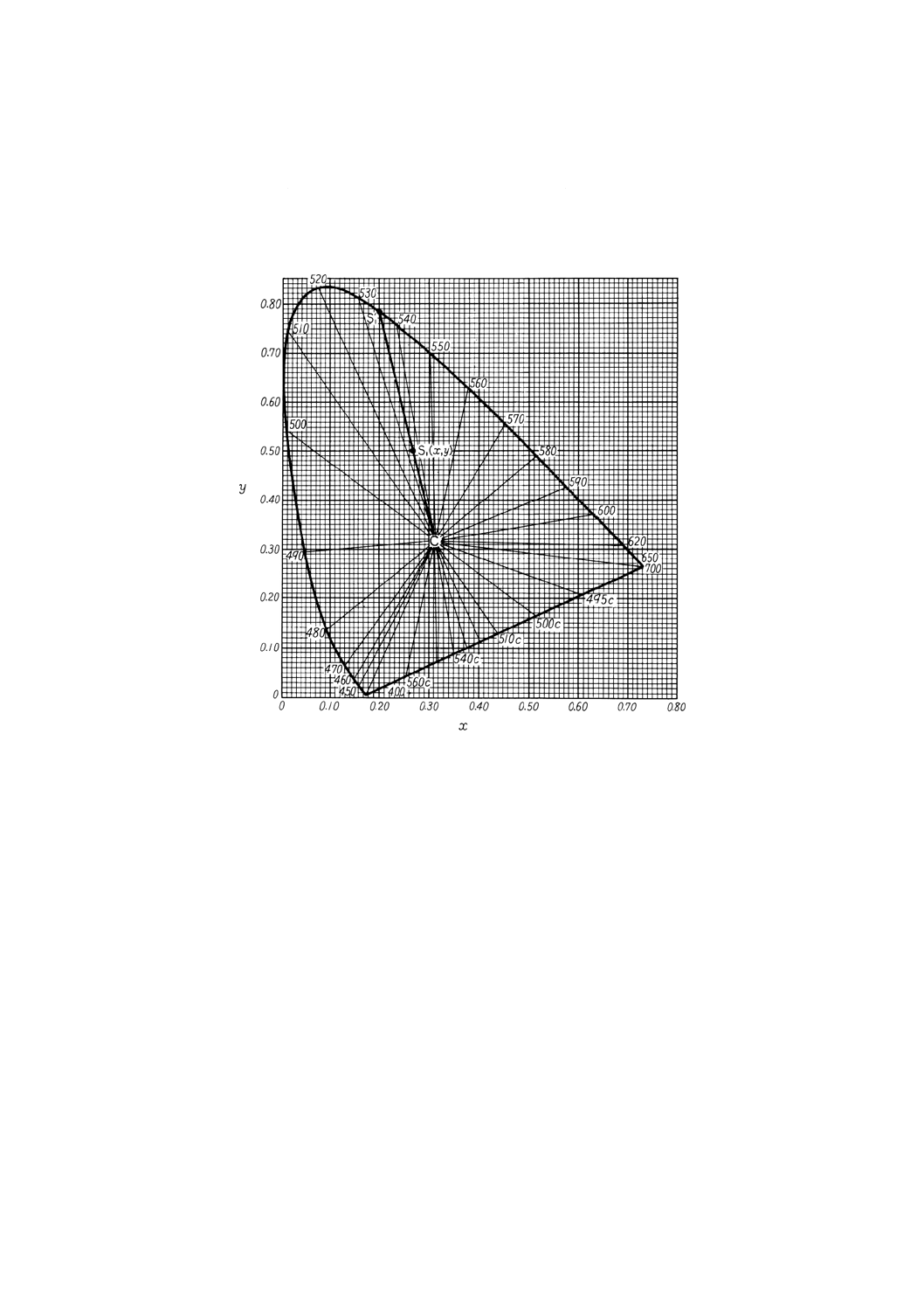
20
K 0101 : 1998
したがって,試料の色は主波長λd=534nm,刺激純度Peは40.7%,刺激値Yは76.50%として
表示される。
なお,試料の色が薄い場合には図10.4,また,試料の色が濃い場合には,図10.5を用いて刺
激純度を求めると便利である。
図10.2 2度視野XYZ系による色度図と主波長との関係図
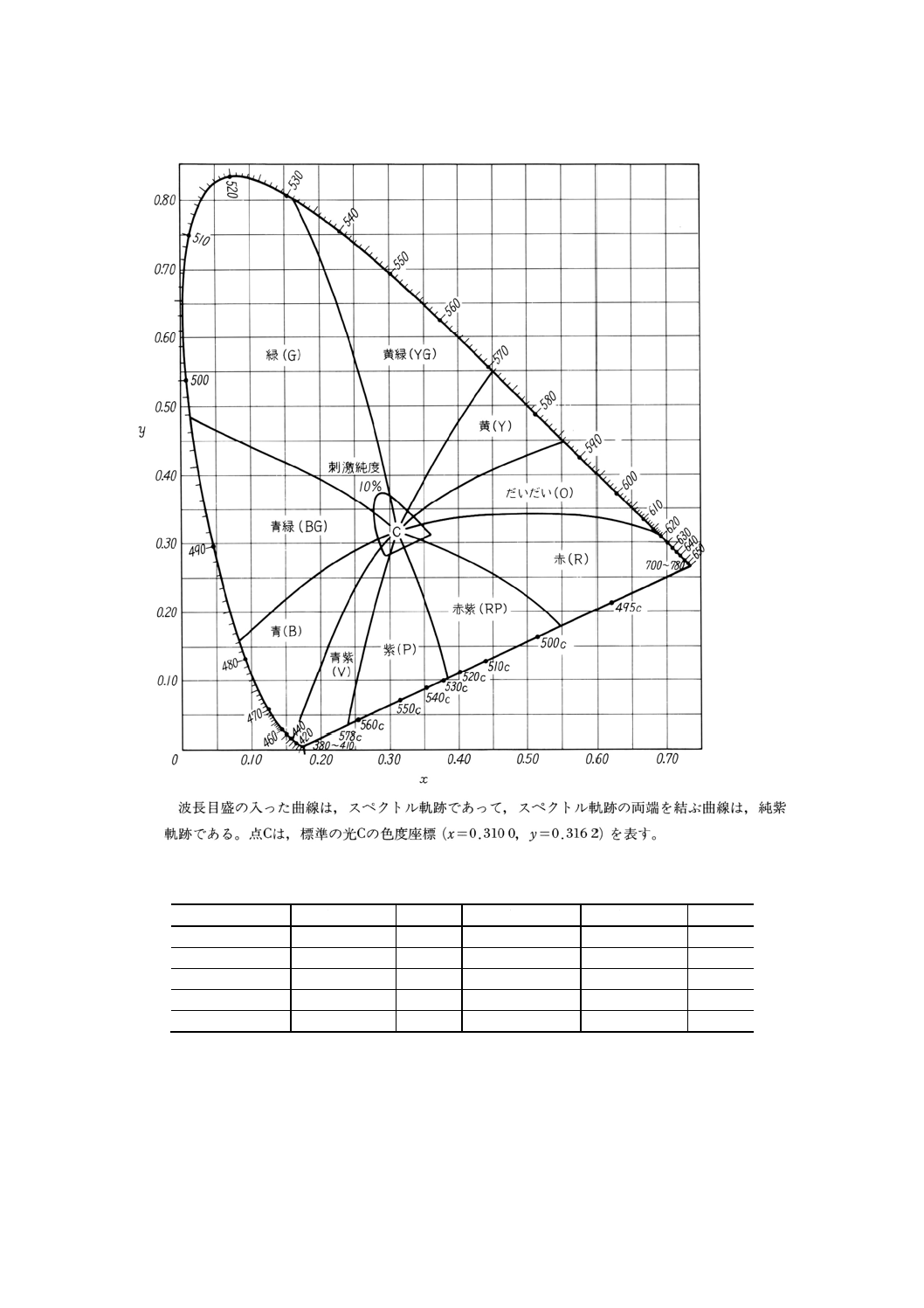
21
K 0101 : 1998
図10.3 2度視野XYZ系による色度図(主波長と色相名)
備考3. 主波長と色相 主波長と色相の関係を表10.2及び図10.3に示す。
表10.2 主波長と色相名
主波長 nm
色相名
略号
主波長 nm
色相名
略号
498c〜700〜618
赤
R
498〜482
青緑
BG
618〜586
だいだい(橙)
O
482〜435
青
B
586〜571
黄
Y
435〜400〜578c
青紫
V
571〜531
黄緑
YG
578c〜528c
紫
P
531〜498
緑
G
528c〜498c
赤紫
RP
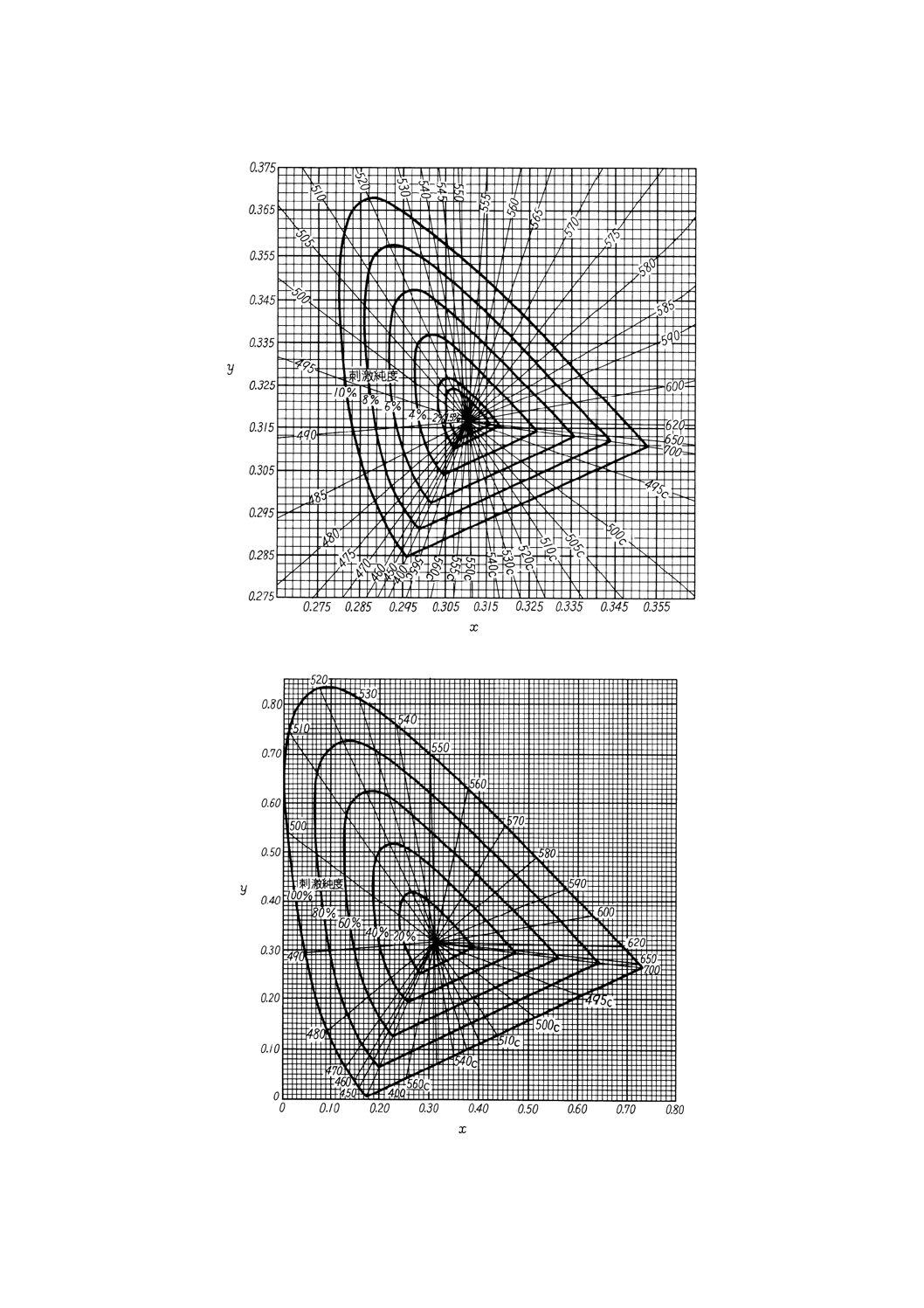
22
K 0101 : 1998
図10.4 刺激純度(10%表示)(試料の色が薄い場合)
図10.5 刺激純度(100%表示)(試料の色が濃い場合)
23
K 0101 : 1998
11. pH pHの測定には,JIS Z 8802によるガラス電極法を適用する。
pHは試料採取後直ちに測定する。
11.1 ガラス電極法 ガラス電極を用いたpH計によってpHを測定する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水。ほう酸塩pH標準液及び炭酸塩pH標準液を調製する
場合は,2.(12)(b)の炭酸を含まない水を用いる。
(b) 二しゅう酸三水素カリウム二水和物 JIS K 8474に規定する二しゅう酸三水素カリウム二水和物
(c) フタル酸水素カリウム JIS K 8809に規定するフタル酸水素カリウムのpH標準液用。
(d) りん酸二水素カリウム JIS K 9007に規定するりん酸二水素カリウムのpH標準液用。
(e) りん酸水素二ナトリウム JIS K 9020に規定するりん酸水素二ナトリウムのpH標準液用。
(f) 四ほう酸ナトリウム JIS K 8866に規定する四ほう酸ナトリウム十水和物のpH標準液用。
(g) 炭酸水素ナトリウム JIS K 8622に規定する炭酸水素ナトリウムのpH標準液用。
(h) 炭酸ナトリウム JIS K 8625に規定する炭酸ナトリウムのpH標準液用。
(2) pH標準液(1) pH標準液は,次のものを用いる。
(a) しゅう酸塩pH標準液 調製pH標準液の場合には,二しゅう酸三水素カリウム二水和物をめのう
乳鉢ですりつぶし,デシケーター中に18時間以上保存し,その12.71gを少量の水に溶かし,全量
フラスコ1 000mlに移し入れ,水を標線まで加える。これを共栓ポリエチレン瓶又は共栓ほうけい
酸ガラス瓶に入れて保存する。規格pH標準液の場合には,JIS K 0018に規定する標準物質‐pH標
準液‐しゅう酸塩の第2種を用いる。
(b) フタル酸塩pH標準液 調製pH標準液の場合には,あらかじめフタル酸水素カリウムを120℃で約
1時間加熱し,デシケーター中で放冷した後,その10.21gをとり,少量の水に溶かし,全量フラス
コ1 000mlに移し入れ,水を標線まで加える。これを共栓ポリエチレン瓶又は共栓ほうけい酸ガラ
ス瓶に入れて保存する。規格pH標準液の場合には,JIS K 0019に規定する標準物質‐pH標準液‐
フタル酸塩の第2種を用いる。
(c) 中性りん酸塩pH標準液 調製pH標準液の場合には,あらかじめりん酸二水素カリウムを105±2℃
で2時間,りん酸水素二ナトリウムは110℃で2時間それぞれ加熱し,デシケーター中で放冷した
後,そのりん酸二水素カリウム3.40gと,そのりん酸水素二ナトリウム3.55gとをとり,少量の水に
溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。これを共栓ポリエチレン瓶又は
共栓ほうけい酸ガラス瓶に入れて保存する。規格pH標準液の場合には,JIS K 0020に規定する標
準物質‐pH標準液‐中性りん酸塩の第2種を用いる。
(d) ほう酸塩pH標準液 調製pH標準液の場合には,四ほう酸ナトリウム十水和物をめのう乳鉢です
りつぶし,臭化ナトリウム溶液(飽和)に,更にJIS K 8514に規定する臭化ナトリウムを加えた溶
液を入れたデシケーター中に放置して恒量とした後,その3.81gをとり,少量の炭酸を含まない水
[2.(12)(b)による]に溶かし,全量フラスコ1 000mlに移し入れ,さきの炭酸を含まない水を標線ま
で加える。これを共栓ポリエチレン瓶又は共栓ほうけい酸ガラス瓶に一杯に満たした状態にして保
存する。規格pH標準液の場合には,JIS K 0021に規定する標準物質‐pH標準液‐ほう酸塩の第2
種を用いる。
(e) 炭酸塩pH標準液 調製pH標準液の場合には,炭酸水素ナトリウムをデシケーター中で約3時間
放置し,その2.10gをとる。別に,炭酸ナトリウムをあらかじめ白金るつぼに入れ,600℃で加熱し
て恒量とし,その2.65gをとる。両者を少量の炭酸を含まない水[2.(12)(b)による]に溶かし,全量
24
K 0101 : 1998
フラスコ1 000mlに移し入れ,さきの炭酸を含まない水を標線まで加える。これを共栓ポリエチレ
ン瓶又は共栓ほうけい酸ガラス瓶に入れ,JIS K 8603に規定するソーダ石灰又はJIS K 8574に規定
する水酸化カリウムなどを入れたデシケーター中に保存する。規格pH標準液の場合には,JIS K
0022に規定する標準物質‐pH標準液‐炭酸塩の第2種を用いる。
注(1) pH標準液は,調製pH標準液又は規格pH標準液の第2種を用いる。
調製pH標準液は,試験担当者が自らそれぞれのpH標準液の調製方法に従って調製したもの。
それぞれの調製pH標準液の各温度でのpH値は表11.1のpH値を適用する。
規格pH標準液は,国立の研究機関が保有する一次標準物質にトレーサブル(上位の標準に
求源性のある。)なpH標準液として体系化されたもの。pH標準液製造業者が調製したpH標準
液を,国の監督指導下にある公的機関(国立の研究機関の所有する一次pH標準液で確定した
二次pH標準液を所有する機関)に提出して検定を受け,合格した市販のpH標準液。
規格pH標準液には第1種と第2種があるが,第1種の規格pH標準液は,pH値を小数第3
位まで保証したもので,この試験に用いる第2種の規格pH標準液は,第1種のpH値の小数第
3位を四捨五入して小数第2位までにしたもの。各規格標準液の第2種の各温度でのpH値は表
11.2に示してある。
表11.1 調整pH標準液の各温度におけるpH値
温度
℃
pH値
しゅう酸塩
フタル酸塩 中性りん酸塩
ほう酸塩
炭酸塩(*)
0
1.67
4.01
6.98
9.46
10.32
5
1.67
4.01
6.95
9.39
(10.25)
10
1.67
4.00
6.92
9.33
10.18
15
1.67
4.00
6.90
9.27
(10.12)
20
1.68
4.00
6.88
9.22
(10.07)
25
1.68
4.01
6.86
9.18
10.02
30
1.69
4.01
6.85
9.14
(9.97)
35
1.69
4.02
6.84
9.10
(9.93)
38
−
−
−
−
9.91
40
1.70
4.03
6.84
9.07
−
45
1.70
4.04
6.83
9.04
−
50
1.71
4.06
6.83
9.01
−
55
1.72
4.08
6.84
8.99
−
60
1.73
4.10
6.84
8.96
−
70
1.74
4.12
6.85
8.93
−
80
1.77
4.16
6.86
8.89
−
90
1.80
4.20
6.88
8.85
−
95
1.81
4.23
6.89
8.83
−
注(*) 括弧内の値は,2次補間値を示す。
表11.2 規格pH標準液の各温度におけるpH値
温度
℃
pH値
しゅう酸塩
フタル酸塩
中性りん酸塩
ほう酸塩
炭酸塩
第2種
第2種
第2種
第2種
第2種
0
1.67
4.00
6.98
9.46
10.32
5
1.67
4.00
6.95
9.40
10.24
10
1.67
4.00
6.92
9.33
10.18
15
1.67
4.00
6.90
9.28
10.12
20
1.68
4.00
6.88
9.22
10.06
25
K 0101 : 1998
温度
℃
pH値
しゅう酸塩
フタル酸塩
中性りん酸塩
ほう酸塩
炭酸塩
第2種
第2種
第2種
第2種
第2種
25(*)
1.68(*)
4.01(*)
6.86(*)
9.18(*)
10.01(*)
30
1.68
4.02
6.85
9.14
9.97
35
1.69
4.02
6.84
9.10.
9.92
38
1.69
4.03
6.84
9.08
−
40
1.69
4.04
6.84
9.07
9.89
45
1.70
4.05
6.83
9.04
9.86
50
1.71
4.06
6.83
9.01
9.83
55
1.72
4.08
6.83
8.98
−
60
1.72
4.09
6.84
8.96
−
70
1.74
4.13
6.84
8.92
−
80
1.77
4.16
6.86
8.88
−
90
1.79
4.20
6.88
8.85
−
95
1.81
4.23
6.89
8.83
−
注(*) (*)印の25℃におけるpH値については,pH標準液ごとに日本工業規格として規定している。
備考1. 表11.1及び表11.2に記載されていない温度におけるpH値は,補間して求める。
2. 各pH標準液は,保存中にpH値が変化することがあるので長期間保存したものは使用しない。特
にほう酸塩pH標準液及び炭酸塩pH標準液は,容易に大気中の二酸化炭素を吸収しpH値が低下
するので注意する。
3. 各pH標準液は,一度使用したもの及び大気中に開放して放置したものは使用しない。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) pH計 JIS Z 8802に規定する形式IIを用いる(2)。
(b) 温度計 JIS B 7411に規定する一般用ガラス製棒状温度計の50度温度計又は100度温度計(3)。
注(2) 試験目的に応じてpH計の形式を選択する。
JIS Z 8802ではpH計の形式を0〜IIIの4段階に区分し,各形式のpH計の繰返し性は1種類
のpH標準液を用いてpH値を測定したとき,形式0では±0.005,形式Iでは±0.02,形式IIで
は±0.05,形式IIIでは±0.1と規定している。
(3) JIS B 7411に規定する一般用ガラス製棒状温度計で,各目盛における許容差±1℃のもの。
(4) pH計の校正 pH計の校正は,次のとおり行う。
(a) pH計の電源を入れ,検出部[ガラス電極(4)(5)及び参照電極(6),温度計など]を取り付ける。
(b) 検出部は,水で繰り返し3回以上洗い,きれいな柔らかい紙でぬぐっておく。
(c) 中性りん酸塩pH標準液をビーカーにとり検出部を浸す。温度補償用ダイヤル又はデジタルスイッ
チの設定のあるものは目盛値を,中性りん酸塩pH標準液の温度に合わせる(7)(8)。
(d) 中性りん酸塩pH標準液の温度に対応するpH値(表11.1又は表11.2)に調整ダイヤル(非対称電
位調整ダイヤル)を調節して合わせる。
(e) 検出部を水で繰り返し3回以上洗い,きれいな柔らかい紙などでぬぐっておく。
(f) 試料のpH値が7以下の場合は,フタル酸塩pH標準液又はしゅう酸塩pH標準液をビーカーにとり
検出部を浸す。スパン調整ダイヤルを調節して使用したpH標準液の温度に対応するpH値(表11.1
又は表11.2)に合わせる(8)。試料のpH値が7を超える場合は(9),ほう酸塩pH標準液又は炭酸塩
pH標準液を用い,同じ操作でpH標準液の温度に対応するpH値に合わせる(8)。
(g) 再び(b)〜(f)の操作を行い,pH値の指示値がpH標準液の温度に対応するpH値に±0.05(10)で一致す
るまでこの操作を繰り返す。
注(4) 長く乾燥状態にあったガラス電極は,あらかじめ水に浸して平衡に達してから使用する。

26
K 0101 : 1998
(5) ガラス電極が汚れている場合は,必要に応じて洗剤や塩酸 (1+20) などで短時間洗い,更に流
水で十分に洗う。電極の取扱いは,取扱説明書による。
(6) 参照電極の汚れの除去はガラス電極と同じ操作で行い,内部液(塩化カリウム溶液)の交換な
どは取扱説明書を参照する。
(7) pH標準液の温度は,できるだけ試料の温度に合わせる。
(8) 校正中は各pH標準液の温度変動は±2℃とする。
(9) 試料のpH値が11以上の場合には,通常のガラス電極ではアルカリ誤差を生じ,測定値が低く
なる。特にアルカリ金属イオン濃度が高い場合は誤差が大きくなるので,備考4.によって測定
する。
(10) pH計形式Iでは±0.02,pH計形式IIIでは±0.1で一致するまで繰り返す。
(5) 操作 操作は,次のとおり行う。
(a) 校正したpH計の検出部を水で繰り返し3回以上洗い,きれいな柔らかい紙などでぬぐっておく。
(b) 試料をビーカーにとり検出部を浸す。温度補償用ダイヤル又はデジタルスイッチの設定のあるもの
は目盛値を試料の温度(11)に合わせた後pH値を測定する。
(c) 検出部を取り出し,水で繰り返し3回以上洗い,きれいな柔らかい紙などでぬぐっておく。
(d) 再び試料をビーカーにとり検出部を浸し,pH値を測定する(11)。
(e) 再び(c)及び(d)の操作を行って3回の測定値が±0.1(12)で一致した測定値を平均して試料のpH値を
算出する。
注(11) pH値は試料の温度によって異なるので,試料の温度変動は±2℃とする。
(12) 緩衝性の低い試料は,容易にpH値が変化するため,pH値が±0.1の繰返し性が得られない場
合がある。この場合は,pH値が±0.2で一致する値を平均してpH値を算出する。
また,大気中の二酸化炭素で容易にpH値が変動する場合には,流液形の電極を使用すると
よい。
備考4. 試料のpH値が11以上で,特にアルカリ金属元素の濃度が高い場合には,アルカリ誤差を生
じやすいため,アルカリ誤差の少ない電極(例えば,リチウム電極など)を用い,炭酸塩を
含まない0.1mol/l水酸化ナトリウム溶液又は25℃の水酸化カルシウム溶液(飽和)を用いて
pH計の校正を行う。
0.1mol/l水酸化ナトリウム溶液又は25℃の水酸化カルシウム溶液(飽和)は,大気中の二
酸化炭素を吸収して容易にpHが低下するので使用の都度調製する。
表11.3に0.1mol/l水酸化ナトリウム溶液及び水酸化カルシウム溶液(飽和)の各温度にお
けるpH値を示す。
表11.3 0.1mol/l水酸化ナトリウム溶液及び水酸化カルシウム溶液(飽和)の各温度におけるpH値
温度
℃
0.1mol/l水酸化
ナトリウム溶液
水酸化カルシウム
溶液(飽和)(*)
温度
℃
0.1mol/l水酸化
ナトリウム溶液
水酸化カルシウム
溶液(飽和)(*)
0
13.8
13.43
35
12.6
12.14
5
13.6
13.21
40
12.4
11.99
10
13.4
13.00
45
12.3
11.84
15
13.2
12.81
50
12.2
11.70
20
13.1
12.63
55
12.0
11.58
25
12.9
12.45
60
11.9
11.45
30
12.7
12.30
注(*) 25℃における水酸化カルシウム溶液(飽和)
27
K 0101 : 1998
12. 電気伝導率 電気伝導率は,溶液がもつ電気抵抗率 (Ω・m) の逆数に相当し,S/mの単位で表す。
また,電気伝導度は,溶液がもつ電気抵抗 (Ω) の逆数に相当し,Sの単位で表す。
水の試験では,温度25℃の値を用いS/m及びSの千分の一を単位とし,それぞれmS/m(1)及びmSで表
す(2)。試料の電気伝導率が1mS/m (25℃) 以下の測定の場合には,JIS K 0552を適用する。
注(1) mS/mは,ミリジーメンス毎メートルと読む。
(2) 従来の単位で表した1μS/cmは0.1mS/mに相当 (1μS/cm=1×10-6S/10-2m=1×10-4S/m=0.1mS/m)
する。
備考1. 従来,水の試験では,電気伝導度及び電気伝導率の単位としてそれぞれμS及びμS/cm,また,
セル定数の単位としてcm-1が用いられていた。
電気伝導度としては,mSの単位で表した数値を1 000倍するとμSの単位で表した数値と
なる。
電気伝導率としては,mS/mの単位で表した数値を10倍するとμS/cmの単位で表した数値
となる。
また,セル定数としては,m-1の単位で表した数値を0.01倍するとcm-1の単位で表した数
値となる。
なお,{ }で表した単位及び数値は従来の単位によるもので,参考として併記した。
(1) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気伝導度計 検出部と指示部からなるもの。検出部は,白金電極面に白金黒めっきを行った電極
を組み入れたセルからなる。セル定数は表12.1に示したものを用意する。指示部は,ホイートスト
ンブリッジ回路などを組み入れたものを用いる。セルは,水中に保存する(3)。
(b) 温度計 JIS B 7411に規定する一般用ガラス製棒状温度計の50度温度計
注(3) セルは,備考2.の方法で定期的に確認する。
(2) 操作 操作は,次のとおり行う。
(a) あらかじめ電気伝導度計の電源を入れておく。試料の電気伝導率に応じて表12.1に示すセル定数を
もった電極を用い,水でセルを2,3回洗う[特に汚れている場合には,塩酸 (1+100) に浸し,更
に流水で十分に洗い,最後に水で2,3回洗う。]。
(b) このセルを試料で2,3回洗った後,試料を満たし,25±0.5℃(4)に保って電気伝導度(5)(6)の測定を
行う。測定値が±3%(7)で一致するまで試料を数回取り替えて測定を繰り返し,その電気伝導度を求
める。
(c) 電気伝導度から,次の式によって試料の電気伝導率 (mS/m) (25℃) を算出する。
L=J×Lx
ここに,
L: 試料の電気伝導率 (mS/m) (25℃)
J: セル定数 (m-1)
Lx: 測定した電気伝導度 (mS)
注(4) 精度を特に必要としない場合には,温度補償回路を組み入れた電気伝導度計を用いるか,温度
換算式を用いてもよい。電気伝導率は,温度によって変化し,1℃の上昇で約2%大きくなる。
ただし,電気伝導率が1mS/m {10μS/cm} 以下になると,水の解離によって生じる水素イオン及
び水酸化物イオンの影響が大きくなるので,この換算式は適用できない。
(5) 電気伝導度計の指示が電気抵抗 (Ω) になっているときは,次の式によって電気伝導率を計算す
28
K 0101 : 1998
る。
3
10
×
=
x
R
J
L
ここに,
L: 試料の電気伝導率 (mS/m) (25℃)
J: セル定数 (m-1) ただし,電気抵抗率 (Ω・m) が直示される場
合は1とする。
Rx: 測定した電気抵抗 (Ω)
(6) 電気伝導度計の指示が電気伝導度 (μS) になっている場合には,次の式によって電気伝導率
(mS/m) を算出する。
L=J'×Lx'×0.1
ここに,
L: 試料の電気伝導率 (mS/m) (25℃)
J': セル定数 (cm-1)
Lx': 測定した電気伝導度 (μS)
(7) 試料の電気伝導率が1mS/m (25℃) 未満の場合には,±3%で一致しないことがあるので,JIS K
0552に従って試験するか,又は流液形のセルを用いる。
表12.1 セル定数と測定範囲
セル定数(*)
測定範囲
m-1
{cm-1}
mS/m
{μS/cm}
1
0.01
2以下
20以下
10
0.1
0.1 〜
200
1〜
200
100
1
1 〜 2 000
10〜
2 000
1 000
10
10 〜 20 000
100〜20 000
5 000
50
100 〜 200 000 1 000〜200 000
注(*) セル定数m-1×0.01=cm-1
備考2. セル定数の測定又はセル定数の確認 セル定数の測定又はセル定数の確認は,試料を試験す
るたびに行う必要はないが,定期的に塩化カリウム標準液 (A〜D) を用いてその数値を確か
める。セル定数の測定及びセル定数の確認は次による。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水。ただし,電気伝導率0.2mS/m {2μS/cm}
(25℃) 以下のもの。調製時に20±2℃に調節して用いる。
(b) 塩化カリウム JIS K 8121に規定する塩化カリウム(電気伝導率測定用)をめのう乳鉢で
粉末にし,500℃で約4時間加熱し,デシケーター中で放冷したもの。
(c) 塩化カリウム標準液 (A) 塩化カリウム74.246gをはかりとり,少量の水に溶かして全量
フラスコ1 000mlに移し入れ,水を標線まで加える。
(d) 塩化カリウム標準液 (B) 塩化カリウム7.437gをはかりとり,少量の水に溶かして全量
フラスコ1 000mlに移し入れ,水を標線まで加える。
(e) 塩化カリウム標準液 (C) 塩化カリウム0.744gをはかりとり,少量の水に溶かして全量
フラスコ1 000mlに移し入れ,水を標線まで加える。
(f) 塩化カリウム標準液 (D) 塩化カリウム標準液 (C) 100mlを全量フラスコ1 000mlにとり,
水を標線まで加える。
これらの塩化カリウム標準液は,ポリエチレン瓶又はほうけい酸ガラス瓶に密栓をして保
存する。塩化カリウム標準液の電気伝導率を表12.2に示す。
29
K 0101 : 1998
表12.2 塩化カリウム標準液 (A〜D) の電気伝導率
塩化カリウム標準液
℃
mS/m
{μS/cm}
A
0
6518
65180
18
9784
97840
25
11134
111340
B
0
714
7140
18
1117
11170
25
1286
12860
C
0
77.4
774
18
122.0
1220
25
140.9
1409
D
25
14.7
147
(2) 操作 操作は,次のとおり行う。
(a) セル定数の測定 セル定数を測定又は確認しようとする場合には,セルを水で2,3回洗い,
次に,塩化カリウム標準液(セル定数に応じ,表12.1の測定範囲の塩化カリウム標準液を
用いる。)で2,3回洗った後,その塩化カリウム標準液を満たす。このセルを25±0.5℃に
保ち,電気伝導度を測定する。同じ塩化カリウム標準液を数回入れ替えて測定を行い,測
定値が±3%で一致するまで繰り返す(±3%で一致しない場合には白金黒電極に,備考3.
によって新たに白金黒めっきを行う。)。
測定された値から次の式によってセル定数を算出する。
XO
O
H
KCl
L
L
L
J
2
+
=
ここに,
J: セル定数 (m-1)
LXO: 測定した電気伝導度 (mS) ,ただし,電気伝導度の指示
がμSになっているときは,μS×1000
1値を用いる。
LKCl: 使用した塩化カリウム標準液のこの温度における電気伝
導率 (mS/m)
O
H
L2: 塩化カリウム標準液の調製に用いた水のこの温度におけ
る電気伝導率 (mS/m)
なお,電気伝導度がμSとなっている場合でセル定数をcm-1の単位で求めたいときは,次
の式による。
′
′
+
′
=
′
XO
O
H
KCl
L
L
L
J
2
ここに,
J': セル定数 {cm-1}
LXO': 測定した電気伝導度 (μS)
LKC1': 使用した塩化カリウム標準液のこの温度における電気
伝導率 {μS/cm}
O
H
L2: 塩化カリウム標準液の調製に用いた水のこの温度にお
ける電気伝導率 {μS/cm}
また,表12.1中で濃度の接近している2種類の塩化カリウム標準液を用いてセル定数を
測定し,もし,その値が±1%で一致しないときは,白金黒電極の場合は備考3.によって,
白金極面の白金黒を溶かし,新たに白金黒めっきを行う。
備考3. 電極の白金黒めっき 電極の白金黒めっきは,次のとおり行う。
(a) 白金黒は,塩酸 (1+11) 中で白金黒電極を陽極として電解すると,容易に取り除くことがで
30
K 0101 : 1998
きる。
(b) この白金電極をヘキサクロロ白金 (IV) 酸 (30g/l) と酢酸鉛 (0.25g/l) からなる電解液に入れ,
直流約6V,電流密度10〜40A/m2 {1〜4mA/cm2} とし,適当な方法で電解液をかき混ぜなが
ら,15〜30kC(クーロン)/m2 {1.5〜3.0C/cm2} を通電する。
(c) 次に,硫酸 (1+360) 中で約30分間,時々電流の方向を変えて通電し,付着,吸蔵したヘキ
サクロロ白金 (IV) 酸及び塩素を除く。
4. 市販の測定装置は,電気伝導率が直接指示されるものが多い。必要に応じて塩化カリウム標
準液で,指示値を確認して用いる。
13. 酸消費量 酸消費量は,水に溶けている炭酸水素塩,炭酸塩,水酸化物などのアルカリを所定のpH
に中和するのに要する水素イオンの量(酸の量)を,試料1lについてのmmol数で表すか,又は水素イオ
ン(酸)に相当する炭酸カルシウムの量に換算して,試料1lについてのmg数で表す。
酸消費量を酸消費量 (pH4.8) と酸消費量 (pH8.3) に区分する。
13.1 酸消費量 (pH4.8) 酸消費量 (pH4.8) は,試料に指示薬としてメチルレッド‐ブロモクレゾールグ
リーン混合溶液を加え,10mmol/l硫酸で滴定して求める。
(1) 試薬 試薬は,次のものを用いる。
(a) メチルレッド-ブロモクレゾールグリーン混合溶液 JIS K 8896に規定するメチルレッド0.02gと
JIS K 8840に規定するブロモクレゾールグリーン0.1gとをJIS K 8102に規定するエタノール (95)
100mlに溶かす。
(b) 50mmol/l硫酸 JIS K 8951に規定する硫酸3mlを,あらかじめ水100mlを入れたビーカーに加えて
よくかき混ぜ,放冷後,水を加えて1lとする。
標定 JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムを600℃で約1時間加熱した後,
デシケーター中で放冷する。その1.06gを1mgのけたまではかりとり,水に溶かして全量フラスコ
200mlに移し入れ,水を標線まで加える。
この20mlをビーカーにとり,指示薬としてメチルレッド‐ブロモクレゾールグリーン混合溶液3
〜5滴を加えた後,この50mmol/l硫酸で滴定する。溶液の色が灰紫になったら,煮沸して二酸化
炭素を追い出し,放冷後,溶液の色が灰紫になるまで滴定を続ける。滴定に要した50mmol/l硫酸
のml数から,次の式によって50mmol/l硫酸のファクター (f) を算出する。
30
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 炭酸ナトリウムの量 (g)
b: 炭酸ナトリウムの純度 (%)
x: 滴定に要した50mmol/l硫酸 (ml)
0.005 30: 50mmol/l硫酸1mlの炭酸ナトリウム相当量 (g)
(c) 10mmol/l硫酸 50mmol/l硫酸200mlを全量フラスコ1 000mlにとり,水を標線まで加える。
(2) 操作 操作は,次のとおり行う。
(a) 試料(濁りがあるときはろ紙5種Bでろ過するか又は遠心分離してその上澄み液を用いる。)100ml
をビーカーにとり,指示薬としてメチルレッド‐ブロモクレゾールグリーン混合溶液3〜5滴を加え
る(1)。
(b) この溶液を緩やかにかき混ぜながら10mmol/l硫酸で,溶液の色が青から灰紫 (pH 4.8) になるまで
31
K 0101 : 1998
滴定する。
(c) 次の式によって酸消費量 (pH 4.8) を算出する。
i)
mmol/lで表す場合
02
.0
1000×
×
×
=
V
f
a
A
ここに,
A: 酸消費量 (pH4.8) (mmol/l)
a: 滴定に要した10mmol/l硫酸 (ml)
f: 10mmol/l硫酸のファクター(2)
V: 試料 (ml)
0.02: 10mmol/l硫酸1mlの水素イオン相当量 (mmol)
ii)
mgCaCO3/lで表す場合
001
.1
1000×
×
×
=
V
f
a
B
ここに,
B: 酸消費量 (pH4.8) (mgCaCO3/l)
a: 滴定に要した10mmol/l硫酸 (ml)
f: 10mmol/l硫酸のファクター(2)
V: 試料 (ml)
1.001: 10mmol/l硫酸1mlの炭酸カルシウム相当量 (mg)
注(1) 残留塩素などの酸化性物質が共存する場合には,チオ硫酸ナトリウム溶液 (0.1mol/l) で還元し
てから加える。
(2) 50mmol/l硫酸のファクターを用いる。
備考1. 着色した試料など,指示薬による変色が明らかでない場合には,指示薬に代えてpH計を使
用し,pH-10mmol/l硫酸の滴定曲線を作成して,pH4.8における10mmol/l硫酸のml数を求め
る。
13.2 酸消費量 (pH 8.3) 酸消費量 (pH 8.3) は,試料に指示薬としてフェノールフタレイン溶液を加え,
10mmol/l硫酸で滴定して求める。
(1) 試薬 試薬は,次のものを用いる。
(a) フェノールフタレイン溶液 (5g/l) JIS K 8799に規定するフェノールフタレイン0.5gをとり,JIS K
8102に規定するエタノール (95) 50mlに溶かし,水を加えて100mlとし,この溶液の色がわずかに
赤に変わるまで20mmol/l水酸化ナトリウム溶液を滴加する。
(b) 10mmol/l硫酸 13.1(1)(c)による。
(2) 操作 操作は,次のとおり行う。
(a) 試料100mlをビーカーにとり,指示薬としてフェノールフタレイン溶液 (5g/l) 3〜5滴を加える。
(b) この溶液を緩やかにかき混ぜながら10mmol/l硫酸で,溶液の赤い色が消えるまで滴定する。
(c) 次の式によって酸消費量 (pH 8.3) を算出する。
i)
mmol/lで表す場合
02
.0
1000×
×
×
=
V
f
a
A
ここに,
A: 酸消費量 (pH 8.3) (mg当量/l)
a: 滴定に要した10mmol/l硫酸 (ml)
f: 10mmol/l硫酸のファクター(2)
V: 試料 (ml)
0.02: 10mmol/l硫酸1mlの水素イオン相当量 (mmol)
32
K 0101 : 1998
ii)
mgCaCO3/lで表す場合
001
.1
1000×
×
×
=
V
f
a
B
ここに,
B: 酸消費量 (pH 8.3) (mgCaCO3/l)
a: 滴定に要した10mmol/l硫酸 (ml)
f: 10mmol/l硫酸のファクター(2)
V: 試料 (ml)
1.001: 10mmol/l硫酸1mlの炭酸カルシウム相当量 (mg)
備考2. 備考1.による。ただし,pH 8.3における10mmol/l硫酸のml数を求める。
14. アルカリ消費量 アルカリ消費量は,水に溶けている強酸,炭酸,有機酸及び水酸化物として沈殿す
る金属元素などを所定のpHまで中和するのに要する水酸化物イオンの量(アルカリの量)を試料1lにつ
いてのmmol数で表すか,又は水酸化物イオンの量(アルカリの量)に相当する炭酸カルシウムの量に換
算して,試料1lについてのmg数で表す。
アルカリ消費量は,アルカリ消費量 (pH 8.3) ,アルカリ消費量 (pH 4.8) 及びアルカリ消費量(遊離酸)
に区分する。
14.1 アルカリ消費量 (pH 8.3) アルカリ消費量 (pH 8.3) は,試料に指示薬としてフェノールフタレイ
ン溶液を加え,20mmol/l水酸化ナトリウム溶液で滴定して求める。
(1) 試薬 試薬は,次のものを用いる。
(a) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(b) 0.1mol/l水酸化ナトリウム溶液 水約30mlをポリエチレン瓶にとり,冷却しながらJIS K 8576に規
定する水酸化ナトリウム約35gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。その上澄み
液5mlをポリエチレン製の気密容器1lにとり,2.(12)(b)の炭酸を含まない水を加えて1lとし,混合
した後,二酸化炭素を遮断して保存する。
標定 JIS K 8005に規定する容量分析用標準物質のアミド硫酸をデシケーター中に2kPa以下で約
48時間放置して乾燥する。その約0.2gを1mgのけたまではかりとり,三角フラスコ200mlに入れ,
水約25mlを加えて溶かす。これに指示薬としてブロモチモールブルー溶液 (1g/l) (1)3〜5滴を加え,
この0.1mol/l水酸化ナトリウム溶液で滴定し,溶液の色が緑になったときを終点とする。次の式に
よって0.1mol/l水酸化ナトリウム溶液のファクター (f) を算出する。
00971
.0
1
100
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の量 (g)
b: アミド硫酸の純度 (%)
x: 滴定に要した0.1mol/l水酸化ナトリウム溶液 (ml)
0.009 71: 0.1mol/l水酸化ナトリウム溶液1mlのアミド硫酸相当
量 (g)
(c) 20mmol/l水酸化ナトリウム溶液 0.1mmol/l水酸化ナトリウム溶液200mlを全量フラスコ1 000ml
にとり,2.(12)(b)の炭酸を含まない水を標線まで加える。この溶液は使用時に調製する。
注(1) JIS K 8842に規定するブロモチモールブルー0.1gをとり,JIS K 8102に規定するエタノール (95)
50mlに溶かし,水で100mlとする。
備考1. アミド硫酸による標定の代わりに,JIS K 8005に規定する容量分析用標準物質の炭酸ナトリ
ウムを用いて標定した50mmol/l硫酸を用いて,次のように標定してもよい。
33
K 0101 : 1998
標定 13.1(1)(b)の50mmol/l硫酸20mlをビーカーにとり,指示薬としてフェノールフタレ
イン溶液 (5g/l) 3〜5滴を加えた後,この0.1mol/l水酸化ナトリウム溶液で溶液の色がわず
かに赤になるまで滴定する。滴定に要した0.1mol/l水酸化ナトリウム溶液のml数から,次
の式によって0.1mol/l水酸化ナトリウム溶液のファクター (f1) を算出する。
x
f
f
×
=20
1
ここに,
f: 50mmol/l硫酸のファクター
x: 滴定に要した0.1mol/l水酸化ナトリウム溶液 (ml)
(2) 器具 器具は,次のとおりとする。
(a) メスシリンダー(有栓形) 100ml又は共栓ガラス円筒100ml
(3) 操作 操作は,次のとおり行う。
(a) 試料容器をなるべく振り動かさないように注意し,試料(2)100mlをメスシリンダー(有栓形)100ml
にとる。
(b) 指示薬としてフェノールフタレイン溶液 (5g/l) 4〜5滴を加えて白紙上に置き,20mmol/l水酸化ナト
リウム溶液で溶液がわずかに赤い色(3)(4)になるまで滴定し,これを予備試験とする。
(c) 別のメスシリンダー(有栓形)100mlに試料100mlを(a)と同じ操作でとり,指示薬としてフェノー
ルフタレイン溶液 (5g/l) 4〜5滴を加え,これに(b)の予備試験で消費した量と同量の20mmol/l水酸
化ナトリウム溶液を一度に加え,栓をして静かに振り混ぜ,溶液の赤い色が消えたら更に滴定を続
け,わずかに赤い色になるまで滴定する。
(d) 次の式によってアルカリ消費量 (pH8.3) を算出する。
i)
mmol/lで表す場合
02
.0
000
1
1
×
×
×
=
V
f
a
A
ここに,
A: アルカリ消費量 (pH 8.3) (mmol/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
0.02: 20mmol/l水酸化ナトリウム溶液1mlの水酸化物イオン相当
量 (mmol)
ii)
mgCaCO3/lで表す場合
001
.1
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量 (pH 8.3) (mgCaCO3/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
1.001: 20mmol/l水酸化ナトリウム溶液1mlの炭酸カルシウム相
当量 (mg)
注(2) 試料に色や濁りがある場合には,試料100mlをメスシリンダー(有栓形)にとり,これを対照
にして滴定する。
(3) 終点における着色の強さは,炭酸水素ナトリウム溶液 (10mmol/l) 100mlをメスシリンダー(有
栓形)100mlにとり,フェノールフタレイン溶液 (5g/l) 4,5滴を加えて振り混ぜたときの色と
等しくなるようにする。
34
K 0101 : 1998
(4) メスシリンダー(有栓形)を白紙上に置き,斜め上から観察する。
(5) 0.1mol/l水酸化ナトリウム溶液のファクターを用いる。
14.2 アルカリ消費量 (pH4.8) アルカリ消費量 (pH4.8) は,試料に指示薬としてメチルレッド‐ブロモ
クレゾールグリーン混合溶液を加え,20mmol/l水酸化ナトリウム溶液で滴定して求める。この測定値には,
鉄及びアルミニウムなどの塩類との反応に要するアルカリの量も含まれる。
(1) 試薬 試薬は,次のものを用いる。
(a) メチルレッド‐ブロモクレゾールグリーン混合溶液 13.1(1)(a)による。
(b) 20mmol/l水酸化ナトリウム溶液 14.1(1)(c)による。
(2) 操作 操作は,次のとおり行う。
(a) 試料(2)100mlをビーカーにとり,指示薬としてメチルレッド‐ブロモクレゾールグリーン混合溶液
3〜5滴を加える。
(b) この溶液を緩やかにかき混ぜながら,20mmol/l水酸化ナトリウム溶液で溶液の色が赤から灰紫
(pH4.8) に変わるまで滴定する。
(c) 次の式によってアルカリ消費量 (pH4.8) を算出する。
i)
mmol/lで表す場合
02
.0
000
1
1
×
×
×
=
V
f
a
A
ここに,
A: アルカリ消費量 (pH 4.8) (mmol/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
00.2: 20mmol/l水酸化ナトリウム溶液1mlの水酸化物イオン相当
量 (mmol)
ii)
mgCaCO3/lで表す場合
001
.1
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量 (pH 4.8) (mgCaCO3/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
1.001: 20mmol/l水酸化ナトリウム溶液1mlの炭酸カルシウム相
当量 (mg)
備考2. 13. の備考1.による。ただし,20mmol/l水酸化ナトリウム溶液のml数を求める。
14.3 アルカリ消費量(遊離酸) アルカリ消費量(遊離酸)は,水に溶けている硫酸,塩酸,硝酸,強
い有機酸などをしゅう酸カリウムの存在で20mmol/l水酸化ナトリウム溶液で滴定し,pH-水酸化ナトリウ
ム溶液 (ml) 滴定曲線から求める。
この測定値には,鉄,アルミニウムなどの塩類によるアルカリ消費量は含まれず,遊離酸に対応するア
ルカリ消費量が求められる。
(1) 試薬 試薬は,次のものを用いる。
(a) しゅう酸カリウム一水和物 JIS K 8522に規定するもの。
(b) 20mmol/l水酸化ナトリウム溶液 14.1(1)(c)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
35
K 0101 : 1998
(a) マグネチックスターラー
(b) pH計 JIS Z 8802に規定する形式IIのもの。
(3) 操作 操作は,次のとおり行う。
(a) 試料100mlをビーカーにとり,液温を約10℃に調節する。
(b) しゅう酸カリウム一水和物約20gを加え,かき混ぜて溶かす。
(c) マグネチックスターラーでかき混ぜ,pH計を用いてpHを測定しながら20mmol/l水酸化ナトリウ
ム溶液で滴定する。
(d) 滴定の終点(変曲点)前後では,20mmol/l水酸化ナトリウム溶液を0.1mlずつ加える。
(e) pHと20mmol/l水酸化ナトリウム溶液 (ml) の滴定曲線を作成して終点を求め,次の式によってア
ルカリ消費量(遊離酸)を算出する。
i)
mmol/lで表す場合
02
.0
000
1
1
×
×
×
=
V
f
a
A
ここに,
A: アルカリ消費量(遊離酸) (mmol/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
0.02: 20mmol/l水酸化ナトリウム溶液1mlの水酸化物イオン相当
量 (mmol)
ii)
mgCaCO3/lで表す場合
001
.1
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(遊離酸) (mgCaCO3/l)
a: 滴定に要した20mmol/l水酸化ナトリウム溶液 (ml)
f1: 20mmol/l水酸化ナトリウム溶液のファクター(5)
V: 試料 (ml)
1.001: 20mmol/l水酸化ナトリウム溶液1mlの炭酸カルシウム相
当量 (mg)
15. 硬度 硬度は,水中のカルシウムイオン及びマグネシウムイオンの量を,これに対応する炭酸カルシ
ウムの量に換算して試料1lについてのmg数で表す。
硬度は,全硬度,カルシウム硬度及びマグネシウム硬度に区分する。場合によって,全硬度,非炭酸塩
硬度及び炭酸塩硬度に区分する。
15.1 全硬度
15.1.1 キレート滴定法
(1) ろ過した試料について50.1(2)において滴定に要する10mmol/l EDTA溶液の量を求め,次の式によって
全硬度を算出する。
001
.1
000
1
×
×
=
V
b
H
ここに,
H: 全硬度 (mgCaCO3/l)
b: 50.1(2)の滴定に要した10mmol/l EDTA溶液 (ml)
V: 試料 (ml)
1.001: 10mmol/l EDTA溶液1mlの炭酸カルシウム相当量 (mg)
36
K 0101 : 1998
備考1. 非炭酸塩硬度及び炭酸塩硬度は,酸消費量 (pH 4.8) と全硬度との関係から,次のように算出
する。
酸消費量 (pH4.8) (mgCaCO3/l) <全硬度 (mgCaCO3/l)
のときは,
非炭酸塩硬度 (mgCaCO3/l) =全硬度 (mgCaCO3/l) −酸消費量 (pH4.8)
(mgCaCO3/l)
酸消費量 (pH4.8) (mgCaCO3/l) ≧全硬度 (mgCaCO3/l)
のときは,
炭酸塩硬度 (mgCaCO3/l) =全硬度 (mgCaCO3/l)
非炭酸塩硬度 (mgCaCO3/l) =0
15.1.2 フレーム原子吸光法
(1) ろ過した試料について49.2によってカルシウムの濃度を,また,50.2によってマグネシウムの濃度を
求め,次の式によって全硬度を算出する。
H= (2.497×CCa) + (4.118×CMg)
ここに,
H: 全硬度 (mgCaCO3/l)
CCa: カルシウムの濃度 (mgCa/l)
2.497: カルシウムの量を炭酸カルシウム相当量に換算する場合
の係数 (100.09/40.08)
CMg: マグネシウムの濃度 (mgMg/l)
4.118: マグネシウムの量を炭酸カルシウム相当量に換算する場
合の係数 (100.09/24.305)
備考2. 備考1.による。
15.1.3 ICP発光分光分析法
(1) ろ過した試料について49.3によってカルシウムの濃度を,また,50.3によってマグネシウムの濃度を
求め,15.1.2(1)の式によって全硬度を算出する。
備考3. 備考1.による。
15.2 カルシウム硬度
15.2.1 キレート滴定法
(1) ろ過した試料について49.1(2)において滴定に要する10mmol/l EDTA溶液の量を求め,次の式によって
カルシウム硬度を算出する。
001
.1
000
1
×
×
=
V
a
HCa
ここに,
HCa: カルシウム硬度 (mgCaCO3/l)
a: 49.1(2)の滴定に要した10mmol/l EDTA溶液 (ml)
V: 試料 (ml)
1.001: 10mmol/l EDTA溶液1mlの炭酸カルシウム相当量 (mg)
15.2.2 フレーム原子吸光法
(1) ろ過した試料について49.2によってカルシウムの濃度を求め,次の式によってカルシウム硬度を算出
する。
HCa=2.497×CCa
ここに,
HCa: カルシウム硬度 (mgCaCO3/l)
CCa: カルシウムの濃度 (mgCa/l)
2.497: カルシウムの量を炭酸カルシウム相当量に換算する場合
37
K 0101 : 1998
の係数 (100.09/40.08)
15.2.3 ICP発光分光分析法
(1) ろ過した試料について49.3によってカルシウムの濃度を求め,15.2.2(1)の式によってカルシウム硬度
を算出する。
15.3 マグネシウム硬度
15.3.1 キレート滴定法
(1) 50.1(2)においてカルシウム及びマグネシウムの滴定に要する10mmol/l EDTA溶液の量を,また,49.1(2)
においてカルシウムの滴定に要する10mmol/l EDTA溶液の量を求め,次の式によってマグネシウム硬
度を算出する。
001
.1
000
1
×
×
−
=
Ca
Mg
V
a
V
b
H
ここに,
HMg: マグネシウム硬度 (mgCaCO3/l)
b: 50.1(2)の滴定に要した10mmol/l EDTA溶液 (ml)
V: 50.1(2)の試料 (ml)
a: 49.1(2)の滴定に要した10mmol/l EDTA溶液 (ml)
VCa: 49.1(2)の試料 (ml)
1.001: 10mmol/l EDTA溶液1mlの炭酸カルシウム相当量 (mg)
15.3.2 フレーム原子吸光法
(1) ろ過した試料について50.2によってマグネシウムの濃度を求め,次の式によってマグネシウム硬度を
算出する。
HMg=4.118×CMg
ここに,
HMg: マグネシウム硬度 (mgCaCO3/l)
CMg: マグネシウムの濃度 (mgMg/l)
4.118: マグネシウムの量を炭酸カルシウム相当量に換算する場
合の係数 (100.09/24.305)
15.3.3 ICP発光分光分析法
(1) ろ過した試料について50.3によってマグネシウムの濃度を求め,15.3.2(1)の式によってマグネシウム
硬度を算出する。
16. 懸濁物質及び蒸発残留物 水中に懸濁している物質及び水を蒸発したときの残留物質を次のように区
分して試験する。
(1) 懸濁物質 試料をろ過したとき,ろ過材上に残留する物質。
(2) 全蒸発残留物 試料を蒸発乾固したときに残留する物質。
(3) 溶解性蒸発残留物 懸濁物質をろ別したろ液を蒸発乾固したときに残留する物質。
(4) 強熱残留物 懸濁物質,全蒸発残留物及び溶解性蒸発残留物のそれぞれを600±25℃で30分間強熱し
たときの残留物で,それぞれの強熱残留物として示す。
(5) 強熱減量 (4)の測定時における減少量で,それぞれの強熱減量として示す。
試験は,試料採取後直ちに行う。直ちに行えない場合には,試料を0〜10℃の暗所に保存し,できるだ
け早く試験する。
16.1 懸濁物質 試料をろ過し,ろ過材上に残留した物質を105〜110℃で乾燥し,質量をはかる。
(1) 器具 器具は,次のとおりとする。
(a) ろ過器(分離形) 図16.1に一例を示す。
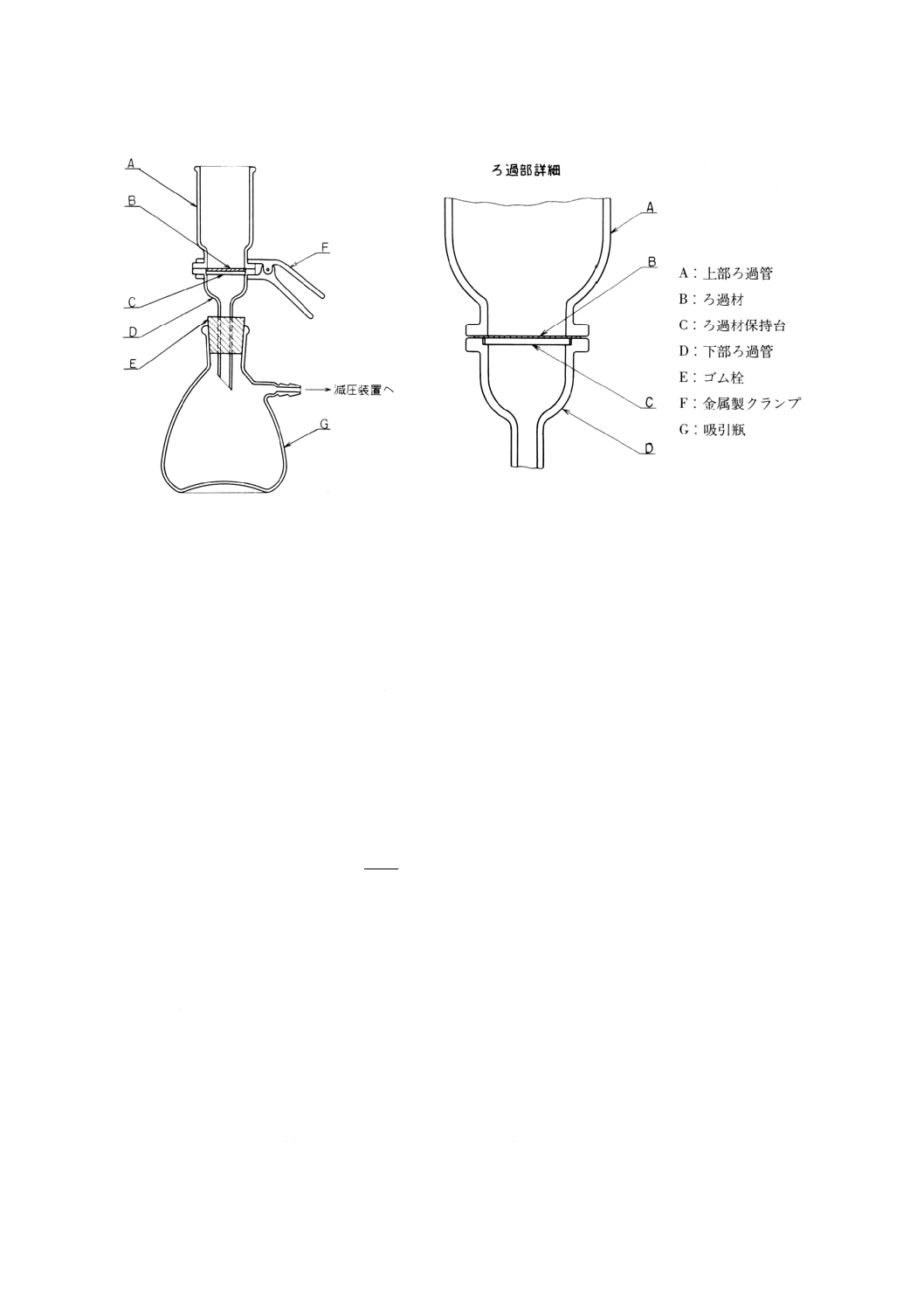
38
K 0101 : 1998
図16.1 ろ過器(分離形)の一例
(b) ろ過材 ガラス繊維ろ紙,有機性ろ過膜又は金属性ろ過膜。孔径1μmで直径25〜50mm。
備考1. ガラス繊維ろ紙は目詰まりが少ないが,ガラス繊維が離脱するおそれがある。有機性ろ過膜
は,種類によって耐薬品,耐熱性に差があるので,取扱いに注意する。
(2) 操作 操作は,次のとおり行う。
(a) ガラス繊維ろ紙を用いる場合はあらかじめろ過器に取り付け,水で十分に吸引洗浄した後,このろ
過材(1)を,時計皿(2)上に置き,105〜110℃で約1時間加熱し(3),デシケーター中で放冷した後,そ
の質量をはかる。
(b) ろ過材をろ過器に取り付け,試料(4)の適量(5)をろ過器に注ぎ入れて吸引ろ過する。試料容器及びろ
過管の器壁に付着した物質は,水でろ過材上に洗い落とし,ろ過材上の残留物質に合わせ,これを
水で数回洗浄する。
(c) 残留物は,ろ過材とともにピンセットなどを用いてろ過器から注意して取り外し,(a)で用いた時計
皿上に移し,105〜110℃で2時間加熱し,先のデシケーター中で放冷した後,その質量をはかる。
(d) 次の式によって懸濁物質 (mg/l) を算出する。
V
b
a
S
000
1
)
(
×
−
=
ここに, S: 懸濁物質 (mg/l)
a: 懸濁物質を含んだろ過材及び時計皿の質量 (mg)
b: ろ過材及び時計皿の質量 (mg)
V: 試料 (ml)
注(1) アクリル樹脂などのバインダー処理を行ったガラス繊維ろ紙,有機性ろ過膜,金属性ろ過膜は
洗浄しなくてよい。
(2) なるべく軽い時計皿を用いるか,又はアルミニウムはくなどの軽い容器を用いる。
(3) 有機性ろ過膜では105〜110℃で加熱すると,変形するものがある。この場合は90℃で加熱する。
(4) 目開き2mmのふるいを通過した試料を用い,十分に振り混ぜて懸濁物質が均一になってから,
手早く採取する。
(5) 乾燥後の懸濁物質の量が2mg以上になるように試料をとる。
39
K 0101 : 1998
備考2. ろ過しにくい試料の場合には,適量をビーカーにとり,その都度よく振り混ぜて,液がろ過
され終わる直前ごとに加え,ろ過速度が極めて遅くなったら試料の追加を止める。ビーカー
の中の残量から試料の量を求める。
3. 試料容器に付着しやすい懸濁物質を含む場合は,適量の試料を採取して,その全量を用いて
試験する。
採取した容器の器壁に付着した懸濁物質はポリスマン(ゴム管付きガラス棒)などで落と
して,ろ過材上に集める。
4. 懸濁物質中の強熱残留物を定量する場合には,有機性ろ過膜を用いる。
5. 油脂,グリース,ワックスなどを含む試料で,これらを除いた懸濁物質を測定する場合には,
試料をろ過した後,ろ過材を取り外すことなく,ろ過管ごと乾燥し,再び吸引瓶に取り付け
た後,ヘキサン10mlずつを数回注ぎ入れ,油脂類を洗って除く。続いて(c)の操作を行う。
16.2 全蒸発残留物
(1) 器具 器具は,次のとおりとする。
(a) 蒸発皿 白金皿,石英ガラス皿又は磁器蒸発皿
(2) 操作 操作は,次のとおり行う。
(a) 適当な材質(6)の蒸発皿を105〜110℃で約1時間加熱し,デシケーター中で放冷した後,その質量を
はかる。加熱,放冷及びひょう量を繰り返して恒量とする。
(b) 試料(4)の適量(7)を蒸発皿に入れ(8),注意して蒸発乾固する(9)。
(c) この蒸発皿を105〜110℃で約2時間加熱し,先のデシケーター中で放冷した後,その質量をはかる。
(d) 次の式によって全蒸発残留物 (mg/l) を算出する。
V
b
a
R
000
1
)
(
×
−
=
ここに, R: 全蒸発残留物 (mg/l)
a: 残留物の入った蒸発皿の質量 (mg)
b: 蒸発皿の質量 (mg)
V: 試料 (ml)
注(6) 試料の性質によって用いる蒸発皿の材質を選ぶ。
例えば,塩化物イオンと強力な酸化性の物質とを一緒に含む試料の場合には,白金皿は用い
ない。
(7) 乾燥後の全蒸発残留物の質量が,5mg以上になるように採取する。
(8) 試料の全量が蒸発皿に入らない場合には,数回に分けて入れる。
(9) 蒸発乾固には,加熱板,沸騰水浴,赤外線ランプなどを用い,蒸発中に沸騰させないようにす
る。また,外部からの汚染に注意する。
備考6. 備考3.による。
7. 引き続き強熱残留物を定量する場合には,蒸発皿はあらかじめ600±25℃で加熱して恒量に
したものを用いる。
16.3 溶解性蒸発残留物
(1) 器具 器具は,次のとおりとする。
(a) 蒸発皿 16.2(1)(a)による。
(b) ろ過器(分離形) 16.1(1)(a)による。
40
K 0101 : 1998
(c) ろ過材 16.1(1)(b)による。
(2) 操作 操作は,次のとおり行う。
(a) ろ過材をろ過器に取り付け,試料をろ過する。
(b) ろ液について16.2(2)(a)〜(d)に準じて操作する。
備考8. 備考1.及び備考4.による。
16.4 強熱残留物
16.4.1 懸濁物質の強熱残留物
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸アンモニウム溶液 (250g/l) JIS K 8545に規定する硝酸アンモニウム25gを水に溶かして
100mlとする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) るつぼ 白金又は磁器るつぼ10〜20ml
(b) 電気炉 600±25℃に調節できるもの。
(3) 操作 操作は,次のとおり行う。
(a) 600±25℃で加熱して恒量にしたるつぼに16.1(2)(c)で得た懸濁物質を,ろ過材とともに移し入れ,
硝酸アンモニウム溶液 (250g/l) を滴加して湿した後(10)電気炉に入れ,徐々に温度を上げ600±25℃
で約30分間加熱して灰化し,デシケーター中で放冷した後,その質量をはかる。
(b) 次の式によって強熱残留物 (mg/l) を算出する。
V
b
a
R
000
1
)
(
×
−
=
ここに, R: 懸濁物質の強熱残留物 (mg/l)
a: 強熱残留物の入ったるつぼの質量 (mg)
b: るつぼの質量 (mg)
V: 試料 (ml)
注(10) 有機性ろ過膜がニトロ化合物の場合には,JIS K 8839に規定する2-プロパノールを滴加して湿し
た後,強熱して灰化するとよい。
備考9. ろ過材にガラス繊維ろ紙を使用した場合には,あらかじめ別の同形のガラス繊維ろ紙を600
±25℃の電気炉で加熱して空試験値を求め,ガラス繊維ろ紙の質量を補正する。
16.4.2 全蒸発残留物の強熱残留物
(1) 操作 操作は,次のとおり行う。
(a) 16.2(2)(c)で得た全蒸発残留物について16.4.1(3)の操作に準じて操作する。
16.4.3 溶解性蒸発残留物の強熱残留物
(1) 操作 操作は,次のとおり行う。
(a) 16.3(2)で得た溶解性蒸発残留物について16.4.1(3)の操作に準じて操作する。
16.5 強熱減量
(1) 操作 操作は,次のとおり行う。
(a) 懸濁物質の強熱減量,全蒸発残留物の強熱減量及び溶解性残留物の強熱減量は,次の式によって算
出する。
E=S−R
ここに, E: それぞれの強熱減量 (mg/l)
S: それぞれの蒸発残留物 (mg/l)
41
K 0101 : 1998
R: それぞれの強熱残留物 (mg/l)
17. 100℃における過マンガン酸カリウムによる酸素消費量 (CODMn) 試料を硫酸酸性とし,酸化剤とし
て過マンガン酸カリウムを加え,沸騰水浴中で30分間反応させ,そのとき消費した過マンガン酸の量を求
め,相当する酸素の量 (mgO/l) で表す。
この試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験
する。
定量範囲:CODMn 0.5〜11mgO/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA4の水(1)(2)
(b) 硫酸 (1+2) 水2容をビーカーにとり,これをかき混ぜながらJIS K 8951に規定する硫酸1容を
徐々に加えた後,薄い紅色を呈するまで過マンガン酸カリウム溶液 (3g/l) を加える。
(c) 硝酸銀溶液 (200g/l) JIS K 8550に規定する硝酸銀20gを水に溶かして100mlとする。着色ガラス
瓶に入れて保存する。
(d) しゅう酸ナトリウム溶液 (12.5mmol/l) JIS K 8528に規定するしゅう酸ナトリウム1.8gを水に溶
かして1lとする。ただし,(e)の5mmol/l過マンガン酸カリウム溶液のモル濃度の2.5倍よりわずか
に高いモル濃度のものを調製する。
(e) 5mmol/l過マンガン酸カリウム溶液 JIS K 8247に規定する過マンガン酸カリウム0.8gを平底フラ
スコにとり,水1 050〜1 100mlを加えて溶かす。これを1〜2時間静かに煮沸した後,一夜放置す
る。
上澄み液をガラスろ過器G4を用いてろ過する(ろ過前後に水洗いしない。)。ろ液は約30分間水
蒸気洗浄した着色ガラス瓶に入れて保存する。
標定 JIS K 8005に規定する容量分析用標準物質のしゅう酸ナトリウムをあらかじめ200℃で約1
時間加熱し,デシケーター中で放冷する。その約0.42gを1mgのけたまではかりとり,少量の水に
溶かして,全量フラスコ250mlに移し入れ,水を標線まで加える。この溶液25mlを三角フラスコ
300mlにとり,水で約100mlとし,硫酸 (1+2) 10mlを加える。液温25〜30℃で,ビュレットでこ
の5mmol/l過マンガン酸カリウム溶液約22mlを一度に加え,紅色が消えるまで放置する。次に,50
〜60℃に加熱し,この5mmol/l過マンガン酸カリウム溶液で滴定する。終点は微紅色を約30秒間保
つときとする。
次の式によって5mmol/l過マンガン酸カリウム溶液のファクター (f) (3)を算出する。
675
001
.0
1
250
25
100
×
×
×
×
=
x
b
a
f
ここに,
a: しゅう酸ナトリウムの量 (g)
b: しゅう酸ナトリウムの純度 (%)
x: 滴定に要した5mmol/l過マンガン酸カリウム溶液 (ml)
0.001 675: 5mmol/l過マンガン酸カリウム溶液1mlのしゅう酸ナ
トリウム相当量 (g)
注(1) 水はCODMn値を与える物質を含んでいてはならない。次のようにしてその適否を確認できる。
水100mlについて(3)(a)〜(d)を行う。このときの滴定に要した5mmol/l過マンガン酸カリウム
溶液をamlとする。別に,水100mlについて加熱を除いた(a)〜(d)の操作を行う。このときの滴
定に要した5mmol/l過マンガン酸カリウム溶液をbmlとする。 (a−b) mlを求める。この値が
42
K 0101 : 1998
0.15〜0.2程度であればよい。これ以上の場合は,水(又は試薬)に有機物が含まれていること
が考えられ,使用に適しない。
(2) 水を蒸留精製する場合は,硫酸 (1+2) を加えて微酸性とし,これに過マンガン酸カリウム溶液
(3g/l) を加えて着色させた後,蒸留するとよい。ただし,蒸留の終わりまで過マンガン酸の着
色をしていること。
(3) ファクターはなるべく1に近い (0.95〜1.05) ものを使用する。
(2) 器具 器具は,次のとおりとする。
(a) 水浴 試料を入れたとき,引き続いて沸騰状態を保てるような,熱容量及び加熱能力が大きなもの。
三角フラスコ300mlが水浴の底に直接接触しないように,底から離して金網などを設ける。
(3) 操作 操作は,次のとおり行う。
(a) 試料(4)の適量(5)を三角フラスコ300mlにとり,水を加えて100mlとし,硫酸 (1+2) 10mlを加え,
振り混ぜながら硝酸銀溶液 (200g/l) 5ml(6)(7)を加える。
(b) 5mmol/l過マンガン酸カリウム溶液10mlを加えて振り混ぜ,直ちに沸騰水浴中に入れ(8)(9),30分間
加熱する(10)。
(c) 水浴から取り出し,しゅう酸ナトリウム溶液 (12.5mmol/l) 10mlを加えて振り混ぜ,よく反応させる
(11)。
(d) 液温を50〜60℃で,5mmol/l過マンガン酸カリウム溶液でわずかに赤い色を呈するまで滴定する。
(e) 別に,水100mlを三角フラスコ300mlにとり,(a)〜(d)の操作を行う(12)。
(f) 次の式によってCODMn (mgO/l) を算出する。
2.0
000
1
)
(
×
×
×
−
=
V
f
b
a
CODMn
ここに, CODMn: 100℃における過マンガン酸カリウムによる酸素消費量
(mgO/l)
a: 滴定に要した5mmol/l過マンガン酸カリウム溶液 (ml)
b: 水を用いた試験の滴定に要した5mmol/l過マンガン酸カ
リウム溶液 (ml)
f: 5mmol/l過マンガン酸カリウム溶液のファクター
V: 試料 (ml)
0.2: 5mmol/l過マンガン酸カリウム溶液1mlの酸素相当量
(mg)
注(4) 懸濁物を含む場合には,よく振り混ぜて均一にした後,手早く採取する。
(5) 30分間加熱した後の5mmol/l過マンガン酸カリウム溶液の残留量が4.5〜6.5mlになるような量。
ただし,試料のCODMnが11mgO/l以下の場合には,100mlとする。試料の適量は(3)の操作によ
って予備試験を行って決める。
CODMnの概略値が分かっている場合には,次の式によって試料の適量 (Vml) を求めることが
できる。
)
/
mgO
(
0.2
000
1
)
5.5
~
3.5
(5.4
Mn
l
COD
V
予想値
試料の
又は
×
×
=
ここに,
V: 試料の採取量 (ml)
4.5(又は3.5〜5.5): 5mmol/l過マンガン酸カリウム溶液の反
応予想量 (ml)
0.2: 5mmol/l過マンガン酸カリウム溶液1ml
の酸素相当量 (mg)
43
K 0101 : 1998
(6) 硝酸銀溶液 (200g/l) に代え,これに対応する硝酸銀の粉末を加えてもよい。
(7) 試料中に塩化物イオンが存在する場合は当量になるまで加え,更に5mlを加える。ただし,塩
化物イオンが多く,硝酸銀溶液 (200g/l) 10ml以上を必要とする場合には,硝酸銀溶液 (500g/l)
を用いて当量よりも2ml過剰に加えるか,又は粉末にした硝酸銀を当量よりも1g過剰に加え,
更に水5mlを加える。
塩化物イオン1gに対する硝酸銀 (AgNO3) の当量は4.8gである。通常の海水[塩化物イオン
(18g/l)]100mlと当量の硝酸銀は8.64gで,添加量は9.6gとなる。
(8) 多数の試料を一度に入れると,水浴の沸騰が止まるおそれがあるだけでなく,取り出したとき
のしゅう酸ナトリウム溶液 (12.5mmol/l) の添加操作の所要時間だけ加熱時間のずれが生じる
おそれがある。その所要時間だけの間隔をおいて入れるとよい。
(9) 三角フラスコが倒れないように,その首に鉛製,鉄製などのリング状のおもりを付ける。
(10) このとき,三角フラスコ300ml中の試料の液面は沸騰水浴の水面下になるように保つ。
(11) 塩化銀に酸化マンガン (IV) が混入し,反応にやや時間を要することがある。
(12) 塩化物イオンの多い試料に硝酸銀溶液 (200g/l) 5ml以上を加えた場合も,この操作では硝酸銀
溶液 (200g/l) 5mlを用いる。
備考 硝酸銀溶液 (200g/l) 5mlに代え,めのう乳鉢でよくすりつぶした硫酸銀の粉末1gを加えてもよ
い。ただし,(e)でも硫酸銀の粉末1gを用いる。塩化物イオンの多い試料では,塩化物イオン
と当量よりも10%多い量に,更に1gを加えた量を加える。
塩化物イオン1gと当量の硫酸銀は4.4gであり,
硫酸銀添加量=[塩化物イオン (g) ×4.4×1.1+1] (g) =[塩化物イオン (g) ×4.84+1] (g)
となる。通常の海水[塩化物イオン (18g/l)]100mlでは9.7gとなる。
18. 二クロム酸カリウムによる酸素消費量 (CODCr) 試料に二クロム酸カリウムと硫酸を加え,還流冷
却器を付けて2時間煮沸した後,消費した二クロム酸の量を求め,相当する酸素の量 (mgO/l) で表す。
この試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験
する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA4の水
(b) 硫酸銀-硫酸溶液 JIS K 8965に規定する硫酸銀11gをJIS K 8951に規定する硫酸1lに溶かす。溶
けるまで1,2日間を要する(加熱して溶かしてもよい。)。
(c) 硫酸水銀 (II) JIS K 8980に規定するもの。
(d) 二クロム酸カリウム溶液 (240
1mol/l) JIS K 8517に規定する二クロム酸カリウム1.23gをとり,水
に溶かして1lとする。
(e) 1, 10-フェナントロリン鉄 (II) 溶液 JIS K 8789に規定する1, 10-フェナントロリン一水和物
(1)1.48gとJIS K 8978に規定する硫酸鉄 (II) 七水和物0.70gとを水に溶かして100mlとする。
(f) 25mmol/l硫酸アンモニウム鉄 (II) 溶液 (2)JIS K 8979に規定する硫酸アンモニウム鉄 (II) 六水和
物10gを水約500mlに溶かし,硫酸20mlを加える。冷却した後,水を加えて1lとする。使用時に
標定する。
標定 JIS K 8005に規定する容量分析用標準物質の二クロム酸カリウムを,あらかじめめのう乳鉢
中で砕き,150℃で約1時間加熱し,デシケーター中で放冷する。K2Cr2O7100%に対してその0.246g

44
K 0101 : 1998
を1mgのけたまではかりとり,少量の水に溶かして全量フラスコ200mlに移し入れ,水を標線まで
加える。この溶液20mlを三角フラスコ300mlにとり,水を加えて約100mlとし,JIS K 8951に規
定する硫酸30mlを加える。冷却した後,指示薬として1, 10-フェナントロリン鉄 (II) 溶液2,3滴
を加えて,この25mmol/l硫酸アンモニウム鉄 (II) 溶液で滴定し,溶液の色が青緑から赤褐色に変
わった点を終点とする。次の式によって25mmol/l硫酸アンモニウム鉄 (II) 溶液のファクター (f) を
算出する。
226
001
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 二クロム酸カリウムの量 (g)
b: 二クロム酸カリウムの純度 (%)
x: 滴定に要した25mmol/l硫酸アンモニウム鉄 (II) 溶液
(ml)
0.001 226: 25mmol/l硫酸アンモニウム鉄 (II) 溶液1mlの二クロ
ム酸カリウム相当量 (g)
注(1) JIS K 8202に規定する塩化1, 10-フェナントロリニウム一水和物を用いる場合には,1.78gをとる。
(2) 25mmol/l硫酸アンモニウム鉄 (II) 溶液1mlは,二クロム酸カリウム溶液 (240
1mol/l) 1mlに相当
する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 還流冷却器 共通すり合わせリービッヒ冷却器又は球管冷却器300mmのもの。
(b) 丸底フラスコ又は三角フラスコ 250〜300ml(a)の還流冷却器と共通すり合わせのもの。
(c) 加熱板又はマントルヒーター
(3) 操作 操作は,次のとおり行う。
(a) 試料(3)の適量(4)をあらかじめ硫酸水銀 (II) 0.4g(5)を入れた丸底フラスコ又は三角フラスコ250mlに
とり,水を加えて20mlとし,よく振り混ぜる。
(b) 二クロム酸カリウム溶液 (240
1mol/l) 10mlを加え,注意してよく振り混ぜながら硫酸銀-硫酸溶液
30mlを加えた後,沸騰石数個を入れる。
(c) 還流冷却器を付けて2時間加熱する。
(d) 冷却した後,水約10mlで還流冷却器を洗って洗液をフラスコに流し入れ,更に水で約140mlに薄
める。
(e) 指示薬として1, 10-フェナントロリン鉄 (II) 溶液2,3滴を加えて,過剰の二クロム酸を25mmol/l
硫酸アンモニウム鉄 (II) 溶液で滴定し,溶液の色が青緑から赤褐色に変わった点を終点とする。
(f) 別に,水20mlをとり,(a)〜(e)の操作を行う。
(g) 次の式によってCODCr (mgO/l) を算出する。
2.0
000
1
)
(
Cr
×
×
×
−
=
V
f
a
b
COD
ここに, CODCr: 二クロム酸カリウムによる酸素消費量 (mgO/l)
a: 滴定に要した25mmol/l硫酸アンモニウム鉄 (II) 溶液
(ml)
b: 水を用いた試験の滴定に要した25mmol/l硫酸アンモニウ
ム鉄 (II) 溶液 (ml)
f: 25mmol/l硫酸アンモニウム鉄 (II) 溶液のファクター
V: 試料 (ml)
0.2: 25mmol/l硫酸アンモニウム鉄 (II) 溶液1mlの酸素相当量
45
K 0101 : 1998
(mg)
注(3) 懸濁物を含む場合には,よく振り混ぜて均一にした後,手早く採取する。
(4) 2時間加熱した後に最初に加えた二クロム酸カリウム溶液の約21が残るような量。
(5) 塩化物イオン40mgをマスキングするが,海水のように塩化物イオンの濃度が高い場合には,
妨害を除去できないので,この方法は適用できない。
備考 この方法では,水銀化合物を使用するので,試験後の廃液の処理について特に注意すること。
19. 生物化学的酸素消費量 (BOD) 生物化学的酸素消費量とは,水中の好気性微生物によって消費され
る溶存酸素の量をいう。試料を希釈水で希釈し,20℃で5日間放置したとき消費された溶存酸素の量で表
す。
この試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験
する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水(1)
(b) 緩衝液 (pH7.2) (A液) JIS K 9017に規定するりん酸水素二カリウム21.75g,JIS K 9007に規定
するりん酸二水素カリウム8.5g,JIS K 9019に規定するりん酸水素二ナトリウム・12水44.6g及び
JIS K 8116に規定する塩化アンモニウム1.7gを水に溶かして1 000mlとする。この緩衝液のpHは
7.2である。
(c) 硫酸マグネシウム溶液(B液) JIS K 8995に規定する硫酸マグネシウム七水和物22.5gを水に溶
かして1lとする。
(d) 塩化カルシウム溶液(C液) JIS K 8123に規定する塩化カルシウム27.5gを水に溶かして1lとす
る。
(e) 塩化鉄 (III) 溶液(D液) JIS K 8142に規定する塩化鉄 (III) 六水和物0.25gを水に溶かして1l
とする。使用時に調製する。
(f) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(g) 水酸化ナトリウム溶液 (40g/l) JIS K 8576に規定する水酸化ナトリウム4gを水に溶かして100ml
とする。
(h) 亜硫酸ナトリウム溶液 (12.5mmol/l) JIS K 8061に規定する亜硫酸ナトリウム1.6gを水に溶かし
て1lとする。使用時に調製する。
(i) よう化カリウム JIS K 8913に規定するもの。
(j) 希釈水 水温を20℃近くに調節し,ばっ気して溶存酸素を飽和させた水(2)1lに対して,A液,B液,
C液及びD液それぞれ1mlずつを加える。この溶液は,pH 7.2である[pH 7.2を示さないときは,
塩酸 (1+11) 又は水酸化ナトリウム溶液 (40g/l) でpH 7.2に調節する。]。希釈水は,培養瓶に詰め
て20℃の恒温槽に5日間放置したとき,初めの溶存酸素の量と5日間後の溶存酸素の量との差が
0.2mgO/l以下であることをあらかじめ確認しておく(3)。
(k) 植種液 下水の上澄み液(4)(5),河川水(6),土壌抽出液(7)などを用いる。
(l) 植種希釈水(8) 試験に際し植種液の適量(9)を希釈水に加えて,植種希釈液を調製する。
注(1) 石英ガラス又はほうけい酸ガラス-1製の蒸留器で精製したもの。
(2) 洗浄して汚染物質を除いた空気を通して,溶存酸素を飽和させるとよい。空気の洗浄は次の方
法による。
46
K 0101 : 1998
空気を活性炭ろ過器[例えば,粒状活性炭をガス乾燥塔 (300mm) に充てんする。]でろ過し,
次に,硫酸酸性にした過マンガン酸カリウム溶液 (5g/l) で洗い,更に水酸化カリウム溶液
(250g/l) で洗う。
(3) 生物化学反応は,含まれている有機物の濃度及び微生物の種類によって異なるため,希釈水に
ついて空試験を行って補正することは困難である。このため,希釈水は5日間の酸素消費量が
0.2mgO/l以下のものを用いる。
(4) 植種液には,家庭生下水がよく用いられる。新鮮な生下水を20℃(又は室温)で24〜36時間
放置後,その上澄み液を用いる。下水中に硝化生物(アンモニウムイオン及び亜硝酸イオンを
酸化する生物)の多いもの及び十分な生物化学的平衡に達していない新鮮な下水は好ましくな
い。
(5) 植種液として下水を用いた場合に,正常なBODを示さない試料には,植種液として土壌抽出液
を用いるか,又は試験室でこの試料にならした微生物を培養し,この培養液を用いる。
(6) 常時この試料の放流を受けている河川の放流地点から500〜1 000m下流の水を植種に用いると
良好な結果を得ることがある。試料中に生物化学的反応に有害な物質が共存しても,その試料
の放流を受けている河川,湖沼などには,耐性をもった生物相が発達していることが多いから
である。
(7) 土壌(植物の生育している土壌)約200gを水2lに加えてかき混ぜた後,その上澄み液を用い
る。
(8) 試料中に好気性の微生物や細菌が存在しない場合,又はその数が不足している場合に,植種希
釈水を用いる。
試料のBODの試験に植種希釈水を用いるときは,次の操作によって植種希釈水の調製に用い
た植種液についての補正(植種補正)を行う。
植種液を希釈水で適当に希釈して数段階の希釈した植種液を調製し,希釈試料と並行して測
定する。希釈した植種液の培養前の溶存酸素の量をB1,5日間放置後の溶存酸素の量をB2とす
るとき
1
2
1
)
(
B
B
B−
×100が40〜70%の範囲内にあるものを選び,植種補正値として (B1−B2) ×f
を用いる[(4)(d)のBODの算出式を参照]。植種希釈水の5日間の溶存酸素の消費量を求めて補
正を行ってはならない。
(9) 微生物が正常な活動をするために,植種希釈水のBODが0.6〜1mgO/lになるように加える。通
常,希釈水1lに対し,下水の上澄み液では5〜10ml,河川水では10〜50ml,土壌抽出液では20
〜30ml程度である。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 培養瓶 正確に容量の分かっている細口共栓ガラス瓶 (100〜300ml) で,共栓は斜めに切り落とし
たもの。図19.1に一例を示す。
47
K 0101 : 1998
図19.1 培養瓶の一例
(b) 恒温器 JIS T 1702に規定するふ(孵)卵器20±1℃に調節できるもの。希釈試料中の藻類による
二酸化炭素同化作用(炭酸同化作用)を防ぐために,光を遮断しておく。同様な仕様の恒温水槽を
使ってもよい。
(3) 試料の前処理 試料の前処理は,次のとおり行う。
試料に酸及びアルカリ,残留塩素などの酸化性物質,過飽和の溶存酸素又は溶存気体が含まれてい
る場合には,次の前処理を行う。
また,前処理によって液量が増加する場合には,増加分について結果を補正する。
(a) アルカリ又は酸を含む試料 塩酸 (1+11) 又は水酸化ナトリウム溶液 (40g/l) を加えて試料のpH
を約7にする。
(b) 残留塩素などの酸化性物質を含む試料 あらかじめ試料100mlにJIS K 9501に規定するアジ化ナト
リウム0.1gとJIS K 8913に規定するよう化カリウム1gとを加えて振り混ぜた後,塩酸 (1+1) を
加えてpHを約1とし,暗所に数分間放置する。遊離したよう素をでんぷん溶液を指示薬として亜
硫酸ナトリウム溶液 (12.5mmol/l) でよう素でんぷんの青い色が消えるまで滴定する。別に,同量の
試料をとり,先の滴定値から求めた計算量の亜硫酸ナトリウム溶液 (12.5mmol/l) を加えて残留塩素
を還元した後,必要ならば水酸化ナトリウム溶液 (40g/l) 又は塩酸 (1+11) を用いてpHを約7に
調節する。
(c) 溶存酸素又は溶存気体が過飽和の試料 冬季に採取した処理水,河川水などの試料の温度が20℃以
下のときは,20℃にしたときに溶存酸素及び溶存気体が過飽和になりやすい。
また,緑藻類の多い河川水及び湖沼水では,二酸化炭素同化作用によって酸素が発生するので,
溶存酸素が過飽和になりやすい。これらの試料では,BOD測定中に溶存酸素が気体となり,培養瓶
外に散逸し,結果が不正確になるから,あらかじめかき混ぜるか,ばっ気するなどの方法によって
溶存酸素及び溶存気体を20℃の飽和量近くに減少させておく。
備考1. 生物化学的処理が済んだ試料には,炭素質の有機物を分解する好気性細菌のほかにアンモニ
アなどの窒素化合物を酸化(硝化)する硝化細菌が繁殖していることがある。このような試
料では,有機物の酸化分解に消費される酸素と,アンモニアなどの窒素化合物の硝化に消費
される酸素の合量が測定される。この硝化による酸素の消費量は,試料中の窒素化合物の量
に対応するものではなく,硝化細菌の数によって変化する。
硝化作用を抑制した状態の生物化学的酸素消費量を測定するには,次の操作を行う。
(4)(a)の希釈試料の調製時に,希釈試料1lについてN-(2-プロペニル)チオ尿素2mg(*)又
48
K 0101 : 1998
は2-クロロ-6-(トリクロロメチル)ピリジンの粉末10mg(2*)が含まれるように添加する。
注(*) N-(2-プロペニル)チオ尿素(N-アリルチオ尿素)溶液 (1mg/ml) [N-(2-プロペニル)チオ尿
素0.1gを水に溶かして100mlとする。0〜10℃の暗所に保存する。]2mlを加える。
(2*) 2-クロロ-6-(トリクロロメチル)ピリジンは,水に溶けにくいので粉末で加える。添加した後
も完全には溶けず一部は浮上することがあるので,培養瓶に移す場合など注意する。水に溶け
やすいように,他の試薬と混合したものがあり,これを用いてもよい。
(4) 操作 操作は,次のとおり行う。
(a) 希釈試料の調製 サイホンを用い,泡が入らないように注意して希釈水又は植種希釈水をメスシリ
ンダー(有栓形)1 000ml[培養瓶が200ml以上の場合には,メスシリンダー(有栓形)2 000mlを
用いる。]に約半分までとる。次に,前処理を行った試料の適量(10)(11)(12)を加え,希釈水又は植種希
釈水を1 000mlの標線[メスシリンダー(有栓形)2 000mlの場合には,2 000mlの標線]まで加え
る。栓をして静かに混ぜ合わせる。試料の量を変えて同じ操作を行うか又は同様の操作によって,
この溶液を希釈して,更に段階的に希釈倍率の異なる希釈試料4〜5種類を調製する(13)(14)(15)。
調製したそれぞれの希釈試料1種類について,培養瓶2〜4本を用意し,サイホンを用いて希釈試
料を移し入れ,十分にあふれさせた後,密栓する。
希釈倍率の異なる各組の培養瓶のうち1本は,培養前の溶存酸素の定量に用い,ほかは20±1℃
に調節した恒温器に入れて5日間培養する(16)(17)。
(b) 培養前の希釈試料の溶存酸素量の測定 希釈試料を調製後15分間放置し(18),溶存酸素を24.2,24.3
又は24.4によって定量する。24.3又は24.4による場合は,メスシリンダー中に残った希釈試料を用
いて溶存酸素の量を測定してもよい。
(c) 培養後の溶存酸素の測定 恒温器又は恒温水槽の中に5日間培養した希釈試料の溶存酸素の量を(b)
と同じ方法で測定する。
(d) BODの算出 培養前後の溶存酸素の量から,次の式によって試料のBOD (mgO/l) を算出する(19)。
i)
植種を行わないとき。
P
D
D
BOD
)
(
2
1−
=
ii)
植種希釈水を用いたとき(19)。
P
f
B
B
D
D
BOD
×
−
−
−
=
)
(
)
(
2
1
2
1
ここに, BOD: 生物化学的酸素消費量 (mgO/l)
D1: 希釈試料を調製してから15分間後の溶存酸素 (mgO/l)
D2: 培養後の希釈試料の溶存酸素 (mgO/l) (20)
P: 希釈試料中の試料の占める割合(試料/希釈試料)
B1: 植種液のBODを測定する際の希釈した植種液の培養前の
溶存酸素 (mgO/l)
B2: 植種液のBODを測定する際の希釈した植種液の培養後の
溶存酸素 (mgO/l)
f:
y
x
x: 試料のBODを測定する際の希釈試料中の植種液 (%)
y: 植種液のBODを測定する際の希釈した植種液中の植種液
(%)
注(10) 試料の正常なBODを得るための希釈試料の溶存酸素の消費量 (D1−D2) は,3.5〜6.2mgO/lの範
49
K 0101 : 1998
囲である。希釈不足のため残留する溶存酸素の量が1mgO/l以下であったり,逆に希釈し過ぎて
溶存酸素の消費量が2mgO/l以下となる場合には,正常なBODを得にくい。
(11) 試料のBODが,経験その他で予想できれば,採取する試料の量は,次のようにして求める。
例えば,20℃における溶存酸素の飽和量は,8.84mgO/lであり,これの40%は約3.5mgO/l,70%
は約6.2mgO/lであるから,希釈水と試料を合わせて1lにする場合には,採取する試料の量 (Vml)
は,次の式で得られる。
)
/
mgO
(
1000
)2.6
~
5.3(
Mn
l
BOD
V
予想値
試料の
×
=
ここに,
V: 試料の採取量 (ml)
3.5: 20℃における溶存酸素の飽和量 (8.84mgO/l) の40%相当量
6.2: 20℃における溶存酸素の飽和量 (8.84mgO/l) の70%相当量
試料のBODが5mgO/l以下の場合には,試料の採取量は800ml以上とする。
また,溶存酸素が十分に含まれていない場合には,ばっ気した後試験する。
(12) 懸濁物を含む試料の場合には,懸濁物が均一になるように混ぜ合わせた後,適量をとる。
(13) メスシリンダー中に残っている希釈試料を基にして,順次希釈倍数の高い希釈試料を調製し続
けると,労力と時間を節約することができる。
(14) 100倍以上に希釈する場合には,一度に希釈せずに,あらかじめ他のメスシリンダー(有栓形)
1 000mlに試料50〜100mlをとり,希釈水又は植種希釈水を1 000mlの標線まで加える。この希
釈した試料を用いて(a)の希釈試料を調製する。
(15) BODが100mgO/l以下の試料の場合には,次の方法によって培養瓶で直接希釈してもよい。
容量の正確に分かっている培養瓶4本を用意し,それぞれにあらかじめ約半量の希釈水又は
植種希釈水を入れておき,次に,希釈倍数に応じて瓶の容量に対する計算量の試料を加え,更
に,瓶内の空間を希釈水又は植種希釈水で満たす。この操作中,泡が入らないように注意する。
(16) 恒温水槽を使用する場合には,培養瓶全体を水に浸す。
(17) 培養瓶を水封して恒温器中におく場合には,水封水が蒸発するから,ときどき補充する。
(18) 硫化物,亜硫酸塩,鉄 (II) などの還元性物質が共存する場合には,15分間の酸素消費量
(Immediate dissolved oxygen demand,以下IDODと略記する。)とBODとを区別する。IDOD
を求めるには,次のように操作する。
あらかじめ試料と希釈水との溶存酸素を測定した後,一定の割合で試料を希釈水で薄め,15
分間放置した後,溶存酸素 (D1) をはかる。初めに測定した試料と希釈水それぞれの溶存酸素
(mgO/l) から希釈試料の溶存酸素 (Dc) を算出し,次の式によって試料のIDOD (mgO/l) を算出
する。
P
D
D
IDOD
c
)
(
1
−
=
ここに, IDOD: 15分間の酸素消費量 (mgO/l)
DC: 培養前の希釈試料水の溶存酸素 (mgO/l) = (S×P) +
(DO×p)
S: 試料の溶存酸素 (mgO/l)
DO: 希釈水の溶存酸素 (mgO/l)
p: 希釈試料中の希釈水の占める割合(希釈水/希釈試料)
P: 希釈試料中の試料の占める割合(試料/希釈試料)
D1: 希釈試料を調製してから15分間放置後の溶存酸素 (mgO/l)
50
K 0101 : 1998
(19) 次のような方法で算出してもよい。
100
)1
(
)
(
)
(
1
2
2
1
1
2
1
−
×
×
×
−
−
×
−
=
n
V
n
B
B
n
D
D
BOD
ここに, D1: 希釈試料を調製してから15分間後の溶存酸素 (mgO/l)
D2: 培養後の希釈試料の溶存酸素 (mgO/l)
n1: 希釈試料の希釈倍数
試料
希釈試料
n2: 植種液のBOD測定時の希釈倍数
測定における植種液
植物液の
植種液
測定における希釈した
植物液の
(ml)
BOD
(ml)
BOD
B1: 植種液のBOD測定における培養前の希釈した植種液の溶存
酸素 (mgO/l)
B2: 植種液のBOD測定における培養後の希釈した植種液の溶存
酸素 (mgO/l)
V: 植種希釈水中に含まれる植種液のパーセント(vol%) 通常
2
2
1
)
(
100
6.0
n
B
B
V
×
−
×
>
になるようにとる。
(20) 5日間の溶存酸素の消費量 (D1−D2) が3.5〜6.2mgO/l以内,
1
2
1
D
D
D−
×100=40〜70%の範囲内
の値のものをとり,BODを算出する。この条件の中央値付近になるのが最も望ましい。ただし,
試料のBODが3.5mgO/l以下のときは,希釈しない場合でも5日間の溶存酸素の消費量は,溶
存酸素の飽和量の40%以上にならない。このような場合には,その値から算出する。
備考2. 植種液の調製方法 微生物を試料にならすための培養は,次の方法で行うとよい。
ガラス水槽(約6l)に試料5lを入れ,塩酸 (1+11) 又は水酸化ナトリウム溶液 (40g/l) を
用いてpHを約7に調節しておく。微生物が豊富に存在する下水,河川水などを植種液とし,
その100〜300ml及び緩衝液(A液)10〜50mlを加える。よくかき混ぜた後,その一部をと
り,CODMn又は有機体炭素の量を測定しておく。次に,24〜48時間連続ばっ気した後,再び
その一部をとり,CODMn又は有機体炭素の量を測定する。前後の測定値に顕著な変化がある
場合には,試料中に生物化学反応が進行しているものと判定し,更にばっ気を続けて試料に
適応する生物を繁殖させる。もし,顕著な変化が生じない場合には,別に試料をとり,希釈
水で適当に希釈した後,前記と同様に植種を行い,24〜48時間連続ばっ気し,CODMn又は有
機体炭素の量の変化及び懸濁物の量の変化などを試験する。その結果,これらに顕著な変化
がある場合には,生物化学反応が活発になっていることの現れである。試料中の有機物の組
成によっては,このような操作を1週間以上試みる必要がある。
また,試料を希釈水で10倍以上に希釈して上記の操作を行った場合,CODMn又は有機体
炭素の量に顕著な変化を生じたら,徐々に試料の比率を増加してみることも必要である。こ
のように試料に適応する微生物を培養し,植種液として用いる。
3. 試料操作の確認の方法 植種液,植種希釈水などの使用の適否,又は試験操作を確認するた
めに,次の方法が推奨される。
グルコース-グルタミン酸混合標準液[JIS K 8824に規定するD (+) -グルコース150mg及
びJIS K 9047に規定するL-グルタミン酸150mgをとり,水に溶かして全量フラスコ1 000ml
に移し入れ,水を標線まで加える。]5〜10mlを容量の正確に分かった培養瓶300ml(培養瓶
が100mlの場合には,前記の31量を用いる。)にとり,植種希釈水を満たして密栓し,BOD
51
K 0101 : 1998
をはかる。この標準液のBODは220±10mgO/lである。もし,この値からの偏差が著しい場
合には,希釈水の水質又は植種液の活性度などに疑問がある。
4. 試料中に銅,クロム,水銀,銀,ひ素などの重金属元素が溶存していると正しい測定値が得
られないことがある。このような場合には,これらの重金属元素によくならした植種液を備
考2.によって培養しておく。
20. 有機体炭素 (TOC) 有機体炭素とは,水中に存在する有機物中の炭素をいう。これの定量には,燃
焼酸化-赤外線式TOC分析法及び燃焼酸化-赤外線式TOC自動計測法を適用する。
この試験は,試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試
験する。
20.1 燃焼酸化-赤外線式TOC分析法 少量の試料を二酸化炭素を除去した空気又は酸素とともに高温の
全炭素測定管に送り込み,有機物中の炭素及び無機物[無機体炭素(主として炭酸塩類)]中の炭素を二酸
化炭素とした後,その濃度を非分散形赤外線ガス分析計で測定して全炭素の量を求める。
別に,試料を有機物が分解されない温度に保った無機体炭素測定管に送り込み,生成した二酸化炭素を
測定し,無機体炭素の量を求める。
全炭素の量から無機体炭素の量を差し引いて有機体炭素の量を算出する。
定量範囲:C 1〜150mg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3又はA4の水(1)(2)(3) で,炭酸を含まない水(4)を用いる。試薬の調製
及び操作には,この水を用いる。(5)によって空試験を行い,使用の適否を確認しておく。
(b) TOC標準液 (1mgC/ml) JIS K 8005に規定する容量分析用標準物質のフタル酸水素カリウムを
120℃で約1時間加熱し,デシケーター中で放冷する。その2.125gをとり,少量の水に溶かして全
量フラスコ1 000mlに移し入れ,水を標線まで加える。
(c) TOC標準液 (0.1mgC/ml) TOC標準液 (1mgC/ml) 10mlを全量フラスコ100mlにとり,水を標線
まで加える。
(d) 無機体炭素標準液 (1mgC/ml) JIS K 8622に規定する炭酸水素ナトリウムをデシケーター中で約
3時間放置し,その3.497gをとる。別に,JIS K 8005に規定する容量分析用標準物質の炭酸ナトリ
ウムを,あらかじめ600℃で約1時間加熱し,デシケーター中で放冷し,その4.412gをとる。両者
を少量の水に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加える。
(e) 無機体炭素標準液 (0.1mgC/ml) 無機体炭素標準液 (1mgC/ml) 10mlを全量フラスコ100mlにとり,
水を標線まで加える。
(f) 全炭素測定管 全炭素定量用触媒を充てんしたもの。
(g) 無機体炭素測定管 無機体炭素定量用触媒を充てんしたもの。
(h) キャリヤーガス 二酸化炭素を除去した空気又はJIS K 1101に規定する酸素
注(1) TOCの濃度をできるだけ低くした水を用いる。精製した水は,容器に入れて保存すると徐々に
汚染されてTOCの濃度が高くなることがあるので,精製後は早く使用することが望ましい。
(2) TOCの濃度をできるだけ低くするには,イオン交換水又は蒸留水を蒸留フラスコにとり,過マ
ンガン酸カリウム溶液 (3g/l) を着色するまで滴加し,水1 000ml当たり硫酸 (1+1) 2〜3mlを
加えて蒸留する(蒸留が終わるまで過マンガン酸カリウムによる着色が残るようにする。)。初
留分(蒸留フラスコ中の水量の約51に相当する。)を捨て,中間の53に相当する留分を採取する。
52
K 0101 : 1998
(3) イオン交換法,蒸留法,逆浸透法,紫外線照射法,活性炭吸着ろ過法,限外ろ過法及び精密ろ
過法などを適宜組み合わせて精製した水も使用できる。
(4) 2.(12)(b)によって精製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロシリンジ 20〜150μl
(b) TOC分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) TOC分析装置を作動できる状態にする。
(b) TOC標準液 (1mgC/ml) 又はTOC標準液 (0.1mgC/ml) の一定量(5)(例えば,20μl)をマイクロシリ
ンジでTOC分析装置の全炭素測定管に注入し,指示値(ピーク高さ)を読み取る。
(c) (b)の操作を数回繰り返して指示値が一定になることを確かめる。
(d) 試料をよく振り混ぜて均一にした後,(b)と同量をマイクロシリンジで全炭素測定管に注入して,指
示値を読み取り,(b)と比較して試料中の概略の全炭素の濃度 (mgC/l) を求める。
注(5) 試料の炭素の濃度が低い場合には,注入量は100〜150μlとし,炭素の濃度が高い場合には,注
入量を少なくするか一定倍率で薄める。
(4) 検量線の作成 検量線の作成は,次のとおり行う。
(a) (3)(d)で求めた試料の概略の炭素の濃度がほぼ中央になるようにTOC標準液 (1mgC/ml) 又はTOC
標準液 (0.1mgC/ml) を全量フラスコ100mlに段階的にとり,水を標線まで加える。
(b) (a)で調製したTOC標準液の最高濃度のものの一定量[例えば,(3)(b)と同量]をマイクロシリンジ
で全炭素測定管に注入して,指示値が最大目盛値の約80%になるようにTOC分析装置の感度及び
標準液の注入量を調節する。
(c) (a)で調製した各濃度のTOC標準液の一定量[(b)で定めた量]を順次マイクロシリンジで全炭素測
定管に注入して,指示値を読み取る。
(d) 空試験として,(c)と同量の水をマイクロシリンジで全炭素測定管に注入して,指示値を読み取り,
(c)の結果を補正し,有機体炭素の量と指示値との関係線を作成して,これを全炭素の検量線とする。
(e) 無機体炭素標準液 (1mgC/ml) 又は無機体炭素標準液 (0.1mgC/ml) を用いて,(a)で段階的に調製し
たTOC標準液と同量の炭素を含むように無機体炭素標準液を段階的に調製する。
(f) (e)で調製した各濃度の無機体炭素標準液の一定量[(b)で定めた量]を順次マイクロシリンジで無機
炭素測定管に注入し,指示値を読み取る。
(g) 空試験として(f)と同量の水をマイクロシリンジで無機体炭素測定管に注入して,指示値を読み取り,
(f)の結果を補正し,無機体炭素の量と指示値との関係線を作成して,これを無機体炭素の検量線と
する。
(5) 操作 操作は,次のとおり行う。
(a) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
(b) 試料の一定量(5)[例えば,(4)(b)と同量]をマイクロシリンジで全炭素測定管に注入し,指示値を読
み取る。
(c) 試料の一定量[例えば,(4)(f)と同量]をマイクロシリンジで無機体炭素測定管に注入し,指示値を
読み取る。
(d) 試料を薄めた場合には,(b)及び(c)の空試験としてそれぞれ同量の水をマイクロシリンジでとり,(b)
53
K 0101 : 1998
及び(c)の操作を行って試料について得た結果を補正する。
(e) あらかじめ作成した全炭素及び無機体炭素の検量線から注入試料中の全炭素及び無機体炭素の量を
求め,それぞれの濃度 (mgC/l) を算出する。
(f) 次の式によって試料の有機体炭素 (TOC) の濃度 (mgC/l) を算出する。
TOC= (Ct−Ci) ×d
ここに, TOC: 有機体炭素 (mgC/l)
Ct: 注入試料中の全炭素 (mgC/l)
Ci: 無機体炭素 (mgC/l)
d: 注入試料の希釈倍数
備考1. 全炭素の量から無機体炭素の量を差し引いて有機体炭素の量を求める方法のほか,あらかじ
め試料に塩酸を加えてpHを2以下にし,JIS K 1107に規定する高純度窒素2級を通気して無機
体炭素を除去した後,その少量を高温の全炭素測定管に送り込み,炭素の定量を行ってこれ
を有機体炭素の量とする方法がある。この方法は,無機体炭素が多い試料の場合は優れてい
る。ただし,揮発性の有機物を含む場合には誤差が大きい。
2. TOC分析装置で有機体炭素を二酸化炭素とする方式には,燃焼法のほかにアンプルを用いる
湿式酸化法がある。この湿式酸化法は,試料3〜10mlをガラス製アンプルにとり,ペルオキ
ソ二硫酸カリウムとりん酸,又は二クロム酸カリウムとりん酸を加えて酸性とした後,酸素
を十分に通気して二酸化炭素を除去する。アンプルを溶封した後,オートクレーブ中で一定
時間加熱して有機物を酸化する。アンプルを装置内で破壊し,生じた二酸化炭素を窒素で二
酸化炭素測定部に導入する。
3. 生成した二酸化炭素の定量には,赤外線分析法のほかに熱伝導度測定法が用いられる。
20.2 燃焼酸化-赤外線式TOC自動計測法 計測器に連続的に供給した試料に酸を加えてpHを2以下にし,
通気して無機体炭素を除去した後,その一定量をキャリヤーガスとともに高温の全炭素測定管に送り込み,
有機物中の炭素を二酸化炭素とし,その濃度を非分散形赤外線ガス分析計で測定して有機体炭素 (TOC)
の濃度を求める。
定量範囲:C 0.05〜150mg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異な
る。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 20.1(1)(a)による。
(b) TOC標準液 (1mgC/ml) 20.1(1)(b)による。
(c) TOC標準液 (0.1mgC/ml) 20.1(1)(c)による。
(d) ゼロ校正液 (a)の水を用いる。
(e) スパン校正液 TOC標準液 (0.1mgC/ml) [又はTOC標準液 (1mgC/ml)]の適量を全量フラスコに
とり,水を標線まで加える。同じ操作で計測器の測定範囲の約80%に相当するTOCの濃度になる
ように調製する。使用時に調製する。
(f) 酸溶液 JIS K 9005に規定するりん酸,JIS K 8180に規定する塩酸又はJIS K 8951に規定する硫酸
でTOCの濃度のできるだけ少ないものを用い,所定の濃度に調製する。
(g) キャリヤーガス 20.1(1)(h)による。
(2) 装置 装置は,次のとおりとする。
(a) TOC自動計測器 JIS K 0805に規定する測定範囲が1 000μgC/l以下又は1mgC/l以上の燃焼酸化-
54
K 0101 : 1998
赤外線式TOC自動計測器
(3) 準備操作 準備操作は,次のとおり行う。
(a) 酸溶液及びキャリヤーガスを,計測器に供給する。
(b) 計測器の暖機運転を行い,各部の機能及び指示記録部を安定させる。
(c) ゼロ校正液及びスパン校正液を用いて計測器を校正する。
(4) 操作 操作は,次のとおり行う。
(a) 試料を,計測器に供給して指示値が安定したことを確認する。
(b) 指示値から試料中の有機体炭素 (TOC) の濃度 (mgC/l) を求める。
備考4. TOC自動計測器で有機体炭素を二酸化炭素とする方式には,燃焼酸化方式のほかに酸化剤
(ペルオキソ二硫酸塩)を添加して高圧高温下で湿式酸化分解(例えば,約2MPa,200℃)
する方式がある。この方式には,試料のpHを2以下の酸性とし,ばっ気して無機体炭素を除
去した後測定する方式と,試料を酸性にし酸化剤を加えて全炭素を定量し,別に,試料を酸
性にし有機物が分解されない温度(約130℃)で無機体炭素を定量して,全炭素の量から無機
体炭素の量を差し引いて有機体炭素の量を求める方式の二種類がある。
21. 全酸素消費量 (TOD) 全酸素消費量とは試料を燃焼させたとき,試料中の有機物の構成元素である
炭素,水素,窒素,硫黄,りんなどによって消費される酸素の量をいう。この試験には燃焼法を適用する。
少量の試料を一定量の酸素を含む不活性気体とともに高温の燃焼管に送り込み,有機物などを燃焼させ
た後,不活性気体中の酸素の濃度を定量し,その減量から全酸素消費量を求める。
この試験は,試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試
験する。
定量範囲:O 10〜500mg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3又はA4の水(1)(2)(3)で,溶存酸素を含まない水(4)を用いる。試薬の調
製及び操作には,この水を用いる。(5)によって空試験を行い,使用の適否を確認しておく。
(b) TOD標準液 (1mgO/ml) JIS K 8005に規定する容量分析用標準物質のフタル酸水素カリウムを
120℃で約1時間加熱し,デシケーター中で放冷する。その0.851gをとり,水に溶かして全量フラ
スコ1 000mlに移し入れ,水を標線まで加える。
(c) TOD標準液 (0.1mgO/ml) TOD標準液 (1mgO/ml) 10mlを全量フラスコ100mlにとり,水を標線
まで加える。使用時に調製する。
(d) キャリヤーガス JIS K 1107に規定する高純度窒素2級及びJIS K 1101に規定する酸素
注(1) TODの濃度をできるだけ低くした水を用いる。精製した水は,容器に入れて保存すると徐々に
汚染されてTODの濃度が高くなることがあるので,精製後は早く使用することが望ましい。
(2) TODの濃度を低くするには,20.の注(2)の蒸留操作を行う。
(3) 20.の注(3)による。
(4) 2.(12)(a)によって精製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロシリンジ 10〜20μl
(b) TOD分析装置
(3) 準備操作 準備操作は,次のとおり行う。
55
K 0101 : 1998
(a) TOD分析装置を作動できる状態にする。
(b) TOD標準液 (1mgO/ml) 又はTOD標準液 (0.1mgO/ml) の一定量(例えば,20μl)をマイクロシリン
ジでTOD分析装置に注入し,指示値(ピーク高さ)が最大目盛値の約80%になるようにTOD分析
装置の感度を調節する。
(c) (b)の操作を数回繰り返して指示値が一定になることを確かめる。
(d) 試料(5)をよく振り混ぜて均一にした後,その定量[(b)で定めた量]をマイクロシリンジで注入して,
指示値を読み取り,(c)と比較して試料の概略の全酸素消費量を求める。
注(5) TODが500mgO/l以上の試料の場合には,水で適当に希釈した後,試験する。
(4) 検量線の作成 検量線の作成は,次のとおり行う。
(a) (3)(d)で求めた試料の概略のTODの値がほぼ中央になるようにTOD標準液 (1mgO/ml) 又はTOD標
準液 (0.1mgO/ml) を全量フラスコ100mlに段階的にとり,水を標線まで加える。
(b) (a)で調製したTOD標準液の最高濃度のものの一定量(例えば,20μl)をマイクロシリンジで注入
し,指示値が最大目盛の約80%になるように感度を調節する。
(c) (a)で調製した各濃度のTOD標準液の一定量[(b)で定めた量]を順次マイクロシリンジで注入し,
指示値を読み取る。
(d) 空試験として(c)と同量の水をマイクロシリンジでとり,(c)と同様に操作して指示値を読み取り,(c)
と指示値を補正してTOD標準液の各酸素相当量と指示値との関係線を作成する。
(5) 操作 操作は,次のとおり行う。
(a) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
(b) 試料(6)の定量[例えば,4.(b)と同量]をマイクロシリンジでTOD分析装置に注入し,指示値を読み
取る。
(c) 試料を薄めた場合には,空試験として(b)と同量の水をマイクロシリンジでとり,(b)の操作を行って
指示値を読み取り,試料について得た結果を補正する。
(d) あらかじめ作成した検量線から,注入した試料中の酸素消費量を求め,注入した試料のTOD (mgO/l)
を算出する。
(e) 次の式によって試料中の全酸素消費量 (TOD) の濃度 (mgO/l) を算出する。
TOD=a×d
ここに, TOD: 全酸素消費量 (mgO/l)
a: 注入試料の酸素消費量 (mgO/l)
d: 注入試料の希釈倍数
注(6) 試料の全酸素消費量が高い場合には,一定倍数に薄める。
備考1. 溶存酸素が共存すると妨害し,特に全酸素消費量が少ない場合には影響が大きい。この場合
は,別に,試料中の溶存酸素の量を測定して補正する。
2. 試料が酸性で硫酸イオンを含む場合には,高温で加熱すると次のように分解して酸素を生じ
負の誤差となる。
2H2SO4→2H2O+2SO2+O2
ただし,試料が蒸発したとき,硫酸がアルカリ金属塩となるような試料は,この反応は生
じない。このため,硫酸イオンが共存する場合には,水酸化ナトリウム溶液 (200g/l) を加え
てpHを約11に調節してから試験する。
56
K 0101 : 1998
3. 硝酸イオンが共存する場合には,高温で加熱すると次のように分解して酸素を生じ,負の誤
差となる。
4NaNO3→2Na2O+4NO+3O2 又は 2NaNO3→Na2O+N2O+2O2
4. 重金属イオンを含む試料を長時間測定すると燃焼管中の触媒が劣化し,酸化率が低下してく
る。このような場合には,触媒の交換又は再生が必要である。
5. 海水など塩類を多量に含む場合には,指示値が元に戻らないことがあるので,安定した指示
値が得られるまで測定を繰り返すか,又は試料を薄めてから試験する。
22. フェノール類及びp-クレゾール類 フェノール類とp-クレゾール類に区分する。
22.1 フェノール類 フェノール類の試験は,前処理(蒸留)した試料について,4-アミノアンチピリン
吸光光度法を適用する。
フェノール類はフェノール分解菌によって分解されやすい。
また,酸化性物質,還元性物質,アルカリなどの作用も受けやすいので,試験は試料採取後直ちに行う。
直ちに行えない場合は,3.3によって保存し,できるだけ早く試験する。
22.1.1 前処理
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水(1)。ほうけい酸ガラス瓶に保存する。
(b) りん酸 (1+9) JIS K 9005に規定するりん酸を用いて調製する。
(c) 硫酸銅 (II) 溶液 JIS K 8983に規定する硫酸銅 (II) 五水和物10gを水に溶かして100mlとする。
(d) メチルオレンジ溶液 (1g/l) JIS K 8893に規定するメチルオレンジ0.1gを熱水100mlに溶かす。
注(1) 石英ガラス製又はほうけい酸ガラス製の蒸留装置を用いて精製したもの。
(2) 装置 装置は,次のとおりとする。
(a) 蒸留装置 すり合わせのもの
(3) 蒸留操作 蒸留操作は,次のとおり行う。
(a) 試料250ml(2)(3)を蒸留フラスコ500mlにとり,メチルオレンジ溶液 (1g/l) 数滴を加え,メチルオレ
ンジが変色するまでりん酸 (1+9) を加えてpH約4の酸性にした後,硫酸銅 (II) 溶液2.5mlを加
える。
(b) 蒸留フラスコを蒸留装置に取り付け,受器にメスシリンダー(有栓形)250mlを用いて蒸留する。
(c) メスシリンダー中の留出液が225mlになったとき,いったん加熱を止める。
(d) 蒸留フラスコ中の試料の沸騰がやんだ後,蒸留フラスコに水25mlを加え,再び蒸留を続けて更に
25mlを留出させ,全留出液量を250mlとする(4)。
注(2) 試料中のフェノール類の濃度が50mg/l以上の場合には,その適量をとり,水を加えて250mlと
する。
試料中のフェノール類の濃度の概略値が分かっている場合には,試料を100ml,硫酸銅 (II) 溶
液の添加量を1mlにして,同じ操作で全留出液量を100mlとしてもよい。
(3) フェノール類の濃度が25μg/l以下の場合には,試料500mlを蒸留フラスコ1lにとり硫酸銅 (II)
溶液5mlを加え,蒸留して450mlが留出したとき,いったん加熱を止め,冷却した後,水50ml
を加え,再び蒸留を続けて更に50mlを留出させ,500mlとする。
(4) 留出液が白濁している場合には,留出液に再びりん酸 (1+9) を加えてpH約4の酸性とし,硫
酸銅 (II) 溶液2.5mlを加え,蒸留操作を繰り返す。再蒸留を行っても白濁が消えない場合には,
57
K 0101 : 1998
22.1.2の備考1.(3)の油分及びタール類の除去に従って処理する。
22.1.2 4-アミノアンチピリン吸光光度法 前処理(蒸留)した試料のpHを約10に調節し,4-アミノアン
チピリン(4-アミノ-1, 2-ジヒドロ-1, 5-ジメチル-2-フェニル-3H-ピラゾール-3-オン)溶液とヘキサシアノ鉄
(III) 酸カリウム溶液を加えて,生成する赤い色のアンチピリン色素の吸光度を測定してフェノール類を定
量する。
この方法では,フェノールのほかo-,m-位置に置換基のあるフェノール誘導体及び多環式化合物にヒド
ロキシル基が置換したものも4-アミノアンチピリンと反応してアンチピリン色素を生成して定量される。
p-位置に置換基があるフェノール誘導体は,4-アミノアンチピリンと反応しにくいので,ほとんど発色し
ない。アンチピリン色素の発色の強さは,置換基の種類,位置,数などによって差がある。
この試験では,フェノール標準液による発色の強さと比較して,フェノールとして表す。
定量範囲:抽出法 C6H5OH 2.5〜50μg,直接法 C6H5OH 50〜500μg,繰返し分析精度:変動係数で
3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 塩化アンモニウム-アンモニア緩衝液 (pH10) JIS K 8116に規定する塩化アンモニウム67.5gを
JIS K 8085に規定するアンモニア水570mlに溶かし,水で1lとする。この溶液は密栓して冷所に保
存する。
(c) 臭素酸カリウム溶液 (601mol/l) JIS K 8530に規定する臭素酸カリウム2.78gとJIS K 8506に規定
する臭化カリウム10gとを水に溶かして1lとする。
(d) 0.1mmol/lチオ硫酸ナトリウム溶液 JIS K 8637に規定するチオ硫酸ナトリウム五水和物26gとJIS
K 8625に規定する炭酸ナトリウム0.2gをとり,水に溶かして1lとし,気密容器に入れて少なくと
も2日間放置する。標定は使用時に行う。
標定 JIS K 8005に規定する容量分析用標準物質のよう素酸カリウムを130℃で約2時間加熱し,
デシケーター中で放冷する。その0.713gをとり,少量の水に溶かし,全量フラスコ200mlに移し入
れ,水を標線まで加える。この20mlを共栓三角フラスコ300mlにとり,JIS K 8913に規定するよ
う化カリウム2g及び硫酸 (1+5) 5mlを加え,直ちに栓をして静かに振り混ぜ,暗所に約5分間放
置する。
水100mlを加え,遊離したよう素をこのチオ硫酸ナトリウム溶液で滴定する。溶液の黄色が薄く
なってから,指示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消え
るまで滴定する。
別に,水について同一条件で空試験を行って補正したml数から次の式によって0.1mol/lチオ硫酸
ナトリウム溶液のファクター (f) を算出する。
567
003
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: よう素酸カリウムの量 (g)
b: よう素酸カリウムの純度 (%)
x: 滴定に要した0.1mol/lチオ硫酸ナトリウム溶液 (ml)
0.003 567: 0.1mol/lチオ硫酸ナトリウム溶液1mlのよう素酸カリ
ウム相当量 (g)
(e) ヘキサシアノ鉄 (III) 酸カリウム溶液 JIS K 8801に規定するヘキサシアノ鉄 (III) 酸カリウムの
大きな結晶9gをとり,少量の水で表面を洗った後,水に溶かして100mlとし,必要があればろ過
58
K 0101 : 1998
する。1週間ごとに調製するが,1週間以内でも色が暗い赤に変わったものは使用しない。
(f) 硫酸ナトリウム JIS K 8987に規定するもの。
(g) よう化カリウム JIS K 8913に規定するもの。
(h) 4-アミノアンチピリン溶液 (20g/l) JIS K 8048に規定する4-アミノアンチピリン(4-アミノ-1, 2-
ジヒドロ-1, 5-ジメチル-2フェニル-3H-ピラゾール-3-オン)2.0gを水に溶かして100mlとする。使用
時に調製する。
(i) でんぷん溶液 (10g/l) JIS K 8659に規定するでんぷん(溶性)1gを水約5mlに混ぜ,熱水100ml
中にかき混ぜながら加え,約1分間煮沸した後,放冷する。使用時に調製する。
(j) クロロホルム JIS K 8322に規定するもの。
(k) フェノール標準液 (1mgC6H5OH/ml) JIS K 8798に規定するフェノール(5)1gを水に溶かして1lと
する。冷暗所に保存する。
標定 共栓三角フラスコ500mlにこの溶液50mlをとり,水約100mlを加える。これに臭素酸カリ
ウム溶液 (601mol/l) 50ml(反応量は約40mlである。)を加え,更に塩酸5mlを加える(このときト
リブロモフェノールの白い沈殿を生じる。)。
密栓して静かに振り混ぜ,褐色の臭素が遊離した後,約10分間放置する。次に,JIS K 8913に規
定するよう化カリウム1gを加え,遊離したよう素を0.1mol/lチオ硫酸ナトリウム溶液で滴定し,溶
液の黄色が薄くなってから,指示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷん
の青い色が消えるまで滴定する。これに要した0.1mol/lチオ硫酸ナトリウム溶液の滴定ml数を(b)
とする。
別に,水100mlに臭素酸カリウム溶液 (601mol/l) 20mlを加えた溶液について,前と同様に操作し
て滴定に要する0.1mol/lチオ硫酸ナトリウム溶液のml数(a)を求める。次の式によってフェノール標
準液の濃度 (mg/ml) を算出する。
569
.1
50
1
)
5.2(
×
×
×
−
=
f
b
a
P
ここに,
P: フェノール標準液 (mgC6H5OH/ml)
f: 0.1mol/lチオ硫酸ナトリウム溶液のファクター
1.569: 0.1mol/lチオ硫酸ナトリウム溶液1mlのフェノール相当量
(mg)
(l) フェノール標準液 (10μgC6H5OH/ml) フェノール10mgに相当する量のフェノール標準液
(1mgC6H5OH/ml) を全量フラスコ1 000mlにとり,水を標線まで加える。使用時に調製する。
(m) フェノール標準液 (1μgC6H5OH/ml) フェノール標準液 (10μgC6H5OH/ml) 50mlを全量フラスコ
500mlにとり,水を標線まで加える。使用時に調製する。
注(5) ガスクロマトグラフ法で試験し,クレゾール類に相当する保持時間にピークのないものを用い
る。
次の操作を行って確認する。
フェノール標準液 (1mgC6H5OH/ml) 10mlを分液漏斗にとり,硫酸 (1+3) でpHを1〜2に調
節し,JIS K 8465に規定する1, 2-ジクロロエタン5mlを加えて振り混ぜ放置する。有機溶媒層
を分離し,硫酸ナトリウムを加え振り混ぜて脱水する。この溶液5μlをガスクロマトグラフ装
置に注入してクロマトグラムを求める。
ガスクロマトグラフの条件
カラム用管 ガラス製 内径3mm,長さ3m
59
K 0101 : 1998
カラム充てん剤 酸洗浄した粒径180〜250μmの耐火れんが(*)にエステル系固定相液体約
2.5%を含浸させたもの。
検出器 水素炎イオン化検出器
キャリヤーガス JIS K 1107に規定する高純度窒素2級を用い,流量は40〜50ml/min
カラム槽温度 180℃
検出器槽温度 195℃
この条件でo-クレゾール,フェノール,p-クレゾール,m-クレゾールの順にピークが出現す
る。
注(*) けい藻土を主成分とした耐火温度1 100℃のれんが。
参考 カラム充てん剤の市販品には,担体としてクロモゾルブW又はこれと同等の性能をもつものに,
エステル系固定相液体を含浸させたKG-02,FAP-Sなどがある。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 200ml
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 22.1.1の前処理を行った試料100ml(6)(フェノールとして2.5〜50μgを含む。)を分液漏斗200mlに
とり,塩化アンモニウム-アンモニア緩衝液 (pH 10) 3mlを加えて振り混ぜ,pHを10±0.2に調節す
る。
(b) 4-アミノアンチピリン溶液 (20g/l) 2mlを加えて振り混ぜ,次に,ヘキサシアノ鉄 (III) 酸カリウム
溶液2mlを加えて十分に振り混ぜた後,約3分間放置する(7)。
(c) クロロホルム10mlを加えて約1分間以上激しく振り混ぜた後静置する。クロロホルム層を乾いた
ろ紙でろ過するか,ビーカーに移した後,硫酸ナトリウム約1gを加えて脱水する。
(d) これを吸収セルに移し,別に,水100mlについて(a)〜(c)によって操作した空試験のクロロホルムを
対照液として波長460nm付近の吸光度を測定する。
(e) 検量線からフェノールの量を求め,試料中のフェノール類の濃度 (mgC6H5OH/l) を算出する。
検量線 フェノール標準液 (1μgC6H5OH/ml) 2.5〜50mlを分液漏斗250mlに段階的にとり,水を加え
て100mlとし,塩化アンモニウム-アンモニア緩衝液 (pH10) 3mlを加えて振り混ぜ,pHを10±0.2
に調節し,(b)〜(d)の操作を行ってフェノール (C6H5OH) の量と吸光度の関係線を作成する。
注(6) 試料100ml中のフェノールの量が2.5μg以下の場合には,試料500mlをとって蒸留し,その全量
を分液漏斗1 000mlにとり,塩化アンモニウム-アンモニア緩衝液 (pH10) 10ml, 4-アミノアン
チピリン溶液 (20g/l) 3ml,ヘキサシアノ鉄 (III) 酸カリウム溶液3mlを加え,十分に振り混ぜた
後3分間放置し,クロロホルム10mlを加えて抽出する。
なお,検量線は同一条件で作成する。
(7) このときの発色が十分に強いときは,この溶液を吸収セルに移し,別に,水100mlについて(a)
及び(b)に従って操作した空試験の溶液を対照液として,波長510mm付近の吸光度を測定する。
検量線からフェノールの量を求め,試料中のフェノール類の濃度 (mgC6H5OH/l) を算出する。
検量線 フェノール標準液 (10μgC6H5OH/ml) 5〜50mlをメスシリンダー(有栓形)100mlに段
階的にとり,水を100mlの標線まで加える。以下,(a)及び(b)の操作を行って,フェノールの量
と吸光度との関係線を作成する。
備考1. この試験では,酸化性物質,還元性物質,金属イオン,芳香族アミン類,油分及びタール類
60
K 0101 : 1998
などが妨害となる。
通常,蒸留操作で大部分の妨害を除くことができるが,酸化性物質,硫黄化合物,油分及
びタール類が試料中に含まれる場合には,次のように処理する。
(1) 酸化性物質 残留塩素のような酸化性物質が含まれていると,酸性でよう化カリウムを加え
たとき,よう素が遊離する。この場合は,試料採取直後に,JIS K 8978に規定する硫酸鉄 (II)
七水和物又はJIS K 8046に規定するメタ亜ひ酸ナトリウムの少過剰量を加えておく。
(2) 硫黄化合物 硫化水素及び亜硫酸イオンが含まれている場合には,試料採取直後にりん酸を
加えてpHを約4とし,注意して試料に空気を吹き込むか,かき混ぜて,硫化水素,二酸化
硫黄を追い出した後,JIS K 8983に規定する硫酸銅 (II) 五水和物を加える。
(3) 油分及びタール類 油分及びタール類が含まれている場合には,試料採取直後に硫酸銅 (II)
五水和物を加えずに,JIS K 8576に規定する水酸化ナトリウム(粒状)を加えてpHを12〜
12.5に調節し,分液漏斗に移してJIS K 8322に規定するクロロホルムを加え,油分及びター
ル類を抽出して,クロロホルム層を捨てる。水層は,水浴上で加熱して残留するクロロホル
ムを除去した後,JIS K 9005に規定するりん酸を加えてpHを4以下に調節して,硫酸銅 (II)
溶液2.5mlを加える。
2. 試料に色や濁りがなく,かつ,この試験に妨害となる成分を含まない場合には,蒸留操作を
省略し,直接試験を行ってよい。
22.2 p-クレゾール類 p-クレゾール類の試験は,前処理(水蒸気蒸留)した試料について,p-ヒドラジノ
ベンゼンスルホン酸吸光光度法を適用する。
22.2.1 p-ヒドラジノベンゼンスルホン酸吸光光度法 フェノール類をpH 8.0でギブス試薬と反応させて
インドフェノールに変え,アスコルビン酸酸性で水蒸気蒸留して,ギブス試薬と反応しないp-クレゾール
類を留出させる。留出したp-クレゾール類に,p-ヒドラジノベンゼンスルホン酸と亜硝酸から生成するジ
アゾ化合物,p-スルホンベンゼンジアゾニウム塩をカップリングさせて生じるアゾ色素の赤い色の吸光度
を測定してp-クレゾール類を定量する。
定量範囲:p-CH3C6H4OH 10〜150μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+17) JIS K 8951に規定する硫酸を用いて調製する。
(b) 水酸化ナトリウム溶液 (100g/l) JIS K 8576に規定する水酸化ナトリウム10gを水に溶かして
100mlとする。
(c) 炭酸ナトリウム JIS K 8625に規定するもの。
(d) 塩化ナトリウム JIS K 8150に規定するもの。
(e) メチルオレンジ溶液 (1g/l) 22.1.1(1)(d)による。
(f) クロロホルム又はジエチルエーテル JIS K 8322に規定するクロロホルム又はJIS K 8103に規定す
るジエチルエーテル。
(g) ギブス試薬 JIS K 8491に規定する2, 6-ジブロモ-N-クロロ-p-ベンゾキノンモノイミン(2, 6-ジブロ
モキノンクロロイミド)0.5gをJIS K 8102に規定するエタノール (95) 50mlに溶かす。使用時に調
製する。
(h) L (+) -アスコルビン酸 JIS K 9502に規定するもの。
(i) アンモニア水 (1+7) JIS K 8085に規定するアンモニア水を用いて調製する。
(j) p-ヒドラジノベンゼンスルホン酸溶液
61
K 0101 : 1998
A液 JIS K 9525に規定するp-ヒドラジノベンゼンスルホン酸0.5水和物1gとJIS K 8625に規定す
る炭酸ナトリウム0.3gとを水80mlに加え,水浴中で加熱して溶かし,これにJIS K 8180に規定す
る塩酸9mlを加え,更に水を加えて100mlとする。室温では結晶が析出するから,約37℃の恒温槽
中に保存する。1週間以上経過したものは使用しない。
B液 A液4mlを全量フラスコ100mlにとり,約10℃に冷却した後,亜硝酸ナトリウム溶液 (10g/l)
(JIS K 8019に規定する亜硝酸ナトリウム1gを水に溶かして100mlとする。)5mlを加えて約10℃
で3〜5分間放置する。これにあらかじめ約10℃に冷却した水を標線まで加える。使用時に調製す
る。
(k) p-クレゾール標準液 (1mgCH3C6H4OH/ml) JIS K 8306に規定するp-クレゾール1.00gをとり,少
量の水に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加える。
(l) p-クレゾール標準液 (0.1mgCH3C6H4OH/ml) p-クレゾール標準液 (1mgCH3C6H4OH/ml) 20mlを全
量フラスコ200mlにとり,水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 水蒸気蒸留装置 小形,すり合わせのもの。
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料500ml(p-クレゾールとして0.1〜1.5mgを含む。)を分液漏斗1 000mlにとり,指示薬としてメ
チルオレンジ溶液 (1g/l) 数滴を加え,溶液の色が赤になるまで硫酸 (1+17) を滴加して酸性[試料
が酸性の場合は,水酸化ナトリウム溶液 (100g/l) で溶液の色が黄色になるまで中和した後,硫酸 (1
+17) を滴加して再び酸性にする。]とし,これに塩化ナトリウム150gとクロロホルム(8)40mlを加
え,激しく振り混ぜて抽出し,クロロホルム層を別の分液漏斗200mlに移す。
(b) 更にクロロホルム25mlを用いて(a)と同様の抽出操作を4回繰り返し,クロロホルム層を先の分液
漏斗に合わせる。
(c) このクロロホルム層に水酸化ナトリウム溶液 (100g/l) 4mlを加えて逆抽出し,更に3mlを加えて逆
抽出を2回繰り返し,逆抽出液を合わせる。
(d) この逆抽出液を沸騰水浴上で加熱して,溶けているクロロホルムを揮散させた後,放冷する。
(e) 水約20mlで全量フラスコ100mlに移し入れる。次に,炭酸ナトリウム2gを加え,硫酸 (1+17) を
滴加してpHを8に調節し,水約20mlとギブス試薬5mlとを加えて24時間放置する(フェノール
類が共存すれば溶液の色は青になる。)。
(f) L (+) -アスコルビン酸1gを加え,水を標線まで加える。
(g) この溶液10mlを蒸留フラスコにとり水蒸気蒸留を行い,メスシリンダー(有栓形)50mlに30ml
を留出させる。
(h) 留出液を水で約40mlに薄めた後,p-ヒドラジノベンゼンスルホン酸溶液のB液5mlを加えて振り
混ぜ,次に,アンモニア水 (1+7) 1mlを加え,水を50mlの標線まで加えて再び振り混ぜ,約5分
間放置する。
(i) 溶液の一部を吸収セルに移し,波長495nm付近の吸光度を測定する。
(j) 空試験として水40mlをとり,(h)及び(i)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
(k) 検量線からp-クレゾール類の量を求め,試料中のp-クレゾール類の濃度 (mgCH3C6H4OH/l) を算出
する。
62
K 0101 : 1998
検量線 p-クレゾール標準液 (0.1mgCH3C6H4OH/ml) 1〜15mlを全量フラスコ100mlに段階的にとり,
(e)の炭酸ナトリウム2gを加える以降の操作,及び(f)の操作を行う。この溶液10mlを蒸留フラスコ
にとり,水蒸気蒸留を行って留出液30mlを得た後,水で約40mlに薄める。次に,p-ヒドラジノベ
ンゼンスルホン酸のB液5mlを加え,(h)〜(j)の操作を行ってp-クレゾール (CH3C6H4OH) の量と吸
光度との関係線を作成する。
注(8) クロロホルムの代わりにJIS K 8103に規定するジエチルエーテルを用いてもよい。この場合に
は,JIS K 8150に規定する塩化ナトリウムを加える必要はない。
23. 界面活性剤 界面活性剤は,陰イオン界面活性剤及び非イオン界面活性剤に区分する。
界面活性剤は,微生物によって容易に分解されるため,試験は試料採取後直ちに行う。直ちに試験が行
えない場合は,3.3によって保存し,できるだけ早く試験する。
23.1 陰イオン界面活性剤 陰イオン界面活性剤の定量には,メチレンブルー吸光光度法,エチルバイオ
レット吸光光度法又は溶媒抽出-フレーム原子吸光法を適用する。
陰イオン界面活性剤には高級アルコール硫酸エステル類,脂肪酸硫酸エステル類及びスルホン酸形陰イ
オン界面活性剤[アルキルアリールスルホン酸塩類(直鎖アルキルベンゼンスルホン酸塩類,LAS),アル
キルスルホン酸塩類,アルケンスルホン酸塩類など]などがある。
23.1.1 メチレンブルー吸光光度法 陰イオン界面活性剤がメチレンブルー[3, 7-ビス(ジメチルアミノ)
フェノチアジン-5-イウムクロリド]と反応して生じるイオン対をクロロホルムで抽出して,その吸光度を
測定し,ドデシル硫酸ナトリウムとして表す。
定量範囲:陰イオン界面活性剤 [NaO3SO (CH2) 11CH3] 2〜50μg,繰返し分析精度:変動係数で5〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(c) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(d) アルカリ性四ほう酸ナトリウム溶液 JIS K 8866に規定する四ほう酸ナトリウム十水和物9.54gを
水に溶かした後,水で500mlとし,これに水酸化ナトリウム溶液 (40g/l) 50mlを加え,水で全量を
1lとする。
(e) メチレンブルー溶液 (0.25g/l) JIS K 8897に規定するメチレンブルー(通常は三水和物)0.3gを水
に溶かして1lとする。
(f) 脱脂綿
(g) クロロホルム JIS K 8322に規定するもの。
(h) 陰イオン界面活性剤標準液 [1mgNaO3SO (CH2) 11CH3/ml] ドデシル硫酸ナトリウム(1)をその
100%(2)に対して1.00gをとり,水に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加え
る。
(i) 陰イオン界面活性剤標準液 [10μgNaO3SO (CH2) 11CH3/ml] 陰イオン界面活性剤標準液
[1mgNaO3SO (CH2) 11CH3/ml] 10mlを全量フラスコ1 000mlにとり,水を標線まで加える。使用時に
調製する。
注(1) 純度及び平均分子量の分かった市販品を用いる。
(2) 純度及び平均分子量を確認する場合には,備考4.による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
63
K 0101 : 1998
(a) 分液漏斗 250ml
(b) 光度計 分光光度計又は光電光度計
(3) 準備操作 準備操作は,次のとおり行う。
(a) 分液漏斗 (A) に水50ml,アルカリ性四ほう酸ナトリウム溶液10ml及びメチレンブルー溶液
(0.25g/l) 5mlを入れる。
分液漏斗 (B) に水100ml,アルカリ性四ほう酸ナトリウム溶液10ml及びメチレンブルー溶液
(0.25g/l) 5mlを入れる。
(b) それぞれにクロロホルム10mlを加え,約30秒間激しく振り混ぜた後,放置してクロロホルム層を
捨てる。この操作を更に1回繰り返す。
(c) 水層にクロロホルム2〜3mlを加え,緩やかに振り混ぜた後,放置してクロロホルム層を捨てる。
この操作をクロロホルム層が無色になるまで繰り返す。
(d) クロロホルムで洗い終わった分液漏斗 (B) 中の水層に硫酸 (1+35) 3mlを加える。
なお,分液漏斗 (A) 及び (B) の脚部がぬれているときには,ろ紙などでふきとる。
(4) 操作 操作は,次のとおり行う。
(a) (3) の準備操作を行った分液漏斗 (A) 中の水層に,試料(3)の適量[NaO3SO (CH2) 11CH3として2〜
50μgを含む。]を加える。ただし,全量が100mlを超えないようにする。
(b) クロロホルム10mlを加えて,緩やかに約1分間振り混ぜて放置し,クロロホルム層を(3)の準備操
作を行った分液漏斗 (B) に移し入れる。
(c) 分液漏斗 (B) を緩やかに約1分間振り混ぜた後放置する。分液漏斗の脚部に脱脂綿を詰め,クロロ
ホルム層を全量フラスコ25mlに移し入れる。
(d) 再び分液漏斗 (A) にクロロホルム10mlを加えて,(b)及び(c)の操作を繰り返して抽出を行い,クロ
ロホルム層を(c)と同様に先の全量フラスコ25mlに合わせ,クロロホルムを標線まで加える。
(e) これを吸収セル(4)に移し,クロロホルムを対照液として波長650nm付近の吸光度を測定する。
(f) 空試験として水50mlを用い,あらかじめ(3)の準備操作を行った分液漏斗に入れ,(a)〜(e)の操作を
行って吸光度を測定し,試料について得た吸光度を補正する。
(g) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度 [mgNaO3SO (CH2)
11CH3/l] を算出する。
検量線 陰イオン界面活性剤標準液 [10μgNaO3SO (CH2) 11CH3/ml] 0.2〜5mlを段階的にとり,あら
かじめ (3) の準備操作を行った分液漏斗に入れ,(a)〜(f)の操作を行って陰イオン界面活性剤
[NaO3SO (CH2) 11CH3] の量と吸光度との関係線を作成する。
注(3) 酸性の場合には,pH計を用いて水酸化ナトリウム溶液 (40g/l) で,また,アルカリ性の場合に
は,硫酸 (1+35) でpHを約7に調節する。
(4) 吸収セル50mmを用いると0.4〜10μgの陰イオン界面活性剤が定量できる。
備考1. 硝酸,シアン化物,チオシアン酸などのイオンが多量に存在すると定量を妨害する。
陽イオン界面活性剤は,陰イオン界面活性剤と強く結合するため,その共存量に応じて負
の誤差を与える。しかし,通常の水ではその量は,陰イオン界面活性剤と比較して非常に少
ない。
2. みずみみず,いとみみずなどがいる底泥付近の水では,正の誤差が生じやすい。
3. スルホン酸形陰イオン界面活性剤(LASなど)を定量するには,次の操作によってアルコー
ル系などの陰イオン界面活性剤を加水分解し,残ったスルホン酸形陰イオン界面活性剤を
64
K 0101 : 1998
(4) の操作で定量して,ドデシル硫酸ナトリウムとして表す。
試料の適量[NaO3SO (CH2) 11CH3として4〜100μgを含む。]をすり合わせ三角フラスコに
とり,JIS K 8180に規定する塩酸25ml及び沸騰石数個を加え,水で液量を約50mlとした後,
還流冷却器を付けて約2時間静かに煮沸する。
放冷後,指示薬としてフェノールフタレイン溶液 (5g/l) [13.2(1)(a)による。]数滴を加え,
溶液の色が微赤になるまで,初めは水酸化ナトリウム溶液 (400g/l) を,中和点近くなってか
らは水酸化ナトリウム溶液 (40g/l) を加えて中和し,水で100mlとする。
以下,(3)及び(4)の操作を行ってスルホン酸形陰イオン界面活性剤の量を求め,試料中のス
ルホン酸形陰イオン界面活性剤の濃度 [mgNaO3SO (CH2) 11CH3/l] を算出する。
4. ドデシル硫酸ナトリウムの純度及び平均分子量の測定は,次の方法による。
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (0.5mol/l) 水1l中にJIS K 8951に規定する硫酸30mlを徐々に加える。
(b) エタノール (95) JIS K 8102に規定するもの。
(c) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(d) ヘキサン JIS K 8848に規定するもの。
(e) 硫酸ナトリウム JIS K 8987に規定するもの。
(f) 1mol/l水酸化ナトリウム溶液 水約60mlをポリエチレン瓶などの耐アルカリ容器にとり,
冷却しながらJIS K 8576に規定する水酸化ナトリウム80g(少量の水で表面を洗う。)を少
量ずつ加えて溶かし,密栓して4〜5日間放置する。その上澄み液50mlをポリエチレン製
の気密容器1lにとり,2.(12)(b)の炭酸を含まない水を加えて1lとし,混合した後,二酸化
炭素を遮断して保存する。
標定 JIS K 8005に規定する容量分析用標準物質のアミド硫酸をデシケーター中に2kPa
以下で約48時間放置して乾燥する。その約2gを1mgのけたまではかりとり,三角フラス
コ200mlに入れ,水約25mlを加えて溶かし,これに指示薬としてブロモチモールブルー
溶液 (1g/l) [14.の注(1)による。]3〜5滴を加え,この1mol/l水酸化ナトリウム溶液で滴
定し,溶液の色が緑になったときを終点とする。次の式によって1mol/l水酸化ナトリウム
溶液のファクター (f) を算出する。
1
097
.0
1
100
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の量 (g)
b: アミド硫酸の純度 (%)
x: 滴定に要した1mol/l水酸化ナトリウム溶液 (ml)
0.097 1: 1mol/l水酸化ナトリウム溶液1mlのアミド硫酸相当量
(g)
(g) 高級アルコール混合標準液 1-デカノール0.50gと,1-ドデカノール0.50g及び1-テトラデ
カノール0.50gをはかりとり,JIS K 8848に規定するヘキサン15mlに溶かす。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 還流冷却器付三角フラスコ 冷却部200〜300mmの還流冷却器(リービッヒ冷却器,球管
冷却器)と,すり合わせ三角フラスコ300ml
(b) 分液漏斗 300ml
(c) ガスクロマトグラフ 次の条件を満たすもの。
65
K 0101 : 1998
カラム用管 ステンレス鋼製 内径3〜4mm,長さ1〜2m
カラム充てん剤 粒径150〜250μmの耐火れんが(*)を担体とし,これにシリコーン系固定
相液体約10%を含浸させたもの。
検出器 水素炎イオン化検出器
キャリヤーガス JIS K 1107に規定する高純度窒素2級又はヘリウム (99.8vol%)
試料気化室温度 290〜300℃
カラム槽温度 170〜180℃
キャリヤーガスの流量は,高級アルコール類が約30分間で流出するように調節する。
注(*) けい藻土を主成分とした耐火温度1 100℃のれんが。
参考 カラム充てん剤の市販品には,耐火れんがとしてクロモソルブW又はこれと同等の性
能をもつものを担体とし,これに固定相液体としてシリコーンSE-30などを含浸させ
たものがある。
(3) 純度の測定 操作は,次のとおり行う。
(a) ドデシル硫酸ナトリウム約4gを1mgのけたまではかりとり,三角フラスコ300mlに入れ
る。
(b) 硫酸 (0.5mol/l) 20mlを加えた後,還流冷却器を付けて水浴上で加熱する。泡立ちに注意し,
時々三角フラスコを軽く揺り動かし,液が透明になるまで加熱する。
(c) 引き続き加熱板上で約2時間,加熱還流する。
(d) 放冷後,冷却器の上部からエタノール (95) 約30mlを注いで内壁を洗い,適量の水で洗っ
た後,冷却器を取り除く。
(e) 水を加えて液量を約100mlとし,指示薬としてフェノールフタレイン溶液 (5g/l) 数滴を加
え,1mol/l水酸化ナトリウム溶液で滴定し,溶液の色が微赤になった点を終点とする。
(f) 別に,同一条件で空試験を行い,次の式によってドデシル硫酸ナトリウムの純度 (%) を算
出する。
000
1
)
(
×
×
×
−
=
S
M
f
b
a
P
ここに,
P: ドデシル硫酸ナトリウムの純度 (%)
a: 滴定に要した1mol/l水酸化ナトリウム溶液 (ml)
b: 空試験に要した1mol/l水酸化ナトリウム溶液 (ml)
f: 1mol/l水酸化ナトリウム溶液のファクター
M: ドデシル硫酸ナトリウムの平均分子量
S: ドデシル硫酸ナトリウムの量 (g)
(4) ドデシル硫酸ナトリウムの平均分子量の測定 操作は,次のとおり行う。
(a) (3)(e)の操作で得られた溶液中から50mlを分液漏斗300mlにとる。
(b) エタノールと水の混合液 (2+1) 100mlとヘキサン50mlとを加えて振り混ぜ,高級アルコ
ール類を抽出する。
(c) 静置してヘキサン層を分離し,水層を別の分液漏斗300mlに移す。
(d) この水層にヘキサン50mlを加え,振り混ぜて抽出し,静置してヘキサン層を分離する。水
層を捨て,ヘキサン層を前のヘキサン層に合わせる。
(e) これに水50mlを加えて振り混ぜ,静置してヘキサン層を分離し,水層を捨てる。この洗浄
操作を再び行い,水層を完全に分離して捨てる。
66
K 0101 : 1998
(f) 硫酸ナトリウム約2gを加えてヘキサン層を脱水した後,磁器蒸発皿100mlに移し入れ,水
浴又は加熱板上でヘキサンを揮散させる。
(g) 残留した高級アルコール類にヘキサン15mlを加えて溶かす(高級アルコール約100g/lを
含むヘキサン溶液になる。)。
(h) ガスクロマトグラフをJIS K 0114に従って最適条件に設定し,高級アルコール混合標準液
1μlをマイクロシリンジでとり,ガスクロマトグラフに注入し,各高級アルコールのクロ
マトグラムを記録し,流出位置を確認する。
(i) 次に,(g)で得た高級アルコール類のヘキサン溶液1μlをマイクロシリンジでとり,ガスク
ロマトグラフに注入し,各高級アルコールのクロマトグラムを記録する。
(j) (i)の操作を3回繰り返して行い,各高級アルコールのピーク面積を測定(半値幅法などで)
し,ピーク面積の平均値を求める。
(k) 炭素数10〜14の各高級アルコールのモル百分率を次の式によって求め,ドデシル硫酸ナト
リウムの平均分子量を算出する。
100
14
14
12
12
10
10
10
10
10
×
+
+
=
C
R
C
R
C
R
C
R
m
100
14
14
12
12
10
10
12
12
12
×
+
+
=
C
R
C
R
C
R
C
R
m
100
14
14
12
12
10
10
14
14
14
×
+
+
=
C
R
C
R
C
R
C
R
m
102
100
100
100
14
14
12
12
10
10
+
×
+
×
+
×
=
m
C
m
C
m
C
M
ここに, m10〜m14: 炭素数10〜14の各高級アルコールのモル百分率 (%)
C10〜C14: 炭素数10〜14の各高級アルコールの分子量 (C10=
158.29, C12=186.34, C14=214.39)
R10〜R14: 炭素数10〜14の各高級アルコールのピーク面積
M: ドデシル硫酸ナトリウムの平均分子量
102: ドデシル硫酸ナトリウムに換算するための補正項
(SO4Naの式量からOHの式量を差し引いた値)
23.1.2 エチルバイオレット吸光光度法 陰イオン界面活性剤がエチルバイオレット【N-〔4-{ビス[4-(ジ
エチルアミノ)フェニル]メチレン}-2, 5-シクロヘキサジエン-1-イリデン〕-N-エチルエタンアミンイウ
ムクロリド】と反応して生じるイオン対をトルエンに抽出して,その吸光度を測定し,ドデシル硫酸ナト
リウムとして表す。
定量範囲:陰イオン界面活性剤 [NaO3SO (CH2) 11CH3] 0.5〜12.5μg,繰返し分析精度:変動係数で5
〜10%
(1) 試薬 試薬は,次のものを用いる。
67
K 0101 : 1998
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸ナトリウム溶液 (1mol/l) JIS K 8987に規定する硫酸ナトリウム142gを水に溶かして1lとす
る。
(c) 酢酸-EDTA緩衝液 (pH5) JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二
水和物7.5gを水に溶かして約700mlとし,これにJIS K 8355に規定する酢酸12.5mlを加え,pH計
を用いてpHが5になるまで水酸化ナトリウム溶液 (2mol/l) を加えた後,水を加えて1lとする。
(d) エチルバイオレット溶液 (1mmol/l) エチルバイオレット(5)0.280gを水に溶かして500mlとする。
(e) トルエン JIS K 8680に規定するもの。
(f) 陰イオン界面活性剤標準液 [1mgNaO3SO (CH2) 11CH3/ml] 23.1.1(1)(h)による。
(g) 陰イオン界面活性剤標準液 [10μgNaO3SO (CH2) 11CH3/ml] 23.1.1(1)(i)による。
(h) 陰イオン界面活性剤標準液 [0.5μgNaO3SO (CH2) 11CH3/ml] 陰イオン界面活性剤標準液
[10μgNaO3SO (CH2) 11CH3/ml] 10mlを全量フラスコ200mlにとり,水を標線まで加える。使用時に調
製する。
注(5) エチルバイオレットは,塩化亜鉛21モルが付加した複塩を用いる。この複塩以外を用いる場合
には,その濃度が1mmol/lになる量をとって調製する。
また,(3)(g)の空試験の操作を行ったときの吸光度の値が大きい(0.04程度以上)場合には,
別のロットのエチルバイオレットを用いて調製し直す。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 200ml
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量[NaO3SO (CH2) 11CH3として0.5〜12.5μgを含む。]を分液漏斗にとり,水を加えて100ml
とする。
(b) これに硫酸ナトリウム溶液 (1mol/l) 5ml,酢酸-EDTA緩衝液 (pH5) 5ml及びエチルバイオレット溶
液 (1mmol/l) 2mlを加える。
(c) トルエン5mlを加え,約10分間振り混ぜる(6)。
(d) 静置し,水層約100mlを捨てる。
(e) 更に静置し,トルエン層が完全に分離したら水層を捨てる。
(f) トルエン層を吸収セルに移し,トルエンを対照液として波長611nm付近の吸光度を測定する。
(g) 空試験として水100mlを用い,(b)〜(f)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
(h) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度 [mgNaO3SO (CH2)
11CH3/l] を算出する。
検量線 陰イオン界面活性剤 [0.5μgNaO3SO (CH2) 11CH3/ml] 1〜25mlを段階的に分液漏斗にとり,
水を加えて100mlとした後,(b)〜(g) の操作を行って陰イオン界面活性剤 [NaO3SO (CH2) 11CH3] の
量と吸光度との関係線を作成する。
注(6) 振り混ぜ時間が吸光度に幾分影響するから,振り混ぜ時間を守る。
備考5. 海水,又は海水が混入した試料のように多量の塩化物イオンが共存すると,その一部がエチ
ルバイオレットとイオン対を生成し,トルエンに抽出されて吸光度を増加させる。このよう
な試料の場合は,(3)(d)及び(e)の操作を行った後も,器壁に付着物が残るが,トルエン層を小
68
K 0101 : 1998
形の分液漏斗に移し入れ,エチルバイオレット(15μmol/l)-硫酸ナトリウム (10g/l) の溶液
[エチルバイオレット (1mmol/l) 7.5mlをとり,硫酸ナトリウム5gを加え,水で500mlとする。]
20mlを加え,約1分間振り混ぜる。静置した後,水層の大部分を捨て,再び静置してトルエ
ン層が完全に分離したら水層を捨てる。以下,(f)〜(h)の操作を行う。
6. 硝酸イオンは1mgNO3-/l程度までは妨害しないが,それ以上共存すると正の誤差を与える。
通常の河川水などに含まれるそのほかのイオンは妨害しない。
陽イオン界面活性剤は,陰イオン界面活性剤と強く結合するため,その共存量に応じて負
の誤差を与える。しかし,通常の水ではその量は,陰イオン界面活性剤と比較して非常に少
ない。
23.1.3 溶媒抽出-フレーム原子吸光法 陰イオン界面活性剤をカリウムを取り込んだジベンゾ-18-クラウ
ン-6とイオン対とし,これを4-メチル-2-ペンタノンに抽出し,抽出溶液中のカリウムをフレーム原子吸光
法で定量し,ドデシル硫酸ナトリウムとして表示する。
定量範囲:陰イオン界面活性剤 [NaO3SO (CH2) 11CH3] 2.5〜50μg,繰返し分析精度:変動係数で2〜
10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸カリウム (20mmol/l) -酢酸アンモニウム (50mmol/l) 混合溶液 JIS K 8962に規定する硫酸カ
リウム3.5gとJIS K 8359に規定する酢酸アンモニウム3.9gとを水に溶かして約700mlとし,pH計
を用いて硫酸 (1+35) を加えpHを5に調節して,水で1lとする。
(c) 硫酸カリウム (4mmol/l) -酢酸アンモニウム (10mmol/l) 混合溶液 硫酸カリウム (20mmol/l) -酢酸
アンモニウム (50mmol/l) 混合溶液200mlを水で薄めて1lとする。
(d) ジベンゾ-18-クラウン-6の4-メチル-2-ペンタノン溶液 (0.5mmol/l) 精製したジベンゾ−18-クラウ
ン−6(7)90mgをJIS K 8903に規定する4-メチル−2-ペンタノン500mlに溶かす。
(e) 陰イオン界面活性剤標準液 [1mgNaO3SO (CH2) 11CH3/ml] 23.1.1(1)(h)による。
(f) 陰イオン界面活性剤標準液 [5μgNaO3SO (CH2) 11CH3/ml] 陰イオン界面活性剤標準液
[1mgNaO3SO (CH2) 11CH3/ml] 5mlを全量フラスコ1 000mlにとり,水を標線まで加える。使用時に調
製する。
注(7) ジベンゾ-18-クラウン-6の精製は,次による。
ジベンゾ-18-クラウン-6約2.5gをJIS K 8858に規定するベンゼン約200ml中に加え,水浴上
で加熱して溶かす。これをガラスろ過器 (1G3) で吸引ろ過する。ろ液が冷えると直ちに結晶が
生成するが,再び水浴上で加熱して溶かし,吸引ろ過する。この操作を結晶が白くなるまで行
った(2,3回)後,ろ液を冷却し,吸引ろ過する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 100ml
(b) フレーム原子吸光分析装置
(c) カリウム中空陰極ランプ
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量[NaO3SO (CH2) 11CH3として2.5〜50μgを含む。]を分液漏斗100mlに取り,硫酸カリウ
ム (20mmol/l) -酢酸アンモニウム (50mmol/l) 混合溶液10mlを加え,水で液量を50mlとする。
(b) ジベンゾ-18-クラウン-6の4-メチル-2-ペンタノン溶液 (0.5mmol/l) 10mlを加え約1分間振り混ぜる。
69
K 0101 : 1998
(c) 静置後水層を捨て,硫酸カリウム (4mmol/l) -酢酸アンモニウム (10mmol/l) 混合溶液25mlを加えて
振り混ぜ,放置し,水層を捨てる。
(d) 4-メチル-2-ペンタノン層をJIS K 0121の6.(操作方法)の操作に従って,アセチレン-空気フレー
ム中に噴霧し,波長766.5nmの指示値(8)を読み取る。
(e) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度 [mgNaO3SO (CH2)
11CH3/l] を算出する。
検量線 陰イオン界面活性剤 [5μgNaO3SO (CH2) 11CH3/ml] 0.5〜10mlを分液漏斗に段階的にとり,
(a)〜(d)の操作を行って陰イオン界面活性剤 [NaO3SO (CH2) 11CH3] の量と指示値との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(8) 吸光度又はその比例値
備考7. カルシウム,マグネシウムは500mg共存しても影響しない。ナトリウムは数10mgの共存で
も,一部がジベンゾ-18-クラウン-6に取り込まれ,陰イオン界面活性剤とイオン対を作って抽
出され,そのまま噴霧すると負の誤差を与えるが,抽出分離後の溶媒層を(3)(c)の硫酸カリウ
ム (4mmol/l) -酢酸アンモニウム (10mmol/l) 混合溶液と振り混ぜることによって,ナトリウ
ムはカリウムに置換され,妨害は除かれる。
陽イオン界面活性剤は陰イオン界面活性剤と強く結合するため,その共存量に応じて負の
誤差を与える。しかし,通常の水ではその量は陰イオン界面活性剤と比較して非常に少ない。
非イオン界面活性剤は400μg程度共存しても妨害しない。
23.2 非イオン界面活性剤 非イオン界面活性剤には,ポリオキシエチレンアルキルエーテル類,ポリオ
キシエチレンアルキルフェノールエーテル類,ポリオキシエチレンアルキルエステル類,ポリオキシエチ
レンソルビタンアルキルエステル類などがある。
非イオン界面活性剤の定量には,前処理(イオン交換分離)を行った試料について,テトラチオシアナ
トコバルト (II) 酸吸光光度法を適用する。
23.2.1 テトラチオシアナトコバルト (II) 酸吸光光度法 非イオン界面活性剤とテトラチオシアナトコバ
ルト (II) 酸アンモニウムとの錯体をベンゼンで抽出して,紫外部の吸光度を測定し,ヘプタオキシエチレ
ンドデシルエーテルとして表示する。
定量範囲: 非イオン界面活性剤 [CH3 (CH2) 11O (CH2CH2O) 7H] 0.1〜2mg,繰返し分析精度:変動係
数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(c) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(d) テトラチオシアナトコバルト (II) 酸アンモニウム溶液 JIS K 9000に規定するチオシアン酸アン
モニウム310gとJIS K 8552に規定する硝酸コバルト (II) 六水和物140gとを水に溶かして500ml
とする。これを分液漏斗1 000mlに移し,JIS K 8858に規定するベンゼン50mlを加えて激しく振り
混ぜて放置する。ベンゼン層を捨て,再びベンゼン50mlを加えて振り混ぜ放置する。ベンゼン層
を捨て,水層を乾いたろ紙でろ過し,ベンゼンの小泡を除く。
(e) 塩化ナトリウム JIS K 8150に規定するもの。
(f) 硫酸ナトリウム JIS K 8987に規定するもの。
(g) エタノール (95) JIS K 8102に規定するもの。
70
K 0101 : 1998
(h) エタノール (1+1) 水1容に,JIS K 8102に規定するエタール (95) 1容を加えて調製する。
(i) ベンゼン JIS K 8858に規定するもの。
(j) 強酸性陽イオン交換樹脂 低架橋度(ジビニルベンゼン含量4〜6%)で粒子径300〜1 180μmのも
の。R-Na+形。次のように精製して用いる。
強酸性陽イオン交換樹脂250mlを内径40〜50mm,高さ約1 000mmのカラム(ガラス製又はアク
リル樹脂製)に水とともに流し入れ気泡が混入しないように充てんする。塩酸 (1+11) 2lを約5l/
(l-樹脂・h)で流した後,水1lを同様に流して洗浄する。次に,水酸化ナトリウム溶液 (40g/l) 1l
を約5l/(l-樹脂・h)で流し,水1lを同様に流して洗浄する。さらに塩酸 (1+11) 1lと水酸化ナト
リウム溶液 (40g/l) 1lとを同様に流して洗浄する。次に,フェノールフタレイン溶液 (5g/l)
[13.2(1)(a)による。]の赤い色がほとんど認められなくなるまで水で洗浄[約20l/(l-樹脂・h)で
流す。]する。
(k) 強塩基性陰イオン交換樹脂(I形) 低架橋度(ジビニルベンゼン含量4〜6%)で粒子径300〜1 180μm
のもの。R−C-形。次のように精製して用いる。
強塩基性陰イオン交換樹脂(I形)500mlを内径40〜50mm,高さ約1 000mmのカラム(ガラス製
又はアクリル樹脂製)に水とともに流し入れ気泡が混入しないように充てんする。水酸化ナトリウ
ム溶液 (40g/l) 2lを約5l/(l-樹脂・h)で流した後,水約2lを同様に流して洗浄する。次に,塩酸 (1
+11) 2lを約5l/(l-樹脂・h)で流した後,水約2lを同様に流して洗浄する。さらに水酸化ナトリウ
ム溶液 (40g/l) 2lと塩酸 (1+11) 2lとを同様に流して洗浄する。次に,メチルレッド-ブロモクレゾ
ールグリーン混合溶液[13.1(1)(a)による。]に対して青い色になるまで水で洗浄[約20l/(l-樹脂・
h)で流す。]する。
(l) 非イオン界面活性剤標準液 [0.1mgCH3 (CH2) 11O (CH2CH2O) 7H/ml] ヘプタオキシエチレンドデ
シルエーテル(9)をその100%に対して0.100gをはかりとり,水に溶かして全量フラスコ1 000mlに
移し入れ,水を標線まで加える。使用時に調製する。
注(9) 品質を確認する場合には,社団法人日本油化学協会で定めた試験方法による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 200ml
(b) イオン交換樹脂カラム 図23.1に一例を示す。
イオン交換樹脂カラムの作り方 強酸性陽イオン交換樹脂と強塩基性陰イオン交換樹脂(I形)と
を体積比で1 : 2になるようにとる。水を加えてよく混合しながら,気泡の混入しないように図23.1
のガラス管に充てんし,イオン交換樹脂柱の高さを約200mmに調節する。エタノール (1+1) 100ml
を流す。このイオン交換樹脂カラムは,数回繰り返し使用してもよい。
71
K 0101 : 1998
図23.1 イオン交換樹脂カラムの一例
(c) 光度計 分光光度計
(d) 吸収セル 石英ガラス製又は同等の品質のもの。
(3) 前処理 前処理は,次のとおり行う。
(a) 試料(10)100mlをとり,エタノール (95) 100mlを加えて振り混ぜる。
(b) この溶液をイオン交換樹脂カラムに10〜15l/(l-樹脂・h)で流し,流出液をビーカー500mlに受け
る。
(c) イオン交換樹脂カラムのイオン交換樹脂柱の上部に液面が近づいたら,エタノール (1+1) 100mlを
少量ずつ加え,イオン交換樹脂カラム内の試料を流出させる。流出液は(b)のビーカー500mlに合わ
せる。
(d) 流出液を水浴上で約30mlになるまで蒸発させる。
(e) 放冷後,この溶液を全量フラスコ100mlに移し入れ,水を標線まで加える。
注(10) 酸性の場合には,水酸化ナトリウム溶液 (40g/l) で,また,アルカリ性の場合には,塩酸 (1+
11) でpH計を用いてpHを約7に調節する。
(4) 操作 操作は,次のとおり行う。
(a) (3)(e)の溶液の適量[CH3 (CH2) 11O (CH2CH2O) 7Hとして0.1〜2mgを含む。]を分液漏斗200mlにと
り,水で100mlとする。
(b) テトラチオシアナトコバルト (II) 酸アンモニウム溶液15mlと塩化ナトリウム(11)35gとを加えて約
1分間振り混ぜた後,約15分間放置する。
(c) ベンゼン(12)25mlを加えて約1分間激しく振り混ぜて放置する。
(d) 水層を捨て,ベンゼン層をビーカーに移し,硫酸ナトリウム約5gを加えて振り混ぜ,脱水する。
(e) これを吸収セルに移し,水100mlについて,(b)〜(d)の操作を行ったベンゼンを対照液とし,波長
322nm付近の吸光度を測定する。
72
K 0101 : 1998
(f) 空試験として(a)と同量の(3)(e)の溶液を分液漏斗200mlにとり,水で100mlとし,(b)のテトラチオ
シアナトコバルト (II) 酸アンモニウム溶液15mlの代わりに水15mlを用い,(b)〜(d)の操作を行っ
た後,ベンゼンを対照液として波長322nm付近の吸光度を測定し,試料について得た吸光度を補正
する。
(g) 検量線から非イオン界面活性剤の量を求め,試料中の非イオン界面活性剤の濃度 [mgCH3 (CH2) 11O
(CH2CH2O) 7H/l] を算出する。
検量線 非イオン界面活性剤標準液 [0.1mgCH3 (CH2) 11O (CH2CH2O) 7H/ml] 1〜20mlを分液漏斗
200mlに段階的にとり,水を加えて100mlとし,(b)〜(e)の操作を行って非イオン界面活性剤 [CH3
(CH2) 11O (CH2CH2O) 7H] の量と吸光度との関係線を作成する。
注(11) 塩化カリウムを用いてもよい。
(12) 1, 2-ジクロロエタンを用いてもよい。
備考8. ポリエチレングリコールが共存すると非イオン界面活性剤として定量値に含まれて誤差とな
るので,JIS K 8810に規定する1-ブタノール又はJIS K 8900に規定する2-ブタノン(エチルメ
チルケトン)であらかじめ抽出除去した後,(3)の前処理を行う。
9. 陰イオン界面活性剤及び陽イオン界面活性剤が共存しない場合には,(3)の前処理を省略する
ことができる。
10. 非イオン界面活性剤の濃度が1mgCH3 (CH2) 11O (CH2CH2O) 7H/l以下の場合には,次のように
濃縮した後,操作する。
試料500mlにつき塩化ナトリウム50gとJIS K 8625に規定する炭酸ナトリウム2.5gを加え
て溶かし,分液漏斗1 000mlに移し,JIS K 8361に規定する酢酸エチル25mlを加えて約2分
間激しく振り混ぜて放置する。分離した酢酸エチル層をビーカーに移し,水層には酢酸エチ
ル25mlを加えて再び抽出を繰り返す。分離した酢酸エチル層を先のビーカーに合わせる。
酢酸エチル層を水浴上で加熱して酢酸エチルを揮発除去し,少量のメタノールを加えて溶か
し,水を加えて一定体積とした後,(3)の前処理を行って定量する。
24. 溶存酸素 溶存酸素の定量には,ウインクラー法,ウインクラー-アジ化ナトリウム変法,ミラー変法
又は隔膜電極法を適用する。この試験は,試料採取後直ちに行う。
24.1 ウインクラー法 硫酸マンガン (II) とアルカリ性よう化カリウムとを加えて生成した水酸化マン
ガン (II) が溶存酸素によって酸化されて水酸化マンガン (III) となり,次に,硫酸を加えて沈殿を溶かし,
遊離したよう素をチオ硫酸ナトリウム溶液で滴定して溶存酸素を定量する方法である。
定量範囲:O 0.1mg/l以上
(1) 試薬 試薬は,次のものを用いる。
(a) アルカリ性よう化カリウム溶液 JIS K 8574に規定する水酸化カリウム700gとJIS K 8913に規定
するよう化カリウム150gとをそれぞれ水に溶かし,これらを混合し,水を加えて1lとする。着色
瓶に入れて保存する。
(b) よう素溶液 (50mmol/l) JIS K 8913に規定するよう化カリウム40gを少量の水に溶かし,これに
JIS K 8920に規定するよう素12.7gを加えて溶かし,水を加えて1lとする。
(c) よう素−アルカリ性よう化カリウム溶液 アルカリ性よう化カリウム溶液125mlを全量フラスコ
250mlにとり,よう素溶液 (50mmol/l) 10ml(1)を加えた後,アルカリ性よう化カリウム溶液を標線ま
で加える。
73
K 0101 : 1998
(d) 硫酸マンガン (II) 溶液 JIS K 8997に規定する硫酸マンガン (II) 五水和物480gを水に溶かして1l
とする。
(e) 硫酸 (3+1) 水250mlをビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸750mlを徐々に加える。室温まで冷却した後,水を加えて1lとする。
(f) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(g) 50mmol/lチオ硫酸ナトリウム溶液 JIS K 8637に規定するチオ硫酸ナトリウム五水和物12.5gと
JIS K 8625に規定する炭酸ナトリウム0.2gとを水に溶かし,水を加えて1lとする。少なくとも2
日間放置する。標定は使用時に行う。
標定 JIS K 8005に規定する容量分析用標準物質のよう素酸カリウムを130℃で約2時間加熱し,
デシケーター中で放冷する。その0.357gをとり,少量の水に溶かし,全量フラスコ200mlに移し入
れ,水を標線まで加える。この20mlを共栓三角フラスコ300mlにとり,JIS K 8913に規定するよ
う化カリウム2g及び硫酸 (1+5) 5mlを加え,直ちに栓をして静かに振り混ぜ,暗所に約5分間放
置する。
水100mlを加え,遊離したよう素をこのチオ硫酸ナトリウム溶液で滴定する。溶液の黄色が薄く
なってから,指示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消え
るまで滴定する。
別に,水について同一条件で空試験を行って補正したml数から,次の式によって50mmol/lチオ
硫酸ナトリウム溶液のファクター (f) を算出する。
783
001
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
x: 滴定に要した50mmol/lチオ硫酸ナトリウム溶液(補正
した値) (ml)
a: よう素酸カリウムの量 (g)
b: よう素酸カリウムの純度 (%)
0.001 783: 50mmol/lチオ硫酸ナトリウム溶液1mlのよう素酸カリ
ウム相当量 (g)
(h) 5mmol/lチオ硫酸ナトリウム溶液 50mmol/lチオ硫酸ナトリウム溶液25mlを全量フラスコ250ml
にとり,水を標線まで加える。使用時に調製し,調製後12時間以上経過したものは使用しない。
注(1) 通常の試験には,アルカリ性よう化カリウム溶液によう素溶液 (50mmol/l) 10mlを加えて調製す
る。必要があれば亜硫酸イオン,硫化物イオンなどの還元性物質の量を求め,これに相当する
よう素の量を算出して添加量を求める。
例えば,亜硫酸イオン (SO32-) 1mg及びヒドラジニウムイオン (N2H5+) 0.2mgに対しては,ア
ルカリ性よう化カリウム溶液によう素溶液 (50mmol/l) 16mlを加える。
(2) 器具 器具は,次のとおりとする。
(a) 試料採取器 図24.1に示すような容量500±5mlのもの2個。
74
K 0101 : 1998
図24.1 試料採取器の一例
(b) マグネチックスターラー
(3) 配管及び装置類からの試料採取 試料採取は,次のとおり行う。
(a) 2個の試料採取器の出口を上方に向け,試料採取器の入口を配管の試料採取口よりも高い位置に支
持できるように組み立て,試料採取器の下端を軟質塩化ビニル管(又は肉厚ゴム管)とY字管とで
試料採取口に接続する(試料採取器の出口には何も接続しない。)。
(b) 試料の温度が室温よりも高い場合には,試料の温度が室温より1〜2℃低くなるように試料採取配管
中に適当な冷却蛇管を設ける(2)。
(c) 2個の試料採取器とも同時に40〜60秒間で満たされるように試料の流量を調節し,試料配管中の元
の試料が完全に入れ代わるように,連続して試料を十分に流す(3)。
(d) 2個の試料採取器のA部のコックを閉じ,直ちに2個ともB部のコックを閉じ,接続管を外し,試
料採取器を逆にして気泡が全くないことを確かめる。もし,少しでも気泡があれば,2個とも試料
をとり直す。
(e) 試料採取器の1個を試験用,他を空試験用にする。
注(2) 冷却蛇管を使用する場合には,冷却水調節用弁を冷却蛇管の入口に設けて冷却水を流してあふ
れさせ,試料の流量調節弁は冷却蛇管の出口に設ける。
75
K 0101 : 1998
(3) 試料配管を断続的に使用する場合にも,試料配管や冷却蛇管中の元の試料を完全に置換するの
に必要な時間だけ試料を流す。
(4) 操作 操作は,次のとおり行う。
(a) 試験用の試料採取器のA部の管の水を切り,A部の管の最上部の基線まで,よう素-アルカリ性よう
化カリウム溶液を満たす。管内に気泡のある場合には,きれいな銅線で除く。
(b) A部のコックを開き,B部のコックで調節しながら液面が下部の基線に一致するまで,よう素-アル
カリ性よう化カリウム溶液を入れ,両方のコックを閉じ,洗浄瓶を用いて試料採取器のA部及びB
部の管内を洗浄する。
(c) 試料採取口を逆にして,(a)の操作に準じてB部の管の最上部の基線まで硫酸マンガン (II) 溶液を
満たし,(b)の操作に準じて硫酸マンガン (II) 溶液を入れる。
(d) 洗浄瓶を用いて試料採取器のA部及びB部の管内を洗浄した後,試料採取器を約1分間繰り返し転
倒させて十分に混ぜ合わせ,しばらく放置する。
(e) 再び試料採取器を転倒して試料採取器中に生じた沈殿を均一に懸濁させ,手早くB部の管の最上部
の基線まで硫酸 (3+1) を満たし,(a)の操作に準じて硫酸 (3+1) を加え,転倒して混ぜ合わせる。
(f) 試薬の添加操作は正確に行い,(a)〜(e)までの操作は,試料採取後15分間以内に行う。
(g) 試料採取器のA部の管から4〜10mlをメスシリンダー10ml中に排出し,その量を記録して捨てる。
(h) 残りの液を磁器蒸発皿に移し,ガラス棒又はマグネチックスターラーでかき混ぜながら5mmol/lチ
オ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから指示薬としてでんぷん溶液 (10g/l)
3mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(i) 空試験用の試料採取容器の試料については(a)及び(b)と同じ操作で,A部の管からよう素−アルカリ
性よう化カリウム溶液を入れ,次に,(e)に準じた操作で,B部の管から硫酸 (3+1) を加える。よ
く混ぜ合わせた後,(c)と同じ操作で,硫酸マンガン (II) 溶液を入れ,十分に混ぜ合わせる。
(j) 試料の場合と同じ磁器蒸発皿を用い,(g)及び(h)の操作で滴定する。
(k) 次の式によって試料中の溶存酸素の濃度 (mgO/l) を算出する。
4
010
.0
04
.0
000
1
−
×
×
×
−
=
f
V
b
V
a
O
b
a
ここに,
O: 溶存酸素 (mgO/l)
a: 滴定に要した5mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した5mmol/lチオ硫酸ナトリウム溶液 (ml)
Va: 滴定した試料 (ml) (試料採取器の容量から滴定前に捨
てた量を差し引いた量)
Vb: 滴定した空試験用試料 (ml) (試料採取器の容量から滴
定前に捨てた量を差し引いた量)
f: 5mmol/lチオ硫酸ナトリウム溶液のファクター(4)
0.04: 5mmol/lチオ硫酸ナトリウム溶液1mlの酸素相当量 (mg)
0.010 4: 添加試薬中の溶存酸素の補正値
注(4) 50mmol/lチオ硫酸ナトリウム溶液のファクターを用いる。
備考1. 溶存酸素が1mgO/l以上ある場合には,25mmol/lチオ硫酸ナトリウム溶液を用いてもよい。
2. 溶存酸素の (mgO/l) を (mlO/l) に換算するには0.700 4を乗じる。
24.2 ウインクラー-アジ化ナトリウム変法 ウインクラー法で妨害となる亜硝酸イオンをアジ化ナトリ
ウムを加えて分解し,溶存酸素を定量する。
定量範囲:O 0.5mg/l以上
76
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) アルカリ性よう化カリウム-アジ化ナトリウム溶液 JIS K 8574に規定する水酸化カリウム350g(又
はJIS K 8576に規定する水酸化ナトリウム250g)とJIS K 8913に規定するよう化カリウム75gを
それぞれ水に溶かし,これを混合し,水を加えて500mlとする。別に,JIS K 9501に規定するアジ
化ナトリウム5gを水20mlに溶かし,これも混合する。遮光したポリエチレン瓶に入れて暗所に保
存する。
(b) 硫酸マンガン (II) 溶液 24.1(1)(d)による。
(c) 硫酸 JIS K 8951に規定するもの。
(d) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(e) 25mmol/lチオ硫酸ナトリウム溶液 24.1(1)(g)の50mmol/lチオ硫酸ナトリウム溶液100mlを全量フ
ラスコ200mlにとり,これにJIS K 8625に規定する炭酸ナトリウム0.1gを水に溶かして加え,更
に水を標線まで加える。この溶液は使用時に調製し,12時間以上経過したものは使用しない。
(2) 器具 器具は,次のとおりとする。
(a) 溶存酸素測定瓶 19.(2)(a)による。
(3) 試料採取 試料採取は,次のとおり行う。
(a) 採水器を使用する場合 バンドーン採水器,絶縁採水器などを用いるときは採水器の取出口に軟質
塩化ビニル管を接続し,この軟質塩化ビニル管を溶存酸素測定瓶の底まで入れ,気泡が生じないよ
うに注意して試料を溶存酸素測定瓶に31ほど手早く流し込み,溶存酸素測定瓶を洗う。同じ操作で,
改めて試料を溶存酸素測定瓶に入れ,瓶の容量の25〜50%の試料をあふれさせてから,静かに軟質
塩化ビニル管を取り出し,気泡が残らないように密栓する。
(b) 配管及び装置類から採取する場合 配管及び装置類に取り付けてある試料採取弁に軟質塩化ビニル
管を付け,約1l/minで連続的に流出させる。軟質塩化ビニル管を溶存酸素測定瓶の底まで入れ,溶
存酸素測定瓶の容量の約5倍量の試料をあふれさせてから,軟質塩化ビニル管を取り出し,気泡が
残らないように密栓する。
(c) 直接採取する場合 河川及び水路,貯留槽などの表面水を溶存酸素測定瓶で直接採取するには,ま
ず,試料で溶存酸素測定瓶をよく洗い,溶存酸素測定瓶を水面下に入れ,満水するまで静かに試料
を流し込んで気泡が残らないように密栓する。バケツなどで採取した場合も同じ操作で流し入れて
密栓する。
(4) 操作 操作は,次のとおり行う。
(a) 溶存酸素測定瓶の栓を取り,これに試料100mlについて硫酸マンガン (II) 溶液1mlとアルカリ性よ
う化カリウム-アジ化ナトリウム溶液1mlとをそれぞれピペットの先端を試料中に挿入して手早く
加え,溶存酸素測定瓶中に空気が残らないように密栓する。
(b) 約1分間転倒を繰り返し,生成した沈殿が瓶の全体に広がるように十分に混ぜ合わせる。
(c) しばらく静置し,沈殿が沈降したら再び(b)の操作を行った後,静置する。
(d) 沈殿が沈降し,上澄み液が瓶全体の21程度になったら静かに開栓し,瓶の首に沿ってピペットで試
料100mlについて硫酸1mlを加え,再び密栓して数回転倒して沈殿を溶かす。
(e) この溶液の適量(全量でもよい。)を分取し,三角フラスコに入れる。
(f) 25mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから指示薬としてでんぷん溶
液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(g) 次の式によって試料中の溶存酸素の濃度 (mgO/l) を算出する。
77
K 0101 : 1998
2.0
1000
1
2
1
×
−
×
×
×
=
v
V
V
V
f
a
O
ここに,
O: 溶存酸素 (mgO/l)
a: 滴定に要した25mmol/lチオ硫酸ナトリウム溶液 (ml)
V1: 共栓を施したときの溶存酸素測定瓶の容量 (ml)
V2: 滴定のため溶存酸素測定瓶から分取した試料 (ml)
v: アルカリ性よう化カリウム-アジ化ナトリウム溶液と硫酸マ
ンガン (II) 溶液の合計量 (ml)
f: 25mmol/lチオ硫酸ナトリウム溶液のファクター(5)
0.2: 25mmol/lチオ硫酸ナトリウム溶液1mlの酸素相当量 (mg)
注(5) 24.1(1)(g)の50mmol/lチオ硫酸ナトリウム溶液のファクターを用いる。
備考3. 酸化性物質を含む試料の場合 水に残留塩素などが含まれる場合には,空試験として別の溶
存酸素測定瓶を用い,(3)の試料採取の操作に従って試料を採取し,アルカリ性よう化カリウ
ム-アジ化ナトリウム溶液と硫酸とを加えて密栓し,転倒を繰り返して混合し,次に,硫酸マ
ンガン (II) 溶液を加えて密栓し,転倒を繰り返して混合する。これについて(4)(e)〜(g)の操
作を行って滴定し,溶存酸素の量を補正する。
4. 還元性物質を含む試料の場合 アルカリ性よう化カリウム-アジ化ナトリウム溶液の代わり
に24.1(1)(c)のよう素−アルカリ性よう化カリウム溶液を用いるとよい。この場合も備考3.
と同じように,別の溶存酸素測定瓶を用いて空試験を行い,溶存酸素の量を補正する。
5. 試料が海水の場合 海水は微生物を含む場合が多いから,反応を速めて手早く試験する。反
応促進のためにアルカリ性よう化カリウム-アジ化ナトリウム溶液と硫酸マンガン (II) 溶液
をそれぞれ2倍量添加する。転倒後の硫酸は2倍量を加える。
6. 試料中に鉄 (III) が共存する場合 硫酸の添加前に,試料100mlについてふっ化カリウム溶
液 (300g/l) 1mlを加えれば,鉄 (III) 100〜200mg/lが含まれていても妨害しない。
24.3 ミラー変法 流動パラフィンで試料と空気を遮断し,酒石酸ナトリウムカリウム-水酸化ナトリウム
溶液と3, 7-ビス(ジメチルアミノ)フェノチアジン-5-イウムクロリド(メチレンブルー)溶液を加え,硫
酸アンモニウム鉄 (II) 溶液で滴定し,溶存酸素を定量する。
定量範囲:O 1mg/l以上
(1) 試薬 試薬は,次のものを用いる。
(a) 酒石酸ナトリウムカリウム-水酸化ナトリウム溶液 JIS K 8536に規定する (+) -酒石酸ナトリウム
カリウム四水和物350g及びJIS K 8576に規定する水酸化ナトリウム100gを水に溶かして,1lとす
る。
(b) メチレンブルー溶液 JIS K 8897に規定するメチレンブルー(通常は三水和物)0.1gを水100mlに
溶かす。
(c) 流動パラフィン JIS K 9003に規定するもの。
(d) 硫酸アンモニウム鉄 (II) 溶液 JIS K 8951に規定する硫酸5mlを水100mlに加え,これにJIS K
8979に規定する硫酸アンモニウム鉄 (II) 六水和物5.4gを加えて溶かし,2.(12)(a)の溶存酸素を含ま
ない水を加えて1lとする。
標定 この溶液の溶存酸素相当量は24.2で溶存酸素の濃度を求めた水を標準とし,(3)の操作に従っ
てこの溶液で滴定し,次の式によって算出する。
b
a
f
1
1000
50×
×
=
78
K 0101 : 1998
ここに,
f: 硫酸アンモニウム鉄 (II) 溶液1mlの溶存酸素相当量 (mgO)
a: 使用した水の溶存酸素 (mgO/l)
b: 滴定に要した硫酸アンモニウム鉄 (II) 溶液 (ml)
この標定は,使用時に行う。
(2) 器具 器具は,次のとおりとする。
(a) 試料採取器 注射筒50mlの先端に内径約1.5mm,長さ250〜300mmのガラス管をすり合わせ又は
ゴム管で接続したもの。
(b) 溶存酸素測定用試験管 外径約30mm,高さ約200mmの試験管
(c) かき混ぜ棒 直径約3mm,全長約250mmのガラス棒で,下部を約10mm折り曲げたもの又は下部
をらせん状にしたもの。
(d) 足長ビュレット 5〜10ml,足の先端が試験管の下部に達するもの。
(3) 操作 操作は次のとおり行う。
(a) 溶存酸素測定用試験管にメチレンブルー溶液2滴,酒石酸ナトリウムカリウム-水酸化ナトリウム溶
液5mlと流動パラフィン約5mlを加える。
(b) 試料採取器に試料を吸引して2回洗った後,気泡が入らないように徐々に吸引して試料50mlを採
取する。このときガラス管部分に試料が満たされた状態に保つ。
(c) 試料採取器のガラス管の先端を静かに流動パラフィン層の下の水層に入れ,流動パラフィン層が乱
れないように注意しながら,試料50mlを注入する。
(d) かき混ぜ棒を静かに入れ,足長ビュレットの先端を流動パラフィン層の下の水層に入れる。
(e) 硫酸アンモニウム鉄 (II) 溶液で,メチレンブルーの青い色が消えるまで滴定する。
(f) 次の式によって試料中の溶存酸素の濃度 (mgO/l) を算出する。
50
1000
×
×
=
f
a
O
ここに, O: 溶存酸素 (mgO/l)
a: 滴定に要した硫酸アンモニウム鉄 (II) 溶液 (ml)
f: 硫酸アンモニウム鉄 (II) 溶液1mlの溶存酸素相当量 (mg)
24.4 隔膜電極法 隔膜電極を用いて,試料中の溶存酸素の量を測定する。
定量範囲:O 0.5mg/l以上,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 亜硫酸ナトリウム溶液 JIS K 8061に規定する亜硫酸ナトリウム約25gを水に溶かし,水を加えて
500mlとする。使用時に調製する。この溶液は,ゼロ調節に用いる(6)。
(b) 溶存酸素飽和水(8) 水酸化カリウム溶液 (250g/l) で洗浄した空気を約1l/minの流量で球形又は板
状のガラスろ過器を用いて水に通気(7)して,溶存酸素を飽和させる(8)。スパン調節操作を行う直前
に調製する。
注(6) JIS K 8129に規定する塩化コバルト (II) 六水和物を微量添加すると,溶存酸素は容易に亜硫酸
ナトリウムによって還元される。
(7) 通常,水200mlの場合には5〜10分間,500mlの場合には10〜20分間通気する。
(8) 溶存酸素飽和水は,試料の温度と±0.5℃で一致する温度のものを調製する。この溶液の溶存酸
素の濃度は表24.1から求める。溶存酸素の濃度は,気圧変動によっても異なるので気圧補正を
行うとよい。
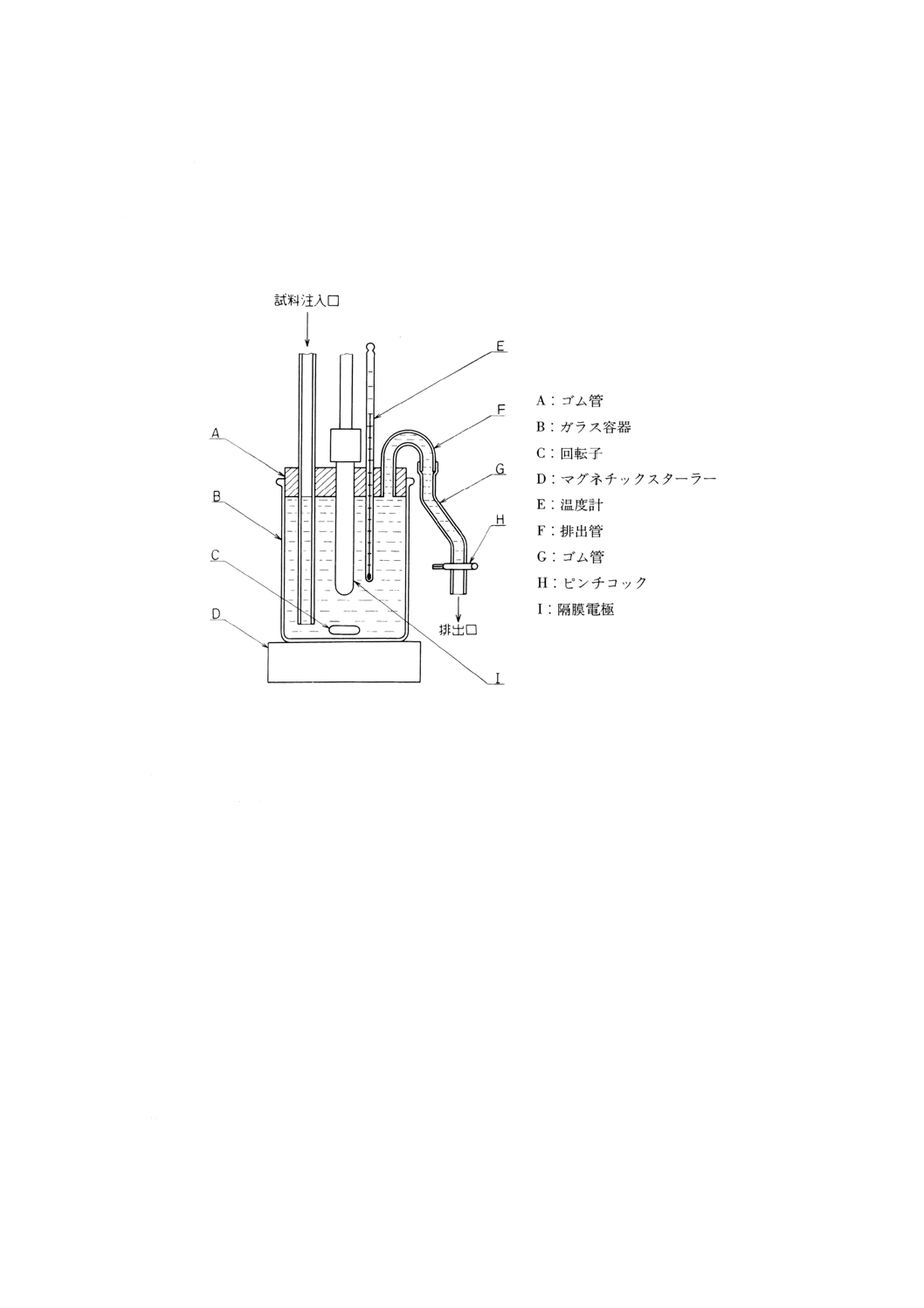
79
K 0101 : 1998
また,塩類の濃度の高い試料の溶存酸素の濃度を測定する場合には,試料の塩類のモル濃度
に合わせたJIS K 8150に規定する塩化ナトリウムを添加した溶存酸素飽和水を調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 溶存酸素測定容器 ガラス容器100〜300mlにゴム栓を付け,栓に試料を採取するための注入管及
び排出するための排出管を取り付けたもの(9)。図24.2に一例を示す。
図24.2 測定容器の一例
(b) 温度計 JIS B 7411に規定する一般用ガラス製棒状温度計の50度温度計
(c) 溶存酸素計 一般に,温度補償回路を組み入れたもの
(d) マグネチックスターラー
注(9) 溶存酸素測定瓶,培養瓶などを用いてもよい。
(3) 準備操作 準備操作は,次のとおり行う。
(a) 溶存酸素計の電極を接続し,約30分間通電しておく。
(b) 測定容器に試料と同じ温度にした亜硫酸ナトリウム溶液を注入し,マグネチックスターラーで静か
にかき混ぜながら電極を挿入し,指示値が安定してから(10),ゼロ調節ダイヤルを回して指示値をゼ
ロに合わせる。
(c) 電極及び温度計を取り出し,水でよく洗い(11),別の測定容器に挿入する。
(d) 注入管の一端から測定容器の底に静かに溶存酸素飽和水(12)を注入し,測定容器の容量の25〜50%を
流出させた後,排出管の先端を閉じる。
(e) マグネチックスターラーでかき混ぜ(13)ながら,溶存酸素計の指示値が安定するのを待つ。温度を読
み取り,対応する溶存酸素飽和量を表24.1から求めスパン調節ダイヤルを回し,指示値を合わせる。
(f) (b)〜(e)の操作を2,3回繰り返して,指示値がそれぞれゼロ及び溶存酸素の飽和量に合致している
ことを確かめる。
注(10) 通常2〜5分間を要する。
(11) 準備操作(b)から(c)に移るときには,電極を特によく洗浄する。
80
K 0101 : 1998
(12) 容器内で通気して溶存酸素飽和水を調製してもよい。
(13) かき混ぜ速度によって指示値に差が生じるので,できるだけスパン調節操作時と同じ条件に保
つ。
(4) 操作 操作は,次のとおり行う。
(a) (3)(d)に準じて,試料を測定容器の底に,泡が入らないように静かに注入する(14)。
(b) マグネチックスターラーでかき混ぜながら(13)温度計の目盛を確認し,次に,溶存酸素計の指示値が
安定するのを待って,指示値 (mgO/l) を読み取る。
注(14) 溶存酸素測定瓶又は培養瓶などを測定容器として用いる場合には,サイホンを用いて静かに試
料を容器に取り,直ちに電極と温度計を挿入して測定する。
備考7. 指示値は,温度1℃の上昇につき約5%増大する。
表24.1 水中の飽和溶存酸素
温度
℃
水中の塩化物イオンmgCl-/l
塩化物イオン
100mgCl-/lごと
に差し引く溶
存酸素 mgO/l
0
5 000
10 000
15 000
20 000
溶存酸素 mgO/l
0
14.16
13.40
12.63
11.87
11.10
0.015 3
1
13.77
13.03
12.29
11.55
10.80
0.014 8
2
13.40
12.68
11.97
11.25
10.52
0.014 4
3
13.04
12.35
11.65
10.95
10.25
0.014 0
4
12.70
12.03
11.35
10.67
9.99
0.013 5
5
12.37
11.72
11.06
10.40
9.74
0.013 1
6
12.06
11.42
10.79
10.15
9.51
0.012 8
7
11.75
11.15
10.52
9.90
9.28
0.012 4
8
11.47
10.87
10.27
9.67
9.06
0.012 0
9
11.19
10.61
10.03
9.44
8.85
0.011 7
10
10.92
10.36
9.79
9.23
8.66
0.011 3
11
10.67
10.12
9.57
9.02
8.47
0.011 0
12
10.43
9.90
9.36
8.82
8.29
0.010 7
13
10.20
9.68
9.16
8.64
8.11
0.010 4
14
9.97
9.47
8.97
8.46
7.95
0.010 1
15
9.76
9.27
8.78
8.29
7.79
0.009 9
16
9.56
9.06
8.60
8.12
7.63
0.009 6
17
9.37
8.90
8.44
7.97
7.49
0.009 4
18
9.18
8.73
8.27
7.82
7.36
0.009 1
19
9.01
8.57
8.12
7.67
7.22
0.008 9
20
8.84
8.41
7.97
7.54
7.10
0.008 7
21
8.68
8.26
7.83
7.40
6.97
0.008 6
22
8.53
8.11
7.70
7.26
6.85
0.008 4
23
8.39
7.98
7.57
7.16
6.74
0.008 2
24
8.25
7.85
7.44
7.04
6.65
0.008 1
25
8.11
7.72
7.32
6.95
6.52
0.007 9
26
7.99
7.60
7.21
6.82
6.42
0.007 8
27
7.87
7.48
7.10
6.71
6.32
0.007 7
28
7.75
7.37
6.99
6.61
6.22
0.007 6
29
7.64
7.26
6.88
6.51
6.12
0.007 6
30
7.53
7.16
6.78
6.41
6.03
0.007 5
31
7.43
7.06
6.66
6.31
5.93
0.007 5
32
7.32
6.96
6.59
6.21
5.84
0.007 4

81
K 0101 : 1998
温度
℃
水中の塩化物イオンmgCl-/l
塩化物イオン
100mgCl-/lごと
に差し引く溶
存酸素 mgO/l
0
5 000
10 000
15 000
20 000
溶存酸素 mgO/l
33
7.23
6.86
6.49
6.12
5.75
0.007 4
34
7.13
6.77
6.40
6.03
5.65
0.007 4
35
7.04
6.67
6.30
5.93
5.56
0.007 4
25. 全炭酸 全炭酸は,炭酸,炭酸水素イオン及び炭酸イオンの合量で二酸化炭素 (CO2) の量として表す。
全炭酸の定量には,塩化ストロンチウム-塩酸滴定法又は赤外線分析法を適用する。
25.1 塩化ストロンチウム-塩酸滴定法 水酸化ナトリウム溶液に試料を加えて,全炭酸を炭酸イオンに変
える。次に,塩化ストロンチウムを加えて炭酸ストロンチウムの沈殿を生成させる。塩酸を加えて過剰の
水酸化ナトリウムを中和し,更に塩酸の一定量を加えて沈殿を溶かす。通気して遊離した二酸化炭素を除
去した後,過剰の塩酸を水酸化ナトリウム溶液で滴定して消費された塩酸の量を求め,全炭酸を定量する。
定量範囲:CO2 1〜40mg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 20.1(1)(a)による。
(b) 塩化ストロンチウム溶液 JIS K 8132に規定する塩化ストロンチウム六水和物17gを水に溶かして
100mlとする。
(c) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(d) 0.1mol/l塩酸 JIS K 8180に規定する塩酸10mlを,あらかじめ水100mlを入れたビーカー1 000ml
にとり,水を加えて1lとする。
標定 JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムを600℃で約1時間加熱し,デ
シケーター中で放冷する。その約1gを1mgのけたまではかりとり,少量の水に溶かして,全量フ
ラスコ200mlに移し入れ,水を標線まで加える。これから20mlをとり,三角フラスコ200mlに入
れ,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液(1)2,3滴を加え,この0.1mol/l
塩酸で溶液の色が灰紫 (pH4.8) になるまで滴定する。
終点付近で煮沸して二酸化炭素を追い出し,放冷後再び灰紫になるまで滴定する。滴定に要した
0.1mol/l塩酸のml数から,次の式によって0.1mol/l塩酸のファクター (f1) を算出する。
30
005
.0
1
200
20
100
1
×
×
×
×
=
x
b
a
f
ここに,
a: 炭酸ナトリウムの量 (g)
b: 炭酸ナトリウムの純度 (%)
x: 滴定に要した0.1mol/l塩酸 (ml)
0.005 30: 0.1mol/l塩酸1mlの炭酸ナトリウム相当量 (g)
注(1) 13.1(1)(a)による。
(e) 40mmol/l塩酸 0.1mol/l塩酸100mlを全量フラスコ250mlにとり,水を標線まで加える。この溶液
のファクターは,0.1mol/l塩酸のファクターを用いる。
(f) 40mmol/l水酸化ナトリウム溶液 水約30mlをポリエチレン瓶にとり,冷却しながら水酸化ナトリ
ウム約35gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。この上澄み液2mlをポリエチ
レン製の気密容器1lにとり,2.(12)(b)の炭酸を含まない水を加えて1lとする。
この溶液は自動ビュレット(容量50ml)付試薬瓶(ポリエチレン製)に入れ,空気の出入口には,
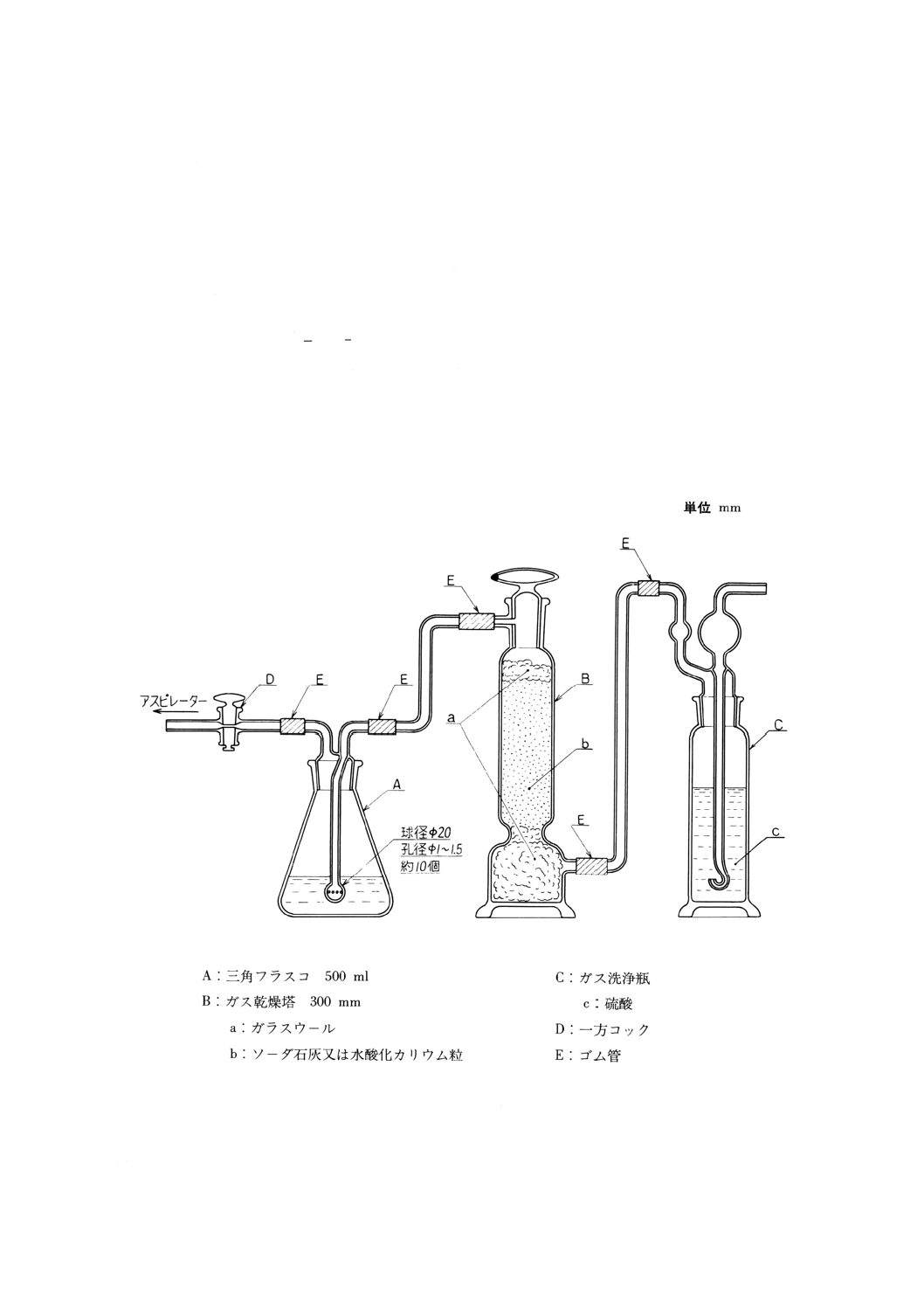
82
K 0101 : 1998
JIS K 8603に規定するソーダ石灰又はJIS K 8574に規定する水酸化カリウム粒を詰めた管を取り付
けて保存する。
標定 (e)の40mmol/l塩酸20mlを三角フラスコ200mlにとり,指示薬としてフェノールフタレイン
溶液 (5g/l) 2,3滴を加え溶液の色が微赤になるまで,この40mmol/l水酸化ナトリウム溶液で滴定
する。
滴定に要した40mmol/l水酸化ナトリウム溶液のml数から,次の式によって40mmol/l水酸化ナト
リウム溶液のファクター (f2) を算出する。
x
f
f
1
2
20×
=
ここに, f1: 40mmol/l塩酸のファクター
x: 滴定に要した40mmol/l水酸化ナトリウム溶液 (ml)
(2) 装置 装置は,次のとおりとする。
(a) 全炭酸測定装置 図25.1に一例を示す。
図25.1 全炭酸測定装置の一例
(3) 操作 操作は,次のとおり行う。
(a) 三角フラスコ500mlに40mmol/l水酸化ナトリウム溶液50mlと指示薬としてフェノールフタレイン
溶液 (5g/l) 0.2mlを加える。
(b) 試料200mlを加え(2)(試料中の全炭酸が多いときは40mg以下になるように適量をとり,水を加え
83
K 0101 : 1998
て200mlにする。),三角フラスコ500mlを図25.1のように接続して数分間放置する。
(c) 塩化ストロンチウム溶液10mlを加え,再び接続して十分に振り混ぜて約10分間放置する。
(d) 40mmol/l塩酸を滴加して約1分間無色を保つまで中和する(3)。
(e) 40mmol/l塩酸50mlを加え,再び接続する。
(f) 空気を1l/minで約5分間通気する。
(g) 三角フラスコ500mlを取り外し,空気導入管を水洗する。次に,40mmol/l水酸化ナトリウム溶液で
徐々に滴定し,溶液の色がわずかに赤になった点を終点とする。
(h) 空試験として試料に代え,水200mlを三角フラスコ500mlにとり,(a)〜(g)の操作を行う。
(i) 次の式によって試料中の全炭酸の濃度 (mgCO2/l) を算出する。
2
880
.0
1000
)
(
2
×
×
×
−
=
V
f
a
b
C
ここに,
C: 全炭酸 (mgCO2/l)
a: 滴定に要した40mmol/l水酸化ナトリウム溶液 (ml)
b: 空試験に要した40mmol/l水酸化ナトリウム溶液 (ml)
f2: 40mmol/l水酸化ナトリウム溶液のファクター
V: 試料 (ml)
0.880 2: 40mmol/l水酸化ナトリウム溶液1mlの二酸化炭素相当量
(mg)
注(2) 三角フラスコ中の溶液のpHは12以下にならないようにする。pHはアルカリブルーpH試験紙を
用いて確認する。試料が酸性の場合は,pH約7になるまで40mmol/l水酸化ナトリウム溶液を加
える。
(3) マグネシウムイオンが多量に共存すると,中和のとき変色が見にくいことがあるので終点が近
づいたら徐々に滴定する。
備考1. この方法は,次の成分ごとに以下に示す限度を超えない場合に適用できる。
Mg2+
200mgMg/l
Fe2+
25mgFe/l
PO43-
5mgPO43-/l
Fe2+
5mgFe/l
共存
PO43-
10mgPO43-/l
Fe3+
2.5mgFe/l
共存
PO43-
5mgPO43-/l
その他,アルミニウム,アンモニウムイオン,シリカなどが多量に共存すると妨害となる。
2. 炭酸,炭酸水素イオン及び炭酸イオンの濃度の算出。
試料中の全炭酸の濃度と試料のpHから,炭酸,炭酸水素イオン及び炭酸イオンのそれぞ
れの濃度は,次の式及び表25.1又は図25.2から算出することができる。
H2CO3=C×a×1.409
HCO3-=C×b×1.387
CO32-=C×c×1.364
ここに, H2CO3: 炭酸 (mgH2CO3/l)
HCO3-: 炭酸水素イオン (mgHCO3-/l)
84
K 0101 : 1998
CO32-: 炭酸イオン (mgCO32-/l)
C: 全炭酸 (mgCO2/l)
a: 全炭酸に対する炭酸のモル比
b: 全炭酸に対する炭酸水素イオンのモル比
c: 全炭酸に対する炭酸イオンのモル比
1.409: 全炭酸 (CO2) の量を炭酸相当量に換算する場合の係数
(62.03/44.01)
1.387: 全炭酸 (CO2) の量を炭酸水素イオン相当量に換算する
場合の係数 (61.02/44.01)
1.364: 全炭酸 (CO2) の量を炭酸イオン相当量に換算する場合
の係数 (60.01/44.01)
表25.1 pHに対する全炭酸の濃度分布
pH
濃度分布 (25℃)
pH
濃度分布 (25℃)
a (H2CO3)
b (HCO3-)
c (CO32-)
a (H2CO3)
b (HCO3-)
c (CO32-)
2.0
1.000 0
−
−
8.0
0.022 6
0.972 8
0.004 6
2.5
0.999 9
0.000 1
−
8.5
0.007 2
0.978 3
0.014 5
3.0
0.999 6
0.000 4
−
9.0
0.002 2
0.953 0
0.044 8
3.5
0.998 6
0.001 4
−
9.5
0.000 6
0.870 1
0.129 3
4.0
0.995 7
0.004 3
−
10.0
0.000 2
0.680 1
0.319 7
4.5
0.986 6
0.013 4
−
10.5
0.000 0
0.402 2
0.507 8
5.0
0.958 7
0.041 3
−
11.0
−
0.175 4
0.824 6
5.5
0.880 0
0.120 0
−
11.5
−
0.063 0
0.937 0
6.0
0.698 8
0.301 2
0.000 0
12.0
−
0.020 8
0.979 2
6.5
0.423 2
0.576 7
0.000 1
12.5
−
0.006 7
0.993 3
7.0
0.188 3
0.811 3
0.000 4
13.0
−
0.002 1
0.997 9
7.5
0.068 3
0.930 3
0.001 4
図25.2 pHに対する全炭酸の濃度分布 (25℃)
表25.1及び図25.2に試料のpH (25℃) における全炭酸に対する炭酸,炭酸水素イオン及び
炭酸イオンのそれぞれのモル比を示す。
25.2 赤外線分析法 有機体炭素 (TOC) を赤外線分析法によって定量するときに測定される無機体炭素
の場合と同様に操作して,全炭酸を定量する。
定量範囲:CO2 3〜450mgCO2/l,繰返し分析精度:変動係数で3〜10%
85
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 水 20.1(1)(a)による。
(b) 炭酸標準液 (0.5mgCO2/ml) JIS K 8622に規定する炭酸水素ナトリウムをデシケーター中で約3
時間放置し,その0.478gをとる。別に,JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウ
ムを,あらかじめ600℃で約1時間加熱し,デシケーター中で放冷し,その0.602gをとる。両者を
少量の水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。
(c) 炭酸標準液 (0.1mgCO2/ml) 炭酸標準液 (0.5mgCO2/ml) 20mlを全量フラスコ100mlにとり,水を
標線まで加える。
(d) 無機体炭素測定管 20.1(1)(g)による。
(e) キャリヤーガス 20.1(1)(h)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロシリンジ 10μl及び150μl
(b) 全炭酸定量装置 20.1(2)(b)による。
(3) 準備操作 準備操作は,次のとおり行う。
(a) 20.1(3)(a)〜(c)に準じ,炭酸標準液 (0.1mgCO2/ml) [又は炭酸標準液 (0.5mgCO2/ml)]20μlをマイク
ロシリンジで全炭酸定量装置に注入して指示計[赤外線ガス分析計(記録計)]が示すピーク高さを
読み取る。
(b) 以下,20.1(3)(d)の操作を行う。
(4) 検量線の作成 検量線の作成は,次のとおり行う。
(a) 20.1(4)(e)〜(g)に準じ,炭酸標準液 (0.1mgCO2/ml) [又は炭酸標準液 (0.5mgCO2/ml)]を用いて検量
線を作成する。
(5) 操作 操作は,次のとおり行う。
(a) 20.1(5)に準じて操作し,検量線の作成に用いた炭酸標準液と同量の試料をマイクロシリンジで全炭
酸定量装置に注入(2,3回繰り返す。)し,対応するピーク高さを読み取る。
(b) あらかじめ作成した検量線から,試料中の全炭酸の濃度 (mgCO2/l) を求める。
備考3. 検量線の作成に標準液として20.1(1)(e)を用いた場合には,次の式によって全炭酸の濃度
(mgCO2/l) を算出する。
全炭酸 (CO2) (mgCO2/l) =無機体炭素 (mgC/l) ×3.664
ここに, 3.664: 炭素の量を全炭酸 (CO2) 相当量に換算する場合の係数
4. 試料中の炭酸,炭酸水素イオン及び炭酸イオンの濃度を算出する場合には,備考2.による。
26. ヘキサン抽出物質 ヘキサン(n-ヘキサン)抽出物質とは試料を微酸性とし,ヘキサン抽出を行った
後,約80℃でヘキサンを揮散させたときに残留する物質をいう。
この試験は,主として揮散しにくい鉱物油及び動植物油脂類の定量を目的とするが,これらのほかヘキ
サンに抽出された揮散しにくいものは,定量値に含まれる。
この試験は,抽出法を適用する。
26.1 試料採取 試料の採取は,次のとおり行う。
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) メチルオレンジ溶液 (1g/l) 22.1.1(1)(d)による。
86
K 0101 : 1998
(2) 器具 器具は,次のとおりとする。
(a) 試料容器 表層の水及び落下する水の場合は,共栓広口ガラス瓶1〜2l。下層の水の場合には,採
水器に装着できる共栓ガラス瓶1〜2l。いずれも使用前にヘキサンでよく洗っておく。
(b) 採水器 ハイロート採水器又はこれに類する適当な採水器。
(3) 採取方法 採取方法は,次のとおりとする。
(a) 落下している水の採取 水路,せき,溝,管などから落下している場合には,試料を直接試料容器
に受け,適当な空間が残る程度に採取をとどめる(1)。
(b) 通水状態の配管装置などからの採取 配管,装置などが通水状態の場合には,試料採取弁を開き,
試料採取配管内に滞留している水の約5倍量を約1l/minで流出させてから試料容器に受け,適当な
空間が残る程度に採取をとどめる(1)(2)(3)(4)。
(c) 深い水路や水槽などからの採取 深い水路や水槽の水を採取する場合には,全層試料を採取できる
採水器を使用し,全層の試料を採取する。ハイロート採取器では,採水器の枠に試料容器を取り付
けて,底部近くに降ろし,採水しながら一定速度で採水器を引き上げ,水面に達したとき適当な空
間が残るように採取する(5)。
(d) 貯留池,湖沼,河川などからの採取 試料容器を取り付けた採水器を用い,任意の深さの試料を採
取するか,又は試験目的よって(c)に準じて採取する。
注(1) この場合は,試料容器を試料で洗わない。
(2) 高温高圧又は負圧状態にある配管装置などから試料採取する場合には,次のようにする。
高温水の場合は,冷却器を試料採取管に設けて室温以下に冷却する。高圧水(圧力が1.96MPa
以上)の場合には,減圧器を設けて減圧した後に採取し,高温であれば冷却器を通して室温以
下に冷却する。負圧水の場合には,昇圧器で大気圧にしてから採取する(負圧水で高温の場合
は,昇圧器の前に冷却器を設けて室温にしてから大気圧にする。)(JIS K 0094の4.3を参照す
る。)。
(3) 装置などが停止状態にあるときは,油状物質が配管や装置中で水と分離していることが多いた
め,通水速度や通水時間によって油状物質の濃度に変動が生じる。試料採取弁や配管中に油状
物質が付着しているおそれがある場合には,試料採取弁を全開して約10分間通水してから,約
1l/minで,更に10分間通水する。この操作を繰り返して洗浄する。
(4) 試料採取直前に流量を変更してはならない。
(5) JIS K 2251(原油及び石油製品-試料採取方法)に規定する全層試料に準じて採取する。
(4) 試料の取扱い 試料の取扱いは,次のとおりとする。
(a) (3)によって採取した試料は,他の容器に移し替え一部を採取してはならない。試験には全量を用い
る。
(b) 試料の量は,試料を入れた容器の質量から試料容器の質量を差し引いて求めるか,又は試料を採取
したときに試料容器の水面の位置に印を付けておき,試験終了時に印のところまで水を入れてその
水の体積を試料の量とする。
(c) 試料を保存したり運搬する必要がある場合には,指示薬としてメチルオレンジ溶液 (1g/l) 数滴を加
え,溶液の色が赤くなるまで塩酸 (1+1) を加えて密栓する(6)。
注(6) 油状物質が浮上している場合には,油状物質が運搬中の振動でにじみやすいので,取扱いに注
意する。
87
K 0101 : 1998
26.2 抽出法 試料をpH 4以下の塩酸酸性にして,ヘキサンで抽出を行い,80℃でヘキサンを揮散させて
残留する物質の質量をはかってヘキサン抽出物質を定量する。
定量範囲:5〜500mg,繰返し分析精度:変動係数で10〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(c) 硫酸ナトリウム JIS K 8987に規定するもの。
(d) メチルオレンジ溶液 (1g/l) 22.1.1(1)(d)による。
(e) ヘキサン JIS K 8848に規定するもの。
(f) 窒素 JIS K 1107に規定する高純度窒素2級
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 200ml及び1 000〜3 000mlで脚部の短いもの。使用前にヘキサンで洗う。コックに滑剤
を塗布しない。
(b) 乾燥器 80±5℃に温度調節できるもの。
(c) 加熱板又はマントルヒーター 80±5℃に温度調節できるもの。温度調節ができる水浴を用いてもよ
い。
(d) 蒸留装置 共通すり合わせで,蒸留フラスコ(容量50〜100ml),トの字形連結管とリービッヒ冷却
器(長さ300mm)を接続できるものを用いる。いずれも使用前にヘキサンで洗っておく。
(e) 蒸発容器 アルミニウムはく皿,白金皿,ビーカー。容量50〜100mlで,できるだけ質量の小さい
もの。いずれも使用前にヘキサンでよく洗い,80±5℃で約30分間加熱してデシケーター中で放冷
した後,0.1mgのけたまで質量を求めておく。
(3) 操作 操作は,次のとおり行う。
(a) 26.1で採取した試料(7)を分液漏斗1 000〜3 000ml(8)に移し,指示薬としてメチルオレンジ溶液 (1g/l)
2, 3滴を加え,溶液の色が赤に変わるまで塩酸 (1+1) を滴加する。
(b) 試料容器を約20mlずつのヘキサンで2回洗い,洗液を分液漏斗1 000〜30 00mlに加える。約2分
間激しく振り混ぜ,放置する(9)。
(c) 水層は試料容器に戻し,更に分液漏斗1 000〜3 000mlを静かに揺り動かして,残った水層をできる
だけ分離して(10)試料容器に戻す。ヘキサン層は分液漏斗200mlに移す。
(d) 試料容器の水層を(a)で使用した分液漏斗1 000〜3 000mlに入れ,再び(b)及び(c)の操作を行ってヘ
キサン層と水層を分離し,ヘキサン層を分液漏斗200mlに合わせる。
(e) 分液漏斗1 000〜3 000mlを少量のヘキサンで洗い,洗液を分液漏斗200mlに合わせる。
(f) 分液漏斗200mlを静かに揺り動かして静置し,ヘキサンを損失しないように注意しながら混入した
水分を十分に分離除去する(10)。
(g) ヘキサン層に水約20mlを加え,約1分間振り混ぜて放置し,水層を捨てる。この洗浄操作を洗液
がメチルオレンジに対して黄色になるまで数回繰り返す。できるだけ水層を除去する。
(h) ヘキサン層に硫酸ナトリウム3〜5gを加えて振り混ぜ,水分を除く(11)。
(i) 分液漏斗200mlの脚部を乾いたろ紙(12)でふきとり,脱脂綿又はろ紙(12)を用いてヘキサン層をろ過
し,蒸留装置の蒸留フラスコに移し入れる(13)。
(j) 分液漏斗200mlを少量のヘキサンで洗い,この洗液も(i)と同じ操作でろ過し,蒸留装置の蒸留フラ
スコに移し入れる。使用した脱脂綿又はろ紙は,ヘキサン約5mlずつで2回洗い,この洗液も蒸留
88
K 0101 : 1998
フラスコに移し入れる。
(k) 蒸留フラスコをマントルヒーターに入れ,トの字形連結管及びリービッヒ冷却器を接続して,マン
トルヒーターの温度を約80℃に調節し,ヘキサンを毎秒約1滴の留出速度で蒸留し,留出するヘキ
サンを受器に受ける(14)。蒸留は,蒸留フラスコ内の液量が約2mlになるまで続ける。トの字形連結
管の上部口からJIS K 1107に規定する高純度窒素2級を室温になるまで送入する。
(l) 蒸留フラスコの残留液を質量既知の蒸発容器に移し入れる。蒸留フラスコを少量のヘキサンで3回
洗い,この洗液も蒸発容器に加える。蒸発容器を約80℃に保った加熱板の上又はマントルヒーター
の中に置いてヘキサンを揮散させる(15)。
(m) 蒸発容器の外側を湿った清浄な布などでぬぐい,次に,乾いた清浄な布などでぬぐって,80±5℃に
調節した乾燥器中に移し,約30分間加熱する。蒸発容器をデシケーター中で約30分間放冷した後,
その質量を0.1mgのけたまではかり,蒸発容器の質量を差し引き,ヘキサン抽出物質の質量 (mg) を
求める。
(n) 空試験として,この試験に使用した全ヘキサンと同量のヘキサンを蒸留フラスコにとり(13),(k)〜
(m)の操作を行って残留物質の質量 (mg) を求める。
(o) 次の式によって試料中のヘキサン抽出物質の濃度 (mg/l) を算出する。
V
b
a
P
000
1
)
(
×
−
=
ここに, P: ヘキサン抽出物質 (mg/l)
a: 試験におけるヘキサン抽出物質の質量 (mg)
b: 空試験における残留物質の質量 (mg)
V: 試料 (ml)
注(7) 試料は通常約1lでよいが,ヘキサン抽出物質が5〜500mgになるように試料を採取し,全量を用
いる。
(8) 試料の量に応じて適当な大きさの分液漏斗を選ぶ。
(9) 試料の性質によっては,エマルションが生成したり,ヘキサン層が濁ることがある。このよう
な試料では,分液漏斗中の水層をできるだけ元の試料容器に戻し,JIS K 8150に規定する塩化
ナトリウム又はJIS K 8960に規定する硫酸アンモニウム約10g(ヘキサンに溶ける物質を含ま
ないもの。)を加え,分液漏斗の口に約300mmの共通すり合わせリービッヒ冷却器又はジムロ
ート冷却器を取り付け,約80℃に保った恒温水浴中に分液漏斗を浸し,約10分間ヘキサンを
還流させるとエマルションがなくなることがある。この加熱還流のほか,分液漏斗中のヘキサ
ン層とエマルション層にJIS K 8150に規定する塩化ナトリウム又はJIS K 8960に規定する硫酸
アンモニウム約10gを加えて振り混ぜた後,少量の水で遠心分離管に移し,8 000min-1以上で約
5分間遠心分離すると,エマルション層はわずかになり,ヘキサン層の分離を容易にすること
ができる。これを分液漏斗に戻し,(c)以下の操作に移る。
(10) 分離する水層が1ml以下になるまで続ける。試料が多量のグリース類又は固体油脂を含む場合
には,水層を分離する前にヘキサンを追加する。
(11) ヘキサン層が濁っていることがある。このような場合には,水層をできるだけ分離した後,硫
酸ナトリウムを加えて脱水すると透明になることがある。JIS K 8150に規定する塩化ナトリウ
ム又はJIS K 8960に規定する硫酸アンモニウムを使用する方が効果的な場合もある。ただし,
ヘキサンに溶ける物質を含む試薬は使用しない。
89
K 0101 : 1998
(12) ヘキサンで十分に洗って抽出物質を除いたもので,ろ過のときにはあらかじめ少量のヘキサン
で潤しておく。
(13) 蒸留フラスコに一度に入りきれないときは,2,3回に分割してヘキサンを留出させる。
(14) 蒸留によって留出したヘキサンは,再蒸留すれば,再使用できる。
(15) 引火しないように十分注意をする。溶媒は揮散廃棄せずに,できるだけ回収する。ヘキサンを
揮散後,蒸発容器中に水分が認められる場合には,アセトンを加えて蒸発を繰り返すと水分は
除去できる。水分中に塩類が残留すると誤差になるので注意する。もし,塩類が残留する場合
には(m)の操作を行い,ヘキサン抽出物質の質量 (mg) を求めた後,ヘキサン抽出物質を少量の
ヘキサンを加えて溶かし分離する。この操作を繰り返し行い,ヘキサン抽出物質を除去した後,
(m)の操作を行って残留物質の質量 (mg) を求めて補正する。
備考1. ヘキサン抽出物質の質量が5mg以下で定量が困難な場合には,JIS K 0102の24.3(抽出容器に
よる抽出法)又は24.4(捕集濃縮・抽出法)によって試験するとよい。
2. 濁りが著しい試料又はエマルションが生成しやすい試料の場合は,ソックスレー抽出法を適
用するとよい。試料に指示薬としてメチルオレンジ溶液 (1g/l) 2,3滴を加え,溶液の色が赤
に変わるまで塩酸 (1+1) を加えてpHを4以下に調節する。次に,ブフナー漏斗にろ紙5種
A2枚を重ねて敷き,これにけい藻土懸濁液約100mlを加えて吸引ろ過し,けい藻土を減圧状
態で水約1lで洗う。このろ過材に酸性にした試料を加えて吸引ろ過し,十分に吸引した後,
ろ過材をろ紙ごと巻いて円筒ろ紙に移し,漏斗の壁,容器,かき混ぜ棒などをヘキサンで洗
ったろ紙でふきとり,同じ円筒ろ紙に入れて80±5℃の乾燥器中で約30分間加熱する。
円筒ろ紙をソックスレー抽出器に移す。試料容器は十分に乾燥した後,ヘキサン約20ml
ずつで2回洗い,洗液を円筒ろ紙に注ぐ。次に,抽出操作に移り,抽出を約20回繰り返す。
以下,26.2(3)(k)〜(o)の操作を行って試料中のヘキサン抽出物質の濃度 (mg/l) を算出する。
27. 欠番
28. 残留塩素 残留塩素とは,塩素剤が水に溶けて生成する次亜塩素酸及びこれがアンモニアと結合して
生じるクロロアミンをいい,前者を遊離残留塩素,後者を結合残留塩素,両者を合わせて残留塩素という。
残留塩素の定量は,濃度が低い場合にはo-トリジン比色法又はジエチル-p-フェニレンジアミン (DPD)
比色法を適用し,濃度が比較的高い場合には,よう素滴定法を適用する。
また,結合残留塩素中のモノクロロアミン及びジクロロアミンなどを定量する場合には,DPD-硫酸アン
モニウム鉄 (II) 滴定法又は電流滴定法を適用する。この試験は試料採取後直ちに行う。
28.1 o-トリジン比色法 試料に3,3-ジメチルベンジジン(o-トリジン)溶液を加え,残留塩素との反応
で生じる黄色を,残留塩素標準比色液と比較して残留塩素を定量する方法である。亜ひ酸ナトリウム溶液
で処理し,残留塩素,遊離残留塩素,結合残留塩素に区別することができる。
定量範囲:Cl 0.01〜2.0mg/l,繰返し分析精度:変動係数で5〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) o-トリジン溶液 JIS K 8669に規定する二塩化3,3´-ジメチルベンジジニウム(o-トリジン二塩酸
塩)0.14gを水50mlに溶かし,塩酸 (3+7) 50ml中にかき混ぜながら加える。着色瓶に入れて保存
する。6か月以上経過したものは使用しない。
90
K 0101 : 1998
(c) りん酸塩緩衝液 (pH6.5) JIS K 9020に規定するりん酸水素二ナトリウムを110℃で約2時間加熱
し,デシケーター中で放冷した後,その22.86gと,JIS K 9007に規定するりん酸二水素カリウム
46.14gとを水に溶かして1lとする。沈殿物を生じた場合には,ろ別する。この溶液200mlをとり水
で1lとする。
(d) クロム酸カリウム-二クロム酸カリウム溶液 JIS K 8312に規定するクロム酸カリウム3.63gとJIS
K 8517に規定する二クロム酸カリウム1.21gとをりん酸塩緩衝液 (pH6.5) に溶かし,全量フラスコ
1 000mlに移し入れ,りん酸緩衝液 (pH6.5) を標線まで加える。
(e) 残留塩素標準比色液 相当する残留塩素の濃度 (mgCl/l) に応じ,クロム酸カリウム-二クロム酸カ
リウム溶液及びりん酸塩緩衝液 (pH6.5) を表28.1に示す割合に比色管100mlにとり,混ぜ合わせる。
暗所に保存し,沈殿が生じた場合には使用しない。
(f) 亜ひ酸ナトリウム溶液 (5g/l) JIS K 8046に規定するメタ亜ひ酸ナトリウム0.5gを水に溶かして
100mlとする。
(2) 器具 器具は,次のとおりとする。
(a) 比色管 100ml,底部から200±1.5mmの高さに100mlの標線を付けた平底のもの。
(b) 比色管立 100ml用,底部及び側面に乳白板を付けたもの。
表28.1 残留塩素標準比色液(液層200mm用)
残留塩素
mgCl/l
クロム酸カリウム-
二クロム酸カリウム
溶液 ml
りん酸塩緩衝液
(pH 6.5) ml
残留塩素
mgCl/l
クロム酸カリウム-
二クロム酸カリウム
溶液 ml
りん酸塩緩衝液
(pH 6.5) ml
0.01
0.18
99.82
0.70
7.48
92.52
0.02
0.28
99.72
0.80
8.54
91.46
0.05
0.61
99.39
0.90
9.60
90.40
0.07
0.82
99.18
1.00
10.66
89.34
0.10
1.13
98.87
1.10
12.22
87.78
0.15
1.66
98.34
1.20
13.35
86.65
0.20
2.19
97.81
1.30
14.48
85.52
0.25
2.72
97.28
1.40
15.60
84.40
0.30
3.25
96.75
1.50
16.75
83.25
0.35
3.78
96.22
1.60
17.84
82.16
0.40
4.31
95.69
1.70
18.97
81.03
0.45
4.84
95.16
1.80
20.09
79.91
0.50
5.37
94.63
1.90
21.22
78.78
0.60
6.42
93.58
2.00
22.34
77.66
(3) 操作 操作は,次のとおり行う。
(a) 比色管にo-トリジン溶液5mlをとり,これに試料(1)の適量(残留塩素0.2mg以下を含む。)を加え,
更に水を100mlの標線まで加え,手早く栓をして振り混ぜる。
(b) 5分間(2)暗所に放置する。
(c) 上方から透視して残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当
する残留塩素の濃度a (mgCl/l) を記録する。
(d) 別の比色管にo-トリジン溶液5mlをとり,これに(a)の操作と同量の試料を加え,手早く栓をして振
り混ぜる。
(e) 5秒間以内に亜ひ酸ナトリウム溶液 (5g/l) 5mlを加えて振り混ぜ,更に水を100mlの標線まで加え
て振り混ぜる。
91
K 0101 : 1998
(f) 残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する残留塩素の濃
度b (mgCl/l) を記録する。
(g) 空試験として比色管100mlに亜ひ酸ナトリウム溶液 (5g/l) 5mlをとり,これに(a)の操作と同量の試
料を加えて振り混ぜる。
(h) o-トリジン溶液5mlを加えて振り混ぜ,更に水を100mlの標線まで加えて振り混ぜる。
(i) 5秒間以内に残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する
残留塩素の濃度c1 (mgCl/l) を記録する。
(j) 更に5分間暗所に放置後,残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,こ
れに相当する残留塩素の濃度c2 (mgCl/l) を記録する。
(k) 次の式によって残留塩素,遊離残留塩素及び結合残留塩素の濃度を算出する。
V
c
a
A
100
)
(
2×
−
=
V
c
b
B
100
)
(
1×
−
=
C=A−B
ここに, A: 残留塩素 (mgCl/l)
a: (c)で求めた残留塩素 (mgCl/l)
c2: (j)で求めた残留塩素 (mgCl/l)
V: 試料 (ml)
B: 遊離残留塩素 (mgCl/l)
b: (f)で求めた残留塩素 (mgCl/l)
c1: (i)で求めた残留塩素 (mgCl/l)
C: 結合残留塩素 (mgCl/l)
注(1) 試料がアルカリ性の場合にはpH計を用い,塩酸 (1+5) を加えてpHを約7にする。
また,発色時のpHは常に1.3以下とする。
(2) 残留塩素のうち結合残留塩素は,最高発色に達するのに0℃で6分間,20℃で3分間,25℃で2
分30秒間必要である。
備考1. 空試験を行わない場合には,鉄0.3mg/l以上,マンガン0.01mg/l以上及び亜硝酸イオン0.3mg/l
以上が含まれていると妨害する。鉄及びマンガンの妨害を防ぐには,試料100mlにつき1, 2-
シクロヘキサンジアミン四酢酸溶液 (10g/l) 3mlを添加する。
2. 市販の残留塩素測定器を用いる場合には,あらかじめ残留塩素標準比色液と比較して,誤り
のないことを確認しておく。
3. この試験に用いる水は,残留塩素のないこと及び塩素を消費しないことを29.(3)によって確
かめておく。
28.2 ジエチル-p-フェニレンジアミン (DPD) 比色法 試料に硫酸N, N-ジエチル-p-フェニレンジアンモ
ニウム (DPD) を加え,残留塩素との反応で生じる桃色から桃赤色を,残留塩素標準比色液と比較して定
量する。
定量範囲:Cl 0.05〜2mg/l,繰返し分析精度:変動係数で5〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) よう化カリウム JIS K 8913に規定するもの。
92
K 0101 : 1998
(c) DPD希釈粉末 硫酸N, N-ジエチル-p-フェニレンジアンモニウム(N, N-ジエチル-p-フェニレンジア
ミン硫酸塩)1.0gをめのう乳鉢中で粉砕し,これにJIS K 8987に規定する硫酸ナトリウム24gを加
えてよく混和し,結晶粒を着色ガラス瓶中に入れ,湿気を避けて,0〜10℃の暗所に保存する。着色
したものは使用しない。
(d) りん酸塩緩衝液 (pH6.5) りん酸二水素カリウム溶液 (0.2mol/l) (JIS K 9007に規定するりん酸二
水素カリウム27.2gを水に溶かして1lとする。)100mlをとり,水酸化ナトリウム溶液 (0.2mol/l) (JIS
K 8576に規定する水酸化ナトリウム8gを水に溶かして1lとする。)をpH計を用いてpH 6.5になる
まで加え,これに1, 2-シクロヘキサンジアミン四酢酸一水和物0.13gを加えて溶かす。
(e) C. I. Acid Red 265溶液 C. I. Acid Red 265[1-(4-メチルベンゼンスルホンアミド)-7-(2-メチルフ
ェニルアゾ)-8-ヒドロキシ-3, 6-ナフタレンジスルホン酸二ナトリウム]を105〜110℃で3〜4時間
加熱し,デシケーター中で放冷する。その0.329gを1mgのけたまではかりとり,少量の水に溶か
して全量フラスコ1 000mlに移し入れ,水を標線まで加える。この溶液50mlを全量フラスコ500ml
にとり,水を標線まで加える。0〜10℃の暗所に保存し,6か月間以上経過したものは使用しない。
(f) DPD残留塩素標準比色液 C. I. Acid Red 265溶液を表28.2に従って全量フラスコ50mlにとり,水
を標線まで加える。これを比色管50mlにそれぞれ移す。密栓して0〜10℃の暗所に保存する。6か
月以上経過したものは使用しない。
表28.2 DPD残留塩素標準比色液(50ml中)
残留塩素
mg/l
C. I. Acid Red 265溶液
ml
0.05
0.5
0.1
1.0
0.2
2.0
0.3
3.0
0.4
4.0
0.5
5.0
0.6
6.0
0.7
7.0
0.8
8.0
0.9
9.0
1.0
10.0
1.2
12.0
1.4
14.0
1.6
16.0
1.8
18.0
2.0
20.0
(2) 器具 器具は,次のとおりとする。
(a) 比色管 50ml,底部から150±1mmの高さに50mlの標線を付けた平底のもの。
(b) 比色管立 50ml用,底部及び側面に乳白板を付けたもの。
(3) 操作 操作は,次のとおり行う。
(a) りん酸塩緩衝液 (pH6.5) 2.5mlを比色管50mlにとり,これにDPD希釈粉末0.5gを加える。次に,
試料(3)の適量(残留塩素0.1mg以下を含む。)を加え,更に水を標線まで加える。
(b) 栓をしてよく振り混ぜ,1分間(4)以内にその発色を側面から透視して,DPD残留塩素標準比色液と
比較し,該当するDPD残留塩素標準比色液から,これに相当する残留塩素の濃度 (mgCl/l) を求め,
93
K 0101 : 1998
これを遊離残留塩素として,試料中の遊離残留塩素の濃度 (mgCl/l) を算出する。
(c) (b) の操作が終了したら,よう化カリウム約0.5gを加え,栓をして振り混ぜて溶かし,約2分間放
置後,その発色を(b)と同様に残留塩素標準比色液と比較し,該当する残留塩素標準比色液からこれ
に相当する残留塩素の濃度 (mgCl/l) を求め,試料中の遊離残留塩素の濃度を算出する。
(d) 結合残留塩素の濃度 (mgCl/l) は,次の式によって算出する。
結合残留塩素の濃度 (mgCl/l) =残留塩素 (mgCl/l) −遊離残留塩素 (mgCl/l)
注(3) 試料の酸性又はアルカリ性が強い場合には,炭酸ナトリウム溶液 (50g/l) 又は塩酸 (1+11) を
用いてpHを約6.5に調節する。
(4) 振り混ぜ時間を含める。DPD希釈粉末中の硫酸ナトリウムは完全に溶けなくてもよい。
28.3 よう素滴定法 残留塩素とよう化カリウムとが反応して遊離するよう素をチオ硫酸ナトリウム溶液
で滴定し,残留塩素を定量する。よう素を遊離させる酸化性物質が共存すると,残留塩素として定量され
る。
定量範囲:Cl 0.1mg以上
(1) 試薬 試薬は次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) よう化カリウム JIS K 8913に規定するもの。
(c) 酢酸 (1+1) JIS K 8355に規定する酢酸を用いて調製する。
(d) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(e) 10mmol/lチオ硫酸ナトリウム溶液 22.1.2(1)(d)の0.1mol/lチオ硫酸ナトリウム溶液25mlを全量フ
ラスコ250mlにとり,水を標線まで加える。この溶液は使用時に調製し,調製後12時間以上経過
したものは,使用しない。ファクターは,0.1mol/lチオ硫酸ナトリウム溶液のものを用いる。
(2) 操作 操作は,次のとおり行う。
(a) 試料(1)の適量(残留塩素0.1〜7mgを含む。)を共栓三角フラスコ500mlにとり,水を加えて約300ml
とし,よう化カリウム1g及び酢酸 (1+1) 5mlを加える。
(b) 栓をして振り混ぜ,暗所に約5分間放置する。
(c) 遊離したよう素を,10mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指
示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(d) 空試験として水100mlをとり,(a)〜(c)の操作を行う。
(e) 次の式によって試料中の残留塩素の濃度 (mgCl/l) を算出する。
5
354
.0
1000
)
(
×
×
×
−
=
V
f
b
a
A
ここに,
A: 残留塩素 (mgCl/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター
V: 試料 (ml)
0.354 5: 10mmol/lチオ硫酸ナトリウム溶液1mlの残留塩素相当
量 (mg)
備考4. 試料の着色や濁りが著しく試験が困難な場合には,JIS K 0102の33.(残留塩素)備考4.の方
法で残留塩素を分離して定量する。

94
K 0101 : 1998
28.4 DPD-硫酸アンモニウム鉄 (II) 滴定法 遊離残留塩素を硫酸N, N-ジエチル-p-フェニレンジアンモニ
ウム (DPD) を指示薬として硫酸アンモニウム鉄 (II) 溶液で滴定して定量し,更によう化カリウムを加え
て結合残留塩素のモノクロロアミン及びジクロロアミンを分別して定量する。
定量範囲:Cl 20〜500μg
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) りん酸塩緩衝液 (pH6.2) JIS K 9020に規定するりん酸水素二ナトリウム24gとJIS K 9007に規定
するりん酸二水素カリウム46gとを水約800mlに溶かし,これにJIS K 8107に規定するエチレンジ
アミン四酢酸二水素二ナトウム二水和物0.8gを水約100mlに溶かして加え,更に水を加えて全量を
1lとする。
(c) DPD溶液 硫酸N, N-ジエチル-p-フェニレンジアンモニウム(N, N-ジエチル-p-フェニレンジアミ
ン硫酸塩)1.1gを硫酸 (1+3) 8mlとJIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリ
ウム二水和物0.2gを加えた水に溶かし,水で全量を1lとする。
この溶液は,着色の共栓ガラス瓶に入れて冷暗所に保存する(5)。
(d) よう化カリウム JIS K 8913に規定するもの。
(e) よう化カリウム溶液 JIS K 8913に規定するよう化カリウム0.5gを水100mlに溶かす。使用時に調
製する。
(f) ジフェニルアミンスルホン酸バリウム溶液 (1g/l) ジフェニルアミンスルホン酸バリウム0.1gを
水100mlに溶かす。
(g)
300
1mol/l二クロム酸カリウム溶液(標定用) JIS K 8005に規定する容量分析用標準物質の二クロ
ム酸カリウムを,あらかじめめのう乳鉢中で砕き,150℃で約1時間加熱し,デシケーター中で放冷
する。K2Cr2O7100%に対してその0.981gをとり,少量の水に溶かし,全量フラスコ1 000mlに移し
入れ,水を標線まで加える。
(h) 2.82mmol/l硫酸アンモニウム鉄 (II) 溶液 JIS K 8979に規定する硫酸アンモニウム鉄 (II) 六水和
物1.11gを硫酸 (1+3) 8mlを加えた水に溶かし,水を加えて全量を1lとする。使用時に標定する。
標定 この2.82mmol/l硫酸アンモニウム鉄 (II) 溶液100mlを三角フラスコにとり,これに硫酸 (1
+5) 10mlとJIS K 9005に規定するりん酸5mlとを加え,指示薬としてジフェニルアミンスルホン
酸バリウム溶液 (1g/l) 2mlを加える。この溶液を300
1mol/l二クロム酸カリウム溶液(標定用)で滴定
し,溶液の色が青紫になり約30秒間保持する点を終点とする。滴定に要した300
1mol/l二クロム酸カ
リウム溶液(標定用)のml数から,次の式によって2.82mmol/l硫酸アンモニウム鉄 (II) 溶液のフ
ァクター (f) を算出する。
100
82
.2
1000
300
6
×
×
×
=
x
f
ここに, x: 滴定に要した300
1mol/l二クロム酸カリウム溶液(標定用)の
ml数
注(5) 代わりに28.2(1)(c)のDPD希釈粉末約0.5gを用いてもよい。
(2) 操作 操作は,次のとおり行う。ただし,残留塩素の定量には(a)〜(c),遊離残留塩素の定量には(d),
モノクロロアミンの定量には(e)及びジクロロアミンの定量は(f)の操作を行う。
(a) 試料(3)の適量(残留塩素Clとして20〜500μgを含む。)を三角フラスコ300mlにとり,水を加えて
95
K 0101 : 1998
約100mlとし,りん酸塩緩衝液 (pH 6.2) 5mlとDPD溶液5ml(6)とを加え,振り混ぜる。
(b) よう化カリウム約1gを加えて溶かし,約2分間静置して赤に発色させる。
(c) 2.82mmol/l硫酸アンモニウム鉄 (II) 溶液で滴定し,赤い色が消えた点を終点する。滴定量をa (ml)
とする。
(d) 別に,(a)で採取した同量の試料を三角フラスコ300mlにとり,水を加えて約100mlとし,りん酸塩
緩衝液 (pH6.2) 5mlとDPD溶液5mlとを加えて振り混ぜる。手早く2.82mmol/l硫酸アンモニウム鉄
(II) 溶液で滴定し,赤い色が消えた点を終点する。滴定量をb (ml) とする。
(e) 次に,よう化カリウム溶液(7)0.1ml(2滴に相当)を加えて振り混ぜ,2.82mmol/l硫酸アンモニウム
鉄 (II) 溶液で滴定し,赤い色が消えた点を終点とする。滴定量をc (ml) とする。
(f) 更によう化カリウム(結晶)約1g(8) 加えて溶かし,約2分間静置して赤い色を発色させる。
(g) 2.82mmol/l硫酸アンモニウム鉄 (II) 溶液で滴定し,赤い色が消えた点を終点する。滴定量をd (ml)
とする。
(h) 次の式によって試料中の残留塩素,遊離残留塩素,モノクロロアミン及びジクロロアミンの濃度
(mgCl/l) を算出する。
1.0
000
1
×
×
×
=
V
f
a
A
1.0
000
1
×
×
×
=
V
f
b
B
1.0
000
1
×
×
×
=
V
f
c
C
1.0
000
1
×
×
×
=
V
f
d
D
ここに,
A: 残留塩素 (mgCl/l)
B: 遊離残留塩素 (mgCl/l)
C: モノクロロアミン (mgCl/l)
D: ジクロロアミン (mgCl/l)
f: 2.82mmol/l硫酸アンモニウム鉄 (II) 溶液のファクター
V: 試料 (ml)
0.1: 2.82mmol/l硫酸アンモニウム鉄 (II) 溶液1mlの残留塩素相
当量 (mg)
注(6) 28.2(1)(c)のDPD希釈粉末を使用する場合は,約0.5gを添加する。
(7) JIS K 8913に規定するよう化カリウムの細かくした結晶を,0.5mg添加してもよい。
(8) ジクロロアミンの濃度が1mg/l以上の場合には,2分間以上静置しても発色が不完全になりやす
い。このような場合には,JIS K 8913に規定するよう化カリウムの添加量を0.5g程度にする。
備考5. 三塩化窒素を含む試料の場合には,ジクロロアミンの定量値に三塩化窒素も含まれる。分別
定量は,次の操作で行う。
試料の適量(残留塩素Clとして20〜500μgを含む。)を三角フラスコ300mlにとり,水を
加えて100mlとし,よう化カリウム溶液0.1ml又はJIS K 8913に規定するよう化カリウムを
細かくし,その結晶約0.5mgを加えて振り混ぜる。別のビーカーにりん酸塩緩衝液 (pH6.2)
5mlとDPD溶液5ml(又はDPD希釈粉末約0.5g)とを入れて混ぜ合わせ,先の三角フラス
コに加える。
96
K 0101 : 1998
2.82mmol/l硫酸アンモニウム鉄 (II) 溶液で手早く滴定し,赤い色が消えた点を終点とする。
この滴定量をe (ml) とする。この滴定値には遊離残留塩素量が含まれる。次の式によって三
塩化窒素の濃度と,ジクロロアミンの濃度を算出する。
1.0
000
1
)
(2
×
×
×
−
=
V
f
a
e
E
1.0
000
1
)
(
×
×
×
−
=
V
f
e
d
D
ここに, E: 三塩化窒素 (mgCl/l) ,その他は(2)(h)による。
6. よう化物イオン又は臭化物イオンが共存する場合には,遊離残留塩素の定量値によう化物イ
オン及び臭化物イオンも含まれる。遊離残留塩素の定量値を補正するには,次の操作で行う。
遊離残留塩素は(2)(d)の操作によって求める。別に,試料100mlを三角フラスコ300mlにと
り,アミノ酢酸溶液[JIS K 8291に規定するアミノ酢酸(グリシン)20gを水100mlに溶か
す。]1mlを加える。
次に,りん酸塩緩衝液 (pH 6.2) 5mlとDPD溶液5mlとを加え,2.82mmol/l硫酸アンモニウ
ム鉄 (II) 溶液で滴定し,溶液の赤い色が消えた点を終点とする。(2)(d)の滴定値からこの滴
定値を差し引いて遊離残留塩素の濃度を算出する。
7. 二酸化塩素 (IV) は,残留塩素や遊離塩素の値に含まれる。
8. 滴定時のpHは6.2〜6.5の範囲に調節する。pHが低いと遊離残留塩素の定量値にモノクロロ
アミンの一部が含まれるようになる。また,pHが高くなると溶存酸素によっても発色する。
9. 測定時の液温が高くなると,遊離残留塩素の定量値に結合残留塩素が含まれるようになり,
DPDの発色も薄くなる傾向がある。
10. この試験では,銅は10mg/l程度までは,りん酸塩緩衝液 (pH6.2) にEDTAが含まれている
ので影響しない。クロム酸イオンが2mg/l以上共存する場合は滴定終点が不明確になるので,
あらかじめ試料に塩化バリウムを加えてクロム酸バリウムの沈殿にして妨害を除く。
11. 電流滴定法 残留塩素をフェニルアルセノオキシド溶液による電流滴定法で定量する。この
方法では,遊離残留塩素,結合残留塩素,モノクロロアミン及びジクロロアミンを分別して
定量することができる。
なお,残留塩素の定量には備考11.の(3)(a)〜(f),遊離残留塩素の定量には備考11.の(3)(g)
〜(i),結合残留塩素のうちモノクロロアミンの定量には備考11.の(3)(g)〜(k),ジクロロアミ
ンの定量には備考11.の(3)(g)〜(m)の操作を行う。鉄,マンガン,亜硝酸イオンなどは妨害し
ない。
定量範囲:Cl 0.04mg以上
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) りん酸塩緩衝液 (pH7) JIS K 9007に規定するりん酸二水素カリウム25.4gとJIS K 9020
に規定するりん酸水素二ナトリウム34.1gをビーカーにとり,水800mlに溶かし,水を加
えて1lとする。
(c) 酢酸-酢酸緩衝液 (pH4) JIS K 8355に規定する酢酸480gとJIS K 8371に規定する酢酸ナ
トリウム三水和物243gとをビーカーにとり,水400mlに溶かし,水を加えて1lとする。
(d) よう化カリウム溶液 (50g/l) JIS K 8913に規定するよう化カリウム5gを水に溶かして
97
K 0101 : 1998
100mlとする。使用時に調製し,着色ガラス瓶に入れる。
(e) 50mmol/lよう素溶液 JIS K 8913に規定するよう化カリウム8gを水約20mlに溶かし,こ
れにJIS K 8920に規定するよう素2.6gを加えて溶かし,水で200mlとし,着色ガラス瓶に
移し入れる。
標定 このよう素溶液20mlを共栓三角フラスコ300mlにとり,0.1mol/lチオ硫酸ナトリウ
ム溶液[22.1.2(1)(d)による。]で滴定し,溶液の黄色が薄くなったら,指示薬としてでん
ぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。滴定
に要した0.1mol/lチオ硫酸ナトリウム溶液のml数 (x) から,次の式によって50mmol/lよ
う素溶液のファクター (f) を算出する。
20
0f
x
f
×
=
ここに, f0: 0.1mol/lチオ硫酸ナトリウム溶液のファクター
(f) 2.82mmol/lよう素溶液(標定用) JIS K 8913に規定するよう化カリウム10gを水約20ml
に溶かし,全量フラスコ200mlに移し入れ,これに50mmol/lよう素溶液56.4/fml(f: 50mmol/l
よう素溶液のファクター)を加えた後,水を標線まで加える。この溶液20mlを全量フラス
コ100mlにとり,水を標線まで加える。使用時に調製する。
(g) 5.64mmol/lフェニルアルセノオキシド溶液 フェニルアルセノオキシド0.8gを水酸化ナト
リウム溶液 (12g/l) 150mlに溶かし,その110mlを水800ml中に入れてよくかき混ぜ,塩酸
(1+11) を用いてpHを6〜7に調節し,クロロホルム1mlを加えた後,水を加えて1lとす
る。
標定 2.82mmol/lよう素溶液 (0.2mgCl/ml) (標定用)5mlをビーカー300mlにとり,水を加
えて200mlとし,電流滴定装置を用いて,このフェニルアルセノオキシド溶液で滴定し,
電流計の指示値が下がらなくなったときを終点とする。これに要したフェニルアルセノオ
キシド溶液のml数 (x) を求め,次の式によってフェニルアルセノオキシド溶液のファク
ター (f1) を算出する。
x
f
5
1=
(2) 装置 装置は,次のとおりとする。
(a) 電流滴定装置
直流電流計 JIS C 1102-1及びJIS C 1102-2に規定する直動式指示電気計器の標準定格値
5μA, 1級電流計
白金電極
参照電極
(b) マグネチックスターラー
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(Clとして0.04〜2mgを含む。)をビーカー300mlにとり,水を加えて約200ml
とする。
(b) よう化カリウム溶液 (50g/l) 1mlを加え,更に酢酸-酢酸塩緩衝液 (pH 4) 1mlを加える(pH
を約4に調節する。)。
(c) 試料中に白金電極と参照電極を浸し,白金電極は直流電流計の正の端子に,参照電極は負
98
K 0101 : 1998
の端子に接続する。
(d) マグネチックスターラーで,空気を引き込まない程度にかき混ぜる。
(e) 5.64mmol/lフェニルアルセノオキシド溶液の一定量ずつを電流計の指示値が下がらなくな
るまで滴加する。
(f) 5.64mmol/lフェニルアルセノオキシド溶液のml数と電流計の指示値を読み取り,滴定曲線
を作成して滴定終点を求め,これをa (ml) とする。
(g) 別に,試料の適量(Clとして0.04〜2mgを含む。)をビーカー300mlにとり,水を加えて約
200mlとする。
(h) りん酸塩緩衝液 (pH 7) 1mlを加える。
(i) (c)〜(f)の操作を行って滴定終点を求め,これをb (ml) とする。
(j) 滴定後の試料に,よう化カリウム溶液 (50g/l) 0.2mlを加える。
(k) 電流計の指示値が変化したら,(c)〜(f)の操作を行って滴定終点を求め,これをc (ml) とす
る。
(l) 次に,滴定後の試料に酢酸−酢酸塩緩衝液 (pH 4) 1mlを加える。
(m) 電流計の指示値が変化したら,(e)及び(f)の操作を行って滴定終点を求め,これをd (ml) と
する。
(n) 次の式によって試料中の残留塩素,遊離残留塩素,モノクロロアミン及びジクロロアミン
の濃度 (mgCl/l) を算出する。
2.0
000
1
1
×
×
×
=
V
f
a
A
2.0
000
1
1
×
×
×
=
V
f
b
B
2.0
000
1
1
×
×
×
=
V
f
c
C
2.0
000
1
1
×
×
×
=
V
f
d
D
ここに,
A: 残留塩素 (mgCl/l)
B: 遊離残留塩素 (mgCl/l)
C: モノクロロアミン (mgCl/l)
D: ジクロロアミン (mgCl/l)
f1: 5.64mmol/lフェニルアルセノオキシド溶液のファクター
V: 試料 (ml)
0.2: 5.64mmol/lフェニルアルセノオキシド溶液1mlの残留塩素相
当量 (mg)
29. 塩素要求量 塩素要求量とは,試料に塩素を加え,一定時間反応させた後の残留塩素が所定の濃度に
なるために必要な塩素の添加量をいう。
塩素の添加量を多くしていくに従い,一定時間後の残留塩素の濃度は増加するが,アンモニウムイオン,
有機体窒素などを多く含む試料では,ある添加量に達すると残留塩素の濃度は減少する。更に塩素を添加
すると残留塩素の濃度が増大し始める。この関係を作図し,残留塩素が所定の濃度になるための塩素添加
濃度を求め,塩素要求量とする。

99
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。ほうけい酸ガラス瓶に保存する。
(b) 塩素標準液 (0.1mgCl/ml) 次亜塩素酸ナトリウム溶液(有効塩素7〜12%)を水で有効塩素約
0.1mgCl/mlになるように薄める。使用時に28.3(2)の操作を行って濃度を測定する。
(2) 器具 器具は,次のとおりとする。
(a) 共栓三角フラスコ 300ml
(3) 操作 操作は,次のとおり行う。
(a) 試料200mlずつを数個の共栓三角フラスコ300mlにとり,これに塩素標準液 (0.1mgCl/ml) (1)1〜20ml
をフラスコの内壁に付着しないように注意しながら,段階的に加え,密栓して振り混ぜた後,暗所
に放置(2)する。
(b) 1時間後28.1,28.2,28.3又は28.4の方法によって,それぞれの残留塩素の濃度を測定する。
(c) 次に,方眼紙上の縦軸に残留塩素の濃度を,横軸に(a)での塩素添加濃度(塩素水添加直後の濃度)
をとり,図29.1のように作図する(3)。
図29.1 作図による塩素要求量の求め方
(d) II形のような場合には,残留塩素濃度が所定の値k(4) (mgCl/l) を示すための塩素添加濃度a (mgCl/l)
を塩素要求量とする。
(e) III形のような場合には,残留塩素濃度が極小値cを示す点を過ぎた後,所定の値k (mgCl/l) を示す
ような塩素添加濃度d (mgCl/l) を塩素要求量とする。
注(1) 塩素標準液の濃度は0.05〜0.1mgCl/mlの範囲内であれば十分である。しかし,塩素要求量が6mg/l
以上の試料の場合は0.2mgCl/mlの濃度が適当である。
(2) できるだけ,採水時の水温と同温度に保持する。
また,放置時間は,塩素注入地点から施設の所定の場所に到達するのに要する時間を原則と
し,一般には1時間程度を採用している。
(3) 塩素と反応する物質を含まない試料は,理論的にはI形を示す。実際の試料はII形又はIII形を
示す。
(4) kの値は,鉄及びマンガンの除去を目的とする場合は,0.5〜1.0 (mgCl/l) の範囲が実際的である。
上水の場合は普通0.1 (mgCl/l) を用いている。
備考1. 上水の試験では塩素要求量は,a, bなどの値からkの値(普通は0.1)を差し引いた値を用
いている。
また,bからk (0.1) を差し引いた値を,塩素消費量と称している。II形の場合は,塩素要
求量と塩素消費量は等しい。
100
K 0101 : 1998
30. 水酸化物イオン (OH-) 水酸化物イオンの定量には中和滴定法を適用する。塩化ストロンチウムの添
加によって,共存する炭酸イオン,りん酸イオンなどをストロンチウム塩として沈殿させた後,フェノー
ルフタレインを指示薬として硫酸で滴定し,水酸化物イオンを定量する。
定量範囲:OH- 0.1mg以上
(1) 試薬 試薬は,次のものを用いる。
(a) 10mmol/l硫酸 13.1(1)(c)による。
(b) 塩化ストロンチウム溶液 JIS K 8132に規定する塩化ストロンチウム六水和物4.5gを水に溶かして
1lとする。
(c) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(d) 窒素 JIS K 1107に規定する高純度窒素2級
(2) 器具 器具は,次のとおりとする。
(a) 共栓三角フラスコ 300ml
(3) 操作 操作は,次のとおり行う。
(a) 試料100ml(OH-として15mg以上を含む場合には,その適量をとり,水を加えて100mlとする。)
を共栓三角フラスコ300mlにとる。
(b) 共存する炭酸イオンとりん酸イオンのそれぞれ1mgにつき塩化ストロンチウム溶液1mlを加え,更
に4mlを過剰に加えて静かに振り混ぜる。
(c) 二酸化炭素を除いた空気又は窒素(1)を通しながら加熱し,約30秒間煮沸した後,水で冷却する。
(d) 冷却後,指示薬としてフェノールフタレイン溶液 (5g/l) 2,3滴を加え,10mmol/l硫酸で溶液の赤い
色が消えるまで滴定する。
(e) 次の式によって試料中の水酸化物イオンの濃度 (mgOH-/l) を算出する。
2
340
.0
1000×
×
×
=
V
f
a
H
ここに,
H: 水酸化物イオン (mgOH-/l)
a: 滴定に要した10mmol/l硫酸 (ml)
f: 10mmol/l硫酸のファクター
V: 試料 (ml)
0.340 2: 10mmol/l硫酸1mlの水酸化物イオン相当量 (mg)
注(1) 水酸化カリウム溶液 (220g/l) と水で洗浄した空気又は窒素を液面に通す。
備考1. 炭酸イオン及びりん酸イオンの濃度が不明のときは,塩化ストロンチウム溶液を十分に過剰
に加える必要がある。
2. 塩化ストロンチウムを過剰に加えると硫酸ストロンチウムの白濁を生じることがあるが,滴
定を妨害しない。
31. ふっ素化合物 ふっ素化合物は,ふっ化物イオン,金属ふっ化物などを総称し,ふっ化物イオンとし
て表す。ふっ化物イオンの定量には,ランタン-アリザリンコンプレキソン吸光光度法又はイオン電極法を
適用する。
31.1 ランタン-アリザリンコンプレキソン吸光光度法 ランタン (III) とアリザリンコンプレキソンとの
錯体がふっ化物イオンと反応して生じる青い色の複合錯体の吸光度を測定して,ふっ化物イオンを定量す
る。
101
K 0101 : 1998
定量範囲:F- 4〜50μg,繰返し分析精度:変動係数で3〜10%
備考1. この方法は,陰イオンの影響は少ないが,陽イオンの影響が多く,特にアルミニウム,カド
ミウム,コバルト,鉄,ニッケル,ベリリウム及び鉛などが妨害するので,あらかじめ蒸留
してふっ化物イオンを分離する。
(1) 試薬 試薬は,次のものを用いる。
(a) 過塩素酸 JIS K 8223に規定するものを,加熱して白煙を発生させた後,放冷したもの。
(b) りん酸 JIS K 9005に規定するもの。
(c) 水酸化ナトリウム溶液 (100g/l) 22.2.1(1)(b)による。
(d) 二酸化けい素 JIS K 8885に規定する二酸化けい素で粒径100〜150μmのもの(1)。
(e) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(f) ランタン-アリザリンコンプレキソン溶液(2) アリザリンコンプレキソン(1, 2-ジヒドロキシアント
ラキノン-3-イルメチルアミン-N, N-二酢酸二水和物)0.192gをアンモニア水 (1+10) 4mlと酢酸アン
モニウム溶液 (200g/l) 4mlに溶かし,これを酢酸ナトリウム溶液(JIS K 8371に規定する酢酸ナト
リウム三水和物41gを水400mlに溶かし,JIS K 8355に規定する酢酸24mlを加えたもの。)中にか
き混ぜながら加える。この溶液をかき混ぜながらJIS K 8034に規定するアセトン400mlを徐々に加
え,更にランタン溶液[酸化ランタン (III) 0.163gを塩酸 (1+5) 10mlに加熱して溶かしたもの。]
を加えてかき混ぜる。放冷後,酢酸又はJIS K 8085に規定するアンモニア水でpH計を用いてpH
を約4.7に調節した後,水を加えて1lとする。
(g) ふっ化物イオン標準液 (0.1mgF-/ml) JIS K 8005に規定する容量分析用標準物質のふっ化ナトリ
ウムを白金皿にとり,500℃で約1時間加熱し,デシケーター中で放冷する。NaF100%に対してそ
の0.221gをとり,少量の水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。ポ
リエチレン瓶に入れて保存する。又はJIS K 0030に規定するふっ化物イオン標準液のF-の100を用
いる。
(h) ふっ化物イオン標準液(2μgF-/ml) ふっ化物イオン標準液 (0.1mgF-, /ml) 10mlを全量フラスコ
500mlにとり,水を標線まで加える。
注(1) 結晶質のものを用いる。品質が分からない場合には,白金るつぼ中で1150℃以上で約1時間加熱
し,デシケーター中で放冷したものを用いる。この場合ふっ化物イオン標準液(2μgF-/ml)10ml
をとり,(3)(b)〜(e)及び(4)(a)〜(e)を行って回収率を確認する。
(2) 市販品を用いてもよい。
参考 市販のアルフッソンを用いる場合は,その2.5gを水に溶かして50mlとする。使用時に調製す
る。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 蒸留装置 図31.1に一例を示す。
(b) 光度計 分光光度計又は光電光度計
(3) 蒸留操作 蒸留操作は,次のとおり行う。
(a) 試料の適量(3)(F-として30μg以上を含む。)を磁器蒸発皿又はビーカーにとり,フェノールフタレ
イン溶液 (5g/l) 2,3滴を加え,水酸化ナトリウム溶液 (100g/l) を滴加して微アルカリ性とした後,
加熱して約30mlに濃縮する。
(b) 図31.1の蒸留フラスコ中に水約10mlで洗い移す。次に,二酸化けい素約1g,りん酸1ml及び過塩
素酸40ml[又はJIS K 8951に規定する硫酸(4)30ml]を加える(5)。受器の全量フラスコ250mlには
102
K 0101 : 1998
水20ml(6)を加え,逆流止めの先端は水面下に保つ。
(c) 蒸留フラスコを直接加熱(7)し,蒸留フラスコ内の液温が約140℃に達してから,水蒸気を通す。
(d) 蒸留温度を145±5℃,留出速度を3〜5ml/minに調節し,受器の液量が約220mlになるまで蒸留を
続ける。
(e) 冷却器と逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器に
加え,更に水を標線まで加える。
注(3) 溶存ふっ化物イオンを試験するときは,試料をろ紙5種Cでろ過し,初めのろ液約50mlを捨て,
その後のろ液を用いる。
(4) JIS K 8951に規定する硫酸をビーカーに入れ,加熱して盛んに白煙を発生させた後,放冷した
もの。
(5) 蒸留フラスコには,沸騰石(直径2〜3mm)を約10個入れる。
(6) 試料中にふっ化物イオン以外のハロゲン化物が多量に含まれる場合には,水酸化ナトリウム溶
液 (40g/l) 数滴とフェノールフタレイン溶液 (5g/l) 数滴を加えておく。受器中の溶液は蒸留が
終わるまで微紅色を保つように,必要に応じて水酸化ナトリウム溶液 (40g/l) を滴加する。
なお,この場合は蒸留が終わった後,留出液に硫酸 (1+35) を微紅色が消えるまで滴加し,
以下,(e)の操作を行う。
(7) 蒸留フラスコ中の液面まで加熱できるようにフレームを調節する。油浴,グリセリン浴などを
用いてもよい。
備考2. 蒸留フラスコに代えて二重管形の蒸留フラスコを用いてもよい。この場合,その外筒にJIS K
9520に規定する1,1,2,2-テトラクロロエタン(沸点:146.3℃)を入れ,外筒を直接加熱
して1,1,2,2-テトラクロロエタンが沸騰し始めてから水蒸気を通す。1,1,2,2-テトラ
クロロエタンを長時間使用すると分解して着色するとともに沸点が降下する。この場合は蒸
留して146℃の留分を用いる。
なお,使用後の1,1,2,2-テトラクロロエタンを処分するときは環境汚染を生じないよ
うに注意する。
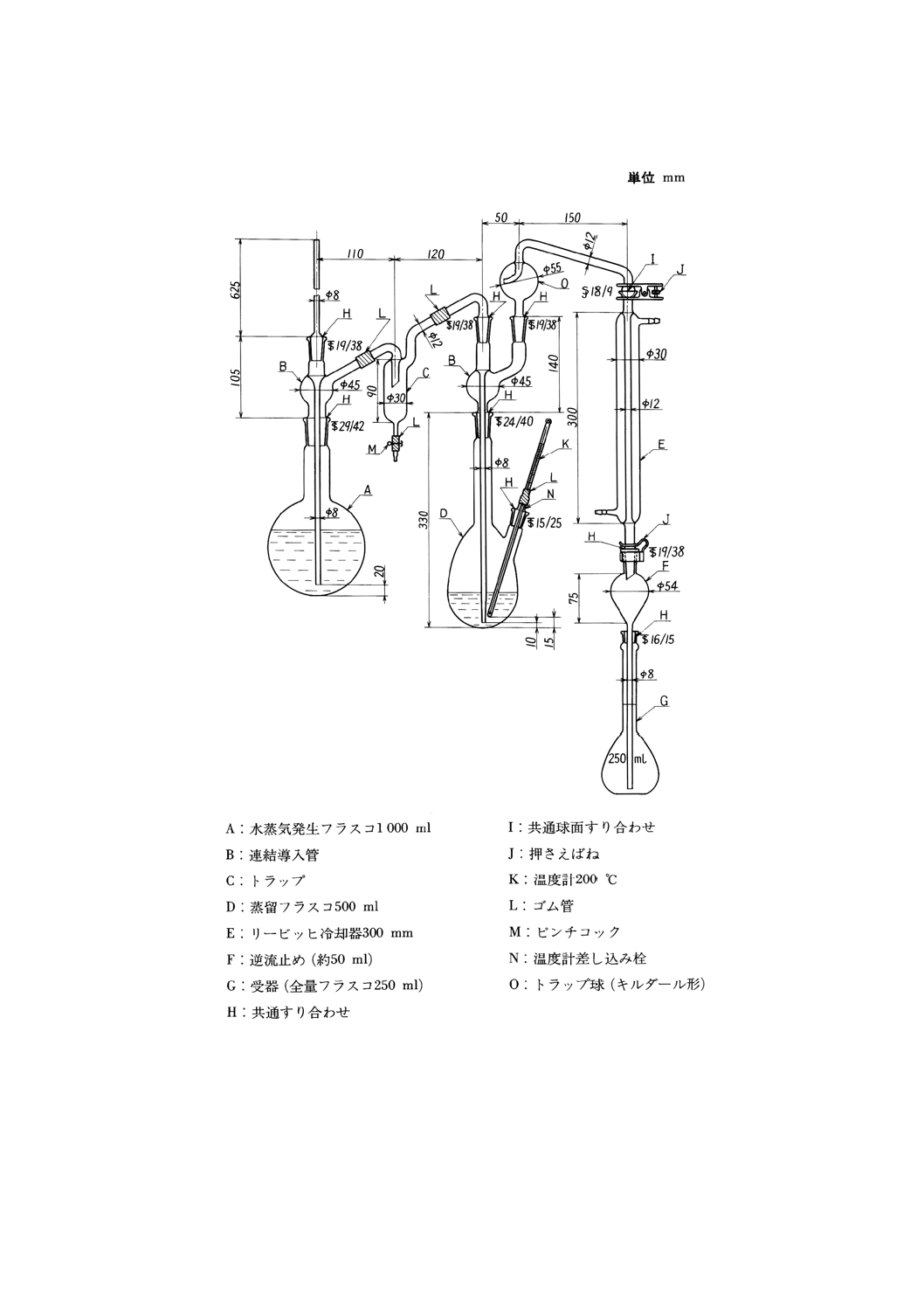
103
K 0101 : 1998
図31.1 蒸留装置の一例
(4) 操作 操作は,次のとおり行う。
(a) (3)の蒸留操作で得た留出液から30ml以下の適量(F-として4〜50μgを含む。)を全量フラスコ50ml
にとる。
(b) ランタン-アリザリンコンプレキソン溶液(8) 20mlを加え,更に水を標線まで加えて振り混ぜ,約1
時間放置する。
104
K 0101 : 1998
(c) 別に,水30mlを全量フラスコ50mlにとり,(b)の操作を行う。
(d) 試料について(b)で得た溶液の一部を吸収セルに移し,(c)の溶液を対照液として波長620nm付近の
吸光度を測定する。
(e) 検量線からふっ化物イオンの量を求め,試料中のふっ化物イオンの濃度 (mgF-/l) を算出する。
検量線 ふっ化物イオン標準液 (2μgF-/ml) 2〜25mlを全量フラスコ50mlに段階的にとり,(a)〜(d)
の操作を行って吸光度を測定し,ふっ化物イオン (F-) の量と吸光度との関係線を作成する。
注(8) 31.1(1)の参考で調製したアルフッソン溶液を用いる場合には,その5mlとJIS K 8034に規定す
るアセトン10mlを試料溶液に加えた後,水を標線まで加える。
31.2 イオン電極法 ふっ素化合物を前処理して蒸留分離し,緩衝液(イオン強度調節液)を加えてpHを
5.0〜5.5に調節し,ふっ化物イオン電極を指示電極として電位を測定し,ふっ化物イオンを定量する。
定量範囲:F- 0.1〜100mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 緩衝液 (pH5.2)(9) JIS K 8150に規定する塩化ナトリウム58gとJIS K 8284に規定するくえん酸水
素二アンモニウム1gとを水500mlに加えて溶かし,JIS K 8355に規定する酢酸50mlを加え,水酸
化ナトリウム溶液 (200g/l) を滴加して,pH計を用いてpHを5.2に調節した後,水を加えて1lとす
る。
(b) ふっ化物イオン標準液 (100mgF-/l) 31.1(1)(g)による。
(c) ふっ化物イオン標準液 (10mgF-/l) ふっ化物イオン標準液 (100mgF-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。使用時に調製する。
(d) ふっ化物イオン標準液 (1mgF-/l) ふっ化物イオン標準液 (10mgF-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(e) ふっ化物イオン標準液 (0.1mgF-/l) ふっ化物イオン標準液 (1mgF-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。使用時に調製する。
注(9) 緩衝液として,次の組成のものを使用してもよい。
(i) 水500mlにJIS K 8355に規定する酢酸57ml,JIS K 8150に規定する塩化ナトリウム58g, 1,
2-シクロヘキサンジアミン四酢酸一水和物4gを加えて溶かし,水酸化ナトリウム溶液
(200g/l) を滴加し,pH計を用いてpHを5.0〜5.5に調節した後,水を加えて1lとする。
(ii) 水500mlにJIS K 8355に規定する酢酸57ml,JIS K 8150に規定する塩化ナトリウム58g,JIS
K 8288に規定するくえん酸三ナトリウム二水和物0.3gを加えて溶かし,水酸化ナトリウム溶
液 (200g/l) を加え,pH計を用いてpHを5.0〜5.5に調節した後,水を加えて1lとする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 最小目盛1mVの高入力抵抗電位差計(例えば,デジタル式pH-mV計,拡大スパン付
pH-mV計及びイオン電極用電位差計など。)。
(b) 指示電極 ふっ化物イオン電極
(c) 参照電極 二重液絡形(又は塩橋)参照電極(ダブルジャンクションのスリーブ形参照電極又はセ
ラミックス形参照電極で抵抗の小さいもの。)。内筒液には塩化カリウム溶液(3mol/l〜飽和)を入
れる。外筒液には塩化カリウム溶液(3mol/l〜飽和)又は硝酸カリウム溶液 (100g/l) を入れる。
(d) マグネチックスターラー 回転による発熱で液温に変化を与えないもの。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) ふっ化物イオン標準液 (0.1mgF-/l) 100mlをビーカー200mlにとり,緩衝液 (pH 5.2) 10mlを加える(10)。
105
K 0101 : 1998
(b) これに,指示電極(11)(12) と参照電極(13)(14)とを浸し,マグネチックスターラー(15)を用いて,泡が電
極に触れない程度に強くかき混ぜる(16)。
(c) 液温をはかり,電位差計で電位を測定する(17)。
(d) ふっ化物イオン標準液 (1mgF-/l) 100ml,ふっ化物イオン標準液 (10mgF-/l) 100ml及びふっ化物イオ
ン標準液 (100mgF-/l) 100mlをそれぞれビーカー200mlにとり,緩衝液 (pH5.2) 10mlを加える。それ
ぞれのふっ化物イオン標準液 (1〜100mgF-/l) の液温を(c)での液温の±1℃に調節し(17),(b)及び(c)
の操作を行って電位を測定する(18)。
(e) 片対数紙の対数軸にふっ化物イオンの濃度を,均等軸に電位をとり,ふっ化物イオンの濃度 (mgF-/l)
と電位との関係線を作成する(18)。
注(10) 緩衝液 (pH5.2) は,測定時においてpHを約5.2に調節し,イオン強度を一定にするためのもの
である。
(11) 指示電極(ふっ化物イオン電極)は,使用時にふっ化物イオン標準液 (0.1mgF-/l) に浸し,指示
値が安定してから電位を測定する。
(12) 指示電極の感応膜に傷がつくと,検量線のこう配(電位こう配)が小さくなり,応答速度も遅
くなるので注意する。
また,指示電極の感応膜が汚れると,応答速度が遅くなるので,エタノール (95) を含ませた
脱脂綿又は柔らかい紙で汚れをふき取り,水で洗浄する。
(13) 参照電極は,抵抗の小さいものを選ぶ。一般にスリーブ形,セラミックス形を用いる。
スリーブ形は抵抗も小さく最適であるが,スリーブを締め過ぎると抵抗が大きくなり,緩す
ぎると外筒液の流出が多くなるので,適度の締め付けが必要である。
セラミックス形は抵抗の大きい製品もあるので,イオン電極用を用いる。セラミックス形は
乾燥したり,汚れると抵抗が大きくなるので注意する。
参照電極は,いずれの場合も外筒液と同じ溶液中に浸しておく。スリーブ形は使用時にスリ
ーブの締め付けを調節する。
(14) 内筒液及び外筒液に塩化カリウム溶液(飽和)を使用する場合には,液温が低下すると塩化カ
リウムの結晶が析出し,固着して抵抗が大きくなることがあるので注意する。
(15) マグネチックスターラーを長時間使用すると,発熱して液温に変化を与えることがあるので,
液温の変化に注意する。
(16) かき混ぜ速度で電位差計の指示が不安定になる場合には,参照電極の抵抗が大きくなっている
ことが多い。
(17) ふっ化物イオン電極の応答時間は,液温10〜30℃の場合には,ふっ化物イオンの濃度が0.1mgF-/l
で約1分間,1mgF-/l以上では約30秒間である。
(18) ふっ化物イオン標準液 (1mgF-/l) とふっ化物イオン標準液 (100mgF-/l) との電位の差は,110〜
120mV (25℃) の範囲に入り,ふっ化物イオンの濃度0.1〜100mgF-/lの間の検量線は直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 31.1(3)の蒸留操作で得た留出液から100mlをビーカー200mlにとり,緩衝液 (pH 5.2) 10mlを加え,
液温を(3)(c)の液温の±1℃に調節する。
(b) (3)(b)及び(c)の操作を行って検量線からふっ化物イオンの濃度を求め,試料中のふっ化物イオンの濃
度 (mgF/l) を算出する。
備考3. イオン濃度計の場合には,ふっ化物イオン標準液 (1mgF-/l) と,ふっ化物イオン標準液
106
K 0101 : 1998
(100mgF-/l) とを用い,(3)(b)及び(c)の操作を行ってイオン濃度計の指示値を1mgF-/lと
100mgF-/lになるように調節する。さらに,ふっ化物イオン標準液 (0.1mgF-/l) とふっ化物イ
オン標準液 (10mgF-/l) を用いてイオン濃度計の指示値を確認する。
4. イオン電極法では,ふっ化物イオンだけが測定できるので,あらかじめふっ素化合物を蒸留
操作ですべてふっ化物イオンにしてから測定する。
主な共存物質の許容限度を最大比率で次に示す。
HCO3-, Cl-, NO3-, I-, Br-,
HPO42-
:103
SO42-:104
OH-及びAl3+, Fe3+は測定を妨害するが,蒸留分離によって除去されるため影響はない。
5. ふっ化物イオン電極による電位差滴定法 31.1(3)の蒸留操作で得た留出液から100mlをビー
カーにとり,(3)(b)及び(c)の操作に準じて電位を測定しながら300
1〜301mol/lの硝酸ランタン
(III) 溶液で滴定して滴定曲線を作図し,滴定終点を求め,ふっ化物イオンの量を算出する。
30
1mol/l硝酸ランタン (III) 溶液1mlはF- 1.899mgに相当する。
32. 塩化物イオン (Cl-) 塩化物イオンの定量には,チオシアン酸水銀 (II) 吸光光度法,硝酸水銀 (II) 滴
定法,硝酸銀滴定法,イオン電極法又はイオンクロマトグラフ法を適用する。
32.1 チオシアン酸水銀 (II) 吸光光度法 試料にチオシアン酸水銀 (II) と硫酸アンモニウム鉄 (III) を
加えたとき,塩化物イオンによって置換されたチオシアン酸イオンと鉄 (III) とが反応して生じるだいだ
い赤の錯体の吸光度を測定して塩化物イオンを定量する。
定量範囲:Cl- 20〜500μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸アンモニウム鉄 (III) 溶液 JIS K 8982に規定する硫酸アンモニウム鉄 (III) ・12水60gを硝
酸 (5mol/l) (JIS K 8541に規定する硝酸380mlに水600mlを加え,室温まで冷却し,更に水を加え
て1lとする。)1lに溶かす。濁りがあればろ過し,着色瓶に入れて保存する。
(b) チオシアン酸水銀 (II) エタノール溶液 JIS K 9519に規定するチオシアン酸水銀 (II) 1.5gをJIS K
8102に規定するエタノール (95) 500mlに溶かし,着色ガラス瓶に入れて保存する。
(c) 塩化物イオン標準液 (1mgCl-/ml) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを
あらかじめ600℃で約1時間加熱し,デシケーター中で放冷する。NaCl100%に対してその1.648g
をとり,少量の水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0029
に規定する塩化物イオン標準液のCl-の1 000を用いる。
(d) 塩化物イオン標準液 (10μgCl-/ml) 塩化物イオン標準液 (1mgCl-/ml) 10mlを全量フラスコ1 000ml
にとり,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水で洗う。
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ紙5種Cでろ過し,初めのろ液約50mlを捨て,次のろ液50ml(Cl-0.25mg以上を含む場合
には適量をとり,水を加えて50mlとする。)をメスシリンダー(有栓形)50mlにとる。
(b) 硫酸アンモニウム鉄 (III) 溶液10mlとチオシアン酸水銀 (II) エタノール溶液5mlとを加え,栓を
107
K 0101 : 1998
してよく振り混ぜる。
(c) 液温を約20℃(1)に保ち約10分間放置する。
(d) 空試験として,水50mlについて,(a)〜(c)の操作を行う。
(e) (c)の溶液を吸収セル(2)に移し,(d)の空試験の溶液を対照液とし,波長460nm付近の吸光度を測定
する。
(f) 検量線から,塩化物イオンの量を求め,試料中の塩化物イオンの濃度 (mgCl-/l) を算出する。
検量線 塩化物イオン標準液 (10μgCl-/ml) 2〜50mlをメスシリンダー(有栓形)50mlに段階的にと
り,水を加えて50mlとした後,(b)〜(e)の操作を行って塩化物イオン (Cl-) の量と吸光度との関係
線を作成する。
注(1) 発色速度は温度によって異なるので,発色時の温度差が±2℃になるようにする。
(2) 光路長20mmの吸収セルを用いる場合にはCl- 10〜250μg,光路長50mmの吸収セルを用いる場
合には,Cl- 5〜100μg,光路長100mmの吸収セルを用いる場合にはCl- 2.5〜50μgの定量ができ
る。
なお,光路長100mmの吸収セルを用いる場合には,試料100mlをとり,これに使用する試薬
は,それぞれ2倍量を用いる。
備考1. 臭化物イオン,よう化物イオン,シアン化物イオンなどは妨害する。チオ硫酸イオン,硫化
物イオン及び亜硫酸イオンも妨害するので,あらかじめ酸化しておく。
2. 塩化物イオンは広く存在するから,手の汗などからの汚染や実験室の空気などからの汚染に
注意する。
3. 水銀化合物を使用するから廃液の処分には特に注意する。
32.2 硝酸水銀 (II) 滴定法 試料のpHを2.5に調節し,硝酸水銀 (II) 溶液で滴定して塩化物イオンを定
量する。
よう化物イオン及び臭化物イオンは塩化物イオンとして定量される。亜硫酸イオン,ヒドラジニウムイ
オン(ヒドラジン),ヒドロキシルアミンなどの還元性物質は妨害するが,過酸化水素で酸化すれば妨害し
ない。クロム (VI) 及び鉄 (III) は,いずれも10mg/l以下であれば妨害しない。
定量範囲:Cl- 0.1〜5mg
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 (1+65) JIS K 8541に規定する硝酸を用いて調製する。
(b) 過酸化水素 (1+1) JIS K 8230に規定する過酸化水素を用いて調製する。
(c) 混合指示薬溶液 JIS K 8489に規定するジフェニルカルバゾン0.50g,JIS K 8844に規定するブロモ
フェノールブルー0.05g及びJIS K 8272に規定するキシレンシアノールFF0.12gをはかりとり,JIS
K 8102に規定するエタノール (95) 100mlに溶かし,着色ガラス瓶に入れ保存する。
(d) 塩化物イオン標準液 (0.5mgCl-/ml) 32.1(1)(c)の塩化ナトリウム標準液 (1mgCl-/ml) 100mlを全量
フラスコ200mlにとり,水を標線まで加える。
(e) 7.05mmol/l硝酸水銀 (II) 溶液 JIS K 8558に規定する硝酸水銀 (II) n水和物約2.5gをとり,JIS K
8541に規定する硝酸0.5mlを含む水20mlを加えて溶かし,ビーカー1 000mlに移し入れ,水で1l
とする。
標定 塩化物イオン標準液 (0.5mgCl-/ml) 20mlをビーカーにとり,水を加えて100mlとし,混合指
示薬溶液5滴を加え,溶液の色が青から青緑になるまで硝酸 (1+65) を滴加し,更に1mlを加えて
pHを約2.5にする。次に,この7.05mmol/l硝酸水銀 (II) 溶液で滴定し,溶液の色が黄緑から灰色
108
K 0101 : 1998
を経て紫になるときを終点とする。別に,水100mlをとり,空試験を行って滴定値を補正する。補
正したml数 (x) から,次の式によって7.05mmol/l硝酸水銀 (II) 溶液のファクター (f) を算出する。
x
f
20
=
(2) 操作 操作は,次のとおり行う。
(a) 試料に濁りがある場合には,ろ紙5種Cでろ過し(3),初めのろ液約50mlを捨て,次のろ液100ml
(Cl- 5mg以上を含む場合には,適量をとり,水を加えて100mlとする。)をビーカーにとる。
(b) 亜硫酸イオン,ヒドラジニウムイオン,ヒドロキシルアミンなどの還元性物質が共存する場合には,
過酸化水素 (1+1) を滴加し,かき混ぜて分解する。
(c) 混合指示薬溶液5滴を加え,溶液の色が明らかに青から青緑ないし黄緑になるまで硝酸 (1+65) を
滴加した後,更に1mlを加える(4)。
(d) 7.05mmol/l硝酸水銀 (II) 溶液で滴定し,溶液の色が黄緑から灰色を経て紫になるときを終点とする。
(e) 空試験として水100mlをとり,(c)及び(d)の操作を行う。
(f) 次の式によって試料中の塩化物イオンの濃度 (mgCl-/l) を算出する。
5.0
000
1
)
(
×
×
×
−
=
V
f
b
a
C
ここに,
C: 塩化物イオン (mgCl-/l)
a: 滴定に要した7.05mmol/l硝酸水銀 (II) 溶液 (ml)
b: 空試験に要した7.05mmol/l硝酸水銀 (II) 溶液 (ml)
f: 7.05mmol/l硝酸水銀 (II) 溶液のファクター
V: 試料 (ml)
0.5: 7.05mmol/l硝酸水銀 (II) 溶液1mlの塩化物イオン相当量
(mg)
注(3) 試料に著しい濁りが認められない場合は,ろ過操作を省略してもよい。
(4) 酸性が強い場合には,水酸化ナトリウム溶液 (40g/l) でpHを約2.5に調節する。
備考4. 備考3.による。
32.3 硝酸銀滴定法 試料のpHを約7に調節し,ウラニン(フルオレセインナトリウム)[9-(2-カルボ
キシフェニル)-6-ヒドロキシ-3H-キサンテン-3-オン二ナトリウム塩(IUPACによる名称)]溶液を指示薬
として,硝酸銀溶液で滴定して塩化物イオンを定量する。
定量範囲:Cl- 1mg以上
備考5. 臭化物イオン,よう化物イオン,シアン化物イオンなどが共存すると,塩化物イオンとして
定量される亜硫酸イオン,チオ硫酸イオン,硫化物イオンも妨害するが,あらかじめ過酸化
水素で酸化すれば妨害しない。
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 (1+65) 32.2(1)(a)による。
(b) 炭酸ナトリウム溶液 (50g/l) JIS K 8625に規定する炭酸ナトリウム5gを水に溶かして100mlと
する。
(c) フルオレセインナトリウム溶液 (2g/l) JIS K 8830に規定するウラニン(フルオレセインナトリウ
ム)0.2gを水に溶かして100mlとする。
(d) デキストリン溶液 JIS K 8646に規定するデキストリン水和物2gを水に溶かし,100mlとする。こ
の溶液は使用時に調製する。
109
K 0101 : 1998
(e) 塩化物イオン標準液 (1mgCl-/ml) 32.1(1)(c)による。
(f) 28.2mmol/l硝酸銀溶液 JIS K 8550に規定する硝酸銀4.8gを水に溶かして1lとする。着色ガラス瓶
に保存する。
標定 塩化物イオン標準液 (1mgCl-/ml) 20mlをビーカーにとり,水を加えて液量を約50mlとし,
これにデキストリン溶液5mlとフルオレセインナトリウム溶液 (2g/l) 1,2滴を加え,静かにかき混
ぜながらこの硝酸銀溶液で滴定する。黄緑の蛍光が消失してわずかに赤くなったときを終点とする。
ここに要したこの硝酸銀溶液のml数 (x) から,次の式によって28.2mmol/l硝酸銀溶液のファクタ
ー (f) を算出する。
x
f
20
=
(2) 操作 操作は,次のとおり行う。
(a) 試料に濁りがある場合には,ろ紙5種Cでろ過し(3),初めのろ液約50mlを捨て,次のろ液50ml(Cl-
20mg以上を含む場合には適量をとり,水を加えて50mlとする。)をビーカーにとる。
(b) 試料が酸性の場合には,炭酸ナトリウム溶液 (50g/l) で,また,アルカリ性の場合には,硝酸 (1+
65) を用いてpHを約7に調節する。
(c) デキストリン溶液5ml及びフルオレセインナトリウム溶液 (2g/l) 1,2滴を加えてかき混ぜる。
(d) 静かにかき混ぜながら28.2mmol/l硝酸銀溶液で滴定する。黄緑の蛍光が消失してわずかに赤くなっ
たときを終点とする。
(e) 次の式によって試料中の塩化物イオンの濃度 (mgCl-/l) を算出する。
1
000
1
×
×
×
=
V
f
a
C
ここに, C: 塩化物イオン (mgCl-/l)
a: 滴定に要した28.2mmol/l硝酸銀溶液 (ml)
f: 28.2mmol/l硝酸銀溶液のファクター
V: 試料 (ml)
1: 28.2mmol/l硝酸銀溶液1mlの塩化物イオン相当量 (mg)
32.4 イオン電極法 試料に酢酸塩緩衝液を加えてpHを約5に調節し,塩化物イオン電極を指示電極とし
て電位を測定し,塩化物イオンを定量する。
備考6. この方法では硫化物イオンなどが妨害する。
定量範囲:Cl- 5〜1 000mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸塩緩衝液 (pH5) JIS K 8548に規定する硝酸カリウム100gとJIS K 8355に規定する酢酸
50mlを水500mlに加えて溶かし,これに水酸化ナトリウム溶液 (100g/l) を加え,pH計を用いてpH
を5に調節し,水を加えて1lとする。
(b) 塩化物イオン標準液 (1 000mgCl-/l) 32.1(1)(c)による。
(c) 塩化物イオン標準液 (100mgCl-/l) 塩化物イオン標準液 (1 000mgCl-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。
(d) 塩化物イオン標準液 (10mgCl-/l) 塩化物イオン標準液 (100mgCl-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(e) 塩化物イオン標準液 (5mgCl-/l) 塩化物イオン標準液 (100mgCl-/l) 10mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
110
K 0101 : 1998
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 31.2(2)(a)による。
(b) 指示電極 塩化物イオン電極
(c) 参照電極 31.2(2)(c)による。ただし,外筒液には硝酸カリウム溶液 (100g/l) を用いる。
(d) マグネチックスターラー 31.2(2)(d)による。
(3) 検量線の作成
(a) 塩化物イオン標準液 (5mgCl-/l) 100mlをビーカー200mlにとり,酢酸塩緩衝液 (pH5) 10ml(5)を加え
る。
(b) 指示電極(6)(7)と参照電極(8)(9)とを浸し,マグネチックスターラー(10)で泡が電極に触れない程度に強
くかき混ぜる(11)。
(c) 液温をはかり,電位差計で電位を測定する(12)。
(d) 塩化物イオン標準液 (10mgCl-/l) 100ml,塩化物イオン標準液 (100mgCl-/l) 100ml及び塩化物イオン
標準液 (1 000mgCl-/l) 100mlをそれぞれビーカー200mlにとり,酢酸塩緩衝液 (pH5) 10ml(5)を加える。
それぞれの塩化物イオン標準液 (10〜1 000mgCl-/l) の液温を(c)の液温の±1℃に調節し,(b)及び(c)
の操作を行って塩化物イオン標準液の電位を測定する。
(e) 片対数紙の対数軸に塩化物イオンの濃度を,均等軸に電位(13)をとり,塩化物イオンの濃度 (mgCl-/l)
と電位との関係線を作成する。
注(5) 酢酸塩緩衝液 (pH5) は,測定時においてpHを約5に調節し,イオン強度を一定にするためのも
のである。
(6) 塩化物イオン標準液 (5mgCl-/l) に浸し,指示値が安定してから電位を測定する。
(7) 31.の注(12)による。
(8) 31.の注(13)による。
(9) 参照電極の内筒液には塩化カリウム溶液(3mol/l〜飽和),外筒液には硝酸カリウム溶液 (100g/l)
を用いる。内筒液に塩化カリウム溶液(飽和)を用いる場合には,液温の低下で塩化カリウム
の結晶が析出し,固着して電気抵抗が大きくなることがあるので注意する。
外筒液の硝酸カリウム溶液 (100g/l) に内筒液の塩化カリウム溶液が混入してくるので,外筒
液も定期的に取り替える。
(10) 31.の注(15)による。
(11) 31.の注(16)による。
(12) 塩化物イオン電極の応答時間は,液温10〜30℃の場合は,塩化物イオンの濃度が5mgCl-/l以上
ならば1分間以内である。
(13) 塩化物イオン標準液 (10mgCl-/l) と塩化物イオン標準液 (1 000mgCl-/l) との電位の差は,110〜
120mV (25℃) の範囲に入り,塩化物イオンの濃度5〜1 000mgCl-/lの間の検量線は直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 試料100ml (14)(15) をビーカー200mlにとり,酢酸塩緩衝液 (pH 5) 10mlを加え,液温を(3)(c)の液温
の±1℃に調節する。
(b) (3)(b)及び(c)の操作を行って検量線から塩化物イオンの濃度を求め,試料中の塩化物イオンの濃度
(mgCl-/l) を算出する。
注(14) 試料が酸性の場合には,水酸化ナトリウム溶液 (40g/l) で,また,アルカリ性の場合には,酢
酸 (1+10) で,あらかじめpHを約5に調節する。
111
K 0101 : 1998
(15) 試料に硫化物イオンが含まれている場合には,あらかじめ,酢酸亜鉛溶液 (100g/l) を加えて硫
化物イオンを固定してろ紙でろ過し,ろ液のpHを約5に調節する。
備考7. イオン濃度計の場合には,塩化物イオン標準液 (10mgCl-/l) と塩化物イオン標準液 (1
000mgCl-/l) とを用い,(3)(b)〜(c)の操作を行ってイオン濃度計の指示値を10mgCl-/lと1
000mgCl-/lになるように調節する。さらに,その他の塩化物イオン標準液 (5mgCl-/l) と塩化
物イオン標準液 (100mgCl-/l) を用いて,イオン濃度計の指示を確認する。
8. 主な共存物質の許容限度を最大比率で次に示す。
NO3-, SO42-, PO43-:104
F-:102
Br-:10-2
I-, CN-, S2-:10-3
9. イオン電極による電位差滴定法 試料100mlをビーカーにとり,試料のpHを7に調節し,
塩化物イオン電極又は銀イオン電極を用い,(3)(b)の操作に準じて電位を測定しながら10〜
100mmol/l硝酸銀溶液で滴定し,滴定曲線を作図して,滴定終点を求める。滴定曲線の変曲
点は,よう化物イオン,臭化物イオン及び塩化物イオンの順になる。それぞれの変曲点から
終点を求め,各イオンの濃度を算出する。
10mmol/l硝酸銀溶液1mlはI- 1.269mg,Br- 0.799mg及びCl- 0.354 5mgに相当する。
32.5 イオンクロマトグラフ法 試料中の塩化物イオンをイオンクロマトグラフ法によって定量する。
定量範囲: Cl- 0.1〜25mg/l (16),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異
なる。)
注(16) サプレッサと組み合わせる方式の場合には,Cl- 0.05〜25mg/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 溶離液(17)は,装置の種類及び分離カラムに充てんした陰イオン交換体の種類によって異な
るので,あらかじめ塩化物イオン,亜硝酸イオン,臭化物イオン,硝酸イオン及び硫酸イオンの分
離を注(21)の操作で確認する。
(c) 再生液 再生液(18)は,サプレッサを用いる場合に使用するが,装置の種類及びサプレッサの種類に
よって異なる。あらかじめ分離カラムと組み合わせて注(21)の操作を行って再生液の性能を確認する。
(d) 塩化物イオン標準液 (1mgCl-/ml) 32.1(1)(c)による。
(e) 塩化物イオン標準液 (0.1mgCl-/ml) 塩化物イオン標準液 (1mgCl-/ml) 10mlを全量フラスコ100ml
にとり,水を標線まで加える。
(f) 陰イオン混合標準液 [ (0.1mgCl-, 0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO4-2) /ml] 32.1(1)(c)の塩
化物イオン標準液 (1mgCl-/ml) 10ml,37.1.2(1)(d)の亜硝酸イオン標準液 (5mgNO2-/ml) 10ml,
34.2(1)(d)の臭化物イオン標準液 (5mgBr-/ml) 10ml,37.2.5(1)(d)の硝酸イオン標準液 (5mgNO3-/ml)
10ml及び42.4(1)(d)の硫酸イオン標準液 (10mgSO42-/ml) 10mlをそれぞれ全量フラスコ100mlにとり,
水を標線まで加える。使用時に調製する。
注(17) 溶離液の調製方法の一例を,次に示す。
サプレッサを用いる場合の例
[炭酸水素ナトリウム溶液 (1.7mmol/l) -炭酸ナトリウム溶液 (1.8mmol/l)] JIS K 8622に規定
する炭酸水素ナトリウム0.143gと,JIS K 8625に規定する炭酸ナトリウム0.191gとをとり,水
112
K 0101 : 1998
に溶かして1lとする。
[炭酸水素ナトリウム溶液 (0.3mmol/l) -炭酸ナトリウム溶液 (2.7mmol/l)] JIS K 8622に規定
する炭酸水素ナトりウム0.025gとJIS K 8625に規定する炭酸ナトリウム0.286gとをとり,水
に溶かして1lとする。
炭酸ナトリウム溶液 (3mmol/l) JIS K 8625に規定する炭酸ナトリウム0.318gをとり,水に溶
かして1lとする。
サプレッサを用いない場合の例
[グルコン酸カリウム溶液 (1.3mmol/l) -四ほう酸ナトリウム溶液 (1.3mmol/l) -ほう酸溶液
(30mmol/l) -アセトニトリル溶液 (100g/l) -グリセリン溶液 (5g/l) ] グルコン酸カリウム0.31g
とJIS K 8866に規定する四ほう酸ナトリウム十水和物0.5gと,JIS K 8863に規定するほう酸
1.86gと,JIS K 8032に規定するアセトニトリル100g (128ml) と,JIS K 8295に規定するグリ
セリン5g (4ml) とを水に溶かして1lとする。
[フタル酸溶液 (2.5mmol/l) -2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール溶液
(2.4mmol/l)] フタル酸0.415gと,JIS K 9704に規定する2-アミノ-2-ヒドロキシメチル−1, 3-
プロパンジオール[トリス(ヒドロキシメチル)アミノメタン]0.291gとを水に溶かして1l
とする。
(18) 再生液の調製方法の例を,次に示す。
硫酸 (12.5mmol/l) 硫酸 (0.5mol/l) (JIS K 8951に規定する硫酸30mlを少量ずつ水500ml中
に加え,冷却した後,水で1lとする。)25mlを水で1lとする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(2.1) イオンクロマトグラフ イオンクロマトグラフには,分離カラムとサプレッサ(19)を組み合わせた方
式のもの,分離カラム単独の方式のものいずれでもよいが,次に掲げる条件を満たすもので,塩化
物イオン,亜硝酸イオン,臭化物イオン,硝酸イオン,硫酸イオンなどが分離定量できるもの。
(a) 分離カラム ステンレス鋼製又は合成樹脂製(20)のものに,強塩基性陰イオン交換体(表層被覆形又
は全多孔性シリカ形など)を充てんしたもの(21)。
(b) 検出器 電気伝導率検出器
(2.2) 記録部 JIS K 0127の4.2(6)による。
注(19) 溶離液中の陽イオンを水素イオンに変換するためのもので,溶離液中の陽イオンの濃度に対し
て十分なイオン交換容量をもつ陽イオン交換膜(膜形,電気透析形がある。)又は同様な性能を
もった陽イオン交換体を充てんしたもの。再生液と組み合わせて用いる。ただし,電気透析形
の場合は,再生液として検出器からの流出液(検出器から排出される溶液)を用いる。
(20) 例えば,四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などがある。
(21) 溶離液を一定の流量(例えば,1〜2ml/min)で流し,陰イオン混合標準液 [ (10μgCl-, 10μgNO2-,
10μgBr-, 10μgNO3-, 10μgSO42-)/ml ] の一定量をイオンクロマトグラフの分離カラムに注入し,
クロマトグラムを求め,それぞれの陰イオンが分離(分離度1.3以上)できるものを用いる。
また,定期的に分離カラムの性能を確認するとよい。
陰イオン混合標準液 [ (10μgCl-, 10μgNO2-, 10μgBr-, 10μgNO3-, 10μgSO42-)/ml ] は,次のよう
に調製する。
塩化物イオン標準液 (1mgCl-/ml) ,亜硝酸イオン標準液 (1mgNO2-/ml) ,臭化物イオン標準
液 (1mgBr-/ml) ,硝酸イオン標準液 (1mgNO3-/ml) 及び硫酸イオン標準液 (1mgSO42-/ml) をそれ
113
K 0101 : 1998
ぞれ5mlずつ全量フラスコ500mlにとり,水を標線まで加える。
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を孔径0.45μmのろ過膜又はろ紙5種C(ろ紙6種)でろ過し,初めのろ液約50mlを捨て,そ
の後のろ液をとる。
(b) 試料の電気伝導率が10mS/m {100μS/cm} (25℃) 以上の場合には,電気伝導率が10mS/m以下になる
ように,水で一定の割合に薄める。
(4) 操作 操作は,次のとおり行う。
(a) イオンクロマトグラフを作動できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜
2ml/min)で流しておく。サプレッサを必要とする装置では再生液を一定の流量で流しておく。
(b) (3)の準備操作を行った試料の一定量(例えば,50〜200μlの一定量)をイオンクロマトグラフに注
入してクロマトグラムを記録する。
(c) クロマトグラム上の塩化物イオンに相当するピークについて,指示値(22)を読み取る。
(d) 試料を薄めた場合には,空試験として試料と同量の水について,(a)〜(c)の操作を行って試料につい
て得た指示値(22)を補正する。
(e) 検量線から塩化物イオンの量を求め,試料中の塩化物イオンの濃度 (mgCl-/l) を算出する。
検量線 塩化物イオン標準液 (0.1mgCl-/ml) (23)0.1〜25mlを段階的に全量フラスコ100mlにとり,水
を標線まで加える。この溶液について(a)〜(c)の操作を行ってそれぞれの塩化物イオンに相当するピ
ークについて,指示値(22)を読み取る。別に,空試験として水について(a)〜(c)の操作を行ってそれ
ぞれの塩化物イオンに相当する指示値を補正した後,塩化物イオン (Cl-) の量と指示値との関係線
を作成する。検量線の作成は,試料の測定時に行う。
注(22) ピーク高さ又はピーク面積
(23) 塩化物イオン以外の陰イオンを同時に試験する場合には,陰イオン混合標準液 [(0.1mgCl-,
0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] を用いる。
備考10. 塩化物イオンの濃度が1mg/lのとき亜硝酸イオンは200mg/l以下ならば妨害しない。
11. 分離カラムは,使用を続けると性能が低下するので,定期的に注(21)の操作を行って確認する。
性能が低下した場合,溶離液の約10倍の濃度のものを調製し,分離カラムに注入し洗浄し
た後,注(21)の操作で確認し,性能が回復しない場合には,新品と取り替える。
試料中の懸濁物及び有機物(たん白質,油類,界面活性剤など。)などによって汚染され性
能が徐々に低下するので,懸濁物を含む試料は(3)の準備操作で除去した後に試験する。
また,有機物を含む試料は限外ろ過膜でろ過し,有機物をできるだけ除去した後,試験す
る。
試料中に分離カラムの充てん剤と親和力の強い陰イオン(例えば,よう化物イオン,クロ
ム酸イオンなど)が存在すると,これらが充てん剤に吸着され,分離性能が徐々に低下する
ので,溶離液の5〜10倍の濃度のものを調製し,試料と同様に分離カラムに注入し洗浄する。
そのほか酸化性物質,還元性物質が共存すると,分離カラムの分離性能を低下させる。こ
のような場合には,試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐこと
ができる。
33. よう化物イオン (I-) よう化物イオンの定量には,よう素抽出吸光光度法又はよう素滴定法を適用す
る。
114
K 0101 : 1998
33.1 よう素抽出吸光光度法 よう化物イオンを硫酸酸性で亜硝酸イオンと反応させ,遊離したよう素を
クロロホルムで抽出し,その吸光度を測定してよう化物イオンを定量する。
定量範囲:I- 0.1〜5mg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+1) 4.4(1)(b)による。
(b) 亜硝酸ナトリウム JIS K 8019に規定するもの。
(c) 尿素溶液 (10g/l) JIS K 8731に規定する尿素1gをとり,水に溶かして100mlとする。
(d) 硫酸ナトリウム JIS K 8987に規定するもの。
(e) クロロホルム JIS K 8322に規定するもの。
(f) よう化物イオン標準液 (1mgI-/ml) JIS K 8913に規定するよう化カリウム1.31gをとり,少量の水
に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加える。
(g) よう化物イオン標準液 (0.1mgI-/ml) よう化物イオン標準液 (1mgI-/ml) 20mlを全量フラスコ
200mlにとり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 100ml
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(1)(2)(I-として0.1〜5mgを含む。)を分液漏斗にとり,水を加えて約50mlとする。
(b) 硫酸 (1+1) [試料がアルカリ性の場合には,硫酸 (1+1) を加えて中和しておく。]1mlと亜硝酸
ナトリウム0.5gを加えて振り混ぜる。
(c) クロロホルム10mlを加え,約2分間激しく振り混ぜた後放置する。
(d) クロロホルム層を別の分液漏斗に移す。再び水層にクロロホルム10mlを加えて抽出し,クロロホ
ルム層は先のクロロホルム層に合わせる。
(e) クロロホルムを入れた分液漏斗に尿素溶液 (10g/l) 50mlを加え,約2分間激しく振り混ぜてクロロ
ホルム層を洗浄する。
(f) 約5分間放置した後,クロロホルム層を硫酸ナトリウム約1gを入れた共栓三角フラスコ30mlに移
し,振り混ぜて脱水する。
(g) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長515nm付近の吸光度を
測定する。
(h) 空試験として水50mlをとり,(b)〜(g)の操作を行って試料について得た吸光度を補正する。
(i) 検量線からよう化物イオンの量を求め,試料中のよう化物イオンの濃度 (mgI-/l) を算出する。
検量線 よう化物イオン標準液 (0.1mgI-/ml) 1〜50mlを段階的に分液漏斗にとり,(a)〜(h)の操作を
行って吸光度を測定し,よう化物イオン (I-) の量と吸光度との関係線を作成する。
注(1) よう化物イオンの濃度が2mgI-/l以下の場合には,試料の適量をとり,水酸化ナトリウム溶液
(200g/l) を加えてアルカリ性として,静かに加熱して濃縮する。濃縮中に濁りが生じた場合に
は,ろ過し,(a)以降の操作を行う。
(2) 有機物が多量に共存する場合には,試料200mlをとり硫酸カリウムアルミニウム溶液(JIS K
8255に規定する硫酸カリウムアルミニウム・12水5gを水に溶かして100mlとする。)2〜3ml
を加えた後,水酸化ナトリウム溶液 (50g/l) を水酸化アルミニウムの沈殿が生成するまで加え
る。約5分間放置した後ろ過し,そのろ液の適量をとり,(a)以降の操作を行う。
115
K 0101 : 1998
備考1. よう素酸イオンが含まれると,硫酸酸性としたとき,よう化物イオンと反応してよう素を生
じるため,よう素酸イオンの一部又は全部がよう化物イオンとして定量される。臭化物イオ
ンは妨害しない。
33.2 よう素滴定法 よう化物イオンをpH 1.3〜2.0で次亜塩素酸で酸化し,よう素酸イオンとする。過剰
の次亜塩素酸を,pH 3〜7でぎ酸ナトリウムで分解した後,よう化カリウムを加え,遊離するよう素をチ
オ硫酸ナトリウム溶液で滴定してよう化物イオンを定量する。
定量範囲:I- 0.1mg以上
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(c) 次亜塩素酸ナトリウム溶液(有効塩素35g/l) 次亜塩素酸ナトリウム溶液(有効塩素7〜12%)の
有効塩素を定量し(3),有効塩素が約35g/lになるように水で薄める。使用時に調製する。
(d) ぎ酸ナトリウム溶液 (400g/l) JIS K 8267に規定するぎ酸ナトリウム40gを水に溶かして100mlと
する。
(e) よう化カリウム JIS K 8913に規定するもの。
(f) メチルオレンジ溶液 (1g/l) 22.1.1(1)(d)による。
(g) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(h) 10mmol/lチオ硫酸ナトリウム溶液 28.3(1)(e)による。
注(3) 次亜塩素酸ナトリウム溶液(有効塩素7〜12%)10mlを全量フラスコ200mlにとり,水を標線ま
で加える。この10mlを共栓三角フラスコ300mlにとり,水を加えて約100mlとする。よう化カ
リウム1, 2g及び酢酸 (1+1) 6mlを加えて密栓し,よく振り混ぜて暗所に約5分間放置した後,
50mmol/lチオ硫酸ナトリウム溶液[24.1(1)(g)による。]で滴定する。溶液の黄色が薄くなった
ら,指示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるま
で滴定する。別に,空試験として水10mlをとり,同じ操作を行って滴定値を補正する。次の式
によって有効塩素量を算出する。
773
001
.0
000
1
10
200
×
×
×
×
=
V
f
a
N
ここに,
N: 有効塩素量 (g/l)
a: 滴定に要した50mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 50mmol/lチオ硫酸ナトリウム溶液のファクター
0.001 773: 50mmol/lチオ硫酸ナトリウム溶液1mlの有効塩素相当
量 (g)
V: 次亜塩素酸ナトリウム溶液(有効塩素7〜12%) (ml)
(2) 操作 操作は,次のとおり行う。
(a) 試料の適量(1)(2)(I-として0.1〜5mgを含む。)を共栓三角フラスコ300mlにとり,指示薬としてメ
チルオレンジ溶液 (1g/l) 1滴を加え,溶液の色が微赤になるまで塩酸 (1+11) を滴加した後,水を
加えて約50mlとする。
(b) 次亜塩素酸ナトリウム溶液(有効塩素35g/l)1mlを加え,塩酸 (1+11) を加えてpHを1.3〜2.0に
調節し,沸騰水浴中に約5分間浸す。
(c) ぎ酸ナトリウム溶液 (400g/l) 5mlを加え(4),再び沸騰水浴中に約5分間浸して過剰の次亜塩素酸を
分解する。
116
K 0101 : 1998
(d) 放冷後,よう化カリウム1gと塩酸 (1+1) 6mlとを加え,密栓して振り混ぜ,暗所に約5分間放置
する。
(e) 遊離したよう素を10mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指示
薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(f) 空試験として水50mlを共栓三角フラスコ300mlにとり,(a)〜(e)の操作を行う。
(g) 次の式によって試料中のよう化物イオンの濃度 (mgI-/l) を算出する。
5
211
.0
000
1
)
(
×
×
×
−
=
V
f
b
a
C
ここに,
C: よう化物イオン (mgI-/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター(5)
V: 試料 (ml)
0.211 5: 10mmol/lチオ硫酸ナトリウム溶液1mlのよう化物イオン
相当量 (mg)
注(4) ぎ酸ナトリウム溶液 (400g/l) による次亜塩素酸の分解はpH 3〜7で行う。pH 2.7以下になると,
よう素酸イオンが還元され,pH7以上になると次亜塩素酸の分解が不完全になる。
(5) 0.1mol/lチオ硫酸ナトリウム溶液のファクターを用いる。
備考2. この方法では鉄 (II,III) は妨害する。マンガン0.2mg以上,ひ酸イオン1mg以上は妨害する。
硫化水素,多量の有機物も妨害する。妨害の除去は次による。
(1) 鉄,マンガン 試料に水酸化ナトリウム溶液 (200g/l) を加えてアルカリ性とし,約1時間放
置した後,ろ過する。ろ液と洗液とを合わせて30〜50mlになるまで沸騰水浴上で濃縮する。
濁り及び沈殿物があるときは,ろ過して洗浄する。ろ液と洗液とを合わせ,(2)(a)以降の操作
を行う。
(2) ひ酸イオン 試料500mlにつき塩化鉄 (III) 溶液[JIS K 8142に規定する塩化鉄 (III) 六水和
物5gを塩酸 (1+1) 10mlに溶かし,水で100mlとする。]1mlを加え,以下,(1)と同様に操
作する。ただし,水酸化ナトリウム溶液 (200g/l) に代え,アンモニア水 (1+1) を用いる。
(3) 硫化水素 試料500mlにつき硫酸亜鉛溶液(JIS K 8953に規定する硫酸亜鉛七水和物10gを
水に溶かして100mlとする。)2〜3mlを加え,よくかき混ぜ,以下,(1)と同様に操作する。
3. この方法では,よう素酸イオンもよう化物イオンとして定量される。
34. 臭化物イオン (Br-) 臭化物イオンの定量には,よう素滴定法又はイオンクロマトグラフ法を適用す
る。
34.1 よう素滴定法 臭化物イオンをpH 6.5〜8.0で次亜塩素酸で酸化し,臭素酸イオンとする。過剰の次
亜塩素酸をpH 3〜7でぎ酸ナトリウムで分解した後,よう化カリウムを加えて遊離するよう素をチオ硫酸
ナトリウム溶液で滴定して臭化物イオンを定量する。よう化物イオンも同じ反応をするので,別に定量し
て差し引く。
定量範囲:Br- 0.1mg以上
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
117
K 0101 : 1998
(c) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(d) りん酸二水素ナトリウム溶液 (500g/l) JIS K 9009に規定するりん酸二水素ナトリウム二水和物
65gを水に溶かし100mlとする。
(e) 次亜塩素酸ナトリウム溶液(有効塩素35g/l) 33.2.(1)(c)による。
(f) ぎ酸ナトリウム (400g/l) 33.2.(1)(d)による。
(g) よう化カリウム JIS K 8913に規定するもの。
(h) メチルオレンジ溶液 (1g/l) 22.1.1(1)(d)による。
(i) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(j) 10mmol/lチオ硫酸ナトリウム溶液 28.3(1)(e)による。
(2) 操作 操作は,次のとおり行う。
(a) 試料の適量(1)(Br-として0.1〜3mgを含む。)を共栓三角フラスコ300mlにとり,指示薬としてメチ
ルオレンジ溶液 (1g/l) 1滴を加え,溶液の色がわずかに赤くなるまで塩酸 (1+11) を滴加した後,
水を加えて約50mlとる。
(b) りん酸二水素ナトリウム溶液 (500g/l) 2mlと次亜塩素酸ナトリウム溶液(有効塩素35g/l)3mlを加
え,水酸化ナトリウム溶液 (40g/l) 又は塩酸 (1+11) を用いてpHを6.5〜8.0に調節した後,約10
分間煮沸する。
(c) ぎ酸ナトリウム溶液 (400g/l) 3ml(2)を加え約5分間煮沸し,過剰の次亜塩素酸を分解する。
(d) 放冷後,よう化カリウム1gと塩酸 (1+1) 6mlを加え,密栓して振り混ぜ,暗所に約5分間放置す
る。
(e) 遊離したよう素を10mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指示
薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(f) 空試験として水50mlを共栓三角フラスコ300mlにとり,(a)〜(e)の操作を行う。
(g) 別に,33.1又は33.2によって試料中のよう化物イオンの濃度 (mgI-/l) を定量する。
(h) 次の式によって試料中の臭化物イオンの濃度 (mgBr-/l) を算出する。
6
629
.0
2
133
.0
000
1
)
(
×
−
×
×
×
−
=
C
V
f
b
a
B
ここに,
B: 臭化物イオン (mgBr-/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター(3)
0.133 2: 10mmol/lチオ硫酸ナトリウム溶液1mlの臭化物イオン相
当量 (mg)
V: 試料 (ml)
C: よう化物イオンの濃度 (mgI-/l)
0.629 6: よう化物イオンの量を臭化物イオン相当量に換算する場
合の係数
90
.
126
904
.
79
注(1) 臭化物イオンの濃度が2mg/l以下の場合には,33.の注(1)と同様に操作する。
(2) ぎ酸ナトリウム溶液 (400g/l) による次亜塩素酸の分解は,pH 3〜7で行う。pH 2.7以下になる
と,臭素酸イオンが還元される。
また,pH 7以上になると次亜塩素酸の分解が不完全になる。
(3) 0.1mol/lチオ硫酸ナトリウム溶液のファクターを用いる。
118
K 0101 : 1998
備考1. この方法では鉄 (II,III) は妨害する。マンガン0.2mg以上,ひ酸イオンは1mg以上共存する
と妨害する。硫化水素及び多量の有機物も妨害する。妨害の除去は33.2の備考2.による。
34.2 イオンクロマトグラフ法 試料中の臭化物イオンをイオンクロマトグラフ法によって定量する。
定量範囲: Br- 0.5〜40mg/ (4),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異な
る。)
注(4) サプレッサと組み合わせる方式の場合には,0.1〜40mg/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 32.5(1)(b)による。
(c) 再生液 32.5(1)(c)による。
(d) 臭化物イオン標準液 (5mgBr-/ml) JIS K 8506に規定する臭化カリウムを105℃で約4時間加熱し,
デシケーター中で放冷する。その0.745gをとり,少量の水に溶かし,全量フラスコ100mlに移し入
れ,水を標線まで加える。
(e) 臭化物イオン標準液 (0.5mgBr-/ml) 臭化物イオン標準液 (5mgBr-/ml) 10mlを全量フラスコ100ml
にとり,水を標線まで加える。この溶液は使用時に調製する。
(f) 陰イオン混合標準液 [(0.1mgCl-, 0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] 32.5(1)(f)によ
る。
(2) 器具及び装置 器具及び装置は32.5(2)による。ただし,紫外吸光検出器を用いてもよい。
(3) 準備操作 準備操作は,32.5(3)による。
(4) 操作 操作は次による。
(a) 32.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上の臭化物イオンに相当するピークについて,指示値(5)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
いて得た指示値(5)を補正する。
(d) 検量線から臭化物イオンの量を求め,試料中の臭化物イオンの濃度 (mgBr-/l) を算出する。
検量線 臭化物イオン標準液 (0.5mgBr-/ml) (6)0.1〜8mlを段階的に全量フラスコ100mlにとり,水
を標線まで加える。この溶液について(a)及び(b)の操作を行ってそれぞれの臭化物イオンに相当する
指示値(5)を読み取る。別に,空試験として水について(a)及び(b)の操作を行ってそれぞれの臭化物イ
オンに相当する指示値を補正した後,臭化物イオン (Br-) の量と指示値(5)との関係線を作成する。
検量線の作成は,試料の測定時に行う。
注(5) ピーク高さ又はピーク面積
(6) 臭化物イオン以外の陰イオンを同時に試験する場合には陰イオン混合標準液 [(0.1mgCl-,
0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] を用いる。
備考2. 臭化物イオンの濃度が1mg/lのとき亜硝酸イオンは200mg/l以下ならば妨害しない。
3. 32.の備考11.による。
35. シアン化合物 シアン化合物は,水中のシアン化物イオンやシアノ錯体などを総称し,シアン化物イ
オンと全シアンに区分する。
シアン化合物は前処理でシアン化物イオンとし,定量には,4-ピリジンカルボン酸-ピラゾロン吸光光度
法又はイオン電極法を適用する。
119
K 0101 : 1998
シアン化合物は変化しやすいから,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によ
って保存し,できるだけ早く試験する。
35.1 前処理 試料を微酸性として通気又は加熱蒸留し,発生するシアン化水素を捕集する。
35.1.1 シアン化物 この前処理ではシアン化物イオン及び生成定数の小さい亜鉛,カドミウムのシアノ錯
体からはほぼ完全に,また,ニッケル,銅などのシアノ錯体からは一部シアン化水素を発生する。鉄 (II),
鉄 (III) のシアノ錯体からはシアン化水素を発生しない。
35.1.1.1 通気法(pH 5.0で発生するシアン化水素) 試料のpHを5.0に調節し,恒温水槽で40℃に保持
しながら,約1.2l/minで通気し,発生したシアン化水素を水酸化ナトリウム溶液に捕集する。
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸 (1+1) JIS K 8355に規定する酢酸を用いて調製する。
(b) 酢酸 (1+49) JIS K 8355に規定する酢酸を用いて調製する。
(c) 水酸化ナトリウム溶液 (200g/l) JIS K 8576に規定する水酸化ナトリウム20gを水に溶かして
100mlとする。
(d) 水酸化ナトリウム溶液 (20g/l) (c)の水酸化ナトリウム溶液 (200g/l) を水で10倍に薄める。
(2) 装置 装置は,次のとおりとする。
(a) 通気装置 図35.1に一例を示す。
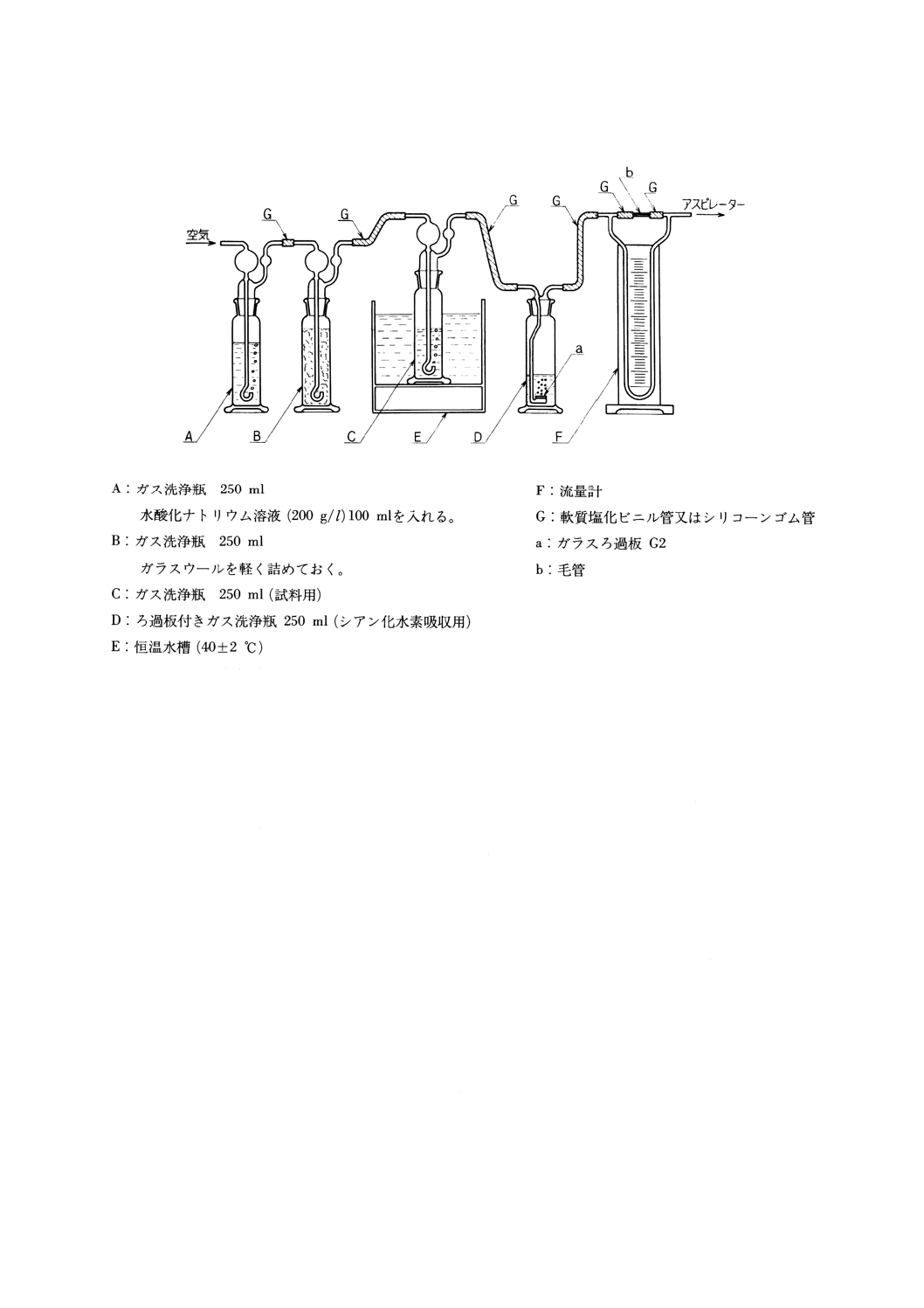
120
K 0101 : 1998
図35.1 通気装置の一例
(3) 通気操作 通気操作は,次のとおり行う。
(a) 通気装置を図35.1のように組み立て,ろ過板付きガラス洗浄瓶 (D) には,シアン化水素吸収用と
して水40mlと水酸化ナトリウム溶液 (20g/l) 20mlを入れる。
(b) あらかじめ試料100mlをビーカー300mlにとり,pH計を用いてpH 5.0±0.2になるまで酢酸 (1+1)
及び酢酸 (1+49) 又は水酸化ナトリウム溶液 (20g/l) を滴加し,その量を求める。
(c) 試料100ml(1)(2)をガス洗浄瓶 (C) に入れ,(b)の操作で求めた酢酸 (1+1) 及び酢酸 (1+49) 又は水
酸化ナトリウム溶液 (20g/l) の量を加え,図35.1のように連結する。
(d) 恒温水槽を40±2℃に保持して,約1.2l/minで1時間通気する。
(e) 通気後ろ過板付きガス洗浄瓶 (D) の中の水酸化ナトリウム溶液(吸収液)を全量フラスコ100ml
に移し入れ,ろ過板付きガス洗浄瓶 (D) を水で洗い,洗液も移し入れて水を標線まで加える。
注(1) 試料の採取量は,35.2〜35.3のそれぞれの方法に記載した定量範囲から求めた最適量。
(2) 試料中に,油脂類,残留塩素などの酸化性物質,硫化物などの還元性物質が含まれている場合
には,あらかじめ備考1.〜3.に示す方法によって除去する。
備考1. 試料中に多量の油脂類が含まれている場合には,あらかじめ酢酸又は水酸化ナトリウムを加
えてpHを6〜7に調節し,分液漏斗に移し入れる。試料の約2vol%量のヘキサン又はクロロホ
ルムを加えて,静かに振り混ぜ,放置して油脂類を分離した後,35.1.1.1の操作を行う。
2. 試料中に残留塩素などの酸化性物質が含まれている場合には,L (+) −アスコルビン酸溶液
(100g/l) [JIS K 9502に規定するL (+) −アスコルビン酸10gを水に溶かして100mlとする。]
又は亜ひ酸ナトリウム溶液 (100g/l) (JIS K 8046に規定するメタ亜ひ酸ナトリウム10gを水
121
K 0101 : 1998
に溶かして100mlとする。)を加えて還元する。
3. 硫化物が含まれている場合には,あらかじめ酢酸亜鉛溶液 (100g/l) [35.1.1.2(1)(d)による。]
2mlを加える。酢酸亜鉛溶液 (100g/l) 1mlは,硫化物イオン約14mgに相当する。
35.1.1.2 加熱蒸留法(pH5.5で酢酸亜鉛の存在下で発生するシアン化水素) 試料に酢酸亜鉛を加え,pH
を5.5に調節して加熱蒸留し,発生するシアン化水素を水酸化ナトリウム溶液に捕集する。
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸 (1+1) 35.1.1.1(1)(a)による。
(b) 酢酸 (1+49) 35.1.1.1(1)(b)による。
(c) 水酸化ナトリウム溶液 (20g/l) 35.1.1.1(1)(d)による。
(d) 酢酸亜鉛溶液 (100g/l) JIS K 8356に規定する酢酸亜鉛二水和物12gを水に溶かして100mlとする。
(2) 装置 装置は,次のとおりとする。
(a) 蒸留装置 図35.2に一例を示す。
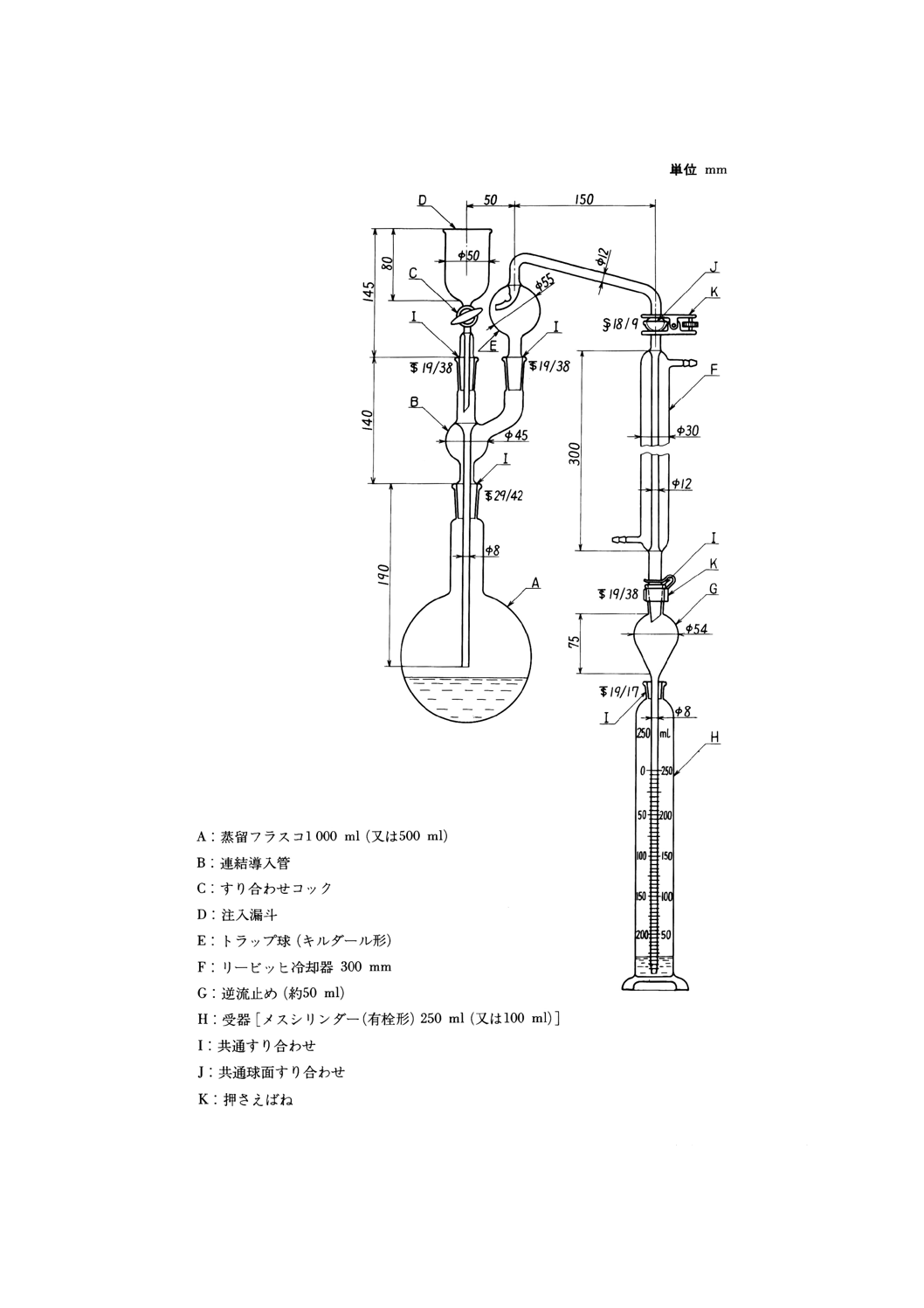
122
K 0101 : 1998
図35.2 蒸留装置の一例
(3) 蒸留操作 蒸留操作は,次のとおり行う。
(a) 試料が強アルカリ性の場合には,試料500ml(3)をビーカー1 000mlにとり,酢酸 (1+1) をpH計を
123
K 0101 : 1998
用いて滴加し,pHを約7とする。この中和に必要な添加量を求める。
(b) これに酢酸亜鉛溶液 (100g/l) 20mlを加え,再び酢酸 (1+49) をpH計を用いて滴加し,pHを5.5に
調節する。この酢酸 (1+49) の添加量を求める(4)。
(c) 試料500mlを蒸留フラスコ1 000mlにとり,沸騰石(5)(粒径2〜3mm)約10個を入れる。
(d) これに(a)で求めた量の酢酸 (1+1) を加え,蒸留フラスコを図35.2のように蒸留装置に接続する。
(e) 蒸留装置の受器には,メスシリンダー(有栓形)250mlを用い,これに水酸化ナトリウム溶液 (20g/l)
20mlを入れ,受器を図35.2のように接続する。
(f) 次に,注入漏斗から,酢酸亜鉛溶液 (100g/l) 20mlを加え,更に(b)で求めた酢酸 (1+49) を加える。
(g) 蒸留フラスコを加熱し,留出速度(6)を2〜3ml/minに調節し,受器の液量が約230mlになるまで蒸留
する(7)。
(h) 冷却器と逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器に
加え,更に水を250mlの標線まで加える。
注(3) 試料の採取量は,35.2〜35.3のそれぞれの方法に記載した定量範囲から求めた最適量。
(4) 各試薬の添加量は,できるだけ正確にする。
(5) 毛管の一端を封じたものでもよい。
(6) 留出速度は3ml/min以上にしない。シアン化水素の回収率が低下する。
(7) 逆流止めの先端は,常に液面下約15mmを保つように,メスシリンダー(有栓形)250mlの高
さを調節する。
備考4. 試料中に多量の油脂類が含まれている場合には,備考1.と同じ操作を行う。
5. 試料中に残留塩素などの酸化性物質が含まれている場合には,備考2.と同じ操作を行う。
6. 試料中に還元性物質が含まれている場合には,そのまま蒸留操作を行い,留出液に対して,
次のような酸化処理を行った後,再び蒸留操作を行って除去する。
35.1.1.2の蒸留操作を行った受器中の留出液と洗液とを再び蒸留フラスコに移し,指示薬と
してフェノールフタレイン溶液 (5g/l) [13.2(1)(a)による。]2,3滴を加えて酢酸 (1+1) で
中和し,更に硝酸 (50mmol/l) 約30mlを加える。次に,過マンガン酸カリウム溶液 (3g/l) を
滴加し,過マンガン酸の微赤になる点,又は酸化マンガン (IV) の褐色の濁りが生成した点
から更に過剰に1mlを加え,水を加えて約300mlとする。蒸留フラスコを図35.2のように蒸
留装置に接続し,受器にはメスシリンダー(有栓形)100mlを用い,これに水酸化ナトリウ
ム溶液 (20g/l) 20mlを入れ,受器を図35.2のように接続する。蒸留フラスコを加熱し,留出
速度を2〜3ml/mimに調節し,受器の液量が約90mlになったら蒸留を止め,冷却器と逆流止
めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器中に加えた
後,水を100mlの標線まで加える。
35.1.2 全シアン(pH 2以下で発生するシアン化水素) 試料にりん酸を加えてpHを2以下にし,エチレ
ンジアミン四酢酸二水素二ナトリウムを加えて加熱蒸留し,発生したシアン化水素を水酸化ナトリウム溶
液に捕集する。
備考7. 前処理によってシアン化物イオン,ほとんどのシアノ錯体中のシアンは留出する。酸化性物
質が共存する状態で蒸留すると,チオシアン酸,2-プロペンニトリル(アクリロニトリル)
などが分解してシアン化水素が発生するので,あらかじめ酸化性物質を還元しておく。
(1) 試薬 試薬は,次のものを用いる。
(a) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
124
K 0101 : 1998
(b) 水酸化ナトリウム溶液 (20g/l) 35.1.1.1(1)(d)による。
(c) アミド硫酸アンモニウム溶液 (100g/l) JIS K 8588に規定するアミド硫酸アンモニウム10gを水に
溶かして100mlとする。
(d) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物10gを水
に溶かして100mlとする。
(e) りん酸 JIS K 9005に規定するもの。
(2) 装置 装置は,次のとおりとする。
(a) 蒸留装置 図35.2に一例を示す。
(3) 蒸留操作 蒸留操作は,次のとおり行う。
(a) 試料の適量(3)を蒸留フラスコ500mlにとり,水を加えて約250mlとする。沸騰石(5)(粒径2〜3mm)
約10個を入れる。指示薬としてフェノールフタレイン溶液 (5g/l) 1滴を加える。
(b) アルカリ性の場合には,溶液の赤い色が消えるまで,りん酸を滴加する(8)。
(c) 次に,アミド硫酸アンモニウム溶液 (100g/l) 1ml(9)を加える。
(d) 蒸留フラスコを図35.2のように接続し,受器にはメスシリンダー(有栓形)100mlを用い,これに
水酸化ナトリウム溶液 (20g/l) 20mlを入れ,図35.2のように接続する。
(e) 注入漏斗から蒸留フラスコにりん酸10mlを加え,次にEDTA溶液10mlを加え,少量の水で注入漏
斗を洗い,洗液を蒸留フラスコに加える。
(f) 数分間放置した後,蒸留フラスコを加熱し,留出速度(6)2〜3ml/minで受器の液量が約90mlになる
まで蒸留する(7)。
(g) 冷却器と逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器に
加えた後,更に水を100mlの標線まで加える。
注(8) りん酸を加えて弱酸性になればよい。
(9) アミド硫酸アンモニウム溶液 (100g/l) の添加は,試料中の亜硝酸イオンの妨害を除くために加
える。これを加えない場合には,亜硝酸イオンが存在すると,加熱蒸留時にEDTAと反応して
シアン化水素を生成する。アミド硫酸アンモニウム溶液 (100g/l) 1mlは,亜硝酸イオン (NO2-)
約40mgに相当する。亜硝酸イオン40mg以上に共存する場合には,その量に応じて添加量を増
加する。
特殊な試料では亜硝酸イオン以外にもEDTAとの反応によってシアン化水素を生成し,アミ
ド硫酸アンモニウム溶液 (100g/l) の添加によってもその妨害を除けないものもある。
また,EDTA以外にも類似の反応をする有機物もある。
備考8. 油脂類の除去は,備考1.の操作を行う。
9. 残留塩素などの酸化性物質が含まれている場合には,備考2.の操作を行う。
10. 試料に硫化物などの還元物質が含まれている場合には,全シアンの蒸留操作を行って得た留
出液について備考6.の操作を行う。
35.2 4-ピリジンカルボン酸-ピラゾロン吸光光度法 前処理して得られたシアン化物イオン溶液の一部を
とり,酢酸で中和した後,クロラミンT溶液を加えて塩化シアンとし,これに4-ピリジンカルボン酸-ピ
ラゾロン溶液を加え,生成する青い色の吸光度を測定してシアン化物イオンを定量する。
定量範囲:CN- 0.5〜9μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸 (1+8) JIS K 8355に規定する酢酸を用いて調製する。
125
K 0101 : 1998
(b) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(c) りん酸塩緩衝液 (pH7.2) JIS K 9020に規定するりん酸水素二ナトリウム17.8gを水約300mlに溶
かし,りん酸二水素カリウム溶液 (200g/l) をpH7.2になるまで加え,水で500mlとする。
(d) クロラミンT溶液 (10g/l) JIS K 8318に規定するp-トルエンスルホンクロロアミドナトリウム三水
和物(クロラミンT)0.62gを水に溶かして50mlとする。この溶液は使用時に調製する。
(e) 4-ピリジンカルボン酸-ピラゾロン溶液 JIS K 9548に規定する3-メチル-1-フェニル-5-ピラゾロン
0.3gをJIS K 8500に規定するN, N-ジメチルホルムアミド20mlに溶かす。別に,4-ピリジンカルボ
ン酸1.5gを水酸化ナトリウム溶液 (40g/l) 約20mlに溶かし,塩酸 (1+10) を滴加してpHを約7
とする(10)。両液を合わせ,水を加えて100mlとする。この溶液は10℃以下の暗所に保存し,20日
間以上経過したものは使用しない。
(f) 0.1mmol/l硝酸銀溶液 JIS K 8550に規定する硝酸銀17gを水に溶かして1lとする。着色ガラス瓶
に移し入れ,保存する。
標定 JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを600℃で約1時間加熱し,デ
シケーター中で放冷する。NaCl100%に対してその1.169gをとり,少量の水に溶かして全量フラス
コ200mlに移し入れ,水を標線まで加える。この20mlをとり,水を加えて液量を約50mlとし,デ
キストリン溶液[32.3(1)(d)による。]5ml及び指示薬としてフルオレセインナトリウム溶液 (2g/l)
[32.3(1)(c)による。]3, 4滴を加え,0.1mol/l硝酸銀溶液で滴定し,黄緑の蛍光が消え,わずかに
赤くなるときを終点とする。次の式によってファクター (f) を算出する。
844
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 塩化ナトリウムの質量 (g)
b: 塩化ナトリウムの純度 (%)
x: 滴定に要した0.1mol/l硝酸銀溶液 (ml)
0.005 844: 0.1mol/l硝酸銀溶液1mlの塩化ナトリウム相当量 (g)
(g) シアン化物イオン標準液 (1mgCN-/ml) JIS K 8443に規定するシアン化カリウム0.63gを少量の水
に溶かし,水酸化ナトリウム溶液 (20g/l) 2.5mlを加え,水で250mlとする。この溶液は使用時に調
製し,その濃度は,次の方法で求める。
この溶液100mlをとり,指示薬としてp-ジメチルアミノベンジリデンロダニンのアセトン溶液
(0.2g/l) [JIS K 8495に規定するp-ジメチルアミノベンジリデンロダニン〔5-(4-ジメチルアミノベ
ンジリデン)-2-チオキソ-4-チアゾリジノン〕20mgをJIS K 8034に規定するアセトン100mlに溶か
す。]0.5mlを加え,0.1mol/l硝酸銀溶液で滴定し,溶液の色が黄色から赤になったときを終点とす
る。次の式によってシアン化物イオン標準液の濃度 (mgCN-/ml) を算出する。
100
1
204
.5
×
×
×
=
f
a
C
ここに,
C: シアン化物イオン標準液 (mgCN-/ml)
a: 滴定に要した0.1mol/l硝酸銀溶液 (ml)
f: 0.1mol/l硝酸銀溶液ファクター
5.204: 0.1mol/l硝酸銀溶液1mlのシアン化物イオン相当量 (mg)
(h) シアン化物イオン標準液 (1μgCN-/ml) シアン化物イオン標準液 (1mgCN-/ml) 10mlを全量フラス
コ1 000mlにとり,水酸化ナトリウム溶液 (20g/l) 100mlを加えた後,水を標線まで加える。その10ml
を全量フラスコ100mlにとり,水を標線まで加える。使用時に調製する。この溶液の濃度は,シア
126
K 0101 : 1998
ン化物イオン標準液 (1mgCN-/ml) の濃度から算出する。
注(10) この溶液に代え,4-ピリジンカルボン酸ナトリウム1.8gを水約50mlに溶かした溶液を用いても
よい。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 35.1で前処理して得られたシアン化物イオン溶液から適量(CN-として0.5〜9μgを含む。)を全量フ
ラスコ50mlにとる。
(b) 指示薬としてフェノールフタレイン溶液 (5g/l) を1滴加え,静かに振り混ぜながら酢酸 (1+8) を
滴加して中和した後,りん酸塩緩衝液 (pH7.2) 10ml(11)を加える。
(c) クロラミンT溶液 (10g/l) 0.5mlを加え,約25℃の水浴中に約5分間放置する。
(d) 4-ピリジンカルボン酸-ピラゾロン溶液10mlを加え,更に水を標線まで加え,密栓して静かに振り
混ぜた後,約25℃の水浴中で約30分間放置する。
(e) この一部を吸収セルに移し,波長638nm付近の吸光度を測定する。
(f) 空試験として水10mlを全量フラスコ50mlにとり,りん酸塩緩衝液 (pH 7.2) 10mlを加えた後,(c)
〜(e)の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
(g) 検量線からシアン化物イオンの量を求め,試料中のシアン化物イオンの濃度 (mgCN-/l) を算出する。
検量線 シアン化物イオン標準液(1μgCN-/ml)0.5〜9mlを全量フラスコ50mlに段階的にとり,水
を加えて約10mlとし,(b)〜(f)の操作を行ってシアン化物イオン (CN-) の量と吸光度との関係線を
作成する。
注(11) 発色時のpHは7〜8の範囲に入らなければならない。
35.3 イオン電極法 前処理して得られたシアン化物イオン溶液 (pH 12〜13) について,シアン化物イオ
ン電極を指示電極として電位を測定し,シアン化物イオンを定量する。
定量範囲:CN- 0.1〜100mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 水酸化ナトリウム溶液 (0.1mol/l) JIS K 8576に規定する水酸化ナトリウム4gを水に溶かして1l
とする。
(b) シアン化物イオン標準液 (100mgCN-/l) 35.2(1)(g)のシアン化物イオン標準液 (1mgCN-/ml) 20ml
を全量フラスコ200mlにとり,水酸化ナトリウム溶液 (0.1mol/l) を標線まで加える(12)。使用時に調
製する。この溶液の濃度は,シアン化物イオン標準液 (1mgCN-/ml) から算出する。
(c) シアン化物イオン標準液 (10mgCN-/l) シアン化物イオン標準液 (100mgCN-/l) 20mlを全量フラス
コ200mlにとり,水酸化ナトリウム溶液 (0.1mol/l) を標線まで加える(12)。使用時に調製する。この
溶液の濃度は,シアン化物イオン標準液 (100mgCN-/l) から算出する。
(d) シアン化物イオン標準液 (1mgCN-/l) シアン化物イオン標準液 (10mgCN-/l) 20mlを全量フラスコ
200mlにとり,水酸化ナトリウム溶液 (0.1mol/l) を標線まで加える(12)。使用時に調製する。この溶
液の濃度は,シアン化物イオン標準液 (10mgCN-/l) から算出する。
(e) シアン化物イオン標準液 (0.1mgCN-/l) シアン化物イオン標準液 (1mgCN-/l) 20mlを全量フラスコ
200mlにとり,水酸化ナトリウム溶液 (0.1mol/l) を標線まで加える(12)。使用時に調製する。この溶
液の濃度は,シアン化物イオン標準液 (1mgCN-/l) から算出する。
注(12) 各シアン化物イオン標準液のpHは,約13になる。
127
K 0101 : 1998
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 31.2(2)(a)による。
(b) 指示電極 シアン化物イオン電極
(c) 参照電極 31.2(2)(c)による。
(d) マグネチックスターラー 31.2(2)(d)による。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) シアン化物イオン標準液 (0.1mgCN-/l) 100mlをビーカー200mlにとり,指示電極(13)(14)と参照電極
(15)(16)とを浸し,マグネチックスターラー(17)で泡が電極に触れない程度に強くかき混ぜる(18)。
(b) 液温をはかり,電位差計で電位を測定する(19)。
(c) シアン化物イオン標準液 (1mgCN-/l) 100ml,シアン化物イオン標準液 (10mgCN-/l) 100ml及びシア
ン化物イオン標準液 (100mgCN-/l) 100mlをそれぞれビーカー200mlにとり,以下,(a)の操作を行う。
(d) それぞれのシアン化物イオン標準液 (1〜100mgCN-/l) の液温を(b)の液温の±1℃に調節し,シアン
化物イオン標準液 (1〜100mgCN-/l) の電位を測定する。
(e) 片対数紙の対数軸にシアン化物イオンの濃度を,均等軸に電位をとり,シアン化物イオンの濃度
(mgCN-/l) と電位との関係線を作成する(20)。
注(13) シアン化物イオン電極は,使用時にシアン化物イオン標準液 (0.1mgCN-/l) に浸しておき,指示
値が安定してから電位を測定する。シアン化物イオン電極の応答時間は,液温10〜30℃の場合,
シアン化物イオンの濃度が0.1mg/lで約1分間,1mg/l以上であれば約30秒間である。
(14) 31.の注(12)による。
(15) 31.の注(13)による。
(16) 31.の注(14)による。
(17) 31.の注(15)による。
(18) 31.の注(16)による。
(19) シアン化物イオン電極には,よう化銀を用いたものが多いので,直射日光を受けると電位が大
幅に変動して,正の誤差を与える。室内照明の影響は少ない。
(20) シアン化物イオン標準液 (0.1mgCN-/l) とシアン化物イオン標準液 (10mgCN-/l) との電位の差
は,110〜120mV (25℃) の範囲に入り,シアン化物イオンの濃度0.1〜100mg/lの間の検量線は
直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 35.1の前処理で得られたシアン化物イオン溶液から100mlをビーカー200mlにとり,液温を(3)(b)
の液温の±1℃に調節し,(3)(a)の操作を行う。
(b) (3)(b)の操作を行って検量線からシアン化物イオンの濃度を求め,試料中のシアン化物イオンの濃度
(mgCN-/l) を算出する。
備考11. イオン濃度計の場合には,シアン化物イオン標準液 (0.1mgCN-/l) 及びシアン化物イオン標準
液 (10mgCN-/l) を用い,(3)(a)及び(b)の操作を行ってイオン濃度計の指示値を0.1mgCN-/lと
10mgCN-/lになるように調節する。更にその他のシアン化物イオン標準液 (1mgCN-/l) とシア
ン化物イオン標準液 (100mgCN-/l) を用いてイオン濃度計の指示を確認する。
12. 硫化物及びメルカプト酢酸(チオグリコール酸)の妨害は前処理で除く。亜硫酸イオンは,
シアン化物イオンの103倍以内であれば妨害しないので備考6.の酸化処理を省略することが
できる。ホルムアルデヒドは負の妨害を与える。
128
K 0101 : 1998
主な共存物質の許容限度を最大比率で次に示す。
Cl-, F-, NO3-, CrO42-, K+, Na+:104
Br-, SCN-, HCO3-, CO32-, SO32-, SO42-, PO43-:103
S2O32-, Ag+:10
I-:0.1
13. イオン電極による電位差滴定法 35.1で前処理して得られたシアン化物イオン溶液のうち
100mlをビーカーにとり,指示電極(シアン化物イオン電極又は銀イオン電極)を用い,(3)
の操作に準じて電位を測定しながら1〜100mmol/l硝酸銀溶液で滴定し,滴定曲線を作図する。
滴定曲線から滴定終点を求め,シアン化物イオンの濃度を算出する。100mmol/l硝酸銀溶液
1mlは,シアン化物イオン5.204mgに相当する。
36. アンモニウムイオン (NH4+) アンモニウムイオンの定量にはインドフェノール青吸光光度法,中和
滴定法,イオン電極法又はイオンクロマトグラフ法を適用する。
試料を凝集沈殿処理又は蒸留処理して妨害物質から分離した後,インドフェノール青吸光光度法又はイ
オン電極法によって定量する。ただし,アンモニウムイオンの濃度が高い場合には,中和滴定法又はイオ
ン電極法を適用する。
また,イオンクロマトグラフ法を適用する場合には,試料の前処理及び3.3の保存処理を行わず,試料
採取後,直ちに試験する。
アンモニウムイオンは変化しやすいから,試験は試料採取後直ちに行う。直ちに試験ができない場合に
は,3.3によって保存し,できるだけ早く試験する。
36.1 前処理 前処理には,凝集沈殿法又は蒸留法を適用する。
36.1.1 凝集沈殿法 硫酸亜鉛又は炭酸ナトリウムと水酸化ナトリウムによる凝集沈殿で濁り,色及び金属
元素を除去する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。試薬の調製及び操作にはこの水を用いる。
(b) 水酸化ナトリウム-炭酸ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム30gを水約60ml
に溶かす。別に,JIS K 8625に規定する炭酸ナトリウム25gを水約100mlに溶かし,両溶液を合わ
せて,水で200mlとする。
(c) 炭酸ナトリウム溶液 (250g/l) JIS K 8625に規定する炭酸ナトリウム25gを水に溶かして100ml
とする。
(d) 水酸化ナトリウム溶液 (250g/l) JIS K 8576に規定する水酸化ナトリウム25gを水に溶かして
100mlとする。
(e) 硫酸亜鉛溶液 JIS K 8953に規定する硫酸亜鉛七水和物10gを水に溶かして100mlとする。
(2) 器具 器具は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗浄する。
(3) 操作 操作は,次のとおり行う。
(3.1) 硫酸亜鉛による凝集沈殿 この方法は,カルシウム及びマグネシウムの比較的少ない試料に適用す
る。この方法で処理しても濁りや色が残る場合には,36.1.2で処理する。
(a) 試料100mlに硫酸亜鉛溶液1mlを加え,よくかき混ぜてから水酸化ナトリウム-炭酸ナトリウム溶液
を加えて(通常0.3〜0.5ml)pHを約10.5に調節し,再びよくかき混ぜ,しばらく放置する。
129
K 0101 : 1998
(b) 上澄み液を遠心分離するか又はろ過(1)し(初めのろ液約25mlは捨てる。),澄明な溶液とし,共栓
三角フラスコに移し入れ密栓する。
(3.2) 炭酸ナトリウムと水酸化ナトリウムによる凝集沈殿 この方法は,カルシウム及びマグネシウムを
比較的多量に含む試料に適用する。この方法で処理しても色や濁りが残る場合には,36.1.2で前処
理する。
(a) 試料[酸性の場合には,水酸化ナトリウム溶液 (250g/l) を用いてpHを約7まで中和する。]に炭酸
ナトリウム溶液 (250g/l) 1mlと水酸化ナトリウム溶液 (250g/l) 0.7mlとを加え(2)よくかき混ぜる。
(b) これをメスシリンダー(有栓形)200mlに移し入れ,水を200mlの標線まで加える。
(c) 栓をしてよく振り混ぜた後,冷暗所に2時間以上放置し,上澄み液をデカンテーションするか又は
ろ過(1)して(初めのろ液約25mlは捨てる。)澄明な溶液とし,密栓する。
注(1) ろ紙は5種Aを用い,使用前に水でよく洗ってから使用する。
(2) この添加量は,カルシウム90mg,マグネシウム40mgに相当する。
36.1.2 蒸留法 試料に酸化マグネシウムを加えて弱いアルカリ性とし,蒸留を行い,留出したアンモニア
を硫酸 (25mmol/l) に吸収捕集する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 36.1.1(1)(a)による。
(b) 硫酸 (25mmol/l) JIS K 8951に規定する硫酸約1.4mlをあらかじめ水100mlを入れたビーカーに加
えてよくかき混ぜ,水を加えて1lとする。
(c) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(d) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(e) 酸化マグネシウム JIS K 8432に規定する酸化マグネシウムを使用前に600℃で約30分間加熱し,
デシケーター中で放冷する。
(2) 装置 装置は,次のとおりとする。
(a) 蒸留装置 図36.1に一例を示す。ガラス器具類は,使用前に(1)(a)の水でよく洗う。
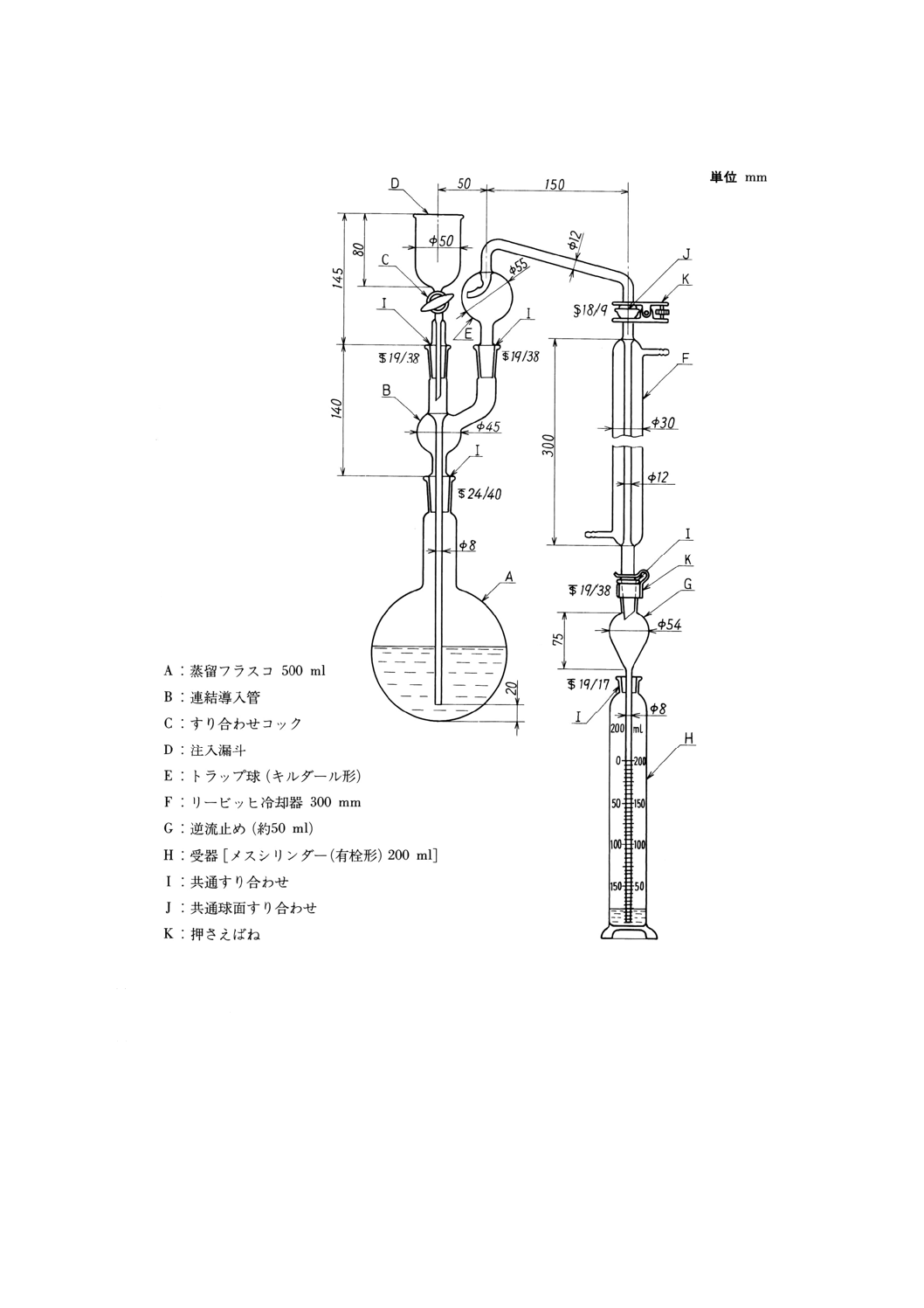
130
K 0101 : 1998
図36.1 蒸留装置の一例
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(3)をとり,中性でない場合には水酸化ナトリウム溶液 (40g/l) 又は硫酸 (1+35) でpH
を約7に調節する。
(b) 蒸留フラスコに移し入れ,酸化マグネシウム0.25g,沸騰石(粒径2〜3mm)数個及び水を加えて液
量を約350mlとする。
(c) 蒸留装置を図36.1のように組み立て,受器のメスシリンダー(有栓形)200ml(4)に硫酸 (25mmol/l)
50mlを入れる。
(d) 蒸留フラスコを加熱し,留出速度5〜7ml/minで蒸留を行う(5)。
131
K 0101 : 1998
(e) 約140mlが留出したら蒸留を止める。
(f) 冷却器と逆流止めを外し,冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液は受器のメス
シリンダー(有栓形)200mlに入れ(6),水を200mlの標線まで加える。
注(3) インドフェノール青吸光光度法で定量する場合には,NH4+として40μg以上,また,中和滴定
法の場合には0.3〜40mg,イオン電極法の場合には40μg以上を含むようにとる。
(4) 留出液を中和滴定法に用いる場合には,受器には三角フラスコ500mlを用い,これに硫酸
(25mmol/l) 50mlを正しく加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液
[13.1(1)(a)による。]5〜7滴を加えておく。
(5) 冷却器の管の先端は,常に液面下約15mmを保つようにする。
(6) 留出液を中和滴定法に用いる場合には,冷却器の内管,逆流止めの内外の洗液は,三角フラス
コ500mlに合わせ,全量を滴定に用いる。
備考1. 蒸留法として水蒸気蒸留法を用いてもよい。その場合は,図36.1の蒸留フラスコに水蒸気を
送るように装置を組み立て,蒸留フラスコを加熱し,沸騰し始めたら,水蒸気を蒸留フラス
コに送り,留出速度3〜5ml/minで蒸留し,約140mlが留出したら蒸留を止める。
36.2 インドフェノール青吸光光度法 アンモニウムイオンが次亜塩素酸イオンの共存のもとで,フェノ
ールと反応して生じるインドフェノール青の吸光度を測定してアンモニウムイオンを定量する。
定量範囲:NH4+5〜100μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物5gを水に
溶かして100mlとする。
(c) 水酸化ナトリウム溶液 (200g/l) 35.1.1.1(1)(c)による。この溶液は使用時に調製する。
(d) ナトリウムフェノキシド溶液 (c)の水酸化ナトリウム溶液 (200g/l) 55mlをビーカーにとり,冷水中
で冷却しながらJIS K 8798に規定するフェノール25gを少量ずつ加えて溶かす。放冷後,JIS K 8034
に規定するアセトン6mlを加え,水で200mlとする。10℃以下の暗所に保存し,5日間以上経過し
たものは使用しない。
(e) 次亜塩素酸ナトリウム溶液(有効塩素10g/l) 次亜塩素酸ナトリウム溶液(有効塩素7〜12%)の
有効塩素の濃度を求め(7),有効塩素が約10g/lになるように水で薄める。使用時に調製する。
(f) アンモニウムイオン標準液 (1mgNH4+/ml) JIS K 8116に規定する塩化アンモニウムをデシケー
ター[JIS K 8228に規定する過塩素酸マグネシウム(乾燥用)を入れたもの。]中に16時間以上放
置し,その2.97gをとり,水に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加える。
又はJIS K 0034に規定する標準物質-標準液-アンモニウムイオンのNH4+1 000を用いる。
(g) アンモニウムイオン標準液 (10μgNH4+/ml) アンモニウムイオン標準液 (1mgNH4+/ml) 10mlを全
量フラスコ1 000mlにとり,水を標線まで加える。使用時に調製する。
注(7) 33.の注(3)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に(1)(a)の水でよく洗う。
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 36.1の前処理を行った試料の適量(NH4+として5〜100μgを含む。)を全量フラスコ50mlにとり(8),
132
K 0101 : 1998
水を加えて約25mlとする。
(b) EDTA溶液1ml(9)とナトリウムフェノキシド溶液10mlを加えて振り混ぜる。
(c) 次亜塩素酸ナトリウム溶液(有効塩素10g/l)5mlを加え,水を標線まで加えた後,栓をして振り混
ぜる。
(d) 液温を20〜25℃に保って約30分間(10)放置する。
(e) この溶液の一部を吸収セルに移し,波長630nm付近の吸光度を測定する。
(f) 空試験として水25mlをとり,(b)〜(e)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
(g) 検量線からアンモニウムイオンの量を求め,試料中のアンモニウムイオンの濃度 (mgNH4+/l) を算
出する。
検量線 アンモニウムイオン標準液 (10μgNH4+/ml) 0.5〜10mlを段階的に全量フラスコ50mlにとり,
水を加えて約25mlとし,(b)〜(f)の操作を行って吸光度を測定し,アンモニウムイオン (NH4+) の
量と吸光度との関係線を作成する。
注(8) 36.1.1(3.2)(b)の操作を行った場合には,塩酸 (1+1) を用いて中和(pH約7まで)する。発色の
強さは,pH 11.5〜12.5のとき最高となる。
(9) EDTA溶液を添加するとインドフェノールの青の発色がやや弱くなるので,検量線作成時にも
同一量のEDTA溶液を添加する。
(10) 液温が20〜25℃のとき約30分間で発色は最高となり,その後約30分間は安定である。
備考2. 微量のアンモニウムイオンを定量する場合には,(3)(b)の操作でナトリウムフェノキシド溶液
10mlに続き,ペンタシアノニトロシル鉄 (III) 酸二ナトリウム溶液[JIS K 8722に規定する
ペンタシアノニトロシル鉄 (III) 酸ナトリウム二水和物0.15gを水に溶かして100mlとする。]
1mlを加えてもよい。
この場合の定量範囲はNH4+として2.5〜50μgとなる。検量線は同一操作で作成する。
3. アンモニウム体窒素で表現する場合は,次の換算式を用いる。
アンモニウム体窒素 (mgNH4+−N/l) =アンモニウムイオン (mgNH4+/l) ×0.776 6
4. 鉄 (II) 及び銅 (II) は,いずれも0.15mg/lまで妨害しないが,それぞれ1mg/lまでは,EDTA
溶液の添加で妨害を除くことができる。
脂肪族アミン類は妨害しないが,芳香族アミン類の一部は次亜塩素酸塩によって酸化され
て着色物質を生じるので妨害する。p-アミノフェノールのような物質は,アルカリ性溶液中
でフェノールと反応してインドフェノール青を生じるので妨害する。p-ヒドロキノンは妨害
しない。ヒドロキシルアミンも妨害するが,JIS K 8230に規定する過酸化水素を定量的に加
えて酸化すれば,妨害を除くことができる。
36.3 中和滴定法 前処理(蒸留)を行って留出したアンモニアを一定量の硫酸 (25mmol/l) 中に吸収させ
た溶液について,50mmol/l水酸化ナトリウム溶液で,残った硫酸を滴定してアンモニウムイオンを定量す
る。
定量範囲:NH4+0.3〜40mg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (25mmol/l) 36.1.2(1)(b)による。
(c) メチルレッド-ブロモクレゾールグリーン混合溶液 13.1(1)(a)による。
133
K 0101 : 1998
(d) 50mmol/l水酸化ナトリウム溶液 14.1(1)(b)の0.1mol/l水酸化ナトリウム溶液100mlを全量フラスコ
200mlにとり,2.(12)(b)の炭酸を含まない水を標線まで加える。使用時に調製する。
(2) 操作 操作は,次のとおり行う。
(a) 36.1.2(3)の前処理で得た留出液の全量を用い,50mmol/l水酸化ナトリウム溶液で,溶液の色が灰紫
(pH 4.8) になるまで滴定する。
(b) 別に,硫酸 (25mmol/l) 50mlを正しく三角フラスコ500mlにとり,メチルレッド-ブロモクレゾール
グリーン混合溶液5〜7滴を加え,50mmol/l水酸化ナトリウム溶液で溶液の色が灰紫 (pH 4.8) にな
るまで滴定し,硫酸 (25mmol/l) 50mlに相当する50mmol/l水酸化ナトリウム溶液のml数を求める。
(c) 次の式によって試料中のアンモニウムイオンの濃度 (mgNH4+/l) を算出する。
902
.0
000
1
)
(
×
×
×
−
=
V
f
a
b
A
ここに,
A: アンモニウムイオン (mgNH4+/l)
b: 硫酸 (25mmol/l) 50mlに相当する50mmol/l水酸化ナトリウ
ム溶液 (ml)
a: 滴定に要した50mmol/l水酸化ナトリウム溶液 (ml)
f: 50mmol/l水酸化ナトリウム溶液のファクター(0.1mol/l水
酸化ナトリウム溶液のファクターを用いる。)
V: 試料 (ml)
0.902: 50mmol/l水酸化ナトリウム溶液1mlのアンモニウムイオ
ン相当量 (mg)
備考5. 36.1.2(3)(c)の硫酸 (25mmol/l) の代わりにほう酸溶液(飽和)を用いてもよい。この場合は次
のように操作する。
三角フラスコ500mlにほう酸溶液(飽和)(JIS K 8863に規定するほう酸50gに水1lを加
えて振り混ぜ,その上澄み液を用いる。)50mlを加え,指示薬としてメチルレッド-ブロモク
レゾールグリーン混合溶液5〜7滴を加え,36.1.2(3)(d)及び(e)の操作を行う。
次に,25mmol/l硫酸[13.1(1)(b)の50mmol/l硫酸100mlを全量フラスコ200mlにとり,水
を標線まで加える。]で溶液の色が灰紫 (pH 4.8) になるまで滴定する。別に,空試験として
ほう酸溶液(飽和)50mlを三角フラスコ500mlにとり,水150mlを加え,指示薬として,メ
チルレッド-ブロモクレゾールグリーン混合溶液5〜7滴を加え,以下,試料の場合と同様に
滴定を行って次の式によって試料中のアンモニウムイオンの濃度 (mgNH4+/l) を算出する。
902
.0
000
1
)
(
×
×
×
−
=
V
f
b
a
A
ここに,
A: アンモニウムイオン (mgNH4+/l)
a: 滴定に要した25mmol/l硫酸 (ml)
b: 空試験に要した25mmol/l硫酸 (ml)
f: 25mmol/l硫酸のファクター(50mmol/l硫酸のファクター
を用いる。)
V: 試料 (ml)
0.902: 25mmol/l硫酸1mlのアンモニウムイオン相当量 (mg)
36.4 イオン電極法 前処理を行った試料に水酸化ナトリウム溶液を加えpHを11〜13に調節してアンモ
ニウムイオンをアンモニアに変え,アンモニア電極を指示電極として電位を測定し,アンモニウムイオン
を定量する。
定量範囲:NH4+0.1〜100mg/l,繰返し分析精度:変動係数で5〜20%
134
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 水酸化ナトリウム溶液 (100g/l) 22.2.1(1)(b)による。
(c) アンモニウムイオン標準液 (100mgNH4+/l) 36.2(1)(f)のアンモニウムイオン標準液 (1mgNH4+
/ml) 20mlを全量フラスコ200mlにとり,水を標線まで加える。
(d) アンモニウムイオン標準液 (10mgNH4+/l) アンモニウムイオン標準液 (100mgNH4+/l) 20mlを全
量フラスコ200mlにとり,水を標線まで加える。
(e) アンモニウムイオン標準液 (1mgNH4+/l) アンモニウムイオン標準液 (10mgNH4+/l) 20mlを全量
フラスコ200mlにとり,水を標線まで加える。使用時に調製する。
(f) アンモニウムイオン標準液 (0.1mgNH4+/l) アンモニウムイオン標準液 (1mgNH4+/l) 20mlを全量
フラスコ200mlにとり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 31.2(2)(a)による。
(b) 指示電極 アンモニア電極
(c) マグネチックスターラー 31.2(2)(d)による。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) アンモニウムイオン標準液 (0.1mgNH4+/l) 100mlを三角フラスコ200ml(11)にとり,水酸化ナトリウ
ム溶液 (100g/l) 1mlを加える(12)。
(b) これに指示電極 (13)(14)を浸し,マグネチックスターラー(15)で泡が電極に触れない程度に強くかき混
ぜる(16)。
(c) 液温をはかり,電位差計で電位を測定する(17)。
(d) アンモニウムイオン標準液 (1mgNH4+/l) 100ml,アンモニウムイオン標準液 (10mgNH4+/l) 100ml及
びアンモニウムイオン標準液 (100mgNH4+/l) 100mlをそれぞれ三角フラスコ200ml(11)にとり,水酸
化ナトリウム溶液 (100g/l) 1mlを加える(12)。液温を(c)での液温の±1℃に調節して(b)及び(c)の操作
を行ってそれぞれのアンモニウムイオン標準液 (1〜100mgNH4+/l) の電位を測定する(18)。
(e) 片対数紙の対数軸にアンモニウムイオンの濃度 (mgNH4+/l) を,均等軸に電位(19)をとって,アンモ
ニウムイオンの濃度と電位との関係線を作成(20)する。
注(11) アンモニアは揮散しやすいため,できるだけ口の狭い容器を用いる。必要に応じて密閉できる
容器を用いるとよい。
(12) pHは約12となる。pH 11以上で,アンモニウムイオンはアンモニアとなる。アンモニアは揮散
しやすいので,水酸化ナトリウム溶液 (100g/l) は電位測定の直前に加える。
(13) 指示電極が隔膜電極の場合には,内部ガラス電極のガラス膜面に隔膜を強く押しつけると隔膜
に傷がつくので注意する。
また,ガラス膜面と隔膜が離れ過ぎると,応答時間が長くなる。
(14) 隔膜電極の隔膜が汚れると,電位が不安定になり,応答時間も長くなる。
(15) 31.の注(14)による。
(16) かき混ぜが強すぎると,泡が膜を覆い誤差を生じるので注意する。
(17) アンモニア電極の応答時間は,液温10〜30℃の場合アンモニウムイオン標準液 (0.1mgNH4+/l)
で3〜5分間,アンモニウムイオン標準液(1mgNH4+/l以上)では2〜3分間である。
(18) 電位の測定は,低濃度から順に行う。高濃度から低濃度へ移ると,応答速度は遅くなるので,
135
K 0101 : 1998
水で洗ってアンモニアを除き,次に,(3)(a)の水酸化ナトリウム溶液 (100g/l) 1ml加えたアンモ
ニウムイオン標準液 (0.1mgNH4+/l) に浸し,指示値が安定してから測定する。
(19) アンモニウムイオン標準液 (0.1mgNH4+/l) とアンモニウムイオン標準液 (10mgNH4+/l) との電
位差は110〜120mV (25℃) の範囲になる。
(20) アンモニウムイオン濃度0.1mg/l付近から100mg/lまで検量線は直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 36.1の前処理を行った試料の適量(NH4+として0.02〜20mgを含む。)をとり,水で約100mlとした
後,前処理が36.1.2によった場合には,水酸化ナトリウム溶液 (100g/l) を加えてpHを約8に調節
し,全量フラスコ200mlに移し入れ,水を標線まで加える。この溶液100mlを三角フラスコ200ml
にとり,水酸化ナトリウム溶液 (100g/l) 1ml加える(12)。
(b) 液温を(3)(c)での液温の±1℃に調節する。
(c) (3)(b)及び(c)の操作を行って検量線からアンモニウムイオンの濃度 (mgNH4+/l) を求め,試料中のア
ンモニウムイオンの濃度 (mgNH4+/l) を算出する。
備考6. イオン濃度計の場合には,アンモニウムイオン標準液 (0.1mgNH4+/l) とアンモニウムイオン
標準液 (10mgNH4+/l) を用い,(3)(a)〜(c)の操作を行ってイオン濃度計の指示値を0.1mgNH4+
/lと10mgNH4+/lになるように調節する。さらにアンモニウムイオン標準液 (1mgNH4+/l) と
アンモニウムイオン標準液 (100mgNH4+/l) を用いてイオン濃度計の指示値を確認する。
7. 試料中のアンモニウムイオンの濃度が0.1mg/lの場合,ヒドラジニウムイオン(ヒドラジン)
は1mg/l以下では影響しないが,10mg/l及び100mg/l共存すれば,それぞれ+35%,+100%
の誤差を生じる。
試料中のアンモニウムイオンの濃度が1mg/l以上の場合には,ヒドラジニウムイオン
100mg/l共存しても影響しない。試料中のアンモニウムイオンの濃度が0.1mg/lの場合,モル
ホリン(テトラヒドロ-1, 4-オキサジン,C4H8ONH)は10mg/lまで共存しても影響しない
が,100mg/l共存すると,+100%の誤差を生じる。
また,試料中のアンモニウムイオンが1mg/l以上の場合には,モルホリンが100mg/l共存
しても影響しない。
36.5 イオンクロマトグラフ法 試料中のアンモニウムイオンをイオンクロマトグラフ法によって定量す
る。この方法を適用する場合には,3.3の保存処理は行わず,試料採取後直ちに試験する。
定量範囲: NH4+ 0.1〜30mg/l(21),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって
異なる。)
注(21) サプレッサと組み合わせる方式の場合も,ほぼ同じである。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 溶離液(22)は,装置の種類及び分離カラムに充てんした陽イオン交換体の種類によって異な
るので,あらかじめ,アンモニウムイオン,ナトリウム,カリウムの分離を注(26)の操作で確認する。
(c) 再生液 再生液(23)は,サプレッサを用いる場合に使用するが,装置の種類及び除去カラムの種類に
よって再生液が異なる。あらかじめ分離カラムと組み合わせて注(26)の操作を行って,再生液の性能
を確認する。
(d) アンモニウムイオン標準液 (1mgNH4+/ml) 36.2(1)(f)による。
(e) アンモニウムイオン標準液 (0.1mgNH4+/ml) アンモニウムイオン標準液 (1mgNH4+/ml) 10mlを
136
K 0101 : 1998
全量フラスコ100mlにとり,水を標線まで加える。
(f) アルカリ金属元素-アンモニウムイオン混合標準液 [ (0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml]
36.2(1)(f)のアンモニウムイオン標準液 (1mgNH4+/ml) 10ml,47.1(1)(a)のナトリウム標準液
(1mgNa/ml) 10ml及び48.1(1)(a)のカリウム標準液 (1mgK/ml) 10mlをそれぞれ全量フラスコ100ml
にとり,水を標線まで加える。使用時に調製する。
注(22) 溶離液の調製方法の例を次に示す。
サプレッサを用いる場合の例
メタンスルホン酸溶液 (20mmol/l) メタンスルホン酸192.3g(約123ml)をとり,水に溶か
して1lとする。この溶液10mlをとり,水で1lとする。
硝酸 (5mmol/l) 硝酸 (0.5mol/l) (JIS K 8541に規定する硝酸36mlを水に溶かして1lとす
る。)10mlを水で1lとする。
サプレッサを用いない場合の例
[2, 6-ピリジンジカルボン酸溶液 (1mmol/l) −酒石酸溶液 (5mmol/l)] 2,6-ピリジンジ
カルボン酸0.16gとJIS K 8532に規定するL (+) −酒石酸0.750gとをとり,水に溶かして
1lとする。
(23) 再生液の調製方法の例を示す。
水酸化テトラメチルアンモニウム溶液 (40mmol/l) 水酸化テトラメチルアンモニウム五水
和物7.25gを水に溶かして1lとする。
水酸化ナトリウム溶液 (50mmol/l) 36.3(1)(d)による。ただし,標定は省略する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(2.1) イオンクロマトグラフ イオンクロマトグラフには,分離カラムとサプレッサ(24)を組み合わせた方
式のもの,分離カラム単独の方式のもの,いずれでもよいが,次に掲げる条件を満たすもので,ア
ンモニウムイオン,ナトリウム及びカリウムが分離定量できるもの。
(a) 分離カラム ステンレス鋼製又は合成樹脂製(25)のものに,陽イオン交換体(表層被覆形又は全多孔
性シリカ形など)を充てんしたもの(26)。
(b) 検出器 電気伝導率検出器
注(24) 溶離液中の陰イオンを水酸化物イオンに変換するためのもので,溶離液中の陰イオンの濃度に
対して十分なイオン交換容量をもった陰イオン交換膜(膜形,電気透析形がある。)又は同様な
性能をもった陰イオン交換体を充てんしたもの。再生液と組み合わせて用いる。ただし,電気
透析形の場合は,検出器からの流出液を用いる。
(25) 例えば,四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などがある。
(26) 溶離液を一定の流量(例えば,1〜2ml/min)で流し,アルカリ金属元素-アンモニウムイオン混
合標準液 [ (10μgNH4+, 10μgNa, 10μgK)/ml ] の一定量をイオンクロマトグラフに注入し,ク
ロマトグラムを求め,それぞれの陽イオンが分離(分離度1.3以上)できるものを用いる。定
期的に分離カラムの性能を確認するとよい。
アルカリ金属元素-アンモニウムイオン混合標準液 [ (10μgNH4+, 10μgNa, 10μgK)/ml ] は,
次のように調製する。
36.2(1)(f)のアンモニウムイオン標準液 (1mgNH4+/ml) 5ml, 47.1(1)(a)のナトリウム標準液 (1
000mgNa/l) 5ml及び48.1(1)(a)のカリウム標準液 (1 000mgK/l) 5mlをそれぞれ全量フラスコ
500mlにとり,水を標線まで加える。
137
K 0101 : 1998
(2.2) 記録部 JIS K 0127の4.2(6)による。
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を孔径0.45μmのろ過膜又はろ紙5種C(又はろ紙6種)でろ過し,初めのろ液約50mlを捨て,
その後のろ液をとる。
(b) 試料の電気伝導率が10mS/m {100μS/cm} (25℃) 以上の場合には,電気伝導率が10mS/m以下になる
ように,水で一定の割合に薄める。
(4) 操作 操作は,次のとおり行う。
(a) イオンクロマトグラフを作動できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜
2ml/min)で流しておく。サプレッサを必要とする装置では再生液を一定の流量で流しておく。
(b) (3)の準備操作を行った試料の一定量(例えば,50〜200μlの一定量)をイオンクロマトグラフに注
入し,クロマトグラムを記録する。
(c) クロマトグラム上のアンモニウムイオンに相当するピークについて,指示値(27)を読み取る。
(d) 試料を薄めた場合には,空試験として試料と同量の水について,(a)〜(c)の操作を行って試料につい
て得た指示値(27)を補正する。
(e) 検量線から,アンモニウムイオンの濃度を求め,試料中のアンモニウムイオンの濃度 (mgNH4+/l) を
算出する。
検量線 アンモニウムイオン標準液 (0.1mgNH4+/ml) (28)0.1〜30mlを段階的に全量フラスコ100ml
にとり,水を標線まで加える。この溶液について(a)〜(c)の操作を行ってそれぞれのアンモニウムイ
オンに相当する指示値(27)を読み取る。別に,空試験として水について(a)〜(c)の操作を行ってそれ
ぞれのアンモニウムイオンに相当する指示値を補正した後,アンモニウムイオン (NH4+) の量と指
示値との関係線を作成する。検量線の作成は測定時に行う。
注(27) ピーク高さ又はピーク面積
(28) ナトリウム及びカリウムを同時に試験する場合には,アルカリ金属元素-アンモニウムイオン混
合標準液 [(0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml] を用いる。
備考8. アンモニウムイオンの濃度が1mg/lのとき,ナトリウム及びカリウムはいずれも100mg/l以下
ならば妨害しない。
9. 分離カラムは,使用を続けると性能が低下するので,定期的に注(26)の操作で確認する。性能
が低下している場合には,溶離液の20〜200倍の濃度のものを調製し,分離カラムに注入し,
洗浄した後,性能を確認する。性能が回復しない場合は,新品と取り替える。
試料中の懸濁物及び有機物などによっても汚染されて性能が徐々に低下する。懸濁物を含
む試料は(3)の準備操作で除去した後,試験する。
また,有機物(たん白質,油脂,界面活性剤など)を含む試料は限外ろ過膜でろ過し,で
きるだけ有機物を除去した後,試験する。
試料中に分離カラムの充てん剤と親和力の強い陽イオン(例えば,カルシウム,マグネシ
ウムなど)が存在すると,これらが充てん剤に吸着され,分離性能が徐々に低下するので,
定期的に溶離液の20〜200倍の濃度のものを調製し,試料と同様に分離カラムに注入し洗浄
する。
そのほか酸化性物質,還元性物質が共存すると,分離カラムの分離性能が低下する。この
ような場合には,試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐことが
できる。
138
K 0101 : 1998
10. カルボン酸形の陽イオン交換カラムと溶離液として,メタンスルホン酸溶液,[2, 6-ピリジン
ジカルボン酸-L (+) -酒石酸]溶液などを溶離液に用いると,カルシウム,マグネシウムなど
2価の陽イオンの溶離及び定量が可能になる。
37. 亜硝酸イオン (NO2-) 及び硝酸イオン (NO3-)
37.1 亜硝酸イオン (NO2-) 亜硝酸イオンの定量には,ナフチルエチレンジアミン吸光光度法又はイオン
クロマトグラフ法を適用する。亜硝酸イオンは変化しやすいから,試験は試料採取後直ちに行う。直ちに
行えない場合には,3.3によって保存し,できるだけ早く試験する。ただし,イオンクロマトグラフ法を適
用する場合には,3.3の保存処理を行わず,試料採取後直ちに試験する。
37.1.1 ナフチルエチレンジアミン吸光光度法 試料にスルファニルアミド(4-アミノベンゼンスルホンア
ミド)を加え,これを亜硝酸イオンによってジアゾ化し,N-1-ナフチルエチレンジアミン(二塩化N-1-ナ
フチルエチレンジアンモニウム)を加えて生じる赤い色のアゾ化合物の吸光度を測定して亜硝酸イオンを
定量する。
定量範囲:NO2- 0.6〜6μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 4-アミノベンゼンスルホンアミド溶液 JIS K 9066に規定するスルファニルアミド(4-アミノベン
ゼンスルホンアミド)2gをJIS K 8180に規定する塩酸60mlと水約80mlに溶かし,更に水を加え
て200mlとする。
(c) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 JIS K 8197に規定するN-1-ナフチルエチレンジ
アミン二塩酸塩(二塩化N-1-ナフチルエチレンジアンモニウム)0.2gを水に溶かして200mlとする。
着色ガラス瓶に入れて保存し,1週間以上経過したものは使用しない。
(d) 亜硝酸イオン標準液 (1mgNO2-/ml) JIS K 8019に規定する亜硝酸ナトリウムを105〜110℃で約4
時間加熱し,デシケーター中で放冷した後,亜硝酸ナトリウムの純度を求め(1),NaNO2 100%に対し
て1.50gに相当する亜硝酸ナトリウムをとり,少量の水に溶かして全量フラスコ1 000mlに移し入
れ,水を標線まで加える。使用時に調製する。又はJIS K 0032に規定する亜硝酸イオン標準液の
NO2- 1 000を用いる。
(e) 亜硝酸イオン標準液 (20μgNO2-/ml) 亜硝酸イオン標準液 (1mgNO2-/ml) 10mlを全量フラスコ
500mlにとり,水を標線まで加える。使用時に調製する。
(f) 亜硝酸イオン標準液 (2μgNO2-/ml) 亜硝酸イオン標準液 (20μgNO2-/ml) 20mlを全量フラスコ
200mlにとり,水を標線まで加える。使用時に調製する。
注(1) JIS K 8019の6.(1)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ紙5種C(又はろ紙6種)を用いてろ過し,初めのろ液約50mlは捨て,その後のろ液(2)
の適量(NO2-として0.6〜6μgを含む。)をメスシリンダー(有栓形)10mlにとり,水を加えて10ml
とする。
(b) 4-アミノベンゼンスルホンアミド溶液1mlを加えて振り混ぜ,約5分間放置した後,二塩化N-1-ナ
フチルエチレンジアンモニウム溶液1mlを加えて振り混ぜ,室温で約20分間放置する。
139
K 0101 : 1998
(c) 溶液の一部を吸収セルに移し,波長540nm付近の吸光度を測定する。
(d) 空試験として水10mlをメスシリンダー(有栓形)10mlにとり,(b)及び(c)の操作を行って吸光度を
測定し,試料について得た吸光度を補正する。
(e) 検量線から亜硝酸イオンの量を求め,試料中の亜硝酸イオンの濃度 (mgNO2-/l) を算出する。
検量線 亜硝酸イオン標準液 (2μgNO2-/ml) 3〜30mlを段階的に全量フラスコ100mlにとり,水を標
線まで加える。その中からそれぞれ10mlをメスシリンダー(有栓形)10mlにとり,(b)〜(d)の操作
を行って亜硝酸イオン (NO2-) の量と吸光度との関係線を作成する。
注(2) ろ過しても,色,濁りが残る場合には,36.1.1(3)(3.1)の硫酸亜鉛による凝集沈殿又は次の硫酸ア
ルミニウム凝集沈殿法によって除去する。
硫酸アルミニウム凝集沈殿法は,試料100mlにつき硫酸カリウムアルミニウム溶液(JIS K
8255に規定する硫酸カリウムアルミニウム・12水5gを水に溶かして100mlとする。)2ml及び
水酸化ナトリウム溶液 (40g/l) を加えて水酸化アルミニウムのフロックを生成させ,数分間放
置した後,ろ過(初めのろ液約20mlは捨てる。)して澄明な溶液とする。
凝集沈殿処理すると水酸化アルミニウムに亜硝酸イオンが一部吸着されて発色が低下するの
で,別に,亜硝酸イオン標準液(2μgNO2-/ml)を段階的にとり,同様に処理したものを用いて検量
線を作成して定量する。
備考1. 一般に,亜硝酸イオンは,残留塩素などの酸化性物質と共存することはないが,亜硝酸イオ
ンが存在しなくても残留塩素及びクロロアミン類(モノクロロアミン,ジクロロアミン,三
塩化窒素)などが存在すると赤に発色して亜硝酸イオンとして誤認されることがある。
試料中の酸化性物質の存在を確認するには,次のように操作する。
試料100mlをとり,ふっ化カリウム溶液 (300g/l) 1mlとJIS K 9501に規定するアジ化ナト
リウム0.5gを加える。塩酸 (1+1) を加えて酸性(pH約1)とし,JIS K 8913に規定するよ
う化カリウム1gを加えてかき混ぜた後,暗所に数分間放置する。これに指示薬としてでんぷ
ん溶液 (10g/l) [22.1.2(1)(i)による。]2mlを加える。よう素でんぷんの青い色が認められる
場合は,酸化性物質が存在する。このような試料について亜硝酸イオンを試験するには,こ
の操作を行った後よう素でんぷんの青い色が消えるまで,亜硫酸ナトリウム溶液 (50mmol/l)
で滴定し,その消費量から,試料中の酸化性物質の量に相当する亜硫酸ナトリウム溶液
(50mmol/l) の量を求め,この量を試料に添加した後,(a)〜(e)の操作を行う。
2. 亜硝酸体窒素で表示する場合は,次の換算式を用いる。
亜硝酸体窒素 (mgNO2-−N/l) =亜硝酸イオン (mgNO2-/l) ×0.3045
37.1.2 イオンクロマトグラフ法 試料中の亜硝酸イオンをイオンクロマトグラフ法で定量する。この方法
を適用する場合には,3.3の保存処理を行わず,試料採取後直ちに試験する。
定量範囲: NO2- 0.5〜40mg/l(3),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって
異なる。)
注(3) サプレッサと組み合わせる方式の場合には,0.1〜40mgNO2-/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 32.5(1)(b)による。
(c) 再生液 32.5(1)(c)による。
(d) 亜硝酸イオン標準液 (5mgNO2-/ml) JIS K 8019に規定する亜硝酸ナトリウムを105〜110℃で約4
140
K 0101 : 1998
時間加熱し,デシケーター中で放冷した後,亜硝酸ナトリウムの純度を求め(1),NaNO2100%に対し
て0.750gに相当する亜硝酸ナトリウムをとり,少量の水に溶かして,全量フラスコ100mlに移し入
れ,水を標線まで加える。使用時に調製する。
(e) 亜硝酸イオン標準液 (0.5mgNO2-/ml) 亜硝酸イオン標準液 (5mgNO2-/ml) 10mlを全量フラスコ
100mlにとり,水を標線まで加える。使用時に調製する。
(f) 陰イオン混合標準液 [(0.1mgCl-, 0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] 32.5(1)(f)による。
(2) 器具及び装置 器具及び装置は,32.5(2)による。ただし,検出器は,紫外吸光検出器を用いてもよい。
(3) 準備操作 準備操作は,32.5(3)による。
(4) 操作 操作は,次のように行う。
(a) 32.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上の亜硝酸イオンに相当するピークについて,指示値(4)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
いて得た指示値(4)を補正する。
(d) 検量線から,亜硝酸イオンの濃度を求め,試料中の亜硝酸イオンの濃度 (mgNO2-/l) を算出する。
検量線 亜硝酸イオン標準液 (0.5mgNO2-/ml) (5)0.1〜8mlを段階的に全量フラスコ100mlにとり,水
を標線まで加える。この溶液について(a)及び(b)の操作を行ってそれぞれの亜硝酸イオンに相当する
指示値(4)を読み取る。別に,空試験として水について(a)及び(b)の操作を行ってそれぞれの亜硝酸イ
オンに相当する指示値(4)を補正した後,亜硝酸イオン (NO2-) の量と指示値との関係線を作成する。
検量線の作成は,試料の測定時に行う。
注(4) ピーク高さ又はピーク面積
(5) 亜硝酸イオン以外の陰イオンを同時に試験する場合には,陰イオン混合標準液 [(0.1mgCl-,
0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] を用いる。
備考3. 亜硝酸イオンの濃度が1mg/lのとき塩化物イオンは50mg/l以下,臭化物イオン200mg/l以下及
び硫酸イオン500mg/l以下ならば妨害しない。
4. 32.の備考11.による。
37.2 硝酸イオン (NO3-) 硝酸イオンの定量には,還元蒸留-インドフェノール青吸光光度法,還元蒸留-
中和滴定法,銅・カドミウムカラム還元-ナフチルエチレンジアミン吸光光度法,ブルシン吸光光度法又は
イオンクロマトグラフ法を適用する。
この試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験
する。
37.2.1 還元蒸留-インドフェノール青吸光光度法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモ
ニウムイオン及び一部の有機体窒素化合物の分解で生じたアンモニアを除去した後,デバルダ合金を加え
て亜硝酸イオン及び硝酸イオンをアンモニアに還元して蒸留し,留出したアンモニアを硫酸 (25mmol/l)
に吸収させる。次に,この留出液中のアンモニウムイオンをインドフェノール青吸光光度法によって定量
し,硝酸イオンと亜硝酸イオンの合量を求め,その値から,別に,亜硝酸イオンを定量して差し引き,硝
酸イオンの量を算出する。
定量範囲:NO3- 17〜340μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (25mmol/l) 36.1.2(1)(b)による。
141
K 0101 : 1998
(c) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(d) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(e) 水酸化ナトリウム溶液 (300g/l) JIS K 8576に規定する水酸化ナトリウム30gを水に溶かして
100mlとする。使用時に調製する。
(f) デバルダ合金 JIS K 8653に規定するもので粉末のもの。
(g) ナトリウムフェノキシド溶液 36.2(1)(d)による。
(h) 次亜塩素酸ナトリウム溶液(有効塩素10g/l) 36.2(1)(e)による。
(i) アンモニウムイオン標準液 (1mgNH4+/ml) 36.2(1)(f)による。
(j) アンモニウムイオン標準液 (10μgNH4+/ml) 36.2(1)(g)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗う。
(b) 蒸留装置 36.1.2(2)(a)による。使用前に水でよく洗う。
(c) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(NO3-として0.14mg以上を含む。)をとり,試料が中性でない場合には,水酸化ナトリ
ウム溶液 (40g/l) 又は硫酸 (1+35) でpHを約7に調節する。
(b) これを水で蒸留フラスコに洗い移し,水酸化ナトリウム溶液 (300g/l) 10mlと沸騰石(粒径2〜3mm)
数個を加え,水を加えて約350mlとし,36.1.2(3)(c)〜(e)の蒸留操作を行って(6)アンモニアを除去し,
冷却器と逆流止めを取り外し,冷却器の内管と逆流止めの内外を水でよく洗う。
(c) 蒸留フラスコ中の残留液を放冷する。
(d) 受器に別のメスシリンダー(有栓形)200mlを用い,硫酸 (25mmol/l) 50mlを入れる。
(e) 蒸留フラスコ中の残留液にデバルタ合金3gを手早く加え,水を加えて約350mlとした後,装置を
組み立てる。
(f) 36.1.2(3)(d)〜(f)の操作を行う(7)。
(g) (f)で得た留出液の適量(NH4+として5〜100μgを含む。)を全量フラスコ50mlに分取し,水を加え
て約25mlとする。
(h) 36.2(3)(b)〜(e)の操作を行って吸光度を測定する。
(i) 空試験として水約100mlを蒸留フラスコにとり,水酸化ナトリウム溶液 (300g/l) 10mlを加えた後,
(d)〜(f)の操作を行う。
(j) (i)で得た留出液について(g)と同量を全量フラスコ50mlに分取し,(h)の操作を行って吸光度を測定
し,試料について得た吸光度を補正する。
(k) 36.2(3)の検量線から,分取した留出液中のアンモニウムイオンの量 (mgNH4+) を求める。
(l) 別に,37.1.1又は37.1.2によって試料中の亜硝酸イオンの濃度 (mgNO2-/l) を求める。
(m) 次の式によって試料中の硝酸イオンの濃度 (mgNO3-/l) を算出する。
348
.1
200
000
1
437
.3
2
1
×
−
×
×
×
=
C
V
V
a
N
ここに,
N: 硝酸イオン (mgNO3-/l)
a: (k)で求めた留出液中のアンモニウムイオンの量 (mgNH4+)
3.437: アンモニウムイオンを硝酸イオンの相当量に換算するとき
142
K 0101 : 1998
の係数
04
.
18
00
.
62
V1: 試料 (ml)
V2: (g)で分取した留出液 (ml)
C: (l)で求めた試料中の亜硝酸イオン (mgNO2-/l)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係数
01
.
46
00
.
62
注(6) この操作では,受器には硫酸 (25mmol/l) に代え,水を入れておいてもよい。
(7) 蒸留の初めに泡立ちが激しいときは加熱を弱め,約10分間経過して泡立ちが静まってから再び
蒸留する。
備考5. (3)(b)の操作で留出したアンモニアには,試料中に存在したアンモニウムイオンのほか,有機
窒素化合物の分解によって生じたものを含む場合があるから,この留出液を用いて試料中の
アンモニウムイオンの定量を行ってはならない。
6. 硝酸体窒素で表示する場合には,次の換算式を用いる。
硝酸体窒素 (mgNO3-N/l) =硝酸イオン (mgNO3-/l) ×0.225 9
37.2.2 還元蒸留-中和滴定法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモニウムイオン及び一
部の有機窒素化合物の分解から生じたアンモニアを除去した後,デバルタ合金を加えて亜硝酸イオン及び
硝酸イオンをアンモニアに還元して蒸留し,留出したアンモニアを一定量の硫酸 (25mmol/l) 中に吸収し,
中和滴定法によって定量して亜硝酸イオンと硝酸イオンに相当する量を求める。別に,亜硝酸イオンを定
量して差し引き,硝酸イオンの量を算出する。
定量範囲:NO3- 1〜140mg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (25mmol/l) 36.1.2(1)(b)による。
(c) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(d) 50mmol/l水酸化ナトリウム溶液 36.3(1)(d)による。
(e) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(f) 水酸化ナトリウム溶液 (300g/l) 37.2.1(1)(e)による。
(g) デバルタ合金 37.2.1(1)(f)による。
(h) メチルレッド-ブロモクレゾールグリーン混合溶液 13.1(1)(a)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗う。
(b) 蒸留装置 36.1.2(2)(a)による。使用前に水でよく洗う。
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(NO3-として1mg以上,NO2-及びNO3-の合量がNO3-として140mg以下を含む。)をとり,
試料が中性でない場合には,水酸化ナトリウム溶液 (40g/l) 又は硫酸 (1+35) でpHを約7に調節
する。
(b) これを水で蒸留フラスコに洗い移し,水酸化ナトリウム溶液 (300g/l) 10mlと沸騰石(粒径2〜3mm)
数個を加え,水を加えて約350mlとし,36.1.2(3)(c)〜(e)の蒸留操作を行って(6)アンモニアを除去し,
冷却器と逆流止めを取り外し,冷却器の内管と逆流止めの内外を水でよく洗う。
143
K 0101 : 1998
(c) 蒸留フラスコ中の残留液を放冷する。
(d) 受器として三角フラスコ500mlを用い,これに硫酸 (25mmol/l) 50mlを正しく加え,指示薬として
メチルレッド-ブロモクレゾールグリーン混合溶液5〜7滴を加えておく。
(e) 蒸留フラスコ中の残留液にデバルタ合金3gを手早く加え,水を加えて約350mlとした後,装置を
組み立てる。
(f) 36.1.2(3)(d)〜(f)の操作を行う(7)。
(g) 留出液の全量を用い,36.3(2)(a)の滴定操作を行う。
(h) 空試験として水約100mlを蒸留フラスコにとり,水酸化ナトリウム溶液 (300g/l) 10mlを加えた後,
(d)〜(f)の操作を行って得られた留出液について(g)と同様に滴定操作を行う。
(i) 別に,37.1.1又は37.1.2によって試料中の亜硝酸イオンの濃度 (mgNO2-/l) を求める。
(j) 次の式によって試料中の硝酸イオンの濃度 (mgNO3-/l) を算出する。
348
.1
1.3
000
1
)
(
1
×
−
×
×
×
−
=
C
V
f
a
b
N
ここに,
N: 硝酸イオン (mgNO3-/l)
a: 滴定に要した50mmol/l水酸化ナトリウム溶液 (ml)
b: 空試験に要した50mmol/l水酸化ナトリウム溶液 (ml)
f: 50mmol/l水酸化ナトリウム溶液のファクター
3.1: 50mmol/l水酸化ナトリウム溶液1mlの硝酸イオン相当量
(mg)
V: 試料 (ml)
C: (i)で求めた試料中の亜硝酸イオン (mgNO2-/l)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係
数
01
.
46
00
.
62
37.2.3 銅・カドミウムカラム還元−ナフチルエチレンジアミン吸光光度法 試料中の硝酸イオンを銅・カ
ドミウムカラムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法によって定量
し,硝酸イオンの濃度を求める。
定量範囲:NO3- 0.8〜8μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(c) 塩化アンモニウム−アンモニア溶液 JIS K 8116に規定する塩化アンモニウム100gを水約700ml
に溶かした後,JIS K 8085に規定するアンモニア水50mlを加え,更に水を加えて1lとする。
(d) カラム活性化液 水約700mlに水酸化ナトリウム溶液 (80g/l) 70mlを加えたものにJIS K 8107に規
定するエチレンジアミン四酢酸二水素二ナトリウム二水和物38g及びJIS K 8983に規定する硫酸銅
(II) 五水和物12.5gを溶かし,更に水酸化ナトリウム溶液 (80g/l) を滴加して溶液のpHを7とした
後,水を加えて1lとする。
(e) 銅・カドミウムカラム充てん剤 粒状カドミウム(粒径0.5〜2mm)約40gを三角フラスコ300ml
にとり,塩酸 (1+5) 約50mlを加えて振り混ぜて,カドミウムの表面を洗浄し,洗液を捨て,水約
100mlずつで5回洗浄する。次に,硝酸 (1+39) 約50mlを加えて振り混ぜてカドミウムの表面を
洗浄し,洗液を捨てる。この操作を2回行った後,水約100mlずつで5回洗浄する。次に,カラム
活性化液200mlを加えて24時間放置し,カドミウムの表面に銅の皮膜を形成させる。この銅・カ
144
K 0101 : 1998
ドミウムカラム充てん剤は,このまま密栓して保存することができる。
なお,この方法で調製したものに代え,市販の銅・カドミウムカラム充てん剤を用いてもよい。
(f) カラム充てん液 塩化アンモニウム-アンモニア溶液を水で10倍に薄める。
(g) 4-アミノベンゼンスルホンアミド溶液 37.1.1(1)(b)による。
(h) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 37.1.1(1)(c)による。
(i) 硝酸イオン標準液 (1mgNO3-/ml) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110℃で
約3時間加熱し,デシケーター中で放冷する。その1.63gをとり,水に溶かして全量フラスコ1 000ml
に移し入れ,水を標線まで加える。0〜10℃の暗所に保存する。又はJIS K 0031に規定する硝酸イ
オン標準液のNO3-1 000を用いる。
(j) 硝酸イオン標準液 (10μgNO3-/ml) 硝酸イオン標準液 (1mgNO3-/ml) 10mlを全量フラスコ1 000ml
にとり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 銅・カドミウムカラム 図37.1(A)に示すようなガラス管の底部にJIS K 8251に規定するガラスウ
ールを詰め,カラム充てん液を満たした後,銅・カドミウムカラム充てん剤を空気に触れないよう
流し入れた後,上部にガラスウールを詰め,円筒形滴下漏斗を取り付ける。次に,円筒形滴下漏斗
から,カラム充てん液100ml,硝酸イオン標準液 (1mgNO3-/ml) をカラム充てん液で100倍に薄め
た溶液200ml,更にカラム充てん液100mlの順で,流量約10ml/minで流下させる。このとき,カラ
ム内の溶液の液面は,充てん剤よりわずかに上部になるようにする。
なお,銅・カドミウムカラムを使用しないときは,銅・カドミウムカラム充てん剤が空気に触れ
ないように,上部までカラム充てん液を入れておく。銅・カドミウムカラムは使用に伴って劣化し,
硝酸イオンの亜硝酸イオンへの還元率が低下するので,必要に応じ,カラム活性化液を用いて再生
する。再生には銅・カドミウムカラムにカラム活性化液を満たし,2〜3時間放置した後,カラム充
てん液で洗浄する(8)。
(b) 光度計 分光光度計又は光電光度計
注(8) 試料について15〜20回使用するごとにカラム活性化液約20mlを銅・カドミウムカラムに流し,
次にカラム充てん液約100mlで洗浄すれば,硝酸イオンの還元率の低下を防ぐことができる。
145
K 0101 : 1998
図37.1 銅・カドミウムカラムの一例
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ紙5種C(又はろ紙6種)でろ過し,初めのろ液約50mlは捨てる(9)(10)。その後のろ液の
適量(NO3-として8μg以上,NO2-及びNO3-の合量がNO3-として80μg以下を含む。)を全量フラス
コ100mlにとる。
(b) これに塩化アンモニウム−アンモニア溶液10mlを加え,更に水を標線まで加えて還元用溶液とす
る。
(c) 上部の円筒形滴下漏斗に還元用溶液を入れ,銅・カドミウムカラム内の液面を銅・カドミウムカラ
ム充てん剤よりわずかに上部に保ちながら約10ml/minで流下させ,流出液約30mlを捨てる。還元
用溶液を追加し,同様に流下させ,その後の30mlをメスシリンダー50mlに集める。
(d) この流出液から10mlを共栓試験管にとり,37.1.1(3)(b)及び(c)の操作を行う。
(e) 空試験として水を全量フラスコ100mlにとり,(b)〜(d)の操作を行って吸光度を測定し,試料につい
て得た吸光度を補正する。
(f) 検量線から(d)での流出液10ml中の硝酸イオンの量を求め試料中の硝酸イオンの濃度[亜硝酸イオ
ン及び硝酸イオンの合量の濃度(硝酸イオン換算量)] (mgNO3-/l) を算出する。
(g) 別に,37.1.1によって試料中の亜硝酸イオンの濃度 (mgNO2-/l) を求める。
146
K 0101 : 1998
(h) 次の式によって試料中の硝酸イオン濃度 (mgNO3-/l) を算出する。
N=a−b×1.348
ここに,
N: 硝酸イオン (mgNO3-/l)
a: (f) で算出した試料中の亜硝酸イオン及び硝酸イオンの
合量 (mgNO3-/l)
b: (g) で求めた試料中の亜硝酸イオン (mgNO2-/l)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係
数
01
.
46
00
.
62
検量線 硝酸イオン標準液 (10μgNO3-/ml) 0.8〜8mlを全量フラスコ100mlに段階的にとり,(b)
〜(e)の操作を行って硝酸イオン (NO3-) の量と吸光度との関係線を作成する。
注(9) 注(2)による。ただし,凝集沈殿処理しても色が残る場合には,この方法を適用することはでき
ない。このような場合には,37.2.1又は37.2.2によって定量する。
(10) 酸化性物質及び還元性物質は妨害するので,あらかじめ除去する。残留塩素などの酸化性物質
が共存する場合には,当量の亜硫酸ナトリウム溶液 (6.3g/l) 又は亜ひ酸ナトリウム溶液[JIS K
8044に規定する三酸化二ひ素0.5gを水酸化ナトリウム溶液 (40g/l) 5mlに溶かした後,塩酸 (1
+11) 6mlを加えて水で100mlとする。]を加えた後,試験する。
また,亜硫酸イオンなどの還元性物質が共存する場合には,弱アルカリ性にして当量の過酸
化水素水 (1+100) を加えた後,試験する。
37.2.4 ブルシン吸光光度法 強い硫酸酸性で,硝酸イオンとブルシンとの反応によって生じる黄色の化合
物の吸光度を測定して硝酸イオンを定量する。
定量範囲:NO3- 5〜100μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (20+3) 水75mlをビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸500mlを徐々に加え,放冷し,密栓して保存する。
(c) プルシン・4-アミノベンゼンスルホン酸溶液 JIS K 8832に規定するブルシンn水和物(2, 3-ジメ
トキシストリキニジン-10-オン・n水和物)1gとJIS K 8586に規定するスルファニル酸(4-アミノ
ベンゼンスルホン酸)0.1gとをJIS K 8180に規定する塩酸3mlに溶かし,水で100mlとする。
(d) 硝酸イオン標準液 (1mgNO3-/ml) 37.2.3(1)(i)による。
(e) 硝酸イオン標準液 (0.1mgNO3-/ml) 硝酸イオン標準液 (1mgNO3-/ml) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 安全ピペット 1ml
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ紙5種C(又はろ紙6種)でろ過し,初めのろ液約50mlは捨てる(9)(10)(11)。その後のろ液
から2ml(NO3-として5〜100μgを含む。)ずつを2個のビーカー50ml(12) (A1) , (B1) にとる。
(b) (A1) にはブルシン・4-アミノベンゼンスルホン酸溶液1mlを安全ピペットを用いて加え, (B1) に
はブルシン・4-アミノベンゼンスルホン酸溶液の代わりに水1mlを加える。
(c) 別に,2個のビーカー50ml (A2) , (B2) に硫酸 (20+3) 10ml (13) ずつをとり,ビーカー (A2) にビ
147
K 0101 : 1998
ーカー (A1) の溶液を注意して移し,よく振り混ぜる。次に,ビーカー (A2) の溶液をビーカー (A1)
に移し,再びよく振り混ぜる。この操作を5回繰り返した後,暗所 (14) に約10分間放置する。
(d) ビーカー (B2) にはビーカー (B1) の溶液を注意して移し,よく振り混ぜる。次に,ビーカー (B2) の
溶液をビーカー (B1) に移し,再びよく振り混ぜる。この操作を5回繰り返した後,暗所に約10分
間放置する。
(e) ビーカー (A2) 及び (B2) に水10mlをそれぞれ加えて振り混ぜ,器壁を洗った後,洗液をそれぞれ
ビーカー (A1) 及び (B1) の溶液に合わせて振り混ぜる。冷暗所に約30分間放置する。
(f) ビーカー (A1) の溶液の一部を吸収セルにとり,ビーカー (B1) の溶液を対照液として波長410nm
付近の吸光度を測定する。
(g) 空試験として水2mlずつ2個のビーカー50ml(12) (A1) , (B1) にとり,(b)〜(f)の操作を行って吸光
度を測定し,試料について得た吸光度を補正する。
(h) 検量線から硝酸イオンの量を求め,試料中の硝酸イオンの濃度 (mgNO3-/l) を算出する。
検量線 硝酸イオン標準液 (0.1mgNO3-/ml) 2.5〜50mlを全量フラスコ100mlに段階的(15)にとり,水
を標線まで加える。その中からそれぞれ2mlをとり,(b)〜(g)の操作を行って硝酸イオン (NO3-) の
量と吸光度との関係線を作成する。
注(11) アルカリ性が強い場合は,硫酸 (1+5) を加えてpHを約7に調節してから試験する。
(12) この試験には,検量線作成時とほぼ同形のビーカー50ml4個を用いる。
(13) 自動ビュレット10mlを用いると操作しやすい。
(14) ビーカーにボール箱又は木箱などをかぶせておくとよい。
(15) 硝酸イオンとブルシンの反応によって生じた黄色は,厳密にはランバート−ベアの法則に従わ
ないので,硝酸イオン標準液の段階を細かくする必要がある。
備考7. 鉄 (II) ,鉄 (III) 及びマンガン (IV) が共存すると正の誤差を生じるが,これらの濃度が
1mg/l以下ならば差し支えない。
8. 硝酸イオンとブルシンによる反応は,温度の上昇とともに速くなる。したがって,検量線作
成時と同一温度で操作する。
37.2.5 イオンクロマトグラフ法 試料中の硝酸イオンをイオンクロマトグラフ法で定量する。この方法を
適用する場合には,3.3の保存処理を行わず,試料採取後直ちに試験する。
定量範囲: NO3- 0.5〜40mg/l(16),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって
異なる。)
注(16) サプレッサと組み合わせる方式の場合には,0.1〜40mgNO3-/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 32.5(1)(b)による。
(c) 再生液 32.5(1)(c)による。
(d) 硝酸イオン標準液 (5mgNO3-/ml) JIS K 8548に規定する硝酸カリウムをあらかじめ105±2℃で約
2時間加熱し,デシケーター中で放冷する。その0.815gをとり,少量の水に溶かして全量フラスコ
100mlに移し入れ,水を標線まで加える。0〜10℃の暗所に保存する。
(e) 硝酸イオン標準液 (0.5mgNO3-/ml) 硝酸イオン標準液 (5mgNO3-/ml) 10mlを全量フラスコ100ml
にとり,水を標線まで加える。使用時に調製する。
(f) 陰イオン混合標準液 [(0.1mgCl-, 0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] 32.5(1)(f)による。
148
K 0101 : 1998
(2) 器具及び装置 器具及び装置は,32.5(2)による。ただし,検出器は,紫外吸光検出器を用いてもよい。
(3) 準備操作 準備操作は,32.5(3)による。
(4) 操作 操作は,次のとおり行う。
(a) 32.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上の硝酸イオンに相当するピークについて,指示値(17)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
いて得た指示値(17)を補正する。
(d) 検量線から硝酸イオンの濃度を求め,試料中の硝酸イオンの濃度 (mgNO3-/l) を算出する。
検量線 硝酸イオン標準液 (0.5mgNO3-/ml) (18)0.1〜8mlを段階的に全量フラスコ100mlにとり,水
を標線まで加え,(a)及び(b)の操作を行ってそれぞれの硝酸イオンに相当する指示値(17)を読み取る。
別に,空試験として水について(a)及び(b)の操作を行ってそれぞれの硝酸イオンに相当する指示値を
補正した後,硝酸イオン (NO3-) の量と指示値との関係線を作成する。検量線の作成は,試料の測
定時に行う。
注(17) ピーク高さ又はピーク面積
(18) 硝酸イオン以外の陰イオンを同時に試験する場合には,陰イオン混合標準液 [(0.1mgCl-,
0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] を用いる。
備考9. 硝酸イオンの濃度が1mg/lのとき臭化物イオンは200mg/l以下及び硫酸イオン500mg/l以下な
らば妨害しない。
10. 32.の備考11.による。
38. 有機体窒素 試料を前処理して有機物を分解し,有機体窒素をアンモニウムイオンに変え,蒸留分離
した後,インドフェノール青吸光光度法又は中和滴定法でアンモニウムイオンを定量し,別に,処理前の
試料中のアンモニウムイオンを定量して差し引き,有機体窒素を求める。
有機体窒素は変化しやすいので,試験は直ちに行う。直ちに行えない場合には,3.3によって保存し,で
きるだけ早く試験する。
38.1 前処理(ケルダール法) 試料に,硫酸銅,硫酸及び硫酸カリウムを加え,加熱して有機物を分解
する。次に,水酸化ナトリウムを加えてアルカリ性とした後,蒸留し,留出したアンモニアを硫酸
(25mmol/l) に吸収させる。
備考1. この方法では,アミノ酸,ポリペプチド,たん白質などは分解しやすいが,ニトロ,ニトロ
ソ,アゾ及び複素環式化合物(特に,ピリジン環をもつ化合物)などは完全には分解できな
い。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 JIS K 8951に規定するもの。
(c) 硫酸 (25mmol/l) 36.1.2(1)(b)による。
(d) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(e) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(f) 水酸化ナトリウム溶液 (500g/l) JIS K 8576に規定する水酸化ナトリウム50gを水に溶かして
100mlとする。使用時に調製する。
(g) 硫酸カリウム JIS K 8962に規定するもの。
149
K 0101 : 1998
(h) 硫酸銅 (II) 五水和物 JIS K 8983に規定するもの。粉末にして用いる。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗う。
(b) ケルダールフラスコ 200ml 使用前に水でよく洗う。
(c) 蒸留装置 36.1.2(2)(a)による。使用前に水でよく洗う。
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(1)をビーカー500mlにとり,硫酸 (1+35) を加えて弱酸性とし,加熱して約30mlになる
まで濃縮する。
(b) 放冷後,少量の水で内容物をケルダールフラスコ200mlに洗い移す。
(c) 硫酸10ml,硫酸カリウム5g及び硫酸銅 (II) 五水和物2gを加え,加熱して硫酸白煙を発生させ,
引き続き約30分間強熱して有機物を分解(2)する。
(d) 放冷後,少量の水を加えて加熱溶解し,水で蒸留フラスコに洗い移して約300mlとする。
(e) 蒸留フラスコを図36.1のように連結し,受器にはメスシリンダー(有栓形)200mlを用い,硫酸
(25mmol/l) 50mlを入れておく(3)。
(f) 蒸留フラスコ上部の注入漏斗から水酸化ナトリウム溶液 (500g/l) 40mlを加えた後,36.1.2(3)(d)〜(f)
の操作を行う。
(g) 空試験として水30mlをとり,(c)〜(f)の操作を行う。
注(1) インドフェノール青吸光光度法で定量する場合には,有機体窒素をNとして32μg以上,中和滴
定法の場合には,有機体窒素をNとして0.23mg以上,有機体窒素とアンモニウムイオンの合量
をNとして30mg以下を含むようにとる。
(2) フラスコ中の溶液は,無色又は淡黄色になる。
(3) 中和滴定法の場合には,メスシリンダー(有栓形)200mlの代わりに三角フラスコ500mlを用
いて,36.1.2(1)(b)の硫酸 (25mmol/l) 50mlを正しく加え,指示薬として13.1(1)(a)のメチルレッ
ド−ブロモクレゾールグリーン混合溶液5〜7滴を加える。
備考2. この方法では,硝酸イオン,亜硝酸イオンは,有機体窒素の定量の妨害にならない。
3. 試料中のアンモニウムイオンをあらかじめ除去した後,有機体窒素を定量する場合には,
36.1.2(3)(a)〜(e)を行い,この残液について(3)(a)〜(f)の前処理を行う。
38.2 インドフェノール青吸光光度法 前処理(ケルダール法)による留出液についてインドフェノール
青吸光光度法によってアンモニウムイオンを定量して,試料中に含まれるアンモニウムイオン及び有機体
窒素から生じたアンモニウムイオンの合量を求め,別に,試料中のアンモニウムイオンを定量して差し引
き,有機体窒素を算出する。
定量範囲:N 4〜80μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) ナトリウムフェノキシド溶液 36.2(1)(d)による。
(c) 次亜塩素酸ナトリウム溶液(有効塩素10g/l) 36.2(1)(e)による。
(d) アンモニウムイオン標準液 (1mgNH4+/ml) 36.2(1)(f)による。
(e) アンモニウムイオン標準液 (10μgNH4+/ml) 36.2(1)(g)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗う。
150
K 0101 : 1998
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 38.1(3)(f)で得た留出液の適量(Nとして4〜80μgを含む。)を全量フラスコ50mlにとり,水を加え
て約25mlとした後,36.2(3)(b)〜(e)の操作を行って吸光度を測定する。
(b) 空試験として38.1(3)(g)で得た流出液から(a)と同量を分取し,(a)の操作を行って吸光度を測定し,
試料について得た吸光度を補正する。
(c) 36.2(3)の検量線から,分取した留出液中のアンモニウムイオンの量 (mg) を求める。
(d) 別に,36.2によって試料中のアンモニウムイオンの濃度 (mgNH4+/l) を求める。
(e) 次の式によって試料中の有機体窒素の濃度 (mgN/l) を算出する。
7766
.0
200
1000
2
1
×
−
×
×
=
A
V
V
a
N
ここに,
N: 有機体窒素 (mgN/l)
a: (c)の留出液中のアンモニウムイオンの量 (mgNH4+)
V1: 38.1(3)(a)で用いた試料 (ml)
V2: (a)で分取した留出液 (ml)
A: (d)で求めた試料中のアンモニウムイオン (mgNH4+/l)
0.7766: アンモニウムイオンを窒素の相当量に換算するときの係
数
04
.
18
01
.
14
38.3 中和滴定法 前処理(ケルダール法)による留出液について中和滴定法によってアンモニアを定量
して,試料中に含まれるアンモニウムイオン及び有機体窒素から生じたアンモニウムイオンの合量を求め,
別に,試料中のアンモニウムイオンを定量して差し引き,有機体窒素を算出する。
定量範囲:N 0.23〜30mg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 50mmol/l水酸化ナトリウム溶液 36.3(1)(d)による。
(2) 操作 操作は,次のとおり行う。
(a) 38.1(3)(f)で得た留出液の全量を用い,36.3(2)(a)の滴定操作を行う。
(b) 空試験として38.1(3)(g)で得た留出液の全量を用いて,(a)と同様の滴定操作を行う。
(c) 別に,36.3によって試料中のアンモニウムイオンの濃度 (mgNH4+/l) を求める。
(d) 次の式によって試料中の有機体窒素の濃度 (mgN/l) を算出する。
6
776
.0
700
.0
000
1
)
(
×
−
×
×
×
−
=
A
V
f
a
b
N
ここに,
N: 有機体窒素 (mgN/l)
b: 空試験に要した50mmol/l水酸化ナトリウム溶液 (ml)
a: 滴定に要した50mmol/l水酸化ナトリウム溶液 (ml)
f: 50mmol/l水酸化ナトリウム溶液のファクター
0.700: 50mmol/l水酸化ナトリウム溶液1mlの窒素相当量 (mg)
V: 38.1(3)(a)で用いた試料 (ml)
A: (c)で求めた試料中のアンモニウムイオン (mgNH4+/l)
0.776 6: アンモニウムイオンを窒素の相当量に換算するときの
係数
04
.
18
01
.
14
151
K 0101 : 1998
備考4. 38.1(3)(e)の硫酸 (25mmol/l) の代わりにほう酸溶液(飽和)を用いてもよい。その場合は36.
の備考5.の操作を行う。ただし,空試験は,水30mlを用いて38.1(3)(c)〜(f)の操作を行って得
られた留出液について,試料と同様に滴定した値を用いる。
39. 全窒素 亜硝酸イオンと硝酸イオンに相当する窒素と,アンモニウムイオンと有機体窒素に相当する
窒素とを求めて合計する総和法,又は全窒素化合物を硝酸イオンに変えた後の紫外線吸光光度法,硫酸ヒ
ドラジニウム還元法,銅・カドミウムカラム還元法若しくは熱分解法を適用する。
窒素化合物は変化しやすいから,試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって
保存し,できるだけ早く試験する。
39.1 総和法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモニウムイオン及び一部の有機窒素化
合物の分解で生じたアンモニアを除いた後,デバルタ合金を加えて亜硝酸イオン及び硝酸イオンを還元し
てアンモニアとし,蒸留によって分離し,インドフェノール青吸光光度法で窒素の量を定量する。別に,
試料に硫酸銅,硫酸カリウム,硫酸を加えて加熱分解して有機体窒素をアンモニウムイオンに変えた後,
アルカリ性として蒸留し,試料中に含まれるアンモニウムイオンとともに蒸留分離し,インドフェノール
青吸光光度法によってその窒素の量を定量する。先に求めた亜硝酸イオン,硝酸イオン相当の窒素量とを
合わせて,全窒素の濃度を算出する。
定量範囲:N 8〜160μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸 (25mmol/l) 36.1.2(1)(b)による。
(c) 硫酸 (1+35) 36.1.2(1)(c)による。
(d) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(e) 水酸化ナトリウム溶液 (300g/l) 37.2.1(1)(e)による。
(f) デバルダ合金 37.2.1(1)(f)による。
(g) 硫酸カリウム JIS K 8962に規定するもの。
(h) 硫酸銅 (II) 五水和物 JIS K 8983に規定するもの。粉末にして用いる。
(i) ナトリウムフェノキシド溶液 36.2(1)(d)による。
(j) 次亜塩素酸ナトリウム溶液(有効塩素10g/l) 36.2(1)(e)による。
(k) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(l) アンモニウムイオン標準液 (1mgNH4+/ml) 36.2(1)(f)による。
(m) アンモニウムイオン標準液 (10μgNH4+/ml) 36.2(1)(g)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具類 使用前に水でよく洗う。
(b) ケルダールフラスコ 200ml 使用前に水でよく洗う。
(c) 蒸留装置 36.1.2(2)(a)による。使用前に水でよく洗う。
(d) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料50ml(1)をとり,中性でない場合には水酸化ナトリウム溶液 (40g/l) 又は硫酸 (1+35) でpHを
約7に調節する。
(b) 37.2.1(3)(b)〜(f)の操作を行う。

152
K 0101 : 1998
(c) 試料について得た留出液から25ml(2)を全量フラスコ50mlにとる。
(d) 36.2(3)(b)〜(e)の操作を行って吸光度を測定する。
(e) 空試験として水約50mlをとり,水酸化ナトリウム溶液 (300g/l) 10mlを加えた後,37.2.1(3)(d)〜(f)
の操作を行い,得られた留出液について(c)及び(d)の操作を行って吸光度を測定し,(d)で得た吸光
度を補正する。
(f) 36.2(3)の検量線から(c)で分取した留出液25ml中のアンモニウムイオンの量 (mg) を求める。
(g) 別に,試料50ml(3)をケルダールフラスコ200mlにとり,38.1(3)(c)〜(f)の操作を行う。
(h) この留出液の25ml(2)を全量フラスコ50mlにとり,36.2(3)(b)〜(e)の操作を行って吸光度を測定する。
(i) 空試験として水50mlをとり,(g)及び(h)の操作を行って(h)で得た吸光度を補正する。
(j) 36.2(3)の検量線から(h)で分取した留出液25ml中のアンモニウムイオンの量 (mg) を求める。
(k) 次の式によって試料中の全窒素の濃度 (mgN/l) を算出する(4)。
7766
.0
25
200
50
000
1
7766
.0
25
200
50
000
1
×
×
×
+
×
×
×
=
b
a
N
ここに,
N: 全窒素 (mgN/l)
a: (f)の操作で得たアンモニウムイオン (mg)
b: (j)の操作で得たアンモニウムイオン (mg)
0.7766: アンモニウムイオンを窒素の相当量に換算するときの
係数
04
.
18
01
.
14
注(1) 全窒素の低濃度のものを定量する必要がある場合は試料の量を増加する。ただし,この場合は
(k)の式中のa×50
1000に代え,a×V
1000を用いる。
ここに,V:試料 (ml)
(2) 留出液200ml中のアンモニウムイオンが0.8mg以上の場合は,留出液の適当量(NH4+が0.4mg
未満になる量)を硫酸 (25mmol/l) 25mlが入った全量フラスコ100mlにとり,水を標線まで加え,
これから25mlを分取する。
(3) 全窒素の低濃度のものを定量する必要がある場合には,試料の量を増加し,38.1(3)(a)及び(b)の
操作を行う。ただし,この操作を行った場合は(k)の計算式中のb×50
1000に代え,b×V
1000を用いる。
ここに,V:試料 (ml)
(4) (c)又は(h)の操作で,注(2)の操作を行った場合には,(k)の算出式中のa又はbに代え,それぞれ
a×c
100又はb×b
100を用いる。ただし,c又はdは,それぞれ注(2)の操作において,全量フラスコ
100mlにとった溶液の量 (ml)。
39.2 紫外線吸光光度法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120℃に加熱して
窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液のpHを2〜3とした後,硝酸イオ
ンによる波長220nmの吸光度を測定して定量する。この方法は,試料中の有機物が分解されやすく,少量
であり,また,試験に影響する量の臭化物イオン,クロムなどを含まない試料に適用する。
定量範囲:N5〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+16) JIS K 8180に規定する塩酸を用いて調製する。
(c) 塩酸 (1+500) JIS K 8180に規定する塩酸を用いて調製する。
(d) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 JIS K 8826に規定する水酸化ナトリウム(窒素
153
K 0101 : 1998
測定用)40gを水500mlに加えた後,JIS K 8253に規定するペルオキソ二硫酸カリウム(窒素・り
ん測定用)15gを溶かす。使用時に調製する(5)。
(e) 窒素標準液 (0.1mgN/ml) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110℃で約3時
間加熱し,デシケーター中で放冷する。その0.722gを少量の水に溶かし,全量フラスコ1 000mlに
移し入れ,水を標線まで加える。0〜10℃の暗所で保存する。
(f) 窒素標準液 (20μgN/ml) 窒素標準液 (0.1mgN/ml) 50mlを全量フラスコ250mlにとり,水を標線ま
で加える。使用時に調製する。
注(5) この溶液の窒素含有量は,0.4mg/l以下でなければならない。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分解瓶 耐圧の四ふっ化エチレン樹脂瓶又は耐熱・耐圧のガラス瓶(容量約100ml)で,高圧蒸気
滅菌器中(約120℃)で使用できるもの(6)。
(b) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので約120℃に加熱できるもの。
(c) 光度計 分光光度計
(d) 吸収セル 石英ガラス製のもの。
注(6) ガラス製アンプル(容量約100ml)で高圧蒸気滅菌器中(約120℃)で使用できるものを用いて
もよい。
(3) 操作 操作は,次のとおり行う。
(a) 試料(7)50mlを分解瓶にとる。
(b) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液10mlを加え,直ちに密栓した後,混合する。
(c) 高圧蒸気滅菌器に入れて加熱し,約120℃に達してから30分間加熱分解を行う。
(d) 分解瓶を高圧蒸気滅菌器から取り出し,放冷する。
(e) 上澄み液(8)25mlをビーカー50mlに分取する。
(f) 塩酸 (1+16) 5ml(9)を加えて溶液のpHを2〜3に調節する。
(g) 溶液の一部を吸収セルに移し,波長220nmにおける吸光度を測定する。
(h) 空試験として水50mlを分解瓶にとり,(b)〜(g)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
(i) 検量線から(e)で分取した溶液中の全窒素の量を求め,次の式によって試料中の全窒素の濃度
(mgN/l) を算出する(10)。
50
1000
25
60×
×
=a
N
ここに, N: 全窒素 (mgN/l)
a: (e)で分取した溶液25ml中の全窒素 (mg)
検量線 窒素標準液 (20μgN/ml) 1〜10mlを段階的に全量フラスコ100mlにとり,それぞれに水を標
線まで加える。その25mlをそれぞれビーカー50mlに分取し,塩酸 (1+500) 5mlを加えた後,一部
を吸収セルに移し,波長220nmの吸光度を測定する。別に,空試験として水25mlをビーカー50ml
にとり,塩酸 (1+500) 5mlを加えた後,波長220nmの吸光度を測定し,窒素標準液について得た吸
光度を補正する。採取した溶液25ml中の窒素 (N) の量と吸光度との関係線を作成する。
注(7) 試料50ml中の全窒素が0.1mg以上の場合には,試料の適量(Nとして0.2mg未満を含む。)を全
量フラスコ100mlにとり,水を標線まで加えたものを用いる。ただし,試料50ml中の全窒素が,
0.1mg以上でpHが5〜9の範囲にない場合には,試料の適量(Nとして0.2mg未満を含む。)を
154
K 0101 : 1998
とり,塩酸 (1+11) 又は水酸化ナトリウム溶液 (40g/l) で中和した後,全量フラスコ100mlに移
し入れ,水を標線まで加えたものを用いる。
(8) 水酸化物の沈殿を含まないように注意する。必要に応じ,孔径1μm以下のガラス繊維ろ紙を用
いてろ過し,初めのろ液5〜10mlを捨てた後のろ液を用いる。
(9) 分解後の溶液に水酸化物の沈殿を生じた場合には,注(8)によってろ過したろ液25mlをとり,水
酸化物の生成量に応じて濃度を低めた塩酸5mlを添加し,pHを2〜3に調節する。
(10) (a)の操作において,注(7)の操作を行った試料を用いた場合には,次の式によって試料中の全窒
素の濃度 (mgN/l) を算出する。
V
a
N
100
50
000
1
25
60
×
×
×
=
ここに, N: 全窒素 (mgN/l)
a: (e)で分取した溶液25ml中の全窒素 (mg)
V: 試料 (ml)
備考1. 試料50ml中の全窒素が0.1mg未満でpHが5〜9の範囲にない場合には,試料の適量 (50〜
100ml) をとり,塩酸 (1+11) 又は水酸化ナトリウム溶液 (40g/l) を用いて中和し(中和に用
いた両溶液の量を求めておく。),この溶液から50mlを分解瓶にとり,(3)(b)〜(e)の操作を行
う。ただし,この場合は,次の式を用いる。
V
b
V
a
N
+
×
×
×
=
50
000
1
25
60
ここに, N: 全窒素 (mgN/l)
a: 前処理後に分取した試料25ml中の全窒素 (mg)
b: 中和に要した塩酸 (1+11) 及び水酸化ナトリウム溶液
(40g/l) (ml)
V: 試料 (ml)
2. (3)(g)で吸収セルに移す溶液の全窒素の濃度が0.4mg/l未満の場合には,吸収セル50mmを用
いる。ただし,空試験,検量線の作成には,いずれにも吸収セル50mmを用いる。検量線は
窒素標準液 (20μgN/ml) を5倍に薄めた窒素標準液 (4μgN/ml) の1〜10mlをとり,(3)の検量
線と同じ操作で作成する。
3. この方法では臭化物イオン10mg/l,クロム0.1mg/l程度で妨害することがある。このような
試料には,この方法は適用しない。
39.3 硫酸ヒドラジニウム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120℃に
加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅を触
媒として硫酸ヒドラジニウムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法
によって定量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である試
料に適用する。
定量範囲:N0.33〜3.3μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 39.2(1)(d)による。
(c) 銅−亜鉛溶液 JIS K 8983に規定する硫酸銅 (II) 五水和物0.08gとJIS K 8953に規定する硫酸亜鉛
七水和物1.76gを水に溶かして200mlとし,その5mlを水で薄めて250mlとする。
155
K 0101 : 1998
(d) 硫酸ヒドラジニウム溶液 (7g/l) JIS K 8992に規定する硫酸ヒドラジニウム3.5gを水に溶かして
500mlとする。
(e) 硫酸ヒドラジニウム溶液 (0.7g/l) 硫酸ヒドラジニウム溶液 (7g/l) を水で10倍に薄める。使用時
に調製する。
(f) 4-アミノベンゼンスルホンアミド溶液 37.1.1(1)(b)による。
(g) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 37.1.1(1)(c)による。
(h) 窒素標準液 (20μgN/ml) 39.2(1)(f)による。
(i) 窒素標準液 (4μgN/ml) 窒素標準液 (20μgN/ml) 20mlを全量フラスコ100mlにとり,水を標線まで
加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分解瓶 39.2(2)(a)による。
(b) 共栓試験管 材質及び形状が同じものを用いる。
(c) 水浴 35±1℃に温度が調節できるもの。
(d) 高圧蒸気滅菌器 39.2(2)(b)による。
(e) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(11)(12)50mlを分解瓶にとる。
(b) 39.2(3)(b)〜(d)の操作を行う。
(c) 上澄み液(8)(13)10mlを共栓試験管にとる。
(d) 銅-亜鉛溶液1mlを加えて振り混ぜた後,硫酸ヒドラジニウム溶液 (0.7g/l) 1mlを加えて振り混ぜ,
35±1℃の水浴中に浸す。
(e) 2時間後水浴から取り出し,室温まで冷却する。
(f) 37.1.1(3)(b)及び(c)の操作を行う。
(g) 空試験として水50mlを分解瓶にとり,(b)〜(f)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
(h) 検量線から分解瓶にとった溶液50ml中の全窒素の量を求め(14),次の式によって試料中の全窒素の
濃度 (mgN/l) を算出する。
50
1000
×
=a
N
ここに, N: 全窒素 (mgN/l)
a: 分解瓶にとった溶液50ml中の全窒素 (mg)
検量線 窒素標準液 (4μgN/ml) 1〜10mlを段階的に全量フラスコ100mlにとり,それぞれに水を標
線まで加える。この溶液について(a)〜(g)の操作を行って分解瓶にとった溶液50ml中の窒素 (N) の
量と吸光度との関係線を作成する。
注(11) 注(7)による。ただし,試料中の全窒素の濃度 (mgN/l) の算出には,次の式を用いる。
V
a
N
100
50
000
1
×
×
=
ここに, N: 全窒素 (mgN/l)
a: 分解瓶にとった溶液50ml中の全窒素 (mg)
V: 試料 (ml)
(12) 備考1.による。ただし,全窒素の濃度 (mgN/l) の算出には,次の式を用いる。
156
K 0101 : 1998
V
b
V
a
N
+
×
×
=
50
000
1
ここに, N: 全窒素 (mgN/l)
a: 分解瓶にとった溶液50ml中の全窒素 (mg)
b: 中和に要した塩酸 (1+11) 及び水酸化ナトリウム溶液
(40g/l) (ml)
V: 試料 (ml)
(13) 分解瓶にとった溶液50ml中の窒素が20μg以上の場合には,上澄み液の適量(Nとして30μg
未満となる量。)を全量フラスコ100mlにとり,水酸化ナトリウム溶液 (40g/l) 5mlを加えた後,
水を標線まで加えたものを用いる。
(14) (c)の操作で注(13)を行った場合には,検量線によって求めた窒素の量 (mg) にc
100を乗じることに
よって,分解瓶にとった試料50ml中の全窒素の量 (mg) を求める。ただし,cは全量フラスコ
100mlにとった上澄み液の量 (ml) 。
備考4. 試料が海水などの場合は,含まれる無機物が硝酸イオンの還元率に影響するので,次の標準
添加法を行う。
試料40mlを分解瓶にとり,水10mlを加える。以下,(3)(b)〜(g)の操作を行って吸光度を
測定し,下記の検量線から試料40ml中の全窒素の量 (mg) を求める。別に,空試験として水
50mlを分解瓶にとり,(3)(b)〜(g)の操作を行って吸光度を測定し,(3)の検量線から,相当す
る窒素の量 (mg) を求める。次の式によって試料中の全窒素の濃度 (mgN/l) を算出する。
40
000
1
)
(
×
−
=
b
a
N
ここに, N: 全窒素 (mgN/l)
a: 試料40ml中の全窒素 (mg)
b: 空試験で得た窒素 (mg)
検量線 窒素標準液 (4μgN/ml) 1〜8mlを段階的に分解瓶にとり,それぞれに試料40mlを加えた後,
水を加えて50mlとし,(3)(b)〜(f)の操作を行って吸光度を測定し,その値から試料40mlを用いて得た
吸光度を差し引いて補正する。
なお,海水など多量のマグネシウムイオンが存在する試料の場合は,分解操作を行った溶液のpH
が低下して,マグネシウムの一部が上澄み液又はろ液に混入し,硝酸イオンの還元率を低下させる。
このため,分解後の溶液に水酸化ナトリウム溶液 (40g/l) を加えてpHを12.6〜12.8とした後の上澄み
液を用いる。この操作を行った場合には,全窒素の算出式については,希釈による補正を行う。
試料40ml中の窒素の量が10μg以上の場合には,試料の適量(Nとして25μg未満を含む。)をと
り,注(7)に準じた操作を行い,これから40mlを分解瓶にとる。
また,試料40ml中の窒素量が10μg未満でpHが5〜9の範囲にない場合には,備考1.に準じた中和
操作を行い,これから40mlを分解瓶にとる。これらの操作を行った場合には,検量線の作成におい
ても,この溶液を用い,全窒素の算出式については,それぞれ希釈に伴う補正を行う。
39.4 銅・カドミウムカラム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120℃
に加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅・
カドミウムカラムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法によって定
量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である試料に適用す
る。
157
K 0101 : 1998
定量範囲:N0.2〜2μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(c) 塩化アンモニウム-アンモニア溶液 37.2.3(1)(c)による。
(d) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 39.2(1)(d)による。
(e) カラム活性化液 37.2.3(1)(d)による。
(f) 銅・カドミウムカラム充てん剤 37.2.3(1)(e)による。
(g) カラム充てん液 37.2.3(1)(f)による。
(h) 4-アミノベンゼンスルホンアミド溶液 37.1.1(1)(b)による。
(i) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 37.1.1(1)(c)による。
(j) 窒素標準液 (0.1mgN/ml) 39.2(1)(e)による。
(k) 窒素標準液 (2μgN/ml) 窒素標準液 (0.1mgN/ml) 10mlを全量フラスコ500mlにとり,水を標線ま
で加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分解瓶 39.2(2)(a)による。
(b) 高圧蒸気滅菌器 39.2(2)(b)による。
(c) 銅・カドミウムカラム 37.2.3(2)(a)による。
(d) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(15)(16)50mlを分解瓶にとる。
(b) 39.2(3)(b)〜(d)の操作を行う。
(c) 分解瓶に塩酸 (1+11) 10mlを加えて振り混ぜた後,溶液を全量フラスコ100ml(17)に移す。
(d) 分解瓶の内壁を少量の水で数回洗浄して洗液を(c)の全量フラスコ100mlに加える。
(e) 塩化アンモニウム-アンモニア溶液10mlを加え,水を標線まで加えて還元用溶液とする。
(f) 37.2.3(3)(c)及び(d)の操作を行う。
(g) 空試験として水50mlを分解瓶にとり,(b)〜(f)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
(h) 検量線から還元用溶液中の全窒素の量を求め,次の式によって試料中の全窒素の濃度 (mgN/l) を算
出する(18)。
50
000
1
×
=a
N
ここに, N: 全窒素 (mgN/l)
a: 還元用溶液100ml中の全窒素 (mg)
検量線 窒素標準液 (2μgN/ml) 1〜10mlを段階的に全量フラスコ100mlにとり,(e)及び(f)の操作を
行って吸光度を測定する。別に,水約50mlを全量フラスコ100mlにとり,(e)及び(f)の操作を行っ
て吸光度を測定し,窒素標準液 (2μgN/ml) について得た吸光度を補正し,窒素 (N) の量と吸光度
との関係線を作成する。
注(15) 注(7)による。ただし,試料中の全窒素の濃度 (mgN/l) の算出には,次の式を用いる。
158
K 0101 : 1998
V
a
N
100
50
000
1
×
×
=
ここに, N: 全窒素 (mgN/l)
a: 還元用溶液100ml中の全窒素 (mg)
V: 試料 (ml)
(16) 備考1.による。ただし,試料中の全窒素の濃度 (mgN/l) の算出には,次の式を用いる。
V
b
V
a
N
+
×
×
=
50
000
1
ここに, N: 全窒素 (mgN/l)
a: 還元用溶液100ml中の全窒素 (mg)
b: 中和に用いた塩酸 (1+11) 及び水酸化ナトリウム溶液
(40g/l) (ml)
V: 試料 (ml)
(17) (a)で分解瓶にとった溶液50ml中の全窒素が20μg以上の場合には,全量フラスコ200〜500ml
を用い,(e)の操作において塩化アンモニウム-アンモニア溶液を最終液量100ml当たり10mlと
なるように添加した後,水を標線まで加えたものを還元用溶液とする。
(18) (c)の操作において注(17)を行った場合は,算出式100cを乗じて補正する。ただし,cは用いた全量
フラスコの容量 (ml) 。
39.5 熱分解法 試料中の窒素化合物を熱分解してアンモニア又は窒素を生成させ,それらを定量する。
若しくは一酸化窒素に変えた後,化学発光によって窒素を定量し,それぞれ全窒素を求める方法である。
定量範囲:N1〜200mg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 全窒素標準液 (0.2mgN/ml) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110℃で約3
時間加熱し,デシケーター中で放冷する。その1.444gを少量の水に溶かして全量フラスコ1 000ml
に移し入れ,水を標線まで加える。0〜10℃の暗所に保存する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロシリンジ 20〜150μl
(b) ホモジナイザー又はミキサー
(c) 全窒素分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 全窒素分析装置を作動できる状態にする。
(b) 全窒素標準液 (0.2mgN/ml) の定量(例えば,20μl)をマイクロシリンジで全窒素分析装置の試料注
入部から注入し,指示値(ピーク高さ)が最大目盛の約80%になるように装置の感度を調節する。
(c) (b)の操作を繰り返し,指示値が一定になることを確かめる。
(d) 試料(19)をよく振り混ぜて均一にした後,その一定量[例えば,(b)と同量]をマイクロシリンジで
試料注入部から注入して指示値を読み取り,(c)と比較して試料中の概略の全窒素の濃度を求める。
注(19) 全窒素の濃度が200mgN/l以上の試料の場合には,水で薄めた後,試験する。
(4) 検量線の作成 検量線の作成は,次のとおり行う。
(a) (3)の準備操作(d)で求めた試料中の概略の全窒素の濃度がほぼ中央になるように,全窒素標準液
(0.2mgN/ml) を全量フラスコ100mlに段階的にとり,水を標線まで加えて各濃度の全窒素標準液を
159
K 0101 : 1998
調製する。
(b) (a)で調製した全窒素標準液の最高濃度のものの一定量[例えば,(3)(b)と同量]をマイクロシリン
ジで試料注入部に注入し,指示値が最大目盛値の80%になるように全窒素分析装置の感度を調節す
る。
(c) 順次(a)で調製した各濃度の全窒素標準液の定量[(b)で定めた量]をマイクロシリンジで試料注入部
から注入し,指示値を読み取る。
(d) 空試験として(c)と同量の水をマイクロシリンジでとり,(c)と同様に操作して指示値を読み取り,(c)
の指示値を補正して窒素 (N) の濃度と指示値との関係線を作成する。
(5) 操作 操作は,次のとおり行う。
(a) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
(b) 試料の一定量[例えば,(4)(b)と同量]をマイクロシリンジで装置の試料注入部に注入し,指示値を
読み取る。
(c) 空試験として(b)と同量の水をマイクロシリンジでとり,(b)と同様に操作して指示値を読み取り,(b)
の指示値を補正する。
(d) あらかじめ作成した検量線から,注入試料中の全窒素の濃度を求め,試料中の全窒素の濃度 (mgN/l)
を算出する。
備考5. 全窒素分析装置には各種のものがある。アンモニアを生成させて定量する方式としては,試
料を水素気流中で熱分解し,全窒素を触媒によってアンモニアとした後,電量滴定法又は電
気伝導度測定法によるものがある。
窒素を生成させて定量する方式としては,試料をヘリウム気流中で熱分解し,触媒によっ
て全窒素を窒素とした後,熱伝導度測定法によるものがある。
また,化学発光方式による方式としては,試料を酸素気流中で熱分解して全窒素を一酸化
窒素とし,更にこれをオゾンと反応させ,二酸化窒素に酸化するとき生じる化学発光(波長
650〜900nm)を測定するものがある。
40. 硫化物イオン (S2-) 硫化物イオンの定量には,メチレンブルー吸光光度法又はよう素滴定法を適用
する。
硫化物イオンは不安定で,酸化されたり,硫化水素として空気中に散逸したりするので,試料採取後直
ちに試験を行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験する。
40.1 メチレンブルー吸光光度法 硫化物イオンが,塩化鉄 (III) の存在の下でN, N'-ジメチル-p-フェニレ
ンジアミンと反応して生成するメチレンブルー[3, 7-ビス(ジメチルアミノ)フェノチアジン-5-イウム]
の吸光度を測定して硫化物イオンを定量する。
定量範囲:S2- 5〜40μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+1) 4.4(1)(b)による。
(b) N, N´-ジメチル−p-フェニレンジアンモニウム溶液 二塩化N, N'-ジメチル-p-フェニレンジアンモ
ニウム0.8gに硫酸 (1+1) を加えて100mlとする。使用時に調製する。
(c) 塩化鉄 (III) 溶液 JIS K 8142に規定する塩化鉄 (III) 六水和物10gを水に溶かして100mlとする。
(d) りん酸水素二アンモニウム溶液 (400g/l) JIS K 9016に規定するりん酸水素二アンモニウム40gを
160
K 0101 : 1998
水に溶かして100mlとする。
(e) 硫化物イオン標準液 (1mgS2-/ml) JIS K 8949に規定する硫化ナトリウム九水和物の結晶7.6gをと
り,少量の水で表面を洗い,これをろ紙上にとって水を除いた後,2.(12)(a)の溶存酸素を含まない
水に溶かして1lとし,気密容器に移し入れ,使用時に標定する。
標定 よう素溶液 (50mmol/l) (1)20mlをとり,共栓三角フラスコ300mlに入れ,塩酸 (1+1) 0.5ml
を加える。次に,この硫化物イオン標準液20mlを全量ピペットでとり,ピペットの先端をこのよ
う素溶液中に入れて加える(2)。直ちに密栓して振り混ぜて数分間放置する。0.1mol/lチオ硫酸ナト
リウム溶液(3)で滴定し,溶液の黄色が薄くなってから,指示薬としてでんぷん溶液 (10g/l) (4)1mlを
加え,生じたよう素でんぷんの青い色が消えるまで滴定する。別に,よう素溶液 (50mmol/l) 20ml
を共栓三角フラスコ300mlにとり,塩酸 (1+1) 0.2mlを加えた後,同様に0.1mol/lチオ硫酸ナトリ
ウム溶液で滴定する。次の式によって硫化物イオン標準液1ml中の硫化物イオンの量を算出する。
603
.1
20
1
)
(
×
×
×
−
=
f
a
b
S
ここに,
S: 硫化物イオン標準液 (mgS2-/ml)
a: 滴定に要した0.1mol/lチオ硫酸ナトリウム溶液 (ml)
b: よう素溶液 (50mmol/l) 20mlに相当する0.1mol/lチオ硫酸
ナトリウム溶液 (ml)
f: 0.1mol/lチオ硫酸ナトリウム溶液のファクター
1.603: 0.1mol/lチオ硫酸ナトリウム溶液1mlの硫化物イオン相当
量 (mg)
(f) 硫化物イオン標準液 (10μgS2-/ml) 使用時に(e)の硫化物イオン標準液 (1mgS2-/ml) 10mlを全量フ
ラスコ1 000mlにとり,2.(12)(a)の溶存酸素を含まない水を標線まで加える。ただし,この溶液の濃
度は,(e)の硫化物イオン標準液 (1mgS2-/ml) の濃度から算出する。
注(1) 24.1(1)(b)による。
(2) 塩酸酸性としたよう素溶液に硫化物イオン標準液 (1mgS2-/ml) を加える。
(3) 22.1.2(1)(d)による。
(4) 22.1.2(1)(i)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(5)(6)(S2-として5〜40μgを含む。)をメスシリンダー(有栓形)50mlにとり,2.(12)(a)
の溶存酸素を含まない水を加えて約40mlとした後,硫酸 (1+1) 1ml(7)を加え,更に同じ水を50ml
の標線まで加える。
(b) N, N´-ジメチル-p-フェニレンジアンモニウム溶液0.5mlを加えて振り混ぜた後,塩化鉄 (III) 溶液
1mlを加え,再び振り混ぜ,約1分間放置する。
(c) りん酸水素二アンモニウム溶液 (400g/l) 1.5mlを加えて振り混ぜた後,約5分間放置する。
(d) 別に,メスシリンダー(有栓形)50mlに硫酸 (1+1) 1mlをとり,水を50mlの標線まで加えた後,
(b)及び(c)の操作を行う。
(e) (c)の溶液を吸収セルにとり,(d)の溶液を対照液として波長670nm付近の吸光度を測定する。
(f) 検量線から硫化物イオンの量を求め,試料中の硫化物イオンの濃度 (mgS2-/l) を算出する。
検量線 硫化物イオン標準液 (10μgS2-/ml) 0.5〜4mlをメスシリンダー(有栓形)50mlに段階的にと
161
K 0101 : 1998
り,(a)〜(e)の操作を行って硫化物イオン (S2-) の量と吸光度との関係線を作成する。
注(5) 溶存の硫化物イオンを定量する場合には,試料採取後直ちにろ紙5種C又は6種を用いてろ過し,
最初のろ液約50mlを捨て,その後のろ液を用いる。
(6) 試料採取後,直ちに試験が行えない場合には,3.3によって試料の保存処理を行うか,備考2.
による硫化亜鉛として固定する保存処理を行う。備考2.による保存処理を行った場合には,
40.2(3)によって,硫化亜鉛から硫化水素として分離した後,試験操作を行う。ただし,この場
合の硫化水素の吸収には,酢酸亜鉛溶液に代え,水酸化ナトリウム溶液 (20mmol/l) を用いる。
(7) 発色の強さは,pH 0.4〜1.0のときに最高になる。試料がアルカリ性の場合には,硫酸 (1+1) で
中和した後,更に硫酸 (1+1) 1mlを加える。
備考1. この方法は,共存物質の妨害が比較的少ないが,酸化性物質及び還元性物質が妨害する。亜
硫酸イオン及びチオ硫酸イオンは,10mg/l以上で妨害する。チオシアン酸イオンは,少量で
も妨害する。
2. 硫化物イオンを硫化亜鉛として固定する操作は,次による。
JIS K 8953に規定する硫酸亜鉛七水和物20gを水100mlに溶かした溶液と炭酸ナトリウム
溶液 (100g/l) を用意し,使用時にその等体積ずつを混合し,塩基性炭酸亜鉛の懸濁液を調製
する。試料容器には,19.(2)(a)の培養瓶を用い,気泡が残らないように注意して試料を採取
した後,塩基性炭酸亜鉛の懸濁液を試料100mlにつき約2mlを加え,気泡が残らないように
注意して密栓し,転倒して混ぜ合わせる。塩基性炭酸亜鉛の懸濁液10mlは,硫化物イオン
約50mgを固定できる。
引き続き溶液をろ紙5種Cでろ過するか又は遠心分離によって沈殿を分離し,この沈殿に
ついて試験を行うことによって試料中に共存する亜硫酸イオン,チオ硫酸イオンなどと分離
定量できる。
なお,試料中の硫化物イオンの濃度を算出するときの試料の量は,培養瓶の容量 (ml) か
ら塩基性炭酸亜鉛の懸濁液の添加量 (ml) を差し引いた値を用いる。
40.2 よう素滴定法 硫化物イオン又は硫化物を含む溶液に一定過剰量のよう素溶液と塩酸を加え,でん
ぷん溶液を指示薬として,残ったよう素をチオ硫酸ナトリウム溶液で滴定して硫化物イオンを定量する。
定量範囲:S2- 0.2mg以上
備考3. 試料に直接滴定操作を行うと,亜硫酸イオン,チオ硫酸イオンなどの還元性物質も硫化物イ
オンと同様に反応して滴定されるので,あらかじめ硫化物イオンの分離を行ってから操作す
る。
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 硫酸 (1+1) 4.4(1)(b)による。
(c) 塩化ヒドロキシルアンモニウム溶液 (100g/l) JIS K 8201に規定する塩化ヒドロキシルアンモニ
ウム10gを水に溶かして100mlとする。
(d) 酢酸亜鉛溶液 JIS K 8356に規定する酢酸亜鉛二水和物24gを水に溶かして100mlとする。
(e) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(f) よう素溶液 (5mmol/l) 24.1(1)(b)のよう素溶液 (50mmol/l) 50mlをとり,水で500mlとする。着色
ガラス瓶に保存する。
(g) 10mmol/lチオ硫酸ナトリウム溶液 28.3(1)(e)による。
162
K 0101 : 1998
(h) 窒素 JIS K 1107に規定する高純度窒素2級
(2) 装置 装置は,次のとおりとする。
(a) 硫化水素発生及び吸収装置 図40.1に一例を示す(8)。
図40.1 硫化水素発生及び吸収装置の一例
注(8) フラスコ (A) は,試料から直接硫化水素を発生させる場合には,1 000mlのものを用い,硫化
亜鉛として固定したものから硫化水素を発生させる場合には,300mlのものを用いる。
(3) 分離操作 分離操作は,次のとおり行う。
(a) 酢酸亜鉛溶液5mlを水で100mlに薄め,三角フラスコ (G1) , (G2) にそれぞれ50mlずつ入れる。
(b) 試料(5)(9)の適量(通常500ml)をフラスコ (A) に入れ,鉄 (III) が含まれている場合には,塩化ヒ
163
K 0101 : 1998
ドロキシルアンモニウム溶液 (100g/l) 1ml又はJIS K 9502に規定するL (+) −アスコルビン酸0.1g
を加える。次に,上部の注入漏斗から硫酸 (1+1) 100mlを加える。
(c) フラスコ (A) を約50℃に加熱し,窒素(又は二酸化炭素)をゆっくりと約20分間通し,硫化水素
を追い出して酢酸亜鉛溶液に吸収させる。
注(9) 備考2.によって硫化亜鉛として固定し,沈殿を分離したものから硫化水素を発生させる場合に
は,沈殿をろ紙とともに水でフラスコ (A) に入れ,水約50mlを加える。注入漏斗から塩酸 (1
+1) 50mlを加え,続いて(c)の操作を行う。
備考4. 泡立ちが激しい試料の場合には,ジフェニルエーテルなどの消泡剤を加えるとよい。
5. 二酸化炭素を約1時間通じれば,加熱しないでも硫化水素を追い出すことができる。
6. 試料から直接硫化水素を発生させる方法では,チオ硫酸イオンのほか鉄,亜鉛などの金属元
素から硫化物イオンを分離できるが,亜硫酸イオンの分離は完全でない。注(9)の方法では,
亜硫酸イオン及びチオ硫酸イオンは,あらかじめ分離されている。
(4) 操作 操作は,次のとおり行う。
(a) 硫化水素を吸収した図40.1の三角フラスコ (G1) , (G2) によう素溶液 (5mmol/l) を過剰になるよ
うに定量を加える。大部分の硫化水素は三角フラスコ (G1) に吸収されているから,よう素溶液
(5mmol/l) の大部分は三角フラスコ (G1) に加えるようにする。
(b) 三角フラスコ (G1) , (G2) に塩酸(10)2.5mlずつを加えて振り混ぜた後,内容物を三角フラスコ500ml
に移し入れる。水で三角フラスコG1,G2をよく洗い,洗液も合わせる。
(c) 10mmol/lチオ硫酸ナトリウム溶液で滴定し,よう素の黄色が薄くなったら,指示薬としてでんぷん
溶液 (10g/l) 約1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(d) 空試験として三角フラスコ500mlに水100mlをとり,酢酸亜鉛溶液5ml及び試験に用いたのと同量
のよう素溶液 (5mmol/l) を加え,さらに塩酸5mlを加えて(c)の操作を行う。
(e) 次の式によって試料中の硫化物イオンの濃度 (mgS2-/l) を算出する。
3
160
.0
000
1
)
(
×
×
×
−
=
V
f
a
b
S
ここに,
S: 硫化物イオン (mgS2-/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター
V: 試料 (ml)
0.160 3: 10mmol/lチオ硫酸ナトリウム溶液1mlの硫化物イオン相
当量 (mg)
注(10) よう素溶液 (5mmol/l) を加えてから塩酸を加える。逆に行うと硫化水素として損失するおそれ
がある。
備考7. 妨害物質の少ない試料で備考2.によってあらかじめ硫化亜鉛として固定した場合には,(3)の
分離操作を行わないで滴定することができる。この場合は,固定で生じた沈殿をろ紙5種C
でろ別して水で洗浄し,ろ紙とともに三角フラスコ300mlに移し,水約100mlを加える。こ
れによう素溶液 (5mmol/l) の定量を加え,次に,塩酸5mlを加えてよく振り混ぜて反応させ,
以下,(c)〜(e)を行って試料中の硫化物イオンの濃度を求める。
なお,硫化亜鉛として固定したときの沈殿が着色している場合には,金属元素の共存が考
えられ,滴定の妨害となることが多いから,注(9)の分離操作によって硫化水素として分離す
164
K 0101 : 1998
る。
41. 亜硫酸イオン (SO32-) 亜硫酸イオンの定量には,よう素滴定法を適用する。
41.1 よう素滴定法 一定量のよう素溶液に酢酸-酢酸ナトリウム緩衝液を加えた後,試料を加え,次に,
過剰のよう素をでんぷん溶液を指示薬としてチオ硫酸ナトリウム溶液で滴定する。別に,同量の試料をと
り,酸性として煮沸して亜硫酸イオンを二酸化硫黄として追い出した後,同一の滴定操作を行い,これを
空試験値として,チオ硫酸イオンなどの還元性物質の影響を補正する。亜硫酸イオンは空気によって酸化
されるので,試験は試料採取後直ちに行う。
定量範囲:SO32-0.2mg以上
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+35) JIS K 8951に規定する硫酸を用いて調製する。
(b) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(c) 酢酸-酢酸ナトリウム緩衝液 (pH3.9) JIS K 8371に規定する酢酸ナトリウム三水和物75gを酢酸
(1+2) 500mlに溶かす。
(d) でんぷん溶液 (10g/l) 22.1.2(1)(i)による。
(e) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(f) よう素溶液 (5mmol/l) 40.2(1)(f)による。
(g) 10mmol/lチオ硫酸ナトリウム溶液 40.2(1)(g)による。
(h) 窒素 JIS K 1107に規定する高純度窒素2級
(2) 操作 操作は,次のとおり行う。
(a) 三角フラスコによう素溶液 (5mmol/l) 20mlをとり,これに酢酸-酢酸ナトリウム緩衝液 (pH3.9) 10ml
を加える。
(b) 試料の適量(SO32-として0.2〜10mgを含む。)をピペットでとり,先端をフラスコの溶液中に浸し,
静かに注入して混ぜ合わせる。
(c) 10mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなったら,指示薬としてでんぷん溶
液 (10g/l) 約1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
(d) 空試験として試験に用いたのと同量の試料を三角フラスコにとり,硫酸 (1+35) 6〜7mlを加え,窒
素(1)を液面に通しながら数分間静かに煮沸して二酸化硫黄を追い出す。窒素を通したまま冷却する。
(e) 冷却後,フェノールフタレイン溶液 (5g/l) を指示薬として,水酸化ナトリウム溶液 (40g/l) で中和
する。よう素溶液 (5mmol/l) 20ml及び酢酸-酢酸ナトリウム緩衝液 (pH 3.9) 10mlを加え,(c)の操作
によって10mmol/lチオ硫酸ナトリウム溶液で滴定する。
(f) 次の式によって試料中の亜硫酸イオンの濃度 (mgSO32-/l) を算出する。
3
400
.0
000
1
)
(
×
×
×
−
=
V
f
a
b
S
ここに,
S: 亜硫酸イオン (mgSO32-/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 空試験に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター
V: 試料 (ml)
0.400 3: 10mmol/lチオ硫酸ナトリウム溶液1mlの亜硫酸イオン相
当量 (mg)
165
K 0101 : 1998
注(1) 窒素中の酸素の除去は,水酸化ナトリウム溶液 (600g/l) 75mlと,水15mlにJIS K 8780に規定す
るピロガロール(1, 2, 3-ベンゼントリオール)5gを溶かした溶液との混液をガス洗浄瓶に入れ,
通気して行う。
備考1. 硫化物イオンは,よう素を消費して妨害し,(2)(d)の空試験を行っても補正されない。硫化物
イオンの妨害を除くには40.の備考2.によって硫化物イオンを硫化亜鉛として固定後,ろ紙5
種Cでろ過(又は遠心分離)し,そのろ液から亜硫酸イオンを定量する。
2. 鉄 (III) イオン及び銅 (II) イオンはよう化物イオンを酸化して妨害する。
3. 配管又は装置から試料を採取する場合には,図41.1に示すような試料採取器を用いると便利
である。この場合には,次のように操作して試料を採取し試験する。
2個の試料採取器の下端に軟質塩化ビニル管とY字管とで試料採取用配管に接続する。試
料採取器の出口を上方に向け,出口には何も接続しない。試料の流量は試料採取用配管出口
で調節する。試料の温度が高い場合には,室温より1〜2℃低くなるように冷却する。
2個の試料採取器とも同時に8〜12秒間程度で満たされるように流量を調節し,配管や試
料採取器が試料で十分に洗浄され内容物が完全に入れ換わるように連結して試料を十分に流
す。
次に,2個の試料採取器の上端にあるコックを閉じ,直ちに下端のコックも閉じ,接続管
を外し,試料採取器を逆にして気泡がないことを確かめる。もし,気泡があれば両方とも試
料を捨てて新しく採取する。2個の試料採取器のうち1個を試験用に,他を空試験用とする。
あらかじめ,よう素溶液 (5mmol/l) 20mlをビーカーにとり,これに酢酸-酢酸ナトリウム緩衝
液 (pH3.9) 10mlを入れ,試験用の試料採取器の両端にある足の部分の試料を捨て,水で洗い
両方の足にJIS K 8102に規定するエタノール (95) 2mlずつを満たす。試料採取器の下方のエ
タノールをできるだけ捨てないようにして,試料採取器の足をビーカーの溶液中に浸して上
方のコックも開き,静かに下方のコックも開きながら試料を注入する。以下,(2)(c)以降によ
って試験する。
166
K 0101 : 1998
図41.1 試料採取器の一例
42. 硫酸イオン (SO42-) 硫酸イオンの定量には,クロム酸バリウム-ジフェニルカルバジド吸光光度法,
クロム酸バリウム吸光光度法,重量法又はイオンクロマトグラフ法を適用する。
42.1 クロム酸バリウム−ジフェニルカルバジド吸光光度法 試料にクロム酸バリウムの酸懸濁液を加え
て硫酸バリウムを沈殿させ,次に,カルシウムイオンを含むアンモニア水とエタノールとを加え,過剰の
クロム酸バリウムを沈殿させ,遠心分離する。硫酸イオンと置換して生じたクロム酸イオンを二クロム酸
イオンに変えジフェニルカルバジド(1, 5-ジフェニルカルボノヒドラジド)を反応させ,生じる赤紫の吸
光度を測定して硫酸イオンを定量する。
定量範囲:SO42-2〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) クロム酸バリウムの酸懸濁液 (A) クロム酸バリウム0.5gを酢酸 (1+15) 100mlと塩酸 (1+500)
100mlとの混合溶液200mlに加え,よく振り混ぜて懸濁液を作り,ポリエチレン瓶に保存する。
クロム酸バリウムは,次の方法で調製する。JIS K 8312に規定するクロム酸カリウム8gを水約
800mlに溶かし,酢酸 (6mol/l) (JIS K 8355に規定する酢酸35mlを水に溶かして100mlとする。)
10mlを加えて約70℃に温める。この溶液を激しくかき混ぜながら,約70℃に温めた塩化バリウム
溶液(JIS K 8155に規定する塩化バリウム二水和物10gを水に溶かして100mlとする。)100mlを1
滴ずつ滴加してクロム酸バリウムを沈殿させ,放置する。上澄み液を捨て,温水約500mlずつで4
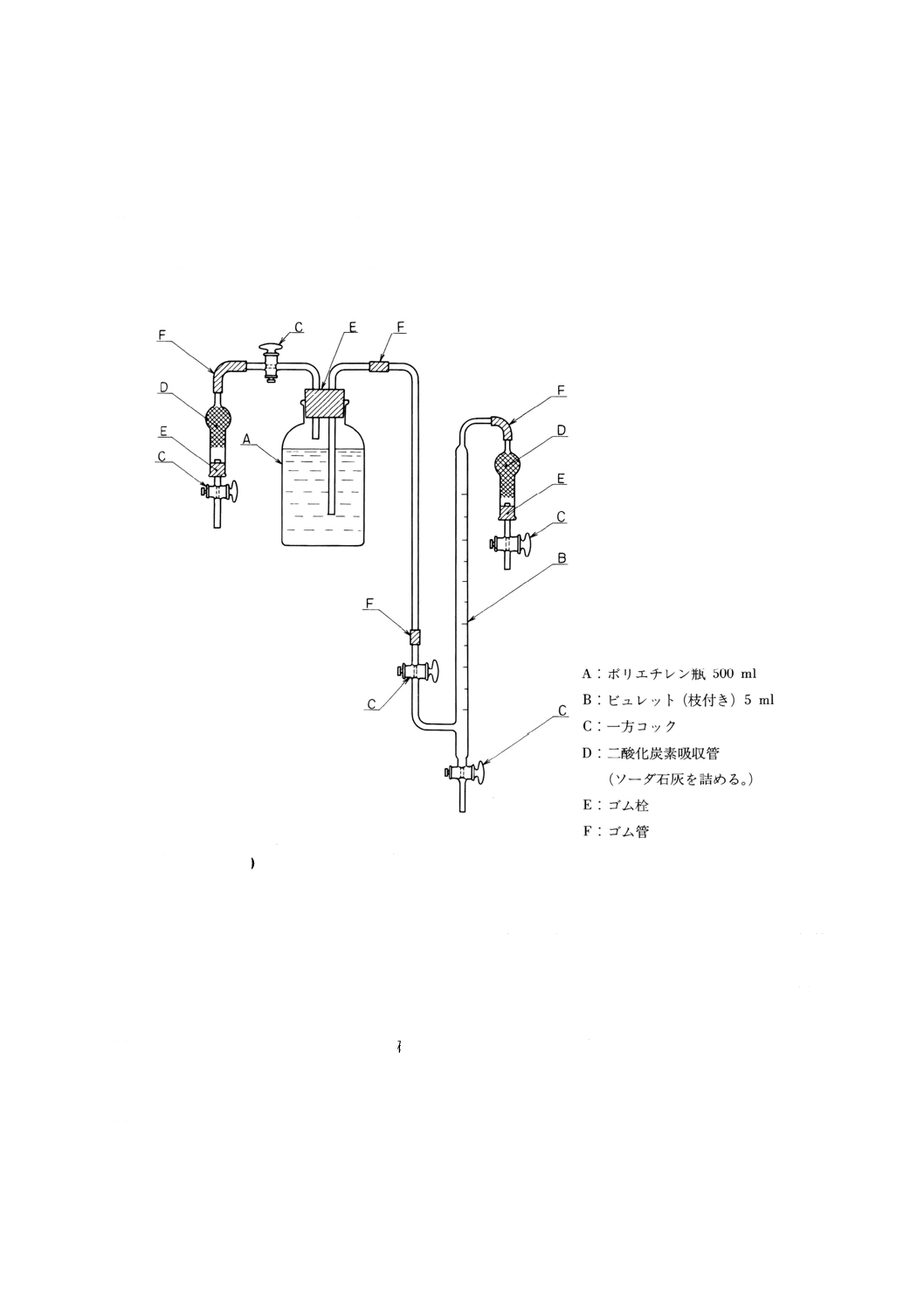
167
K 0101 : 1998
回デカンテーションする。沈殿を遠沈管に移し,遠心分離によって冷水で2〜3回洗浄する。この沈
殿をガラスろ過器に移して吸引ろ過し,105〜110℃で約1時間加熱し,デシケーター中で放冷した
後,めのう乳鉢ですりつぶす。
(c) カルシウムを含むアンモニア水 JIS K 8122に規定する塩化カルシウム二水和物1.85gを,アンモ
ニア水 (3+4) 500mlに溶かし,ポリエチレン瓶に入れ,空気中の二酸化炭素が入らないような方法
で保存する。図42.1のように保存すると便利である。
図42.1 カルシウムを含むアンモニア水の保存の一例
(d) エタノール (95) JIS K 8102に規定するもの。
(e) ジフェニルカルバジド溶液 JIS K 8488に規定する1, 5-ジフェニルカルボノヒドラジド(ジフェニ
ルカルバジド)1gをエタノール (95) 100mlに溶かす。この溶液は使用時に調製する。
(f) 硫酸イオン標準液 (1mgSO42-/ml) JIS K 8962に規定する硫酸カリウムを約700℃で約30分間加熱
し,デシケーター中で放冷する。その1.815gをとり,少量の水に溶かして全量フラスコ1 000mlに
移し入れ,水を標線まで加える。又はJIS K 0028に規定する硫酸イオン標準液のSO42-1 000を用い
る。
(g) 硫酸イオン標準液 (10μgSO42-/ml) 硫酸イオン標準液 (1mgSO42-/ml) 10mlをとり,全量フラスコ1
000mlに入れ,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラスろ過器 ブフナー漏斗形 3G4
(b) 遠心分離器
168
K 0101 : 1998
(c) 遠沈管 共栓付き 20〜30ml
(d) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料10ml(SO42-として2〜50μgを含む。)を遠沈管にとり,20〜30℃(1)に保つ。これに20〜30℃に
保ったクロム酸バリウムの酸懸濁液 (A) 4mlを加えて振り混ぜ,2〜3分間放置する。
(b) カルシウムを含むアンモニア水の上澄み液1mlを図42.1のビュレット又はピペットで静かに加えて
混ぜ,さらにエタノール (95) 10mlを加えて1分間振り混ぜた後,約10分間放置する。
(c) これを遠心分離した後,上澄み液をガラスろ過器に移し,弱く吸引ろ過する。
(d) ろ液にジフェニルカルバジド溶液2mlと塩酸 (1+1) 1.4mlを加えて振り混ぜ,約5分間放置して発
色させる。
(e) 別に,空試験として水10mlをとり,試料と同時に(a)〜(d)の操作を行う。
(f) (d)で得た溶液の一部を吸収セルに移し,(e)の溶液を対照液として波長540nm付近の吸光度を測定
する。
(g) 検量線から硫酸イオンの量を求め,試料中の硫酸イオンの濃度 (mgSO42-/l) を算出する。
検量線 硫酸イオン標準液 (10μgSO42-/ml) 0.2〜5mlを遠沈管に段階的にとり,水で10mlとした後,
(a)〜(f)の操作を行って硫酸イオン (SO42-) の量と吸光度との関係線を作成する。
注(1) 反応時の液温が20〜30℃の範囲で同一の検量線が得られる。
42.2 クロム酸バリウム吸光光度法 試料にクロム酸バリウムの酸懸濁液を加えて硫酸バリウムを沈殿さ
せ,次に,カルシウムイオンを含むアンモニア水とエタノールとを加え,過剰のクロム酸バリウムを沈殿
させ,遠心分離する。硫酸イオンと置換して生じたクロム酸イオンの黄色の吸光度を測定して硫酸イオン
を定量する。
定量範囲:SO42- 50〜500μg,繰返し分析精度:変動係数で5〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) クロム酸バリウムの酸懸濁液 (B) クロム酸バリウム(2)2.5gを,酢酸 (1+15) 100mlと塩酸 (1+
500) 100mlとの混合溶液200mlに加え,よく振り混ぜて懸濁液を作り,ポリエチレン瓶に保存する。
注(2) 42.1(1)(b)で調製したクロム酸バリウム。
(b) カルシウムを含むアンモニア水 42.1(1)(c)による。
(c) エタノール (95) JIS K 8102に規定するもの。
(d) 硫酸イオン標準液 (0.1mgSO42-/ml) 42.1(1)(f)の硫酸イオン標準液 (1mgSO42-/ml) 20mlを全量フラ
スコ200mlにとり,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 遠心分離器
(b) 遠沈管 共栓付き 20〜30ml
(c) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料10ml(SO42-として50〜500μgを含む。)を遠沈管にとり,20〜30℃(1)に保つ。これに20〜30℃
に保ったクロム酸バリウムの酸懸濁液 (B) 4mlを加えて振り混ぜ,2〜3分間放置する。
(b) カルシウムを含むアンモニア水の上澄み液1mlを図42.1のビュレット又はピペットで静かに加えて
混ぜ,さらにエタノール (95) 10mlを加えて1分間振り混ぜた後,約10分間放置する。
(c) これを遠心分離して,その上澄み液を吸収セルにとり,波長370nm付近の吸光度を測定する。
169
K 0101 : 1998
(d) 空試験として水10mlをとり,(a)〜(c)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
(e) 検量線から硫酸イオンの量を求め,試料中の硫酸イオンの濃度 (mgSO42-/l) を算出する。
検量線 硫酸イオン標準液 (0.1mgSO42-/ml) 0.5〜5mlを遠沈管に段階的にとり,水で10mlとした後,
(a)〜(d)の操作を行って硫酸イオン (SO42-) の量と吸光度との関係線を作成する。
備考1. 硝酸,炭酸及び炭酸水素の各イオンは,それぞれ50mg/l以上共存すると妨害する。りん酸,
ひ酸,セレン酸及びバナジン酸の各イオンと鉛は微量でも妨害するので,前処理を行って除
去する。
炭酸イオン及び炭酸水素イオンの除去は,塩酸を加えて煮沸して行う[あらかじめ,試料
の一部をとり,メチルレッド-ブロモクレゾールグリーン混合液(*)を指示薬として塩酸で中和
して中和に必要な塩酸 (1+100) の量を求め,試料に同量の塩酸 (1+100) を加える。塩酸が
過剰にならないようにする。]。
りん酸イオンを含む場合には,試料10mlに塩化カルシウム溶液 (11g/l) 2ml及び水酸化ナ
トリウム溶液 (10g/l) と炭酸ナトリウム溶液 (13g/l) との等容混合液1mlを加える。約10分
間放置後,遠心分離し,その上澄み液の一定量をとり,塩酸 (1+100) で中和した後,水浴
中で約10分間加熱して二酸化炭素を除く。冷却後,水で20mlに薄め,その10mlをとり,(3)
の操作によって硫酸イオンを定量する。このりん酸イオンの除去操作を行った場合の検量線
は,硫酸イオン標準液について同様に操作して作成する。
注(*) 13.1(1)(a)による。
42.3 重量法 硫酸イオンを硫酸バリウムとして沈殿させ,その質量をはかって定量する。
定量範囲:SO42- 10mg以上,繰返し分析精度:変動係数で2%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するもの。
(b) 塩酸 (1+50) JIS K 8180に規定する塩酸を用いて調製する。
(c) 塩化バリウム溶液 (100g/l) JIS K 8155に規定する塩化バリウム二水和物11.7gを水に溶かして
100mlとする。
(d) 硝酸銀溶液 (10g/l) JIS K 8550に規定する硝酸銀1gを水に溶かして100mlとする。
(2) 操作 操作は,次のとおり行う。
(a) 試料の適量(SO42-として10mg以上を含む。)を磁器蒸発皿にとり,塩酸3mlを加えた後,沸騰水浴
上で蒸発乾固し,更に約20分間加熱する。
(b) 放冷後,塩酸2mlで湿し,次に,温水20〜30mlを加え,数分間加熱した後,ろ紙5種Bでろ過し,
塩酸 (1+50) で数回洗う。
(c) ろ液に水を加えて100mlとし,水浴上で加熱し,絶えずかき混ぜながら,これに温塩化バリウム溶
液 (100g/l) を滴加し,沈殿が生じなくなったら,更にその添加量の20〜50%を過剰に加える。
(d) 沸騰水浴上で20〜30分間加熱した後,3〜4時間放置する。
(e) ろ紙6種又は5種Cを用いてろ過し,ろ液に塩化物イオンの反応を認めなくなるまで水で洗う[硝
酸銀溶液 (10g/l) で確かめる。]。
(f) 沈殿はろ紙とともに,あらかじめ800℃で恒量とした磁器るつぼに入れ,乾燥後,徐々に加熱して
ろ紙をいったん炭化した後,灰化する。
(g) 引き続き,800℃で約30分間強熱し,デシケーター中で放冷した後,その質量をはかる。
170
K 0101 : 1998
(h) (g)の操作を繰り返して恒量とする。
(i) 次の式によって試料中の硫酸イオンの濃度 (mgSO42-/l) を算出する。
6
411
.0
000
1
)
(
×
×
×
−
=
V
f
a
b
S
ここに,
S: 硫酸イオン (mgSO42-/l)
a: 硫酸バリウムの質量 (mg)
V: 試料 (ml)
0.411 6: 硫酸バリウム1mgの硫酸イオン相当量 (mg)
42.4 イオンクロマトグラフ法 試料中の硫酸イオンをイオンクロマトグラフ法で定量する。
定量範囲: SO42- 1〜100mg/l(3),繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異
なる。)
注(3) サプレッサと組み合わせる方式の場合には,0.2〜100mgSO42-/l
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3又はA4の水
(b) 溶離液 32.5(1)(b)による。
(c) 再生液 32.5(1)(c)による。
(d) 硫酸イオン標準液 (10mgSO42-/ml) JIS K 8962に規定する硫酸カリウムをあらかじめ約700℃で
約30分間加熱し,デシケーター中で放冷する。その1.815gをとり,少量の水に溶かして,全量フ
ラスコ100mlに移し入れ,水を標線まで加える。
(e) 硫酸イオン標準液 (1mgSO42-/ml) 硫酸イオン標準液 (10mgSO42-/ml) 10mlを全量フラスコ100ml
にとり,水を標線まで加える。使用時に調製する。
(f) 陰イオン混合標準液 [(0.1mgCl-, 0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] 32.5(1)(f)による。
(2) 器具及び装置 器具及び装置は,32.5(2)による。
(3) 準備操作 準備操作は,32.5(3)による。
(4) 操作 操作は,次のとおり行う。
(a) 32.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上の硫酸イオンに相当するピークについて,指示値(4)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
いて得た指示値(4)を補正する。
(d) 検量線から硫酸イオンの濃度を求め,試料中の硫酸イオンの濃度 (mgSO42-/l) を算出する。
検量線 硫酸イオン標準液 (1mgSO42-/ml) (5)0.1〜10mlを段階的に全量フラスコ100mlにとり,水を
標線まで加え,(a)及び(b)の操作を行ってそれぞれの硫酸イオンに相当する指示値(4)を読み取る。別
に,空試験として水について(a)及び(b)の操作を行ってそれぞれの硫酸イオンに相当する指示値を補
正した後,硫酸イオン (SO42-) の量と指示値との関係線を作成する。検量線の作成は,試料測定時
に行う。
注(4) ピーク高さ又はピーク面積
(5) 硫酸イオン以外の陰イオンを同時に試験する場合には,陰イオン混合標準液 [(0.1mgCl-,
0.5mgNO2-, 0.5mgBr-, 0.5mgNO3-, 1mgSO42-) /ml] を用いる。
備考2. 硫酸イオンの濃度が1mg/lのとき臭化物イオンは200mg/l以下及び硝酸イオン400mg/l以下な
171
K 0101 : 1998
らば妨害しない。
3. 32.の備考11.による。
43. りん化合物及び全りん りん化合物は,りん酸,ポリりん酸,動物質及び植物質中のりんなど,水中
に存在するりん化合物のりんを意味し,りん酸イオン,加水分解性りん,全りんに区分する。
また,ろ過した試料について試験することによって,それぞれを溶存及び懸濁のものに区別できる。
りん化合物は変化しやすいから,試験は試料採取後直ちに行う。直ちに行えない場合には,3.3によって
保存し,できるだけ早く試験する。
43.1 りん酸イオン (PO43-)
43.1.1 モリブデン青(アスコルビン酸還元)吸光光度法 りん酸イオンが七モリブデン酸六アンモニウム
及びタルトラトアンチモン (III) 酸カリウムと反応して生成するヘテロポリ化合物をL (+) −アスコルビ
ン酸で還元し,生成したモリブデン青の吸光度を測定してりん酸イオンを定量する。
定量範囲:PO43-2.5〜75μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) アスコルビン酸溶液 (72g/l) JIS K 9502に規定するL (+) −アスコルビン酸7.2gを水に溶かして
100mlとする。0〜10℃の暗所に保存する。着色した溶液は使用しない。
(c) モリブデン酸アンモニウム溶液 JIS K 8905に規定する七モリブデン酸六アンモニウム四水和物6g
とJIS K 8533に規定するビス[ (+) −タルトラト]二アンチモン (III) 酸二カリウム三水和物0.24g
を水約300mlに溶かし,これに硫酸 (2+1) 120mlを加え,次に,JIS K 8588に規定するアミド硫酸
アンモニウム(1)5gを加えて溶かした後,水を加えて500mlとする。
(d) モリブデン酸アンモニウム-アスコルビン酸混合溶液 モリブデン酸アンモニウム溶液とアスコル
ビン酸溶液 (72g/l) とを体積比で5 : 1の割合になるように混合する。使用時に調製する。
(e) りん酸イオン標準液 (0.1mgPO43-/ml) JIS K 9007に規定するりん酸二水素カリウム(pH標準液
用)を105+2℃で約2時間加熱し,デシケーター中で放冷する。その0.143 3gをとり,少量の水に
溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。0〜10℃の暗所に保存する。又は
JIS K 0033に規定する標準物質-標準液-りん酸イオンのPO43-100を用いる。
(f) りん酸イオン標準液 (5μgPO43-/ml) りん酸イオン標準液 (0.1mgPO43-/ml) 10mlを全量フラスコ
200mlにとり,水を標線まで加える。使用時に調製する。
注(1) 試料中に硝酸イオン,亜硝酸イオンが共存しない場合には,アミド硫酸アンモニウムを添加し
なくてもよい(備考3.参照)。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(2)(3)の適量(PO43-として2.5〜75μgを含む。)をメスシリンダー(有栓形)25mlにとり,水を
25mlの標線まで加える。
(b) モリブデン酸アンモニウム-アスコルビン酸混合溶液2mlを加えて振り混ぜた後,20〜40℃(4)で,約
15分間放置する。
(c) 溶液の一部を吸収セルに移し,波長880nm(5)付近の吸光度を測定する(6)。
(d) 空試験として水25mlをとり,(b)及び(c)の操作を行って吸光度を測定し,試料について得た吸光度
172
K 0101 : 1998
を補正する。
(e) 検量線からりん酸イオンの量を求め,試料中のりん酸イオンの濃度 (mgPO43-/l) を算出する(7)。
検量線 りん酸イオン標準液 (5μgPO43-/ml) 0.5〜15mlをメスシリンダー(有栓形)25mlに段階的に
とり,(a)〜(d)の操作を行ってりん酸イオン (PO43-) の量と吸光度との関係線を作成する。
注(2) 溶存のりん酸イオンを定量する場合には,3.3によってろ過した試料を用いる。
(3) 試料が酸性の場合は,指示薬としてp-ニトロフェノール溶液 (1g/l) [43.2(1)(g)による。]2,3
滴を加え,水酸化ナトリウム溶液 (40g/l) を用いてわずかに黄色になるまで中和する。ただし,
このときアルミニウムなどの水酸化物の沈殿が生じる場合には,沈殿が生じる直前でとどめる。
(4) 検量線作成時と同じ発色温度となるようにする。
(5) 光度計が波長880nm付近の吸光度の測定ができない場合には,波長710nm付近の吸光度を測定
する。
(6) 試料に濁り又は色がある場合は,(a)と同量の試料をとり,モリブデン酸アンモニウム-アスコル
ビン酸混合溶液2mlに代えモリブデン酸アンモニウム溶液2mlを用い,(a)及び(b)の操作を行っ
てこの溶液を対照液として吸光度を測定するか,この溶液の吸光度を測定して試料について得
た吸光度を補正する。ただし,これらの場合は試料に対しての(d)による空試験の補正は行わな
い。
なお,この操作による場合,濁りの著しい試料では誤差が大きくなる。
(7) りんの濃度 (mgP/l) で表示する場合には,次の換算式を用いる。
りんの濃度 (mgP/l) =りん酸イオンの濃度 (mgPO43-/l) ×0.3261
備考1. 試料中にひ素 (V) が含まれるときは,りん酸イオンと同様に発色するので,次の操作でその
妨害を除去する。
硫酸酸性二亜硫酸ナトリウム-チオ硫酸ナトリウム溶液2.5mlを試料の適量に加え,1〜2分
間放置し,注(3)によって酸を中和した後,(a)〜(e)の操作を行う。
硫酸酸性二亜硫酸ナトリウム-チオ硫酸ナトリウム溶液は,次のように調製する。
JIS K 8501に規定する二亜硫酸ナトリウム14gを水100mlに溶かし,その40mlに硫酸 (1
+1) 20mlを加え,更にJIS K 8637に規定するチオ硫酸ナトリウム五水和物0.56gを水に溶か
して40mlとした溶液を加える(この溶液は使用時に調製する。)。
2. 塩化物イオン及び硫酸イオンが多量(4%程度)に共存しても妨害しない。したがって,多量
の塩類を含む試料及び前処理によって多量の塩類が生じた試料に適している。ただし,多量
のアンモニウムイオン及びカリウムイオンが共存すると,濁りを生じて妨害となる。
3. モリブデン酸アンモニウム溶液中にアミド硫酸アンモニウムを添加しない場合は,硝酸イオ
ン数g,亜硝酸イオン0.25mgが存在するとモリブデン青は,試薬添加後約15分間で急激に
退色する。更にこれらが多量に共存すれば最高の発色に達しなくなる。アミド硫酸アンモニ
ウムを添加したモリブデン酸アンモニウム溶液を用いるときは,硝酸イオンは妨害しない。
また,亜硝酸イオンも約20mgまでは妨害しない。亜硝酸イオンがこれ以上のときは,その
量に比例してアミド硫酸アンモニウムを追加する。アミド硫酸アンモニウムは別の溶液とし
て添加してもよい。
4. 鉄 (III) 30mg以上は,モリブデン青を退色させる。アスコルビン酸溶液の添加量を増せば,
妨害を抑制できる。
5. りん酸塩が懸濁物として含まれるときは,試薬添加後約15分間経過しても徐々に吸光度が増
173
K 0101 : 1998
加することがある。
6. りん酸イオンの濃度が低い試料の場合には,試料の量とモリブデン酸アンモニウム-アスコル
ビン酸混合溶液の量を増加してモリブデン青を発色させ,2, 6-ジメチル-4-ヘプタノン[ジイ
ソブチルケトン (DIBK) ]で抽出して定量できる。
7. 次のような操作で発色させ,定量することもできる。
試料の適量(PO43-として5〜150μgを含む。)を全量フラスコ50mlにとり,水を加えて約
40mlとし,モリブデン酸アンモニウム-アスコルビン酸混合溶液3.5mlを加え,水を標線まで
加えて振り混ぜた後,20〜40℃で約15分間放置して発色させ,波長880nm付近(又は710nm
付近)の吸光度を測定する。水を用いて空試験を行って吸光度を補正する。
検量線は,りん酸イオン標準液 (5μgPO43-/ml) 1〜30mlについて試料と同様に操作して作成
する。
43.1.2 モリブデン青[塩化すず (II) 還元]吸光光度法 りん酸イオンと七モリブデン酸六アンモニウム
(モリブデン酸アンモニウム)とが反応して生成するヘテロポリ化合物を塩化すず (II) で還元し,生成し
たモリブデン青の吸光度を測定してりん酸イオンを定量する。
定量範囲:PO43- 5〜150μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) モリブデン酸アンモニウム溶液 JIS K 8905に規定する七モリブデン酸六アンモニウム四水和物
15gを水に溶かし,これを硫酸(水600ml中にJIS K 8951に規定する硫酸182mlを静かにかき混ぜ
ながら加えて放冷したもの。)中にかき混ぜながら加え,次にJIS K 8588に規定するアミド硫酸ア
ンモニウム(1)10gを加えて溶かした後,水を加えて1lとする。
(c) 塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物1gをJIS K 8180に規定する塩酸
5mlに溶かし(必要ならば加熱する。),水を加えて50mlとし,JIS K 8580に規定するすずの粒状の
もの2,3個を加え,着色ガラス瓶に入れて保存する。濁りを生じたものは使用しない。
(d) りん酸イオン標準液 (10μgPO43-/ml) 43.1.1(1)(e)のりん酸イオン標準液 (0.1mgPO43-/ml) 20mlを全
量フラスコ200mlにとり,水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(2)(3)の適量(PO43-として5〜150μgを含む。)を全量フラスコ50mlにとり,水を加えて約40ml
とする。
(b) モリブデン酸アンモニウム溶液5mlを加えて振り混ぜた後,塩化すず (II) 溶液0.25mlを加え,水
を標線まで加えて振り混ぜ,約15分間放置する。
(c) 溶液の一部を吸収セルに移し,波長700nm付近の吸光度を測定する(8)。
(d) 空試験として水40mlをとり,(b)及び(c)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
(e) 検量線からりん酸イオンの量を求め,試料中のりん酸イオンの濃度 (mgPO43-/l) を算出する(7)。
検量線 りん酸イオン標準液 (10μgPO43-/ml) 0.5〜15mlを全量フラスコ50mlに段階的にとり,(a)
〜(d)の操作を行って吸光度を測定し,りん酸イオン (PO43-) の量と吸光度との関係線を作成する。
注(8) 試料に濁り又は色がある場合は,(a)と同量の試料をとり,塩化すず (II) 溶液の添加を除いた(a)
174
K 0101 : 1998
及び(b)の操作を行ってこの溶液を対照液として吸光度を測定するか,この溶液の吸光度を測定
して試料について得た吸光度を補正する。ただし,これらの場合は,試料に対しての(d)による
空試験の補正は行わない。
なお,この操作による場合は,濁りの著しい試料では誤差が大きくなる。
備考8. 試料中にひ素 (V) が含まれる場合は,備考1.によって硫酸酸性二亜硫酸ナトリウム-チオ硫酸
ナトリウム溶液を調製し,この溶液5mlを試料の適量に加えた後1〜2分間放置し,注(3)によ
って酸を中和した後,水で約40mlとし,(b)〜(e)の操作を行う。
9. 塩化物イオン,よう化物イオン,臭化物イオンが存在すると,発色が幾分弱められる。それ
ぞれCl-75mg,I-6mg,Br-25mgで吸光度の減少はそれぞれ約5%である。このため試料中に多
量にハロゲン化物イオンが共存する場合には,検量線の作成時に試料と同量のハロゲン化物
イオンを加えるか,又はハロゲン化物のほとんど影響のない43.1.1の方法を用いるとよい。
10. 硫酸イオンが多量に共存すると発色の強さは幾分増加する。モリブデン酸アンモニウム溶液
に含まれる以外の硫酸イオン500mgのとき,吸光度は約3%,1gのとき約5%増加する。し
たがって,多量の硫酸イオンが含まれる場合には,備考9.のハロゲン化物イオンの場合と同
様に,試料と同量の硫酸イオンを共存させて検量線を作成する。又は,43.1.1の方法を用い
る。
11. 亜硫酸イオン150mgが共存すると約5%の正の誤差を生じる。
12. 備考3.による。
13. 鉄 (III) 2mgが共存するとモリブデン青は,試薬添加後約15分間で退色し始める。この妨害
を防ぐには,アンモニア水 (1+50) を用いて試料のpHを約2[水酸化鉄 (III) の沈殿が生成
する直前,水酸化鉄 (III) が生じた場合は,硝酸 (1+25) を滴加して沈殿を溶かす。この場
合,硝酸 (1+25) の過剰量は1mlを超えないようにする。]とし,これによう化カリウム-亜
硫酸ナトリウム溶液(JIS K 8913に規定するよう化カリウム2g及びJIS K 8061に規定する
亜硫酸ナトリウム10gを水に溶かして100mlとする。)1mlを加える。このとき溶液は強い赤
褐色となるが,この色が消えるまで放置する[5〜10分間で消えるが,鉄 (III) の多いときは
沸騰水中に30〜60秒間浸してもよい。]。この溶液について(a)以下の操作を行い,りん酸イ
オンを定量する。
なお,このよう化カリウム-亜硫酸ナトリウム溶液の添加は,硝酸イオン,亜硝酸イオンの
影響も除去する。
14. シリカは,りん酸イオンの500倍量が存在しても誤差は+5%である。
15. 備考5.による。
16. りん酸イオンの濃度が低い試料の場合には,発色させたモリブデン青を1-ブタノールで抽出
して定量できる。
試料の適量(PO43-として2〜40μgを含む。)を分液漏斗100mlにとり,液量を50ml(分液
漏斗にあらかじめ印を付けておく。)とする。硫酸 (1+50) 1mlとJIS K 8810に規定する1-
ブタノール15mlを加えて振り混ぜた後,水層を別の分液漏斗100mlに移す。水層にモリブ
デン酸アンモニウム溶液6.5mlを加えて振り混ぜ,次に,塩化すず (II) 溶液0.25mlを加えて
振り混ぜ,10〜15分間放置する。1-ブタノール10mlを加えて振り混ぜてモリブデン青を抽
出し,静置後水層を捨て,1-ブタノール層の一部を吸収セルに移し,1-ブタノールを対照液
として波長730nm付近の吸光度を測定する。
175
K 0101 : 1998
検量線はりん酸イオン標準液 (5μgPO43-/ml) を5倍に薄めて調製したりん酸イオン標準液
(1μgPO43-/ml) 2〜40mlについて試料と同様に操作して作成する。
なお,この方法では1-ブタノールによる1回目の抽出で水層に1-ブタノールが飽和すると
ともに,試料中の着色妨害物質が抽出される。ひ素 (V) が共存する場合は,備考8.に準じる。
ただし,硫酸酸性二亜硫酸ナトリウム-チオ硫酸ナトリウム溶液5mlを加えた後,約2時間以
上放置する必要がある。その他の妨害物質などについては,備考3.,備考13.,備考14.,備
考15.を参照。
43.2 加水分解性りん 試料を酸性として煮沸したとき,加水分解によってりん酸イオンとなるものをい
う。
試料に硫酸-硝酸の混酸を加え,煮沸してりん酸イオンとした後定量し,この値から加水分解前のりん酸
イオンを差し引き,りん酸イオンに換算した値で表示する。
定量範囲: 43.1.1によるときPO43- 2.5〜75μg,43.1.2によるときPO43- 5〜150μg,繰返し分析精度:
変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硫酸−硝酸の混液 JIS K 8951に規定する硫酸300mlを水約600ml中に注意してかき混ぜながら加
えた後,放冷する。これにJIS K 8541に規定する硝酸4mlと水を加えて全量を1lとする。
(c) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(d) アスコルビン酸溶液 (72g/l) 43.1.1(1)(b)による(9)。
(e) モリブデン酸アンモニウム溶液 43.1.1(1)(c)による(10)。
(f) モリブデン酸アンモニウム-アスコルビン酸混合溶液 43.1.1(1)(d)による(9)。
(g) p-ニトロフェノール溶液 (1g/l) JIS K 8721に規定するp-ニトロフェノール0.1gを水に溶かして
100mlとする。
(h) りん酸イオン標準液 (5μgPO43-/ml) 43.1.1(1)(f)による。
注(9) 43.1.2の方法を用いる場合には,この溶液は必要なく,43.1.2(1)(c)の塩化すず (II) 溶液を調製す
る。
(10) 43.1.2の方法を用いる場合には,この溶液は必要なく,43.1.2(1)(b)のモリブデン酸アンモニウム
溶液を調製する。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(3)(11)の適量(PO43-として1mg以下を含む。)をビーカー200mlにとり,水を加えて50〜100ml
とした後,硫酸-硝酸の混酸1mlを加える。
(b) 静かに煮沸する。液量が25ml以下になったら水を加え,液量を25〜50mlに保って,約90分間煮
沸(12)する。
(c) 放冷後,ろ紙5種Bを用いてろ過し,温水で3〜4回洗う。
(d) ろ液及び洗液を合わせ,指示薬としてp-ニトロフェノール溶液 (1g/l) 数滴を加え,溶液がわずかに
黄色になるまで水酸化ナトリウム溶液 (40g/l) を滴加(3)した後,全量フラスコ100mlに移し入れ,
水を標線まで加える。
(e) この溶液の適量をとり,43.1.1(又は43.1.2)(13)によってりん酸イオンの量を求め,試料中のりん
176
K 0101 : 1998
酸イオンの濃度 (mgPO43-/l) に換算し,この値から別に43.1.1(又は43.1.2)によって定量した試料
中のりん酸イオンの濃度 (mgPO43-/l) を差し引いて加水分解性りんとし,りん酸イオンの濃度
(mgPO43-/l) で表す(7)。
注(11) 溶存の加水分解性りんを定量する場合には,注(2)によってろ過した試料を用いる。
(12) 二りん酸イオン,トリポリりん酸イオンなどは約1時間でりん酸イオンになる。
(13) 発色時の溶液に多量の塩類が含まれている場合には,その影響をほとんど受けない43.1.1の方
法が適している。43.1.2の方法を用いる場合は備考9.及び備考10.を参照。
43.3 全りん ペルオキソ二硫酸カリウム分解,硝酸-過塩素酸分解又は硝酸-硫酸分解によって,試料中の
有機物などを分解し,この溶液についてりん酸イオンを定量し,これを全りんとしてりんの濃度で表示す
る。
43.3.1 ペルオキソ二硫酸カリウム分解法 試料にペルオキソ二硫酸カリウムを加え,高圧蒸気滅菌器中で
加熱して有機物などを分解し,この溶液についてりん酸イオンを定量して全りんの濃度を求める。
定量範囲:P1.25〜25μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) アスコルビン酸溶液 (72g/l) 43.1.1(1)(b)による。
(c) ペルオキソ二硫酸カリウム溶液 (40g/l) JIS K 8253に規定するペルオキソ二硫酸カリウム(窒
素・りん測定用)4gを水に溶かして100mlとする。
(d) モリブデン酸アンモニウム溶液 43.1.1(1)(c)による。ただし,アミド硫酸アンモニウムは加えなく
てもよい(14)。
(e) モリブデン酸アンモニウム−アスコルビン酸混合溶液 43.1.1(1)(d)による。
(f) りん標準液 (50μgP/ml) JIS K 9007に規定するりん酸二水素カリウム(pH標準液用)を105±2℃
で約2時間加熱し,デシケーター中で放冷する。その0.220gをとり,少量の水に溶かして全量フラ
スコ1 000mlに移し入れ,水を標線まで加える。0〜10℃の暗所に保存する。
(g) りん標準液 (5μgP/ml) りん標準液 (50μgP/ml) 20mlを全量フラスコ200mlにとり,水を標線まで
加える。使用時に調製する。
注(14) 全りんの測定では前処理による分解によって亜硝酸イオンは存在しなくなるから,アミド硫酸
アンモニウムを添加する必要がない。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分解瓶 耐熱・耐圧性のガラス瓶(容量約100ml)で,高圧蒸気滅菌器中(約120℃)で使用でき
るもの(15)。
(b) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので,約120℃に加熱できるもの。
(c) 光度計 分光光度計又は光電光度計
注(15) ガラス製アンプル(容量約100ml)で,高圧蒸気滅菌器中(約120℃)で使用できるものを用い
てもよい。
(3) 操作 操作は,次のとおり行う。
(a) 試料(16)50mlを分解瓶にとる。
(b) ペルオキソ二硫酸カリウム溶液 (40g/l) 10mlを加え,密栓して混合する。
(c) 高圧蒸気滅菌器に入れて加熱し,約120℃に達してから30分間加熱分解する。
(d) 分解瓶を取り出し,放冷後(17),上澄み液(18)(19)25mlを共栓試験管に分取する。
177
K 0101 : 1998
(e) 43.1.1(3)(b)及び(c)の操作を行って吸光度を測定する(20)。
(f) 空試験として水50mlを分解瓶にとり,(b)〜(e)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
(g) 検量線から(d)で分取した溶液25ml中のりんの量を求め,次の式によって試料中の全りんの濃度
(mgP/l) を算出する(21)。
50
000
1
25
60×
×
=a
P
ここに, P: 全りん (mgP/l)
a: (d)で分取した溶液25ml中の全りん (mg)
検量線 りん標準液 (5μgP/ml) 1〜20mlを段階的に全量フラスコ100mlにとり,それぞれ水を標線
まで加える。その25mlをそれぞれ共栓試験管にとり,43.1.1(3)(b)及び(c)の操作を行って吸光度を
測定する。別に,空試験として水25mlを共栓試験管にとり,43.1.1(3)(b)及び(c)の操作を行って吸
光度を測定し,りん標準液 (5μgP/ml) について得た吸光度を補正する。採取した溶液25ml中のり
ん (P) の量と吸光度との関係線を作成する。
注(16) 試料50ml中の全りんが60μg以上の場合には,試料の適量(Pとして0.12mg未満を含む。)を全
量フラスコ100mlにとり,水を標線まで加えたものを用いる。ただし,試料50ml中の全りんが
60μg以上で,pHが5〜9の範囲にない場合には,試料の適量(Pとして0.12mg未満を含む。)を
とり,硫酸 (1+35) 又は水酸化ナトリウム溶液 (40g/l) を用いて中和した後,全量フラスコ
100mlに移し入れ,水を標線まで加えたものを用いる。
(17) 塩化物イオンを含む試料の場合は,塩素が生成してモリブデン青の発色を妨害することがある
ので,分解後の溶液に亜硫酸水素ナトリウム溶液 (50g/l) 1mlを加える。
(18) 上澄み液に濁りが認められる場合には,ろ紙5種C又は孔径1μm以下のガラス繊維ろ紙を用い
てろ過し,初めのろ液5〜10mlを捨てた後のろ液を用いる。
(19) 分解後の溶液中に金属水酸化物の沈殿が認められる場合には,これらが溶ける点まで硫酸 (1+
35) 又は必要に応じ水酸化ナトリウム溶液 (40g/l) を加えたものを用いる(加えた両溶液の量を
求めておく。)。
なお,金属水酸化物の沈殿を溶かした後の溶液に濁りが認められる場合には,更に注(18)の操
作を行う。
(20) (d)の操作における上澄み液にひ素 (V) が含まれる場合には,分解後の溶液に,備考1.によって
調製した硫酸酸性二亜硫酸ナトリウム-チオ硫酸ナトリウム溶液5mlを加え,1〜2分間放置後,
注(3)に準じて酸を中和した後この上澄み液25mlをとり,(e)の操作を行う。この操作を行った場
合は,(g)の式の2560に代え,
25
60c
+を用いる。ただし,cは注(20)において用いた,硫酸酸性二亜硫
酸ナトリウム-チオ硫酸ナトリウム溶液,水酸化ナトリウム溶液 (40g/l) 及び硫酸 (1+35) の合
量 (ml) 。
(21) (a)の操作において,注(16)の操作を行った場合には,次の式によって試料中の全りんの濃度
(mgP/l) を算出する。
V
a
P
100
50
000
1
25
60
×
×
×
=
ここに, P: 全りん (mgP/l)
a: (d)で分取した溶液25ml中の全りん (mg)
V: 試料 (ml)
178
K 0101 : 1998
また,(d)において,注(17)又は注(19)の操作,若しくは注(17)と注(19)の操作を併せて行った場合
には,(g)の式又は上記の式中の2560に代えてそれぞれ2561,
25
)
60
(
b
+又は
25
)
61
(
b
+を用いる。ただし,bは
注(19)において添加した硫酸 (1+35) 又は水酸化ナトリウム溶液 (40g/l) (ml) 。
備考17. 試料のpHが5〜9の範囲にない場合には,注(16)によって中和してから分解操作を行うが,試
料50ml中の全りんが60μg未満の場合は,試料の適量 (50〜100ml) をとり,硫酸 (1+35) 及
び水酸化ナトリウム溶液 (40g/l) で中和した後,この溶液から50mlを分解瓶にとる。ただし,
この場合は中和に要した両溶液のml数を求めておき,試料中の全りんの濃度 (mgP/l) の算
出には(3)(g)の式に代え,次の式を用いる。
V
b
V
a
P
+
×
×
×
=
50
000
1
25
60
ここに, P: 全りん (mgP/l)
a: (d)で分取した溶液25ml中の全りん (mg)
b: 中和に要した硫酸 (1+35) 及び水酸化ナトリウム溶液
(40g/l) (ml)
V: 試料 (ml)
18. (3)(a)の操作で分解瓶にとった試料中のりん濃度が低く0.1mg/l未満の場合には,(3)(e)での吸
光度の測定に光路長50mmの吸収セルを用いる。
また,空試験及び検量線の作成における吸光度の測定にも光路長50mmの吸収セルを用い
る。
なお,この場合,検量線の作成には,(1)(g)のりん標準液 (5μgP/ml) に代え,これを10倍
に薄めて調製したりん標準液 (0.5μgP/ml) を用いる。
19. りんの濃度が低く十分な定量精度が得にくい試料については,次の加熱濃縮操作を行うか,
又は備考20.のモリブデン青の溶媒抽出を行う。
試料100〜250mlをビーカー200〜500mlにとり,硫酸 (2+1) 1,2滴を加えた後,加熱板上
で加熱して液量が50ml以下になるまで濃縮する。この溶液を水酸化ナトリウム溶液 (40g/l)
で中和した後,分解瓶(あらかじめ50mlの位置に印を付けたもの。)に移し,水を加えて50ml
とし,(3)(b)以下の操作を行う。ただし,この場合,試料中の全りんの濃度の算出には,(3)(g)
の式中の50
1000に代え,V
1000を用いる。ただし,Vは試料の量 (ml) 。
20. (3)の操作に準じて発色させたモリブデン青を2, 6-ジメチル-4-ヘプタノン[ジイソブチルケト
ン (DIBK)]で抽出することによって微量のりんを定量できる。
(3)(a)〜(c)の操作を行った後,分解瓶中の溶液(16)を分液漏斗100mlに移し,分解瓶は水10ml
で洗浄し,洗液を分液漏斗に合わせる。これにモリブデン酸アンモニウム-アスコルビン酸混
合溶液5.5mlを加え20〜40℃(4)で約15分間放置する。分液漏斗に2, 6-ジメチル-4-ヘプタノ
ン5mlを加えて約5分間振り混ぜる。静置後水層を捨て,2, 6-ジメチル-4-ヘプタノン層(水
滴などによる濁りがあれば,乾燥したろ紙で手早くろ過する。)の一部を吸収セルに移し,波
長640nm付近の吸光度を測定する。
空試験として水50mlを分解瓶にとり,試料の場合と同じ操作を行って吸光度を測定し,試
料について得た吸光度を補正する。検量線から試料中の全りんの量を求め,次の式によって
試料中の全りんの濃度 (mgP/l) を算出する。
V
a
P
000
1
×
=
179
K 0101 : 1998
ここに, P: 全りん (mgP/l)
a: 測定した全りん (mg)
V: 試料 (ml)
検量線 (1)(g)のりん標準液 (5μgP/ml) を10倍に薄めてりん標準液 (0.5μgP/ml) を調製し,その1
〜25mlを段階的に全量フラスコ100mlにとり,それぞれ水を標線まで加える。この50mlをそれぞ
れ分液漏斗100mlにとり,水20mlを加えた後,試料についてと同じ操作を行って吸光度を測定す
る。空試験として水70mlを分液漏斗100mlにとり,同様の操作を行って吸光度を測定し,りん標
準液 (0.5μgP/ml) について得た吸光度を補正し,採取した溶液50ml中のりん (P) の量と吸光度と
の関係線を作成する。
43.3.2 硝酸−過塩素酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸,過塩素酸を加え,再び加熱して有
機物などを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は多量の
有機物を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
定量範囲:P1.25〜25μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硝酸 JIS K 8541に規定するもの。
(c) 過塩素酸 JIS K 8223に規定するもの。
(d) アスコルビン酸溶液 (72g/l) 43.1.1(1)(b)による。
(e) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(f) 水酸化ナトリウム溶液 (200g/l) 35.1.1.1(1)(c)による。
(g) モリブデン酸アンモニウム溶液 43.1.1(1)(c)による。
(h) モリブデン酸アンモニウム-アスコルビン酸混合溶液 43.1.1(1)(d)による。
(i) p-ニトロフェノール溶液 (1g/l) 43.2(1)(g)による。
(j) りん標準液 (5μgP/ml) 43.3.1(1)(g)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料50ml(22)をビーカーにとる。
(b) 硝酸を加えて弱酸性とし,加熱板上で静かに加熱して15〜20mlに濃縮する。
(c) これに硝酸2〜5mlを加えて再び加熱し,約10mlになるまで濃縮した後,更に硝酸2mlを加えて加
熱し,約10mlになるまで濃縮し,放冷する。
(d) 過塩素酸5ml(23)を少量ずつ加える。加熱板上で加熱を続け,過塩素酸の白煙が発生し始めたらビー
カーを時計皿で覆い,過塩素酸の白煙がビーカー内壁を還流する状態に保つ(24)(25)。
(e) 放冷後,水約30mlを加える(26)。
(f) この溶液に指示薬としてp-ニトロフェノール溶液 (1g/l) 数滴を加え,初めに水酸化ナトリウム溶液
(200g/l) を,次に,水酸化ナトリウム溶液 (40g/l) を加えて溶液がわずかに黄色になるまで中和する
(27)。
(g) 溶液を全量フラスコ50mlに移し入れ,水を標線まで加える。
(h) この溶液25mlを共栓試験管に分取し(28),43.1.1(3)(b)及び(c)の操作を行って吸光度を測定する。
(i) 空試験として(a)で採取した試料と同量の水をビーカーにとり,(b)〜(h)の操作を行って吸光度を測
180
K 0101 : 1998
定し,試料について得た吸光度を補正する。
(j) 検量線から(h)で分取した溶液25ml中のりんの量を求め,次の式によって試料中の全りんの濃度
(mgP/l) を算出する(29)。
V
a
P
000
1
25
50×
×
=
ここに, P: 全りん (mgP/l)
a: (h)で分取した溶液25ml中の全りん (mg)
V: 試料 (ml)
検量線 43.3.1(3)(g)の検量線と同じ操作によって作成する。
注(22) 試料中の全りんの濃度が低い場合には,50ml以上としてもよい。また,多量の塩化物イオンを
含む試料で全りんの濃度が高い場合には,50ml未満としてもよい。
(23) 試料に多量の塩化物イオンが含まれる場合には,塩化物イオンの当量よりも多い量を更に加え
る。
(24) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこと
に注意する。
(i) 酸化されやすい有機物は,過塩素酸を加える前に,(b)及び(c)の操作によって十分に分解して
おく。
(ii) 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
(iii) 必ず過塩素酸と硝酸を共存させた状態で加熱分解を行う。
(iv) 濃縮液を乾固させない。
(25) この操作によっても有機物が分解されず,溶液に色が残った場合には,硝酸2mlを加えて加熱
する操作を繰り返す。
(26) 必要に応じ加熱して可溶性塩を溶かす。加熱しても不溶解物が残った場合には,ろ紙5種C又
は孔径1μm以下のガラス繊維ろ紙を用いて溶液をろ過し,次に,ろ紙を少量の水で洗浄し,ろ
液及び洗液を合わせる。
(27) 中和するときに金属水酸化物の沈殿が認められる場合には,水酸化ナトリウム溶液 (40g/l) の
添加は,沈殿の生じる直前でとどめる。必要に応じ硫酸 (1+35) を用いて調節する。
(28) 分取する溶液25ml中の全りんが25μg以上になる場合には,この溶液の適量(りん含有量25μg
未満となる量。)をメスシリンダー(有栓形)50mlに分取し,水を加えて25mlとする。
(29) (h)において注(28)の操作を行った場合には,(j)の式中2550に代えてb50を用いる。ただし,bはメス
シリンダー(有栓形)に分取した溶液の量 (ml) 。
43.3.3 硝酸-硫酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸及び硫酸を加え,更に加熱して有機物な
どを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は多量の有機物
を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
定量範囲:P1.25〜25μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 硝酸 JIS K 8541に規定するもの。
(c) 硫酸 JIS K 8951に規定するもの。
(d) アスコルビン酸溶液 (72g/l) 43.1.1(1)(b)による。
181
K 0101 : 1998
(e) 水酸化ナトリウム溶液 (40g/l) 19.(1)(g)による。
(f) 水酸化ナトリウム溶液 (200g/l) 35.1.1.1(1)(c)による。
(g) モリブデン酸アンモニウム溶液 43.1.1(1)(c)による。
(h) モリブデン酸アンモニウム-アスコルビン酸混合溶液 43.1.1(1)(d)による。
(i) p-ニトロフェノール溶液 (1g/l) 43.2(1)(g)による。
(j) りん標準液 (5μgP/ml) 43.3.1(1)(g)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 43.3.2(3)(a)及び(b)の操作を行う。
(b) (a)の操作を行った後の溶液に硫酸 (1+1) 2ml(23)及び硝酸2〜5mlを加え,加熱して硫酸の白煙が発
生するまで濃縮し,更に加熱して硫酸の白煙を短時間強く発生させた後,放冷する。
(c) この溶液に硝酸5mlを加えて再び加熱し,硫酸の白煙が発生するまで加熱する(25)。
(d) 放冷後,水約30mlを加え,約10分間静かに煮沸する(26)。
(e) 以下,43.3.2(3)(f)〜(h)の操作を行う。
(f) 空試験として(a)で採取した試料と同量の水をビーカーにとり,43.3.2(3)(b)の操作を行った後,(b)
〜(e)の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
(g) 検量線から(e)で分取した溶液25ml中のりんの量を求め,43.3.2(3)(j)の式によって試料中の全りんの
濃度 (mgP/l) を算出する。
検量線 43.3.1(3)(g)の検量線と同じ操作によって作成する。
44. シリカ (SiO2) 水中のシリカはイオン状シリカ(イオン状けい酸),溶存及びコロイド状シリカ及び
全シリカに区分し,いずれも酸化けい素 (IV) (SiO2) として表示する。
44.1 イオン状シリカ イオン状シリカは,七モリブデン酸六アンモニウムと反応してヘテロポリ化合物
の黄色を生成するシリカをいう。
44.1.1 モリブデン黄吸光光度法 イオン状シリカが,七モリブデン酸六アンモニウムと反応して生成する
ヘテロポリ化合物の黄色の吸光度を測定してシリカを定量する。
定量範囲:SiO20.1〜1mg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用い,ポりエチレン瓶に保存する。
(a) 水 JIS K 0557に規定するA3の水。試薬の調製及び操作にはすべてこの水を用いる。
(b) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(c) モリブデン酸アンモニウム溶液 (100g/l) JIS K 8905に規定する七モリブデン酸六アンモニウム
四水和物21.2gを水に溶かして200mlとする。
(d) しゅう酸溶液 JIS K 8519に規定するしゅう酸二水和物20gを水に溶かして200mlとする。
(e) シリカ標準液 (1mgSiO2/ml) 砂状の石英(99.9%以上)を,めのう乳鉢ですりつぶし,700〜800℃
で約1時間加熱した後,デシケーター中で放冷する。その0.500gを白金るつぼにとり,JIS K 8005
に規定する容量分析用標準物質の炭酸ナトリウム4gを加え,十分に混合した後,加熱して約40分
間融解する。放冷後,融成物を水に溶かして全量フラスコ500mlに移し入れ,水を標線まで加える。
(f) シリカ標準液 (0.1mgSiO2/ml) シリカ標準液 (1mgSiO2/ml) 20mlを全量フラスコ200mlにとり,水
を標線まで加える。
182
K 0101 : 1998
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ過し(1),その50ml(2)(SiO2として0.1〜1mgを含む。)をメスシリンダー(有栓形)50ml
にとり,液温を約20℃に調節する。
(b) 塩酸 (1+1) 1mlとモリブデン酸アンモニウム溶液 (100g/l) 2mlとを加えて振り混ぜ(3),5分間放置
する。
(c) しゅう酸溶液1.5ml(4)を加えて振り混ぜ,1分間放置する(5)。
(d) 直ちに溶液の一部を吸収セルに移し,波長410〜450nmの吸光度を測定する。
(e) 空試験として水約50mlをとり,(a)〜(d)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
(f) 検量線からシリカの量を求め,試料中のシリカの濃度 (mgSiO2/l) を算出する。
検量線 シリカ標準液 (0.1mgSiO2/ml) 1〜10mlをメスシリンダー(有栓形)50mlに段階的にとり,
水を50mlの標線まで加えた後,液温を約20℃に調節し,(b)〜(e)の操作を行ってシリカ (SiO2) の
量と吸光度との関係線を作成する。
注(1) ろ紙5種C(又はろ紙6種)又は孔径0.45〜1μmのろ過材を用いてろ過する。初めのろ液約50ml
は捨て,その後のろ液を用いる。
(2) シリカの濃度が高いときは試料の適量をとり,水で50mlに薄める。
(3) このときのpHは1.1〜1.6になる。
(4) りん酸イオンが共存しない場合には,しゅう酸溶液は添加しない。ただし,検量線の作成も同
様にする。
(5) しゅう酸溶液を加えた場合には,放置時間を正しく守る。放置時間が長くなると,シリカによ
るへテロポリ化合物の黄色も退色する。
44.1.2 モリブデン青吸光光度法 イオン状シリカが,七モリブデン酸六アンモニウムと反応して生成する
ヘテロポリ化合物をL (+) −アスコルビン酸で還元してモリブデン青に変え,その吸光度を測定してシリ
カを定量する。
定量範囲:SiO210〜100μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用い,ポリエチレン瓶に保存する。
(a) 水 44.1.1(1)(a)による。
(b) 塩酸 (1+1) 44.1.1(1)(b)による。
(c) 硫酸 (1+5) 水5容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加える。
(d) モリブデン酸アンモニウム溶液 (100g/l) 44.1.1(1)(c)による。
(e) しゅう酸溶液 44.1.1(1)(d)による。
(f) アスコルビン酸溶液 (100g/l) JIS K 9502に規定するL (+) −アスコルビン酸10gを水に溶かして
100mlとする。10℃以下の暗所に保存し,着色した溶液は使用しない。
(g) シリカ標準液 (10μgSiO2/ml) 44.1.1(1)(f)のシリカ標準液 (0.1mgSiO2/ml) 20mlを全量フラスコ
200mlにとり,水を標線まで加える。使用時に調製する。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
183
K 0101 : 1998
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ過し(1),その50ml(2)(SiO2として10〜100μgを含む。)をメスシリンダー(有栓形)50ml
にとり,液温を約20℃に調節する。
(b) 塩酸 (1+1) 1ml[又は硫酸 (1+5) 1ml]とモリブデン酸アンモニウム溶液 (100g/l) 2mlとを加えて
振り混ぜ(3)5分間放置する。
(c) しゅう酸溶液1.5mlを加えて振り混ぜ,1分間放置する(5)。
(d) アスコルビン酸溶液 (100g/l) 1mlを加えて振り混ぜ,約10分間放置する。
(e) 溶液の一部を吸収セルに移し,波長815nm付近の吸光度を測定する。
(f) 空試験として水50mlをとり,液温を約20℃に調節し,(b)〜(e)の操作を行って吸光度を測定し,試
料について得た吸光度を補正する。
(g) 検量線からシリカの量を求め,試料中のシリカの濃度 (mgSiO2/l) を算出する。
検量線 シリカ標準液 (10μgSiO2/ml) 1〜10mlをメスシリンダー(有栓形)50mlに段階的にとり,
水を50mlの標線まで加えた後,液温を約20℃に調節し,(b)〜(f)の操作を行って吸光度を測定し,
シリカ (SiO2) の量と吸光度との関係線を作成する。
備考1. シリカの濃度が低く,モリブデン青の発色が弱い場合には,光路長20mm又は50mmの吸収
セルを用いて吸光度を測定してもよい。ただし,空試験値も大きくなる。このような場合に
は,44.1.3を適用するとよい。
44.1.3 モリブデン青抽出吸光光度法 イオン状のシリカが,七モリブデン酸六アンモニウムと反応して生
成するヘテロポリ化合物をL (+) −アスコルビン酸で還元してモリブデン青に変え,これを1-ブタノール
に抽出し,有機層の吸光度を測定してシリカを定量する。この方法は,シリカの濃度が低い試料に適用す
る。
定量範囲:SiO20.5〜10μg,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用い,ポリエチレン瓶に保存する。
(a) 水 JIS K 0557に規定するA3の水(ステンレス鋼製又は銅製の蒸留装置を用いて蒸留する。)。こ
の試験に用いる試薬の調製及び操作にはこの水を用いる。
(b) 硫酸 (2.5mol/l) −モリブデン酸アンモニウム (188g/l) 混合溶液 JIS K 8951に規定する硫酸140ml
を水約300mlに冷却しながら混合する。これにJIS K 8905に規定する七モリブデン酸六アンモニウ
ム四水和物200gを水約500mlに溶かした溶液を加え,全量フラスコ1 000mlに移し入れ,水を標線
まで加える。
(c) 硫酸 (2+1) 水1容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸2容を徐々に加える。
(d) アスコルビン酸溶液 (100g/l) 44.1.2(1)(f)による。
(e) 硫酸ナトリウム JIS K 8987に規定するもの。
(f) 1-ブタノール JIS K 8810に規定するもの。
(g) シリカ標準液 (1mgSiO2/ml) 44.1.1(1)(e)による。ただし,水は(a)のものを用いる。
(h) シリカ標準液 (50μgSiO2/ml) シリカ標準液 (1mgSiO2/ml) 25mlを全量フラスコ500mlにとり,水
を標線まで加える。使用時に調製する。
(i) シリカ標準液 (1μgSiO2/ml) シリカ標準液 (50μgSiO2/ml) 10mlを全量フラスコ500mlにとり,水
を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
184
K 0101 : 1998
(a) 分液漏斗 300ml プラスチック製のもの。
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料200ml(SiO2として0.5〜10μgを含む。)を分液漏斗にとる。
(b) 硫酸 (2.5mol/l) −モリブデン酸アンモニウム (188g/l) 混合溶液4mlを加えて振り混ぜた後,液温を
約25℃に保ち20分間放置する。
(c) 硫酸 (2+1) 25mlを加えて振り混ぜた後,直ちにアスコルビン酸溶液 (100g/l) 2mlを加えて振り混ぜ
(6),10分間放置する。
(d) 1-ブタノール25mlを加え,約2分間振り混ぜてモリブデン青を抽出する。
(e) 静置後,1-ブタノール層を共栓試験管10mlに入れ,硫酸ナトリウムを加えて脱水する。
(f) これを吸収セル20mm(7)に移し,1-ブタノールを対照液として波長800nm付近の吸光度を測定する。
(g) 次の操作によって,加えた硫酸 (2.5mol/l) −モリブデン酸アンモニウム (188g/l) 混合溶液に基づく
空試験値を求め,試料について得た吸光度を補正する。
分液漏斗 (A) 及び (B) にそれぞれ水200mlをとり,硫酸 (2.5mol/l) −モリブデン酸アンモニウ
ム (188g/l) 混合溶液を (A) には4ml, (B) には8mlを加えて振り混ぜる。液温を約25℃に保って
20分間放置する。次に,(c)〜(f)の操作を行って (A) , (B) それぞれについての吸光度a,bを測
定する。次の式によって硫酸 (2.5mol/l) −モリブデン酸アンモニウム (188g/l) 混合溶液による空試
験値cを算出する。
c=b−a
(h) 検量線からシリカの量を求め,試料中のシリカの濃度 (μgSiO2/l) を算出する。
検量線 シリカ標準液 (1μgSiO2/ml) 0.5〜10mlを分液漏斗に段階的にとり,水(8)を加えて200mlと
した後,(b)〜(f)の操作を行う。別に,この操作に用いた水200mlをとり,(b)〜(f)の操作を行ってシ
リカ標準液について得た吸光度を補正し,シリカ (SiO2) の量と吸光度との関係線を作成する。
注(6) 硫酸 (2+1) を加えて振り混ぜたら,直ちにアスコルビン酸溶液 (100g/l) を加えて振り混ぜる。
(7) 試料中のシリカの濃度が10μgSiO2/l以上の場合には,吸収セル10mmを用いてもよい。ただし,
空試験及び検量線の作成における吸光度の測定にも吸収セル10mmを用いる。検量線の作成に
は,シリカ標準液 (1μgSiO2/ml) 0.5〜10mlに代え,シリカ標準液 (50μgSiO2/ml) を25倍に薄め
て調製したシリカ標準液 (2μgSiO2/ml) 1〜10mlを用いる。
(8) シリカ標準液の調製に用いたのと同じ水を用いる。
備考2. 試料中のシリカの濃度が高く (20〜400μgSiO2/l) ,(a)〜(c)の操作を行った溶液の着色が強い
場合には,その溶液について,吸収セル50mmを用い波長815nmの吸光度を測定してもよい。
空試験としては1-ブタノールによる抽出を除いた(g)の操作を行い,吸収セル50mmを用いて
波長815nmの吸光度を測定し,(g)と同様に空試験値を算出する。
また,検量線はシリカ標準液 (50μgSiO2/ml) を5倍に薄めて調製したシリカ標準液
(10μgSiO2/ml) 0.4〜8mlについて作成する。この場合,試料50mlを用い,この操作に準じた
操作を行ってもよい。
44.2 溶存及びコロイド状シリカ 溶存及びコロイド状シリカは,試料をろ過したときろ液に含まれるシ
リカをいう。試料をろ過し,炭酸水素ナトリウムを加え,煮沸してシリカをイオン状とした後,モリブデ
ン黄吸光光度法又はモリブデン青吸光光度法で定量する。
(1) 試薬 試薬は,次のものを用いる。
185
K 0101 : 1998
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) 44.1.1(1)(b)による。
(c) 炭酸水素ナトリウム JIS K 8622に規定するもので,SiO2の含量0.002%以下のもの。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 四ふっ化エチレン樹脂製ビーカー 200ml
(b) 白金皿
(c) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料をろ過し(1),その適量(溶存及びコロイド状シリカ2mg以下を含む。)を四ふっ化エチレン樹
脂製ビーカー200ml(又は白金皿)にとり,水を加えて50〜100mlとする。
(b) 試料100mlについて炭酸水素ナトリウム0.20gを加え,沸騰水浴中に入れ約20分間加熱する。
(c) 放冷後,塩酸 (1+1) で中和しpHを5とした後,全量フラスコ100mlに移し入れ,水を標線まで加
える。
(d) この溶液の適量(9)を分取し,44.1.1の方法によってモリブデン黄を,又は44.1.2の方法によってモ
リブデン青を発色させて吸光度を測定する。
(e) 空試験として試料と同量の水をとり,(b)〜(d)の操作を行って吸光度を測定し,試料について得た吸
光度を補正する。
(f) 44.1.1(3)(f)又は44.1.2(3)(g)によって試料中の溶存及びコロイド状シリカの濃度 (mgSiO2/l) を算出す
る。
注(9) 溶存及びコロイド状シリカがモリブデン黄吸光光度法の場合には,SiO2 0.1〜1mg,モリブデン
青吸光光度法の場合には,SiO2 10〜100μgになるようにとる。
44.3 全シリカ 全シリカの試験には,水中のシリカをすべてイオン状に変えた後,モリブデン黄吸光光
度法,モリブデン青吸光光度法又は重量法を適用する。
44.3.1 炭酸ナトリウムによる融解 試料に炭酸ナトリウムを加えて蒸発乾固し,融解してシリカをイオン
状とした後,モリブデン黄吸光光度法又はモリブデン青吸光光度法で定量する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) 44.1.1(1)(b)による。
(c) 炭酸ナトリウム JIS K 8005に規定するもので,SiO2の含量0.001%以下のもの。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 四ふっ化エチレン樹脂製ビーカー 200ml(又は白金皿)
(b) 白金るつぼ
(c) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(全シリカ2mg以下を含む。)を四ふっ化エチレン樹脂製ビーカー200ml(又は白金皿)
にとり,炭酸ナトリウム0.20gを加え,加熱蒸発して約5mlに濃縮する。
(b) 内容物を少量の水で白金るつぼに移し,再び蒸発して乾固する。
(c) 静かに加熱して有機物を炭化した後,灰化し,強熱して融解し放冷する。
(d) 水を加え,加熱して融成物を溶かし,放冷後ビーカーに移し,塩酸 (1+1) で中和してpHを約5と
する。
186
K 0101 : 1998
(e) 濁りがある場合には,ろ過して水で洗浄し,ろ液と洗液を全量フラスコ100mlに移し入れ,水を標
線まで加える。
(f) この溶液の適量(10)を分取し,44.1.1の方法によってモリブデン黄を,又は44.1.2の方法によってモ
リブデン青を発色させて吸光度を測定する。
(g) 空試験として試料と同量の水をとり,(a)〜(f)の操作を行って吸光度を測定し,試料について得た吸
光度を補正する。
(h) 44.1.1(3)(f)又は44.1.2(3)(g)によって試料中の全シリカの濃度 (mgSiO2/l) を算出する。
注(10) 全シリカが,モリブデン黄吸光光度法の場合はSiO2 0.1〜1mg,モリブデン青吸光光度法の場合
はSiO2 10〜100μgになるようにとる。
44.3.2 重量法 試料に塩酸と過塩素酸を加えて加熱し,過塩素酸の白煙を発生させ,シリカを脱水して不
溶性にする。水を加えて塩類を溶かした後,シリカをろ別し,加熱して恒量とした後,硫酸とふっ化水素
酸でシリカを揮散させ,その減量からシリカを定量する。
定量範囲:SiO25mg以上,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) 44.1.1(1)(b)による。
(c) 塩酸 (1+50) JIS K 8180に規定する塩酸を用いて調製する。
(d) 硫酸 (1+2) 水2容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加える。
(e) 過塩素酸 JIS K 8223に規定するもの。
(f) ふっ化水素酸 JIS K 8819に規定するもの。
(2) 器具 器具は,次のとおりとする。
(a) 白金るつぼ
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(SiO2 5mg以上を含む。)をビーカーにとり,塩酸 (1+1) 10mlと過塩素酸15mlを加え
る。
(b) 加熱蒸発し,過塩素酸の濃い白煙が発生し始めたら時計皿でビーカーを覆い,引き続き約15分間加
熱し,放冷する。
(c) 温水100mlを加えて可溶性の塩類を溶かし,ろ紙5種Bでろ過し,残留物及びろ紙を温塩酸 (1+
50) で数回洗浄し,次に,温水で数回洗浄する。
(d) 残留物はろ紙とともに約1 000℃で恒量とした白金るつぼに入れ,乾燥後,徐々に加熱してろ紙をい
ったん炭化した後,灰化し,約1 000℃で加熱して恒量とする。
(e) 硫酸 (1+2) 数滴を加えて残留物を湿し,ふっ化水素酸約5mlを加え,注意して加熱し,シリカを
揮散させ,引き続き加熱乾燥後,約1 000℃で加熱しで恒量とする。
(f) 空試験として,あらかじめ約1 000℃で恒量とした白金るつぼに硫酸 (1+2) 数滴と,ふっ化水素酸
5mlとを加えて(e)の操作を行ってふっ化水素酸の強熱残分を求め,ふっ化水素酸の空試験値とする。
(g) 次の式によって試料中のシリカの濃度 (mgSiO2/l) を算出する。
V
W
W
W
S
000
1
)]
(
[
3
2
1
×
−
−
=
ここに,
S: 全シリカ (mgSiO2/l)
187
K 0101 : 1998
W1: (d)で得た残留物の質量 (mg)
W2: (e)で得た残留物の質量 (mg)
W3: ふっ化水素酸の空試験値 (mg)
V: 試料 (ml)
備考3. 試料中に有機物が多量に含まれている場合には,試料にJIS K 8541に規定する硝酸を加えて
酸性とした後加熱し,蒸発して液量が少量となったとき,硝酸10〜20mlを加えて加熱する。
放冷後,過塩素酸5mlを加え,(3)(b)以降の操作を行って定量する。
45. ほう素 (B) ほう素の定量には,メチレンブルー吸光光度法,アゾメチンH吸光光度法又はICP発
光分光分析法を適用する。
45.1 メチレンブルー吸光光度法 ほう素化合物に硫酸とふっ化水素酸を加えてテトラフルオロほう酸イ
オンとした後,メチレンブルー[3, 7-ビス(ジメチルアミノ)フェノチアジン-5-イウムクロリド]を加え,
生成するイオン会合体を1, 2-ジクロロエタンで抽出し,その吸光度を測定してほう素を定量する。
定量範囲:B0.1〜1μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用い,ポリエチレン瓶に保存する。
(a) 水 JIS K 0557に規定するA3の水(ただし,石英ガラス又は金属製の蒸留器を用いて調製したも
の。)
(b) 硫酸 (3+97) JIS K 8951に規定する硫酸を用いて調製する。
(c) ふっ化水素酸 (1+9) JIS K 8819に規定するふっ化水素酸を用いて調製する。
(d) 硫酸銀溶液 (0.3g/l) JIS K 8965に規定する硫酸銀0.15gを水に溶かして500mlとする。
(e) メチレンブルー溶液 (0.4g/l) JIS K 8897に規定するメチレンブルー(通常は三水和物)0.48gを水
に溶かして100mlとする。この溶液10mlを全量フラスコ100mlにとり,水を標線まで加える。
(f) 1, 2-ジクロロエタン JIS K 8465に規定するもの。
(g) ほう素標準液 (0.1mgB/ml) JIS K 8863に規定するほう酸0.572gをとり,水に溶かし,全量フラ
スコ1 000mlに移し入れ,水を標線まで加える。
(h) ほう素標準液 (1μgB/ml) ほう素標準液 (0.1mgB/ml) 10mlを全量フラスコ1 000mlにとり,水を標
線まで加える。
(i) ほう素標準液 (0.1μgB/ml) ほう素標準液 (1μgB/ml) 20mlを全量フラスコ200mlにとり,水を標線
まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具 石英ガラス又はソーダ石灰ガラス製のもの。
(b) 分液漏斗 ポリエチレン製 50ml
(c) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(1)(2)の適量(Bとして0.1〜1μgを含む。)を分液漏斗にとり,水で15mlとし,硫酸 (3+97) 3ml,
メチレンブルー溶液 (0.4g/l) 3ml及び1, 2-ジクロロエタン10mlを加えて約1分間振り混ぜ,放置す
る(3)(4)。
(b) 1, 2-ジクロロエタン層を捨て(5),水層にふっ化水素酸 (1+9) 3mlを加えて約1時間放置する。
(c) 1, 2-ジクロロエタン10mlを加え,約1分間激しく振り混ぜ,放置する。
(d) 1, 2-ジクロロエタン層を別の分液漏斗に移し,硫酸銀溶液 (0.3g/l) 5mlを加えて約1分間振り混ぜ,
188
K 0101 : 1998
1, 2-ジクロロエタン層を洗い,放置する。
(e) 1, 2-ジクロロエタン層の一部を吸収セルに入れ,1, 2-ジクロロエタンを対照液として波長660nm付
近の吸光度を測定する。
(f) 空試験として水15mlをとり,(a)〜(e)の操作を行って試料について得た吸光度を補正する。
(g) 検量線からほう素の量を求め,試料中のほう素の濃度 (mgB/l) を算出する。
検量線 ほう素標準液 (0.1μgB/ml) 1〜10mlを分液漏斗50mlに段階的にとり,(a)〜(f)の操作を行い,
ほう素 (B) の量と吸光度との関係線を作成する。
注(1) 有機物が多量に共存する場合には,試料の一定量を白金皿にとり,JIS K 8625に規定する炭酸
ナトリウム0.1gを加えて蒸発乾固した後,融解する。放冷後,水を加え,加熱して融成物を溶
かし,硫酸 (3+97) を加えて中和した後,液量を一定とする。
この溶液の適量(Bとして0.1〜1μgを含む。)を分液漏斗にとり,水を加えて15mlとし,硫
酸 (3+97) 3mlとふっ化水素酸 (1+9) 3mlとを加えて振り混ぜ,約1時間放置する。次に,メ
チレンブルー溶液 (0.4g/l) 3mlを加えて振り混ぜた後,1, 2-ジクロロエタン10mlを加えて約1
分間激しく振り混ぜ,ほう素のイオン会合体を抽出する。以下,(d)以降の操作を行う。
(2) 試料が中性でない場合には,硫酸 (3+97) 又は水酸化ナトリウム溶液 (40g/l) で中和する。
(3) ふっ化物イオンが共存する場合は(a)の操作を行うと,ほう素が抽出され失われるから,注(1)の
操作を行う。
(4) 1, 2-ジクロロエタン層と水層とが分かれるには,かなりの時間を要する。
(5) この抽出で,試料中の陰イオン界面活性剤などが除かれる。
備考1. クロム酸イオンは妨害するが,過酸化水素 (1+100) 数滴を加えた後,煮沸して過剰の過酸
化水素を分解すれば妨害しない。
2. 分液漏斗及び吸収セルなどにメチレンブルーが付着したときは,エタノールで洗う。
45.2 アゾメチンH吸光光度法 ほう酸が,pH約6でアゾメチンH[8-N-(2-ヒドロキシベンジリデン)
-アミノ-1-ヒドロキシ-3, 6-ナフタレンジスルホン酸]と反応して生成する黄色の錯体の吸光度を測定して
ほう素を定量する。
定量範囲:B5〜25μg,繰返し分析精度:変動係数で3〜10%
備考3. この方法は,汚濁の少ない試料に適用する。
(1) 試薬 試薬は,次のものを用い,ポリエチレン瓶に保存する。
(a) 水 45.1(1)(a)による。
(b) アゾメチンH溶液 アゾメチンH一ナトリウム塩[8-N-(2-ヒドロキシベンジリデン)-アミノ-1-
ヒドロキシ-3, 6-ナフタレンジスルホン酸一ナトリウム塩]1.0gとJIS K 9502に規定するL (+)−ア
スコルビン酸3.0gとを少量の水に溶かした後,全量フラスコ100mlに移し入れ,水を標線まで加え
る。この溶液はポリエチレン瓶に保存する。4〜6℃の暗所に保存すれば1週間は安定である。
(c) 緩樹液 (pH 5.9) JIS K 8359に規定する酢酸アンモニウム250g,JIS K 8951に規定する硫酸15ml,
JIS K 9005に規定するりん酸5ml,JIS K 8283に規定するくえん酸一水和物1.0g及びJIS K 8107に
規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物1.0gを水250ml中に加え,加熱して
溶かす。
(d) アゾメチンH混合溶液 アゾメチンH溶液と緩衝液 (pH 5.9) の等体積を混合する。使用時に調製
する。
(e) ほう素標準液 (1μgB/ml) 45.1(1)(h)による。
189
K 0101 : 1998
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具 45.1(2)(a)による。
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料(6)の適量(Bとして5〜25μgを含む。)(7)をポリエチレンビーカー100mlにとり,水を加えて
25mlとする。
(b) アゾメチンH混合溶液10mlを加え,20℃の暗所に約2時間放置する。
(c) 溶液の一部を吸収セル(7)に移し,410nm付近の吸光度を測定する。
(d) 空試験として水25mlをポリエチレンビーカー100mlにとり,(b)及び(c)の操作を行って吸光度を測
定し,試料について得た吸光度を補正する。
(e) 検量線からほう素の量を求め,試料中のほう素の濃度 (mgB/l) を算出する。
検量線 ほう素標準液 (1μgB/ml) 5〜25mlをポリエチレンビーカー100mlに段階的にとり,(a)〜(d)
の操作を行ってほう素 (B) の量と吸光度との関係線を作成する。
注(6) 懸濁物が含まれる場合には,ろ過又は遠心分離によって除去する。
(7) 吸収セル50mmを用いれば,ほう素1〜5μgが定量できる。
備考4. この方法では,ナトリウム,カリウム,カルシウム,マグネシウム,亜鉛,りん酸塩,硫酸
塩,硝酸塩は妨害しない。
鉄,マンガン,アルミニウム,銅,クロム,ベリリウム,チタン,バナジウム,ジルコニ
ウムは正の誤差を与える。
45.3 ICP発光分光分析法 試料を誘導結合プラズマ中に噴霧し,ほう素による発光を波長249.773nmで
測定してほう素を定量する。
定量範囲:B20〜8 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用い,ポリエチレン瓶に保存する。
(a) 水 45.1(1)(a)による。
(b) ほう素標準液 (20μgB/ml) 45.1(1)(g)のほう素標準液 (0.1mgB/ml) 50mlを全量フラスコ250mlにと
り,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ガラス器具 45.1(2)(a)による。
(b) ICP発光分光分析装置
(3) 操作 操作は,次のとおり行う。
(a) 試料(6)をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導入部を通してプラズマ
中に噴霧し,波長249.773nmの発光強度を測定する(8)(9)(10)。
(b) 空試験として水について(a)の操作を行って試料について得た発光強度を補正する。
(c) 検量線からほう素の量を求め,試料中のほう素の濃度 (μgB/l) を算出する。
検量線 ほう素標準液 (20μgB/ml) 0.1〜40mlを全量フラスコ100mlに段階的にとり,水を標線まで
加える。この溶液について(a)の操作を行う。別に,空試験として水について(a)の操作を行って標準
液について得た発光強度を補正し,ほう素 (B) の量と発光強度との関係線を作成する。検量線の作
成は試料測定時に行う。
注(8) 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では内標準法によることができ
る。内標準法を用いるときは,試料の適量を全量フラスコ100mlにとり,イットリウム溶液
190
K 0101 : 1998
(50μgY/ml) [酸化イットリウム (III) 0.318gをとり,JIS K 9901に規定する高純度試薬-硝酸5ml
を加え,加熱して溶かし,窒素酸化物を追い出し,冷却後,全量フラスコ250mlに移し入れ,
水を標線まで加える。この溶液10mlを全量フラスコ200mlにとり,水を標線まで加える。]10ml
を加えた後,水を標線まで加える。この溶液について(3)(a)の噴霧操作を行って波長249.773nm
と同時に371.029nm(イットリウム)の発光強度を測定し,ほう素とイットリウムとの発光強度
の比を求める。
別に,ほう素標準液 (20μgB/ml) 0.1〜40mlを全量フラスコ100mlに段階的にとり,イットリ
ウム溶液 (50μgY/ml) 10mlをそれぞれ加え,水を標線まで加える。この溶液について(3)(a)の噴
霧操作を行って波長249.773nmと同時に371.029nmの発光強度を測定し,ほう素の濃度に対す
るほう素とイットリウムとの発光強度比の関係線を作成し,検量線とする。この検量線から,
試料について得た発光強度比に相当するほう素の量を求め,試料中のほう素の濃度 (μgB/l) を
算出する。
(9) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(10) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
備考5. ほう素を含む溶液をプラズマ中に噴霧した場合には,メモリー効果が他の元素の場合より大
きいため,次の溶液を噴霧する前に,水を十分な時間噴霧して前の試料の影響を除去する。
46. ひ素 (As) ひ素の定量には,ジエチルジチオカルバミド酸銀吸光光度法,水素化物発生原子吸光法
又は水素化物発生ICP発光分光分析法を適用する。
46.1 ジエチルジチオカルバミド酸銀吸光光度法 ひ素を過マンガン酸カリウムで酸化した後,水酸化鉄
(III) と共沈させ分離,濃縮する。沈殿を硫酸と塩酸で溶かした後,よう化カリウム,塩化すず (II) 及び亜
鉛を加えて水素化ひ素を発生させ,これをジエチルジチオカルバミド酸銀(N, N-ジエチルカルバモジチオ
酸銀)のクロロホルム溶液に吸収させ,生成する赤紫の吸光度を測定してひ素を定量する。
定量範囲:As2〜10μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 JIS K 8180に規定するひ素分析用のもの。試薬の調製及び操作にはこれを用いる。
(b) 塩酸 (1+1) (a)の塩酸を用いて調製する。
(c) 硝酸 JIS K 8541に規定するもの。
(d) 硫酸 (1+5) JIS K 8951に規定する硫酸を用いて調製する。
(e) アンモニア水 (1+2) JIS K 8085に規定するアンモニア水を用いて調製する。
(f) 過マンガン酸カリウム溶液 (3g/l) JIS K 8247に規定する過マンガン酸カリウム0.3gを水に溶か
して100mlとする。
(g) 過酸化水素 (1+30) JIS K 8230に規定する過酸化水素を用いて調製する。
(h) 鉄 (III) 溶液 (10mgFe/ml) JIS K 8142に規定する塩化鉄 (III) 六水和物5g又はJIS K 8982に規
定する硫酸アンモニウム鉄 (III) ・12水9gと(a)の塩酸5mlとを水に溶かして100mlとする。
(i) よう化カリウム溶液 (200g/l) JIS K 8913に規定するよう化カリウム20gを水に溶かして100mlと
する。
191
K 0101 : 1998
(j) 塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物40gを(a)の塩酸に溶かし,塩酸
で100mlとする。JIS K 8580に規定するすずの粒状2, 3個を加えて着色ガラス瓶に保存する。使
用時に適量をとり,水で10倍に薄める。
(k) 酢酸鉛 (II) 溶液 (100g/l) JIS K 8374に規定する酢酸鉛 (II) 三水和物12gをJIS K 8355に規定
する酢酸1,2滴と水に溶かして100mlとする。
(l) 亜鉛 JIS K 8012に規定するひ素分析用のもの。ただし,JIS Z 8801に規定する試験用ふるいでふ
るい分け,目開き1 400μmのふるいを通り1 000μmのふるいに止まるものを用いる。
(m) ジエチルジチオカルバミド酸銀溶液 JIS K 9512に規定するN, N-ジエチルジチオカルバミド酸銀
0.25gとJIS K 8832に規定するブルシンn水和物(2, 3-ジメトキシストリキニジン-10-オンn水和物)
0.1gとをJIS K 8322に規定するクロロホルムに溶かし,クロロホルムで100mlとする。
(n) メタクレゾールパープル溶液 (1g/l) JIS K 8889に規定するメタクレゾールパープル0.1gをJIS
K 8102に規定するエタノール (95) 50mlに溶かし,水を加えて100mlとする。
(o) クロロホルム JIS K 8322に規定するもの。
(p) ひ素標準液 (0.1mgAs/ml) JIS K 8005に規定する容量分析用標準物質の酸化ひ素 (III) (三酸化二
ひ素)を105℃で約2時間加熱し,デシケーター中で放冷する。As2O3100%に対してその0.132gを
とり,水酸化ナトリウム溶液 (40g/l) 2mlに溶かした後,水を加えて500mlとし,次に,硫酸 (1+
10) を加えて微酸性とし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0026
に規定する標準物質-標準液-ひ素のAs100を用いる。
(q) ひ素標準液 (1μgAs/ml) ひ素標準液 (0.1mgAs/ml) 10mlを全量フラスコ1 000mlにとり,水を標線
まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 水素化ひ素発生装置 図46.1及び図46.2に一例を示す。
192
K 0101 : 1998
図46.1 水素化ひ素発生装置の一例
193
K 0101 : 1998
図46.2 水素化ひ素発生装置の一例
(b) 光度計 分光光度計又は光電光度計
(3) 濃縮操作 準備操作は,次のとおり行う。
(a) 試料の適量(Asとして2〜10μgを含む。)をとり,試料1lにつき硝酸3mlと過マンガン酸カリウム
溶液 (3g/l) を滴加して着色させる。
(b) これを加熱煮沸し,過マンガン酸の色が消えたときは,再び過マンガン酸カリウム溶液 (3g/l) を滴
加して煮沸する(1)。
(c) 過酸化水素 (1+30) のできるだけ少量を滴加して過剰の過マンガン酸を分解する。
(d) 鉄 (III) 溶液 (10mgFe/ml) 2ml,指示薬としてメタクレゾールパープル溶液 (1g/l) 2,3滴を加え,液
温を約80℃とし,かき混ぜながらアンモニア水 (1+2) を溶液の色が紫になるまで滴加する(2)。
(e) 沈殿が沈降した後,小形のろ紙5種Aでろ過する。
注(1) 共沈を妨害する有機物で,分解しにくい場合には,4.3又は4.4を行って分解した後,(d)の操作
に移る。
(2) ひ素の水酸化鉄 (III) による共沈にはpH 9〜10が適している。
(4) 操作 操作は,次のとおり行う。
(a) (3)(e)で得た沈殿をなるべく少量の水で水素化ひ素発生瓶に洗い入れる。ろ紙に付着した沈殿は温硫
酸 (1+5) 18mlと塩酸 (1+1) 2mlに溶かし,ろ紙を洗浄し,これらの溶液は水素化ひ素発生瓶に入
194
K 0101 : 1998
れて先の沈殿を溶かし,続いて水を加えて液量を約40mlとする。
(b) よう化カリウム溶液 (200g/l) 15mlと塩化すず (II) 溶液5mlとを加えて振り混ぜ,約10分間放置す
る。
(c) 水素化ひ素発生瓶,導管及びジエチルジチオカルバミド酸銀溶液5mlを入れた水素化ひ素吸収管を
連結した後,水素化ひ素発生瓶に亜鉛約3gを手早く投入する(3)。水素化ひ素発生瓶を約25℃の水
浴中に入れ,約1時間放置してジエチルジチオカルバミド酸銀溶液に水素化ひ素を吸収,発色させ
る。
(d) この溶液にクロロホルムを加えて5mlとする。
(e) 溶液の一部を吸収セルに移し,クロロホルムを対照液として波長510nm付近の吸光度を測定する。
(f) 空試験として鉄 (III) 溶液 (10mgFe/ml) 2mlを水素化ひ素発生装置の水素化ひ素発生瓶にとり,硫酸
(1+5) 18mlと塩酸 (1+1) 2mlを加え,水で約40mlとした後,(b)〜(d)の操作を行って吸光度を測定
し,試料について得た吸光度を補正する。
(g) 検量線(4)からひ素の量を求め,試料中のひ素の濃度 (μgAs/l) を算出する。
検量線 ひ素標準液 (1μgAs/ml) 2〜10mlを水素化ひ素発生瓶に段階的にとり,鉄 (III) 溶液
(10mgFe/ml) 2ml,硫酸 (1+5) 18ml及び塩酸 (1+1) 2mlを加え,液量を水で40mlとする。(b)〜(f)
の操作を試料と同時に行って吸光度を測定し,ひ素 (As) の量と吸光度との関係線を作成する。
注(3) 図46.2の水素化ひ素発生装置を用いるときは,亜鉛投入管に亜鉛約3gを入れ,吸収管を連結し
た後,亜鉛投入管を回転して亜鉛を試料中に添加する。
(4) 検量線は傾斜が変動するので,必ず試験時に作成する。
46.2 水素化物発生原子吸光法 試料を前処理してひ素を水素化ひ素とし,水素-アルゴンフレーム中に導
き,ひ素による原子吸光を波長193.7nmで測定してひ素を定量する。
定量範囲:As5〜50μg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 46.1(1)(a)による。
(b) 塩酸 (1+1) (a)の塩酸を用いて調製する。
(c) 硝酸 JIS K 8541に規定するもの。
(d) 硫酸 (1+1) 4.4(1)(b)による。
(e) 過マンガン酸カリウム溶液 (3g/l) 46.1(1)(f)による。
(f) よう化カリウム溶液 (200g/l) 46.1(1)(i)による。
(g) 塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物10gを(a)の塩酸100mlに溶かす。
(h) 亜鉛粉末 JIS K 8013に規定するひ素分析用を用いる。
(i) 鉄 (III) 溶液 (10mgFe/ml) 46.1(1)(h)による。
(j) アルゴン JIS K 1105に規定するアルゴン2級
(k) ひ素標準液 (0.1μgAs/ml) 46.1(1)(q)のひ素標準液 (1μgAs/ml) 10mlを全量フラスコ100mlにとり,
(b)の塩酸 (1+1) 2mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 水素化ひ素発生装置 図46.3に一例を示す。
(b) 原子吸光分析装置 バックグラウンド補正が可能なもの。
(c) ひ素中空陰極ランプ
195
K 0101 : 1998
図46.3 水素化ひ素発生装置構成の一例
(3) 操作 操作は,次のとおり行う。
(a) 試料(5)の適量(Asとして0.1〜1μgを含む。)をビーカー(6)100mlにとり,硫酸 (1+1) 1ml及び硝酸
2mlを加え,更に過マンガン酸カリウム溶液 (3g/l) を溶液が着色するまで滴加する。
(b) 加熱板上で加熱(7)して硫酸の白煙を発生させる(8)。
(c) 室温まで放冷した後,塩酸 (1+1) 4mlを加え,静かに加熱して残留物を溶かした後放冷し,水素化
ひ素発生装置の反応容器に移し入れ,水を加えて20mlとする。
(d) よう化カリウム溶液 (200g/l) 2ml,塩化すず (II) 溶液2ml及び鉄 (III) 溶液 (10mgFe/ml) 1mlを加え
て振り混ぜ,約15分間放置する。
(e) 水素化ひ素発生装置と原子吸光分析装置を連結し,系内の空気をアルゴンで置換した後,亜鉛粉末
1.0gを手早く(9)(10)反応容器中に加え,マグネチックスターラーを作動して水素化ひ素を発生させる
(11)。
(f) コックを回転して,水素化ひ素を水素-アルゴン(12)フレーム中に導き,波長193.7nmの指示値(13)を
読み取る。
(g) 空試験として試料と同量の水をとり,(a)〜(f)の操作を行って指示値を読み取り,試料について得た
指示値を補正する。
(h) 検量線からひ素の量を求め,試料中のひ素の濃度 (μgAs/l) を算出する。
検量線 ひ素標準液 (0.1μgAs/ml) 1〜10mlを段階的にとり,(c)〜(g)の操作を行ってひ素 (As) の量
と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 有機物,硝酸塩,亜硝酸塩を含まない試料は(a)〜(c)の代わりに,次のように操作してもよい。
試料の適量(Asとして0.1〜1μgを含む。)をビーカー100mlにとり,塩酸 (1+1) 4mlを加え,
沸騰しない程度に数分間加熱した後,冷却する。反応容器に移し入れ,水を加えて20mlにする。
また,多量の有機物を含む場合は(a)及び(b)の代わりに,次のように操作するとよい。
試料の適量に硫酸 (1+1) 1ml,硝酸2ml及びJIS K 8223に規定する過塩素酸3mlを加え,加
熱して白煙を発生させ,有機物を分解する。
(6) ほうけい酸ガラスには,ひ素を含むものがあるので注意する。四ふっ化エチレン樹脂製ビーカ
ーを用いるとよい。
(7) 加熱中に過マンガン酸の色が消えたときは,過マンガン酸カリウム溶液 (3g/l) を追加する。
(8) 硝酸が存在すると水素化ひ素の発生が阻害されるので,十分に硫酸の白煙を発生させて硝酸を
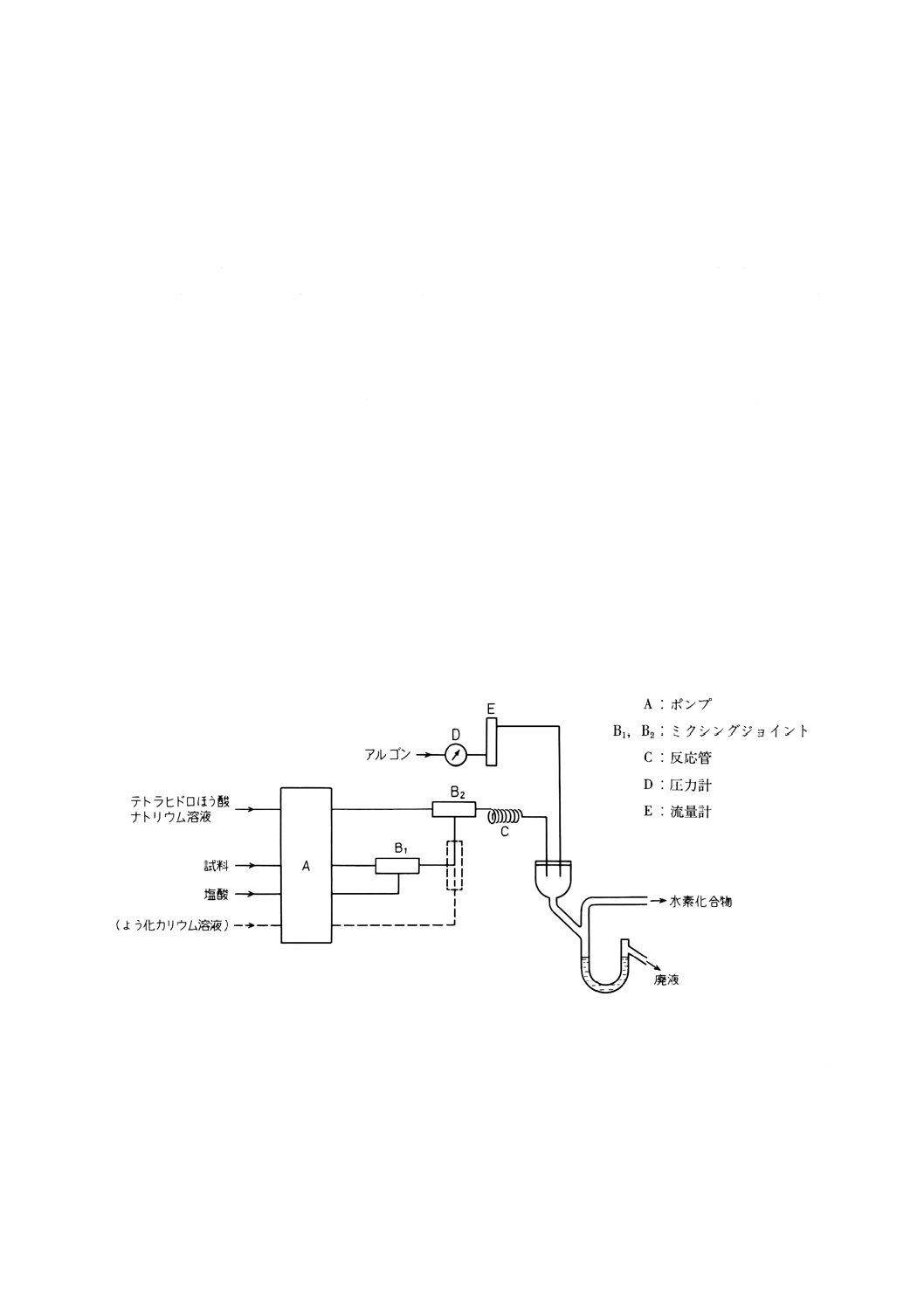
196
K 0101 : 1998
除去する。
(9) 水素化ひ素は亜鉛粉末添加直後に急激に発生するので,逃がさないように注意する。
(10) 亜鉛粉末中には微量のひ素が含まれているので亜鉛粉末の添加量を一定にする。このため,(1)
亜鉛粉末にバインダーを加えて成形した錠剤を添加,(2)亜鉛粉末に水を加えて濃い懸濁液とし,
スポイトで添加,(3)一定量の亜鉛粉末をオブラートで包んで添加するなどの方法を用いる。
亜鉛粉末の代わりにテトラヒドロほう酸ナトリウムを用いてもよい。この場合,塩化すず (II)
溶液及び鉄 (III) 溶液は添加しない(テトラヒドロほう酸ナトリウムによる水素化ひ素の発生条
件は,水素化ひ素発生装置によって異なる。)。
また,水素-アルゴンフレームの代わりに加熱吸収セルを用いてもよい。
テトラヒドロほう酸ナトリウムの添加方法も錠剤添加又は溶液にして少量ずつ添加する方法
などがある。添加時の酸濃度も発生装置によって異なるので注意する。通常,酸濃度は1mol/l
程度で行うが,(3)(b)における硫酸の残留量に応じて(c)の塩酸 (1+1) の添加量を少なめにする。
(11) 捕集した気体の量を知るには圧力による方法,体積による方法などがある。
(12) アルゴンの代わりに,JIS K 1107に規定する高純度窒素2級を用いてもよい。
(13) 吸光度又はその比例値
備考1. 水素化ひ素だけを濃縮するには,ガラスビーズを充てんしたU字管を液体窒素中に浸したコ
ールドトラップを用い水素化ひ素を固定する。U字管を引き上げて,両端を閉じた状態で室
温に戻し,気化した水素化ひ素をアルゴンでフレーム中に送り込む。
2. 図46.3の代わりに,テトラヒドロほう酸ナトリウムを用いた連続式の水素化物発生装置を用
いて定量してもよい。図46.4に一例を示す。この場合の操作は,次による。
図46.4 連続式水素化物発生装置の一例
46.2(3)(a)及び(b)の操作を行った試料を放冷した後,全量フラスコ20mlに移し入れ,水を
標線まで加える。装置にアルゴンを流しながら,この溶液と塩酸 (1〜6mol/l) ,テトラヒド
ロほう酸ナトリウム溶液[テトラヒドロほう酸ナトリウム5gを水酸化ナトリウム溶液
(0.1mol/l) 500mlに溶かしたもの。]を,定量ポンプを用いてそれぞれ1〜10ml/minの流量(装
置によって,試料,塩酸,テトラヒドロほう酸ナトリウム溶液の流量,塩酸及びテトラヒド
ロほう酸ナトリウム溶液の濃度は異なる。)で連続的に装置内に導入し,水素化ひ素を発生さ
197
K 0101 : 1998
せる。
発生した水素化ひ素と廃液を分離した後,水素化ひ素を含む気体を水素-アルゴンフレーム
又は加熱吸収セルに導入し,波長193.7nmの指示値を読み取る。空試験として水について
46.2(3)(a)及び(b)の操作を行った後,試料と同様に操作して指示値を読み取り,試料について
得た指示値を補正する。検量線からひ素の量を求め,試料中のひ素の濃度 (μgAs/l) を算出す
る。
鉄,ニッケル,コバルトはそれぞれ,ひ素の5,10,80倍量程度を超えて共存すると水素
化ひ素の発生を阻害する。しかし,試料を処理後,全量フラスコに移し入れ20mlにすると
きに,よう化カリウム溶液 (100g/l) 4mlを添加するか,又は他の溶液とともによう化カリウ
ム溶液 (20〜100g/l) を1〜10ml/minの流量(濃度,流量は,装置によって異なる。)で水素
化ひ素発生装置に導入することによって鉄による妨害は1 000倍量共存する場合でも除去で
きる。
検量線 ひ素標準液 (0.1μgAs/ml) 1〜10ml(*)を段階的に全量フラスコ20mlにとり,水を標線
まで加える。以下,試料の場合と同様に操作してひ素 (As) の量と指示値との関係線を作成
する。検量線の作成は試料測定時に行う。
注(*) 加熱吸収セルの場合には,水素-アルゴンフレームと比べて10〜50倍程度(装置,操
作条件によって異なる。)感度がよいので,ひ素標準液 (0.1μg/ml) の量を適宜減らす。
46.3 水素化物発生ICP発光分光分析法 試料を前処理してひ素を水素化ひ素とし,試料導入部を通して
誘導結合プラズマ中に噴霧し,ひ素による発光を波長193.696nmで測定してひ素を定量する。
定量範囲:As1〜10μg/l,繰返し分析精度:変動係数で3〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 46.1(1)(a)による。
(b) 硝酸 JIS K 8541に規定するもの。
(c) 硫酸 (1+1) 4.4(1)(b)による。
(d) 臭化カリウム溶液 (1mol/l) JIS K 8506に規定する臭化カリウム11.9gを水に溶かして100mlとす
る。
(e) テトラヒドロほう酸ナトリウム溶液 (10g/l) テトラヒドロほう酸ナトリウム5gを水酸化ナトリウ
ム溶液 (0.1mol/l) に溶かして500mlとする。使用時に調製する。
(f) アルゴン 46.2(1)(j)による。
(g) ひ素標準液 (0.1μgAs/ml) 46.2(1)(k)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 水素化ひ素発生装置 連続式のもの。
(b) ICP発光分光分析装置
(3) 操作 操作は,次のとおり行う。
(a) 試料(14)の適量(Asとして0.05〜0.5μgを含む。)をビーカーにとり,硫酸 (1+1) 1ml及び硝酸2ml
を加え,加熱板上で加熱して硫酸の白煙を発生させる。
(b) 室温まで放冷した後,水8ml,塩酸8ml及び臭化カリウム溶液 (1mol/l) 8mlを加え,約50℃で約50
分間加熱する。
(c) 室温まで冷却した後,この溶液を全量フラスコ50mlに移し入れ,水を標線まで加える。
(d) 水素化ひ素発生装置とICP発光分光分析装置を接続し,アルゴンを流しながら,(c)の溶液,テトラ
198
K 0101 : 1998
ヒドロほう酸ナトリウム溶液 (10g/l) (15)及び塩酸 (1〜6mol/l) (15)を定量ポンプで連続的に導入(16)し,
水素化ひ素を発生させる。
(e) 水素化ひ素を試料導入部を通してプラズマ中に噴霧し,波長193.696nmの発光強度を測定する。
(f) 空試験として試料と同量の水をとり,(a)〜(e)の操作を行い,試料について得た発光強度を補正する。
(g) 検量線からひ素の量を求め,試料中のひ素の濃度 (μgAs/l) を算出する。
検量線 ひ素標準液 (0.1μgAs/ml) 0.5〜5mlを全量フラスコ50mlに段階的にとり,前処理を行った
試料と同じ酸の濃度になるように酸及び臭化カリウム溶液 (1mol/l) を加えた後,水を標線まで加え
る。(d)〜(f)の操作を行ってひ素の量と発光強度との関係線を作成する。検量線の作成は試料測定時
に行う。
注(14) 試料に有機物が多量に含まれる場合には,(a)の操作で硫酸 (1+1) 1ml,硝酸2ml及びJIS K 8223
に規定する過塩素酸3mlを加えて加熱し,白煙を発生させて有機物を分解する。
(15) 装置によって塩酸及びテトラヒドロほう酸ナトリウム溶液の濃度は異なる。
(16) 装置によって試料,塩酸,テトラヒドロほう酸ナトリウム溶液の流量が異なる。
備考3. 水素化物を発生させるときに副生する水素がプラズマに導入されることによってプラズマが
不安定になる場合があるので,特に導入初期には水素の量が多くなり過ぎないように注意す
る。
4. 共存する酸及び塩の影響を受けやすいので注意する。影響がある場合には,水素化物発生原
子吸光法に準じて,干渉抑制剤を用いる。
5. 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定す
る標準添加法を用いるとよい。この場合は,試料の種類によらずバックグラウンド補正を行
う必要がある。
47. ナトリウム (Na) ナトリウムの定量には,フレーム光度法,フレーム原子吸光法,イオン電極法又
はイオンクロマトグラフ法を適用する。
47.1 フレーム光度法 試料をアセチレン-空気フレーム又は水素-酸素フレームなどの中に噴霧し,このと
き生じる波長589.0nmの輝線の強さを測定してナトリウムを定量する。
定量範囲:Na30〜300μg/l,0.3〜3mg/l,3〜30mg/l,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) ナトリウム標準液 (1 000mgNa/l) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを
600℃で約1時間加熱し,デシケーター中で放冷する。NaCl 100%に対してその2.542gをとり,少量
の水に溶かして全量フラスコ1 000mlに移し入れ,水を標線まで加える。ポリエチレン瓶に保存す
る。
(b) ナトリウム標準液 (3〜30mgNa/l) ナトリウム標準液 (1 000mgNa/l) を段階的にとり,これを水で
薄めて3〜30mgNa/lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,30〜300μgNa/l又は0.3〜3mgNa/lの標準液を調製する。
(2) 装置 装置は,次のとおりとする。
(a) フレーム光度計
(3) 操作 操作は,次のとおり行う。
(a) ナトリウム標準液 (30mgNa/l) (2)をフレーム光度計のフレーム中に噴霧し,波長589.0nmの指示値が
100を示すように調節する。
199
K 0101 : 1998
(b) 水を噴霧して指示値がゼロを示すように調節する。
(c) ナトリウム標準液 (3〜30mgNa/l) (1)を順次噴霧し,ナトリウム (Na) の濃度と指示値との関係線を
作成し,検量線とする。
(d) 試料(3)(4)(ナトリウム濃度が30mg/l以上の場合は薄める。)を噴霧して指示値を読み取り,検量線
から試料中のナトリウムの濃度 (mg/l) を求める。
注(2) 低い濃度の測定用には,3mgNa/l又は0.3mgNa/lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むナトリウム標準液 (3〜30mgNa/l) を調製し,検量線を作成する。
備考1. カリウム及びカルシウムが共存すると正の誤差を生じる。
また,リチウム,バリウム,遊離酸,りん酸塩,ほう酸塩,しゅう酸塩,シリカ,グルコ
ース,ゼラチンなどが共存すると負の誤差を生じる。マグネシウムと硫酸イオンはほとんど
干渉しない。
多量のけい酸塩が共存する場合には,試料の適量を石英ガラスビーカー又は白金皿にとり,
塩酸 (1+1) を加えて酸性とした後,蒸発乾固する。放冷後,塩酸 (1+1) 5滴及び少量の水
を加え,加熱して溶かし,ろ過し,ろ液を水で定量とする。
47.2 フレーム原子吸光法 試料をアセチレン−空気フレーム中に噴霧し,ナトリウムによる原子吸光を
波長589.0nmで測定してナトリウムを定量する。
定量範囲:Na0.05〜4mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) ナトリウム標準液 (0.1mgNa/ml) 47.1(1)(a)のナトリウム標準液 (1 000mgNa/l) 10mlを全量フラス
コ100mlにとり,水を標線まで加える。使用時に調製する。
(b) ナトリウム標準液 (10μgNa/ml) ナトリウム標準液 (0.1mgNa/ml) 20mlを全量フラスコ200mlにと
り,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置
(b) ナトリウム中空陰極ランプ
(3) 操作 操作は,次のとおり行う。
(a) 試料(3)をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,波長589.0nmの指示
値(5)を読み取る。
(b) 空試験として水について,(a)の操作を行って試料について得た指示値を補正する。
(c) 検量線からナトリウムの量を求め,試料中のナトリウムの濃度 (μgNa/l) を算出する。
検量線 ナトリウム標準液 (10μgNa/ml) 0.5〜40mlを全量フラスコ100mlに段階的にとり,水を標
線まで加える。この溶液について(a)の操作を行う。別に,空試験として水について(a)の操作を行っ
て標準液について得た指示値を補正し,ナトリウム (Na) の量と指示値との関係線を作成する。検
量線の作成は試料測定時に行う。
注(5) 吸光度又はその比例値。
47.3 イオン電極法 試料のpHを10.2〜10.6に調節し,ナトリウムイオン電極を指示電極として電位を測
定し,ナトリウムを定量する。
定量範囲:Na1〜100mg/l,繰返し分析精度:変動係数で5〜20%
200
K 0101 : 1998
(1) 試薬 試薬は,次のものを用いる。
(a) トリス緩衝液 JIS K 9704に規定する2-アミノ-2-ヒドロキシメチル-1, 3-プロパンジオール[トリス
(ヒドロキシメチル)アミノメタン]60gをとり,水に溶かして1lとする。
(b) ナトリウム標準液 (100mgNa/l) 47.1(1)(a)のナトリウム標準液 (1 000mgNa/l) 20mlを全量フラス
コ200mlにとり,水を標線まで加える。
(c) ナトリウム標準液 (10mgNa/l) ナトリウム標準液 (100mgNa/l) 20mlを全量フラスコ200mlにとり,
水を標線まで加える。
(d) ナトリウム標準液 (1mgNa/l) ナトリウム標準液 (10mgNa/l) 20mlを全量フラスコ200mlにとり,
水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 電位差計 31.2(2)(a)よる。
(b) 指示電極 ナトリウム電極[ガラス膜(6)電極]
(c) 参照電極 31.2(2)(c)による。ただし,外筒液には硝酸アンモニウム溶液 (100g/l) 又は硝酸カリウム
溶液 (100g/l) を用いる。
(d) マグネチックスターラー 31.2(2)(d)による。
注(6) 液体膜のナトリウム電極を用いる場合は,備考4.による。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) ナトリウム標準液 (1mgNa/l) 100mlをビーカー200mlにとり,トリス緩衝液10ml(7)を加える。
(b) これに指示電極(8)(9)と参照電極(10)(11)とを浸し,マグネチックスターラー(12)で泡が電極と触れない
程度に強くかき混ぜる(13)。
(c) 液温をはかり,電位差計で電位を測定する(14)。
(d) ナトリウム標準液 (10mgNa/l) 及びナトリウム標準液 (100mgNa/l) 100mlをビーカー200mlにとり,
トリス緩衝液10ml(7)をそれぞれ加える。それぞれの液温を(c)での液温の±1℃に調節し,(b)及び(c)
の操作を行ってナトリウム標準液 (10〜100mgNa/l) の電位を測定する(14)。
(e) 片対数紙の対数軸にナトリウムの濃度 (mgNa/l) を,均等軸に電位 (mV) をとって,ナトリウムの
濃度と電位との関係線を作成する(15)。
注(7) トリス緩衝液は,測定時のpHを10.2〜10.6に調節し,イオン強度を一定にするためのものであ
る。
(8) ナトリウムイオン電極は,使用時にナトリウム標準液 (1mgNa/l) に浸し,指針が安定してから
電位を測定する。
(9) 31.の注(12)による。
(10) 31.の注(13)による。
(11) 31.の注(14)による。
(12) 31.の注(15)による。
(13) 31.の注(16)による。
(14) ナトリウムイオン電極の応答時間は,液温10〜30℃の場合,2〜3分間である。
(15) ナトリウム標準液1mgNa/lと100mgNa/lとの電位の差は,100〜120mV (25℃) の範囲に入る。
また,ナトリウムの濃度1〜100mgNa/lの間の検量線は直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 試料100ml(16)をビーカー200mlにとり,トリス緩衝液10ml(7)を加える。液温を(3)(c)での液温の±1℃
201
K 0101 : 1998
に調節する。
(b) (3)(b)及び(c)の操作を行い,検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度
(mgNa/l) を算出する。
注(16) 試料が酸性又は強アルカリ性で,トリス緩衝液を加えてもpHが10.2〜10.6に入らない場合には,
あらかじめ試料を全量フラスコ200mlに入れ,これをアンモニア水 (1+1) 又は塩酸 (1+2) で,
pHを約10に調節し,水を標線まで加える。これから100mlをとり,(a)以降の操作を行って測定
したナトリウムの濃度 (mgNa/l) を希釈割合に応じて補正し,試料中のナトリウムの濃度
(mgNa/l) を求める。
備考2. イオン濃度計の場合には,ナトリウム標準液 (1mgNa/l) とナトリウム標準液 (100mgNa/l) を
用いて(3)(a)〜(c)の操作を行ってイオン濃度計の指針をそれぞれ1mgNa/lと100mgNa/lになる
ように調節する。さらにナトリウム標準液 (10mgNa/l) を用いてイオン濃度計の指示を確認
する。
3. ナトリウムイオン電極(ガラス膜)における主な共存物質の許容限度を最大比率で次に示す。
Li+ : 4.5×10, K+ : 4×102, NH4+ : 1.3×103, Ag+ : 0.2
4. 液体膜のナトリウムイオン電極を用いる場合は,参照電極の外筒液に酢酸リチウム溶液
(1mol/l) 又は酢酸リチウム緩衝液(酢酸リチウム二水和物102gを水約500mlに溶かし,これ
にJIS K 8355に規定する酢酸2mlと水を加えて全量1lとする。pH 6〜7になる。)を用いる。
検量線は,ナトリウム標準液100mlにつき酢酸リチウム緩衝液10mlを加え,pHを6〜7に
調節し,(3)(b)以降の操作を行って作成する。測定は,検量線の作成と同じ条件になるように
操作する。
この場合の共存物質の許容限度を最大比率で次に示す。
K+ : 1.25×102, Li+ : 6.25×103, NH4+ : 1.4×103, Ca2+ : 1.5×104
また,界面活性剤が共存するとドリフトが生じるので注意する。
47.4 イオンクロマトグラフ法 試料中のナトリウムをイオンクロマトグラフ法によって定量する。
定量範囲:Na 0.1〜30mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 36.5 (1)(b)による。
(c) 再生液 36.5 (1)(c)による。
(d) ナトリウム標準液 (1mgNa/ml) 47.1(1)(a)による。
(e) ナトリウム標準液 (0.1mgNa/ml) ナトリウム標準液 (1mgNa/ml) 10mlを全量フラスコ100mlにと
り,水を標線まで加える。
(f) アルカリ金属元素-アンモニウムイオン混合標準液 [(0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml]
36.5(1)(f)による。
(2) 器具及び装置 器具及び装置は,36.5(2)による。
(3) 準備操作 準備操作は,36.5(3)による。
(4) 操作 操作は,次のとおり行う。
(a) 36.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上のナトリウムに相当するピークについて,指示値(17)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
202
K 0101 : 1998
いて得た指示値を補正する。
(d) 検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度 (mgNa/l) を算出する。
検量線 ナトリウム標準液 (0.1mgNa/ml) (18)0.1〜30mlを段階的に全量フラスコ100mlにとり,水を
標線まで加え,(a)及び(b)の操作を行ってそれぞれのナトリウムに相当する指示値を読み取る。別に,
空試験として水について(a)及び(b)の操作を行ってそれぞれのナトリウムに相当する指示値を補正
した後,ナトリウム (Na) の量と指示値との関係線を作成する。検量線の作成は測定時に行う。
注(17) ピーク高さ又はピーク面積
(18) アンモニウムイオン及びカリウムを同時に試験する場合には,アルカリ金属元素-アンモニウム
イオン混合標準液 [(0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml] を用いる。
備考5. ナトリウムの濃度が1mg/lのときアンモニウムイオン及びカリウムはいずれも100mg/l以下で
あれば妨害しない。
6. 36.の備考9.による。
7. 36.の備考10.による。
48. カリウム (K) カリウムの定量には,フレーム光度法,フレーム原子吸光法又はイオンクロマトグラ
フ法を適用する。
48.1 フレーム光度法 試料をアセチレン-空気フレーム又は水素−酸素フレームなどの中に噴霧し,この
とき生じる波長766.5nm又は769.9nmの輝線の強さを測定してカリウムを定量する。
定量範囲:K 40〜400μg/l,0.4〜4mg/l,4〜40mg/l,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) カリウム標準液 (1 000mgK/l) JIS K 8121に規定する塩化カリウムを500〜600℃で約1時間加熱
し,デシケーター中で放冷する。その1.907gをとり,少量の水に溶かして全量フラスコ1 000mlに
移し入れ,水を標線まで加える。ポリエチレン瓶に保存する。
(b) カリウム標準液 (4〜40mgK/l) カリウム標準液 (1 000mgK/l) を段階的にとり,これを水で薄めて
4〜40mgK/lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,40〜400μgK/l又は0.4〜4mgK/lの標準液を調製する。
(2) 装置 装置は,次のとおりとする。
(a) フレーム光度計
(3) 操作 操作は,次のとおり行う。
(a) カリウム標準液 (40mgK/l) (2)をフレーム光度計のフレーム中に噴霧し,波長766.5nm又は769.9nm
の指示値が100を示すように調節する。
(b) 水を噴霧して指示値がゼロを示すように調節する。
(c) カリウム標準液 (4〜40mgK/l) (1)を順次噴霧し,カリウム (K) の濃度と指示値との関係線を作成し,
検量線とする。
(d) 試料(3)(4)(カリウム濃度が40mg/l以上の場合は薄める。)を噴霧して指示値を読み取り,検量線か
ら試料中のカリウムの濃度 (mgK/l) を求める。
注(2) 低い濃度の測定用には,4mgK/l又は0.4mgK/lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むカリウム標準液 (4〜40mgK/l) を調製し,検量線を作成する。
203
K 0101 : 1998
備考1. ナトリウムが共存すると正の誤差を生じる。その他の共存物質による影響は,47.の備考1.に
よる。
48.2 フレーム原子吸光法 試料をアセチレン−空気フレーム中に噴霧し,カリウムによる原子吸光を波
長766.5nmで測定してカリウムを定量する。
定量範囲:K0.05〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) カリウム標準液 (0.1mgK/ml) 48.1(1)(a)のカリウム標準液 (1 000mgK/l) 10mlを全量フラスコ
100mlにとり,水を標線まで加える。使用時に調製する。
(b) カリウム標準液 (10μgK/ml) カリウム標準液 (0.1mgK/ml) 20mlを全量フラスコ200mlにとり,水
を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置
(b) カリウム中空陰極ランプ
(3) 操作 操作は,次のとおり行う。
(a) 試料(3)をJIS K 0121の6.(測定方法)の操作に従って,フレーム中に噴霧し,波長766.5nmの指示
値(5)を読み取る。
(b) 空試験として水について,(a)の操作を行って試料について得た指示値を補正する。
(c) 検量線からカリウムの量を求め,試料中のカリウムの濃度 (mgK/l) を算出する。
検量線 カリウム標準液 (10μgK/ml) 0.5〜50mlを全量フラスコ100mlに段階的にとり,水を標線ま
で加える。この溶液について(a)の操作を行う。別に,空試験として水について(a)の操作を行って標
準液について得た指示値を補正し,カリウム (K) の量と指示値との関係線を作成する。検量線の作
成は試料測定時に行う。
注(5) 吸光度又はその比例値。
48.3 イオンクロマトグラフ法 試料中のカリウムをイオンクロマトグラフ法によって定量する。
定量範囲:K0.1〜30mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA2又はA3の水
(b) 溶離液 36.5(1)(b)による。
(c) 再生液 36.5(1)(c)による。
(d) カリウム標準液 (1mgK/ml) 48.1(1)(a)による。
(e) カリウム標準液 (0.1mgK/ml) カリウム標準液 (1mgK/ml) 10mlを全量フラスコ100mlにとり,水
を標線まで加える。
(f) アルカリ金属元素-アンモニウムイオン混合標準液 [(0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml] 36.5(1)(f)
による。
(2) 器具及び装置 器具及び装置は,36.5(2)による。
(3) 準備操作 準備操作は,36.5(3)による。
(4) 操作 操作は,次のとおり行う。
(a) 36.5(4)(a)及び(b)の操作を行う。
(b) クロマトグラム上のカリウムに相当するピークについて,指示値(6)を読み取る。
(c) 試料を薄めた場合には,空試験として試料と同量の水について,(a)及び(b)の操作を行って試料につ
204
K 0101 : 1998
いて得た指示値を補正する。
(d) 検量線から,カリウムの濃度を求め,試料中のカリウムの濃度 (mgK/l) を算出する。
検量線 カリウム標準液 (0.1mgK/ml) (7)0.1〜30mlを段階的に全量フラスコ100mlにとり,水を標
線まで加え,(a)及び(b)の操作を行ってそれぞれのカリウムに相当する指示値を読み取る。別に,空
試験として水について(a)及び(b)の操作を行ってそれぞれのカリウムに相当する指示値を補正した
後,カリウム (K) の量と指示値との関係線を作成する。検量線の作成は測定時に行う。
注(6) ピーク高さ又はピーク面積
(7) アンモニウムイオン及びナトリウムを同時に試験する場合には,アルカリ金属元素-アンモニウ
ムイオン混合標準液 (0.1mgNH4+, 0.1mgNa, 0.1mgK) /ml ] を用いる。
備考2. カリウムの濃度が1mg/lのときアンモニウムイオン及びナトリウムは,いずれも100mg/l以下
であれば妨害しない。
また,アミン類のうち,メタンアミン(モノメチルアミン)が共存するとカリウムのピー
クと重なり妨害する。
3. 36.の備考9.による。
4. 36.の備考10.による。
49. カルシウム (Ca) カルシウムの定量には,キレート滴定法,フレーム原子吸光法又はICP発光分光
分析法を適用する。
49.1 キレート滴定法 試料をpH 12以上とし,指示薬としてHSNN[2-ヒドロキシ-1-〔(2'-ヒドロキシ−
4'-スルホ-1'-ナフタレニル)アゾ〕-3-ナフタレンカルボン酸(IUPACによる名称)]を加え,エチレンジア
ミン四酢酸二水素二ナトリウム溶液で滴定してカルシウムを定量する。
定量範囲:Ca 0.2〜5mg
(1) 試薬 試薬は,次のものを用いる。
(a) 水酸化カリウム溶液 JIS K 8574に規定する水酸化カリウム250gを水に溶かして500mlとする。
ポリエチレン瓶に保存する。
(b) シアン化カリウム溶液 (100g/l) JIS K 8443に規定するシアン化カリウム10gを水に溶かして
100mlとする。ポリエチレン瓶に保存する。
(c) 塩化ヒドロキシルアンモニウム溶液 (100g/l) 40.2(1)(c)による。
(d) HSNN溶液 JIS K 8776に規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-
ナフトエ酸0.5gをJIS K 8891に規定するメタノール100mlに溶かし,JIS K 8201に規定する塩化ヒ
ドロキシルアンモニウム0.5gを加える。着色ガラス瓶に保存する。
(e) 10mmol/l EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物
を80℃で約5時間加熱し,デシケーター中で放冷する。その3.722gをとり,少量の水に溶かし,全
量フラスコ1 000mlに移し入れ,水を標線まで加える。この溶液1mlは,Ca 0.400 8mgに相当する。
(2) 操作 操作は,次のとおり行う。
(a) 試料(1)の適量(Caとして5mg以下を含む。)をビーカーにとり,水を加えて約50mlとする。
(b) 水酸化カリウム溶液4mlを加え,よくかき混ぜた後,約5分間放置する(2)。
(c) シアン化カリウム溶液 (100g/l) 0.5ml及び塩化ヒドロキシルアンモニウム溶液 (100g/l) 0.5mlを加え
てかき混ぜる。
(d) 指示薬としてHSNN溶液(3) 5,6滴を加え(4),10mmol/l EDTA溶液で,溶液の色が赤紫から青にな
205
K 0101 : 1998
るまで滴定する。
(e) 次の式によって試料中のカルシウムの濃度 (mgCa/l) を算出する。
8
400
.0
000
1
×
×
=
V
a
C
ここに,
C: カルシウム (mgCa/l)
a: 滴定に要した10mmol/l EDTA溶液 (ml)
V: 試料 (ml)
0.400 8: 10mmol/l EDTA溶液1mlのカルシウム相当量 (mg)
注(1) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
また,この試験に影響を与える有機物及び着色物質を含む場合には,4.の操作を行った後,
中和する。ただし,4.4の方法は適用しない。
(2) 放置したときに生じる沈殿の量が多いと,終点が不明りょうになる。このような場合には,1
回目の滴定で,おおよその滴定量を求めておき,別のビーカーに同量の試料をとり,1回目の
滴定に要した10mmol/l EDTA溶液の量よりも1ml少ない量の10mmol/l EDTA溶液を加え,水酸
化カリウム溶液4mlを加えてよく振り混ぜた後,約5分間放置する。次に,シアン化カリウム
溶液 (100g/l) 0.5mlと塩化ヒドロキシルアンモニウム溶液 (100g/l) 0.5mlを加えて振り混ぜる。
これに指示薬としてHSNN溶液5, 6滴を加え,再び10mmol/l EDTA溶液で,溶液の色が青と
なるまで滴定する。
(3) HSNN溶液の代わりにJIS K 8776に規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチ
ルアゾ)-3-ナフトエ酸0.5gとJIS K 8962に規定する硫酸カリウム50gとを均一になるまでよく
すりつぶした粉末約0.1gを加えてもよい。
(4) この指示薬は,添加後放置すると酸化されるため,終点の変色が不明りょうになる。
49.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,カルシウムによる原子吸光を波
長422.7nmで測定してカルシウムを定量する。
定量範囲:Ca 0.2〜4mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) ランタン (III) 溶液 (50gLa/l) 酸化ランタン (III) 29gを少量ずつ塩酸 (1+1) 500mlに加えて溶か
す。
(c) カルシウム標準液 (0.5mgCa/ml) JIS K 8617に規定する炭酸カルシウムを180℃で約1時間加熱
し,デシケーター中で放冷する。その1.249gをとり,水約50mlに分散させ,これに塩酸 (1+1) 20ml
を加えて溶かす。沸騰しない程度に数分間加熱して二酸化炭素を除く。放冷後,全量フラスコ1 000ml
に移し入れ,水を標線まで加える。
(d) カルシウム標準液 (20μgCa/ml) カルシウム標準液 (0.5mgCa/ml) 10mlを全量フラスコ250mlにと
り,塩酸 (1+1) 5ml加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) カルシウム中空陰極ランプ
(3) 操作 操作は,次のとおり行う。
(a) 試料(5)の適量(Caとして20〜400μgを含む。)を全量フラスコ100mlにとり,塩酸 (1+1) 2mlを加
206
K 0101 : 1998
えた後,水を標線まで加える。
(b) この溶液10mlを乾いたビーカーにとり,ランタン (III) 溶液 (50gLa/l) 1mlを加える。
(c) (b)の溶液をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,波長422.7nmの指
示値(6)を読み取る。
(d) 空試験として試料と同量の水をとり,(a)〜(c)の操作を行って試料について得た指示値を補正する。
(e) 検量線からカルシウムの量を求め,試料中のカルシウムの濃度 (mgCa/l) を算出する。
検量線 カルシウム標準液 (20μgCa/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,試料と同
じ酸の濃度になるように塩酸 (1+1) を加えた後,水を標線まで加える。この溶液について(b)及び
(c)の操作を行う。別に,空試験として水について試料と同じ酸の濃度になるように塩酸 (1+1) を
加えた後,(b)及び(c)の操作を行って標準液について得た指示値を補正し,カルシウム (Ca) の量と
指示値との関係線を作成する。検量線の作成は試料測定時に行う。
注(5) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(6) 吸光度又はその比例値
備考1. りん酸塩,硫酸塩,アルミニウムなどは妨害するが,ランタン (III) 溶液 (50gLa/l) を加える
ことによって妨害を抑制することができる。
2. 多量のマグネシウム(1 000mg/l以上)の共存は,負の誤差を与える。
49.3 ICP発光分光分析法 試料を試料導入部を通して誘導結合プラズマ中に噴霧し,カルシウムによる
発光を波長393.367nmで測定してカルシウムを定量する。
定量範囲: Ca10〜200μg/l,0.2〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) 49.2(1)(a)による。
(b) カルシウム標準液 (20μgCa/ml) 49.2(1)(d)による。
(c) 混合標準液 [(20μgCa, 10μgMg, 20μgAl) /ml] 49.2(1)(c)のカルシウム標準液 (0.5mgCa/ml) 20ml,
50.2(1)(c)のマグネシウム標準液 (0.5mgMg/ml) 10ml,59.2(1)(b)のアルミニウム標準液 (0.5mgAl/ml)
20mlを全量フラスコ500mlにとり,塩酸 (1+1) 10mlを加え,水を標線まで加える。使用時に調製
する。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 操作 操作は,次のとおり行う。
(a) 試料(5)の適量(Caとして1〜500μgを含む。)を全量フラスコ100mlにとり,塩酸の濃度が約0.1mol/l
になるように塩酸 (1+1) を加え,水を標線まで加える。
(b) (a)の溶液をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導入部を通してプラ
ズマ中に噴霧し,波長393.367nmの発光強度を測定する(7)(8)(9)。
(c) 空試験として塩酸 (1+1) を水で薄めて塩酸の濃度を約0.1mol/lとした溶液について(b)の操作を行
って試料について得た発光強度を補正する。
(d) 検量線からカルシウムの量を求め,試料中のカルシウムの濃度 (mgCa/l) を算出する。
検量線 カルシウム標準液 (20μgCa/ml) 0.05〜1ml(又は1〜25ml)(10)を全量フラスコ100mlに段階
的にとり,試料と同じ酸の濃度になるように塩酸 (1+1) を加えた後,水を標線まで加える。この
溶液について(b)の操作を行う。別に,空試験として水について試料と同じ酸の濃度になるように塩
207
K 0101 : 1998
酸 (1+1) を加えた後,(b)の操作を行って標準液について得た発光強度を補正し,カルシウム (Ca)
の量と発光強度との関係線を作成する。検量線の作成は試料測定時に行う。
注(7) 波長の異なる2本のスペクトル線の同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,試料の適量を全量フラスコ100mlにとり,イットリウム溶液 (50μgY/ml)
[45.の注(8)による。]10mlを加えた後,塩酸の濃度が約0.1mol/lになるように塩酸 (1+1) を加
え,水を標線まで加える。この溶液について,(3)(b)の噴霧操作を行って波長393.367nmと同時
に371.029nmの発光強度を測定し,カルシウムとイットリウムとの発光強度の比を求める。
別に,カルシウム標準液 (20μgCa/ml) 0.05〜1ml(又は1〜25ml)を全量フラスコ100mlに段
階的にとり,イットリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,塩酸の濃度が約0.1mol/lに
なるように塩酸 (1+1) を加え,水を標線まで加える。この溶液について(3)(b)の噴霧操作を行
って波長393.367nmと同時に371.029nm(イットリウム)の発光強度を測定し,カルシウムの
濃度に対するカルシウムとイットリウムの発光強度比の関係線を作成し,検量線とする。この
検量線から,試料について得た発光強度比に相当するカルシウムの量を求め,試料中のカルシ
ウムの濃度 (mgCa/l) を算出する。
(8) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(9) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(10) マグネシウム及びアルミニウムを同時に試験する場合には,混合標準液 [(20μgCa, 10μgMg,
20μgAl) /ml] を用いて,それぞれの金属元素の試験条件で検量線を作成するとよい。
50. マグネシウム (Mg) マグネシウムの定量には,キレート滴定法,フレーム原子吸光法又はICP発光
分光分析法を適用する。
50.1 キレート滴定法 試料に緩衝液を加えてpH 10に調節し,指示薬としてエリオクロムブラックT[3-
ヒドロキシ-4-〔(1-ヒドロキシ-2-ナフタレニル)アゾ〕-7-ニトロ-1-ナフタレンスルホン酸ナトリウム(IUPAC
による名称)]を加え,エチレンジアミン四酢酸二水素二ナトリウム溶液で滴定し,カルシウムとマグネシ
ウムの合量に対する滴定量を求め,カルシウムに対する滴定量を差し引き,マグネシウムを定量する。
定量範囲:MgとCaの合量がCaとして0.15〜5mg
(1) 試薬 試薬は,次のものを用いる。
(a) シアン化カリウム溶液 (100g/l) 49.1(1)(b)による。
(b) 塩化ヒドロキシルアンモニウム溶液 (100g/l) 40.2(1)(c)による。
(c) 塩化アンモニウム-アンモニア緩衝液 (pH10) 22.1.2(1)(b)による。
(d) エリオクロムブラックT溶液 (5g/l) JIS K 8736に規定するエリオクロムブラックT[1-(1-ヒド
ロキシ−2-ナフチルアゾ)-6-ニトロー2-ナフトール-4-スルホン酸ナトリウム]0.5gをJIS K 8891に
規定するメタノール100mlに溶かし,JIS K 8201に規定する塩化ヒドロキシルアンモニウム0.5gを
加える。着色ガラス瓶に入れて保存する。
(e) 10mmol/lEDTA液 49.1(1)(e)による。この溶液1mlは,Mg 0.243 1mgに相当する。
(2) 操作 操作は,次のとおり行う。
(a) 試料(1)の適量(MgとCaとの合量がCaとして5mg以下を含む。)をビーカーにとり,水を加えて
208
K 0101 : 1998
約50mlとする。
(b) シアン化カリウム溶液 (100g/l) 0.5ml,塩化ヒドロキシルアンモニウム溶液 (100g/l) 数滴及び塩化ア
ンモニウム-アンモニア緩衝液 (pH 10) 1mlを加える。
(c) 指示薬としてエリオクロムブラックT溶液 (5g/l) 2,3滴を加える。
(d) 10mmol/lEDTA溶液で,溶液の赤みが消えて青になるまで滴定(2)する。
(e) 別に,同量の試料をとり,49.1(2)(a)〜(d)の操作を行ってカルシウムの量に相当する10mmol/lEDTA
溶液の滴定量 (ml) を求める。
(f) 次の式によって試料中のマグネシウムの濃度 (mgMg/l) を算出する。
1
243
.0
000
1
1
×
×
−
=
ca
V
b
V
a
M
001
.1
000
1
2
×
×
−
=
ca
V
b
V
a
M
ここに,
M1: マグネシウム (mgMg/l)
a: 滴定に要した10mmol/lEDTA溶液 (ml)
b: 49.1(2)で滴定に要した10mmol/lEDTA溶液 (ml)
V: 試料 (ml)
Vca: 49.1(2)での試料 (ml)
0.243 1: 10mmol/lEDTA溶液1mlのマグネシウム相当量 (mg)
M2: マグネシウムの炭酸カルシウム相当量 (mgCaCO3/l)
1.001: 10mmol/lEDTA溶液1mlの炭酸カルシウム相当量 (mg)
注(1) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
また,この試験に影響を与える有機物及び着色物質を含む場合には,4.の操作を行った後,
中和する。ただし,4.4の方法は適用しない。
(2) エリオクロムブラックTの変色は遅いので,変色点近くではよくかき混ぜながら徐々に滴定す
る。
50.2 フレーム原子吸光法 試料をアセチレン−空気フレーム中に噴霧し,マグネシウムによる原子吸光
を波長285.2nmで測定してマグネシウムを定量する。
定量範囲:Mg20〜400μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) 49.2(1)(a)による。
(b) ランタン (III) 溶液 (50gLa/l) 49.2(1)(b)による。
(c) マグネシウム標準液 (0.5mgMg/ml) JIS K 8432に規定する酸化マグネシウムを700〜800℃で約
30分間加熱し,デシケーター中で放冷する。その0.829gを塩酸 (1+1) 20mlに溶かして全量フラス
コ1 000mlに移し入れ,水を標線まで加える。
(d) マグネシウム標準液 (2μgMg/ml) マグネシウム標準液 (0.5mgMg/ml) 10mlを全量フラスコ100ml
にとり,水を標線まで加える。この溶液10mlを全量フラスコ250mlにとり,塩酸 (1+1) 5mlを加
えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) マグネシウム中空陰極ランプ
(3) 操作 操作は,次のとおり行う。
209
K 0101 : 1998
(a) 試料(3)の適量(Mgとして2〜40μgを含む。)を全量フラスコ100mlにとり,塩酸 (1+1) 2mlを加え
た後,水を標線まで加える。
(b) この溶液10mlを乾いたビーカーにとり,ランタン (III) 溶液 (50gLa/l) 1mlを加える。
(c) (b)の溶液をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,波長285.2nmの指
示値(4)を読み取る。
(d) 空試験として試料と同量の水をとり,(a)〜(c)の操作を行って試料について得た指示値を補正する。
(e) 検量線からマグネシウムの量を求め,試料中のマグネシウムの濃度 (μgMg/l) を算出する。
検量線 マグネシウム標準液 (2μgMg/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,試料と
同じ酸の濃度になるように塩酸 (1+1) を加えた後,水を標線まで加える。この溶液について(b)及
び(c)の操作を行う。別に,空試験として水について試料と同じ酸の濃度になるように塩酸 (1+1) を
加えた後,(b)及び(c)の操作を行って標準液について得た指示値を補正し,マグネシウム (Mg) の量
と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 吸光度又はその比例値
備考1. アルミニウムは少量 (2mg/l) でも妨害するが,ランタン (III) 溶液 (50gLa/l) を添加すること
によって抑制することができる。
50.3 ICP発光分光分析法 試料を試料導入部を通して誘導結合プラズマ中に噴霧し,マグネシウムによ
る発光を波長279.553nmで測定してマグネシウムを定量する。
定量範囲:Mg5〜100μg/l,0.1〜3mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によ
って異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) 49.2(1)(a)による。
(b) マグネシウム標準液 (10μgMg/ml) 50.2(1)(c)のマグネシウム標準液 (0.5mgMg/ml) 5mlを全量フラ
スコ250mlにとり,塩酸 (1+1) 5mlを加えた後,水を標線まで加える。
(c) 混合標準液 [(20μgCa, 10μgMg, 20μgAI) /ml] 49.3(1)(c)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 操作 操作は,次のとおり行う。
(a) 試料(3)の適量(Mgとして0.5〜300μgを含む。)を全量フラスコ100mlにとり,塩酸の濃度が約0.1mol/l
になるように塩酸 (1+1) を加え,水を標線まで加える。
(b) (a)の溶液をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導入部を通してプラ
ズマ中に噴霧し,波長279.553nmの発光強度を測定する(5)(6)(7)。
(c) 空試験として塩酸 (1+1) を水で薄めて塩酸の濃度を約0.1mol/lとした溶液について(b)の操作を行
って試料について得た発光強度を補正する。
(d) 検量線からマグネシウムの量を求め,試料中のマグネシウムの濃度 (μgMg/l) を算出する。
検量線 マグネシウム標準液 (10μgMg/ml) 0.05〜1ml(又は1〜30ml)(8)を全量フラスコ100mlに段
階的にとり,試料と同じ酸の濃度になるように塩酸 (1+1) を加えた後,水を標線まで加える。こ
の溶液について(b)の操作を行う。別に,空試験として水について試料と同じ酸の濃度になるように
塩酸 (1+1) を加えた後,(b)の操作を行って標準液について得た発光強度を補正し,マグネシウム
(Mg) の量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
210
K 0101 : 1998
注(5) 波長の異なる2本のスペクトル線の同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,試料の適量を全量フラスコ100mlにとり,イットリウム溶液 (50μgY/ml)
[45.の注(8)による。]10mlを加えた後,塩酸の濃度が約0.1mol/lになるように塩酸 (1+1) を加
え,水を標線まで加える。この溶液について(3)(b)の噴霧操作を行って波長279.553nmと同時に
371.029nm(イットリウム)の発光強度を測定し,マグネシウムとイットリウムとの発光強度の
比を求める。
別に,マグネシウム標準液 (10μgMg/ml) 0.05〜1ml(又は1〜30ml)を全量フラスコ100mlに
段階的にとり,イットリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,塩酸の濃度が約0.1mol/l
になるように塩酸 (1+1) を加え,水を標線まで加える。この溶液について(3)(b)の噴霧操作を
行って波長279.553nmと同時に371.029nmの発光強度を測定し,マグネシウムの濃度に対する
マグネシウムとイットリウムの発光強度比の関係線を作成し,検量線とする。この検量線から,
試料について得た発光強度比に相当するマグネシウムの量を求め,試料中のマグネシウムの濃
度 (μgMg/l) を算出する。
(6) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(7) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(8) カルシウム及びアルミニウムを同時に試験する場合には,混合標準液 [(20μgCa, 10μgMg,
20μgAl) /ml] を用いて,それぞれの金属元素の試験条件で検量線を作成するとよい。
51. 銅 (Cu) 銅の定量には,ジエチルジチオカルバミド酸吸光光度法,フレーム原子吸光法,電気加熱
原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
51.1 ジエチルジチオカルバミド酸吸光光度法 試料中に共存する金属元素のマスキング剤として,くえ
ん酸塩及びエチレンジアミン四酢酸二水素二ナトリウムを加え,アンモニア水でpHを約9とした後,N, N-
ジエチルジチオカルバミド酸ナトリウム(ジエチルカルバモジチオ酸ナトリウム)を加え,生成する黄褐
色の銅錯体を酢酸ブチルで抽出し,その吸光度を測定して銅を定量する。
定量範囲:Cu2〜30μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) アンモニア水 (1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
(b) 硫酸ナトリウム JIS K 8987に規定するもの。
(c) くえん酸水素二アンモニウム溶液 (100g/l) JIS K 8284に規定するくえん酸水素二アンモニウム
10gを水に溶かして100mlとする。
くえん酸水素二アンモニウム中に銅が含まれるときは,次の操作によって精製する。
くえん酸水素二アンモニウム10gを水80mlに溶かし,アンモニア水 (1+1) を加えてpHを約9
とした後,水を加えて100mlとする。これを分液漏斗に入れ,(e)のジエチルジチオカルバミド酸ナ
トリウム溶液 (10g/l) 2ml及び(g)の酢酸ブチル10mlを加え,激しく振り混ぜて放置する。水層を乾
いたろ紙でろ過し,酢酸ブチルの小滴を除いたろ液を用いる。
(d) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物2gを水に
溶かして100mlとする。
211
K 0101 : 1998
(e) ジエチルジチオカルバミド酸ナトリウム溶液 (10g/l) JIS K 8454に規定するN, N-ジエチルジチオ
カルバミド酸ナトリウム三水和物1.3gを水に溶かして100mlとする。着色瓶に保存し,2週間以上
経過したものは使用しない。
(f) メタクレゾールパープル溶液 (1g/l) 46.1(1)(n)による。
(g) 酢酸ブチル JIS K 8377に規定するもの。
(h) 銅標準液 (0.1mgCu/ml) JIS K 8005に規定する容量分析用標準物質の銅を塩酸 (1+3) で洗い,
水洗し,JIS K 8101に規定するエタノール (99.5) で洗い,次に,JIS K 8103に規定するジエチルエ
ーテルで洗った後,直ちにデシケーター中に入れ12時間以上放置する。Cu 100%に対してその0.100g
をとり,硝酸 (1+1) 20ml中に加え,煮沸して溶かし,窒素酸化物を追い出し,放冷後,全量フラ
スコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 8983に規定する硫酸銅 (II) 五水和物
0.393gをとり,硝酸 (1+1) 20mlを加えて溶かし,全量フラスコ1 000mlに移し入れ,水を標線ま
で加える。若しくはJIS K 0010に規定する標準物質-標準液-銅のCu 100を用いる。
(i) 銅標準液 (1μgCu/ml) 銅標準液 (0.1mgCu/ml) 10mlを全量フラスコ1 000mlにとり,硝酸 (1+1)
20mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った試料の適量(1)(Cuとして2〜30μgを含む。)を分液漏斗にとり,指示薬としてメ
タクレゾールパープル溶液 (1g/l) 2,3滴を加えた後,くえん酸水素二アンモニウム溶液 (100g/l) 5ml
及びEDTA溶液1mlを加える。
(b) アンモニア水 (1+1) を加えて溶液の色が薄い紫(2)になるまで(pH約9)中和し,水を加えて50ml(3)
とする。
(c) ジエチルジチオカルバミド酸ナトリウム溶液 (10g/l) 2mlを加えて混合し,次に,酢酸ブチル(4)10ml
を加え,約3分間激しく振り混ぜて放置する。
(d) 水層を捨て,酢酸ブチル層を硫酸ナトリウム約1gを入れた共栓試験管に移し,振り混ぜる(5)。
(e) その一部を吸収セルに移し,酢酸ブチルを対照液として波長440nm付近の吸光度を測定する。
(f) 空試験として水約20mlをとり,(a)〜(e)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
(g) 検量線から銅の量を求め,試料中の銅の濃度 (mgCu/l) を算出する。
検量線 銅標準液 (1μgCu/ml) 2〜30mlを分液漏斗に段階的にとり,(a)〜(f)の操作を行って銅 (Cu)
の量と吸光度との関係線を作成する。
注(1) 有機物及び濁りを含まない試料で銅の濃度が低い場合には,試料250mlまでの適量をとり,4.1
を行い,(a)〜(f)に準じて操作する。この場合は,試料は前処理した全量を用い,試薬は(a)〜(d)
におけるものと同量を用いる。検量線は試料と同様に操作して作成する。
(2) (a)におけるメタクレゾールパープル溶液 (1g/l) を加えずに,pH計又はpH試験紙を用いてもよ
い。
(3) 分液漏斗にあらかじめ印を付けておく。
(4) 抽出溶媒としてクロロホルム又はベンゼンなどを用いてもよい。ただし,試料中にある種の陰
イオン界面活性剤(例えば,スルホン酸形のもの),タンニンなどが含まれるときは,銅の抽出
212
K 0101 : 1998
が不完全になる。
(5) 乾いたろ紙でろ過するか,又は分液漏斗の脚部に乾いた脱脂綿を詰めてろ過してもよい。
備考1. EDTA溶液を加えない場合には,ジエチルジチオカルバミド酸ナトリウムは多くの金属元素
と反応する。しかし,水銀,ひ素,鉛,すず,アンチモンなど,大部分の金属錯体は無色で
ある。鉄,ニッケル,コバルトなどの錯体は有色であるが,この方法では,EDTA溶液によ
ってマスキングされる。
2. ビスマスは銅とともに抽出され黄色を示すが,銅の量の2倍量以下のときは,ほとんど影響
しない。
2倍量以上を含むときは,(3)の操作によって測定した吸光度をA1とし,別に,銅の試験に
用いた試料と同量の試料をとり,操作(a)の後に,シアン化カリウム溶液 (50g/l) 3mlを加え,
銅をシアノ錯体とした後,(b)〜(f)の操作を行ってビスマス錯体だけを抽出し,その吸光度を
A2とする。銅による吸光度はA1−A2である。
3. 試料を4.1によって前処理する場合,シアン化合物を含むときは十分に加熱する。
51.2 フレーム原子吸光法 試料を前処理した後,アセチレン−空気フレーム中に噴霧し,銅による原子
吸光を波長324.8nmで測定して銅を定量する。
定量範囲:Cu0.2〜4mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 銅標準液 (10μgCu/ml) 51.1(1)(h)の銅標準液 (0.1mgCu/ml) 50mlを全量フラスコ500mlにとり,硝
酸 (1+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) 銅中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考4. 銅の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,次のように
して行うか,又は備考5.による。
試料500ml(又は100〜500mlの一定量)をビーカーにとり,JIS K 8180に規定する塩酸
10mlを加え,約5分間煮沸する。放冷後,分液漏斗1 000ml(又は200〜500ml)に移し入れ,
くえん酸水素二アンモニウム溶液 (100g/l) 10ml及び指示薬としてメタクレゾールパープル
溶液 (1g/l) 2,3滴を加えた後,アンモニア水 (1+1) を溶液の色がわずかに紫になるまで加
える。
ジエチルジチオカルバミド酸ナトリウム溶液 (10g/l) 5mlを加えて振り混ぜた後,JIS K
8377に規定する酢酸ブチル10〜20mlを加え,約1分間激しく振り混ぜ,静置する。酢酸ブ
チル層を分離し,ビーカー100mlに入れる。水層に酢酸ブチル5mlを加え抽出操作を繰り返
す。抽出した酢酸ブチル層は先のビーカーに合わせる。加熱して酢酸ブチルを揮散させた後,
JIS K 8541に規定する硝酸2mlとJIS K 8223に規定する過塩素酸2mlを加えて加熱し,有機
物を分解する。ほとんど乾固した後,放冷する。残留物を硝酸 (1+15) 10mlに溶かし,これ
を銅の定量に用いる(亜鉛,カドミウム,ニッケル,鉛などの定量にも使用できる。)。
なお,抽出した酢酸ブチル層に酢酸ブチルを加えて液量を一定量にしたもの,又は抽出条
件を一定にして,1回抽出を行った酢酸ブチル層をそのまま噴霧して原子吸光分析すること
213
K 0101 : 1998
もできる。JIS K 8377に規定する酢酸ブチルに代え,JIS K 8903に規定する4-メチル-2-ペン
タノン又は2, 6-ジメチル-4-ヘプタノン(ジイソブチルケトン,DIBK)を用いてもよい。
2, 6-ジメチル-4-ヘプタノンを用いる場合は,2, 6-ジメチル-4-ヘプタノンは水との相互溶解
がほとんどないので、その添加量は少なくしてもよい。
5. 試料200mlをとり,備考4.と同様に酸処理し,pHを3.5〜4.0に調節する。硫酸アンモニウム
溶液(飽和)20mlを加える。1-ピロリジンカルボジチオ酸アンモニウム(ピロリジン-N-ジチ
オカルバミド酸アンモニウム) (APDC) 溶液 (10g/l) 5mlを加え,静かに振り混ぜた後,約3
分間放置する。次に,JIS K 8903に規定する4-メチル-2-ペンタノン10mlを加え,約3分間
激しく振り混ぜ,静置する。有機層を分離し,ビーカー100mlに入れる。水層に4-メチル−
2-ペンタノン5mlを加え,抽出操作を繰り返す。抽出した有機層は先のビーカーに合わせる。
この有機層を備考4.と同様に処理し,これを銅の定量に用いる(亜鉛,カドミウム,ニッケ
ル,鉛などの定量にも使用できる。)。
なお,抽出した4-メチル-2-ペンタノン層に同じ溶媒を加えて一定量としたもの,又は抽出
条件を一定にして,1回抽出を行った4-メチル-2-ペンタノン層をそのまま噴霧して原子吸光
分析することもできる。
4-メチル-2-ペンタノンの代わりに2, 6-ジメチル-4-ヘプタノンを用いてもよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料を,JIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長324.8nmの指示値(6)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線から銅の量を求め,試料中の銅の濃度 (mgCu/l) を算出する。
検量線 銅標準液 (10μgCu/ml) 2〜40ml(7)を全量フラスコ100mlに段階的にとり,(3)(a)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(8)。この溶液について(a)の操作
を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた
後,(a)の操作を行って標準液について得た指示値を補正し,銅 (Cu) の量と指示値との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(6) 吸光度又はその比例値。
(7) 準備操作として溶媒抽出を適用するときは,銅標準液 (10μgCu/ml) の量を適宜減らす。
(8) 備考4.又は備考5.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2, 6-
ジメチル-4-ヘプタノン層をそのまま噴霧する場合の検量線は,銅標準液 (10μgCu/ml) を適当な
濃度 (0.1〜1μgCu/ml) に薄め,その2〜40mlを段階的にとり,500ml(又は100〜500mlの一定
量)とした後,試料と同様に備考4.及び(4)(a)と(b)を行って銅 (Cu) の量と指示値との関係線を
作成する。
51.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,銅による原子吸光を波長
324.8nmで測定して銅を定量する。
定量範囲:Cu5〜100μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考6. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
214
K 0101 : 1998
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) 銅標準液 (1μgCu/ml) 51.1(1)(i)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) 銅中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計5〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(600
〜1 000℃,30〜40秒間)し,次に,原子化(9)(2 200〜2 700℃,3〜6秒間)し,波長324.8nmの指
示値(6)を読み取る(10)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行い,
試料について得た指示値を補正する。
(c) 検量線から銅の量を求め,試料中の銅の濃度 (μgCu/l) を算出する。
検量線 銅標準液 (1μgCu/ml) 0.5〜10mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った試料
と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作を行
う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,
(a)の操作を行って標準液について得た指示値を補正し,銅 (Cu) の量と指示値との関係線を作成す
る。検量線の作成は,試料測定時に行う。
注(9) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(10) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
51.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銅
による発光を波長324.754nmで測定して銅を定量する。
定量範囲:Cu20〜5000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 銅標準液 (10μgCu/ml) 51.2(1)(a)による。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.1(1)(h)の銅標準
液 (0.1mgCu/ml) 50ml,52.1(1)(a)の亜鉛標準液 (0.1mgZn/ml) 50ml,53.1(1)(a)のカドミウム標準液
(0.1mgCd/ml) 40ml,54.1(1)(i)のニッケル標準液 (0.1mgNi/ml) 50ml,56.1(1)(a)の鉛標準液
(0.1mgPb/ml) 50ml,58.1(1)(d)のマンガン標準液 (0.1mgMn/ml) 50ml,60.1(1)(g)の鉄標準液
(1mgFe/ml) 5mlをそれぞれ全量フラスコ500mlにとり,硝酸 (1+1) 3mlを加え,水を標線まで加え
る。使用時に調製する。
215
K 0101 : 1998
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考7. 準備操作(前処理)を行った試料のナトリウム,カリウム,マグネシウム,カルシウムなど
の濃度が高く,銅の濃度が低い場合には,次のように操作する。
試料500ml(又は100〜500mlの一定量)をビーカーにとり,JIS K 8180に規定する塩酸
5mlを加え,約5分間煮沸する。放冷後,酢酸-酢酸ナトリウム緩衝液 (pH 5) (JIS K 8371
に規定する酢酸ナトリウム三水和物19.2gとJIS K 8355に規定する酢酸3.4mlとを水に溶か
して1lとする。)10mlを加え,アンモニア水 (1+1) 又は硝酸 (1+10) でpHを5.2に調節す
る。この溶液を分液漏斗1 000ml(又は200〜500ml)に移し,1-ピロリジンカルボジチオ酸
アンモニウム溶液 (20g/l) 2ml,ヘキサメチレンアンモニウム-ヘキサメチレンカルバモジチオ
酸(ヘキサメチレンアンモニウム-ヘキサメチレンジチオカルバミド酸)のメタノール溶液
(20g/l) 2mlを加えて混合した後,JIS K 8271に規定するキシレンの一定量 (5〜20ml) を加え
て約5分間激しく振り混ぜて静置する。水層を捨てキシレン層を共栓試験管に入れる。
また,この溶液は亜鉛,カドミウム,ニッケル,鉛,マンガン,鉄,バナジウムのそれぞ
れの定量及び銅との同時定量に用いることができる。
なお,この操作に用いる酢酸-酢酸ナトリウム緩衝液 (pH5) は使用前に1-ピロリジンカル
ボジチオ酸アンモニウム溶液,ヘキサメチレンアンモニウム-ヘキサメチレンカルバモジチオ
酸のメタノール溶液及びキシレンを加えて振り混ぜ,精製したものとする。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長324.754nmの発光強度を測定する(11)(12)(13)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線から銅の量を求め,試料中の銅の濃度 (μgCu/l) を算出する。
検量線 銅標準液 (10μgCu/ml) 0.2〜50ml(14)(15)を全量フラスコ100mlに段階的にとり,(3)(a)を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
えた後,(a)の操作を行って標準液について得た発光強度を補正し,銅 (Cu) の量と発光強度との関
係線を作成する。検量線の作成は,試料測定時に行う。
注(11) 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることができ
る。内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イッ
トリウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度にな
るように酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
324.754nmと同時に371.029nm(イットリウム)の発光強度を測定し,銅とイットリウムとの発
光強度の比を求める。
別に,銅標準液 (10μgCu/ml) 0.2〜50mlを全量フラスコ100mlに段階的にとり,イットリウム
溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加えた後,
水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長324.754nmと同時に
216
K 0101 : 1998
371.029nmの発光強度を測定し,銅の濃度に対する銅とイットリウムとの発光強度比の関係線
を作成し,検量線とする。この検量線から,試料について得た発光強度比に相当する銅の量を
求め,試料中の銅の濃度 (μgCu/l) を算出する。
(12) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(13) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(14) 備考7.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は,銅標準液
(10μgCu/ml) を適当な濃度 (0.1〜1μgCu/ml) に薄め,その0.2〜50mlを段階的にとり,500ml(又
は100〜500mlの一定量)とした後,試料と同様に備考7.及び(4)(a)と(b)の操作を行って銅 (Cu)
の量と発光強度との関係線を作成する。
(15) 亜鉛,カドミウム,ニッケル,鉛,マンガン,鉄を同時に試験する場合には,混合標準液 [(10μgCu,
10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条
件で検量線を作成するとよい。
51.5 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,銅と内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,銅のイオンの電
流と内標準物質のイオンの電流との比を求めて銅を定量する。
定量範囲: Cu 0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件
によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 51.3(1)(a)による。
(b) 硝酸 (1+1) 51.3(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (16) 45.の注(8)のイットリウム溶液 (50μgY/ml) 20mlを全量フラスコ1
000mlにとり,硝酸 (1+1) 1.5mlを加え,水を標線まで加える。使用時に調製する。
(d) 銅標準液 (1μgCu/ml) 51.1(1)(i)による。
(e) 銅標準液 (50ngCu/ml) 51.1(1)(i)の銅標準液 (1μgCu/ml) 50mlを全量フラスコ1 000mlにとり,硝
酸 (1+1) 3mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.1(1)(h)の銅標準液
(0.1mgCu/ml) 10ml,52.1(1)(a)の亜鉛標準液 (0.1mgZn/ml) 10ml,53.1(1)(a)のカドミウム標準液
(0.1mgCd/ml) 10ml,56.1(1)(a)の鉛標準液 (0.1mgPb/ml) 10ml,58.1(1)(d)のマンガン標準液
(0.1mgMn/ml) 10ml,61.1.1(1)(f)のクロム標準液 (0.1mgCr/ml) 10mlをそれぞれ全量フラスコ1 000ml
にとり,硝酸 (1+1) 1.5mlを加え,水を標線まで加える。使用時に調製する。
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.2(1)(a)の銅標準液
(10μgCu/ml) 5ml,52.1(1)(b)の亜鉛標準液 (10μgZn/ml) 5ml,53.1(1)(b)のカドミウム標準液
(10μgCd/ml) 5ml, 56.3(1)(a)の鉛標準液 (10μgPb/ml) 5ml,58.2(1)(a)のマンガン標準液 (10μgMn/ml)
5ml,61.1.2(1)(a)のクロム標準液 (10μgCr/ml) 5mlをそれぞれ全量フラスコ1 000mlにとり,硝酸 (1
+1) 1.5mlを加え,水を標線まで加える。使用時に調製する。
注(16) 内標準物質として用いる。イットリウムのほかにインジウム (In) ,ビスマス (Bi) などが用い
られる。これらの調製方法は,次による。
217
K 0101 : 1998
インジウム溶液 (1μgIn/ml) インジウム0.250gをとり,JIS K 9901に規定する高純度試薬-
硝酸10mlを加え,加熱して溶かし,窒素酸化物を追い出し,放冷後,全量フラスコ250ml
に移し入れ,水を標線まで加える。使用時に,この溶液1mlを全量フラスコ1 000mlにとり,
硝酸 (1+1) 3mlを加え,水を標線まで加える。
ビスマス溶液 (1μgBi/ml) 酸化ビスマス (III) 0.279gをとり,硝酸 (1+1) 10mlを加え,加
熱して溶かし,放冷後,全量フラスコ250mlに移し入れ,水を標線まで加える。使用時に,
この溶液1mlを全量フラスコ1 000mlにとり,硝酸 (1+1) 20mlを加え,水を標線まで加え
る。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考8. イオン源として,誘導結合プラズマと同等の性能をもつものを用いてもよい。
9. 試料の噴霧に超音波ネブライザー又はこれと同等の性能をもったものを用いてもよい。この
場合は,定量下限値を1けた程度下げることができる。ただし,メモリー効果に注意し,十
分に洗浄を行う。
10. サンプリングコーン及びスキマーコーンの材質からの汚染が認められないことを確認するこ
と。
(3) 準備操作 準備操作は,次のとおり行う(17)。
(a) 試料を4.5によって処理する。
(b) (a)で処理した試料の適量(Cuとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イットリ
ウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を加え
た後,水を標線まで加える。
注(17) 分析者からの汚染がないように注意する。JIS T 9107に規定する手術用ゴム手袋(打ち粉のない
もの)などを用いるとよい。
(4) 操作 操作は,次のとおり行う(18)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧して銅とイットリウムの質量/荷電数(19)における指示値(20)を読み取り,銅の指示値とイット
リウムの指示値との比を求める。
(b) 空試験として,(3)(b)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って銅の指
示値とイットリウムの指示値との比を求め,試料について得た銅とイットリウムとの指示値の比を
補正する。
(c) 検量線から銅の量を求め,試料中の銅の濃度 (μgCu/l) を算出する。
検量線 銅標準液(50ngCu/ml又は1μgCu/ml)1〜50ml(21)を全量フラスコ100mlに段階的にとり,
イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝酸 (1+1) を
加えた後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全量フラ
スコに100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように
硝酸 (1+1) を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値の比を
補正し,銅 (Cu) の量に対する指示値とイットリウムの指示値との比の関係線を作成する。検量線
の作成は,試料測定時に行う。
注(18) 妨害物質の存在が不明の場合には,定量に先だってICP質量分析計による定性分析を行うこと
によって目的とする元素及び内標準物質の測定質量数に対する妨害を推定することができる。
218
K 0101 : 1998
妨害が認められる場合には,内標準物質の変更,試料の希釈又は前処理を行って妨害の軽減を
図る。
(19) 質量数を設定するには,表51.1を参考にするとよい。安定同位体がある場合,複数の同位体の
質量/荷電数を用いて測定を行うことによってスペクトル干渉による妨害を推定することがで
きる。スペクトル干渉による影響が無視できない場合は,試料をさらに薄めて測定する。それ
でも影響を受ける場合は,適切な分離法を用いて妨害となるマトリックスを除去した後,測定
を行う。
表51.1 測定質量数の一例
元素名
質量数
銅
63, 65
亜鉛
66, 68, 67
カドミウム
111, 114
鉛
208, 206, 207
マンガン
55
クロム
53, 52, 50
イットリウム 89
インジウム
115
ビスマス
209
(20) 目的元素の質量/荷電数におけるイオン電流又はその比例値
(21) 銅,亜鉛,カドミウム,鉛,マンガン,クロムを同時に定量する場合には,混合標準液 [(1μgCu,
1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 又は混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb,
50ngMn, 50ngCr) /ml] を用いてそれぞれの金属元素の試験条件で検量線を作成するとよい。
備考11. 注(18)の操作で,妨害物質の影響が無視できる試料の場合は,内標準物質の添加を省略して検
量線法によって定量してもよい。
52. 亜鉛 (Zn) 亜鉛の定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法及び
ICP質量分析法を適用する。
52.1 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,亜鉛による原子
吸光を波長213.9nmで測定して亜鉛を定量する。
定量範囲:Zn 0.05〜2mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 亜鉛標準液 (0.1mgZn/ml) JIS K 8005に規定する容量分析用標準物質の亜鉛を塩酸 (1+3) で洗
い,水洗し,JIS K 8101に規定するエタノール (99.5) で洗い,次に,JIS K 8103に規定するジエチ
ルエーテルで洗った後,直ちにデシケーター中に入れ12時間以上放置する。Zn 100%に対してその
0.100gをとり,硝酸 (1+1) 20ml中に加え,煮沸して溶かし,窒素酸化物を追い出し,放冷後,全
量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0011に規定する亜鉛標準液のZn
100を用いる。
(b) 亜鉛標準液 (10μgZn/ml) 亜鉛標準液 (0.1mgZn/ml) 50mlを全量フラスコ500mlにとり,硝酸 (1
+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) 亜鉛中空陰極ランプ
219
K 0101 : 1998
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考1. 亜鉛の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,51.の備考
4.又は備考5.による。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長213.9nmの指示値(1)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度 (mgZn/l) を算出する。
検量線 亜鉛標準液 (10μgZn/ml) 0.5〜20ml(2)を全量フラスコ100mlに段階的にとり,(3)(a)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(3)。この溶液について(a)の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
えた後,(a)の操作を行って標準液について得た指示値を補正し,亜鉛 (Zn) の量と指示値との関係
線を作成する。検量線の作成は,試料測定時に行う。
注(1) 吸光度又はその比例値。
(2) 準備操作として溶媒抽出を適用するときは,亜鉛標準液 (10μgZn/ml) の量を適宜減らす。
(3) 備考1.によって準備操作を行って酢酸ブチル層,4-メチル-2-ペンタノン層又は2, 6-ジメチル-4-
ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
亜鉛標準液 (10μgZn/ml) を適当な濃度 (0.1〜1μg/ml) に薄め,その0.5〜20mlを段階的にと
り約100mlとした後,試料と同様に備考1.及び(4)(a)と(b)を行って亜鉛 (Zn) の量と指示値との
関係線を作成する。
52.2 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,亜鉛による原子吸光を波長
213.9nmで測定して亜鉛を定量する。
定量範囲:Zn 1〜20μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考2. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) 亜鉛標準液 (1μgZn/ml) 52.1(1)(b)の亜鉛標準液 (10μgZn/ml) 10mlを全量フラスコ100mlにとり,
(b)の硝酸 (1+1) 2mlを加えた後,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) 亜鉛中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計5〜50μl又は自動注
入装置
220
K 0101 : 1998
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3) の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(600
〜1 000℃,30〜40秒間)し,次に,原子化(4)(2 200〜2 700℃,3〜6秒間)し,波長213.9nmの指
示値(1)を読み取る(5)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度 (μgZn/l) を算出する。
検量線 亜鉛標準液 (1μgZn/ml) 0.1〜2mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作を
行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,
(a)の操作を行って標準液について得た指示値を補正し,亜鉛 (Zn) の量と指示値との関係線を作成
する。検量線の作成は,試料測定時に行う。
注(4) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(5) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
52.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,亜
鉛による発光を波長213.856nmで測定して亜鉛を定量する。
定量範囲:Zn10〜6 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異な
る。)
(1) 試薬 試薬は,次のものを用いる。
(a) 亜鉛標準液 (10μgZn/ml) 52.1(1)(b)による。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考3. 準備操作を行った試料のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,亜鉛の濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長213.856nmの発光強度を測定する(6)(7)(8)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度 (μgZn/l) を算出する。
検量線 亜鉛標準液 (10μgZn/ml) (9)(10)0.1〜60mlを全量フラスコ100mlに段階的にとり,(3)(a)を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a) の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
221
K 0101 : 1998
えた後,(a)の操作を行って標準液について得た発光強度を補正し,亜鉛 (Zn) の量と発光強度との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(6) 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることができ
る。内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イッ
トリウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度にな
るように酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
213.856nmと同時に371.029nm(イットリウム)の発光強度を測定し,亜鉛とイットリウムとの
発光強度の比を求める。
別に,亜鉛標準液 (10μgZn/ml) 0.1〜60mlを全量フラスコ100mlに段階的にとり,イットリウ
ム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加えた
後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長213.856nmと同時に
371.029nmの発光強度を測定し,亜鉛の濃度に対する亜鉛とイットリウムとの発光強度比の関
係線を作成し,検量線とする。この検量線から,試料について得た発光強度の比に相当する亜
鉛の量を求め,試料中の亜鉛の濃度 (μgZn/l) を算出する。
(7) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(8) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(9) 備考3.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は亜鉛標準液
(10μgZn/ml) を適当な濃度 (0.1〜1μgZn/ml) に薄め,その0.1〜60mlを段階的にとり,500ml(又
は100〜500mlの一定量)とした後,試料と同様に備考3.及び(4)(a)と(b)の操作を行い,亜鉛 (Zn)
の量と発光強度との関係線を作成する。
(10) 銅,カドミウム,ニッケル,鉛,マンガン,鉄を同時に試験する場合には,混合標準液[(10μgCu,
10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条
件で検量線を作成するとよい。
52.4 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,亜鉛と内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,亜鉛のイオン
の電流と内標準物質のイオンの電流との比を求めて亜鉛を定量する。
定量範囲: Zn0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 52.2(1)(a)による。
(b) 硝酸 (1+1) 52.2(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (11) 51.5(1)(c)による。
(d) 亜鉛標準液 (1μgZn/ml) 52.2(1)(c)による。
(e) 亜鉛標準液 (50ngZn/ml) 亜鉛標準液 (1μgZn/ml) 50mlを全量フラスコ1 000mlにとり,硝酸 (1+
1) 3mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.5(1)(g)による。
222
K 0101 : 1998
注(11) 51.の注(16)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考4. 51.の備考8.による。
5. 51.の備考9.による。
6. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(12)。
(a) 試料を4.5によって処理する。
(b) (3)(a)で処理した試料の適量(Znとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イッ
トリウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を
加えた後,水を標線まで加える。
注(12) 51.の注(17)による。
(4) 操作 操作は,次のとおり行う(13)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧して亜鉛とイットリウムの質量/荷電数(14)における指示値(15)を読み取り,亜鉛の指示値とイ
ットリウムの指示値との比を求める。
(b) 空試験として,(3)(a)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って亜鉛の
指示値とイットリウムの指示値との比を求め,試料について得た亜鉛とイットリウムとの指示値の
比を補正する。
(c) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度 (μgZn/l) を算出する。
検量線 亜鉛標準液(50ngZn/ml又は1μgZn/ml)1〜50ml(16)を全量フラスコ100mlに段階的にとり,
イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ条件になるように硝酸 (1+1) を加え
た後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全量フラスコ
100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ条件になるように硝酸 (1+1)
を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値の比を補正し,亜鉛
(Zn) の量に対する指示値とイットリウムの指示値との比の関係線を作成する。検量線の作成は,試
料測定時に行う。
注(13) 51.の注(18)による。
(14) 51.の注(19)による。
(15) 51.の注(20)による。
(16) 51.の注(21)による。
備考7. 51.の備考11.による。
53. カドミウム (Cd) カドミウムの定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分
光分析法又はICP質量分析法を適用する。
53.1 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,カドミウムによ
る原子吸光を波長228.8nmで測定してカドミウムを定量する。
定量範囲:Cd50〜2 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異な
る。)
(1) 試薬 試薬は,次のものを用いる。
223
K 0101 : 1998
(a) カドミウム標準液 (0.1mgCd/ml) カドミウム(99.9%以上)0.100gをとり,硝酸 (1+1) 20mlに溶
かす。煮沸して窒素酸化物を追い出し,放冷後,全量フラスコ1 000mlに移し入れ,水を標線まで
加える。又はJIS K 0012に規定するカドミウム標準液のCd 100を用いる。
(b) カドミウム標準液 (10μgCd/ml) カドミウム標準液 (0.1mgCd/ml) 50mlを全量フラスコ500mlにと
り,硝酸 (1+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) カドミウム中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考1. カドミウムの濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,51.
の備考4.又は備考5.による。
2. 試料中に多量の鉄又はマンガンが含まれている場合は,次の方法によってカドミウムを分離
濃縮する。
イオン交換樹脂による分離
(a) 試料の適量にJIS K 8180に規定する塩酸を加えて約2mol/lの塩酸酸性とする。これをイオン
交換カラム[I形の強塩基性陰イオン交換樹脂を塩化物イオン形に調製したものをカラム(例
えば,内径10mm,長さ200mm)に充てんしたもの。]に約3ml/minで流してカドミウムをク
ロロ錯体として吸着させた後,塩酸 (1+9) で洗浄する。
(b) 受器を代え,硝酸 (1+12) で溶離し,溶離液を一定液量とする。
3. 試料中に多量の亜鉛,銅などが含まれている場合には,試料の適量に臭化水素酸を加えて約
0.5mol/lの臭化水素酸溶液とし,その50mlに対してトリオクチルアミン(N, N-ジオクチル−
1-オクタンアミン)の4-メチル-2-ペンタノン溶液 (1vol%) 10mlを加えて振り混ぜ,カドミウ
ムを抽出する。抽出した4-メチル-2-ペンタノン層をそのまま噴霧して原子吸光分析に用いる。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長228.8nmの指示値(1)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って,
試料について得た指示値を補正する。
(c) 検量線からカドミウムの量を求め,試料中のカドミウムの濃度 (mgCd/l) を算出する。
検量線 カドミウム標準液 (10μgCd/ml) 0.5〜20ml(2)を全量フラスコ100mlに段階的にとり,(3)(a)
を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(3)。この溶液につい
て(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度条件になるよ
うに酸を加えた後,(a)の操作を行って標準液について得た指示値を補正し,カドミウム (Cd) の量
と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(1) 吸光度又はその比例値。
(2) 準備操作として溶媒抽出を適用するときは,カドミウム標準液 (10μgCd/ml) の量を適宜減らす。
(3) 備考1.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2, 6-ジメチル-4-
ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
カドミウム標準液 (10μgCd/ml) を適当な濃度 (0.1〜1μgCd/ml) に薄め,その0.5〜20mlを段
224
K 0101 : 1998
階的にとり,約100mlとした後,試料と同様に備考1.及び(4)(a)と(b)を行ってカドミウム (Cd) の
量と指示値との関係線を作成する。
備考4. アルカリ金属のハロゲン化物が多量に存在すると,その分子吸収,光散乱などによって正の
誤差を生じる。このような場合には,バックグラウンド補正を行うか,又はあらかじめカド
ミウムを分離する。
53.2 電気加熱原子吸光法 試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム
(II) を加え電気加熱炉で原子化し,カドミウムによる原子吸光を波長228.8nmで測定してカドミウムを定
量する。
定量範囲:Cd0.5〜10μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考5. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) 硝酸パラジウム (II) 溶液 (10μgPd/ml) 硝酸パラジウム (II) 0.108gを硝酸 (1+1) 10mlを加えて
溶かし,全量フラスコ500mlに移し入れ,水を標線まで加える。この溶液20mlを全量フラスコ200ml
にとり,水を標線まで加える。
(d) カドミウム標準液 (1μgCd/ml) 53.1(1)(b)のカドミウム標準液 (10μgCd/ml) 10mlを全量フラスコ
100mlにとり,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(e) カドミウム標準液 (0.1μgCd/ml) カドミウム標準液 (1μgCd/ml) 10mlを全量フラスコ100mlにと
り,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) カドミウム中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜500μl又は自動
注入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料15mlずつをそれぞれ全量フラスコ20mlにとり,カドミウム標準液
(0.1μgCd/ml) を加えないものと,0.1〜2mlの範囲で段階的に3段階以上添加したものとを調製し,
それぞれの溶液の酸の濃度が同じになるように硝酸 (1+1) を加えた後,水を標線まで加える。
(b) (a)の操作を行った試料の100μl以上の一定量をマイクロピペットで小形の容器にとり,これと同体
積の硝酸パラジウム (II) 溶液 (10μgPd/ml) を加え,よく混ぜ合わせる。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(500〜800℃,
30〜40秒間)し,次に,原子化(4)(1 600〜2 200℃,3〜6秒間)し,波長228.8nmの指示値(1)を読
225
K 0101 : 1998
み取る(5)。
(d) 空試験として以下の操作を行う。(3)の準備操作での試料と同量の水をとり,試料と同様に(3)の操作
を行った後,その15mlを全量フラスコ20mlにとる。次に,(4)(a)の溶液の酸の濃度と同じになるよ
うに,硝酸 (1+1) を加えた後,水を標線まで加える。この溶液について(b)及び(c)の操作を行って
試料について得た指示値を補正する。
(e) カドミウムの添加量と指示値との関係線を作成し,カドミウムの量を求め,試料中のカドミウムの
濃度 (μgCd/l) を算出する。
注(4) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(5) 引き続いて少なくとも(c)の操作を3回繰り返し,指示値が合うことを確認する。
53.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,カ
ドミウムによる発光を波長214.438nmで測定してカドミウムを定量する。
定量範囲:Cd8〜2000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) カドミウム標準液 (8μgCd/ml) 53.1(1)(a)のカドミウム標準液 (0.1mgCd/ml) 40mlを全量フラスコ
500mlにとり,硝酸 (1+1) 10mlを加えた後,水を標線まで加える。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考6. 準備操作を行った溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,カドミウムの濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長214.438nmの発光強度を測定する(6)(7)(8)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からカドミウムの量を求め,試料中のカドミウムの濃度 (μgCd/l) を算出する。
検量線 カドミウム標準液 (8μgCd/ml) 0.1〜25ml(9)(10)を全量フラスコ100mlに段階的にとり,(3)(a)
を行った試料と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について(a)の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
えた後,(a)の操作を行って標準液について得た発光強度を補正し,カドミウム (Cd) の量と発光強
度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(6) 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることができ
る。内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イッ
トリウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度にな
るように酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
214.438nmと同時に371.029nm(イットリウム)の発光強度を測定し,カドミウムとイットリウ
ムとの発光強度の比を求める。
226
K 0101 : 1998
別に,カドミウム標準液 (8μgCd/ml) 0.1〜25mlを全量フラスコ100mlに段階的にとり,イッ
トリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を
加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長214.438nmと
同時に371.029nmの発光強度を測定し,カドミウムの濃度に対するカドミウムとイットリウム
との発光強度比の関係線を作成し,検量線とする。この検量線から,試料について得た発光強
度の比に相当するカドミウムの量を求め,試料中のカドミウムの濃度 (μgCd/l) を算出する。
(7) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(8) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(9) 備考6.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は,カドミウム
標準液 (8μgCd/ml) を適当な濃度 (0.1〜0.8μgCd/ml) に薄め,その0.1〜25mlを段階的にとり,
500ml(又は100〜500mlの一定量)とした後,試料と同様に備考6.及び(4)(a)と(b)の操作を行っ
てカドミウム (Cd) の量と発光強度との関係線を作成する。
(10) 銅,亜鉛,ニッケル,鉛,マンガン,鉄を同時に試験する場合には,混合標準液 [(10μgCu, 10μgZn,
8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条件で検量
線を作成するとよい。
53.4 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,カドミウムと内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,カドミ
ウムのイオンの電流と内標準物質のイオンの電流との比を求めてカドミウムを定量する。
定量範囲: Cd0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 53.2(1)(a)による。
(b) 硝酸 (1+1) 53.2(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (11) 51.5(1)(c)による。
(d) カドミウム標準液 (1μgCd/ml) 53.2(1)(d)による。
(e) カドミウム標準液 (50ngCd/ml) カドミウム標準液 (1μgCd/ml) 50mlを全量フラスコ1 000mlにと
り,硝酸 (1+1) 3mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による。
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.5(1)(g)による。
注(11) 51.の注(16)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考7. 51.の備考8.による。
8. 51.の備考9.による。
9. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(12)。
(a) 試料を4.5によって処理する。
227
K 0101 : 1998
(b) (3)(a)で処理した試料の適量(Cdとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イッ
トリウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を
加えた後,水を標線まで加える。
注(12) 51.の注(17)による。
(4) 操作 操作は,次のとおり行う(13)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧してカドミウムとイットリウムの質量/荷電数(14)における指示値(15)を読み取り,カドミウム
の指示値とイットリウムの指示値との比を求める。
(b) 空試験として,(3)(a)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行ってカドミ
ウムの指示値とイットリウムの指示値との比を求め,試料について得たカドミウムとイットリウム
との指示値の比を補正する。
(c) 検量線からカドミウムの量を求め,試料中のカドミウムの濃度 (μgCd/l) を算出する。
検量線 カドミウム標準液(50ngCd/ml又は1μgCd/ml)1〜50ml(16)を全量フラスコ100mlに段階的
にとり,イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝酸 (1
+1) を加えた後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全
量フラスコ100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるよ
うに硝酸 (1+1) を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値の
比を補正し,カドミウム (Cd) の量に対する指示値とイットリウムの指示値との比の関係線を作成
する。検量線の作成は,試料測定時に行う。
注(13) 51.の注(18)による。
(14) 51.の注(19)による。
(15) 51.の注(20)による。
(16) 51.の注(21)による。
備考10. 51.の備考11.による。
54. ニッケル (Ni) ニッケルの定量には,ジメチルグリオキシム吸光光度法,フレーム原子吸光法又は
ICP発光分光分析法を適用する。
54.1 ジメチルグリオキシム吸光光度法 試料にくえん酸塩を加え,アンモニア水で微アルカリ性とした
後,2, 3-ブタンジオンジオキシム(ジメチルグリオキシム)を加えて生成したニッケル錯体をクロロホル
ムで抽出し,これを希塩酸で逆抽出する。抽出液に臭素及びアンモニア水を加えてニッケルを酸化し,再
び2, 3-ブタンジオンジオキシムを加えて生じる赤褐色のニッケル錯体の吸光度を測定してニッケルを定量
する。
定量範囲:Ni2〜50μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+20) JIS K 8180に規定する塩酸を用いて調製する。
(b) アンモニア水 (1+1) 及び (1+5) JIS K 8085に規定するアンモニア水を用いて調製する。
(c) 臭素水(飽和) JIS K 8529に規定する臭素3〜4mlを水100mlに加えて激しく振り混ぜ,放置後
その上澄み液を用いる。
(d) くえん酸水素二アンモニウム溶液 (100g/l) JIS K 8284に規定するくえん酸水素二アンモニウム
10gを水約80mlに溶かし,アンモニア水 (1+1) を滴加してpHを約7に調節した後,水を加えて
228
K 0101 : 1998
100mlとする。
(e) フェノールフタレイン溶液 (5g/l) 13.2(1)(a)による。
(f) ジメチルグリオキシムのエタノール溶液 (10g/l) JIS K 8498に規定するジメチルグリオキシム1g
をJIS K 8102に規定するエタノール (95) に溶かして100mlとする。
(g) ジメチルグリオキシムの水酸化ナトリウム溶液 (10g/l) JIS K 8498に規定するジメチルグリオキ
シム1gを水酸化ナトリウム溶液 (10g/l) に溶かし,水酸化ナトリウム溶液 (10g/l) を加えて100ml
とする。不溶解物があるときはろ過する。
(h) クロロホルム JIS K 8322に規定するもの。
(i) ニッケル標準液 (0.1mgNi/ml) JIS K 9062に規定するニッケル(99.9%以上)0.100gをとり,硝酸
(1+1) 20mlに溶かし,加熱して窒素酸化物を追い出し,放冷後,全量フラスコ1 000mlに移し入れ,
水を標線まで加える。又はJIS K 8990に規定する硫酸ニッケル (II) アンモニウム六水和物0.673g
をとり,水と硝酸 (1+1) 10mlに溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。
若しくはJIS K 0013に規定するニッケル標準液のNi100を用いる。
(j) ニッケル標準液 (5μgNi/ml) ニッケル標準液 (0.1mgNi/ml) 50mlを全量フラスコ1 000mlにとり,
硝酸 (1+1) 20mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った試料の適量(1)(Niとして2〜50μgを含む。)を分液漏斗にとり,くえん酸水素二
アンモニウム溶液 (100g/l) 5mlと指示薬としてフェノールフタレイン溶液 (5g/l) (2)2,3滴を加え,
次に,アンモニア水 (1+5) を溶液の色がわずかに赤になるまで滴加する。更にアンモニア水 (1+
5) 2,3滴及び水を加えて約100mlとする。
(b) ジメチルグリオキシムのエタノール溶液 (10g/l) 2mlとクロロホルム10mlとを加え,約1分間激し
く振り混ぜて放置した後,クロロホルム層を別の分液漏斗に入れる。水層にクロロホルム5mlを加
え,約1分間激しく振り混ぜて抽出し,放置後,クロロホルム層をとり,先の分液漏斗に合わせる。
この操作を更に1回繰り返す。
(c) クロロホルム層を入れた分液漏斗にアンモニア水 (1+50) 10〜20mlを加え,約30秒間振り混ぜて
放置した後,クロロホルム層を別の分液漏斗に移す。
(d) クロロホルム層を入れた分液漏斗に塩酸 (1+20) 10mlを加え,約1分間激しく振り混ぜ,ニッケル
を逆抽出する。放置後,クロロホルム層を別の分液漏斗に入れる。クロロホルム層には,再び塩酸 (1
+20) 5mlを加え,逆抽出を繰り返す。クロロホルム層は捨て,水層は先の水層に合わせて全量フラ
スコ25mlに入れる。
(e) 臭素水(飽和)2mlを加えて振り混ぜ,約1分間放置する。次に,アンモニア水 (1+1) を加えて中
和し,更にアンモニア水 (1+1) 2mlを過剰に加え,流水で室温以下に冷却する。
(f) ジメチルグリオキシムの水酸化ナトリウム溶液 (10g/l) 2mlを加えて振り混ぜ,ニッケルを発色させ
た後,水を標線まで加える。
(g) この溶液の一部を吸収セルに移し,波長450nm付近の吸光度を測定する。
(h) 空試験として水約50mlをとり,(a)〜(g)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
229
K 0101 : 1998
(i) 検量線からニッケルの量を求め,試料中のニッケルの濃度 (μgNi/l) を算出する。
検量線 ニッケル標準液 (5μgNi/ml) 0.4〜10mlを全量フラスコ25mlに段階的にとり,(e)〜(h)の操
作を行ってニッケル (Ni) の量と吸光度との関係線を作成する。
注(1) 有機物及び濁りを含まない試料でニッケルの濃度が低い場合は,試料500mlまでの適量をとり,
4.1を行い,(a)〜(h)に準じて操作して定量できる。この場合には,試料は前処理した全量を用い,
試薬は(a)〜(f)におけるものと同量を用いる。検量線は試料と同様に操作して作成する。
(2) リトマス試験紙を用いてもよい。
54.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,ニッケルによる
原子吸光を波長232.0nmで測定してニッケルを定量する。
定量範囲:Ni0.3〜6mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) ニッケル標準液 (10μgNi/ml) 54.1(1)(i)のニッケル標準液 (0.1mgNi/ml) 50mlを全量フラスコ
500mlにとり,硝酸 (1+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) ニッケル中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考1. ニッケルの濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,次に
よって行うか,又は備考2.及び備考3.による。
試料100mlをビーカーにとり,JIS K 8180に規定する塩酸5mlを加え,約5分間煮沸した
後,放冷する。54.1(3)(a)〜(c)に準じて操作し,ニッケルをジメチルグリオキシム錯体として,
クロロホルム層に抽出する。クロロホルム層を合わせ,塩酸 (1+20) 10mlを加え,振り混ぜ
てニッケルを逆抽出する。水層を分離した後,クロロホルム層に塩酸 (1+20) 5mlを加えて
逆抽出を行う。逆抽出液を合わせ,全量フラスコ25mlに移し入れ,水を標線まで加える。
これをニッケルの定量に用いる。
2. 51.の備考4.に準じる。
3. 51.の備考5.に準じる。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長232.0nmの指示値(3)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からニッケルの量を求め,試料中のニッケルの濃度 (mgNi/l) を算出する。
検量線 ニッケル標準液 (10μgNi/ml) 3〜60ml(4)を全量フラスコ100mlに段階的にとり,(3)(a)を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
えた後,(a)の操作を行って標準液について得た指示値を補正し,ニッケル (Ni) の量と指示値との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(3) 吸光度又はその比例値。
230
K 0101 : 1998
(4) 準備操作として溶媒抽出法を適用するときは,ニッケル標準液の量を適宜減らす。
54.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ニ
ッケルによる発光を波長221.647nmで測定してニッケルを定量する。
定量範囲:Ni40〜2 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) ニッケル標準液 (10μgNi/ml) 54.2(1)(a)による。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考4. 準備操作を行った溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,ニッケルの濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分析の定量分析)に従って,試料導入部
を通してプラズマ中に噴霧し,波長221.647nmの発光強度を測定する(5)(6)(7)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からニッケルの量を求め,試料中のニッケルの濃度 (μgNi/l) を算出する。
検量線 ニッケル標準液 (10μgNi/ml) 0.4〜20ml(8)(9)を全量フラスコ100mlに段階的にとり,(3)(a)
を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について
(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸
を加えた後,(a)の操作を行って標準液について得た発光強度を補正し,ニッケル (Ni) の量と発光
強度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 波長の異なる2本のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリ
ウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよ
うに酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
221.647nmと同時に371.029nm(イットリウム)の発光強度を測定し,ニッケルとイットリウム
との発光強度の比を求める。
別に,ニッケル標準液 (10μgNi/ml) 0.4〜20mlを全量フラスコ100mlに段階的にとり,イット
リウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加
えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長221.647nmと同
時に371.029nmの発光強度を測定し,ニッケルの濃度に対するニッケルとイットリウムとの発
光強度比の関係線を作成し,検量線とする。この検量線から,試料について得た発光強度比に
相当するニッケルの量を求め,試料中のニッケルの濃度 (μgNi/l) を算出する。
(6) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(7) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
231
K 0101 : 1998
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(8) 備考4.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は,ニッケル標
準液 (10μgNi/ml) を適当な濃度 (0.2〜1μgNi/ml) に薄め,その0.4〜20mlを段階的にとり,500ml
(又は100〜500mlの一定量)とした後,試料と同様に備考4.及び(4)(a)と(b)の操作を行ってニ
ッケル (Ni) の量と発光強度の関係線を作成する。
(9) 銅,亜鉛,カドミウム,鉛,マンガン,鉄を同時に試験する場合には,混合標準液 [(10μgCu, 10μgZn,
8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条件で検量
線を作成するとよい。
55. すず (Sn) すずの定量には,フェニルフルオロン吸光光度法,ケルセチン吸光光度法又はICP発光
分光分析法を適用する。
55.1 フェニルフルオロン吸光光度法 すずを過マンガン酸カリウムですず (IV) とし,くえん酸,ポリビ
ニルアルコールの存在のもとで,フェニルフルオロン(2, 3, 7-トリヒドロキシ-9-フェニル-6-フルオロン)
を加えて黄色の錯体を生成させ,その吸光度を測定して定量する。
定量範囲:Sn3〜40μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(b) 塩酸 (1+4) JIS K 8180に規定する塩酸を用いて調製する。
(c) 硫酸 (1+1) 4.4(1)(b)による。
(d) L (+) -アスコルビン酸 JIS K 9502に規定するもの。
(e) くえん酸溶液 (100g/l) JIS K 8283に規定するくえん酸一水和物11gを水に溶かして100mlとする。
(f) アンモニア水 (1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
(g) 過マンガン酸カリウム溶液 (3g/l) 46.1(1)(f)による。
(h) ポリビニルアルコール溶液 (5g/l) JIS K 9550に規定するポリビニルアルコール(けん化度約
80mol%のもの。)0.5gを水に溶かして100mlとする。
(i) ブロモクレゾールグリーン溶液 (0.5g/l) JIS K 8840に規定するブロモクレゾールグリーン0.05g
をJIS K 8102に規定するエタノール (95) 20mlに溶かし,水を加えて100mlとする。
(j) フェニルフルオロン溶液 (0.1g/l) JIS K 9547に規定するフェニルフルオロン0.05gを,JIS K 8102
に規定するエタノール (95) 100mlに塩酸 (1+2) 10mlを加えた溶液に溶かし,エタノール (95) を
加えて500mlとする。
(k) すず標準液 (0.1mgSn/ml) JIS K 8580に規定するすず0.100gをビーカーにとり,硫酸10mlを加
え,時計皿で覆い,加熱して溶かす。放冷後,塩酸 (1+4) 200mlを加えて溶かし,室温に冷却する。
全量フラスコ1 000mlに移し入れ,塩酸 (1+4) を標線まで加える。
(l) すず標準液 (2μgSn/ml) すず標準液 (0.1mgSn/ml) 10mlを全量フラスコ500mlにとり,塩酸 (1+
10) を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った試料の適量(Snとして7.5〜100μgを含む。)をとり,硫酸 (1+1) 5ml(1)を加え,
加熱して硫酸白煙を発生させた後,乾固近くまで濃縮する。
232
K 0101 : 1998
(b) 放冷後,塩酸 (1+4) 10mlを徐々に加え,加熱して溶かし,室温まで冷却した後,全量フラスコ50ml
に移し入れ,水を標線まで加える。
(c) この溶液20mlずつを全量フラスコ50ml及びビーカー100mlにそれぞれとる。
(d) ビーカー中の溶液に水を加えて約50mlとし,指示薬としてブロモクレゾールグリーン溶液 (0.5g/l) 2,
3滴を加え,アンモニア水 (1+1) で溶液の色が緑になるまで滴定する。中和に要したアンモニア水
(1+1) の量を記録する。
(e) 全量フラスコ50ml中の溶液に過マンガン酸カリウム溶液 (3g/l) を溶液の色が微赤になるまで滴加
し,約5分間放置してすずを酸化する。
(f) L (+) -アスコルビン酸約0.1gを加えて振り混ぜ,過剰の過マンガン酸を還元する。
(g) くえん酸溶液 (100g/l) 5mlと塩酸 (1+11) 1.5ml及び(d)で中和に要したのと同量のアンモニア水 (1
+1) を加える(pHは1.5〜2.0になる。)。
(h) ポリビニルアルコール溶液 (5g/l) 5mlとフェニルフルオロン溶液 (0.1g/l) 5mlを加えて振り混ぜた
後,水を標線まで加え,約20分間放置する。
(i) 空試験として塩酸 (1+4) 10mlを全量フラスコ50mlにとり,水を標線まで加える。以下,(c)〜(h)
の操作を行う。
(j) (h)で得た溶液の一部を吸収セルに入れ,(i)で得た溶液を対照液として波長510nm付近の吸光度を測
定する。
(k) 検量線からすずの量を求め,試料中のすずの濃度 (μgSn/l) を算出する。
検量線 すず標準液 (2μgSn/ml) 1.5〜20mlを全量フラスコ50ml及びビーカー100mlに段階的にとり,
(d)〜(j)の操作を行ってすず (Sn) の量と吸光度との関係線を作成する。
注(1) 試料の前処理に硫酸を用いた場合には加えない。
備考1. 有機物及び濁りを含まない試料で,すずの濃度が低い場合には,試料500mlまでの適量をと
り,次のように酸化マンガン (IV) (二酸化マンガン)共沈法で濃縮する。
試料100mlにつき硝酸3mlと硝酸マンガン (II) 溶液[JIS K 8568に規定する硝酸マンガン
(II) 六水和物16gを水に溶かして100mlとする。]2mlを加え,静かに加熱して煮沸する。引
き続き溶液100mlにつき過マンガン酸カリウム溶液 (30g/l) (JIS K 8247に規定する過マン
ガン酸カリウム3gを水に溶かして100mlとする。)2mlを加え,5〜10分間静かに煮沸して
酸化マンガン (IV) の沈殿を生成させる。約20分間放置した後,ろ紙5種Bを用いてろ別し,
温水で数回洗浄する。沈殿は元のビーカーに移し入れ,ろ紙に付着した沈殿は硝酸 (1+1)
10mlと過酸化水素 (1+30) をろ紙上に交互に滴加して溶かし,ろ紙は温水で洗浄する。
この洗液は元のビーカーに合わせ,加熱して酸化マンガン (IV) を溶かし,引き続き加熱
して過酸化水素を分解した後,水を加えて約200mlとする。この溶液に硝酸マンガン (II) 溶
液1mlを加え加熱し,静かに煮沸する。過マンガン酸カリウム溶液 (30g/l) 2mlを加え,煮沸
を5〜10分間続けて,再び酸化マンガン (IV) を沈殿させる。これをろ過して温水で洗う。
沈殿は元のビーカーに移し入れ,ろ紙に付着している沈殿は,温硫酸 (1+3) 10mlに過酸化
水素水 (1+30) 5mlを加えた混合溶液を滴加して溶かし,ろ液を元のビーカーに受ける。ろ
紙は水で洗い洗液は合わせる。加熱して沈殿を溶かすとともに蒸発して硫酸白煙を発生させ
て乾固近くまで濃縮する。放冷後,塩酸 (1+4) 10mlを加えて加熱し,残留物を溶かす。放
冷後,この溶液を全量フラスコ50mlに移し入れ,水を標線まで加え,以下,(3)(c)〜(j)の操
作を行う。
233
K 0101 : 1998
2. 妨害元素は,ゲルマニウム,ジルコニウム,アンチモン,ビスマス,鉄などである。ただし,
鉄は,L (+) -アスコルビン酸で還元しておけば,約10mgまでは妨害しない。アンチモンは,
酸化して5価にしておき,ビスマスは発色させた後,JIS K 8107に規定するエチレンジアミ
ン四酢酸二水素二ナトリウム二水和物0.3gを加えて錯体とすれば,それぞれ0.5mgまで妨害
しない。
55.2 ケルセチン吸光光度法 すずを過マンガン酸カリウムですず (IV) とし,過剰の過マンガン酸を還元
する。ケルセチン[2-(3, 4-ジヒドロキシフェニル)-3, 5, 7-トリヒドロキシ-4H-ベンゾピラン-4-オン]を
加え,黄色のすず-ケルセチン錯体を生成させ,4-メチル-2-ペンタノンで抽出し,その吸光度を測定して定
量する。
定量範囲:Sn2〜20μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) 硫酸 (1+1) 4.4(1)(b)による。
(c) 硫酸 (1+19) JIS K 8951に規定する硫酸を用いて調製する。
(d) L (+) -アスコルビン酸 JIS K 9502に規定するもの。
(e) 過マンガン酸カリウム溶液 (3g/l) 46.1(1)(f)による。
(f) 硫酸ナトリウム JIS K 8987に規定するもの。
(g) チオ尿素溶液 (50g/l) JIS K 8635に規定するチオ尿素10gを水に溶かして200mlとする。
(h) 4-メチル-2-ペンタノン JIS K 8903に規定するもの。
(i) ケルセチン溶液 (1g/l) ケルセチン0.2gをJIS K 8101に規定するエタノール (99.5) 約100mlに溶
かし,JIS K 8180に規定する塩酸10mlを加えた後,エタノール (99.5) で200mlとする。使用時に
調製する。
(j) すず標準液 (5μgSn/ml) 55.1(1)(k)のすず標準液 (0.1mgSn/ml) 10mlを全量フラスコ200mlにとり,
塩酸 (1+10) を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 100ml
(b) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った試料の適量(Snとして2〜20μgを含む。)をとり,硫酸 (1+1) 5ml(1)を加え,加
熱して硫酸白煙を発生させた後,乾固近くまで濃縮する。
(b) 放冷後,硫酸 (1+19) 5mlを加え加熱して溶かし,過マンガン酸カリウム溶液 (3g/l) を溶液の色が
微赤になるまで滴加し,約5分間放置してすずを酸化する。
(c) L (+) -アスコルビン酸0.1gを加えて振り混ぜ,過剰の過マンガン酸を還元する。
(d) 分液漏斗100mlに少量の水とともに移し入れ,チオ尿素溶液 (50g/l) 20ml及び塩酸 (1+1) 15mlを
加えて振り混ぜる。ケルセチン溶液 (1g/l) 20mlを加えて再び振り混ぜ,約15分間放置する。
(e) 4-メチル−2-ペンタノン15mlを加えて約1分間激しく振り混ぜ,すず-ケルセチン錯体を抽出する。
(f) 水層を捨て,有機層に硫酸 (1+19) 25mlを加えて約30秒間振り混ぜる。
(g) 水層を捨て,有機層に硫酸ナトリウム約5gを加えて振り混ぜる。
(h) 空試験として,硫酸 (1+19) 5mlをとり,(b)〜(g)の操作を行う。
(i) (g)の有機層の一部を吸収セルにとり,(h)の有機層を対照液として波長440nm付近の吸光度を測定
234
K 0101 : 1998
する。
(j) 検量線からすずの量を求め,試料中のすずの濃度 (μgSn/l) を算出する。
検量線 すず標準液 (5μgSn/ml) 0.4〜4mlを段階的にとり,硫酸 (1+19) 5mlを加え,加熱して液量
を約5mlとし,過マンガン酸カリウム溶液 (3g/l) を溶液の色が微赤になるまで加える。以下,(c)
〜(i)の操作を行ってすず (Sn) の量と吸光度との関係線を作成する。
備考3. 有機物及び濁りを含まない試料で,すずの濃度が低い場合には,備考1.によって酸化マンガ
ン (IV) 共沈法で分離濃縮し,以下,(3)(b)以降の操作を行って定量する。
55.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,す
ずによる発光を波長189.989nmで測定してすずを定量する。
定量範囲:Sn0.4〜2mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) すず標準液 (10μgSn/ml) 55.1(1)(k)のすず標準液 (0.1mgSn/ml) 10mlを全量フラスコ100mlにとり,
塩酸 (1+10) を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長189.989nmの発光強度を測定する(2)(3)(4)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からすずの量を求め,試料中のすずの濃度 (mgSn/l) を算出する。
検量線 すず標準液 (10μgSn/ml) 4〜20mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作を
行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,
(a)の操作を行って標準液について得た発光強度を補正し,すず (Sn) の量と発光強度との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(2) 波長の異なる2本のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリ
ウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよ
うに酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
189.989nmと同時に371.029nm(イットリウム)の発光強度を測定し,すずとイットリウムとの
発光強度の比を求める。
別に,すず標準液 (10μgSn/ml) 4〜20mlを全量フラスコ100mlに段階的にとり,イットリウ
ム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加えた
後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長189.989nmと同時に
371.029nmの発光強度を測定し,すずの濃度に対するすずとイットリウムとの発光強度比の関
係線を作成し,検量線とする。この検量線から,試料について得た発光強度比に相当するすず
の量を求め,試料中のすずの濃度 (mgSn/l) を算出する。
235
K 0101 : 1998
(3) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(4) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
56. 鉛 (Pb) 鉛の定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP
質量分析法を適用する。
56.1 フレーム原子吸光法 試料を前処理した後,アセチレン−空気フレーム中に噴霧し,鉛による原子
吸光を波長283.3nmで測定して鉛を定量する。
定量範囲:Pb1〜20mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 鉛標準液 (0.1mgPb/ml) JIS K 8701に規定する鉛(99.9%以上)0.100gをとり,硝酸 (1+3) 40ml
を加えて溶かし,加熱して窒素酸化物を追い出し,放冷後,全量フラスコ1 000mlに移し入れ,水
を標線まで加える。又はJIS K 8563に規定する硝酸鉛 (II) 0.160gをとり,硝酸 (1+1) 20mlと適量
の水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。若しくはJIS K 0015に規
定する鉛標準液のPb 100を用いる。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) 鉛中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5(1)によって処理する。
注(1) 水溶液をそのまま噴霧する場合は,硫酸イオンが存在すると鉛に負の誤差を与えるので,硫酸
を用いる前処理は行わない。
備考1. 鉛の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,51.の備考4.
又は備考5.に準じる。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長283.3nmの指示値(2)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線から鉛の量を求め,試料中の鉛の濃度 (mgPb/l) を算出する。
検量線 鉛標準液 (0.1mgPb/ml) 1〜20ml(3)を全量フラスコ100mlに段階的にとり,(3)(a)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(4)。この溶液について(a)の操作
を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた
後,(a)の操作を行って標準液について得た指示値を補正し,鉛 (Pb) の量と指示値との関係線を作
成する。検量線の作成は,試料測定時に行う。
注(2) 吸光度又はその比例値。
(3) 準備操作として溶媒抽出を適用するときは,鉛標準液 (0.1mgPb/ml) の量を適宜減らす。
(4) 備考1.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2, 6-ジメチル−
236
K 0101 : 1998
4-ヘプタノン層をそのまま噴霧する場合の検量線の作成には鉛標準液 (0.1mgPb/ml) を適当な
濃度 (1〜5μgPb/ml) に薄め,その1〜20mlを段階的にとり,約500ml(又は100〜500mlの一定
量)とした後,試料と同様に(3)の準備操作を行って鉛 (Pb) の量と指示値との関係線を作成す
る。
56.2 電気加熱原子吸光法 試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム
(II) を加え,電気加熱炉で原子化し,鉛による原子吸光を波長283.3nmで測定して鉛を定量する。
定量範囲:Pb5〜100μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考2. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) 硝酸パラジウム (II) 溶液 (10μgPd/ml) 53.2(1)(c)による。
(d) 鉛標準液 (1μgPb/ml) 56.1(1)(a)の鉛標準液 (0.1mgPb/ml) 10mlを全量フラスコ1 000mlにとり,硝
酸 (1+1) 20mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) 鉛中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜500μl又は自動
注入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料15mlずつをそれぞれ全量フラスコ20mlにとり,鉛標準液 (1μgPb/ml) を
加えないものと,0.1〜2mlの範囲で段階的に3段階以上添加したものとを調製し,それぞれの溶液
の酸の濃度が同じになるように硝酸 (1+1) を加えた後,水を標線まで加える。
(b) (a)の操作を行った試料の100μl以上の定量をマイクロピペットで小形の容器にとり,これと同体積
の硝酸パラジウム (II) 溶液 (10μgPd/ml) を加え,よく混ぜ合わせる。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(500〜800℃,
30〜40秒間)し,次に,原子化(5)(1 800〜2 500℃,3〜6秒間)し,波長283.3nmの指示値(2)を読
み取る(6)。
(d) 空試験として以下の操作を行う。(3)の準備操作での試料と同量の水をとり,試料と同様に(3)の操作
を行った後,その15mlを全量フラスコ20mlにとる。次に,(4)(a)の溶液の酸の濃度と同じになるよ
うに,硝酸 (1+1) を加えた後,水を標線まで加える。この溶液について(b)及び(c)の操作を行って
試料について得た指示値を補正する。
(e) 鉛の添加量と指示値との関係線を作成し,鉛の量を求め,試料中の鉛の濃度 (μgPb/l) を算出する。
237
K 0101 : 1998
注(5) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(6) 引き続いて少なくとも(c)の操作を3回繰り返し,指示値が合うことを確認する。
56.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,鉛
による発光を波長220.351nmで測定して鉛を定量する。
定量範囲:Pb0.1〜2mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 鉛標準液 (10μgPb/ml) 56.1(1)(a)の鉛標準液 (0.1mgPb/ml) 10mlを全量フラスコ100mlにとり,硝
酸 (1+1) 10mlを加え水を標線まで加える。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考3. 準備操作を行った溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,鉛の濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分析の定量分析)に従って,試料導入部
を通してプラズマ中に噴霧し,波長220.351nmの発光強度を測定する(7)(8)(9)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線から鉛の量を求め,試料中の鉛の濃度 (mgPb/l) を算出する。
検量線 鉛標準液 (10μgPb/ml) 1〜20ml(10)(11)を全量フラスコ100mlに段階的にとり,(3)(a)を行った
試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作
を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた
後,(a)の操作を行って標準液について得た発光強度を補正し,鉛 (Pb) の量と発光強度との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(7) 波長の異なる2本のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
内標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリ
ウム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよ
うに酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
220.351nmと同時に371.029nm(イットリウム)の発光強度を測定し,鉛とイットリウムとの発
光強度の比を求める。
別に,鉛標準液 (10μgPb/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,イットリウム
溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加えた後,
水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長220.351nmと同時に
371.029nmの発光強度を測定し,鉛の濃度に対する鉛とイットリウムとの発光強度比の関係線
を作成し,検量線とする。この検量線から,試料について得た発光強度比に相当する鉛の量を
求め,試料中の鉛の濃度 (mgPb/l) を算出する。
(8) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
238
K 0101 : 1998
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(9) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(10) 備考3.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は,鉛標準液
(10μgPb/ml) を適当な濃度 (0.1〜1μgPb/ml) に薄め,その1〜20mlを段階的にとり,500ml(又
は100〜500mlの一定量)とした後,試料と同様に備考3.及び(4)(a)と(b)の操作を行って鉛 (Pb)
の量と発光強度との関係線を作成する。
(11) 銅,亜鉛,カドミウム,ニッケル,マンガン,鉄を同時に試験する場合には,混合標準液 [(10μgCu,
10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条
件で検量線を作成するとよい。
56.4 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,鉛と内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,鉛のイオンの電
流と内標準物質のイオンの電流との比を求めて鉛を定量する。
定量範囲: Pb 0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 56.2(1)(a)による。
(b) 硝酸 (1+1) 56.2(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (12) 51.5(1)(c)による。
(d) 鉛標準液 (1μgPb/ml) 56.2(1)(d)による。
(e) 鉛標準液 (50ngPb/ml) 鉛標準液 (1μgPb/ml) 50mlを全量フラスコ1 000mlにとり,硝酸 (1+1) 3ml
を加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による。
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.5(1)(g)による。
注(12) 51.の注(16)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考4. 51.の備考8.による。
5. 51.の備考9.による。
6. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(13)。
(a) 試料を4.5によって処理する。
(b) (3)(a)で処理した試料の適量(Pbとして0.01〜50μgを含む。)を全量フラスコ100mlにとり,イット
リウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を加
えた後,水を標線まで加える。
注(13) 51.の注(17)による。
(4) 操作 操作は,次のとおり行う(14)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧して鉛とイットリウムの質量/荷電数(15)における指示値(16)を読み取り,鉛の指示値とイット
239
K 0101 : 1998
リウムの指示値との比を求める。
(b) 空試験として,(3)(a)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って鉛の指
示値とイットリウムの指示値との比を求め,試料について得た鉛とイットリウムとの指示値の比を
補正する。
(c) 検量線から鉛の量を求め,試料中の鉛の濃度 (μgPb/l) を算出する。
検量線 鉛標準液(50ngPb/ml又は1μgPb/ml)1〜50ml(17)を段階的に全量フラスコ100mlにとり,
イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝酸 (1+1) を
加えた後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全量フラ
スコ100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように酸
を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値を補正し,鉛 (Pb) の
量に対する鉛の指示値とイットリウムの指示値との比の関係線を作成する。検量線の作成は,試料
測定時に行う。
注(14) 51.の注(18)による。
(15) 51.の注(19)による。
(16) 51.の注(20)による。
(17) 51.の注(21)による。
備考7. 51.の備考11.による。
57. 水銀 (Hg) 水銀の定量には,還元気化原子吸光法又は加熱気化原子吸光法を適用する。
57.1 還元気化原子吸光法 試料を過マンガン酸カリウムで前処理した後,塩化すず (II) で水銀 (II) を還
元する。この溶液に通気して発生する水銀蒸気による原子吸光を波長253.7nmで測定し,水銀を定量する。
定量範囲:Hg 0.5〜10μg/l,繰返し分析精度:変動係数で4〜20%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 JIS K 8541に規定する硝酸で水銀の含量が0.1μg/l以下のもの。
(b) 硫酸 (1+1) 4.4(1)(b)による。ただし,硫酸は水銀の含量が1μg/l以下のものを用いる。
(c) 過マンガン酸カリウム溶液 (50g/l) JIS K 8247に規定する過マンガン酸カリウム(1)50gを水に溶
かしてガラスろ過器でろ過した後,水で1lとする。着色ガラス瓶に保存する。
(d) ペルオキソ二硫酸カリウム溶液 (50g/l) JIS K 8253に規定するペルオキソ二硫酸カリウム50gを
水に溶かして1lとする(2)。
(e) 塩化ヒドロキシルアンモニウム溶液 (80g/l) JIS K 8201に規定する塩化ヒドロキシルアンモニウ
ム8gを水に溶かして100mlとする。この溶液の水銀の含有量は1μg/l以下とする。精製の必要があ
る場合には,この溶液を分液漏斗に移し,ジチゾン-クロロホルム溶液 (50mg/l) (JIS K 8490に規
定するジチゾン5mgをJIS K 8322に規定するクロロホルム100mlに溶かす。ただし,この溶液中
の水銀含有量は0.5μg/l以下とする。)を少量加えて振り混ぜ,放置した後,クロロホルム層を捨て
る。この操作をクロロホルム層が変色しなくなるまで繰り返す。水層を乾いたろ紙でろ過しクロロ
ホルムの小滴を除く。
(f) 塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物10gに硫酸 (1+20) 60mlを加え,
かき混ぜながら加熱して溶かす。放冷後,水を加えて100mlにする。この溶液の水銀含有量は1μg/l
以下とする。精製の必要がある場合には,JIS K 1107に規定する高純度窒素2級を通気する。1週
間以上経過したものは使用しない。
240
K 0101 : 1998
(g) 水銀標準液 (0.5mgHg/ml) JIS K 8139に規定する塩化水銀 (II) 0.339gをとり,少量の水に溶かし,
全量フラスコ500mlに移し入れ,硝酸 (1+1) 5mlを加えた後,水を標線まで加える。ほうけい酸ガ
ラス瓶に保存する。
(h) 水銀標準液 (10μgHg/ml) 水銀標準液 (0.5mgHg/ml) 10mlを全量フラスコ500mlにとり,硝酸 (1
+1) 5mlを加え,水を標線まで加える。ほうけい酸ガラス瓶に保存する。1か月間以上経過したも
のは使用しない。
(i) 水銀標準液 (0.1μgHg/ml) 水銀標準液 (10μgHg/ml) 10mlを全量フラスコ1 000mlにとり,硝酸 (1
+1) 10mlを加えた後,水を標線まで加える。使用時に調製する。
注(1) 原子吸光分析用試薬など,水銀含量の少ないものを用いる。
(2) JIS K 8252に規定するペルオキソ二硫酸アンモニウムを用いてもよい。いずれも溶液中の水銀
は1μg/l以下とする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 原子吸光分析装置又は水銀用原子吸光分析装置
(b) 水銀還元気化装置(3) 原子吸光分析装置と併用する。
(c) 水銀中空陰極ランプ又は水銀ランプ
注(3) 還元容器,吸収セル,空気ポンプ,流量計,乾燥管及び連結管から構成される。図57.1及び図
57.2に構成例を示す。
なお,各構成部分の詳細は,次のとおりである。
還元容器 ガラス瓶(又は三角フラスコ)300〜350ml(250mlの位置に印を付けておく。)
又は図57.3に示すように分液漏斗形 (300ml) のものを用いる。
吸収セル 長さ100〜300mm程度の石英ガラス製のもの又はガラス製,プラスチック製(水
銀蒸気を吸着しないもの。)で,両端に石英ガラス窓を付けたもの。
空気ポンプ 0.5〜3l/minの送気能力をもつダイアフラムポンプ又は同じ性能をもつ空気ポン
プ。水銀蒸気に接する部分が金属製の場合はコロジオンなどを塗布しておく。
流量計 0.5〜5l/minの流量が測定できるもの。
乾燥管 乾燥管又はU字管。JIS K 8228に規定する過塩素酸マグネシウム(乾燥用),JIS K
8124に規定する塩化カルシウム(乾燥用)などの乾燥剤を充てんしておくか,又はコールド
トラップで代用してもよい。吸収セルの部分に小形電球を点灯するなどして吸収セル内の温
度が周囲の温度よりも約10℃高くなるようにしておけば,乾燥管を用いなくてもよい。
連結管 軟質塩化ビニル管
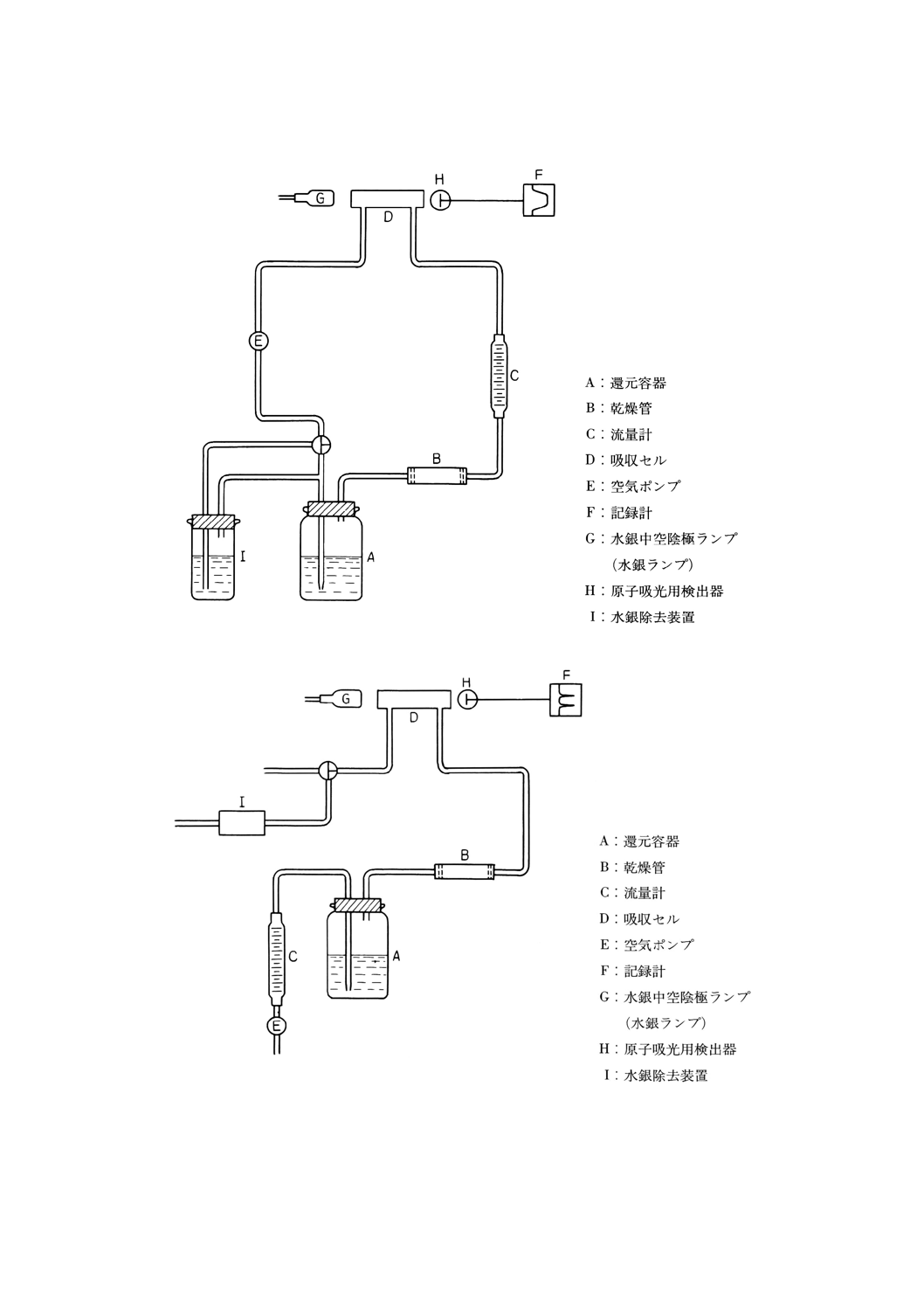
241
K 0101 : 1998
図57.1 密閉循環方式の構成の一例
図57.2 開放送気方式の構成の一例
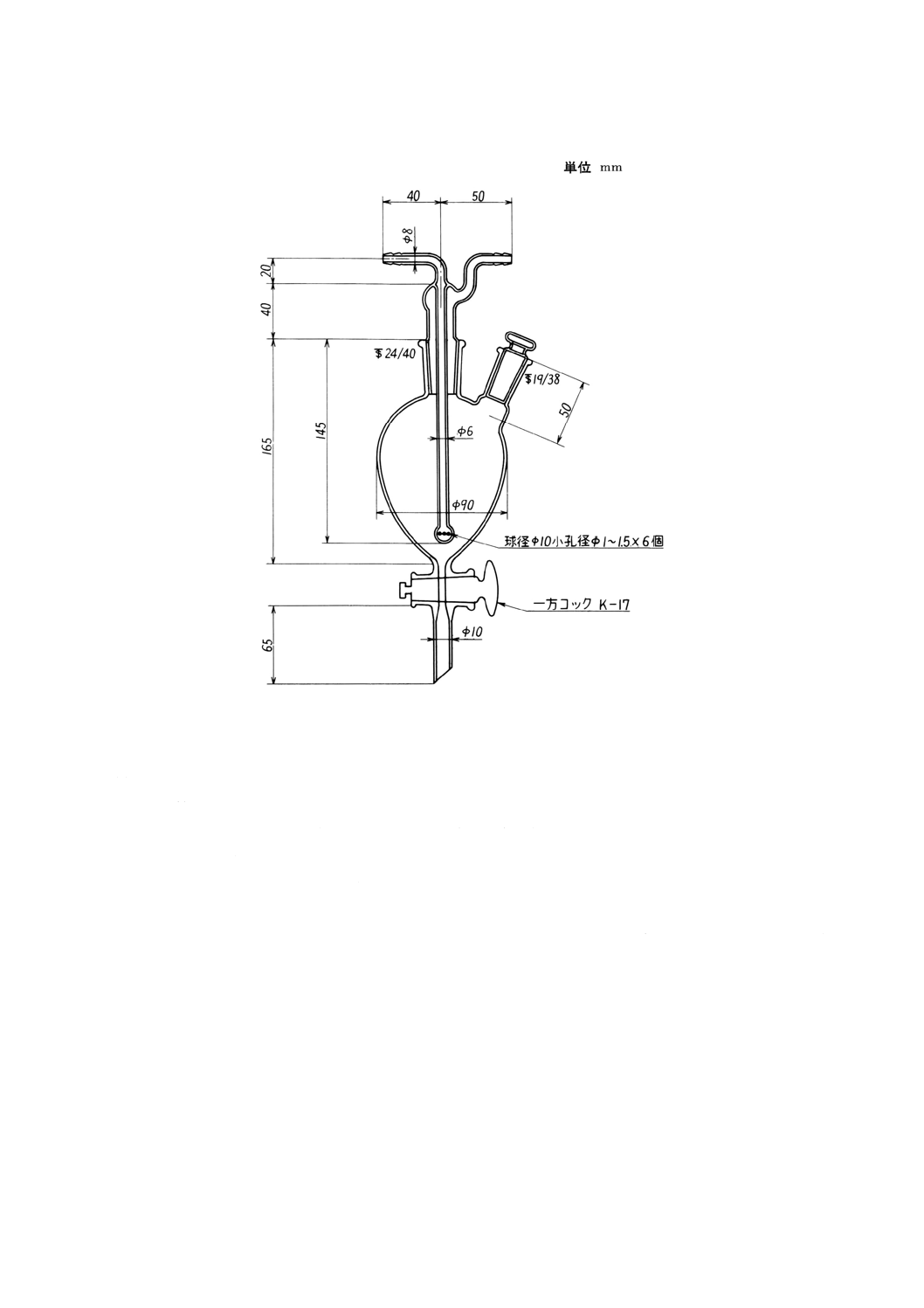
242
K 0101 : 1998
図57.3 還元容器の一例
(3) 操作 操作は,次のとおり行う。
(a) 試料(4)の適量(Hgとして0.1〜2μgを含む。)を三角フラスコ300ml(5)にとり,水を加えて約150ml
とする。
(b) 硫酸 (1+1) 20ml,硝酸5ml及び過マンガン酸カリウム溶液 (50g/l) 20ml(6)を加えて振り混ぜ,約15
分間放置する。
(c) 過マンガン酸カリウムの色が消えたときは,溶液の赤い色が約15分間持続するまで,過マンガン酸
カリウム溶液 (50g/l) を少量ずつ加える。
(d) ペルオキソ二硫酸カリウム溶液 (50g/l) 10mlを加え,約95℃の水浴中に三角フラスコ300mlを浸し
て約2時間加熱する。
(e) 室温まで冷却し,塩化ヒドロキシルアンモニウム溶液 (80g/l) 10mlを添加して過剰の過マンガン酸
を還元する。
(f) 直ちに溶液を還元容器に移し(7),水で250mlとした後,通気回路を組み立てる。
(g) 手早く塩化すず (II) 溶液10mlを加え,あらかじめ設定した最適流量(8)で空気ポンプを作動し,空
気を循環(9)させる。
(h) 波長253.7nmの指示値(10)を読み取る。
(i) バイパスコック(11)を回して指示値が元に戻るまで通気を続ける。
(j) 空試験とし試料と同量の水をとり,(b)〜(d)と同量の試薬を加えた後,(e)〜(i)の操作を行って指示値
を読み取り,試料について得た指示値を補正する。
243
K 0101 : 1998
(k) 検量線から水銀の量を求め,試料中の全水銀の濃度 (μgHg/l) を算出する。
検量線 水銀標準液 (0.1μgHg/ml) 1〜20mlを還元容器300mlに段階的にとり,硫酸 (1+1) 20mlと
水を加えて200mlとした後,(f)〜(i)の操作を行う。別に,空試験として水200mlを還元容器300ml
にとり,硫酸 (1+1) 20mlを加えた後,(f)〜(i)の操作を行って水銀標準液について得た指示値を補
正し,水銀 (Hg) の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(4) 有機物その他の妨害物質が含まれていない試料については(a)〜(e)を省略し,試料を直接還元容
器にとり,硫酸 (1+1) 20mlを加えた後(f)〜(k)の操作を行ってもよい。この場合は検量線も同
様に作成する。
(5) 還元容器用の三角フラスコ又はガラス瓶を用いてもよい。
(6) 有機物の少ない試料では適宜減量する。
(7) 分解に還元容器を用いた場合は,そのまま連結する。
(8) 最適流量は装置によって異なるので,あらかじめ最適条件を求めておく。
(9) 開放送気方式の場合は,還元容器の通気管にコックを付け,塩化すず (II) 溶液添加後,約2分
間激しく振り混ぜた後,装置に連結し,ポンプの作動と同時にコックを開く。最適送気量は,
あらかじめ求めておくが,通常は1〜1.5l/minである。
(10) 吸光度又はその比例値。開放送気方式の場合は,ピーク高さ又はピーク面積を測定する。
(11) 過マンガン酸カリウム溶液 (50g/l) を含む硫酸 (1+4) を入れたガス洗浄瓶を通して大気中に
放出する。
備考1. 塩化物イオンを多量に含む試料では,過マンガン酸カリウム処理においては塩化物イオンが
酸化されて塩素となり,波長253.7nmの光を吸収して正の誤差を生じる。この場合は,塩化
ヒドロキシルアンモニウム溶液 (80g/l) を過剰に加え,塩素を十分に還元しておく。還元容
器中に存在する塩素は,窒素などの送入によってあらかじめ追い出しておく。
2. ベンゼン,アセトンなどは波長253.7nmの光を吸収して正の誤差を生じる。この種の揮発性
有機物を含む試料に対しては,過マンガン酸カリウムによる前処理を行った後,次のいずれ
かの操作を適用する。
(1)少量のヘキサンと振り混ぜて揮発性有機物を抽出除去する。(2)重水素ランプなどによる
バックグラウンド補正を行う。(3)水銀中空陰極ランプと重水素ランプを用いて指示値の差を
求めておき,次に,塩化すず (II) 溶液の添加を省略して同様の測定を行い,両指示値の差と
して水銀を定量する。(4)水銀をジチゾン錯体として抽出分離した後,加熱気化法によって測
定する。
57.2 加熱気化原子吸光法 試料を過マンガン酸カリウムで前処理した後,硫酸酸性溶液から水銀をジチ
ゾン錯体として有機溶媒に抽出する。有機溶媒を揮散させた後,残留物を加熱して水銀蒸気を発生させ,
その原子吸光を波長253.7nmで測定して水銀を定量する。
定量範囲:Hg0.5〜10μg/l,繰返し分析精度:変動係数で4〜20%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 57.1(1)(a)による。
(b) 硫酸 (1+1) 57.1(1)(b)による。
(c) 過マンガン酸カリウム溶液 (50g/l) 57.1(1)(c)による。
(d) ペルオキソ二硫酸カリウム溶液 (50g/l) 57.1(1)(d)による。
(e) 塩化ヒドロキシルアンモニウム溶液 (80g/l) 57.1(1)(e)による。
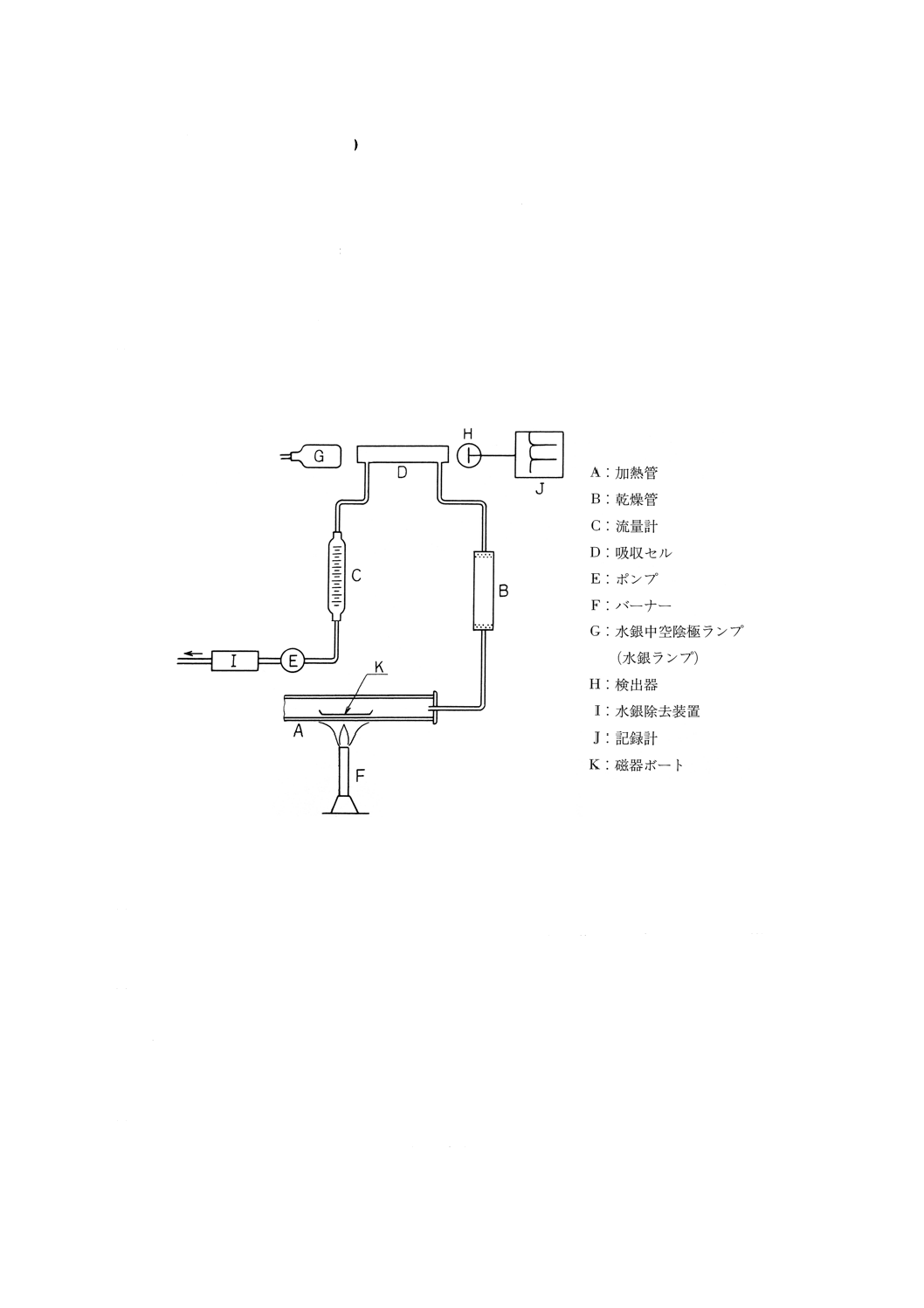
244
K 0101 : 1998
(f) ジチゾン-クロロホルム (0.1g/l) JIS K 8490に規定するジチゾン10mgをJIS K 8322に規定するク
ロロホルム100mlに溶かす。ただし,溶液中の水銀の含量は1μg/l以下とする。
(g) 2, 3-ジメルカプト−1-プロパノール溶液 (0.1vol%) 2, 3-ジメルカプト-1-プロパノール0.1mlをJIS
K 8322に規定するクロロホルム10mlと混合する。使用時にクロロホルムで10倍に薄める。
(h) 水銀標準液 (0.1μgHg/ml) 57.1(1)(i)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 500ml
(b) 原子吸光分析装置又は水銀用原子吸光分析装置
(c) 水銀加熱気化装置 原子吸光分析装置と併用する。図57.4に加熱気化法の装置の構成の一例を示す。
(d) 水銀中空陰極ランプ又は水銀ランプ
図57.4 加熱気化法の装置の構成の一例
(3) 操作 操作は,次のとおり行う。
(a) 試料(4)の適量(Hgとして0.1〜2μgを含む。)を三角フラスコ300mlにとり,水を加えて約150ml
とする。
(b) 57.1(3)(b)〜(e)の操作を行う。
(c) この溶液を分液漏斗に移し,ジチゾン-クロロホルム溶液 (0.1g/l) 5mlを加えて約2分間激しく振り
混ぜた後,放置する。
(d) クロロホルム層を分離し試験管に移す。
(e) 水層に再びジチゾン-クロロホルム溶液 (0.1g/l) 5mlを加えて約2分間激しく振り混ぜた後,放置す
る。
(f) クロロホルム層を先の試験管に合わせ,これに水5mlを加えて振り混ぜた後,放置する。
(g) 水層をスポイトで取り除く。
(h) このクロロホルム溶液の一定量(2.5ml以下)を磁器ボートに移す。
(i) 2, 3-ジメルカプト-1-プロパノール溶液 (0.1vol%) 0.1mlを加え,通気してクロロホルムを揮散させる。
245
K 0101 : 1998
(j) 磁器ボートを水銀加熱気化装置の加熱管に挿入し,吸引(12)と加熱を同時に開始する。
(k) 波長253.7nmの指示値(10)を読み取る。
(l) 空試験とし試料と同量の水をとり,(b)と同量の試薬を加えた後,(c)〜(k)の操作を行って試料につ
いて得た指示値を補正する。
(m) 検量線から水銀の量を求め,試料中の水銀の濃度 (μgHg/l) を算出する。
検量線 水銀標準液 (0.1μgHg/ml) 1〜20mlを分液漏斗に段階的にとり,硫酸 (1+1) 20mlを加え,
水で200mlとした後,(c)〜(k)の操作を行う。別に,空試験として水200ml及び硫酸 (1+1) 20mlを
分液漏斗にとり,(c)〜(k)の操作を行って水銀標準液について得た指示値を補正し,水銀 (Hg) の量
と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(12) 最適な吸引量を求めておく。通常は1〜1.5l/minである。
58. マンガン (Mn) マンガンの定量には,過よう素酸吸光光度法,フレーム原子吸光法,電気加熱原子
吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
58.1 過よう素酸吸光光度法 試料を硫酸酸性とした後,過よう素酸カリウムを加え,加熱して赤紫の過
マンガン酸イオンを生成させ,その吸光度を測定してマンガン定量する。
定量範囲:Mn40〜500μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+1) 4.4(1)(b)による。
(b) りん酸 JIS K 9005に規定するもの。
(c) 過よう素酸カリウム JIS K 8249に規定するもの。
(d) マンガン標準液 (0.1mgMn/ml) JIS K 8247に規定する過マンガン酸カリウム0.288gをとり,水
150mlに硫酸 (1+1) 10mlを加えた溶液に溶かす。亜硫酸水素ナトリウム溶液 (100g/l) (JIS K 8059
に規定する亜硫酸水素ナトリウム10gを水に溶かして100mlとする。)を滴加し,かき混ぜて脱色
した後,煮沸して過剰の二酸化硫黄を追い出す。放冷後,全量フラスコ1 000mlに移し入れ,水を
標線まで加える。又はマンガン(99.9%以上)0.100gをとり,硫酸 (1+3) 20mlに加熱して溶かし,
放冷後,全量フラスコ1 000mlに移し入れ,水を標線まで加える。若しくはJIS K 0027に規定する
標準物質-標準液-マンガンのMn100を用いる。
(e) マンガン標準液 (20μgMn/ml) マンガン標準液 (0.1mgMn/ml) 50mlを全量フラスコ250mlにとり,
水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った(1)試料の適量(2)(Mnとして40〜500μgを含む。)をとり,硫酸 (1+1) 10ml(3)を加
え,加熱して硫酸白煙を発生させ,ハロゲン化物を除去する。
(b) 放冷後,水約20mlとりん酸1mlとを加え,加熱して内容物を溶かす。不溶解物がある場合には,
ろ別し,ろ紙と沈殿を温水で洗い,ろ液と洗液を合わせ,水を加えて約45mlにする。
(c) 過よう素酸カリウム0.5g(4)を加え,沸騰水浴中で30分間加熱し(5)発色させる。
(d) 流水で冷却した後,全量フラスコ50mlに移し入れ,水を標線まで加える。
(e) この溶液の一部を吸収セルに移し,波長525nm付近又は545nm付近の吸光度を測定する。
(f) 空試験として水約30mlをとり,硫酸 (1+1) 10mlとりん酸1mlを加えた後,(c)〜(e)の操作を行っ
246
K 0101 : 1998
て吸光度を測定し,試料について得た吸光度を補正する。
(g) 検量線からマンガンの量を求め,試料中のマンガンの濃度 (μgMn/l) を算出する。
検量線 マンガン標準液 (20μgMn/ml) 2〜25mlをビーカー100mlに段階的にとり,水を加えて液量
を約30mlとし,硫酸 (1+1) 10mlとりん酸1mlとを加えた後,(c)〜(f)の操作を行ってマンガン (Mn)
の量と吸光度との関係線を作成する。
注(1) 試料中にこの試験に影響する有機物及び妨害物質が含まれていない場合には,前処理を省略し
てもよい。
(2) 試料は最大500mlとする。
(3) 前処理操作に硫酸を用いた場合には,硫酸は加えない。ただし,試料中の硫酸は約5mlになる
ように調節する。
(4) 過よう素酸カリウムの代わりに,硝酸銀溶液 (5g/l) (JIS K 8550に規定する硝酸銀0.5gを水に
溶かして100mlとする。)2mlとペルオキソ二硫酸アンモニウム溶液 (200g/l) (JIS K 8252に規
定するペルオキソ二硫酸アンモニウム20gを水に溶かして100mlとする。)5mlを加えて約1分
間煮沸して発色させてもよい。この場合は,発色時の硫酸の濃度は約0.5mol/lになるようにす
る。この方法で塩化銀の沈殿が生じたら,これが消えるまで硝酸酸性硝酸水銀 (II) 溶液 (50g/l)
[JIS K 8558に規定する硝酸水銀 (II) n水和物5gを硝酸 (1+2) 20mlに溶かし,水で100mlと
する。]を滴加し,更に数滴過剰に加えて塩化物イオンを固定する。
(5) 加熱時間が長すぎると,生成した過マンガン酸イオンが分解するおそれがあるから,加熱時間
は正しく守る。
備考1. 溶存マンガンを定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種C
を用いる。)の適量(Mnとして40〜500μgを含む。)を用い,(3)(a)以降の操作を行う。
2. マンガンの濃度が低い場合には,次の鉄共沈法で濃縮して定量できる。
試料500mlまでの適量をとり,約90℃に加熱し,硫酸アンモニウム鉄 (III) 溶液 (2mgFe/ml)
[JIS K 8982に規定する硫酸アンモニウム鉄 (III) ・12水1.8gをとり,硝酸 (1+6) 10mlと
水に溶かして100mlとする。]5mlとJIS K 8230に規定する過酸化水素5〜10mlを加え,こ
の溶液をかき混ぜながらアンモニア水 (1+1) 又は水酸化ナトリウム溶液 (100g/l) を加えて,
水酸化鉄 (III) の沈殿を生成させる。沈殿が沈降した後,ろ紙5種Aを用いてろ過し,温水
で洗う。沈殿をできるだけ元のビーカーに移し,ろ紙に付着した沈殿は,少量の過酸化水素 (1
+10) を加えた硫酸 (1+9) 50mlで溶かし,ろ紙は水で洗う。ろ液と洗液は元のビーカーに
受け,加熱して沈殿を溶かすとともに過酸化水素を分解して液量を約40mlに濃縮し,りん
酸1mlを加えた後,(3)(c)〜(g)の操作を行ってマンガンを定量する。
58.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,マンガンによる
原子吸光を波長279.5nmで測定してマンガンを定量する。
定量範囲:Mn0.1〜4mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) マンガン標準液 (10μgMn/ml) 58.1(1)(d)のマンガン標準液 (0.1mgMn/ml) 50mlを全量フラスコ
500mlにとり,硝酸 (1+1) 10mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) マンガン中空陰極ランプ
247
K 0101 : 1998
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考3. 溶存マンガンを定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種C
を用いる。)の適量をとり,4.5によって処理する。
4. マンガンの濃度が低い場合には,備考2.に準じて処理し,マンガンを濃縮分離する。沈殿は
少量の過酸化水素 (1+10) を加えた塩酸 (1+2) の少量に溶かし,ろ紙は温水で洗浄する。
ろ液と洗液を合わせ,0.1〜1mol/lの塩酸酸性溶液の一定量とする。又は51.の備考5.に準じ
て操作してもよい。この場合には,pH4.5〜5.0で抽出する。
なお,マンガンの1-ピロリジンカルボジチオ酸錯体(APDC錯体)は水層に移行しやすい
ので,抽出及び層分離は手早く行う。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長279.5nmの指示値(6)を読み取る。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からマンガンの量を求め,試料中のマンガンの濃度 (mgMn/l) を算出する。
検量線 マンガン標準液 (10μgMn/ml) 1〜40mlを全量フラスコ100mlに段階的にとり,(3)(a)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操
作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度条件になるように酸を
加えた後,(a)の操作を行って標準液について得た指示値を補正し,マンガン (Mn) の量と指示値と
の関係線を作成する。検量線の作成は,試料測定時に行う。
注(6) 吸光度又はその比例値。
備考5. シリカを多量に含む場合には,干渉抑制剤としてカルシウム(又はマグネシウム)を200mg/l
程度加えておくとよい。
58.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,マンガンによる原子吸光を波
長279.5nmで測定してマンガンを定量する。
定量範囲:Mn1〜30μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考6. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) マンガン標準液 (1μgMn/ml) 58.2(1)(a)のマンガン標準液 (10μgMn/ml) 10mlを全量フラスコ
100mlにとり,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) マンガン中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
248
K 0101 : 1998
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考7. 溶存マンガンを定量する場合には,備考3.による。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(500
〜800℃,約30秒間)し,次に,原子化(7)(2 000〜2 700℃,4〜6秒間)し,波長279.5nmの指示
値(6)を読み取る(8)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からマンガンの量を求め,試料中のマンガンの濃度 (μgMn/l) を算出する。
検量線 マンガン標準液 (1μgMn/ml) 0.1〜3mlを全量フラスコ100mlに段階的にとり,(3)(a)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操
作を行う。別に,空試験として水について(3)(a)を行った試料と同じ条件になるように酸を加えた後,
(a)の操作を行って標準液について得た指示値を補正し,マンガン (Mn) の量と指示値との関係線を
作成する。検量線の作成は,試料測定時に行う。
注(7) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても変わることがある。
(8) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
58.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,マ
ンガンによる発光を波長257.610nmで測定してマンガンを定量する。
定量範囲: Mn10〜200μg/l,0.2〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) マンガン標準液 (10μgMn/ml) 58.2(1)(a)による。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考8. 溶存マンガンを定量する場合には,備考3.による。
9. 準備操作を行った試料のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,マンガンの濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長257.610nmの発光強度を測定する(9)(10)(11)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
249
K 0101 : 1998
(c) 検量線からマンガンの量を求め,試料中のマンガンの濃度 (μgMn/l) を算出する。
検量線 マンガン標準液 (10μgMn/ml) 0.1〜2ml(又は2〜50ml)(12)(13)を全量フラスコ100mlに段
階的にとり,(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
この溶液について(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃
度になるように酸を加えた後,(a)の操作を行って標準液について得た発光強度を補正し,マンガン
(Mn) の量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(9) 波長の異なる2本のスペクトル線を同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリウ
ム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
257.610nmと同時に371.029nm(イットリウム)の発光強度を測定し,マンガンとイットリウム
との発光強度の比を求める。
別に,マンガン標準液 (10μgMn/ml) 0.1〜2ml(又は2〜50ml)を全量フラスコ100mlに段階
的にとり,イットリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度に
なるように酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波
長257.610nmと同時に371.029nmの発光強度を測定し,マンガンの濃度に対するマンガンとイ
ットリウムとの発光強度比の関係線を作成し,検量線とする。この検量線から,試料について
得た発光強度比に相当するマンガンの量を求め,試料中のマンガンの濃度 (μgMm/l) を算出す
る。
(10) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(11) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(12) 備考9.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線は,マンガン標
準液 (10μgMn/ml) を適当な濃度 (0.1〜1μgMn/ml) に薄め,その0.2〜4ml(又は4〜100ml)を
段階的にとり,500ml(又は100〜500mlの一定量)とした後,試料と同様に備考9.及び(4)(a)
と(b)の操作を行ってマンガン (Mn) の量と発光強度の関係線を作成する。
(13) 銅,亜鉛,カドミウム,ニッケル,鉛,鉄を同時に試験する場合には,混合標準液 [(10μgCu, 10μgZn,
8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条件で検量
線を作成するとよい。
58.5 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,マンガンと内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,マンガン
のイオンの電流と内標準物質のイオンの電流との比を求めてマンガンを定量する。
定量範囲: Mn0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 58.3(1)(a)による。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
(c) イットリウム溶液 (1μgY/ml) (14) 51.5(1)(c)による。
250
K 0101 : 1998
(d) マンガン標準液 (1μgMn/ml) 58.3(1)(c)による。
(e) マンガン標準液 (50ngMn/ml) マンガン標準液 (1μgMn/ml) 50mlを全量フラスコ1 000mlにとり,
硝酸 (1+1) 1.5mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による。
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.5(1)(g)による。
注(14) 51.の注(16)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考10. 51.の備考8.による。
11. 51.の備考9.による。
12. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(15)。
(a) 試料を4.5によって処理する。
(b) (3)(a)で処理した試料の適量(Mnとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イッ
トリウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を
加えた後,水を標線まで加える。
注(15) 51.の注(17)による。
備考13. 溶存マンガンを定量する場合には,備考3.によって処理した後,(b)を行う。
(4) 操作 操作は,次のとおり行う(16)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧してマンガンとイットリウムの質量/荷電数(17)における指示値(18)を読み取り,マンガンの指
示値とイットリウムの指示値との比を求める。
(b) 空試験値として,(3)(a)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行ってマン
ガンの指示値とイットリウムの指示値との比を求め,試料について得たマンガンとイットリウムと
の指示値を補正する。
(c) 検量線からマンガンの量を求め,試料中のマンガンの濃度 (μgMn/l) を算出する。
検量線 マンガン標準液(50ngMn/ml又は1μgMn/ml)1〜50ml(19)を全量フラスコ100mlに段階的に
とり,イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝酸 (1
+1) を加えた後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全
量フラスコ100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるよ
うに硝酸 (1+1) を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値を
補正し,マンガン (Mn) の量に対するマンガンの指示値とイットリウムの指示値との比の関係線を
作成する。検量線の作成は,試料測定時に行う。
注(16) 51.の注(18)による。
(17) 51.の注(19)による。
(18) 51.の注(20)による。
(19) 51.の注(21)による。
備考14. 51.の備考11.による。
251
K 0101 : 1998
59. アルミニウム (Al) アルミニウムの定量には,キノリノール吸光光度法,フレーム原子吸光法,電
気加熱原子吸光法又はICP発光分光分析法を適用する。
59.1 キノリノール吸光光度法 微酸性にした試料に,塩化ヒドロキシルアンモニウムと1, 10-フェナント
ロリンを加えて鉄をマスキングした後,8-キノリノール及び酢酸アンモニウムを加え,生成する錯体をク
ロロホルムで抽出する。シアン化カリウムを含む塩化アンモニウム溶液で洗浄して,アルミニウムととも
に抽出された銅,ニッケル,コバルトなどを除去した後,アルミニウム錯体の吸光度を測定してアルミニ
ウムを定量する。
定量範囲:Al5〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+2) JIS K 8180に規定する塩酸を用いて調製する。
(b) アンモニア水 (1+2) JIS K 8085に規定するアンモニア水を用いて調製する。
(c) 硫酸ナトリウム JIS K 8987に規定するもの。
(d) 塩化ヒドロキシルアンモニウム溶液 (100g/l) 40.2(1)(c)による。
(e) 酢酸アンモニウム溶液 (150g/l) JIS K 8359に規定する酢酸アンモニウム15gを水に溶かして
100mlとする。この溶液を分液漏斗に入れ,8-キノリノールクロロホルム溶液[JIS K 8775に規定
する8-キノリノール2gをJIS K 8322に規定するクロロホルム100mlに溶かす。]5mlを加え,激し
く振り混ぜた後放置し,クロロホルム層を捨てる。この操作を,クロロホルム層が着色しなくなる
まで繰り返す。次に水層にクロロホルム5mlを加え,激しく振り混ぜた後放置し,クロロホルム層
を捨てる。この操作を,水層に黄色が認められなくなるまで繰り返す。水層は乾いたろ紙でろ過し,
溶液中のクロロホルムの小滴を除く。
(f) シアン化カリウム-塩化アンモニウム溶液 JIS K 8443に規定するシアン化カリウム1.0gを水に溶
かして500mlとする。これにJIS K 8116に規定する塩化アンモニウムを少量ずつ溶かし,pHを9.0
〜9.5に調節する。この溶液は(e)の酢酸アンモニウム溶液と同様に操作して8-キノリノールクロロ
ホルム溶液及びクロロホルムで洗浄し,精製する。
(g) 1, 10-フェナントロリン溶液 (1g/l) JIS K 8202に規定する塩化1, 10-フェナントロリニウム一水和
物1.3gを水に溶かして1lとする。又はJIS K 8789に規定する1, 10-フェナントロリン一水和物1.1g
をJIS K 8102に規定するエタノール (95) 100mlに溶かし,水を加えて1lとする。
(h) 8-キノリノール溶液 (10g/l) JIS K 8775に規定する8-キノリノール2gにJIS K 8355に規定する酢
酸5mlを加え,わずかに加熱して溶かした後,水を加えて200mlとする。
(i) クロロホルム JIS K 8322に規定するもの。
(j) アルミニウム標準液 (0.1mgAl/ml) JIS K 8255に規定する硫酸カリウムアルミニウム・12水[ビ
ス(硫酸)カリウムアルミニウム−水 (1/12) ]1.76gをとり,塩酸 (1+1) 20mlに溶かし,全量フ
ラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 8069に規定するアルミニウム(99.9%
以上)0.100gをとり,塩酸 (1+1) 20ml中に加熱して溶かし,放冷後,全量フラスコ1 000mlに移
し入れ,水を標線まで加える。
(k) アルミニウム標準液 (1μgAl/ml) アルミニウム標準液 (0.1mgAl/ml) 10mlを全量フラスコ1 000ml
にとり,塩酸 (1+1) 20mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 200ml
(b) 光度計 分光光度計又は光電光度計
252
K 0101 : 1998
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作を行った(1)試料の適量(2)(Alとして5〜50μgを含む。)をとり,塩化ヒドロキシルアンモ
ニウム溶液 (100g/l) 1mlと1, 10-フェナントロリン溶液 (1g/l) 5mlを加えて振り混ぜ,アンモニア水
(1+1) を滴加してpHを約3.5(3)に調節する。
(b) 水を加えて液量約80mlとした後,約15分間放置する。
(c) 8-キノリノール溶液 (10g/l) 3mlと酢酸アンモニウム溶液 (150g/l) 10mlとを加え,アンモニア水 (1
+2) を滴加してpHを5.2〜5.5(4)に調節する。
(d) この溶液を分液漏斗に移し,水を加えて液量を約100mlとした後,クロロホルム10mlを加え,約1
分間激しく振り混ぜて放置する。
(e) クロロホルム層を分離して,別の分液漏斗に移し入れ,シアン化カリウム-塩化アンモニウム溶液
25mlを加え,振り混ぜて放置する。
(f) クロロホルム層を共栓試験管30mlに入れ,硫酸ナトリウム約1gを加えて軽く振り混ぜて水分を除
く。
(g) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長390nm付近の吸光度を
測定する。
(h) 空試験として水約70mlをとり,(a)〜(g)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
(i) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度 (μgAl/l) を算出する。
検量線 アルミニウム標準液 (1μgAl/ml) 5〜50mlを分液漏斗に段階的にとり,水を加えて液量約
70mlとし,以下,(a)〜(h)の操作を行ってアルミニウム (Al) の量と吸光度との関係線を作成する。
注(1) 4.のうち4.3の方法は用いない。有機物が少ない試料の場合には,試料100mlにつき塩酸5mlを加
え,静かに加熱して液量が約51になるまで濃縮してもよい。
(2) 一般に試料は50〜100mlとし,最大500mlまで用いてもよい。
(3) ブロモフェノールブルー試験紙を用いる。
(4) ブロモクレゾールグリーン試験紙を用いる。pHが5.2〜5.5の範囲にならないときは,塩酸 (1
+2) 又はアンモニア水 (1+2) を用いて調節する。
備考1. ふっ化物イオンが試料に含まれる場合には,ふっ化物イオン0.5mgに対し硫酸ベリリウム
36mgを加えておけば,妨害を防ぐことができる。
2. クロムが存在する場合には,pH5.2〜5.5における抽出は,できるだけ低温で行う。氷水で冷
却するとよい。
3. マンガンが多量に含まれる場合には,錯体を抽出したクロロホルム溶液を塩化ヒドロキシル
アンモニウムを加えたpH7以下の酢酸-酢酸アンモニウム溶液で洗浄してマンガンを除去す
る。
4. チタン,モリブデンなどが含まれている場合には,(3)(e)の操作で,銅,ニッケル,コバルト
などを除いた後,さらに,pH10のアンモニアアルカリ性塩化アンモニウム溶液 (50g/l) 25ml
にJIS K 8230に規定する過酸化水素2mlを加えた溶液でクロロホルム層を洗浄する。
5. この方法では,鉄0.45mgまでの存在は影響しない。
6. アルミニウムと鉄を同時に定量する場合には,次のように操作する。
(3)(a)の塩化ヒドロキシルアンモニウム溶液 (100g/l) 1mlと,1, 10-フェナントロリン溶液
(1g/l) 5mlを加える操作を省略し,以下,(b)〜(g)の操作を行って波長390nm付近の吸光度A
253
K 0101 : 1998
と,470nm付近の吸光度Bを測定する。
別に,空試験として水約80mlをとり,(c)〜(g)の操作を行って試料について得た吸光度A
及び吸光度Bを補正し,それぞれ吸光度A´及び吸光度B´とする。
波長470nm付近の鉄 (III) の検量線から吸光度B´に相当する鉄 (III) の量を求め,鉄の濃
度 (mgFe/l) を算出する。
また,吸光度B´に相当する鉄 (III) の量を波長390nm付近の鉄 (III) の検量線に適用して,
波長390nm付近での鉄 (III) による吸光度Cを求める。吸光度A´から吸光度Cを差し引き,
波長390nm付近のアルミニウムによる吸光度Dを求める。
吸光度Dを用い,波長390nm付近のアルミニウムの検量線からアルミニウムの量を求め,
試料中のアルミニウムの濃度 (mgAl/l) を算出する。
検量線 アルミニウム標準液 (1μgAl/ml) 5〜50mlを分液漏斗に段階的にとり,水を加えて液
量約80mlとし,(c)〜(f)の操作を行って波長390nm付近の吸光度を測定する。別に,
鉄 (III) 標準液 (10μgFe/ml) (*) 0.5〜10mlを分液漏斗に段階的にとり,水を加えて
約80mlとし,(c)〜(f)の操作を行って波長470nm付近及び波長390nm付近の吸光度
を測定する。空試験として水約80mlをとり,(c)〜(f)の操作を行って波長470nm付
近及び波長390nm付近の吸光度を測定し,アルミニウム標準液及び鉄 (III) 標準液
について得た吸光度を補正する。アルミニウム (Al) の量と波長390nm付近の吸光
度,鉄 (Fe) の量と波長470nm付近の吸光度及び波長390nm付近の吸光度との関係
線を作成する。
注(*) 鉄 (III) 標準液 (10μgFe/ml) JIS K 8982に規定する硫酸アンモニウム鉄 (III) ・12
水[ビス(硫酸)鉄 (III) アンモニウム−水 (1/12)]8.63gをとり,硫酸 (1+1) 20ml
と水を加えて溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。この
溶液を鉄 (III) 標準液 (1mgFe/ml) とし,その10mlを全量フラスコ1 000mlにとり,
硫酸 (1+1) 10mlを加えた後,水を標線まで加える。
59.2 フレーム原子吸光法 試料を前処理した後,アセチレン-一酸化二窒素フレーム中に噴霧し,アルミ
ニウムによる原子吸光を波長309.3nmで測定してアルミニウムを定量する。
定量範囲:Al5〜100mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩化カリウム溶液 (100g/l) JIS K 8121に規定する塩化カリウム10gを水に溶かして100mlとする。
(b) アルミニウム標準液 (0.5mgAl/ml) JIS K 8255に規定する硫酸カリウムアルミニウム・12水[ビ
ス(硫酸)カリウムアルミニウム‐水 (1/12)]8.794gをとり,塩酸 (1+1) 20mlに溶かし,全量フ
ラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 8069に規定するアルミニウム(99.9%
以上)0.500gをとり,塩酸 (1+1) 30ml中に入れ加熱して溶かし,放冷後,全量フラスコ1 000ml
に移し入れ,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) アルミニウム中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
254
K 0101 : 1998
(a) (3)の準備操作を行った試料の適量(Alとして0.5〜10mgを含む。)を全量フラスコ100mlにとり,
塩酸1mlを加え,水を標線まで加える。
(b) この溶液50mlを乾いたビーカーにとり,塩化カリウム溶液 (100g/l) 2mlを加える。
(c) (b)の試料をJIS K 0121の6.(操作方法)の操作に従って,アセチレン−一酸化二窒素フレーム(5)
中に噴霧し,波長309.3nmにおける指示値(6)を読み取る。
(d) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)〜(c)の操作を行
って指示値を読み取り,試料について得た指示値を補正する。
(e) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度 (mgAl/l) を算出する。
検量線 アルミニウム標準液 (0.5mgAl/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,(3)(a)
を行った試料と同じ酸の濃度になるように酸及び塩酸を加えた後,水を標線まで加える。この溶液
について(b)及び(c)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃
度になるように酸及び塩酸を加えた後,(b)及び(c)の操作を行って標準液について得た指示値を補正
し,アルミニウム (Al) の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(5) 多燃料フレームの方が高感度が得られる。
(6) 吸光度又はその比例値。
59.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,アルミニウムによる原子吸光
を波長309.3nmで測定してアルミニウムを定量する。
定量範囲:Al20〜200μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考7. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) アルミニウム標準液 (1μgAl/ml) 59.1(1)(j)のアルミニウム標準液 (0.1mgAl/ml) 5mlを全量フラス
コ500mlにとり,硝酸 (1+1) 10mlを加え,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの。
(c) アルミニウム中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(600
〜1 000℃,30〜40秒間)し,次に,原子化(7)(2 200〜3 000℃,3〜6秒間)し,波長309.2nmの指
示値(6)を読み取る(8)。
255
K 0101 : 1998
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度 (μgAl/l) を算出する。
検量線 アルミニウム標準液 (1μgAl/ml) 2〜20mlを全量フラスコ100mlに段階的にとり,(3)(a)を
行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)
の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を
加えた後,(a)の操作を行って標準液について得た指示値を補正し,アルミニウム (Al) の量と指示
値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(7) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(8) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
59.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ア
ルミニウムによる発光を波長309.271nmで測定してアルミニウムを定量する。
定量範囲:Al80〜4 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) アルミニウム標準液 (20μgAl/ml) 59.2(1)(b)のアルミニウム標準液 (0.5mgAl/ml) 10mlを全量フラ
スコ250mlにとり,塩酸 (1+1) 5mlを加えた後,水を標線まで加える。
(c) 混合標準液 [(20μgCa, 10μgMg, 20μgAl) /ml] 49.3(1)(c)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考8. 準備操作を行った溶液のナトリウム,カリウム,カルシウム,マグネシウムなどの濃度が高
くアルミニウムの濃度が低い場合には,試料100mlをとり,59.1(3)(a)〜(e)のクロロホルムを
JIS K 8051に規定する3-メチル−1-ブタノール(イソアミルアルコール)に代えた操作を行っ
て,3-メチル-1-ブタノール層を共栓試験管に移し入れる。この場合は,59.1(3)(f)の硫酸ナト
リウムを加えて水分を除く操作を省略してよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長309.271nmの発光強度を測定する(9)(10)(11)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度 (μgAl/l) を算出する。
検量線 アルミニウム標準液 (20μgAl/ml) 0.4〜20ml(12)(13)を全量フラスコ100mlに段階的にとり,
(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液につ
いて(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるよう
に酸を加えた後,(a)の操作を行って標準液について得た発光強度を補正し,アルミニウム (Al) の
量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(9) 波長の異なる2本のスペクトル線を同時測定が可能な装置では内標準法によることができる。内
256
K 0101 : 1998
標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリウ
ム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
309.271nmと同時に371.029nm(イットリウム)の発光強度を測定し,アルミニウムとイットリ
ウムとの発光強度の比を求める。
別に,アルミニウム標準液 (20μgAl/ml) 0.4〜20mlを全量フラスコ100mlに段階的にとり,イ
ットリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸
を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長309.271nm
と同時に371.029nmの発光強度を測定し,アルミニウムの濃度に対するアルミニウムとイット
リウムとの発光強度比の関係線を作成し,検量線とする。この検量線から,試料について得た
発光強度比に相当するアルミニウムの量を求め,試料中のアルミニウムの濃度 (μgAl/l) を算出
する。
(10) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(11) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(12) 備考8.によって準備操作を行い,3-メチル-1-ブタノール層をそのまま噴霧する場合の検量線は,
アルミニウム標準液 (20μgAl/ml) を適切な濃度 (1〜4μgAl/ml) に薄め,その1〜5mlを段階的
にとり,水で100mlとした後,試料と同様に備考8.及び(4)(a)と(b)の操作を行ってアルミニウム
(Al) の量と発光強度との関係線を作成する。
(13) カルシウム及びマグネシウムを同時に試験する場合には,混合標準液 [(20μgCa, 10μgMg,
20μgAl) /ml] を用いて,それぞれの金属元素の試験条件で検量線を作成するとよい。
60. 鉄 (Fe) 鉄の定量には,フェナントロリン吸光光度法,フレーム原子吸光法,電気加熱原子吸光法
又はICP発光分光分析法を適用する。
60.1 フェナントロリン吸光光度法 微酸性溶液中で塩化ヒドロキシルアンモニウムと1, 10-フェナント
ロリンを加えた後,酢酸アンモニウムを加えてpHを4〜5に調節し,生成するだいだい赤の鉄 (II) 錯体
の吸光度を測定して鉄を定量する。
定量範囲:Fe20〜500μg,繰返し分析精度:変動係数で2〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製する。
(c) アンモニア水 (1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
(d) 塩化ヒドロキシルアンモニウム溶液 (100g/l) 40.2(1)(c)による。
(e) 1, 10-フェナントロリン溶液 (1g/l) 59.1(1)(g)による。
(f) 酢酸アンモニウム溶液 (500g/l) JIS K 8359に規定する酢酸アンモニウム500gを水に溶かして1lと
する。
(g) 鉄標準液 (1mgFe/ml) 鉄(99.5%以上)1.000gをとり,塩酸 (1+1) 30ml中に入れ,加熱して溶か
し,放冷後,全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 8979に規定する
257
K 0101 : 1998
硫酸アンモニウム鉄 (II) 六水和物[ビス(硫酸)鉄 (II) アンモニウム六水和物]7.02gをとり,塩
酸 (1+1) 20mlと適量の水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。若
しくはJIS K 0016に規定する鉄標準液のFe 1 000を用いる。
(h) 鉄標準液 (10μgFe/ml) 鉄標準液 (1mgFe/ml) 10mlを全量フラスコ1 000mlにとり,塩酸 (1+1)
20mlを加えた後,水を標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作(1)(2)を行った試料の適量(Feとして20〜500μgを含む。)をビーカーにとり,硝酸 (1+1) 1
〜2mlを加えて煮沸する。
(b) 水を加えて50〜100mlとした後,アンモニア水 (1+1) を加えて微アルカリ性とする。これを数分
間煮沸して沈殿を生成させ(3)(4),しばらく放置する。
(c) 沈殿が沈降した後,ろ紙5種Aでろ過し,温水で数回洗う。沈殿は元のビーカーに洗い入れ,塩酸
(1+1) 4mlを加え,加熱して溶かし,先のろ紙でろ過し,同時にろ紙に付着している水酸化鉄 (III) を
溶かす。ろ紙は温水で数回洗う。
(d) ろ液と洗液を合わせ,水を加えて液量を約70mlとした後,塩化ヒドロキシルアンモニウム溶液
(100g/l) 1ml(5)を加えて振り混ぜる。
(e) 1, 10-フェナントロリン溶液 (1g/l) 5mlを加えて振り混ぜ,続いて酢酸アンモニウム溶液 (500g/l)
10ml(6)を加えて再び振り混ぜ,放冷する。
(f) 全量フラスコ100mlに移し入れ,水を標線まで加えて約20分間放置する。
(g) この溶液の一部を吸収セルに移し,波長510nm付近の吸光度を測定する。
(h) 空試験として水約50mlをとり,硝酸 (1+1) 1〜2mlを加えた後,(b)〜(g)の操作を行って吸光度を
測定し,試料について得た吸光度を補正する。
(i) 検量線から鉄の量を求め,試料中の鉄の濃度 (mgFe/l) を算出する。
検量線 鉄標準液 (10μgFe/ml) 2〜50mlを全量フラスコ100mlに段階的にとり,塩酸 (1+1) 4mlを
加え,(d)〜(h)の操作を行って鉄 (Fe) の量と吸光度との関係線を作成する。
注(1) 試料に有機物及び懸濁物が少なく,また,妨害物質が共存しない場合には,試料100mlにつき
塩酸 (1+1) 4mlを加えて液量が約32になるまで煮沸し,(d)以降の操作を行って定量してもよい。
(2) 4.のうち,4.2の塩酸又は硝酸による分解を適用した場合に,加熱濃縮して液量が約61になった
とき不溶解物が残るときは次のような操作を行う。
加熱濃縮して液量が約61になったとき,更に塩酸10mlを加えて加熱し,ほとんど蒸発乾固す
る。これに塩酸 (1+1) 4mlと少量の水を加えて溶かす。残留物が多い場合には,ろ紙5種Bで
ろ過し,温水で洗う。ろ液と洗液は合わせて保存する。残留物はろ紙とともに白金るつぼに入
れて加熱し,炭化した後,灰化する。
灰化物が着色していないときは,灰化物中には鉄は含まれていないと認めてよい。褐色に着
色している場合には,灰化物を硫酸 (2+1) (JIS K 8951に規定する硫酸を用いて調製する。)
数滴で湿し,JIS K 8819に規定するふっ化水素酸2〜3mlを加え,静かに加熱してシリカを揮散
させ,引き続き加熱して乾燥後,約10分間加熱する。放冷後,JIS K 8783に規定する二硫酸カ
リウム又はJIS K 8972に規定する硫酸水素カリウム2gを加えて融解を行う。放冷後,融解物
を塩酸 (1+1) に溶かし,先のろ液と洗液に合わせ,(b)以降の操作を行って定量する。
258
K 0101 : 1998
なお,けい酸塩類(粘土類)及びシリカの少ない試料の場合は,ふっ化水素酸によるシリカ
の除去を省略して二硫酸カリウム又は硫酸水素カリウムによる融解を行うとよい。
(3) 鉄の量が極めて微量(Fe20μg以下)の場合には,捕集剤としてJIS K 8255に規定する硫酸カリ
ウムアルミニウム・12水0.1gを加えて溶かし,再びアンモニア水 (1+1) を加えて微アルカリ
性とし,水酸化アルミニウムを生成させ,鉄を捕集してろ過する。この際沈殿は塩酸に溶けに
くくなるので,塩酸の添加量を多くし,液量が約5mlになるまで加熱濃縮する。次に,水で液
量を約70mlとし,JIS K 8536に規定する酒石酸ナトリウムカリウム四水和物0.1gを加える。
以下,(d)以降の操作を行う。
(4) 発色を妨害する物質が含まれていない場合には,(d)以降の操作を行って定量してもよい(備考
4.参照)。
(5) JIS K 9502に規定するL (+) -アスコルビン酸0.1gを加えてもよい。
(6) 発色時のpHは約4.8になる。塩酸の濃度が高い場合には,アンモニア水 (1+1) で中和し,発
色時のpHを4〜5に調節する。
また,pH調節の操作は,(3)の操作順序に従い,1, 10-フェナントロリン溶液 (1g/l) を加えた
後に行う。
備考1. 溶存鉄を定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種Cを用い
る。)の適量(Feとして20〜500μgを含む。)をとり,硝酸 (1+1) 1〜2mlを加えて煮沸する。
以下,(3)(b)〜(i)によって定量する。妨害物質が共存しない場合には,(3)(b)及び(c)を省略し,
(3)(d)以降の操作を行って定量してもよい。
2. 懸濁鉄を求めるには,鉄(全鉄)から溶存鉄を差し引く。
3. 鉄 (II) を定量する場合には,次のように操作する。
3.2によってろ過したろ液の適量(Feとして20〜50μg以下を含む。)を全量フラスコ100ml
にとる。1, 10-フェナントロリン溶液 (1g/l) 5mlを加えた後,酢酸アンモニウム溶液 (500g/l)
を加えてpHを約5に調節し,2.(12)(a)の溶存酸素を含まない水を標線まで加え,約20分間
放置する。以下,(g)〜(i)の操作を行って鉄の量を求め,試料中の鉄 (II) の濃度 [mgFe (II) /l]
を算出する。
なお,鉄 (II) は大気中の酸素によって容易に酸化されるから,この試験は,試料採取後直
ちに行うが,全操作を直ちに行えない場合には,採取現場で発色までの操作を行って持ち帰
った後,吸光度を測定する。
4. 鉄をあらかじめ水酸化物として分離しないで試験する場合には,水銀,銅,カドミウム,ニ
ッケル,コバルト,亜鉛などが妨害する。ただし,カドミウム50mg/l,亜鉛10mg/l,水銀1mg/l
までは妨害しない。pH3.5で発色させれば,銅は10mg/l,コバルトは10mg/lまでは妨害しな
い。ニッケルが10mg/l程度共存する場合は,EDTA溶液(JIS K 8107に規定するエチレンジ
アミン四酢酸二水素二ナトリウム二水和物3.7gを水100mlに溶かす。)5mlを添加し,約10
分間煮沸すれば妨害しない。
また,多量の亜鉛が共存するときは,pH9で1, 10-フェナントロリン溶液 (1g/l) を多量に
加えて発色させれば妨害を除くことができる。りん酸イオンが多量に共存すると妨害するが,
発色時のpHを5〜7に調節し,約2時間放置した後,吸光度を測定すれば妨害は少なくなる。
60.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,鉄による原子吸
光を波長248.3nmで測定して鉄を定量する。
259
K 0101 : 1998
定量範囲:Fe0.3〜6mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 鉄標準液 (10μgFe/ml) 60.1(1)(h)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) 鉄中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考5. 溶存鉄を定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種Cを用い
る。)の適量をとり,4.5によって処理する。
6. 懸濁鉄は,備考2.による。
7. 鉄の濃度が低い試料で,しかも干渉物質がほとんど含まれていない場合には,試料100mlを
とり,JIS K 8541に規定する硝酸2mlを加えて煮沸した後,60.1(3)(b)及び(c)に準じて操作し,
分離濃縮する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム中に噴霧し,
波長248.3nmの指示値(7)を読み取る。
(b) 空試験として(3)の準備操作における試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行
って指示値を読み取り,試料について得た指示値を補正する。
(c) 検量線から鉄の量を求め,試料中の鉄の濃度 (mgFe/l) を算出する。
検量線 鉄標準液 (10μgFe/ml) 3〜60mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った試料
と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作を行
う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,
(a)の操作を行って標準液について得た指示値を補正し,鉄 (Fe) の量と指示値との関係線を作成す
る。検量線の作成は,試料測定時に行う。
注(7) 吸光度又はその比例値
備考8. シリカを多量に含む試料の場合には,干渉抑制剤としてカルシウム(又はマグネシウム)を
200mg/l程度加えておく。
60.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,鉄による原子吸光を波長
248.3nmで測定して鉄を定量する。
定量範囲:Fe5〜100μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考9. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) 鉄標準液 (1μgFe/ml) 60.1(1)(h)の鉄標準液 (10μgFe/ml) 10mlを全量フラスコ100mlにとり,硝酸
(1+1) 2mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
260
K 0101 : 1998
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) 鉄中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考10. 溶存鉄を定量する場合には,備考5.による。
11. 備考6.による。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(600
〜1 000℃,30〜40秒間)し,次に,原子化(8)(2 200〜2 800℃,3〜6秒間)し,波長248.3nmの指
示値(7)を読み取る(9)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線から鉄の量を求め,試料中の鉄の濃度 (μgFe/l) を算出する。
検量線 鉄標準液 (1μgFe/ml) 0.5〜10mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った試料
と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作を行
う。別に,空試験として水について(3)(a)を行った試料と同じ条件になるように酸を加えた後,(a)
の操作を行って標準液について得た指示値を補正し,鉄 (Fe) の量と指示値との関係線を作成する。
検量線の作成は,試料測定時に行う。
注(8) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(9) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
60.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,鉄
による発光を波長238.204nmで測定して鉄を定量する。
定量範囲: Fe20〜200μg/l,0.2〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 鉄標準液 (10μgFe/ml) 60.1(1)(h)による。
(b) 混合標準液 [(10μgCu, 10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] 51.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考12. 溶存鉄を定量する場合には,備考5.による。
13. 備考6.による。
14. 準備操作を行った溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
261
K 0101 : 1998
く,鉄の濃度が低い場合には,51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長238.204nmの発光強度を測定する(10)(11)(12)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線から鉄の量を求め,試料中の鉄の濃度 (μgFe/l) を算出する。
検量線 鉄標準液 (10μgFe/ml) 0.2〜2ml(又は2〜50ml)(13)(14)を全量フラスコ100mlに段階的にと
り,(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液
について(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になる
ように酸を加えた後,(a)の操作を行って標準液について得た発光強度を補正し,鉄 (Fe) の量と発
光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(10) 波長の異なる2本のスペクトル線を同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリウ
ム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
238.204nmと同時に371.029nm(イットリウム)の発光強度を測定し,鉄とイットリウムとの発
光強度の比を求める。
別に,鉄標準液 (10μgFe/ml) 0.2〜2ml(又は2〜50ml)を全量フラスコ100mlに段階的にとり,
イットリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように
酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長238.204nm
と同時に371.029nmの発光強度を測定し,鉄の濃度に対する鉄とイットリウムとの発光強度比
の関係線を作成し,検量線とする。この検量線から,試料について得た発光強度比に相当する
鉄の量を求め,試料中の鉄の濃度 (μgFe/l) を算出する。
(11) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(12) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(13) 備考14.によって準備操作を行ってキシレン層をそのまま噴霧する場合の検量線は鉄標準液
(10μgFe/ml) を適当な濃度 (0.1〜1μgFe/ml) に薄め,その0.2〜2ml(又は2〜50ml)を段階的に
とり,500ml(又は100〜500mlの一定量)とした後,試料と同様に備考14.及び(4)(a)と(b)の操
作を行って鉄 (Fe) の量と発光強度の関係線を作成する。
(14) 銅,亜鉛,カドミウム,ニッケル,鉛,マンガンを同時に試験する場合には,混合標準液 [(10μgCu,
10μgZn, 8μgCd, 10μgNi, 10μgPb, 10μgMn, 10μgFe) /ml] を用いて,それぞれの金属元素の試験条
件で検量線を作成するとよい。
61. クロム (Cr) 全クロムとクロム (VI) に区分する。
61.1 全クロム 全クロムの定量には,ジフェニルカルバジド吸光光度法,フレーム原子吸光法,電気加
熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
262
K 0101 : 1998
61.1.1 ジフェニルカルバジド吸光光度法 クロム (III) を過マンガン酸カリウムで酸化してクロム (VI)
とした後,1, 5-ジフェニルカルボノヒドラジド(ジフェニルカルバジド)を加え,生成する赤紫の錯体の
吸光度を測定して全クロムを定量する。
定量範囲:Cr2〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+9) JIS K 8951に規定する硫酸を用いて調製する。
(b) 過マンガン酸カリウム溶液 (3g/l) 46.1(1)(f)による。
(c) 亜硝酸ナトリウム溶液 (20g/l) JIS K 8019に規定する亜硝酸ナトリウム2gを水に溶かして100ml
とする。使用時に調製する。
(d) 尿素溶液 (200g/l) JIS K 8731に規定する尿素20gを水に溶かして100mlとする。
(e) ジフェニルカルバジド溶液 (10g/l) JIS K 8488に規定する1, 5-ジフェニルカルボノヒドラジド
(ジフェニルカルバジド)0.5gをJIS K 8034に規定するアセトン25mlに溶かし,水を加えて50ml
とする。冷暗所に保存する。1週間以上経過したものは使用しない。
(f) クロム標準液 (0.1mgCr/ml) JIS K 8005に規定する容量分析用標準物質の二クロム酸カリウムを
150℃で約1時間加熱し,デシケーター中で放冷する。K2Cr2O7 100%に対してその0.283gをとり,
水に溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0024に規定する
標準物質-標準液-クロムのCr 100を用いる。
(g) クロム標準液 (2μgCr/ml) クロム標準液 (0.1mgCr/ml) 20mlを全量フラスコ1 000mlにとり,水を
標線まで加える。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 4.の操作(1)を行った試料の適量(2)(Crとして2〜50μgを含む。)をビーカーにとり,硫酸 (1+9) 3ml(3)
を加え,加熱して硫酸の白煙を軽く発生させる(4)(5)。放冷後,水約30mlを加え,残留物を加熱し
て溶かす。
(b) 溶液を静かに加熱し,過マンガン酸カリウム溶液 (3g/l) を1滴ずつ加えて着色させる。引き続き加
熱し,溶液の赤い色が消えそうになったら,更に過マンガン酸カリウム溶液 (3g/l) を滴加し,常に
赤い色を保つようにして数分間煮沸を続ける。
(c) 流水で冷却し,尿素溶液 (200g/l) 10mlを加え,激しくかき混ぜながら亜硝酸ナトリウム溶液 (20g/l)
(6)1滴ずつ加えて溶液の赤い色を消し,過剰の過マンガン酸及び酸化マンガン (IV) を分解する。
(d) 全量フラスコ50mlに移し入れ,液温を約15℃(7)に保ち,ジフェニルカルバジド溶液 (10g/l) 1mlを
加え,直ちに振り混ぜる。水を標線まで加えて振り混ぜ,約5分間放置する(8)。
(e) 溶液の一部を吸収セルに移し,波長540nm付近の吸光度を測定する。
(f) 空試験として水約30mlをとり,硫酸 (1+9) 3mlを加えた後,(d)及び(e)の操作を行って吸光度を測
定し,試料について得た吸光度を補正する。
(g) 検量線からクロムの量を求め,試料中の全クロムの濃度 (μgCr/l) を算出する。
検量線 クロム標準液 (2μgCr/ml) 1〜25mlを段階的にビーカーにとり,それぞれに硫酸 (1+9) 3ml
を加え,水を加えて液量を約30mlとした後,(b)〜(f)の操作を行ってクロム (Cr) の量と吸光度との
関係線を作成する。
注(1) 4.のうち4.3の方法は用いない。
263
K 0101 : 1998
(2) クロムの濃度が低く,有機物及び懸濁物がほとんど含まれていない場合には,試料500mlまで
の適量をとり,試料100mlにつきJIS K 8951に規定する硫酸2mlを加えて加熱し,煮沸して放
冷する。硫酸アンモニウム鉄 (II) 溶液 (5mgFe/ml) [JIS K 8979に規定する硫酸アンモニウム
鉄 (II) 六水和物3.5gを硫酸数滴を含む水100mlに溶かす。]1mlを加え,よくかき混ぜ,更に
JIS K 8541に規定する硝酸2mlを加え,しばらく煮沸して鉄 (II) を酸化する。この溶液をしば
らく放置した後,アンモニア水 (1+4) を加えて中和し,アンモニア臭を認めなくなるまで煮沸
し,約80℃に約20分間保ち,沈殿を熟成させる。沈殿をろ紙5種Aを用いてろ別し,温硝酸
アンモニウム溶液 (10g/l) で数回洗った後,硫酸 (1+15) 5mlを加えて溶かし,ろ紙は温水で洗
い,(b)〜(f)の操作を行う。ただし,検量線を作成する場合は,標準液にそれぞれ鉄 [Fe (III)] 5mg
を加える。
(3) ジフェニルカルバジドによるクロム (VI) の発色には,硫酸の濃度は0.1mol/l程度が適当である。
(4) 前処理で多量の硫酸が添加されている場合には,加熱蒸発し,硫酸の白煙を発生させて硫酸を
除去した後,硫酸 (1+9) 3mlを加える。この硫酸の白煙の発生に当たって強熱してはならない。
硫酸クロム (III) の無水物を生成して不溶解性となる。硫酸ナトリウム20mg程度を加えておく
と安全である。
(5) 前処理で硫酸白煙の発生を行った場合には,この操作は省略してよい。
(6) 亜硝酸ナトリウム溶液 (20g/l) の代わりにアジ化ナトリウム溶液 (50g/l) を用いることができ
る。この場合,アジ化ナトリウム溶液 (50g/l) を注意して滴加し,よく振り混ぜて過マンガン
酸を分解し,続いて2〜3分間煮沸して過剰のアジ化ナトリウムを分解する。
(7) 液温は,発色に影響するので,約15℃に保つことが必要である。
(8) 発色は2〜3分間ほどで最高になり,その後徐々に退色するが,5〜15分間はほとんど変化しな
い。
備考1. 試料が鉄を含むとき,鉄が多量になるに従って吸光度が低くなるが,発色液50ml中約1mgで
一定となる(約20%低値を示す。)。ただし,ジフェニルカルバジド溶液を加える前に,二り
ん酸ナトリウム溶液(JIS K 8785に規定する二りん酸ナトリウム十水和物5gを水に溶かし
100mlとする。)2mlを加えると,鉄2.5mgまでは影響しない。
鉄がクロムより少ない場合には無視してもよい。
2. この方法では,モリブデン,水銀,バナジウムなどが影響する。ただし,モリブデンは0.1mg
まで影響しない。水銀は,塩化物イオンの添加によって妨害しなくなる。
また,バナジウムは発色後,10〜15分間経過してから吸光度を測定すれば,その影響は無
視できる。
3. 鉄その他の妨害が多い場合には,試料の適量(Crとして2〜50μgを含む。)を分液漏斗にと
り,試料20mlにつき硫酸 (1+1) 5mlを加え,硫酸の濃度を約1.8mol/lとし,これに過マン
ガン酸カリウム溶液 (3g/l) を滴加し,わずかに着色させる。これに,クペロン溶液 (50g/l)
[JIS K 8289に規定するクペロン(N-ニトロソ-N-フェニルヒドロキシルアミンアンモニウム
塩)(N-ヒドロキシ-N-ニトロソベンゼンアミンアンモニウム塩,IUPACによる名称)5gを水
に溶かして100mlとする。]5mlとJIS K 8322に規定するクロロホルム10mlを加えて約30
秒間激しく振り混ぜ,鉄その他を抽出し静置する。
クロロホルム層を分離し,水層に再びクペロン溶液 (50g/l) 1mlとクロロホルム10mlを加
えて再び抽出し,クロロホルム層を分離する。水層をビーカー100mlに移し,加熱蒸発して
264
K 0101 : 1998
軽く乾燥する。これに少量の硫酸と硝酸とを加え,再び蒸発乾固して有機物を分解する。硫
酸 (1+1) 0.3mlと水約30mlに溶かす。過マンガン酸カリウム溶液 (3g/l) でクロムを酸化し
た後,(3)によって操作する。
61.1.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレームなど中に噴霧し,クロムに
よる原子吸光を波長357.9nmで測定して全クロムを定量する。
定量範囲:Cr0.2〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) クロム標準液 (10μgCr/ml) 61.1.1(1)(f)のクロム標準液 (0.1mgCr/ml) 50mlを全量フラスコ500ml
にとり,硝酸 (1+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) クロム中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5又は備考4.によって処理する(1)(4)。
備考4. クロムの濃度が低い試料で,有機物及び懸濁物をほとんど含まない場合の準備操作は,次に
よる。
試料の適量をとり,注(2)に準じて操作し,水酸化鉄 (III) にクロムを共沈させる。沈殿を
ろ紙5種Aでろ別し,少量の硝酸 (1+2) に加熱して溶かし,ろ紙は温水で洗浄する。ろ液
と洗液を合わせ,0.1〜1mol/lの塩酸又は硝酸酸性溶液の一定量とする。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0121の6.(操作方法)の操作に従って,フレーム(9)中に噴霧し,
波長357.9nmの指示値(10)を読み取る。
(b) 空試験として(3)の準備操作における試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行
って指示値を読み取り,試料について得た指示値を補正する。
(c) 検量線からクロムの量を求め,試料中の全クロムの濃度 (mgCr/l) を算出する。
検量線 クロム標準液 (10μgCr/ml) 2〜50mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った
試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操作
を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加えた
後,(a)の操作を行って標準液について得た指示値を補正し,クロム (Cr) の量と指示値との関係線
を作成する。検量線の作成は,試料測定時に行う。
注(9) 少燃料のアセチレン-空気,又はアセチレン-一酸化二窒素フレームを用いる。
(10) 吸光度又はその比例値。
備考5. クロムの濃度が低い試料で,抽出を妨害する物質を含まない場合には,次の操作で定量して
もよい。
試料の適量(Crとして5〜100μgを含む。)をビーカー100mlにとり,硫酸 (1+2) 2mlを加
え過マンガン酸カリウム溶液 (3g/l) 数滴を加えて加熱する。過マンガン酸の色が消えそうに
なったら,更に過マンガン酸カリウム溶液 (3g/l) を滴加し,常に溶液の色が微赤を保つよう
に数分間煮沸を続ける。流水で冷却し,これを分液漏斗に移し,水を加えて約100mlとする。
N, N-ジオクチルオクタンアミン(トリオクチルアミン)の酢酸ブチル溶液 (30g/l) 20mlを加
え,約10分間激しく振り混ぜた後,放置する。酢酸ブチル層をそのままフレーム中に噴霧し
265
K 0101 : 1998
てクロムを定量する。
なお,検量線の作成にはクロム標準液 (10μgCr/ml) を適当に薄めて用いる。JIS K 8377に
規定する酢酸ブチルの代わりにJIS K 8903に規定する4-メチル-2-ペンタノンを用いてもよ
い。
6. アセチレン-空気フレームでは,多燃料フレームにすると感度は高くなるが,鉄,ニッケルな
ど共存物による妨害も大きくなる。この場合,硫酸ナトリウム,二硫酸カリウム又は二ふっ
化水素アンモニウムを1%程度共存させるとよい。
アセチレン−一酸化二窒素フレームでは妨害の大部分は消滅する。
61.1.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,クロムによる原子吸光を波長
357.9nmで測定して全クロムを定量する。
定量範囲:Cr5〜100μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考7. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) クロム標準液 (1μgCr/ml) 61.1.2(1)(a)のクロム標準液 (10μgCr/ml) 10mlを全量フラスコ100mlに
とり,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) クロム中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5又は備考4.によって処理する(1)。ただし,最終溶液は0.1〜1mol/lの硝酸酸性とする。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(500
〜600℃,30〜40秒間)し,次に,原子化(11)(2 400〜2 900℃,5〜10秒間)し,波長357.9nmの指
示値(10)を読み取る(12)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からクロムの量を求め,試料中の全クロムの濃度 (μgCr/l) を算出する。
検量線 クロム標準液 (1μgCr/ml) 0.5〜10mlを全量フラスコ100mlに段階的にとり,(3)(a)を行った
試料と同じ酸の濃度になるように硝酸を加えた後,水を標線まで加える。この溶液について(a)の操
作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように硝酸を加
えた後,(a)の操作を行って標準液について得た指示値を補正し,クロム (Cr) の量と指示値との関
266
K 0101 : 1998
係線を作成する。検量線の作成は,試料の測定時に行う。
注(11) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(12) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
61.1.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ク
ロムによる発光を波長206.149nmで測定して全クロムを定量する。
定量範囲:Cr20〜4 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) クロム標準液 (10μgCr/ml) 61.1.2(1)(a)による。
(b) 混合標準液 [(10μgCr, 10μgV) /ml] 61.1.1(1)(f)のクロム標準液 (0.1mgCr/ml) 10ml,62.1(1)(g)のバナ
ジウム標準液 (0.1mgV/ml) 10mlをそれぞれ全量フラスコ100mlにとり,硝酸 (1+1) 2mlを加え,
水を標線まで加える。使用時に調製する。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する(1)。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長206.149nmの発光強度を測定する(13)(14)(15)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からクロムの量を求め,試料中の全クロムの濃度 (mgCr/l) を算出する。
検量線 クロム標準液 (10μgCr/ml) 0.2〜40ml(16)を全量フラスコ100mlに段階的にとり,(3)(a)を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の
操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加
えた後,(a)の操作を行って標準液について得た発光強度を補正し,クロム (Cr) の量と発光強度と
の関係線を作成する。検量線の作成は,試料測定時に行う。
注(13) 波長の異なる2本のスペクトル線を同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリウ
ム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
206.149nmと同時に371.029nm(イットリウム)の発光強度を測定し,クロムとイットリウムと
の発光強度の比を求める。
別に,クロム標準液 (10μgCr/ml) 0.2〜40mlを全量フラスコ100mlに段階的にとり,イットリ
ウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を加え
た後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長206.149nmと同時
に371.029nmの発光強度を測定し,クロムの濃度に対するクロムとイットリウムとの発光強度
比の関係線を作成し,検量線とする。この検量線から,試料について得た発光強度比に相当す
るクロムの量を求め,試料中のクロムの濃度 (μgCr/l) を算出する。
(14) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
267
K 0101 : 1998
標準添加法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正
を行う必要がある。
(15) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(16) バナジウムを同時に試験する場合には,混合標準液 [(10μgCr, 10μgV) /ml] を用いてバナジウム
の試験条件で検量線を作成するとよい。
備考8. ナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高く,クロムの濃度が低い
場合には,試料の適量(Crとして0.4〜80μgを含む。)をビーカー100mlにとり,備考5.に準
じて操作するとよい。このときプラズマトーチとしては有機溶媒用のトーチを用いる。
61.1.5 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,クロムと内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,クロムのイ
オンの電流と内標準物質のイオンの電流との比を求めて全クロムを定量する。
定量範囲: Cr0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件に
よって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 61.1.3(1)(a)による。
(b) 硝酸 (1+1) 61.1.3(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (17) 51.5(1)(c)による。
(d) クロム標準液 (1μgCr/ml) 61.1.3(1)(c)による。
(e) クロム標準液 (50ngCr/ml) クロム標準液 (1μgCr/ml) 50mlを全量フラスコ1 000mlにとり,硝酸
(1+1) 3mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による。
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.1(1)(g)による。
注(17) 51.の注(16)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考9. 51.の備考8.による。
10. 51.の備考9.による。
11. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(18)。
(a) 試料を4.5によって処理する(1)。
(b) (3)(a)で処理した試料の適量(Crとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イット
リウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を加
えた後,水を標線まで加える。
注(18) 51.の注(17)による。
(4) 操作 操作は,次のとおり行う(19)。
(a) ICP質量分析装置を作動できる状態にし,(3)(b)の溶液を試料導入部を通して誘導結合プラズマ中に
噴霧してクロムとイットリウムの質量/荷電数(20)における指示値(21)を読み取り,クロムの指示値
とイットリウムの指示値との比を求める。
(b) 空試験値として,(3)(a)での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行ってクロ
268
K 0101 : 1998
ムの指示値とイットリウムの指示値との比を求め,試料について得たクロムとイットリウムとの指
示値の比を補正する。
(c) 検量線からクロムの量を求め,試料中の全クロムの濃度 (μgCr/l) を算出する。
検量線 クロム標準液(50ngCr/ml又は1μgCr/ml) 1〜50ml(22)を全量フラスコ100mlに段階的にとり,
イットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝酸 (1+1) を
加えた後,水を標線まで加える。この溶液について(a)の操作を行う。別に,空試験として全量フラ
スコ100mlにイットリウム溶液 (1μgY/ml) 1mlを加え,(3)(b)の試料と同じ酸の濃度になるように硝
酸 (1+1) を加え,水を標線まで加えた後,(a)の操作を行って標準液について得た指示値を補正し,
クロムの量 (μgCr) に対するクロムの指示値とイットリウムの指示値との比の関係線を作成する。
検量線の作成は,試料測定時に行う。
注(19) 51.の注(18)による。
(20) 51.の注(19)による。
(21) 51.の注(20)による。
(22) 51.の注(21)による。
備考12. 51.の備考11.による。
61.2 クロム (VI) [Cr (VI)] クロム (VI) の定量には,ジフェニルカルバジド吸光光度法,フレーム原子
吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
61.2.1 ジフェニルカルバジド吸光光度法 試料に1, 5-ジフェニルカルボノヒドラジド(ジフェニルカルバ
ジド)を加え,生成する赤紫の錯体の吸光度を測定してクロム (VI) を定量する。
定量範囲:Cr (VI) 2〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 硫酸 (1+9) JIS K 8951に規定する硫酸を用いて調製する。
(b) エタノール (95) JIS K 8102に規定するもの。
(c) ジフェニルカルバジド溶液 (10g/l) 61.1.1(1)(e)による。
(d) クロム (VI) 標準液[2μgCr (VI) /ml] 61.1.1(1)(g)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量[Cr (VI) として2〜50μgを含む。]を2個のビーカー (A) , (B) にとり,試料が酸性
の場合には,水酸化ナトリウム溶液 (40g/l) で,また,アルカリ性のときは硫酸 (1+35) で中和す
る。
(b) ビーカー (A) の溶液は全量フラスコ50ml (A) に移し入れ,硫酸 (1+9) 2.5mlを加える。
(c) ビーカー (B) の溶液に硫酸 (1+9) 2.5mlを加え,次にエタノール (95) を少量加え,煮沸してクロ
ム (VI) をクロム (III) に還元し,過剰のエタノールを追い出す。放冷後,全量フラスコ50ml (B) に
移し入れる。
(d) 全量フラスコ (A) 及び (B) を約15℃に保ち,それぞれにジフェニルカルバジド溶液 (10g/l) 1mlず
つを加え,直ちに振り混ぜ,水を標線まで加え,約5分間放置する。
(e) 全量フラスコ (A) の一部を吸収セルに移し,全量フラスコ (B) の溶液を対照液として波長540nm
付近の吸光度を測定する。
(f) 検量線からクロム (VI) の量を求め,試料中のクロム (VI) の濃度 [mgCr (VI) /l] を算出する。
269
K 0101 : 1998
検量線 クロム (VI) 標準液[2μgCr (VI) /ml]1〜25mlを段階的にとり,(b)〜(d)の操作における全
量フラスコ (A) に対するのと同じ操作を行う。この溶液の一部を吸収セルに移し,水約30mlにつ
いて(c)及び(d)の操作における全量フラスコ (B) に対するのと同じ操作を行った溶液を対照液とし,
波長540nm付近の吸光度を測定し,クロム [Cr (VI) ] の量と吸光度との関係線を作成する。
備考13. 試料が着色していたり,酸性にしたとき,クロム (VI) を還元する物質が共存するときは,
定量は困難である。ただし,クロム (III) が含まれていない試料は61.1に従って定量する。
14. 備考2.による。
61.2.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気などのフレーム中に噴霧し,クロム
(VI) による原子吸光を波長357.9nmで測定してクロム (VI) を定量する。
定量範囲: Cr (VI) 0.2〜5mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異
なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) クロム標準液 (10μgCr/ml) 61.1.2(1)(a)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックグラウンド補正が可能なもの。
(b) クロム中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料の適量(懸濁物を含む場合には,ろ紙5種C又は孔径0.45μmのろ過材でろ過し,最初のろ液
約50mlを捨て,その後のろ液を用いる。)をとり,JIS K 8180に規定する塩酸又はJIS K 8541に規
定する硝酸を加えて0.1〜1mol/lの酸性溶液とする。ただし,試料中にクロム (III) が含まれている
場合には,備考15.の(2)によって操作する。
備考15. クロム (VI) の濃度が低い試料で妨害物質を含まない場合には,次のように操作する。
(1) 試料中にクロム (III) が含まれていないときは,備考4.又は備考5.に準じて操作する。
(2) 試料中にクロム (III) が含まれるときは,次による。
500ml以下の試料をとり,硫酸アンモニウム鉄 (III) 溶液[JIS K 8982に規定する硫酸アン
モニウム鉄 (III) ・12水5gを硫酸 (1+1) 1mlに溶かし,水で100mlにする。]1mlを加えてか
き混ぜる。アンモニア水 (1+4) を加えて微アルカリ性とした後,アンモニア臭がほとんど
なくなるまで静かに煮沸する。沸騰近くの温度に保って沈殿を熟成させた後,ろ紙5種Aで
ろ過し,温硝酸アンモニウム溶液 (10g/l) で洗浄する。ろ液と洗液を合わせ,塩酸又は硝酸
を加えて0.1〜1mol/lの酸性溶液とする。
(4) 操作 操作は,次のとおり行う。
(a) 61.1.2(4)に準じて操作する。
61.2.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,クロムによる原子吸光を波長
357.9nmで測定してクロム (VI) を定量する。
定量範囲: Cr (VI) 5〜100μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異
なる。)
備考16. 備考7.による。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 61.1.3(1)(a)による。
(b) 硝酸 (1+1) 61.1.3(1)(b)による。
270
K 0101 : 1998
(c) クロム標準液 (1μgCr/ml) 61.1.3(1)(c)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) クロム中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料の適量をとり,61.2.2(3)(a)又は備考15.によって処理する。ただし,硝酸を用い,最終溶液は
0.1〜1mol/lの硝酸酸性溶液とする。
(4) 操作 操作は,次のとおり行う。
(a) 61.1.3(4)に準じて操作する。
61.2.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ク
ロムによる発光を波長206.149nmで測定してクロム (VI) を定量する。
定量範囲: Cr (VI) 20〜4 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって
異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) クロム標準液 (10μgCr/ml) 61.1.2(1)(a)による。
(b) 混合標準液 [(10μgCr, 10μgV) /ml] 61.1.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 操作は,次のとおり行う。
(a) 試料の適量をとり,61.2.2(3)(a)又は備考15.によって処理する。ただし,硝酸を用い,最終溶液は
0.1〜0.5mol/lの硝酸酸性溶液とする。
(4) 操作 操作は,次のとおり行う。
(a) 61.1.4(4)に準じて操作する。
61.2.5 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,クロムと内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,クロムのイ
オンの電流と内標準物質のイオンの電流との比を求めてクロム (VI) を定量する。
定量範囲: Cr (VI) 0.5〜25μg/l,10〜500μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条
件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 水 61.1.3(1)(a)による。
(b) 硝酸 (1+1) 61.1.3(1)(b)による。
(c) イットリウム溶液 (1μgY/ml) (17) 51.5(1)(c)による。
(d) クロム標準液 (1μgCr/ml) 61.1.3(1)(c)による。
(e) クロム標準液 (50ngCr/ml) クロム標準液 (1μgCr/ml) 50mlを全量フラスコ1 000mlにとり,硝酸
(1+1) 3mlを加え,水を標線まで加える。使用時に調製する。
(f) 混合標準液 [(1μgCu, 1μgZn, 1μgCd, 1μgPb, 1μgMn, 1μgCr) /ml] 51.5(1)(f)による。
271
K 0101 : 1998
(g) 混合標準液 [(50ngCu, 50ngZn, 50ngCd, 50ngPb, 50ngMn, 50ngCr) /ml] 51.5(1)(g)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP質量分析装置
備考17. 51.の備考8.による。
18. 51.の備考9.による。
19. 51.の備考10.による。
(3) 準備操作 準備操作は,次のとおり行う(18)。
(a) 61.2.3(3)(a)又は備考15.によって処理する。ただし,硝酸を用い,最終溶液は0.1〜0.5mol/lの硝酸酸
性溶液とする。
(b) (3)(a)で処理した試料の適量(Crとして0.05〜50μgを含む。)を全量フラスコ100mlにとり,イット
リウム溶液 (1μgY/ml) 1mlを加え,硝酸の最終濃度が0.1〜0.5mol/lとなるように硝酸 (1+1) を加
えた後,水を標線まで加える。
(4) 操作 操作は,次のとおり行う。
(a) 61.1.5(4)に準じて操作する。
62. バナジウム (V) バナジウムの定量には,N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法,
フレーム原子吸光法,電気加熱原子吸光法又はICP発光分光分析法を適用する。
62.1 N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法 試料を過マンガン酸カリウムで酸化し
てバナジウム (V) とし,これにN-ベンゾイル−N-フェニルヒドロキシルアミン(N-ヒドロキシ-N-フェニ
ルベンザミド,IUPACによる名称)を加えて生成した赤紫のバナジウム錯体を塩酸酸性溶液からクロロホ
ルムで抽出し,その吸光度を測定してバナジウムを定量する。
定量範囲:V2〜50μg,繰返し分析精度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 塩酸 (2+1) JIS K 8180に規定する塩酸を用いて調製する。
(b) 硝酸 JIS K 8541に規定するもの。
(c) 過塩素酸 JIS K 8223に規定するもの。
(d) 銅 (II) 溶液 (10g/l) JIS K 8660に規定する銅(99.9%以上)1gを硝酸 (1+1) 10mlに加え,加熱
して溶かし,過塩素酸20mlを加えて加熱蒸発して白煙を十分に発生させる。放冷後,水で薄めて
100mlとする。
(e) BPHAクロロホルム溶液 (2g/l) JIS K 9569に規定するN-ベンゾイル−N-フェニルヒドロキシル
アミン0.2gをJIS K 8322に規定するクロロホルム100mlに溶かす。
(f) 過マンガン酸カリウム溶液 (3g/l) 46.1(1)(f)による。
(g) バナジウム標準液 (0.1mgV/ml) JIS K 8747に規定するバナジン (V) 酸アンモニウム0.230gをと
り,硫酸 (1+1) 10mlと熱水200mlに溶かす。放冷後,全量フラスコ1 000mlに移し入れ,水を標
線まで加える。
(h) バナジウム標準液 (2μgV/ml) バナジウム標準液 (0.1mgV/ml) 20mlを全量フラスコ1 000mlにと
り,硫酸 (1+1) 10mlを加えた後,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 100ml
(b) 光度計 分光光度計又は光電光度計
272
K 0101 : 1998
(3) 操作 操作は,次のとおり行う。
(a) 試料の適量(Vとして2〜50μgを含む。)をとり,硝酸5mlと過塩素酸3mlとを加えて加熱蒸発し,
過塩素酸の白煙を十分に発生させて乾固近くまで蒸発濃縮する。
(b) 放冷後,水10mlを加えて溶かし,銅 (II) 溶液 (10g/l) 1mlを加えた後,過マンガン酸カリウム溶液
(3g/l) を溶液の色が薄い赤となるまで滴加し,更に1滴を過剰に加えて約5分間放置し,バナジウ
ムを酸化する。
(c) 分液漏斗100mlに移し入れ,水で約30ml(1)とする。
(d) BPHAクロロホルム溶液 (2g/l) 10mlを加える。次に,塩酸 (2+1) 40mlを加えて,過剰の過マンガ
ン酸を還元し,直ちに約30秒間振り混ぜてバナジウム錯体を抽出する。
(e) 放置した後,クロロホルム層を乾いたろ紙でろ過する。
(f) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長530nm付近の吸光度を
測定する。
(g) 空試験として水約10mlをとり,(b)〜(f)の操作を行って試料について得た吸光度を補正する。
(h) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度 (μgV/l) を算出する。
検量線 バナジウム標準液 (2μgV/ml) 1〜25mlを分液漏斗に段階的にとり,(b)〜(g)の操作を行って
バナジウム (V) の量と吸光度との関係線を作成する。
注(1) あらかじめ分液漏斗に目印を付けておく。
62.2 フレーム原子吸光法 試料を前処理した後,アセチレン-一酸化二窒素フレーム中に噴霧し,バナジ
ウムによる原子吸光を波長318.4nmで測定してバナジウムを定量する。
定量範囲:V1〜20mg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) 硝酸 JIS K 8541に規定するもの。
(b) 硝酸アルミニウム溶液 (400g/l) JIS K 8544に規定する硝酸アルミニウム九水和物70gをとり,少
量の水を加え,加熱して溶かす。放冷後,水を加えて100mlにする。
(c) バナジウム標準液 (0.1mgV/ml) 62.1(1)(g)による。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) フレーム原子吸光分析装置 バックがラウンド補正が可能なもの。
(b) バナジウム中空陰極ランプ
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の適量(Vとして0.1〜2mgを含む。)を全量フラスコ100mlにとり,硝
酸1mlを加えた後,水を標線まで加える。
(b) この溶液50mlを乾いたビーカーにとり,硝酸アルミニウム溶液 (400g/l) 1mlを加える。
(c) (b)の溶液をJIS K 0121の6.(操作方法)の操作に従って,アセチレン-一酸化二窒素フレーム中に
噴霧し(2),波長318.4nmの指示値(3)を読み取る。
(d) 空試験として試料と同量の水をとり,(3)及び(4)(a)〜(c)の操作を行って試料について得た指示値を
補正する。
(e) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度 (mgV/l) を算出する。
検量線 バナジウム標準液 (0.1mgV/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,(3)(a)を行
273
K 0101 : 1998
った試料と同じ酸の濃度になるように酸及び硝酸を加えた後,水を標線まで加える。この溶液につ
いて(b)及び(c)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度に
なるように酸及び硝酸を加えた後,(b)及び(c)の操作を行って標準液について得た指示値を補正し,
バナジウム (V) の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(2) 多燃料フレームの方が高感度が得られる。感度の最も高い部分は,フレームのごく限られた位
置であるから,この位置を求めておく。
(3) 吸光度又はその比例値。
62.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,バナジウムによる原子吸光を
波長318.4nmで測定してバナジウムを定量する。
定量範囲:V10〜200μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
備考1. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
(b) 硝酸 (1+1) JIS K 9901に規定する高純度試薬-硝酸を用いて調製する。
(c) バナジウム標準液 (1μgV/ml) 62.1(1)(g)のバナジウム標準液 (0.1mgV/ml) 2mlを全量フラスコ
200mlにとり,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(b) 発熱体 黒鉛製又は耐熱金属製のもの
(c) バナジウム中空陰極ランプ
(d) フローガス JIS K 1105に規定するアルゴン2級
(e) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計10〜50μl又は自動注
入装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,
JIS K 0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,30〜40秒間)した後,灰化(500
〜600℃,30〜40秒間)し,次に,原子化(4)(2 400〜3 000℃,5〜10秒間)し,波長318.4nmの指
示値(3)を読み取る(5)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た指示値を補正する。
(c) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度 (μgV/l) を算出する。
検量線 バナジウム標準液 (1μgV/ml) 1〜20mlを全量フラスコ100mlに段階的にとり,(3)(a)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について(a)の操
作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸を加え
た後,(a)の操作を行って標準液について得た指示値を補正し,バナジウム (V) の量と指示値との
関係線を作成する。検量線の作成は,試料測定時に行う。
274
K 0101 : 1998
注(4) 乾燥,灰化,原子化の条件は装置によって異なる。試料の注入量及び共存する塩類の濃度によ
っても異なることがある。
(5) 引き続いて少なくとも(a)の操作を3回繰り返し,指示値が合うことを確認する。
62.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,バ
ナジウムによる発光を波長309.311nmで測定してバナジウムを定量する。
定量範囲:V20〜2 000μg/l,繰返し分析精度:変動係数で2〜10%(装置,測定条件によって異なる。)
(1) 試薬 試薬は,次のものを用いる。
(a) バナジウム標準液 (10μgV/ml) 62.1(1)(g)のバナジウム標準液 (0.1mgV/ml) 10mlを全量フラスコ
100mlにとり,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(b) 混合標準液 [(10μgCr, 10μgV) /ml] 61.1.4(1)(b)による。
(2) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(3) 準備操作 準備操作は,次のとおり行う。
(a) 試料を4.5によって処理する。
備考2. 準備操作を行った溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高
く,バナジウムの濃度が低い場合には51.の備考7.に準じた操作を行うとよい。
(4) 操作 操作は,次のとおり行う。
(a) (3)の準備操作を行った試料をJIS K 0116の5.8(ICP発光分光分析の定量分析)に従って,試料導
入部を通してプラズマ中に噴霧し,波長309.311nmの発光強度を測定する(6)(7)(8)。
(b) 空試験として(3)の準備操作での試料と同量の水をとり,試料と同様に(3)及び(4)(a)の操作を行って
試料について得た発光強度を補正する。
(c) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度 (μgV/l) を算出する。
検量線 バナジウム標準液 (10μgV/ml) 0.2〜20ml(9)(10)を全量フラスコ100mlに段階的にとり,(3)(a)
を行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液について
(a)の操作を行う。別に,空試験として水について(3)(a)を行った試料と同じ酸の濃度になるように酸
を加えた後,(a)の操作を行って標準液について得た発光強度を補正し,バナジウム (V) の量と発
光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
注(6) 波長の異なる2本のスペクトル線を同時測定が可能な装置では内標準法によることができる。内
標準法を用いるときは,(3)(a)で処理した試料の適量を全量フラスコ100mlにとり,イットリウ
ム溶液 (50μgY/ml) [45.の注(8)による。]10mlを加え,(4)(a)の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長
309.311nmと同時に371.029nm(イットリウム)の発光強度を測定し,バナジウムとイットリウ
ムとの発光強度の比を求める。
別に,バナジウム標準液 (10μgV/ml) 0.2〜20mlを全量フラスコ100mlに段階的にとり,イッ
トリウム溶液 (50μgY/ml) 10mlをそれぞれ加え,(4)(a)の試料と同じ酸の濃度になるように酸を
加えた後,水を標線まで加える。この溶液について(4)(a)の噴霧操作を行って波長309.311nmと
同時に371.029nmの発光強度を測定し,バナジウムの濃度に対するバナジウムとイットリウム
との発光強度比の関係線を作成し,検量線とする。この検量線から,試料について得た発光強
度比に相当するバナジウムの量を求め,試料中のバナジウムの濃度 (μgV/l) を算出する。
(7) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の5.8.3(2)に規定する
275
K 0101 : 1998
標準添加法を用いるとよい。ただし,この場合は試料の種類によらずバックグラウンド補正を
行う必要がある。
(8) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度,正確さを確認してあれば,他の波長を用いてもよい。
(9) 備考2.によって準備操作を行い,キシレン層をそのまま噴霧する場合の検量線はバナジウム標
準液 (10μgV/ml) を適当な濃度 (0.1〜1μgV/ml) に薄め,その0.1〜20mlを段階的にとり,500ml
(又は100〜500mlの一定量)とした後,試料と同様に備考2.及び(4)(a)と(b)の操作を行ってバ
ナジウム (V) の量と発光強度の関係線を作成する。
(10) クロムを同時に試験する場合には,混合標準液 [(10μgCr, 10μgV) /ml] を用いてバナジウムの試
験条件で検量線を作成するとよい。
63. 細菌試験 細菌の試験は,一般細菌,従属栄養細菌及び大腸菌群について行う。この試験は試料採取
後直ちに行う。直ちに試験ができない場合は0〜5℃(凍結させないこと。)の暗所に保存して,できるだ
け早く試験する。
63.1 試料の採取及び細菌の捕集 試料中の細菌が多いと予想される場合には,採水瓶又は採水器を用い
て試料を採取する。
また,試料中の細菌が少ないと予想される場合には,試料の一定量を孔径0.45μmのろ過膜でろ過し,
細菌をろ過膜に捕集する。
(1) 器具 器具は,次のとおりとする。
(a) 採水瓶 共栓ガラス瓶100ml 採水瓶(1)は,栓部と首部を金属はく又は硫酸紙などで覆って
63.2(3)(a)の乾熱滅菌又は63.2(3)(b)の高圧蒸気滅菌を行う。又は滅菌済みの細菌試験用ポリエチレン
瓶100mlを用いてもよい。試料採取時まで汚染されないように注意する。
(b) 採水器 ハイロート採水器 採水器は携帯箱に収めて63.2(3)(a)の乾熱滅菌を行う。図63.1に一例
を示す。
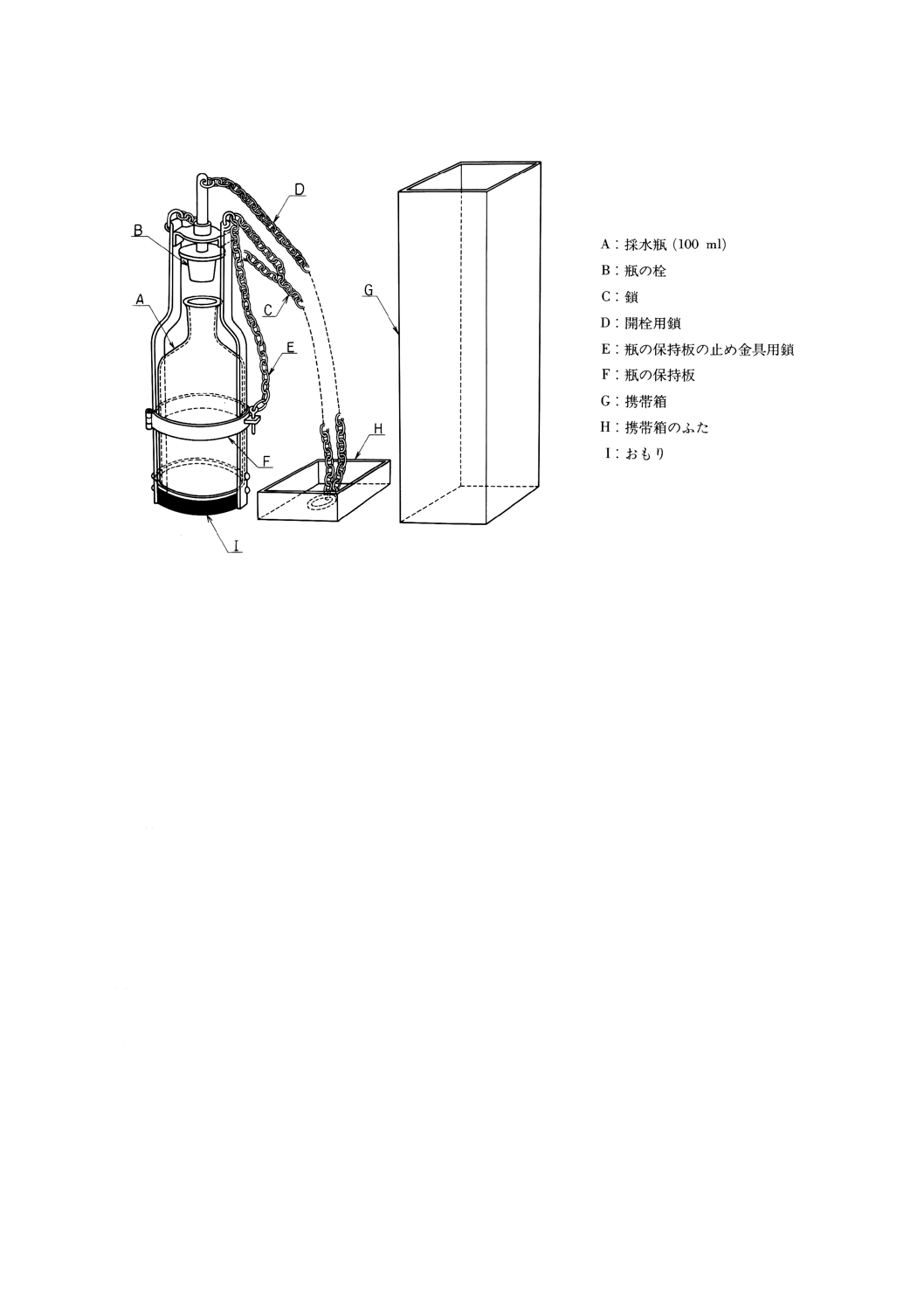
276
K 0101 : 1998
図63.1 採水器の一例
注(1) 残留塩素などの酸化性物質を含む試料を採取する場合には,採水瓶にJIS K 8637に規定するチ
オ硫酸ナトリウム五水和物(粉末にしたもの)20〜50mgを入れ,63.2(3)(b)の高圧蒸気滅菌又は
オキシラン(エチレンオキシド)滅菌をしておく。
(2) 操作 試料の採取及び細菌の捕集は,次のとおり行う。
(2.1) 試料中の細菌が多いと予想される場合には,試料を採水瓶に採取する。
(a) 表層水の採取 河川,水路などの表層水は,採水瓶に直接採取する。
(b) 各深度の水の採取 一定の深さの水は,ハイロート採水器を用いて採取する。
(c) ポンプによる採取 ポンプによって採水する。あらかじめポンプの吐出口を滅菌した後,吐出口に
滅菌した軟質塩化ビニル管を取り付け,十分に放水した後,採水瓶に採取する。
(d) 給水栓からの採取 あらかじめ63.2(3)(d)の火炎滅菌に準じて給水栓口を滅菌した後,栓を開き,配
管中の水を十分に放出した後,採水瓶に採取する。
(e) 配管装置からの採取 (d)と同様に操作して採水瓶に採取する。
(2.2) 試料中の細菌が少ないと予想される場合には,試料の一定量を孔径0.45μmのろ過膜でろ過し,細
菌をろ過膜に捕集する。
(a) 工程水など,細菌が少ないと予想される配管装置から細菌を捕集する場合には,JIS K 0550の4.4
の操作に従って,細菌を直接ろ過膜に捕集する。
(b) (a)の操作で細菌が捕集できない場合には,JIS K 0550の4.4の備考1.の操作に従って細菌ろ過膜に
捕集する。
(c) 試料が容器に入れられている場合には,JIS K 0550の4.4の備考2.の操作に従って細菌ろ過膜に捕
集する。
63.2 一般細菌 一般細菌は,標準寒天培地を用いて36±1℃で24±2時間培養したとき,培地上に集落を
形成した生菌をいい,試料1ml中の個数で表示する。
(1) 試薬及び培地 試薬及び培地は,次のものを用いる。
277
K 0101 : 1998
(a) 水 JIS K 0557に規定するA2又はA3の水。この試験に用いる試薬及び培地の調製には,この水を
用いる。
(b) 希釈水 生理食塩水(JIS K 8150に規定する塩化ナトリウム8.5gを水に溶かして1lとする。)又は
りん酸塩緩衝液 (pH7.2) [JIS K 9007に規定するりん酸二水素カリウム34gを水約500mlに溶かし,
これに水酸化ナトリウム溶液 (1mol/l) を滴加してpHを7.2に調節し,2.(12)(b)の炭酸を含まない水
を加えて全量を1lとする。この溶液1.25mlをとり,水を加えて1lとする。]を用いる。あらかじめ
(3)(b)の高圧蒸気滅菌を約15分間行う。
(c) 標準寒天培地(2) ペプトン(カゼインのパンクレアチン水解物を用いる。)5g,酵母エキス(粉末)
2.5g,JIS K 8824に規定するD (+) -グルコース1g及びJIS K 8263に規定する寒天(粉末)15gを
水1lに加え,加熱して溶かす。滅菌後のpHが7.0±0.1になるように調節し,試験管又はフラスコ
に移し入れ,(3)(b)の高圧蒸気滅菌を約15分間行った後,冷暗所に保存する。
注(2) 市販の培地を使用してもよい(備考1.参照)。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) メスピペット 1ml硫酸紙などに包むか,ピペット滅菌箱に先端を先にして入れ,(3)(a)の乾熱滅菌
をしておく。
(b) 希釈瓶 使用水量の2倍以上の容量の共栓ガラス瓶又は綿栓した試験管若しくは三角フラスコ。い
ずれの場合も,(3)(a)の乾熱滅菌をしておく。あらかじめ,9ml又は99mlの目盛りを付けておくと,
希釈水を入れる場合に便利である。
(c) ペトリ皿 ガラス製の直径約90mm,高さ約15mmのもの。硫酸紙などに包んで,ペトリ皿滅菌箱
に入れて(3)(a)の乾熱滅菌をしておく。又はJIS K 0950に規定するプラスチック製滅菌シャーレの
90B。
(d) 三角フラスコ 300〜500ml及び1 000〜2 000ml。培地及び希釈水の調製に用いる。綿栓をして(3)(a)
の乾熱滅菌又は(3)(b)の高圧蒸気滅菌をしておく。
(e) 綿栓 脱脂をしていない,繊維の長い良質の木綿わたを,試験管類及びフラスコ類の綿栓として用
いるか,又は合成樹脂製,金属製及びシリコーン製の栓を用いてもよい。
(f) 集落計数器 1.5倍の拡大鏡を備えたもの。
(g) 培養器(ふ卵器) JIS T 1702に規定するふ卵器 36±1℃に調節できるもの。
(h) 乾熱滅菌器 160〜200℃に調節できるもの。
(i) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので121℃以上に加熱でき,196kPaの器
内圧力で使用できるもの。
(j) 平圧蒸気滅菌器 100℃に加熱でき,大気圧 (101.325kPa) で使用できるもの。(i)の高圧蒸気滅菌器
を器内圧力を101.325kPa,100℃で使用してもよい。
(3) 器具などの滅菌操作 器具などの滅菌操作は,次のとおり行う。
(a) 乾熱滅菌 ガラス製及び金属製器具類の滅菌に用いる。170℃で1時間滅菌する。
(b) 高圧蒸気滅菌 培地,希釈水,JIS K 8637に規定するチオ硫酸ナトリウム五水和物を入れた採水瓶,
使用済み培地などの滅菌に用いる。121℃で30分間滅菌する。
(c) 蒸気(間欠蒸気)滅菌 糖類を加えた培地など高圧蒸気滅菌ができない場合に用いる。器内圧
101.325kPa,100℃で約30分間ずつ3回(3日間)連続して滅菌する。高圧蒸気滅菌器を利用できる。
(d) 火炎滅菌 試験管及びフラスコの口部などの滅菌に用いる。試験管及びフラスコは,培養操作の前
後に斜めに持って回しながら口部を火炎の中にしばらく入れて滅菌する。
278
K 0101 : 1998
(4) 消毒操作 消毒操作は,次のとおり行う。
(a) 試験操作の前後には,手指及び実験台を消毒する。手指の消毒には,クレゾール石けん溶液 (10g/l) ,
陽性石けん溶液 (1〜10g/l) ,消毒用アルコール[エタノール (80vol%) ]を用いる。
実験台は,陽性石けん溶液 (10g/l) ,フェノール溶液 (30〜50g/l) などを噴霧するか,これらを含
ませた布でぬぐって消毒する。
(b) 使用済みのピペット,採水瓶,希釈瓶などの器具はクレゾール石けん溶液 (30〜50g/l) などの消毒
液中に1日間浸した後,消毒液が完全に除去されるまで,よく水洗する。
(c) 培養試験後の試験管,ペトリ皿類は培地ごと(3)(b)の高圧蒸気滅菌を行った後,培地を捨ててからよ
く水洗する。
(5) 試料の希釈 試料の希釈は,次のとおり行う。
(a) 試料中の一般細菌数が,試料1ml中に300個以上あると予想される場合には,試料を十分に振り混
ぜて均一にした後,その1mlをメスピペットでとり,希釈水9ml又は99mlを入れた希釈瓶に加え
てよく振り混ぜる。
(b) 次に,その1mlをとり(a)と同じ操作で数段階の希釈を行い,培養後30〜300個の範囲で集落が得ら
れるような希釈試料を調製する。
なお,メスピペットは,その都度滅菌済みのものを用いる。
(6) 操作 操作は,次のとおり行う。
(a) 63.1(2)によって採取した試料又は(5)で調製した希釈試料1mlずつをメスピペットを用いてそれぞれ
2枚以上のペトリ皿にとる。
(b) 標準寒天培地を水浴中で加熱して溶かした後,約50℃に保っておき,その約15mlを無菌的にそれ
ぞれのペトリ皿に加え,固まらないうちに,揺り動かしてよく混合する。
(c) ペトリ皿全体に培地と試料の混合物が広がったら,水平の状態で放置し,培地が固まってから,ペ
トリ皿にふたをし,逆さにして培養器に入れる。
(d) 36±1℃で24±2時間培養する。
(e) 培地上に発生した集落を集落計数器を用いて数え,平均値を求めて試料1ml中の個数(個/ml)で表
す。希釈試料の場合は,30〜300個の集落数が得られたものを取り出し,希釈倍数を乗じて試料1ml
中の個数を求める。
菌数が100以上の場合には,有効数字2けたに数値を丸める。
備考1. 培地の取扱い
(1) 調製した培地の保存及び無菌試験 滅菌した培地は,水分の蒸発を防いで冷暗所に保存す
る。長時間経過したものは用いない。使用前に一夜培養器に入れ,雑菌が混入していない
ことを確認する。
(2) 使用済み培地の処理 細菌培養後の使用済み培地は,必ずペトリ皿のまま,高圧蒸気滅菌
してから廃棄する。
63.3 従属栄養細菌 従属栄養細菌は,標準寒天培地を用いて25±1℃で5日間培養したとき,培地上に集
落を形成する生菌をいい,試料1ml中又は1l中の個数で表示する。
(1) 試薬及び培地 試薬及び培地は,次のものを用いる。
(a) 水 63.2(1)(a)による。
(b) 希釈水 63.2(1)(b)による。
(c) 標準寒天培地(2) 63.2(1)(c)による。
279
K 0101 : 1998
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ピペット 1〜10ml。硫酸紙などに包むか,ピペット滅菌箱に先端を先にして入れ,(3)(a)の乾熱滅
菌をしておく。
(b) メスシリンダー 容量100〜1 000ml。口部を硫酸紙などに包んで,(3)(b)の高圧蒸気滅菌又は(3)(c)
の蒸気滅菌をしておく。
(c) ピンセット 先端が滑らかなもの。使用直前に(3)(d)の火炎滅菌をする。
(d) 双眼実体顕微鏡 6〜80倍のもの。
(e) 集落計数器 63.2(2)(f)による。
(f) ろ過装置(分離形) 16.1(1)(a)による。ろ過装置の各部を硫酸紙などに包んで,材質に適した滅菌
をしておく。
(g) ろ過膜 孔径0.45μm,直径約50mmの有機性のもので,ろ過膜に方眼紙と同様な画線が印刷された
もの。
硫酸紙などに包み,ガラス製ペトリ皿に入れて(3)(b)の高圧蒸気滅菌を行う。又は滅菌済みの市販
品を用いてもよい。ろ過膜の取扱には,ピンセットを用いる。
(h) ペトリ皿 63.2(2)(c)による。
(i) 培養器(ふ卵器) JIS T 1702に規定するふ卵器 25±1℃に調節できるもの。
(j) 乾熱滅菌器 63.2(2)(h)による。
(k) 高圧蒸気滅菌器 63.2(2)(i)による。
(3) 器具などの滅菌操作 器具などの滅菌操作は,次のとおり行う。
(a) 乾熱滅菌 63.2(3)(a)による。
(b) 高圧蒸気滅菌 63.2(3)(b)による。
(c) 蒸気(間欠蒸気)滅菌 63.2(3)(c)による。
(d) 火炎滅菌 63.2(3)(d)による。
(4) 消毒方法 63.2(4)による。
(5) 操作 操作は,次のとおり行う。
(5.1) 試料中の細菌数が多いと予想される場合
(a) 63.2(6)(a)〜(c)の操作を行う。
(b) 25±1℃で5日間培養する。
(c) 培地上に発生した集落を集落計数器を用いて数え,平均値を求めて試料1ml中の個数(個/ml)で表
す。希釈試料の場合は,30〜300個の集落数が得られたものを取り出す。
(d) 次の式によって試料1ml中の従属栄養細菌数を算出する。
V
n
a
1
×
=
ここに, a: 従属栄養細菌数(個/ml)
n: 培地上の集落数(個)
V: 試料 (ml)
菌数が100以上の場合には,有効数字2けたに数値を丸める。
(5.2) 試料中の細菌数が少ないと予想される場合
(a) 63.1(2.2)によって細菌をろ過膜に捕集(3)する。
(b) 標準寒天培地を水浴中で加熱して溶かした後,約50℃に保ちながら,その約15mlを無菌的にペト
280
K 0101 : 1998
リ皿にとる。水平に放置し,培地を固まらせる。
(c) ろ過装置から,ピンセットを用いてろ過膜を外して(b)のペトリ皿の培地上に捕集面を上にして密着
させる。このときろ過膜と培地の間に気泡が入らないように注意する。
(d) ペトリ皿にふたをし,逆さにして培養器に入れ,25±1℃で5日間培養する。
(e) 培養後,双眼実体顕微鏡(又は集落計数器)を用いてろ過膜上に発生した集落を計数する。
(f) 次の式によって試料1l中の従属栄養細菌数を算出する。
V
n
a
000
1
×
=
ここに, a: 従属栄養細菌数(個/l)
n: 有機性ろ過膜上の集落数(個)
V: 試料 (ml)
注(3) 試料の量は,培養後の集落数が30〜300個になるようにするが,試料のろ過量を段階的(例えば,
10,100,1 000mlなど)に調節するとよい。
備考2. 培地の取扱いは,備考1.による。
3. 標準寒天培地の代わりにM-TGE寒天培地(M-TGE液体培地)又は標準液体培地を用いても
よい。
M-TGE寒天培地は,次のように調製する。
トリプトン5.0g,肉エキス6.0g,JIS K 8824に規定するD (+) -グルコース2.0g及びJIS K
8263に規定する寒天(粉末)15gを水1lに加え,加熱して溶かし,滅菌後のpHが7.0±0.1
になるように調節する。次に,三角フラスコに入れて(3)(b)の高圧蒸気滅菌を約15分間行っ
た後,冷暗所に保存する。液体培地を調製する場合には,寒天の添加を省略する。
液体培地を用いて試験する場合には,次のように操作する。
ピンセットを用いて,吸収パッド(液体培地1.8〜2.2mlを含むことができるもの。)を1
枚ずつペトリ皿に入れ,吸収パッドが標準液体培地又はM-TGE液体培地で十分に潤うよう
に,標準液体培地又はM-TGE液体培地を加える(通常は2ml程度でよい。)。ピンセットを
用いて細菌を捕集したろ過膜を外してペトリ皿の吸収パッドの上に,捕集面を上にして密着
させる。この際,ろ過膜と吸収パッドとの間に気泡が残らないように注意する。以下,(c)〜
(f)の操作を行う。
63.4 大腸菌群 大腸菌群は,ラクトースを分解して気体と酸を生成する好気性(又は通性嫌気性)の菌
をいい,ラクトース-ブイヨン培地による推定試験とブリリアントグリーン-ラクトース胆汁ブイヨン培地
による確定試験によって判定する。
(1) 試薬及び培地 試薬及び培地は,次のものを用いる。
(a) 水 63.2(1)(a)による。
(b) 2倍濃厚ラクトース-ブイヨン培地(2倍濃厚LB培地)(2) 肉エキス3g,ペプトン5g,JIS K 8728
に規定するラクトース一水和物5gを水500mlに加え,加熱して溶かした後,滅菌後のpHが7.0±
0.2になるように調節し,これにブロモチモールブルー溶液 (2g/l) (4)12mlを加える。これを(2)(b)の
ダーラム発酵管(中試験管)に約10mlずつ移し入れ,(3)(b)の高圧蒸気滅菌を約15分間行って手早
く冷水中に浸して冷却した後,冷暗所に保存する。ダーラム管内に気泡が残留したものは使用しな
い。
(c) プリリアントグリーン-ラクトース胆汁ブイヨン培地(BGLB培地)(2) ペプトン10g,JIS K 8728
281
K 0101 : 1998
に規定するラクトース一水和物10gを,水約500mlに溶かし,別に,乾燥した牛胆汁(粉末)20g
を水約200mlに溶かしたもの (pH7.0〜7.5) を加え,更に水を加えて約970mlとし,pHを7.4に調
節する。次に,ブリリアントグリーン溶液 (1g/l) (5)13.3ml及び水を加えて1lとし,脱脂綿などでろ
過して,(2)(b)のダーラム発酵管(小試験管)に3〜4mlずつ移し入れ,(3)(b)の高圧蒸気滅菌を約
15分間行って手早く冷水中に浸し,冷却した後,冷暗所に保存する。ダーラム管内に気泡が残留し
たものは使用しない。
注(4) JIS K 8842に規定するブロモチモールブルー0.2gを水約50ml中に加え,水酸化ナトリウム溶液
(0.1mol/l) 5mlを加えた後,約50℃に加熱して溶かし,放冷後,水を加えて100mlとする。
(5) ブリリアントグリーン0.1gを水に溶かして100mlとする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) メスピペット 10ml 硫酸紙などに包んで,ピペット滅菌箱に先端を先にして入れ,(3)(a)の乾熱滅
菌を行う。
(b) ダーラム発酵管 小試験管(外径約11mm,高さ約150mm)又は中試験管(外径約15mm,高さ約
150mm)に,外径約10mm,高さ約20mmのダーラム管(ガラス管の一端を封じたもの)の管口を
下にして入れ,綿栓又はキャップをしたもの。
図63.2 ダーラム発酵管(中試験管)の一例
(c) 白金耳 直径0.7〜0.8mm,長さ約80mmの白金又はニクロム線の先端を内径2〜5mmの輪にして,
他端を柄に挿入したもの。使用の前後には(3)(d)の火炎滅菌を行う。
(d) 培養器(ふ卵器) 63.2(2)(g)による。
(e) 乾熱滅菌器 63.2(2)(h)による。
(f) 高圧蒸気滅菌器 63.2(2)(i)による。
(3) 器具などの滅菌操作 器具などの滅菌操作は,次のとおり行う。
(a) 乾熱滅菌 63.2(3)(a)による。
(b) 高圧蒸気滅菌 63.2(3)(b)による。
(c) 蒸気(間欠蒸気)滅菌 63.2(3)(c)による。
(d) 火炎滅菌 63.2(3)(d)による。細菌の移植時に使用する白金耳の滅菌は,白金耳を火炎中に入れて赤
熱するまで加熱する。
(4) 消毒方法 63.2(4)による。
(5) 推定試験 推定試験は,次のとおり行う。
(a) 63.1によって採取した試料10mlずつをメスピペットで5本の2倍濃厚ラクトース-ブイヨン培地を
282
K 0101 : 1998
入れたダーラム発酵管(中試験管)に加える。
(b) これを培養器に入れ,36±1℃で24±2時間培養する。
(c) 気体の発生を認め,かつ,培地の色が黄色に変わったものを推定試験陽性とし,(6)の確定試験を行
う。
(d) 気体の発生が認められないものは,更に培養を続けて48±3時間まで延長する。
(e) 気体の発生を認め,かつ,培地の色が黄色に変わったものを推定試験陽性とし,(6)の確定試験を行
う。気体の発生が認められないものは,大腸菌群陰性と判定する。
(6) 確定試験 確定試験は,次のとおり行う。
(a) 推定試験陽性のものは,その1白金耳量の菌液をブリリアントグリーン-ラクトース胆汁ブイヨン培
地(BGLB培地)を入れたダーラム発酵管(小試験管)に移植する。
(b) これを培養器に入れ,36±1℃で24±2時間又は48±4時間培養する。
(c) 気体の発生が認められないものは大腸菌群陰性,気体の発生が認められたものは確定試験陽性と判
定(6)する。
注(6) 確定試験陽性と認められたダーラム発酵管の数から最確数表によって,大腸菌群数を推定する
ことができる。
63.5 ふん便性大腸菌群 ふん便性大腸菌群は,大腸菌群のうちEC培地又はM-FC培地で44.5±0.2℃で
24±2時間培養したとき,気体を発生するか,集落を形成するものをいう。
(1) 試薬及び培地 試薬及び培地は,次のものを用いる。
(a) 水 63.2(1)(a)による。
(b) EC培地(2) トリプトース20g,JIS K 8728に規定するラクトース一水和物5g,胆汁酸塩 (No.3) 1.5g,
JIS K 9007に規定するりん酸二水素カリウム1.5g,JIS K 9017に規定するりん酸水素二カリウム4g
及びJIS K 8150に規定する塩化ナトリウム5gを水1lに溶かし,滅菌後のpHが6.9になるように調
節した後,63.4(2)(b)のダーラム発酵管(中試験管)に約10mlずつ移し入れ,(3)(b)の高圧蒸気滅菌
を15〜20分間行って手早く冷水中に浸して冷却し,冷暗所に保存する。ダーラム管内に気泡が残留
しているものは使用しない。
(c) M−FC培地(2) トリプトース10g,プロテオーゼペプトン(又はポリペプトン)5g,酵母エキス
(粉末)3g,JIS K 8728に規定するラクトース一水和物5g,胆汁酸塩 (No.3) 1.5g,JIS K 8150に規
定する塩化ナトリウム5g,アニリンブルー0.1g及びJIS K 8263に規定する寒天(粉末)15gを水1l
に加え,加熱して溶かし,約45℃に冷却しpHを7.4に調節する。直ちにその15〜20mlをペトリ皿
に流し込み,固まらせておく。この培地は(3)(b)の高圧蒸気滅菌は行わず,96時間以内に使用する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) ピペット 63.3(2)(a)による。
(b) メスシリンダー 63.3(2)(b)による。
(c) ピンセット 63.3(2)(c)による。
(d) 双眼実体顕微鏡 63.3(2)(d)による。
(e) 集落計数器 63.2(2)(f)による。
(f) ろ過装置(分離形) 63.3(2)(f)による。
(g) ろ過膜 63.3(2)(g)による。
(h) ペトリ皿 63.2(2)(c)による。
(i) ダーラム発酵管 63.4(2)(b)による。
283
K 0101 : 1998
(j) 白金耳 63.4(2)(c)による。
(k) 培養器(ふ卵器) JIS T 1702に規定するふ卵器 44.5±0.2℃に調節できるもの。
(l) 乾熱滅菌器 63.2(2)(h)による。
(m) 高圧蒸気滅菌器 63.2(2)(i)による。
(3) 器具などの滅菌操作 器具などの滅菌操作は,次のとおり行う。
(a) 乾熱滅菌 63.2(3)(a)による。
(b) 高圧蒸気滅菌 63.2(3)(b)による。
(c) 蒸気(間欠蒸気)滅菌 63.2(3)(c)による。
(d) 火炎滅菌 63.4(3)(d)による。
(4) 消毒方法 63.2(4)による。
(5) 操作 操作は,次のとおり行う。
(5.1) EC培地を用いる場合
(a) 63.4(5)(a)〜(e)の推定試験を行って陽性と判定したダーラム発酵管から,その1白金耳量をEC培地
に移植する。
(b) これを培養器に入れ,44.5±0.2℃で24±2時間培養する。
(c) 気体の発生が認められたものを,ふん便性大腸菌群陽性とする。気体の発生が認められないものは,
ふん便性大腸菌群陰性と判定する。
(5.2) M-FC培地を用いる場合
(a) JIS K 0550の4.4の操作を行ってろ過膜に細菌を捕集(3)する。また,試料が容器に入れられている
場合には,ろ過装置(分離形)を用いてろ過膜に細菌を捕集(3)する。
(b) M-FC寒天培地を水浴中で加熱して溶かした後,約50℃に保ちながら,その約15mlを無菌的にペト
リ皿にとる。水平に放置し,培地を固まらせる。
(c) ろ過装置から,ピンセットを用いてろ過膜を外して,(b)のペトリ皿の培地上に捕集面を上にして密
着させる。このときろ過膜と培地の間に気泡が入らないように注意する。
(d) ペトリ皿にふたをし,逆さにして培養器に入れ,44.5±0.2℃で24±2時間培養する。
(e) 双眼実体顕微鏡(又は集落計数器)を用いてろ過膜上に発生した青い集落を確認すればふん便性大
腸菌群陽性と判定する。これを計数してふん便性大腸菌群数とする。
備考4. 培地の取扱いは,備考1.による。
64. 生物試験 生物試験は,特殊な細菌類,藻類,原生動物などに区分し,光学顕微鏡を用いて試験する。
なお,特殊な細菌類については,必要に応じて培養試験を並行して行う。
この試験は原則として,試料採取後直ちに行う。直ちに試験ができない場合には,ホルムアルデヒドを
加え0〜5℃(凍結させない。)の暗所に保存しできるだけ早く試験する。
64.1 生物試験 生物試験は,主として光学顕微鏡を用いて観察し,生物分類図鑑などの資料によってそ
れぞれの属,種を決定する。
なお,肉眼的な大形生物についてもこの試験に含める。
(1) 試薬 試薬は,次のものを用いる。
(a) 固定用ホルムアルデヒド液 JIS K 8872に規定するホルムアルデヒド液(ホルマリン)。
(2) 器具 器具は,次のとおりとする
(a) 試料容器 共栓広口瓶。容量100〜500mlのガラス製又はポリエチレン製のもの。
284
K 0101 : 1998
(b) 駒込ピペット 1,5及び10ml
(c) メスピペット 1,5及び10ml
(d) メスシリンダー 10〜50ml及び100〜500ml
(e) 三角フラスコ 63.2(2)(d)による。
(f) 希釈瓶 63.2(2)(b)による。
(g) ピンセット 先端がへら状のもの
(h) プランクトンネット 網目をふるい絹NXX13番又はポリアミド繊維(ナイロン)NXX13番で張っ
たもの。図64.1に一例を示す。
図64.1 プランクトンネットの一例
(i) 遠心分離器 回転数3 000〜4 000min-1が得られるもの。
(j) 沈殿管 0.1〜0.2mlごとに標線の入った容量10〜30mlで先端を細くしたもの。
(k) 遠心分離管 1ml又は10mlごとに標線の入った容量10ml又は50mlのガラス製のもの。
(l) スライドガラス JIS R 3703に規定する品質2種の標準形 (76.0×26.0mm) 。
(m) 界線入りスライドガラス 1mmの間隔で界線が刻まれている75×36mmのもの。図64.2に一例を
示す。
図64.2 界線入りスライドガラスの一例
(n) カバーガラス JIS R 3702に規定する品質2種(18×18mm,24×24mm又は32×24mm)のもの。
(o) ペトリ皿 63.2(2)(c)による。
(p) 双眼実体顕微鏡 63.3(2)(d)による。
(q) 光学顕微鏡及びその附属品 光学顕微鏡は,総合倍率100〜1 000倍が得られるもので,次のものか
285
K 0101 : 1998
らなる。
(i)
光学顕微鏡 JIS B 7132に規定する液浸系レンズ用生物顕微鏡。微細な藻類,鉄バクテリアその
他の細菌などの観察には,位相差装置を用いると便利である。
(ii)
対物レンズ JIS B 7147に規定する液浸対物レンズ。呼び倍率90以上のもの。
(iii) 接眼レンズ JIS B 7148に規定する呼び倍率5〜15のもの。
(iv)
十字標本移動器(メカニカルステージ) 光学顕微鏡に取り付け,界線入りスライドガラスを前
後左右に移動できるもの。
(3) 試料採取 試料の採取は,次のとおり行う。
(a) 給水栓又は吐水口からの採取 生物の量が多い場合には,試料を100〜500mlを試料容器に採取す
る。生物の量が少ないときは,一定量の試料をプランクトンネットでろ過して集め,水とともに試
料容器に移す。
(b) 水槽,貯水池,冷却塔などからの採取 (a)と同様に操作して採取する。水中に浮遊するフロック,
壁面や構造物などに付着したり,水底に沈殿しているものは,駒込ピペット,ピンセット,ハンド
ネットなどを用いて水とともに試料容器中に採取する。
さびこぶなどは,へらでできるだけ崩さないように注意して水とともに試料容器に採集する。
(c) 取水口,導水路などからの採取 (b)と同様に操作して採取する。直ちに試験ができない場合は,こ
れに固定用ホルムアルデヒド液を,試料1lにつき約90mlを加える。
(4) 操作 操作は,次のとおり行う。
(a) (3)で採取した試料の少量をスライドガラスにとり,カバーガラスを載せて光学顕微鏡で観察し,主
体となる生物から順次にその種(属)を記録していく。生物量の少ない試料の場合には,一定量の
試料を遠心分離管にとり,遠心分離して濃縮したものについて操作する。
(b) 定量的に採取した試料は,沈殿管又はメスシリンダーに移して十分に沈降させた後,その沈殿物の
量を読み取る。
(c) 次に,(b)の試料をよくかき混ぜた後,一定量を界線入りスライドガラスにとり,(a)と同じ操作で光
学顕微鏡で観察しながら生物ごとに計数し,試料1ml又は1l中の生物数を算出する。
(d) 大形の生物は,試料をペトリ皿に移し,形状,構造などの特徴を実体顕微鏡を用いて観察する。
64.2 細菌類 細菌類の試験は,光学顕微鏡で観察し細胞の形態,大きさ,さや(鞘)の有無,鉄の沈着
などによって,ズーグレア,硫酸塩還元菌,硫黄バクテリア,スフェロチルス,鉄バクテリア,真菌など
に大別する。必要に応じて培養試験を併用する。
鉄バクテリア以外は,有機物汚染の進んだ水域で一般に見られるもので,汚染指標生物とされている。
(1) 試薬及び培地 試薬及び培地は,次のものを用いる。
(a) 水 63.2(1)(a)による。
(b) 標準寒天培地(1) 63.2(1)(c)による。
(c) 改良ISA培地(1) トリプトン1.0g,乳酸ナトリウム (70%) 5g,JIS K 8061に規定する亜硫酸ナトリ
ウム0.5g,くえん酸鉄 (III) アンモニウム0.5g,JIS K 8995に規定する硫酸マグネシウム七水和物
2g及びJIS K 8978に規定する硫酸鉄 (II) 七水和物0.5gを水1lに溶かし,滅菌後のpHが7.3にな
るように調節し,(3)(b)の高圧蒸気滅菌を約15分間行って手早く冷水中で冷却し,なるべく早く使
用する。
(d) ストーク培地(Stork's培地)(1) JIS K 8824に規定するD (+) -グルコース1g,JIS K 8995に規定
する硫酸マグネシウム七水和物0.2g,JIS K 8142に規定する塩化鉄 (III) 六水和物10mg,ペプトン
286
K 0101 : 1998
1g,JIS K 8122に規定する塩化カルシウム二水和物50mg及びJIS K 8263に規定する寒天(粉末)
12.5gを水1lに加え,加熱して溶かし,滅菌後のpHが7.2になるように調節し,(3)(b)の高圧蒸気
滅菌を約15分間行う。
(e) ワックスマン培地(Waksman培地)(1) ペプトン5g,JIS K 8824に規定するD (+) -グルコース10g,
JIS K 9007に規定するりん酸二水素カリウム1g,JIS K 8995に規定する硫酸マグネシウム七水和物
0.5g及びJIS K 8263に規定する寒天(粉末)15gを水1lに加え,加熱して溶かし,pHを3.8〜4.0
に調節して(3)(b)の高圧蒸気滅菌を110℃で約15分間行う。
なお,細菌の発育を抑制するためには培地1lにつきローズベンガル(アシッドレッド94)35mg
を加え,更に培養するときにオーレオマイシンを培地1lにつき35mgを加える。
(f) ツァペック・ドックス培地(Czapek Dox培地)(1) JIS K 8383に規定するスクロース30g,JIS K
8562に規定する硝酸ナトリウム3g,JIS K 9017に規定するりん酸水素二カリウム1g,JIS K 8121
に規定する塩化カリウム0.5g,JIS K 8995に規定する硫酸マグネシウム七水和物0.5g,JIS K 8978
に規定する硫酸鉄 (II) 七水和物0.01g及びJIS K 8263に規定する寒天(粉末)15gを水1lに加え,
加熱して溶かし,滅菌後のpHが7.3になるように調節し,(3)(b)の高圧蒸気滅菌を約15分間行う。
(g) ポテトグルコース寒天培地(1) ポテトエキス4g,JIS K 8824に規定するD (+) -グルコース20g及
びJIS K 8263に規定する寒天(粉末)15gを水1lに加え,加熱して溶かし,滅菌後のpHが6.0〜7.0
になるように調節した後,(3)(b)の高圧蒸気滅菌を約15分間行う。
注(1) 63.の注(2)による(備考1.参照)。
備考1. 63.の備考1.による。
(2) 器具 器具は,次のものを用いる。
(a) メスピペット 64.1(2)(c)による。
(b) 駒込ピペット 64.1(2)(b)による。
(c) メスシリンダー 10〜25ml及び50〜500ml
(d) 三角フラスコ 63.2(2)(d)による。
(e) 希釈瓶 63.2(2)(b)による。
(f) ペトリ皿 63.2(2)(c)による。
(g) ピンセット 64.1(2)(g)による。
(h) カバーガラス 64.1(2)(n)による。
(i) スライドガラス 64.1(2)(l)による。
(j) 界線入りスライドガラス 64.1(2)(m)による。
(k) 双眼実体顕微鏡 64.1(2)(p)による。
(l) 光学顕微鏡及びその附属品 64.1(2)(q)による。
(m) 培養器 63.2(2)(g)による。ただし,温度が25〜37℃に任意に調節できるもの。
(3) 器具などの滅菌操作 器具などの滅菌操作は,次のとおり行う。
(a) 乾熱滅菌 63.2(3)(a)による。
(b) 高圧蒸気滅菌 63.2(3)(b)による。
(c) 蒸気(間欠蒸気)滅菌 63.2(3)(c)による。
(d) 火炎滅菌 63.2(3)(d)による。
(4) 消毒方法 63.2(4)による。
(5) 操作 操作は,次のとおり行う。
287
K 0101 : 1998
(5.1) ズーグレア (Zoogloea) ズーグレアは,大きさ1× (2.0〜4.0) μmのかん(桿)菌が多数集合した
もので,ゼラチン状物質に包まれて樹枝状,手指状又は不定形の菌体集落(大きさ500〜1 000μm)
を形成したものである。スライム及びフロックの主体になっていることが多い。
Zoogloea ramigera, Zoogloea filipenduiaの種が知られている。
(a) 試料の一定量を界線入りスライドガラスにとり,64.1(4)(a)と同様に操作して,光学顕微鏡を用いて
観察する。
(b) 各視野に出現するズーグレアの数を計数し,試料1ml中の数を算出する。
(5.2) 硫酸塩還元菌 硫酸塩還元菌は,大きさ (0.5〜1.0) × (2.5〜5.0) μmの湾曲したかん(桿)状又は
らせん状を呈し,活発に運動する菌体である。
(a) 試料の一定量を界線入りスライドガラスにとり,64.1(4)(a)と同様に操作して,光学顕微鏡を用いて
観察する。
(b) 各視野に出現する硫酸塩還元菌を生物図鑑などの資料を参照して,硫酸塩還元菌の種とその数を次
のように記録する(2)。
+
極めてわずかに出現(出現率10%以下)
++
わずかに出現(出現率20〜30%)
+++
普通に出現(出現率40〜60%)
++++
多く出現(出現率70〜80%)
+++++
極めて多く出現(出現率90%以上)
(c) 培養は,共栓試験管に改良ISA培地(3)を入れ(共栓試験管の容量の約21量),これに試料の適量を加
え,更に改良ISA培地を満杯になるまで加え,気泡が残らないように注意しながら密栓する。これ
を30℃で5〜7日間嫌気培養(4)を行う。
(d) 培地の底部又は培地全体(3)が黒に変色したものを硫酸塩還元菌陽性と判定する。
注(2) スライドガラスの界線ごとに硫酸塩還元菌を類別して計数する。この操作を3回繰り返して行っ
た後,次の式によって試料1ml中の硫酸塩還元菌の種とその数を算出する。
n
a
a
a
A
3
10
)
(
3
2
1
×
+
+
=
ここに,
A: ズーグレアの数(固体数/ml)
a1, a2, a3: 各回で計数したズーグレアの固体数
n: 試料の濃縮倍数
(3) (1)(c)の改良ISA培地を調製する際に,JIS K 8263に規定する寒天(粉末)15gを加えて調製し
た改良ISA寒天培地を用いて試験してもよい。この場合は,63.2(6)(a)〜(c)の操作に準じて操作
し,培養すれば,黒い色の集落を形成するので菌数を求めることができる。
(4) 嫌気ジャーを用いるとよい。
備考2. 硫酸塩還元菌は,有機物汚染を受け,嫌気性状態になっている水中及び底泥中に生息し,硫
酸塩を還元して硫化水素を生成する偏性嫌気性菌である。工業用水からもしばしば検出され,
金属類の腐食に関与することがある。Desulfovibrio属がよく検出される。
(5.3) スフェロチルス (Sphaerotilus) スフェロチルスは,透明なさや(鞘)の中に1× (2.0〜6.0) μmの
円筒状のかん菌が一列に並んだ糸状体を形成している。この糸状体は,偽分岐することが大きな特
徴である。
(a) 試料の一定量を界線入りスライドガラスにとり,64.1(4)(a)と同様に操作して,光学顕微鏡を用いて
288
K 0101 : 1998
観察する。
(b) 各視野に出現するスフェロチルスを生物図鑑などの資料を参照して,スフェロチルスの種とその数
を(5.2)(b)のように記録する(5)。
(c) 培養は,ストークス培地又は標準寒天培地を用いて,63.2(6)(a)〜(c)の操作に準じて操作し,25℃で
5日間培養する。
(d) 形成した集落をとって,光学顕微鏡で観察し,透明なさやの存在を確認した場合には,スフェロチ
ルス陽性とする。
注(5) スライドガラスの界線ごとにスフェロチルスを類別して計数する。この操作を3回繰り返して行
った後,注(2)の式によって試料1ml中のスフェロチルスの種とその数を算出する。
備考3. スフェロチルスは,通常,汚水細菌又はミズワタと呼ばれるもので,有機物を多量に含む水
及び工業用水中でスライムやフロックを形成することが多い。Sphaerotilus natansが代表的な
ものとして知られている。
(5.4) 鉄バクテリア 鉄バクテリアは,特徴的な形態を示す茶褐色の菌体で,幅1〜1.5μmのリボン状を
呈するものは,ガリオネラ (Gallionella ferruginea) である。また,幅約2μmの長い直線状のさやに
長かん菌が一列に並んでいるのはレプトスリックス (Leptothrix ochracea) である。これらが代表的
な鉄バクテリアである。
(a) 試料の一定量を界線入りスライドガラスにとり,64.1(4)(a)と同様に操作して,光学顕微鏡を用いて
観察する。
(b) 各視野に出現する鉄バクテリアの種を生物図鑑などの資料を参照して,その種とその数を(5.2)(b)の
ように記録する(6)。
注(6) スライドガラスの界線ごとに鉄バクテリアを類別して計数する。この操作を3回繰り返して行っ
た後,注(2)の式によって試料1ml中の鉄バクテリアの種とその数を算出する。
備考4. 鉄バクテリアは,地下水,伏流水及びゆう(湧)水中に生息するもので,鉄 (II) を酸化して
鉄 (III) とし,赤水の生成及びスライムの形成の原因となる。
(5.5) 硫黄バクテリア 硫黄バクテリアは,その細胞の大きさは3× (2.5〜5) μmで,分岐しない長い糸状
体を形成する。この糸状体は,灰白色の薄い膜(くもの巣状)状となる。スフェロチルス及びオシ
ラトリア(らん藻類)と形態が類似しているが,糸状体の中に硫黄粒が多数認められるので,区別
ができる。
(a) 試料の一定量を界線入りスライドガラスにとり,64.1(4)(a)と同様に操作して,光学顕微鏡を用いて
観察する。
(b) 各視野に出現する硫黄バクテリアの種を生物図鑑などの資料を参照して,その種とその数を(5.2)(b)
のように記録する(7)。
注(7) スライドガラスの界線ごとに硫黄バクテリアを類別して計数する。この操作を3回繰り返して行
った後,注(2)の式によって試料1ml中の硫黄バクテリアの種とその数を算出する。
備考5. 工業用水で障害を生じる代表的なものは,ベギアトア (Beggiatoa alba) である。
(5.6) 真菌 真菌の試験は,ポテトグルコース寒天培地,ワックスマン寒天培地又はツァペック・ドック
ス寒天培地を用いて培養し,胞子を形成させ,菌糸,分生子(柄)などとともにこれらの着生状態
を光学顕微鏡で観察して,生物分類図鑑などから属,種を同定する。
(a) 64.1(3)で採取した試料の適量をとり,63.2(6)(a)〜(c)の操作を行う。ただし,標準寒天培地に代え,
ポテトグルコース寒天培地,ワックスマン寒天培地又はツァペック・ドックス寒天培地を用いる。
289
K 0101 : 1998
(b) 20〜25℃で5〜7日間培養する。
(c) 真菌特有の着色した綿状の集落が培地上に発育する。光学顕微鏡を用いて,胞子の形状,菌糸,分
生子(柄)などを観察して,生物分類図鑑などを参照して属,種とその数を(5.2)(b)のように記録す
る(8)。
注(8) スライドガラスの界線ごとに真菌を類別して計数する。この操作を3回繰り返して行った後,注
(2)の式によって試料1ml中の真菌の種とその数を算出する。
備考6. 真菌類は,種が非常に多く,水中ばかりでなくあらゆる環境中に存在しているので,培養す
る場合には,環境からの汚染を受けないように細心の注意が必要である。
真菌類は,べん(鞭)毛菌類,藻菌類,子のう(嚢)菌類,接合菌類,不完全菌類などに
大別されるが,水中では胞子を形成しないものが多いので,光学顕微鏡で菌糸だけが観察さ
れても種を同定することができない。そのため寒天培地で培養して試験する。
64.3 藻類 藻類の試験は,光学顕微鏡を用いて観察し,藻類図鑑などの資料を参照して,緑藻類,らん
(藍)藻類,けい藻類,紅藻類などに区分し,それぞれの属,種を決める。
(1) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 駒込ピペット 64.1(2)(b)による。
(b) 遠心分離器 64.1(2)(i)による。
(c) 遠心分離管 64.1(2)(k)による。
(d) カバーガラス 64.1(2)(n)による。
(e) スライドガラス 64.1(2)(l)による。
(f) 界線入りスライドガラス 64.1(2)(m)による。
(g) 光学顕微鏡及びその附属品 64.1(2)(q)による。
(2) 操作 操作は,次のとおり行う。
(a) 64.1(4)(a)と同様に操作して,光学顕微鏡を用いて観察する。
(b) 細胞の大きさ,形状及び配列,色素体の色調及び形状などから,藻類図鑑などの資料を参考にして,
各視野に出現する藻類の属及び種を同定し,その数を(5.2)(b)のように記録する。
(c) 定量的に採取した試料の場合には,試料1ml中又は1l中のそれぞれの藻類数を算出する(9)。
注(9) スライドガラスの界線ごとに藻類を類別して計数する。この操作を3回繰り返して行った後,注
(2)の式によって試料1ml中又は1l中のそれぞれの藻類数を算出する。
64.4 動物 水中の動物は,顕微鏡的な小形のものから,肉眼的なものまで幅広く存在するので,それぞ
れの特徴をよく光学顕微鏡を用いて観察し,生物図鑑などを参照して,それぞれの属及び種を同定する。
(1) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 駒込ピペット 64.1(2)(b)による。
(b) 遠心分離器 64.1(2)(i)による。
(c) 遠心分離管 64.1(2)(k)による。
(d) カバーガラス 64.1(2)(n)による。
(e) 界線入りスライドガラス 64.1(2)(m)による。
(f) 光学顕微鏡及びその附属品 64.1(2)(q)による。
(2) 操作 操作は,次のとおり行う。
(a) 64.1(4)(a)及び(b)の操作を行った試料をよくかき混ぜた後,その一定量(例えば,0.1ml)を界線入
りスライドガラスにとり,カバーガラス (24×32mm) をかけ,光学顕微鏡を用いて総合倍率200〜
290
K 0101 : 1998
400倍で観察する。
(b) 各視野に出現する動物の種(属)とその数を(5.2)(b)のように記録し,試料1ml中又は1l中のそれぞ
れの動物の種(属)とその数を算出する(10)。
注(10) スライドガラスの界線ごとに動物を類別して計数する。この操作を3回繰り返して行った後,注
(2)の式によって試料1ml中のそれぞれの動物数を算出する。
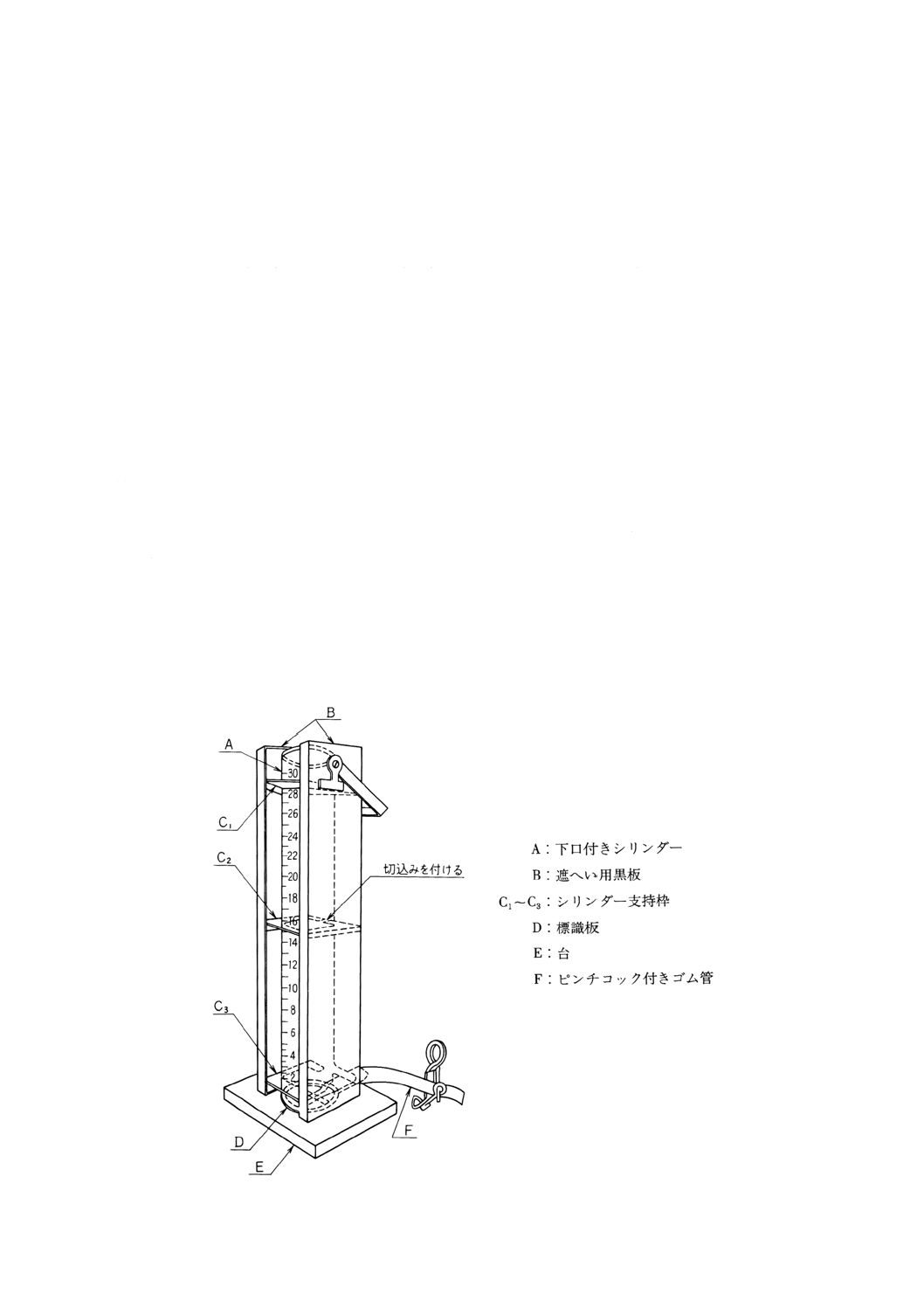
291
K 0101 : 1998
附属書(参考) 補足
参考は,本体の規定に関する事柄を,本体に準じる形で補足したものであって規定の一部ではない。
I.
透視度 試料の透明度を示すもので,透視度計に試料を入れて上部から透視し,底部に置いた標識板
の二重十字が,初めて明らかに識別できるときの水層の高さをはかり,10mmを1度として表す。
測定範囲:1〜30度
(1) 器具 器具は,次のとおりとする。
(a) 透視度計 参考図1に示す。標識板上側から50mmの高さまでは5mmごとに,50〜300mmまでは
10mmごとに目盛を施した下口付のガラス製のもの。底部に参考図2のような標識板を入れて用い
る。
(2) 操作 操作は,次のとおり行う。
(a) よく振り混ぜた試料を透視度計に満たし,上部から底部を透視し,標識板の二重十字が初めて明ら
かに識別できるまで,下口から試料を速やかに流出させたとき(1)の水面の目盛を読む。
(b) (a)の操作を2,3回繰り返し,水面の目盛を読み取り平均値を求め,透視度として度で表す。
注(1) 懸濁物の多い試料の場合には,透視度計の底部に沈積することがあり,誤差の原因となるので
注意する。
備考 同じ照度でも光源の違いによって彩度が異なる場合は透視度が変わる。光源は昼光とし,直射
日光を避ける。
参考図1 透視度計
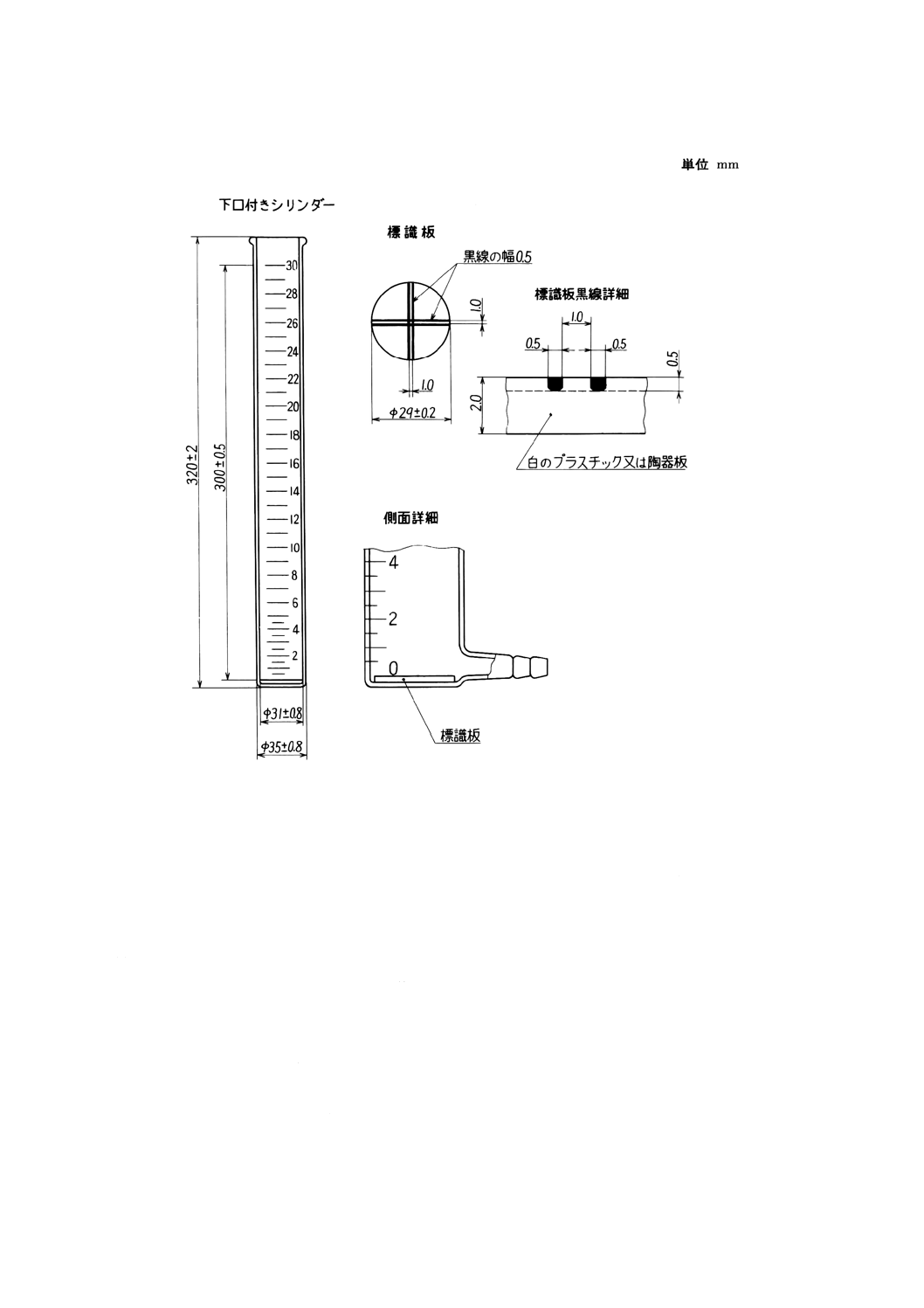
292
K 0101 : 1998
参考図2 透視度計詳細図
II. アルカリ性過マンガン酸カリウムによる酸素消費量 (CODOH) 試料をアルカリ性とし,酸化剤とし
て過マンガン酸カリウムを加え,沸騰水浴中で20分間反応させ,その時消費した過マンガン酸カリウムの
量を求め,相当する酸素の量 (mgO/l) で表す。
この試験は試料採取後直ちに行う。直ちに行えない場合は本体の3.3に従って保存し,できるだけ早く
試験する。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 本体17.(1)(a)による。
(b) 水酸化ナトリウム溶液 (100g/l) 本体22.2.1(1)(b)による。
(c) 硫酸 (2+1) 水1容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸2容を徐々に加える。
(d) アジ化ナトリウム溶液 (40g/l) JIS K 9501に規定するアジ化ナトリウム4gを水に溶かして100ml
とする。
(e) でんぷん溶液 (10g/l) 本体22.1.2(1)(i)による。
(f) よう化カリウム溶液 (100g/l) JIS K 8913に規定するよう化カリウム10gを水に溶かして100mlと
する。使用時に調製する。
293
K 0101 : 1998
(g) 過マンガン酸カリウム溶液 (2mmol/l) JIS K 8247に規定する過マンガン酸カリウム0.32gを平底
フラスコにとり,水1 050〜1 100mlを加えて溶かす。これを1〜2時間静かに煮沸した後,約16時
間放置する。
上澄み液をガラスろ過器G4を用いてろ過する(ろ過前後に水洗しない。)。ろ液は約30分間水蒸
気洗浄した着色ガラス瓶に入れて保存する。
(h) 10mmol/lチオ硫酸ナトリウム溶液 本体28.3(1)(e)による。
(2) 器具 器具は,次のとおりとする。
(a) 共栓三角フラスコ 200ml
(b) 水浴 本体17.(2)(a)による。
(3) 操作 操作は,次のとおり行う。
(a) 試料(1)の適量(2)を共栓三角フラスコ200mlにとり,水を加えて50mlとし,水酸化ナトリウム溶液
(100g/l) 1mlを加える。
(b) 過マンガン酸カリウム溶液 (2mmol/l) 10mlを加えて振り混ぜ,直ちに沸騰水浴中に入れ,20分間加
熱する。このときフラスコ中の試料の液面は沸騰水浴中の水面下で,かつ,共栓三角フラスコが水
浴の底に直接接しないように保つ。
(c) 水浴から取り出し,冷水で室温まで冷却した後,アジ化ナトリウム溶液 (40g/l) 1mlとを加えて振り
混ぜる。
(d) よう化カリウム溶液 (100g/l) 1ml及び硫酸 (2+1) 0.5mlを加え(3),栓をして振り混ぜ,暗所に約5
分間放置する。
(e) 遊離したよう素を,10mmol/lチオ硫酸ナトリウム溶液で滴定し,溶液の色が薄い黄色になったら,
指示薬としてでんぷん溶液 (10g/l) 1mlを加え,生じたよう素でんぷんの青い色が消えるまで滴定す
る。
(f) 別に,水50mlを共栓三角フラスコ200mlにとり,水酸化ナトリウム溶液 (100g/l) 1mlを加え,(b)
〜(e)の操作を行う。
(g) 次の式によってCODOH (mgO/l) を算出する。
08
.0
000
1
)
(
OH
×
×
×
−
=
V
f
a
b
COD
ここに, CODOH: アルカリ性過マンガン酸カリウム溶液による酸素消費
量 (mgO/l)
a: 滴定に要した10mmol/lチオ硫酸ナトリウム溶液 (ml)
b: 水を用いた試験に要した10mmol/lチオ硫酸ナトリウム
溶液 (ml)
f: 10mmol/lチオ硫酸ナトリウム溶液のファクター(4)
0.08: 10mmol/lチオ硫酸ナトリウム溶液1mlの酸素相当量
(mg)
V: 試料 (ml)
注(1) 懸濁物を含む場合は,よく振り混ぜて均一にした後,採取する。
(2) 20分間加熱したときに最初に加えた過マンガン酸カリウム溶液 (2mmol/l) の約21が残るような
量。
ただし,アルカリ性過マンガン酸カリウムによる酸素消費量が8mgO/l以下の場合には,50ml
とする。
試料の適量は,(3)の操作に従って予備試験を行って決める。
294
K 0101 : 1998
CODOHの概略値が分かっている場合には,次の式によって試料の適量 (Vml) を求めることが
できる。
)
(mgO/
08
.0
000
1
5
OH
l
COD
V
予想値
試料の
×
×
=
ここに,
V: 試料の採取量 (ml)
5: 過マンガン酸カリウム溶液 (2mmol/l) の反応予想量 (ml)
0.08: 過マンガン酸カリウム溶液 (2mmol/l) 1mlの酸素相当量
(mg)
(3) 鉄を含むときは,硫酸 (2+1) の添加前にふっ化カリウム溶液 (300g/l) 1mlを加える。
(4) 本体22.1.2(1)(d)の0.1mol/lチオ硫酸ナトリウム溶液のファクターを用いる。
III. 陽イオン界面活性剤 陽イオン界面活性剤には,脂肪族アミン塩類,第四アンモニウム化合物,アル
キルピリジウム塩類などがある。陽イオン界面活性剤の定量には,オレンジII吸光光度法を適用するが,
通常の水の中では,陽イオン界面活性剤は陰イオン界面活性剤と安定なイオン対を作って存在するため,
直接は定量できない。このため,前処理(イオン交換分離)を行って陰イオン界面活性剤を除去した試料
について,オレンジII吸光光度法を適用する。
オレンジII吸光光度法 陽イオン界面活性剤が陰イオン性のオレンジII[4-(2-ヒドロキシ-1-ナフタレニ
ル)アゾベンゼンスルホン酸ナトリウム]と反応して生じるイオン対をクロロホルムに抽出し,吸光度を
測定してテトラデシルジメチルベンジルアンモニウムクロリドとして表示する。
定量範囲 : 陽イオン界面活性剤 [CH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl] 20〜350μg,繰返し分析精
度:変動係数で3〜10%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩化ナトリウム JIS K 8150に規定するもの。
(c) 硫酸ナトリウム JIS K 8987に規定するもの。
(d) 酢酸−酢酸ナトリウム緩衝液 (pH3.5) JIS K 8355に規定する酢酸6mlとJIS K 8371に規定する酢
酸ナトリウム三水和物0.9gを水に溶かして1lとする。
(e) オレンジII溶液 オレンジII[4-(2-ヒドロキシ−1-ナフタレニル)アゾベンゼンスルホン酸ナト
リウム五水和物]0.1gを水に溶かして100mlとする。
(f) メタノール溶液 (50vol%) JIS K 8891に規定するメタノールを用いて調製する。
(g) クロロホルム JIS K 8322に規定するもの。
(h) 強塩基性陰イオン交換樹脂(I形) 本体23.2.1(1)(k)による。使用時にJIS K 8891に規定するメタ
ノールで3,4回,次に,水で3,4回洗浄する。
(i) 陽イオン界面活性剤標準液 [0.1mgCH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl/ml] テトラデシルジメチ
ルベンジルアンモニウムクロリドをその100%に対して0.100gをはかりとり,水に溶かして全量フ
ラスコ1 000mlに移し入れ,水を標線まで加える。使用時に調製する。
(j) 陽イオン界面活性剤標準液[10μgCH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl/ml] 陽イオン界面活性剤
標準液 [0.1mgCH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl/ml] 20mlを全量フラスコ200mlにとり,水を標線
まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 円筒形滴下漏斗 100ml
295
K 0101 : 1998
(b) 分液漏斗 200ml
(c) 陰イオン交換樹脂カラム 内径約12mm,長さ約300mmのコック付ガラス管の下部にJIS K 8251
に規定するガラスウールを詰め,これに強塩基性陰イオン交換樹脂(I形)約25mlに水を加えてよ
く混合しながら,気泡が混入しないように充てんし,上部にガラスウールを詰め,水面がガラスウ
ールの上約10mmになるように調節する。これを陰イオン交換樹脂カラムとする。上部に円筒形滴
下漏斗100mlを付ける。
この陰イオン交換樹脂カラムは,繰り返し約20回使用できる。ただし,再生して再使用はできな
い。
(d) 光度計 分光光度計又は光電光度計
(3) 前処理 前処理は,次のとおり行う。
(a) 試料(1)100ml[CH3 (CH2) 12CH2N (CH3) 2CH2C6H5Clとして20〜350μgを含む]を陰イオン交換樹脂カ
ラムの上部の円筒形滴下漏斗にとり,陰イオン交換樹脂カラムに流量約1ml/minで流下させ(2),流
出液を三角フラスコ300mlに受ける。
(b) 陰イオン交換樹脂カラムの上部の円筒形滴下漏斗中の試料がなくなる寸前に流下を止め,メタノー
ル (50vol%) 50mlを上部の円筒形滴下漏斗に加え,再び約1ml/minで流下させ(2),流出洗は,先の
三角フラスコ300mlに合わせる。
注(1) 試料が酸性の場合には水酸化ナトリウム溶液 (40g/l) で,また,アルカリ性の場合には塩酸 (1
+11) で中和する。
(2) 液面を陰イオン交換樹脂カラムの上部のガラスウールの上10mm程度に保つようにする。
(4) 操作 操作は,次のとおり行う。
(a) (3)(b)で得た流出液を分液漏斗に移し入れ,酢酸-酢酸ナトリウム緩衝溶液 (pH3.5) 30ml,塩化ナト
リウム1.5g及びオレンジII溶液10mlを加えて振り混ぜる。
(b) クロロホルム10mlを加え,約3分間激しく振り混ぜた後,放置する。
(c) クロロホルム層をメスシリンダー(有栓形)20mlに移す。
(d) 水層にクロロホルム10mlを加え,再び約3分間激しく振り混ぜた後,放置する。
(e) クロロホルム層を(c)のメスシリンダー(有栓形)20mlに合わせ,クロロホルムを20mlの標線まで
加える。
(f) 硫酸ナトリウム約3gを加えて振り混ぜて水分を除く。
(g) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長485nm付近の吸光度を
測定する。
(h) 空試験として水100mlとメタノール (50vol%) 50mlとを分液漏斗にとり,(a)〜(g)の操作を行って試
料について得た吸光度を補正する。
(i) 検量線から陽イオン界面活性剤の量を求め,試料中の陽イオン界面活性剤の濃度 [mgCH3 (CH2)
12CH2N (CH3) 2CH2C6H5Cl/l] を算出する。
検量線 陽イオン界面活性剤標準液 [10μgCH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl/ml] 2〜35mlを段階
的に分液漏斗にとり,水を加えて100mlとし,これにメタノール (50vol%) 50mlを加え,(a)〜(h)
の操作を行って陽イオン界面活性剤 [CH3 (CH2) 12CH2N (CH3) 2CH2C6H5Cl] の量と吸光度との関係
線を作成する。
備考 陽イオン界面活性剤の100倍以上の陰イオン界面活性剤が共存しても分離できる。硫酸イオン,
硝酸イオン,炭酸イオン及びりん酸イオンが共存しても妨害しない。
296
K 0101 : 1998
IV. 油類 塩酸酸性でトリクロロトリフルオロエタンによる抽出操作を行うと,試料中の炭化水素,炭化
水素誘導体,動植物油脂類,脂肪酸などが抽出される。このトリクロロトリフルオロエタン溶液について
赤外線吸収を測定し,2, 2, 4-トリメチルペンタン(イソオクタン)・ヘキサデカン(セタン)・ベンゼン混
合標準物質(OCB混合標準物質)の対応量で表す。
1. 試料採取 本体26.1による。ただし,試料容器はトリクロロトリフルオロエタンで洗浄したものを用
い,採取した試料は塩酸を加えてpH4以下とし,0〜10℃で保存する。
2. トリクロロトリフルオロエタン抽出−赤外線分析法 試料を塩酸酸性にして,トリクロロトリフルオ
ロエタンで抽出し,このトリクロロトリフルオロエタン溶液について,波長3.4μm付近の吸収を測定し,
これを油類としてOCB混合標準物質の対応量に換算して表す。
定量範囲:OCB 0.2mg以上,繰返し分析精度:変動係数で3〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+1) JIS K 8180に規定する塩酸を用いて調製する。
(c) 硫酸ナトリウム JIS K 8987に規定するもの。
(d) メチルオレンジ溶液 (1g/l) 本体22.1.1(1)(d)による。
(e) トリクロロトリフルオロエタン 1, 1, 2-トリクロロ−1, 2, 2-トリフルオロエタン,赤外線スペクト
ル測定用のもので,波長3.4μm付近の吸収が少ないもの。
(f) OCB混合標準物質 JIS K 9703に規定する2, 2, 4-トリメチルペンタン,ヘキサデカン及びJIS K
8858に規定するベンゼンをそれぞれ37.5,37.5及び25.0の体積比で混合する。
(g) OCB混合標準液 (1mgOCB/ml) OCB混合標準物質0.200gをとり,トリクロロトリフルオロエタ
ンに溶かし,全量フラスコ200mlに移し入れ,トリクロロトリフルオロエタンを標線まで加える。
(h) OCB混合標準液 (0.1mgOCB/ml) OCB混合標準物質 (1mgOCB/ml) 10mlを全量フラスコ100ml
にとり,トリクロロトリフルオロエタンを標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 本体26.2(2)(a)による。ただし,使用前にトリクロロトリフルオロエタンで洗う。
(b) 赤外線分析計 波長3.4μm付近の赤外線吸収を測定できる赤外分光光度計又は非分散形赤外線分析
計。
(c) 吸収セル 石英ガラス製又は同等の品質のもの50mm。
(3) 操作 操作は,次のとおり行う。
(a) 本体26.1で採取した試料(OCBとして0.2mg以上を含む。)を分液漏斗1 000mlに移し(1),指示薬
としてメチルオレンジ溶液 (1g/l) 2,3滴を加え,溶液の色が赤に変わるまで塩酸 (1+1) を滴加す
る。
(b) 試料容器をトリクロロトリフルオロエタン20mlずつで2回洗い,洗液を分液漏斗1 000mlに加える。
約5分間激しく振り混ぜ,放置する。
(c) トリクロロトリフルオロエタン層を分液漏斗250mlに移し入れ,更に分液漏斗1 000mlを静かに揺
り動かして水層とトリクロロトリフルオロエタン層(2)(3)を分離して,トリクロロトリフルオロエタ
ン層を先の分液漏斗250ml中のトリクロロトリフルオロエタン層に合わせる。
297
K 0101 : 1998
(d) 分液漏斗1 000ml中の水層にトリクロロトリフルオロエタン20mlを加え,再び抽出操作を行ってト
リクロロトリフルオロエタン層は先の分液漏斗250ml中のトリクロロトリフルオロエタン層に合わ
せる。
(e) この分液漏斗250mlに水約20mlを加え,約1分間振り混ぜた後,放置する。トリクロロトリフル
オロエタン層を別の分液漏斗250mlに移し,水層は捨てる。この操作を数回繰り返し,洗液が中性
になるまで洗浄する。水を十分に分離した後,トリクロロトリフルオロエタン層に硫酸ナトリウム
3〜5gを加えて振り混ぜ,脱水する。
(f) 分液漏斗250mlの脚部を乾いたろ紙でふきとり,ろ紙5種B(4)を用いてトリクロロトリフルオロエ
タン層をろ過し,全量フラスコ100mlに移し入れ,トリクロロトリフルオロエタンを標線まで加え
る。
(g) トリクロロトリフルオロエタン層を吸収セルに移し,トリクロロトリフルオロエタンを対照液とし
て,波長3.4μm付近(5)における透過パーセントを測定し,吸光度に換算する。
(h) 検量線からOCB混合標準物質の対応量を求めて,油類とし,試料中の油類の濃度 (mgOCB/l) を算
出する。
検量線 OCB混合標準液 (0.1mgOCB/ml) 2〜40mlを全量フラスコ100mlに段階的にとり,トリクロ
ロトリフルオロエタンを標線まで加える。トリクロロトリフルオロエタンを対照液として波長
3.4μm付近(5)における透過パーセントを測定し,吸光度に換算し,OCB混合標準物質の量と吸光度
との関係線を作成する。
注(1) 低沸点の炭化水素を含む場合には,20℃以下で操作する。
(2) 試料によっては,トリクロロトリフルオロエタン層にエマルションが生じることがある。この
ような場合には,本体26.の注(9)の操作に準じてエマルションを壊す。
(3) トリクロロトリフルオロエタン層が黄色から淡黄色に着色している場合には,無色になるまで
抽出を繰り返す。
(4) トリクロロトリフルオロエタンでよく洗って抽出物質を除いたもの。
(5) 波長3.3〜3.6μm付近の3本の吸収ピークの透過パーセントをそれぞれ吸光度に換算し,その平
均値を用いる。又は非分散形赤外線分析計を用いてもよい。
V. 炭化水素及び動植物油脂類 試料を塩酸酸性とし,トリクロロトリフルオロエタンで抽出する。この
トリクロロトリフルオロエタン溶液の一部をとり波長3.4μm付近の吸収を測定し,これを炭化水素及び動
植物油脂類の合量としてOCB混合標準物質の対応量として求める。残ったトリクロロトリフルオロエタ
ン溶液を活性けい酸マグネシウムカラムに通し,動植物油脂類を吸着除去した後のトリクロロトリフルオ
ロエタン溶液について波長3.4μm付近の吸収を測定し,これを炭化水素としてOCB混合標準物質の対応
量として表す。
動植物油脂類は,炭化水素及び動植物油脂類の合量から炭化水素の量を差し引いて求める。
(1) 試薬 試薬は,次のものを用いる。
(a) 水 JIS K 0557に規定するA3の水
(b) 塩酸 (1+11) JIS K 8180に規定する塩酸を用いて調製する。
(c) 硫酸ナトリウム JIS K 8987に規定するもの。
(d) メチルオレンジ溶液 (1g/l) 本体22.1.1(1)(d)による。
(e) トリクロロトリフルオロエタン 1, 1, 2-トリクロロ−1, 2, 2-トリフルオロエタン,赤外線スペクト
298
K 0101 : 1998
ル測定用のもので,波長3.4μm付近に吸収が少ないもの。
(f) 活性けい酸マグネシウム(1) 粒径150〜250mm,使用前にトリクロロトリフルオロエタンで洗い,
150℃で約2時間加熱してデシケーター中で放冷したもの。
(g) OCB混合標準物質 JIS K 9703に規定する2, 2, 4-トリメチルペンタン,ヘキサデカン及びJIS K
8858に規定するベンゼンをそれぞれ37.5,37.5及び25.0の体積比で混合する。
(h) OCB混合標準液 (1mgOCB/ml) OCB混合標準物質0.200gをとり,トリクロロトリフルオロエタ
ンに溶かし,全量フラスコ200mlに移し入れ,トリクロロトリフルオロエタンを標線まで加える。
(i) OCB混合標準液 (0.1mgOCB/ml) OCB混合標準液 (1mgOCB/ml) 10mlを全量フラスコ100mlにと
り,トリクロロトリフルオロエタンを標線まで加える。
注(1) フロリジルなどの名で市販されている。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 250ml及び1 000〜3 000mlで脚部の短いもの。使用前にトリクロロトリフルオロエタン
で洗う。コックに滑剤を塗布しない。
(b) 活性けい酸マグネシウムカラム 内径約10mm,長さ約150mmのガラス管にコックを付けたものに
JIS K 8251[ガラスウール(試薬)]に規定するガラスウールを詰め,その上120〜130mmに活性け
い酸マグネシウムを気泡が混入しないようにトリクロロトリフルオロエタンとともに充てんする(2)。
コックに滑剤を塗布しない。
(c) 赤外線分析計 波長3.4μm付近の赤外線吸収を測定できる赤外分光光度計又は非分散形赤外線分析
計。
(d) 吸収セル 石英ガラス製又は同等の品質のもの。
注(2) 極性基をもつ物質80〜100mgを吸着する能力がある。使用した活性けい酸マグネシウムは,約
600℃で約4時間強熱すれば繰り返し使用できる。
(3) 試料採取 本体26.1による。ただし,試料容器はトリクロロトリフルオロエタン [(1)(e)] で洗浄した
ものを用い,採取した試料は塩酸を加えてpHを4以下とし,10℃以下で保存する。
(4) 操作 操作は,次のとおり行う。
(a) (3)で採取した試料(OCBとして0.2mg以上を含む。)を分液漏斗1 000〜3 000mlに移し,指示薬と
してメチルオレンジ溶液 (1g/l) 数滴を加え,溶液の色が赤に変わるまで塩酸 (1+1) を滴加する。
(b) 以下,IV.2(4)(b)〜(h)の操作を行って試料中の炭化水素及び動植物油脂類の濃度 (mgOCB/l) を算出
する。
(c) 残りのトリクロロトリフルオロエタン層を活性けい酸マグネシウムカラムに注ぎ入れて約2ml/min
で流出させる。
(d) 最初の流出液約10ml(3)を捨て,その後の流出液25mlをビーカー[(1)(e)のトリクロロトリフルオロ
エタンで洗浄し,乾燥したもの。]にとる。
(e) 流出液の一部を吸収セルにとり,トリクロロトリフルオロエタンを対照液として,波長3.4μm付近
(4)における透過パーセントを測定し,吸光度に換算する。
(f) 検量線からOCB混合標準物質の対応量を求め,炭化水素の量とし,試料中の炭化水素の濃度
(mgOCB/l) を算出する。
(g) 動植物油脂類は(b)で求めた炭化水素及び動植物油脂類の濃度 (mgOCB/l) から,(f)で求めた炭化水
素の濃度 (mgOCB/l) を差し引いて,試料中の動植物油脂類の濃度 (mgOCB/l) とする。
検量線 OCB混合標準液 (0.1mgOCB/ml) 2〜40mlを全量フラスコ100mlに段階的にとり,トリク
299
K 0101 : 1998
ロロトリフルオロエタンを標線まで加える。トリクロロトリフルオロエタンを対照液として波長
3.4μm付近(4)における透過パーセントを測定して吸光度に換算し,OCB混合標準物質の量と吸光度
との関係線を作成する。
注(3) このときカラム中のトリクロロトリフルオロエタンがトリクロロトリフルオロエタン抽出液と
完全に置換すること。
(4) 波長3.3〜3.6μmの3本の吸収ピークの透過パーセントをそれぞれ吸光度に換算し,その平均値
を用いる。又は非分散形赤外線分析計を用いてもよい。
備考 不揮発性炭化水素の量を求めるには,次のように操作する。
(e)の流出液の一定量をビーカーにとり,加熱してトリクロロトリフルオロエタンとともに揮
発性炭化水素を揮散させ,引き続き80℃で30分間加熱し,デシケーター中で放冷する。
トリクロロトリフルオロエタンの少量を加えて残留物を溶かし,トリクロロトリフルオロエ
タンを揮散させる前の体積になるようにトリクロロトリフルオロエタンを加える。この一部を
吸収セルにとり,トリクロロトリフルオロエタンを対照液として波長3.4μm付近における透過
パーセントを測定し,吸光度に換算する。
検量線から,OCB混合標準物質の対応量を求め,不揮発性炭化水素の量とし,試料中の不揮
発性炭化水素の濃度 (mgOCB/l) を算出する。
VI. よう化物イオンのイオン電極法 酢酸塩緩衝液を加えてpHを約5に調節し,よう化物イオン電極を
指示電極として電位を測定し,よう化物イオンを定量する。
定量範囲:I-0.1〜1 000mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸塩緩衝液 (pH5) 本体32.4(1)(a)による。
(b) よう化物イオン標準液 (1 000mgI-/l) 本体33.1(1)(f)による。
(c) よう化物イオン標準液 (100mgI-/l) よう化物イオン標準液 (1 000mgI-/l) 20mlを全量フラスコ
200mlにとり,水を標線まで加える。使用時に調製する。
(d) よう化物イオン標準液 (10mgI-/l) よう化物イオン標準液 (100mgI-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。使用時に調製する。
(e) よう化物イオン標準液 (1mgI-/l) よう化物イオン標準液 (10mgI-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(f) よう化物イオン標準液 (0.1mgI-/l) よう化物イオン標準液 (1mgI-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 本体31.2(2)(a)による。
(b) 指示電極 よう化物イオン電極
(c) 参照電極 本体32.4(2)(c)による。
(d) マグネチックスターラー 本体31.2(2)(d)による。
(3) 検量線の作成 検量線は,次のとおり作成する。
(a) よう化物イオン標準液 (0.1mgI-/l) 100mlをビーカー200mlにとり,酢酸塩緩衝液 (pH5) 10ml(1)を加
える。
(b) これに指示電極(2)(3)と参照電極(4)(5)とを浸し,マグネチックスターラー(6)で泡が電極に触れない程
300
K 0101 : 1998
度にして強くかき混ぜる(7)。
(c) 液温をはかり,電位差計で電位を測定する(8)。
(d) よう化物イオン標準液 (1mgI-/l) 100ml,よう化物イオン標準液 (10mgI-/l) 100ml,よう化物イオン標
準液 (100mgI-/l) 100ml及びよう化物イオン標準液 (1 000mgI-/l) 100mlをそれぞれビーカー200mlに
とり,酢酸塩緩衝液 (pH5) 10ml(1)を加える。それぞれのよう化物イオン標準液の液温を(c)での液温
の±1℃に調節し,(b)及び(c)の操作を行ってよう化物イオン標準液 (1〜1 000mgI-/l) による電位を
測定する(8)。
(e) 片対数紙の対数軸によう化物イオンの濃度を,均等軸に電位をとり,よう化物イオン (I-) の濃度
(mgI-/l) と電位との関係線を作成する(9)。
注(1) 酢酸塩緩衝液は,測定時のpHを5に調節し,イオン強度を一定にするためのものである。
(2) よう化物イオン電極は,使用時に,よう化物イオン標準液 (0.1mgI-/l) に浸し,指示値が安定し
てから電位を測定する。
(3) 本体31.の注(12)による。
(4) 本体31.の注(13)による。
(5) 本体31.の注(14)による。
(6) 本体31.の注(15)による。
(7) 本体31.の注(16)による。
(8) よう化物イオン電極の応答時間は,液温10〜30℃の場合,よう化物イオンの濃度0.1〜1mgI-/l
で約2〜3分間,10mgI-/l以上は1分間程度である。
(9) よう化物イオン標準液 (0.1mgI-/l) とよう化物イオン標準液 (10mgI-/l) との電位の差は,110〜
120mV (25℃) の範囲に入り,よう化物イオンの濃度0.1〜1 000mg/lの間の検量線は直線となる。
(4) 操作 操作は,次のとおり行う。
(a) 試料100ml(10)をビーカーにとり,酢酸塩緩衝液 (pH5) 10mlを加え,液温を(3)(c)の液温の±1℃に調
節する。
(b) (3)(b)及び(c)の操作を行って検量線からよう化物イオンの濃度を求め,試料中のよう化物イオンの濃
度 (mgI-/l) を算出する。
注(10) 試料が酸性又はアルカリ性で,酢酸塩緩衝液 (pH5) を加えてもpH5にならない場合には,あら
かじめ,酢酸 (1+2) 又は水酸化ナトリウム溶液 (100g/l) を加えてpH5に調節した後,水を加
えて一定量とする。
備考1. イオン濃度計の場合には,よう化物イオン標準液((1mgI-/l及び100mgI-/l)を用いて(3)(a)〜
(c)の操作を行い,イオン濃度計の指示値を1mgI-/lと100mgI-/lとになるように調節する。さら
によう化物イオン標準液(0.1mgI-/l及び10mgI-/l)及びよう化物イオン標準液 (1 000mgI-/l) を
用いてイオン濃度計の指示値を確認する。
2. 硫化物イオンは,あらかじめ酢酸亜鉛溶液 (100g/l) を加え,生じた沈殿をろ紙5種Cでろ別
し,ろ液を用いて試験する。
主な共存物質の許容限度を,最大比率で次に示す。
F-, Cl-, Br-, NO3-, SO42-, CO32-: 104
PO43-: 103
S2O32-: 10
CN-: 10-1
301
K 0101 : 1998
3. イオン電極による電位差滴定法 試料100mlをビーカーにとり,試料のpHを7に調節し,
指示電極(よう化物イオン電極又は銀イオン電極)を用い,(3)(b)の操作に準じて電位を測定
しながら10〜100mmol/l硝酸銀溶液で滴定して,滴定曲線を作図し,滴定終点を求める。滴
定曲線の変曲点は,よう化物イオン,臭化物イオン及び塩化物イオンの順になる。それぞれ
の変曲点から終点を求め,各イオンの濃度を算出する。
10mmol/l硝酸銀溶液1mlはI-1.269mg,Br-0.799mg及びCl-0.355mgに相当する。
VII. 臭化物イオンのイオン電極法 酢酸塩緩衝液を加えてpH約5に調節し,臭化物イオン電極を指示電
極として電位を測定し,臭化物イオンを定量する。
定量範囲:Br-0.5〜1 000mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 酢酸塩緩衝液 (pH5) 本体32.4(1)(a)による。
(b) 臭化物イオン標準液 (1 000mgBr-/l) JIS K 8506に規定する臭化カリウムを110℃で約4時間加熱
し,デシケーターで放冷する。その1.49gをとり,水に溶かし,全量フラスコ1 000mlに移し入れ,
水を標線まで加える。
(c) 臭化物イオン標準液 (100mgBr-/l) 臭化物イオン標準液 (1 000mgBr-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。
(d) 臭化物イオン標準液 (10mgBr-/l) 臭化物イオン標準液 (100mgBr-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。
(e) 臭化物イオン標準液 (1mgBr-/l) 臭化物イオン標準液 (10mgBr-/l) 20mlを全量フラスコ200mlにと
り,水を標線まで加える。
(f) 臭化物イオン標準液 (0.5mgBr-/l) 臭化物イオン標準液 (10mgBr-/l) 10mlを全量フラスコ200mlに
とり,水を標線まで加える。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 本体31.2(2)(a)による。
(b) 指示電極 臭化物イオン電極
(c) 参照電極 本体32.4(2)(c)による。
(d) マグネチックスターラー 本体31.2(2)(d)による。
(3) 検量線の作成 検量線は,次のとおり作成する。
(a) 臭化物イオン標準液 (0.5mgBr-/l) 100mlをビーカー200mlにとり,酢酸塩緩衝液 (pH5) 10ml(1)を加え
る。
(b) これに指示電極(臭化物イオン電極)(2)(3)と参照電極(4)(5)とを浸し,マグネチックスターラー(6)で
泡が電極に触れない程度にして強くかき混ぜる(7)。
(c) 液温をはかり,電位差計で電位を測定する(8)。
(d) 臭化物イオン標準液 (1mgBr-/l) 100ml,臭化物イオン標準液 (10mgBr-/l) 100ml,臭化物イオン標準
液 (100mgBr-/l) 100ml及び臭化物イオン標準液 (1 000mgBr-/l) 100mlをそれぞれビーカー200mlにと
り,酢酸塩緩衝液 (pH5) 10ml(1)を加える。それぞれの臭化物イオン標準液の液温を(c)での液温の±
1℃に調節して(b)及び(c)の操作を行い,臭化物イオン標準液 (1〜1 000mgBr-/l) による電位を測定す
る(8)。
(e) 片対数紙の対数軸に臭化物イオンの濃度を,均等軸に電位をとり,臭化物イオンの濃度 (mgBr-/l) と
302
K 0101 : 1998
電位との関係線を作成する(9)。
注(1) 酢酸塩緩衝液は,測定時のpHを5に調節し,イオン強度を一定にするためのものである。
(2) 臭化物イオン電極は,使用時に,臭化物イオン標準液 (0.5mgBr-/l) に浸し,指示値が安定して
から電位を測定する。
(3) 本体31.の注(12)による。
(4) 本体31.の注(13)による。
(5) 本体31.の注(14)による。
(6) 本体31.の注(15)による。
(7) 本体31.の注(16)による。
(8) 臭化物イオン電極の応答時間は,液温10〜30℃の場合,臭化物イオンの濃度0.5mg/lで約1分
間程度である。
(9) 臭化物イオン標準液1mgBr-/lと100mgBr-/lとの電位の差は,110〜120mV (25℃) の範囲に入り,
臭化物イオンの濃度0.5mg/lから1 000mg/lの間の検量線は直線となる。
(4) 操作 操作は,次のとおり行う。
(a) 試料100ml(10)をビーカーにとり,酢酸塩緩衝液 (pH5) 10mlを加え,液温を(3)(c)の液温の±1℃に調
節する。
(b) (3)(b)及び(c)の操作を行い,検量線から臭化物イオンの濃度を求め,臭化物イオンの濃度 (mgBr-/l)
を算出する。
注(10) 試料が酸性又はアルカリ性で,酢酸塩緩衝液 (pH5) を加えてもpH5にならない場合には,あら
かじめ,酢酸 (1+2) 又は水酸化ナトリウム溶液 (100g/l) を加えてpHを5に調節し,水を加え
て100mlにする。
備考1. イオン濃度計の場合には,臭化物イオン標準液(1mgBr-/l及び100mgBr-/l)を用いて(3)(a)〜(c)
の操作を行い,イオン濃度計の指示値を1.0mgBr-/lと100mgBr-/lになるように調節する。さら
にその他の臭化物イオン標準液(0.5mgBr-/l及び10mgBr-/l)及び臭化物イオン標準液 (1
000mgBr-/l) を用いてイオン濃度計の指示値を確認する。
2. 硫化物イオン及びシアン化物イオンは,妨害するので,あらかじめこれらを分離しておく。
硫化物イオンは,あらかじめ酢酸亜鉛緩衝溶液 (100g/l) を加えて硫化物イオンを固定し,ろ
紙5種Cでろ過し,ろ液を用いて試験する。
主な共存物質の許容限度を,最大比率で次に示す。
F-, NO3-, SO42-: 104
Cl-: 102
I-: 10-2
3. イオン電極による電位差滴定法 参考VI.の備考3.による。
VIII. 硝酸イオンのイオン電極法 試料にりん酸塩緩衝液を加えて,pHを6.8に調節し,硝酸イオン電極
を指示電極として電位を測定し,硝酸イオンを定量する。
定量範囲:NO3-0.5〜1 000mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) りん酸塩緩衝液 (pH6.8) JIS K 9007に規定するりん酸二水素カリウム17gとJIS K 9020に規定す
るりん酸水素二ナトリウム17.8gとを,水に溶かして1lとする。
(b) 硝酸イオン標準液 (1 000mgNO3-/l) 本体37.2.3(1)(i)による。
303
K 0101 : 1998
(c) 硝酸イオン標準液 (100mgNO3-/l) 硝酸イオン標準液 (1 000mgNO3-/l) 20mlを全量フラスコ200ml
にとり,水を標線まで加える。使用時に調製する。
(d) 硝酸イオン標準液 (10mgNO3-/l) 硝酸イオン標準液 (100mgNO3-/l) 20mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(e) 硝酸イオン標準液 (1mgNO3-/l) 硝酸イオン標準液 (10mgNO3-/l) 20mlを全量フラスコ200mlにと
り,水を標線まで加える。使用時に調製する。
(f) 硝酸イオン標準液 (0.5mgNO3-/l) 硝酸イオン標準液 (10mgNO3-/l) 10mlを全量フラスコ200mlに
とり,水を標線まで加える。使用時に調製する。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 本体31.2(2)(a)による。
(b) 指示電極 硝酸イオン電極
(c) 参照電極 本体32.4(2)(c)による。外筒液には硫酸カリウム溶液 (0.25mol/l) を用いる。
(d) マグネチックスターラー 本体31.2(2)(d)による。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) 硝酸イオン標準液 (0.5mgNO3-/l) 100mlをビーカー200mlにとり,りん酸塩緩衝液 (pH6.8) 5ml(1)を
加える。
(b) これに指示電極(2)(3)と参照電極(4)(5)とを浸し,マグネチックスターラー(6)で泡が電極に触れない程
度に強くかき混ぜる(7)。
(c) 液温をはかり,電位差計で電位を測定する(8)。
(d) 硝酸イオン標準液 (1mgNO3-/l) 100ml,硝酸イオン標準液 (10mgNO3-/l) 100ml,硝酸イオン標準液
(100mgNO3-/l) 100ml及び硝酸イオン標準液 (1 000mgNO3-/l) 100mlをそれぞれビーカー200mlにとり,
りん酸塩緩衝液 (pH6.8) 5ml加える。それぞれの硝酸イオン標準液の液温を(c)の液温の±1℃に調節
して(b)及び(c)の操作を行い,硝酸イオン標準液 (1〜1 000mgNO3-/l) の電位を測定する。
(e) 片対数紙の対数軸に硝酸イオンの濃度を,均等軸に電位をとり,硝酸イオンの濃度 (mgNO3-/l) と電
位との関係線を作成する(9)。
注(1) りん酸塩緩衝液 (pH6.8) の添加は,測定時のpHに調節し,更にイオン強度を一定にするため
である。
(2) 硝酸イオン電極は,使用時に,硝酸イオン標準液 (0.5mgNO3-/l) に浸し,指示値が安定してか
ら電位を測定する。
(3) 本体31.の注(12)による。
(4) 本体31.の注(13)による。
(5) 参照電極の内筒液には,塩化カリウム溶液(3mol/l〜飽和溶液),外筒液には,硫酸カリウム溶
液 (0.25mol/l) を用いる。内筒液に塩化カリウム飽和溶液を使用する場合には,液温の低下で塩
化カリウムの結晶が析出して電極に固着し,そのために抵抗が大きくなる。
(6) 本体31.の注(15)に同じ。
(7) 本体31.の注(16)に同じ。
(8) 硝酸イオン電極の応答時間は,液温10〜30℃の場合,硝酸イオン濃度0.5〜10mgNO3-/lで約3
分間,10mgNO3-/l以上で約30秒間である。
(9) 硝酸イオン標準液の1mgNO3-/lと100mgNO3-/lとの電位の差は,110〜120mV (25℃) の範囲に入
り,硝酸イオンの濃度0.5mgNO3-/lから1 000mgNO3-/lの間の検量線は直線となる。
304
K 0101 : 1998
(4) 操作 操作は,次のとおり行う。
(a) 試料100ml(10)をビーカー200mlにとり,りん酸塩緩衝液 (pH6.8) 5mlを加え,液温を(3)(c)の液温の
±1℃に調節する。
(b) (3)(b)及び(c)の操作を行い,検量線から硝酸イオンの濃度を求め,試料中の硝酸イオンの濃度
(mgNO3-/l) を算出する。
注(10) 試料が酸性又はアルカリ性で,りん酸塩緩衝液を加えてもpH6.8にならない場合には,試料の
適量をとり,硫酸 (1+5) 又は水酸化ナトリウム溶液 (40g/l) であらかじめ中和した後,一定の
液量にする。
備考1. イオン濃度計の場合には,硝酸イオン標準液 (1〜100mgNO3-/l) を用いて(3)(a)〜(c)の操作を
行い,イオン濃度計の指示値を1mgNO3-/lと100mgNO3-/lになるように調節する。さらに硝酸
イオン標準液(0.5mgNO3-/l及び10mgNO3-/l)及び硝酸イオン標準液 (1 000mgNO3-/l) を用い
てイオン濃度計の指示値を確認する。
2. 主な共存物質の許容限度を,最大比率で次に示す。
SO42-, HPO42- : 3.3×104
HCO3-, CN- : 1×102
H2PO4- : 2×104
NO2- : 25
F- : 1.6×104
Br- : 7.7
PO43- : 1×104
ClO3- : 4×10-1
CO32- : 5×103
I- : 5×10-2
CH3COO- : 2.5×103
ClO4- : 1×10-3
Cl- : 2×102
:
IX. 硫化物イオンのイオン電極法 試料に水酸化ナトリウム緩衝液を加え,pH約13の強アルカリ性にし,
硫化物イオン電極を指示電極として硫化物イオンによる電位を測定し,硫化物イオンを定量する。
定量範囲:S2-0.1〜100mg/l,繰返し分析精度:変動係数で5〜20%
(1) 試薬 試薬は,次のものを用いる。
(a) 水酸化ナトリウム緩衝液 (pH13) JIS K 8576に規定する水酸化ナトリウム40gを水約400mlに溶
かし,冷却後,JIS K 9502に規定するL (+) -アスコルビン酸10gとJIS K 8107に規定するエチレン
ジアミン四酢酸二水素二ナトリウム二水和物9.3gとを加えて溶かし,更にJIS K 8295に規定するグ
リセリン500mlを加え,水を加えて1lとする。
(b) 硫化物イオン標準液 (100mgS2-/l) 本体40.1(1)(e)の硫化物イオン標準液 (1 000mgS2-/l) 20mlを全
量フラスコ200mlにとり,水酸化ナトリウム緩衝液 (pH13) 20ml(1)(2)を加え,水を標線まで加える。
使用時に調製する。この溶液の濃度は本体40.1(1)(e)硫化物イオン標準液 (1 000mgS2-/l) の濃度から
算出する。
(c) 硫化物イオン標準液 (10mgS2-/l) 硫化物イオン標準液 (100mgS2-/l) 20mlを全量フラスコ200mlに
とり,水酸化ナトリウム緩衝液 (pH13) 20ml(1)(2)を加え,水を標線まで加える。使用時に調製する。
この溶液の濃度は本体40.1(1)(e)の硫化物イオン標準液 (1 000mgS2-/l) の濃度から算出する。
(d) 硫化物イオン標準液 (1mgS2-/l) 硫化物イオン標準液 (10mgS2-/l) 20mlを全量フラスコ200mlにと
り,水酸化ナトリウム緩衝液 (pH13) 20ml(1)(2)を加え,水を標線まで加える。使用時に調製する。
この溶液の濃度は本体40.1(1)(e)の硫化物イオン標準液 (1 000mgS2-/l) の濃度から算出する。
(e) 硫化物イオン標準液 (0.1mgS2-/l) 硫化物イオン標準液 (1mgS2-/l) 20mlを全量フラスコ200mlにと
り,水酸化ナトリウム緩衝液 (pH13) 20ml(1)(2)を加え,水を標線まで加える。使用時に調製する。
305
K 0101 : 1998
この溶液の濃度は本体40.1(1)(e)の硫化物イオン標準液 (1 000mgS2-/l) の濃度から算出する。
注(1) 水酸化ナトリウム緩衝液 (pH13) の添加量は,硫化物イオン標準液100ml中に水酸化ナトリウ
ム緩衝液 (pH13) 10mlが含まれるように添加する。
(2) 測定時のpHは12以上の一定のアルカリの濃度とする。
(2) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 電位差計 本体31.2(2)(a)による。
(b) 指示電極 硫化物イオン電極(3)
(c) 参照電極 本体32.4(2)(c)による。
(d) マグネチックスターラー 本体31.2(2)(d)による。
注(3) 硫化物イオン電極には,硫化銀の固体膜電極が用いられている。
(3) 検量線の作成 検量線の作成は,次のとおり行う。
(a) 硫化物イオン標準液 (0.1mgS2-/l) 100mlをビーカー200mlにとり,水10ml(4)を加える。
(b) これに指示電極(5)(6)と参照電極(7)(8)とを浸し,マグネチックスターラー(9)で泡が電極に触れない程
度に強くかき混ぜる(10)。
(c) 液温をはかり,電位差計で電位を測定する(11)。
(d) 硫化物イオン標準液 (1mgS2-/l) 100ml,硫化物イオン標準液 (10mgS2-/l) 100ml及び硫化物イオン標
準液 (100mgS2-/l) 100mlをそれぞれビーカー200mlにとり,水10ml(4)を加える。それぞれの硫化物
イオン標準液の液温を(d)の液温の±1℃に調節して(b)及び(c)の操作を行い,硫化物イオン標準液 (1
〜100mgS2-/l) の電位を測定する。
(e) 片対数紙の対数軸に硫化物イオンの濃度を,均等軸に電位をとり,硫化物イオンの濃度 (mgS2-/l) と
電位との関係線を作成する(12)。
注(4) 水10mlは,試料の測定時試料100ml当たり水酸化ナトリウム緩衝液 (pH13) 10mlを添加するの
で,この液量と合わせるために添加する。
(5) 硫化物イオン電極は,使用時に硫化物イオン標準液 (0.1mgS2-/l) に浸し,指示値が安定してか
ら電位を測定する。
(6) 指示電極(硫化物イオン電極)の感応膜が汚れると,検量線の電位こう配が小さくなり応答速
度も遅くなる。感応膜の汚れは,細かい研磨紙(1 000番程度)に水数滴を滴加し,感応膜を垂
直に保って円を描くようにして磨き,柔らかい紙でぬぐい取り,水で洗浄する。
(7) 本体31.の注(13)による。
(8) 本体31.の注(14)による。
(9) 本体31.の注(15)による。
(10) 本体31.の注(16)による。
(11) 応答時間は,硫化物イオン標準液 (0.1mgS2-/l) で約3分間,1mgS2-/lで1〜2分間,100mgS2-/l
で約1分間である。
(12) 硫化物イオン標準液 (0.1mgS2-/l) は約−680mV,硫化物イオン標準液 (100mgS2-/l) では約−
770mVになり,この間の検量線は直線になる。
(4) 操作 操作は,次のとおり行う。
(a) 試料100mlをビーカー200mlにとり,水酸化ナトリウム緩衝液 (pH13) 10mlを加え,液温を(3)(c)の
液温の±1℃に調節する。
(b) (3)(b)及び(c)の操作を行って検量線から硫化物イオンの量を求め,試料中の硫化物イオンの濃度
306
K 0101 : 1998
(mgS2-/l) を算出する。
備考1. イオン電極法では,懸濁状の硫化物は定量できない。
2. 主な共存物質の許容限度を,最大比率で次に示す。
Cl-, Br-, I-, NO3-:104
X. 硫酸イオンの硫酸バリウム比濁法 試料に安定剤と塩化バリウムを加え,一定の条件の下で硫酸イオ
ンと反応させて硫酸バリウムとし,比濁して硫酸イオンを定量する。
定量範囲:SO42-1〜5mg,繰返し分析精度:変動係数で10%
(1) 試薬 試薬は,次のものを用いる。
(a) グリセリン溶液 (1+1) JIS K 8295に規定するグリセリンを用いて調製する。
(b) 塩化ナトリウム溶液 JIS K 8150に規定する塩化ナトリウム240gをJIS K 8180に規定する塩酸
20mlと水に溶かして1lとする。
(c) 塩化バリウム JIS K 8155に規定する塩化バリウム二水和物をふるい分け,目開き710μmのふるい
を通り,500μmのふるいに止まるもの。
(d) 硫酸イオン標準液 (1mgSO42-/ml) 本体42.1(1)(f)による。
(2) 装置 装置は,次のとおりとする。
(a) 光度計 分光光度計又は光電光度計
(b) マグネチックスターラー
(3) 操作 操作は,次のとおり行う。
(a) ろ過した試料(1)の適量(SO42-として1〜5mgを含む。)を2個のコニカルビーカー100mlにとり,水
を加えてそれぞれ50mlとする。
(b) それぞれにグリセリン溶液 (1+1) 10mlと塩化ナトリウム溶液5mlとを加え(2),マグネチックスタ
ーラーを用いてかき混ぜる。
(c) 一方のコニカルビーカーに塩化バリウム0.3gを加え(2),それぞれ1分間引き続いてかき混ぜた後,
4分間放置し,再び15秒間かき混ぜる。
(d) 直ちにこの液を吸収セルに移し,塩化バリウムを加えない方を対照液とし,1分間以内に波長450nm
付近の吸光度(3)を測定する。
(e) 空試験として水50mlずつを2個のコニカルビーカーにとり,(b)〜(d)の操作を行って試料について
得た吸光度を補正する。
(f) 検量線から硫酸イオンの量を求め,試料中の硫酸イオンの濃度 (mgSO42-/l) を算出する。
検量線 硫酸イオン標準液 (1mgSO42-/ml) 1〜5mlを段階的にとり,水を加えて50mlとし,(b)〜(e)
の操作を行って硫酸イオン (SO42-) の量と吸光度との関係線を作成する。
注(1) 酸性又はアルカリ性の試料の場合は,pHを約7に調節する。
(2) このときのpHは1.4〜1.6になる。
(3) 見掛けの吸光度
307
K 0101 : 1998
付表1 引用規格
JIS B 7132 液浸系レンズ用生物顕微鏡
JIS B 7147 生物顕微鏡用対物レンズ
JIS B 7148 顕微鏡用接眼レンズ
JIS B 7411 一般用ガラス製棒状温度計
JIS C 1102-1 直動式指示電気計器 第1部:定義及び共通する要求事項
JIS C 1102-2 直動式指示電気計器 第2部:電流計及び電圧計に対する要求事項
JIS K 0010 標準物質−標準液−銅
JIS K 0011 亜鉛標準液
JIS K 0012 カドミウム標準液
JIS K 0013 ニッケル標準液
JIS K 0015 鉛標準液
JIS K 0016 鉄標準液
JIS K 0018 標準物質−pH標準液−しゅう酸塩
JIS K 0019 標準物質−pH標準液−フタル酸塩
JIS K 0020 標準物質−pH標準液−中性りん酸塩
JIS K 0021 標準物質−pH標準液−ほう酸塩
JIS K 0022 標準物質−pH標準液−炭酸塩
JIS K 0024 標準物質−標準液−クロム
JIS K 0026 標準物質−標準液−ひ素
JIS K 0027 標準物質−標準液−マンガン
JIS K 0028 硫酸イオン標準液
JIS K 0029 塩化物イオン標準液
JIS K 0030 ふっ化物イオン標準液
JIS K 0031 硝酸イオン標準液
JIS K 0032 亜硝酸イオン標準液
JIS K 0033 標準物質−標準液−りん酸イオン
JIS K 0034 標準物質−標準液−アンモニウムイオン
JIS K 0050 化学分析方法通則
JIS K 0094 工業用水・工場排水の試料採取方法
JIS K 0102 工場排水試験方法
JIS K 0114 ガスクロマトグラフ分析通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0117 赤外分光分析方法通則
JIS K 0121 原子吸光分析通則
JIS K 0122 イオン電極測定方法通則
JIS K 0127 イオンクロマトグラフ分析通則
JIS K 0211 分析化学用語(基礎部門)
308
K 0101 : 1998
JIS K 0215 分析化学用語(分析機器部門)
JIS K 0550 超純水中の細菌数試験方法
JIS K 0552 超純水の電気伝導率試験方法
JIS K 0557 用水・排水の試験に用いる水
JIS K 0805 有機体炭素 (TOC) 自動計測器
JIS K 0950 プラスチック製滅菌シャーレ
JIS K 0970 プッシュボタン式液体用微量体積計
JIS K 1101 酸素
JIS K 1105 アルゴン
JIS K 1107 高純度窒素
JIS K 2251 原油及び石油製品−試料採取方法
JIS K 8005 容量分析用標準物質
JIS K 8012 亜鉛(試薬)
JIS K 8013 亜鉛粉末(試薬)
JIS K 8019 亜硝酸ナトリウム(試薬)
JIS K 8032 アセトニトリル(試薬)
JIS K 8034 アセトン(試薬)
JIS K 8044 三酸化二ひ素(試薬)
JIS K 8046 メタ亜ひ酸ナトリウム(試薬)
JIS K 8048 4-アミノアンチピリン(試薬)
JIS K 8051 3-メチル−1-ブタノール(試薬)
JIS K 8059 亜硫酸水素ナトリウム(試薬)
JIS K 8061 亜硫酸ナトリウム(試薬)
JIS K 8069 アルミニウム(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8101 エタノール (99.5) (試薬)
JIS K 8102 エタノール (95) (試薬)
JIS K 8103 ジエチルエーテル(試薬)
JIS K 8107 エチレンジアミン四酢酸二水素二ナトリウム二水和物(試薬)
JIS K 8116 塩化アンモニウム(試薬)
JIS K 8121 塩化カリウム(試薬)
JIS K 8122 塩化カルシウム二水和物(試薬)
JIS K 8123 塩化カルシウム(試薬)
JIS K 8124 塩化カルシウム(乾燥用)(試薬)
JIS K 8129 塩化コバルト (II) 六水和物(試薬)
JIS K 8132 塩化ストロンチウム六水和物(試薬)
JIS K 8136 塩化すず (II) 二水和物(試薬)
JIS K 8139 塩化水銀 (II) (試薬)
JIS K 8142 塩化鉄 (III) 六水和物(試薬)
JIS K 8150 塩化ナトリウム(試薬)
309
K 0101 : 1998
JIS K 8155 塩化バリウム二水和物(試薬)
JIS K 8159 塩化マグネシウム六水和物(試薬)
JIS K 8163 ヘキサクロロ白金 (IV) 酸カリウム(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8197 N-1-ナフチルエチレンジアミン二塩酸塩(試薬)
JIS K 8201 塩酸ヒドロキシルアンモニウム(試薬)
JIS K 8202 塩化1, 10-フェナントロリニウム一水和物(試薬)
JIS K 8223 過塩素酸(試薬)
JIS K 8228 過塩素酸マグネシウム(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8249 過よう素酸カリウム(試薬)
JIS K 8252 ペルオキソ二硫酸アンモニウム(試薬)
JIS K 8253 ペルオキソ二硫酸カリウム(試薬)
JIS K 8255 硫酸カリウムアルミニウム・12水(試薬)
JIS K 8263 寒天(試薬)
JIS K 8267 ぎ酸ナトリウム(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8272 キシレンシアノールFF(試薬)
JIS K 8283 くえん酸一水和物(試薬)
JIS K 8284 くえん酸水素二アンモニウム(試薬)
JIS K 8288 くえん酸三ナトリウム二水和物(試薬)
JIS K 8289 クペロン(試薬)
JIS K 8291 グリシン(試薬)
JIS K 8295 グリセリン(試薬)
JIS K 8306 p-クレゾール(試薬)
JIS K 8312 クロム酸カリウム(試薬)
JIS K 8318 p-トルエンスルホンクロロアミドナトリウム三水和物(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8356 酢酸亜鉛二水和物(試薬)
JIS K 8359 酢酸アンモニウム(試薬)
JIS K 8361 酢酸エチル(試薬)
JIS K 8371 酢酸ナトリウム三水和物(試薬)
JIS K 8374 酢酸鉛 (II) 三水和物(試薬)
JIS K 8377 酢酸ブチル(試薬)
JIS K 8383 スクロース(試薬)
JIS K 8410 酸化カルシウム(試薬)
JIS K 8432 酸化マグネシウム(試薬)
JIS K 8443 シアン化カリウム(試薬)
310
K 0101 : 1998
JIS K 8454 N, N-ジエチルジチオカルバミド酸ナトリウム三水和物(試薬)
JIS K 8465 1, 2-ジクロロエタン(試薬)
JIS K 8474 二しゅう酸三水素カリウム二水和物(試薬)
JIS K 8488 1, 5-ジフェニルカルボノヒドラジド(試薬)
JIS K 8489 ジフェニルカルバゾン(試薬)
JIS K 8490 ジチゾン(試薬)
JIS K 8491 2, 6-ジブロモ−N-クロロ−p-ベンゾキノンモノイミン(試薬)
JIS K 8495 p-ジメチルアミノベンジリデンロダニン(試薬)
JIS K 8498 ジメチルグリオキシム(試薬)
JIS K 8500 N, N-ジメチルホルムアミド(試薬)
JIS K 8501 二亜硫酸ナトリウム(試薬)
JIS K 8506 臭化カリウム(試薬)
JIS K 8514 臭化ナトリウム(試薬)
JIS K 8517 二クロム酸カリウム(試薬)
JIS K 8519 しゅう酸二水和物(試薬)
JIS K 8522 しゅう酸カリウム一水和物(試薬)
JIS K 8528 しゅう酸ナトリウム(試薬)
JIS K 8529 臭素(試薬)
JIS K 8530 臭素酸カリウム(試薬)
JIS K 8532 L (+) −酒石酸(試薬)
JIS K 8533 ビス[ (+) −タルトラト]二アンチモン (III) 酸二カリウム三水和物(試薬)
JIS K 8536 (+) −酒石酸ナトリウムカリウム四水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8544 硝酸アルミニウム九水和物(試薬)
JIS K 8545 硝酸アンモニウム(試薬)
JIS K 8548 硝酸カリウム(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8552 硝酸コバルト (II) 六水和物(試薬)
JIS K 8558 硝酸水銀 (II) n水和物(試薬)
JIS K 8562 硝酸ナトリウム(試薬)
JIS K 8563 硝酸鉛 (II) (試薬)
JIS K 8568 硝酸マンガン (II) 六水和物(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8580 すず(試薬)
JIS K 8586 スルファニル酸(試薬)
JIS K 8588 アミド硫酸アンモニウム(試薬)
JIS K 8603 ソーダ石灰(試薬)
JIS K 8617 炭酸カルシウム(試薬)
JIS K 8622 炭酸水素ナトリウム(試薬)
311
K 0101 : 1998
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8635 チオ尿素(試薬)
JIS K 8637 チオ硫酸ナトリウム五水和物(試薬)
JIS K 8646 デキストリン水和物(試薬)
JIS K 8653 デバルダ合金(試薬)
JIS K 8659 でんぷん(溶性)(試薬)
JIS K 8660 銅(試薬)
JIS K 8669 二塩化3, 3'-ジメチルベンジジニウム(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8701 鉛(試薬)
JIS K 8721 p-ニトロフェノール(試薬)
JIS K 8722 ペンタシアノニトロシル鉄 (III) 酸ナトリウム二水和物(試薬)
JIS K 8728 ラクトース一水和物(試薬)
JIS K 8731 尿素(試薬)
JIS K 8736 エリオクロムブラックT(試薬)
JIS K 8747 バナジン (V) 酸アンモニウム(試薬)
JIS K 8775 8-キノリノール(試薬)
JIS K 8776 2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-ナフトエ酸(試薬)
JIS K 8780 ピロガロール(試薬)
JIS K 8783 二硫酸カリウム(試薬)
JIS K 8785 二りん酸ナトリウム十水和物(試薬)
JIS K 8789 1, 10-フェナントロリン一水和物(試薬)
JIS K 8798 フェノール(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8801 ヘキサシアノ鉄 (III) 酸カリウム(試薬)
JIS K 8809 フタル酸水素カリウム(試薬)
JIS K 8810 1-ブタノール(試薬)
JIS K 8819 ふっ化水素酸(試薬)
JIS K 8824 D (+) グルコース(試薬)
JIS K 8826 水酸化ナトリウム(窒素測定用)(試薬)
JIS K 8830 ウラニン(試薬)
JIS K 8832 ブルシンn水和物(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS K 8840 ブロモクレゾールグリーン(試薬)
JIS K 8842 ブロモチモールブルー(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8847 ヘキサメチレンテトラミン(試薬)
JIS K 8848 ヘキサン(試薬)
JIS K 8858 ベンゼン(試薬)
JIS K 8863 ほう酸(試薬)
312
K 0101 : 1998
JIS K 8866 四ほう酸ナトリウム十水和物(試薬)
JIS K 8872 ホルムアルデヒド液(試薬)
JIS K 8885 二酸化けい素(試薬)
JIS K 8889 メタクレゾールパープル(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8893 メチルオレンジ(試薬)
JIS K 8896 メチルレッド(試薬)
JIS K 8897 メチレンブルー(試薬)
JIS K 8900 2-ブタノン(試薬)
JIS K 8903 4-メチル-2-ペンタノン(試薬)
JIS K 8905 七モリブデン酸六アンモニウム四水和物(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8920 よう素(試薬)
JIS K 8949 硫化ナトリウム九水和物(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8953 硫酸亜鉛七水和物(試薬)
JIS K 8960 硫酸アンモニウム(試薬)
JIS K 8962 硫酸カリウム(試薬)
JIS K 8965 硫酸銀(試薬)
JIS K 8972 硫酸水素カリウム(試薬)
JIS K 8978 硫酸鉄 (II) 七水和物(試薬)
JIS K 8979 硫酸アンモニウム鉄 (II) 六水和物(試薬)
JIS K 8980 硫酸水銀 (II) (試薬)
JIS K 8982 硫酸アンモニウム鉄 (III) ・12水(試薬)
JIS K 8983 硫酸銅 (II) 五水和物(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 8990 硫酸ニッケル (II) アンモニウム六水和物(試薬)
JIS K 8992 硫酸ヒドラジニウム(試薬)
JIS K 8995 硫酸マグネシウム七水和物(試薬)
JIS K 8997 硫酸マンガン (II) 五水和物(試薬)
JIS K 9000 チオシアン酸アンモニウム(試薬)
JIS K 9003 流動パラフィン(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9007 りん酸二水素カリウム(試薬)
JIS K 9009 りん酸二水素ナトリウム二水和物(試薬)
JIS K 9016 りん酸水素二アンモニウム(試薬)
JIS K 9017 りん酸水素二カリウム(試薬)
JIS K 9019 りん酸水素二ナトリウム・12水(試薬)
JIS K 9020 りん酸水素二ナトリウム(試薬)
JIS K 9047 L-グルタミン酸(試薬)
313
K 0101 : 1998
JIS K 9062 ニッケル(試薬)
JIS K 9066 スルファニルアミド(試薬)
JIS K 9501 アジ化ナトリウム(試薬)
JIS K 9502 L (+) -アスコルビン酸(試薬)
JIS K 9512 N, N-ジエチルジチオカルバミド酸銀(試薬)
JIS K 9519 チオシアン酸水銀 (II) (試薬)
JIS K 9520 1, 1, 2, 2-テトラクロロエタン(試薬)
JIS K 9525 p-ヒドラジノベンゼンスルホン酸0.5水和物(試薬)
JIS K 9547 フェニルフルオロン(試薬)
JIS K 9548 3-メチル-1-フェニル-5-ピラゾロン(試薬)
JIS K 9550 ポリビニルアルコール(試薬)
JIS K 9569 N-ベンゾイル-N-フェニルヒドロキシルアミン(試薬)
JIS K 9703 2, 2, 4-トリメチルペンタン(試薬)
JIS K 9704 2-アミノ-2-ヒドロキシメチル-1, 3-プロパンジオール(試薬)
JIS K 9901 高純度試薬−硝酸
JIS K 9902 高純度試薬−塩酸
JIS K 9904 高純度試薬−過塩素酸
JIS K 9905 高純度試薬−硫酸
JIS P 3801 ろ紙(化学分析用)
JIS R 1301 化学分析用磁器るつぼ
JIS R 1302 化学分析用磁器蒸発ざら
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS R 3702 顕微鏡用カバーガラス
JIS R 3703 顕微鏡用スライドガラス
JIS T 1702 ふ(孵)卵器
JIS T 7322 医療用高圧蒸気滅菌装置
JIS T 7324 医療用小形高圧蒸気滅菌器
JIS T 9107 手術用ゴム手袋
JIS Z 0701 包装用シリカゲル乾燥剤
JIS Z 8719 条件等色指数−照明光条件等色度の評価方法
JIS Z 8801 試験用ふるい
JIS Z 8802 pH測定方法
314
K 0101 : 1998
平成7年度 JIS K 0101 改正原案作成委員会 構成表
小委
員会
幹事
会
氏名
所属
(委員長)
○
○
並 木 博
工学院大学工学部
○
佐 藤 寿 邦
横浜国立大学工学部
○
○
岡 林 哲 夫
工業技術院標準部繊維化学規格課
相 澤 徹
通商産業省環境立地局産業施設課
○
飯 島 孝
環境庁水質保全局水質規制課
○
中 村 進
工業技術院物質工学技術研究所計測化学部
中 村 和 憲
工業技術院生命工学工業技術研究所
○
○
田 尾 博 明
工業技術院資源環境技術総合研究所水圏環
境保全部
田 中 宏 明
建設省土木研究所下水道部
柴 田 康 行
国立環境研究所化学環境部
土 屋 悦 輝
東京都立衛生研究所環境保健部
渡 辺 真利代
東京都立衛生研究所環境保健部
○
○
日 野 隆 信
千葉県衛生研究所
○
小 倉 光 夫
神奈川県環境科学センター水質環境部
○
西 尾 高 好
財団法人日本環境衛生センター東日本支局
環境科学部
○
○
坂 本 勉
財団法人日本規格協会技術部
因 幸二郎
財団法人日本規格協会技術部
○
浅 田 正 三
財団法人日本品質保証機構環境計画センタ
ー
○
○
梅 崎 芳 美
社団法人産業環境管理協会
○
横 倉 清 治
社団法人日本環境測定分析協会
(三菱マテリアル株式会社)
神 代 啓
社団法人日本化学工業協会
○
池 田 久 幸
社団法人日本分析機器工業会
(横河アナリティカルシステムズ株式会
社)
長 澤 忠 彦
社団法人日本鉄鋼連盟
(住友金属工業株式会社)
○
池 田 修 一
社団法人日本下水道協会(東京都下水道局)
土 屋 徳 之
石油連盟(興亜石油株式会社)
松 谷 成 晃
日本石鹸洗剤工業会(ライオン株式会社)
波多江 正 和
日本製紙連合会技術環境部
狩 野 久 直
日本練水株式会社研究所
久 島 俊 和
オルガノ株式会社総合研究所
○
川 瀬 晃
セイコー電子工業株式会社科学機器事業部
○
○
米 倉 茂 男
元東京都立工業技術センター
岩 崎 岩 次
社団法人日本工業用水協会
(事務局)
納 征 治
社団法人日本工業用水協会
飛 渡 祥 弘
社団法人日本工業用水協会
本 郷 秀 昭
社団法人日本工業用水協会
○は小委員会又は幹事会委員を兼任
(文責 並木 博)