K 0083:2017
(1)
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 共通事項························································································································· 3
4 対象物質及び分析方法の種類 ······························································································ 3
5 試料採取方法 ··················································································································· 5
5.1 カドミウム,鉛,ニッケル,マンガン,バナジウム,クロム,ベリリウム,ひ素及びセレンの
試料採取方法 ················································································································· 5
5.2 水素化ひ素,セレン化水素などのガス状化合物の試料採取方法 ·············································· 10
5.3 二酸化セレンなどのガス状セレン化合物の試料採取方法 ······················································· 12
6 試料溶液の調製 ··············································································································· 14
6.1 カドミウム,鉛,ニッケル,マンガン及びバナジウムの試料溶液の調製 ·································· 14
6.2 クロム試料溶液の調製 ··································································································· 15
6.3 ベリリウム試料溶液の調製 ····························································································· 17
6.4 ひ素及びセレンの試料溶液の調製····················································································· 18
6.5 ガス状ひ素化合物及びガス状セレン化合物(水素化ひ素,セレン化水素など)の試料溶液の調製 · 19
6.6 燃焼排ガス中ガス状セレン化合物(二酸化セレンなど)の試料溶液の調製 ······························· 19
7 カドミウムの分析方法 ······································································································ 20
7.1 一般 ··························································································································· 20
7.2 フレーム原子吸光法 ······································································································ 20
7.3 電気加熱原子吸光法 ······································································································ 22
7.4 ICP発光分光分析法······································································································· 23
7.5 ICP質量分析法 ············································································································ 25
8 鉛の分析方法 ·················································································································· 27
8.1 一般 ··························································································································· 27
8.2 フレーム原子吸光法 ······································································································ 27
8.3 電気加熱原子吸光法 ······································································································ 28
8.4 ICP発光分光分析法······································································································· 29
8.5 ICP質量分析法 ············································································································ 30
9 ニッケルの分析方法 ········································································································· 31
9.1 一般 ··························································································································· 31
9.2 ジメチルグリオキシム吸光光度法····················································································· 31
9.3 フレーム原子吸光法 ······································································································ 32
9.4 電気加熱原子吸光法 ······································································································ 33
9.5 ICP発光分光分析法······································································································· 34
9.6 ICP質量分析法 ············································································································ 35
K 0083:2017 目次
(2)
ページ
10 マンガンの分析方法 ······································································································· 36
10.1 一般 ·························································································································· 36
10.2 過よう素酸吸光光度法 ·································································································· 36
10.3 フレーム原子吸光法 ····································································································· 37
10.4 電気加熱原子吸光法 ····································································································· 38
10.5 ICP発光分光分析法 ····································································································· 39
10.6 ICP質量分析法 ··········································································································· 40
11 バナジウムの分析方法 ···································································································· 41
11.1 一般 ·························································································································· 41
11.2 N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法 ···················································· 41
11.3 フレーム原子吸光法 ····································································································· 42
11.4 電気加熱原子吸光法 ····································································································· 43
11.5 ICP発光分光分析法 ····································································································· 44
11.6 ICP質量分析法 ··········································································································· 44
12 クロムの分析方法 ·········································································································· 45
12.1 一般 ·························································································································· 45
12.2 ジフェニルカルバジド吸光光度法 ··················································································· 46
12.3 フレーム原子吸光法 ····································································································· 47
12.4 電気加熱原子吸光法 ····································································································· 48
12.5 ICP発光分光分析法 ····································································································· 49
12.6 ICP質量分析法 ··········································································································· 50
13 ベリリウムの分析方法 ···································································································· 51
13.1 一般 ·························································································································· 51
13.2 フレーム原子吸光法 ····································································································· 51
13.3 電気加熱原子吸光法 ····································································································· 52
13.4 ICP発光分光分析法 ····································································································· 53
13.5 ICP質量分析法 ··········································································································· 54
14 ひ素の分析方法 ············································································································· 55
14.1 一般 ·························································································································· 55
14.2 ジエチルジチオカルバミド酸銀吸光光度法 ······································································· 55
14.3 水素化物発生原子吸光法 ······························································································· 58
14.4 電気加熱原子吸光法 ····································································································· 61
14.5 水素化物発生ICP発光分光分析法 ·················································································· 62
14.6 ICP質量分析法 ··········································································································· 63
15 セレンの分析方法 ·········································································································· 65
15.1 一般 ·························································································································· 65
15.2 3,3'-ジアミノベンジジン吸光光度法 ················································································ 65
15.3 水素化合物発生原子吸光法 ···························································································· 66
15.4 電気加熱原子吸光法 ····································································································· 68
K 0083:2017
(3)
ページ
15.5 ジアミノナフタレン蛍光光度法 ······················································································ 69
15.6 水素化合物発生ICP発光分光分析法 ··············································································· 70
15.7 ICP質量分析法 ··········································································································· 71
15.8 水素化合物発生ICP質量分析法 ····················································································· 72
16 計算 ···························································································································· 73
附属書A(規定)マイクロ波加熱圧力容器による試料の前処理方法··············································· 75
附属書B(規定)サイドストリームサンプリングによる排ガス中のセレンの試料採取方法 ················· 76
K 0083:2017 目次
K 0083:2017 目次
(4)
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人産業
環境管理協会(JEMAI)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日本工
業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS K 0083:2006は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
K 0083:2017
排ガス中の金属分析方法
Methods for determination of metals in flue gas
1
適用範囲
この規格は,燃料及びその他の物の燃焼,金属の製錬・加工,理化学的処理などに伴って,煙道,煙突,
ダクトなどから排出されるガス中の金属のうち,粒子状物質中のカドミウム,鉛,ニッケル,マンガン,
バナジウム,クロム,ベリリウム,ひ素及びセレン並びにガス状物質中のひ素及びセレンを分析する方法
について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0095 排ガス試料採取方法
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0120 蛍光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 0133 高周波プラズマ質量分析通則
JIS K 0557 用水・排水の試験に用いる水
JIS K 0901 気体中のダスト試料捕集用ろ過材の形状,寸法並びに性能試験方法
JIS K 0970 ピストン式ピペット
JIS K 1105 アルゴン
JIS K 1107 窒素
JIS K 8005 容量分析用標準物質
JIS K 8012 亜鉛(試薬)
JIS K 8019 亜硝酸ナトリウム(試薬)
JIS K 8027 アセチルアセトン(試薬)
JIS K 8034 アセトン(試薬)
JIS K 8044 三酸化二ひ素(試薬)
JIS K 8059 亜硫酸水素ナトリウム(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8102 エタノール(95)(試薬)
JIS K 8107 エチレンジアミン四酢酸二水素二ナトリウム二水和物(試薬)
2
K 0083:2017
JIS K 8136 塩化すず(II)二水和物(試薬)
JIS K 8142 塩化鉄(III)六水和物(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8201 塩化ヒドロキシルアンモニウム(試薬)
JIS K 8223 過塩素酸(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8231 過酸化ナトリウム(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8249 過よう素酸カリウム(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8284 くえん酸水素二アンモニウム(試薬)
JIS K 8289 クペロン(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8371 酢酸ナトリウム三水和物(試薬)
JIS K 8374 酢酸鉛(II)三水和物(試薬)
JIS K 8377 酢酸ブチル(試薬)
JIS K 8454 N,N-ジエチルジチオカルバミド酸ナトリウム三水和物(試薬)
JIS K 8464 シクロヘキサン(試薬)
JIS K 8488 1,5-ジフェニルカルボノヒドラジド(試薬)
JIS K 8498 ジメチルグリオキシム(試薬)
JIS K 8509 臭化水素酸(試薬)
JIS K 8529 臭素(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8544 硝酸アルミニウム九水和物(試薬)
JIS K 8562 硝酸ナトリウム(試薬)
JIS K 8563 硝酸鉛(II)(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8580 すず(試薬)
JIS K 8598 セレン(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8660 銅(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8701 鉛(試薬)
JIS K 8731 尿素(試薬)
JIS K 8747 バナジン(V)酸アンモニウム(試薬)
JIS K 8785 二りん酸ナトリウム十水和物(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8819 ふっ化水素酸(試薬)
JIS K 8821 ふっ化ナトリウム(試薬)
3
K 0083:2017
JIS K 8832 ブルシンn水和物(試薬)
JIS K 8842 ブロモチモールブルー(試薬)
JIS K 8889 メタクレゾールパープル(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8903 4-メチル-2-ペンタノン(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8960 硫酸アンモニウム(試薬)
JIS K 8982 硫酸アンモニウム鉄(III)・12水(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9062 ニッケル(試薬)
JIS K 9501 アジ化ナトリウム(試薬)
JIS K 9502 L(+)-アスコルビン酸(試薬)
JIS K 9512 N,N-ジエチルジチオカルバミド酸銀(試薬)
JIS K 9569 N-ベンゾイル-N-フェニルヒドロキシルアミン(試薬)
JIS K 9901 高純度試薬−硝酸
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
JIS Z 8801-2 試験用ふるい−第2部:金属製板ふるい
JIS Z 8801-3 試験用ふるい−第3部:電成ふるい
JIS Z 8808 排ガス中のダスト濃度の測定方法
3
共通事項
共通事項は,次による。
a) 試料ガス採取方法 試料ガス採取方法に共通する一般事項は,JIS K 0095及びJIS Z 8808による。
b) 化学分析法 化学分析法に共通する一般事項は,JIS K 0050による。
c) 原子吸光法及び電気加熱原子吸光法 原子吸光法及び電気加熱原子吸光法に共通する一般事項は,JIS
K 0121による。
d) 吸光光度法 吸光光度法に共通する一般事項は,JIS K 0115による。
e) 誘導結合プラズマ発光分光分析法 誘導結合プラズマ発光分光分析法(以下,ICP発光分光分析法と
いう。)に共通する一般事項は,JIS K 0116による。
f)
蛍光光度法 蛍光光度法に共通する一般事項は,JIS K 0120による。
g) 高周波プラズマ質量分析法 高周波プラズマ質量分析法(以下,ICP質量分析法という。)に共通する
一般事項は,JIS K 0133による。
h) 試薬 試薬は,日本工業規格(以下,JISという。)に規定するもので,試験に支障のないものを用い
る。JISに規定のない場合は,試験に支障のないものを用いる。
なお,複数の等級が存在する場合は,試料溶液(空試験溶液も含む。)と検量線用標準液の調製に用
いる酸との純度は同一にする。
4
対象物質及び分析方法の種類
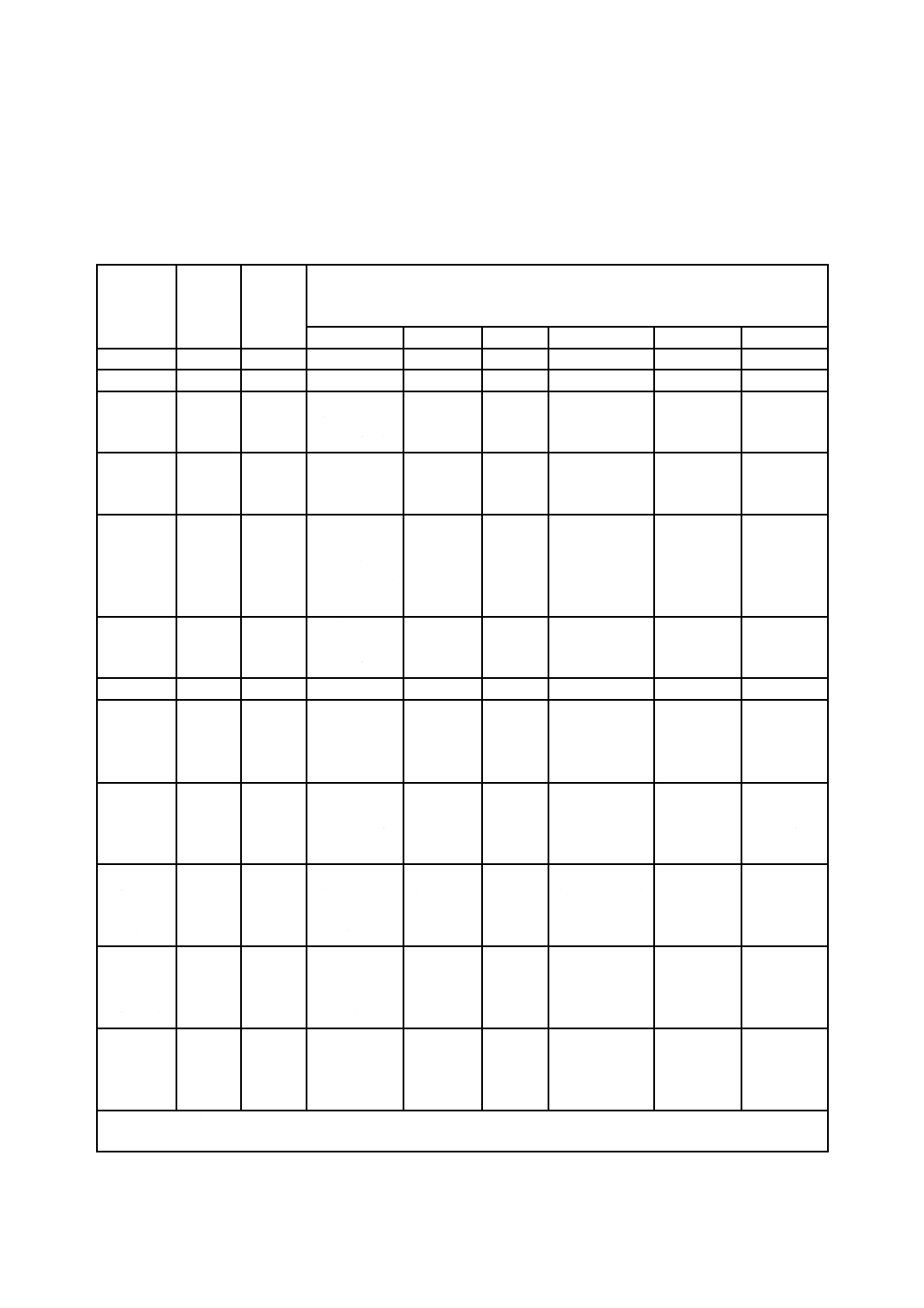
この規格の対象物質及び分析方法は,表1のとおりとする。
4
K 0083:2017
試料採取方法は,通常,5.1に規定する方法を用いるが,対象物質がひ素又はセレンで,分析試料が主に
水素化物などのガス状成分の場合には,5.2に規定する方法を用い,対象物質がセレンで,分析試料が主に
酸化物のガス状成分の場合には,5.3に規定する方法を用いる。
表1−対象物質,試料採取方法,試料溶液調製方法及び分析方法
対象物質 試料採取
方法
試料溶液
の調製方
法
分析方法
箇条番号 箇条番号
吸光光度法
FAAS
ETAAS
ICP-AES
ICP-MS
蛍光光度法
カドミウム
5.1
6.1
−
○
○
○
○
−
鉛
5.1
6.1
−
○
○
○
○
−
ニッケル
5.1
6.1
○(ジメチル
グリオキシム
吸光光度法)
○
○
○
○
−
マンガン
5.1
6.1
○(過よう素
酸吸光光度
法)
○
○
○
○
−
バナジウム
5.1
6.1
○(N-ベンゾ
イル-N-フェ
ニルヒドロキ
シルアミン吸
光光度法)
○
○
○
○
−
クロム
5.1
6.2
○(ジフェニ
ルカルバジド
吸光光度法)
○
○
○
○
−
ベリリウム
5.1
6.3
−
○
○
○
○
−
ひ素
5.1
6.4
○(ジエチル
ジチオカルバ
ミド酸銀吸光
光度法)
○
(HG/AAS)
○
○
(HG/ICP-AES)
○
−
セレン
5.1
6.4
○(3,3'-ジア
ミノベンジジ
ン吸光光度
法)
○
(HG/AAS)
○
○
(HG/ICP-AES)
○
(ICP-MS,
HG/ICP-MS)
○(ジアミノ
ナフタレン
蛍光光度法)
ガス状ひ素
化合物(水
素化ひ素な
ど)
5.2
6.5
○(ジエチル
ジチオカルバ
ミド酸銀吸光
光度法)
○
(HG/AAS)
○
○
(HG/ICP-AES)
○
−
ガス状セレ
ン化合物
(セレン化
水素など)
5.2
6.5
○(3,3'-ジア
ミノベンジジ
ン吸光光度
法)
○
(HG/AAS)
○
○
(HG/ICP-AES)
○
(ICP-MS,
HG/ICP-MS)
○(ジアミノ
ナフタレン
蛍光光度法)
ガス状セレ
ン化合物
(二酸化セ
レンなど)
5.3
6.6
○(3,3'-ジア
ミノベンジジ
ン吸光光度
法)
○
(HG/AAS)
○
○
(HG/ICP-AES)
○
(ICP-MS,
HG/ICP-MS)
○(ジアミノ
ナフタレン
蛍光光度法)
HG:水素化(合)物発生法,FAAS:フレーム原子吸光法,ETAAS:電気加熱原子吸光法,ICP-AES:ICP発光分
光分析法,ICP-MS:ICP質量分析法
5
K 0083:2017
5
試料採取方法
5.1
カドミウム,鉛,ニッケル,マンガン,バナジウム,クロム,ベリリウム,ひ素及びセレンの試料
採取方法
5.1.1
測定位置の選定
測定位置は,JIS Z 8808の5.1(測定位置)による。
5.1.2
測定点の選定
測定点は,JIS Z 8808の5.3(測定点)による。
5.1.3
試料採取装置
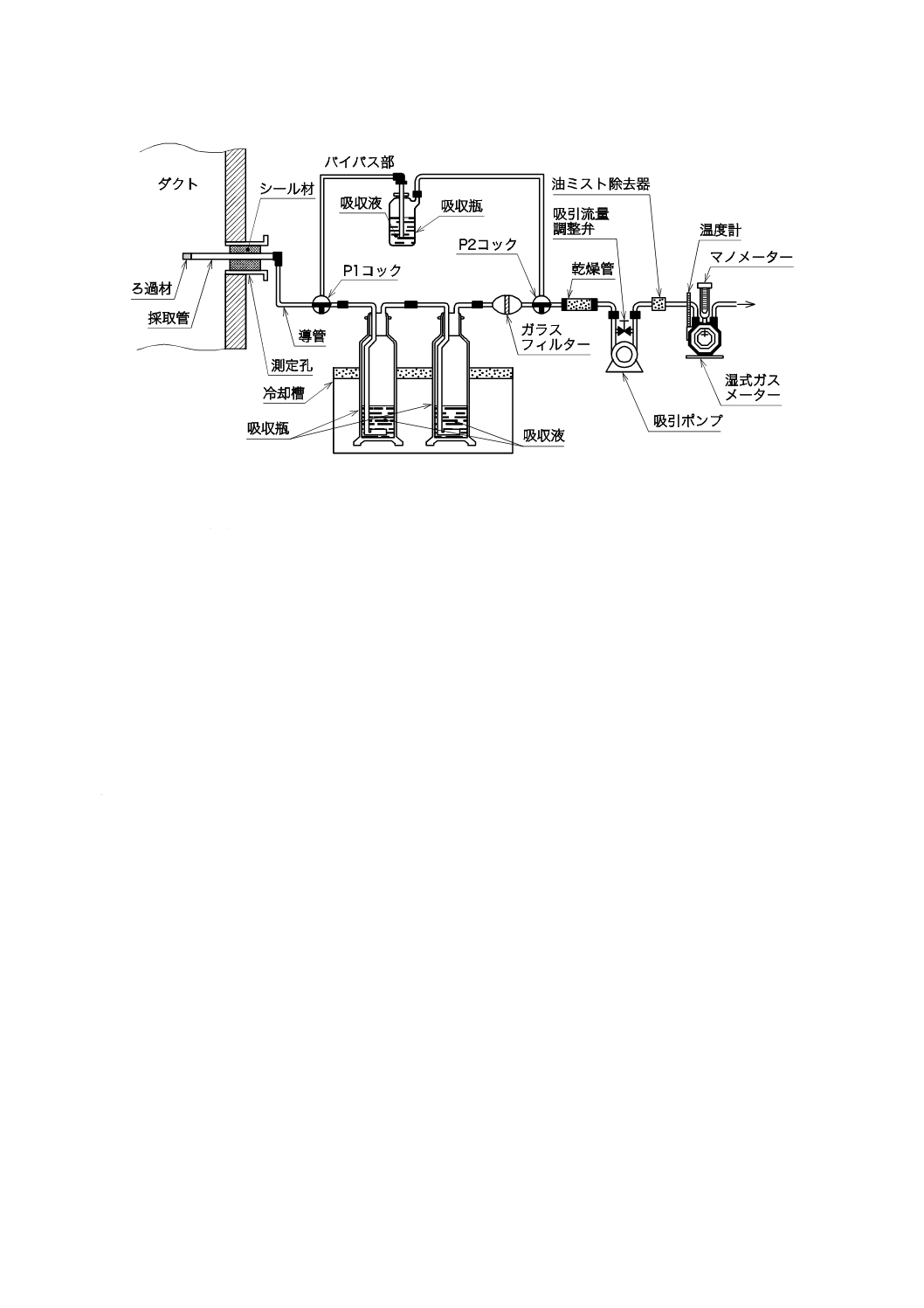
試料採取装置は,JIS Z 8808の9.1(ダスト試料採取装置の種類)a) に規定する普通形試料採取装置又
はb) に規定する平衡形試料採取装置に,ガス吸収部を接続したものを用いる。
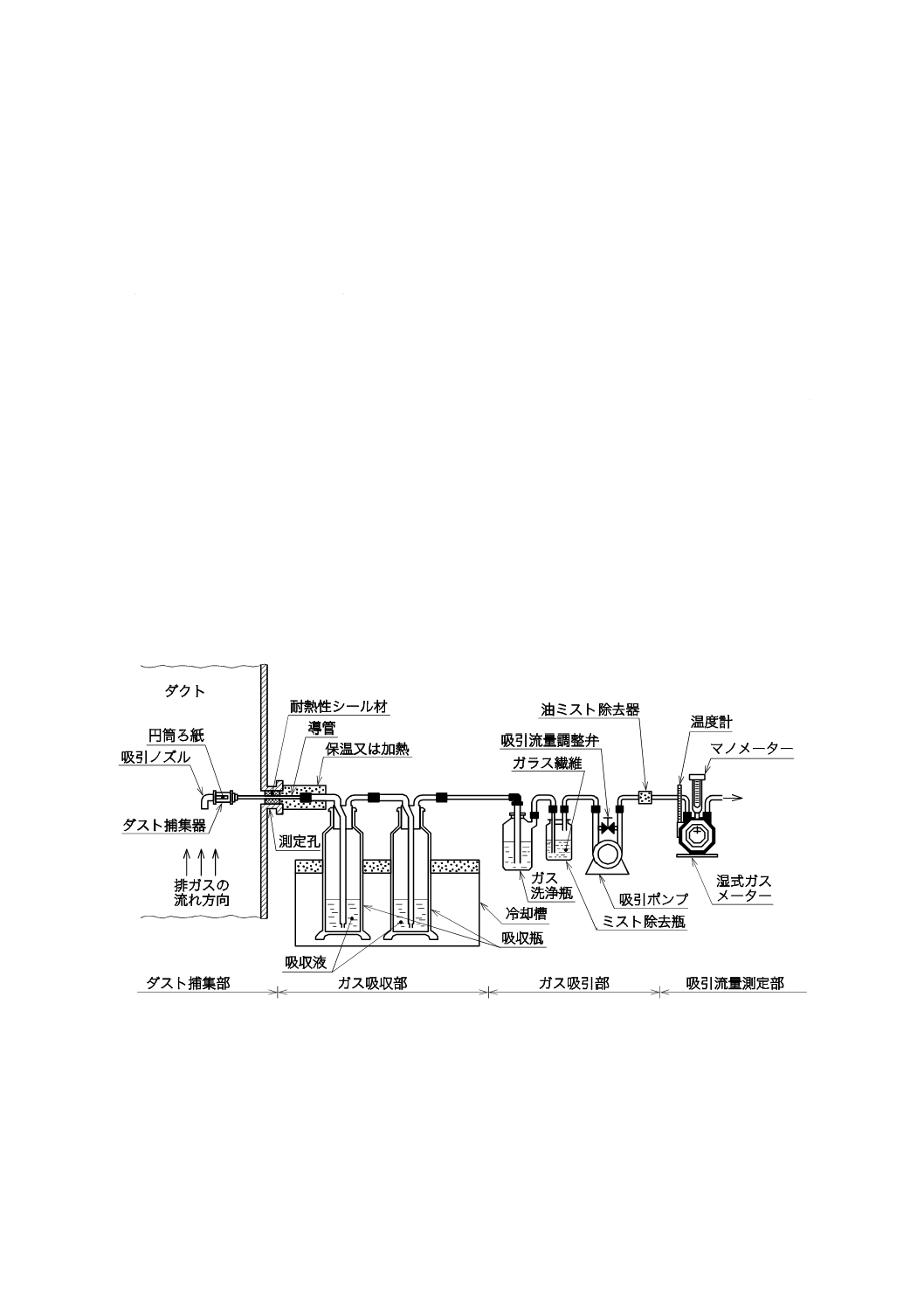
ガス吸収部は,分析成分が揮散してガス化したものを捕集する部分で,図1〜図4に示すように,ダス
ト捕集部とガス吸引部との間に接続する。ただし,排ガス温度が予想される化合物の融点より十分低く,
ヒューム及びミスト状の化合物が漏れ出していないことがこの規格の試料採取方法で確認された場合は,
ガス吸収部は接続しなくてもよい。
a) 試料採取装置 試料採取装置は,ダスト捕集部,ガス吸収部,ガス吸引部及び吸引流量測定部で構成
する。ダスト捕集部は,ダスト捕集器の位置によって1形と2形とに区別し,1形はダスト捕集器を
ダクト内に置き,2形はダスト捕集器をダクト外に置く。いずれを用いてもよい。普通形試料採取装
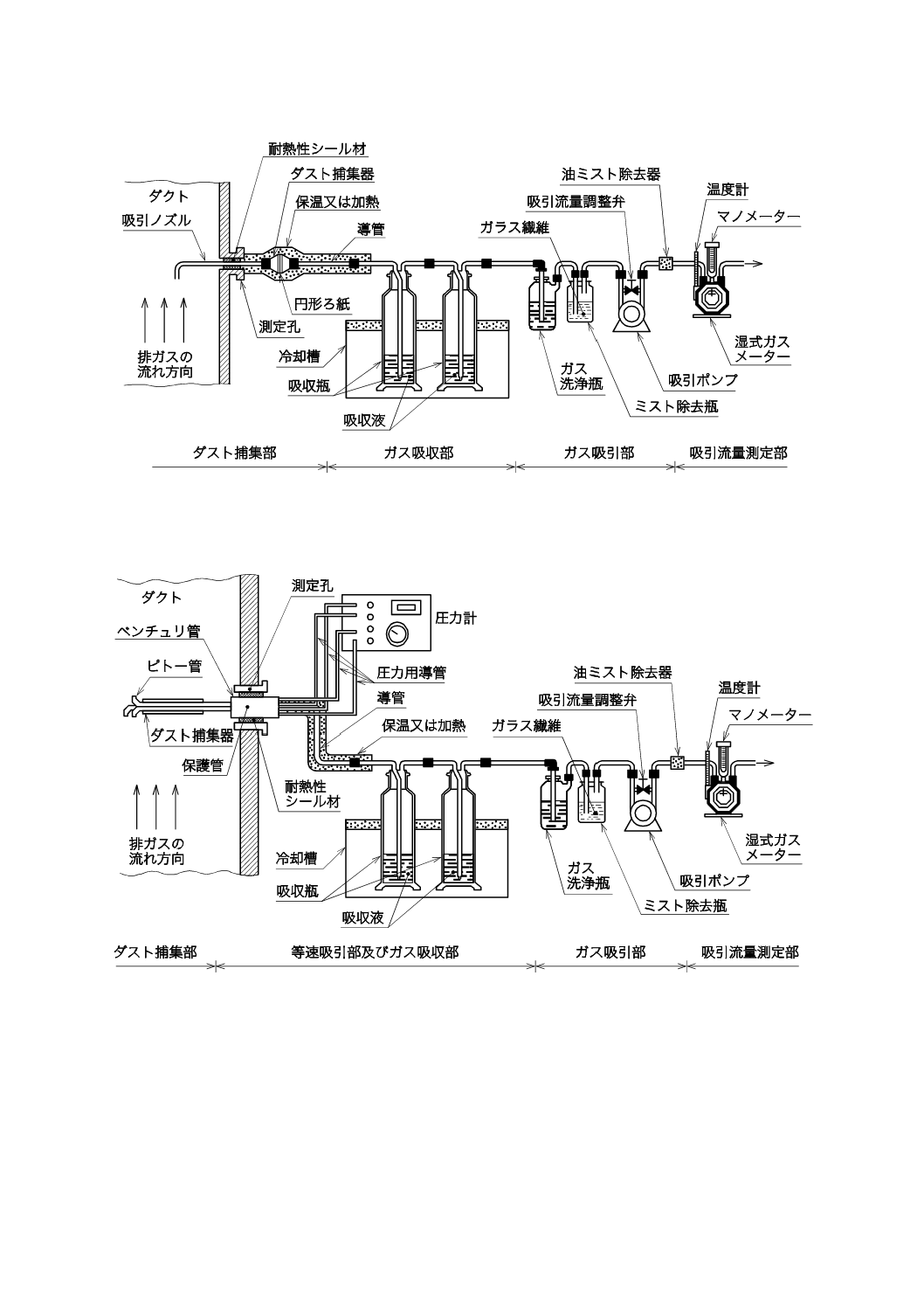
置の構成例を,1形の場合は図1に,2形の場合は図2に示す。また,平衡形試料採取装置の構成例を,
手動採取の場合は図3に,自動採取の場合は図4に示す。
図1−普通形試料採取装置の構成例(1形)(一例)
6
K 0083:2017
図2−普通形試料採取装置の構成例(2形)(一例)
図3−平衡形手動試料採取装置(動圧式)の構成例(一例)
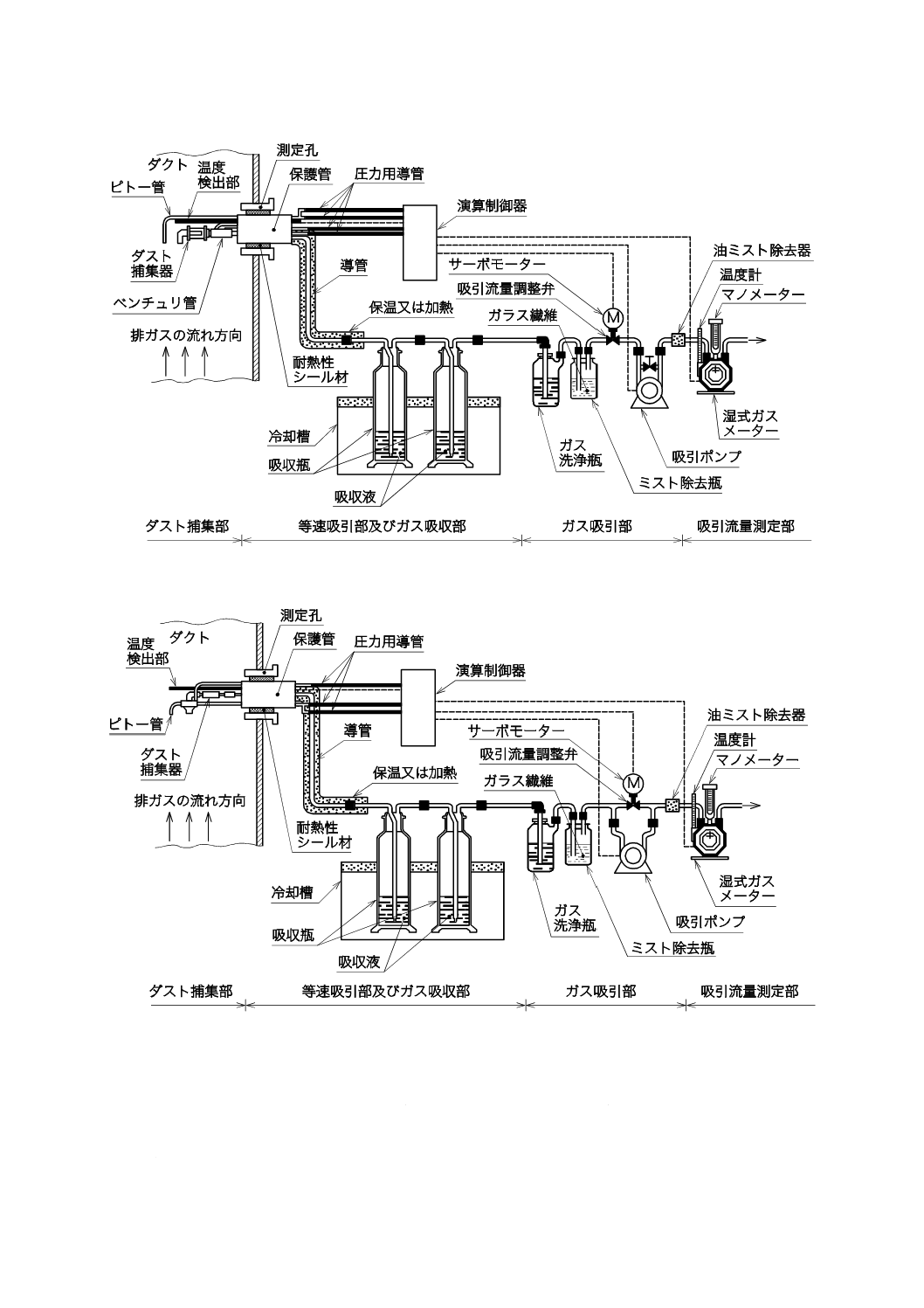
7
K 0083:2017
a) 動圧式の構成例(一例)
b) 静圧式の構成例(一例)
図4−平衡形自動試料採取装置の構成例
b) ダスト捕集部 JIS Z 8808の9.3.1.2(ダスト捕集部)及び9.3.2.2(ダスト捕集部)の規定による。た
だし,ステンレス鋼を用いた吸引ノズルは,成分分析上の妨害となるおそれがあるので用いない。
1) ダスト捕集器 ダスト捕集器は,JIS Z 8808の9.3.1.2 b)(ダスト捕集器)に規定するろ紙を用いる
8
K 0083:2017
ダスト捕集器による。ただし,ろ紙は,JIS K 0901に規定するダスト試料捕集用ろ過材の性能試験
方法によって捕集率,圧力損失,吸湿率及び試験項目の含有量が明らかなものを選定する。
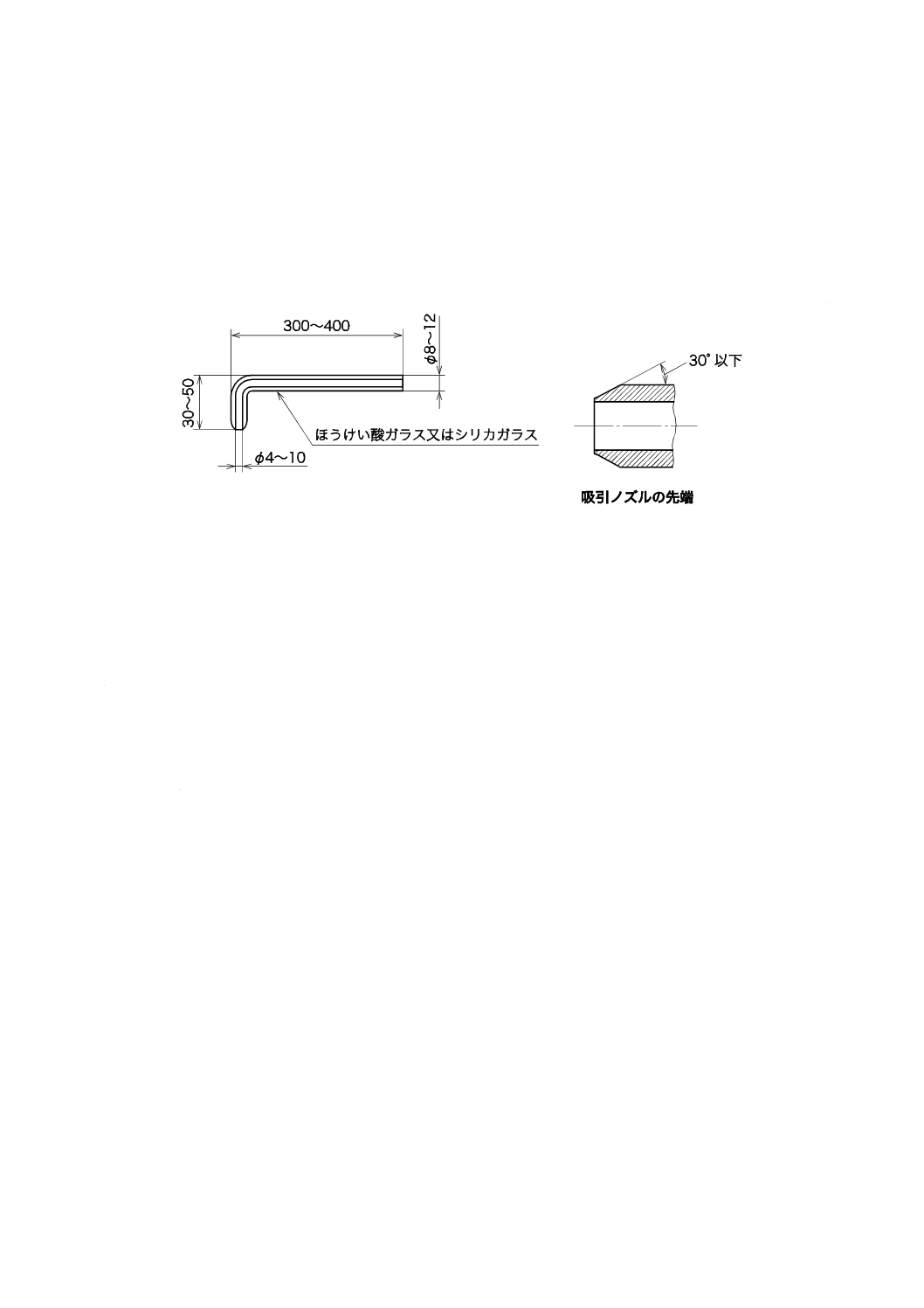
2) 吸引ノズル 吸引ノズルの例を図5に示す。この場合,材質はJIS K 0095の6.2(材質)に,構造
はJIS Z 8808の9.3.1.2 a)(吸引ノズル)による。吸引ノズルは,あらかじめ水で洗浄し,乾燥して
保存する。
単位 mm
図5−吸引ノズル(一例)
3) 導管 導管は,ダスト捕集器からガス吸収瓶までの間を接続するもので,材質はJIS K 0095の6.6
(導管)及びJIS K 0095の6.2(材質)によって,通常ほうけい酸ガラス製のものを用いる。
4) 導管,ダスト捕集器などの保温・加熱 排ガス中の水分が導管などに凝縮しないように,必要に応
じて導管,ダスト捕集器などを保温又は加熱する。
c) ガス吸収部 ガス吸収部は,吸収瓶及び冷却槽で構成する。
1) 試薬 試薬は,次による。
硝酸(1+13)JIS K 8541に規定する硝酸又はそれ以上の純度のものを用いて調製する。
塩酸(1+11)JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
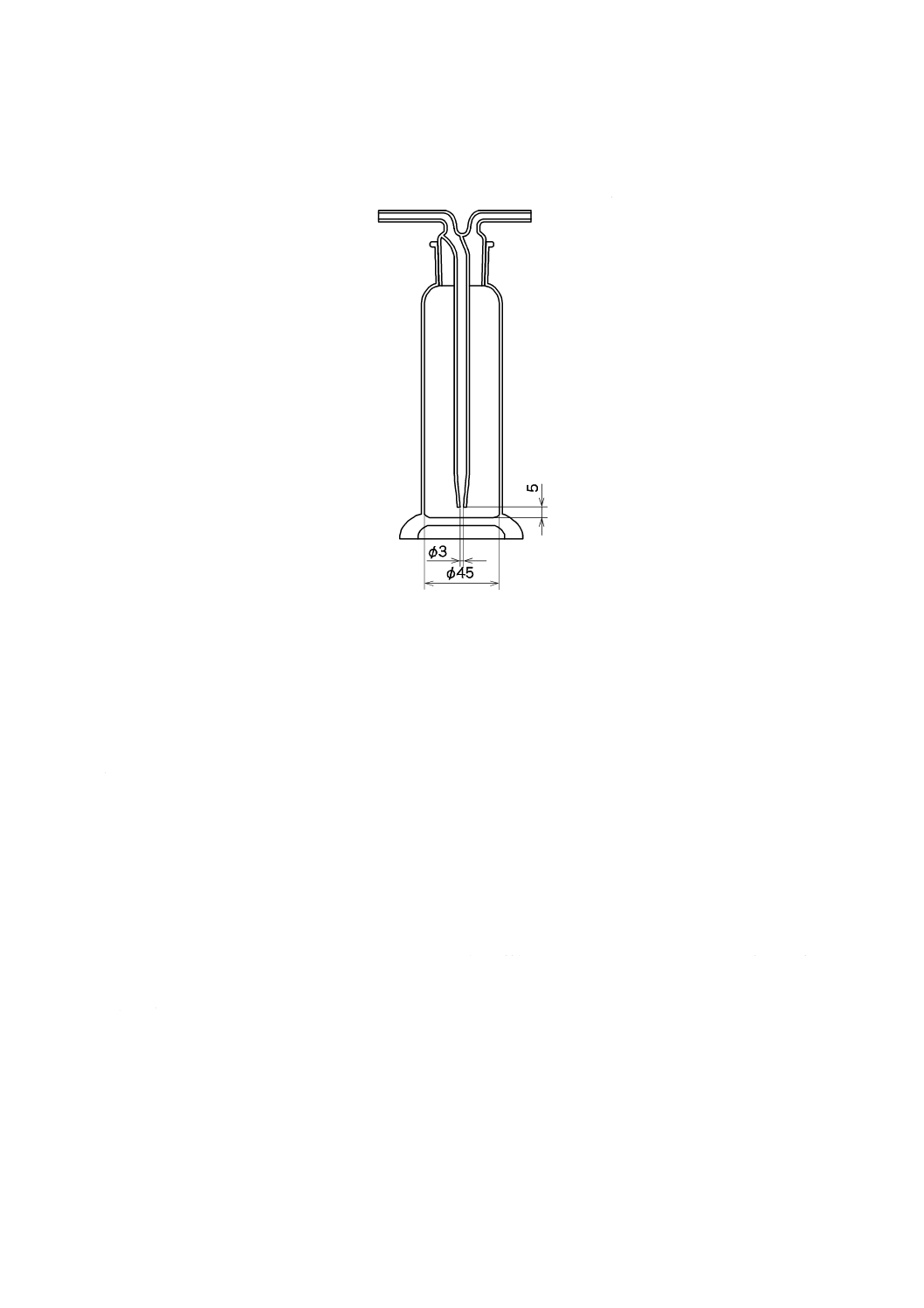
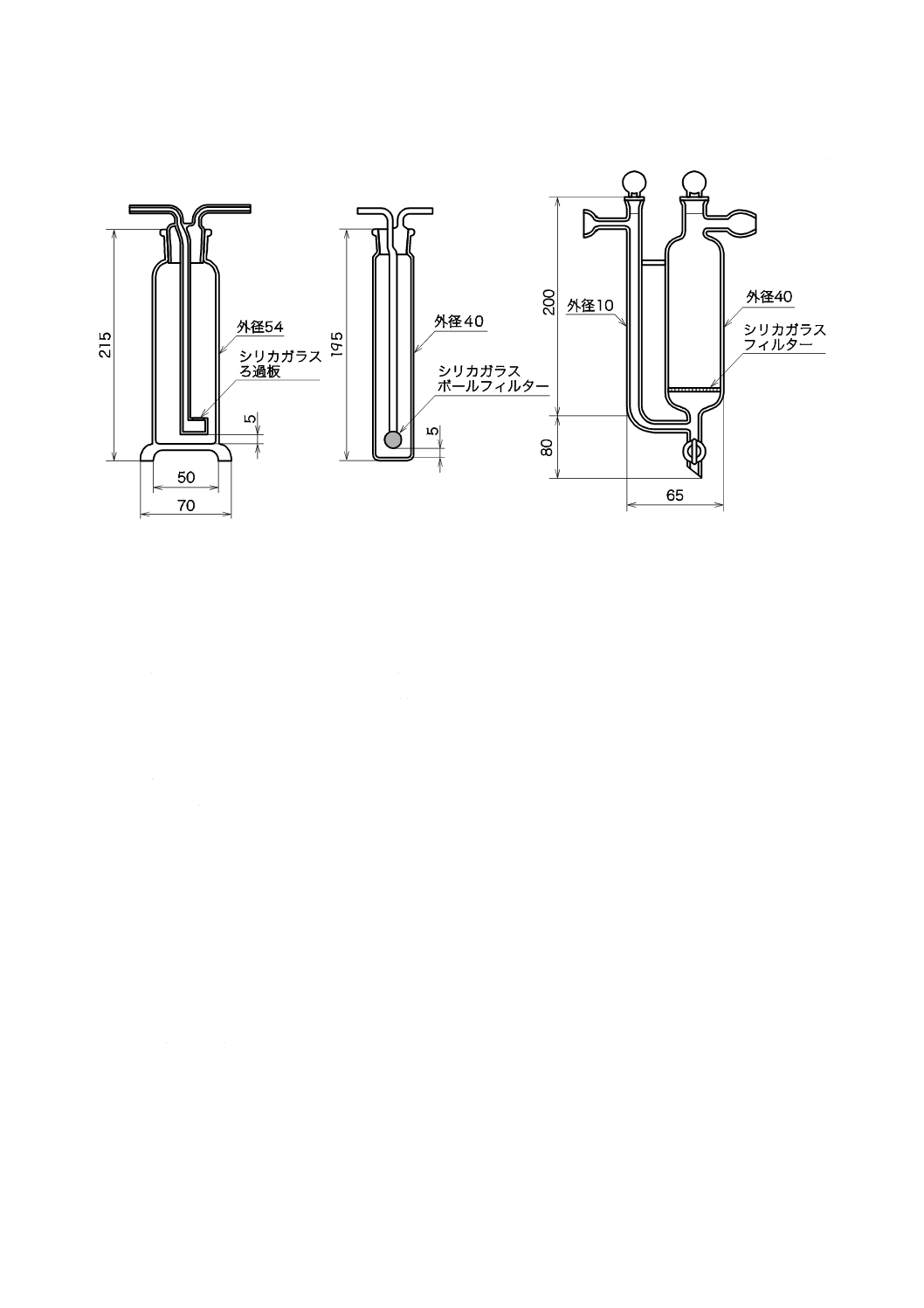
2) 吸収瓶 吸収瓶は,250〜300 mLとし,その一例を図6に示す(材質はほうけい酸ガラスとする)。
吸収瓶はあらかじめ硝酸(1+13)又は塩酸(1+11),及び水で洗浄して乾燥しておく。吸収瓶は2
本以上を用意する。第3の吸収瓶を使う場合には,ガス,ヒューム及びミスト状の化合物が漏れ出
していないことを第1及び第2吸収瓶と同様の定量操作を行うことによって確認する。第3の吸収
瓶の定量値が空試験値よりも大きくなった場合には,その結果を第1,第2吸収瓶の値に加える。
また,ヒューム及びミスト状の化合物が漏れ出している場合は,必要に応じて吸収瓶の後段にバッ
クアップフィルタを設置して,ヒューム及びミスト状の化合物を捕集し,同様に定量する。吸収液
は分析項目によって次のものを用いる。
2.1) カドミウム,鉛,ニッケル,マンガン,バナジウム,クロム及びベリリウムの吸収液 それぞれ
の吸収瓶に硝酸(1+13)又は塩酸(1+11)を,50 mLずつ入れる。試験方法としてICP質量分
析法を用いる場合は,吸収液に硝酸(1+13)を用いる。
2.2) ひ素の吸収液 それぞれの吸収瓶に水を50 mLずつ入れる。
なお,水素化ひ素などのガス状ひ素化合物を含む排ガス試料に対しては,5.2による。
2.3) セレンの吸収液 それぞれの吸収瓶に硝酸(1+13)を50 mLずつ入れる。
なお,セレン化水素などのガス状セレン化合物を含む排ガス試料に対しては,5.2による。二酸
9
K 0083:2017
化セレンなどのガス状セレン化合物を含む排ガス試料に対しては,5.3による。
単位 mm
図6−吸収瓶(一例)
3) 冷却槽 冷却槽は,吸収瓶を収納し,吸収液を0〜10 ℃に保つものとする。一般に氷を入れて冷却
する。
5.1.4
試料の採取
試料の採取は,JIS Z 8808の箇条10(ダスト試料の採取方法)の規定による。
a) 試料の採取方法の種類 試料の採取方法は,JIS Z 8808の10.2.1(ダスト試料の採取方法の種類)に
規定する方法のうち,各点採取法又は移動採取法のいずれかを用いる。一般には移動採取法を用いる。
b) 試料採取の準備
1) 等速吸引のための吸引流量の計算 普通形試料採取装置を用いる場合は,JIS Z 8808の10.3.1(等
速吸引のための吸引流量の計算)に規定する計算式を用いる。
一般に,この計算式に用いるガスメータの圧力,温度などは,排ガスの水分量を測定したときの
値を利用するが,ガス吸収部を設置した場合の冷却効果の影響を考慮して,高温の排ガスの水分量
を測定するときには,JIS Z 8808の7.1.2 a) 2)(吸湿管)の後にガス吸収部を設置した状態で測定す
る。
2) ダスト捕集器の準備 ダスト捕集器の準備は,JIS Z 8808の10.3.2(ダスト捕集器の準備)による。
分析と同時にダスト濃度を測定する場合以外は,ろ紙の乾燥,ひょう量などの操作を行う必要はな
い。
3) ガス吸収瓶の準備 ガス吸収瓶には,分析成分によって,5.1.3 c) に規定する吸収液50 mLずつを
入れ,ダスト捕集器を通ったガスを導入し,ガス中の分析成分を吸収液に捕集するように準備する。
吸収瓶の容量は,等速吸引流量によって決定する。吸引流量が多くなれば,さらに,大容量の瓶と
交換し,吸収瓶のノズルが吸収液の液面に浸るように吸収液の量を調整する。
10
K 0083:2017
注記 発泡が激しい場合には消泡板を用いてもよい。
c) 試料の採取 試料の採取は,図1〜図4に示す試料採取装置を用い,JIS Z 8808の10.4(ダスト試料
の採取)によって行う。
d) 吸引ガス量の測定 試料採取のため吸引したガス量は,JIS Z 8808の10.5(吸引ガス量の測定方法)
によって測定する。
5.2
水素化ひ素,セレン化水素などのガス状化合物の試料採取方法
5.2.1
測定位置の選定
測定位置は,JIS K 0095の5.1(採取位置)による。
5.2.2
測定点の選定
測定点は,JIS K 0095の5.2(採取点)による。
5.2.3
試料採取装置
試料採取装置は,次による。
a) 試薬 試薬は,次による。
1) 過マンガン酸カリウム溶液(5 g/L) JIS K 8247に規定する過マンガン酸カリウム0.5 gを水に溶か
して100 mLとする。
2) 臭素飽和臭化水素酸 JIS K 8509に規定する臭化水素酸1容に水12容を加えた後,JIS K 8529に規
定する臭素を加えて臭素を飽和させたもの。
b) 試料採取装置の構成 試料採取装置の構成は,JIS K 0095の6.1(装置の構成)による。試料採取装置
の構成例を図7に示す。
c) 採取管 採取管は,JIS K 0095の6.3(採取管)に規定するものを用いる。材質は,JIS K 0095の6.2
(材質)によって,通常,ほうけい酸ガラス,シリカガラス,四ふっ化エチレン樹脂などを用いる。
採取管は,あらかじめ水で洗浄し,乾燥して保存する。試料の採取において,水分が凝縮するおそれ
がある場合には,採取管からコック(P1)までの間を保温又は加熱する。また,採取管から吸収瓶ま
での距離が短い場合は,図7のP1コックからP2コックまでの間のバイパスを付けなくてもよい。
d) ろ過材 試料ガス中にばいじんなどが混入することを防ぐため,採取管の先端又は途中に適切なろ過
材を入れる。通常は,シリカウール,無アルカリガラスウールなどを用いる。
11
K 0083:2017
図7−試料採取装置の構成例(一例)
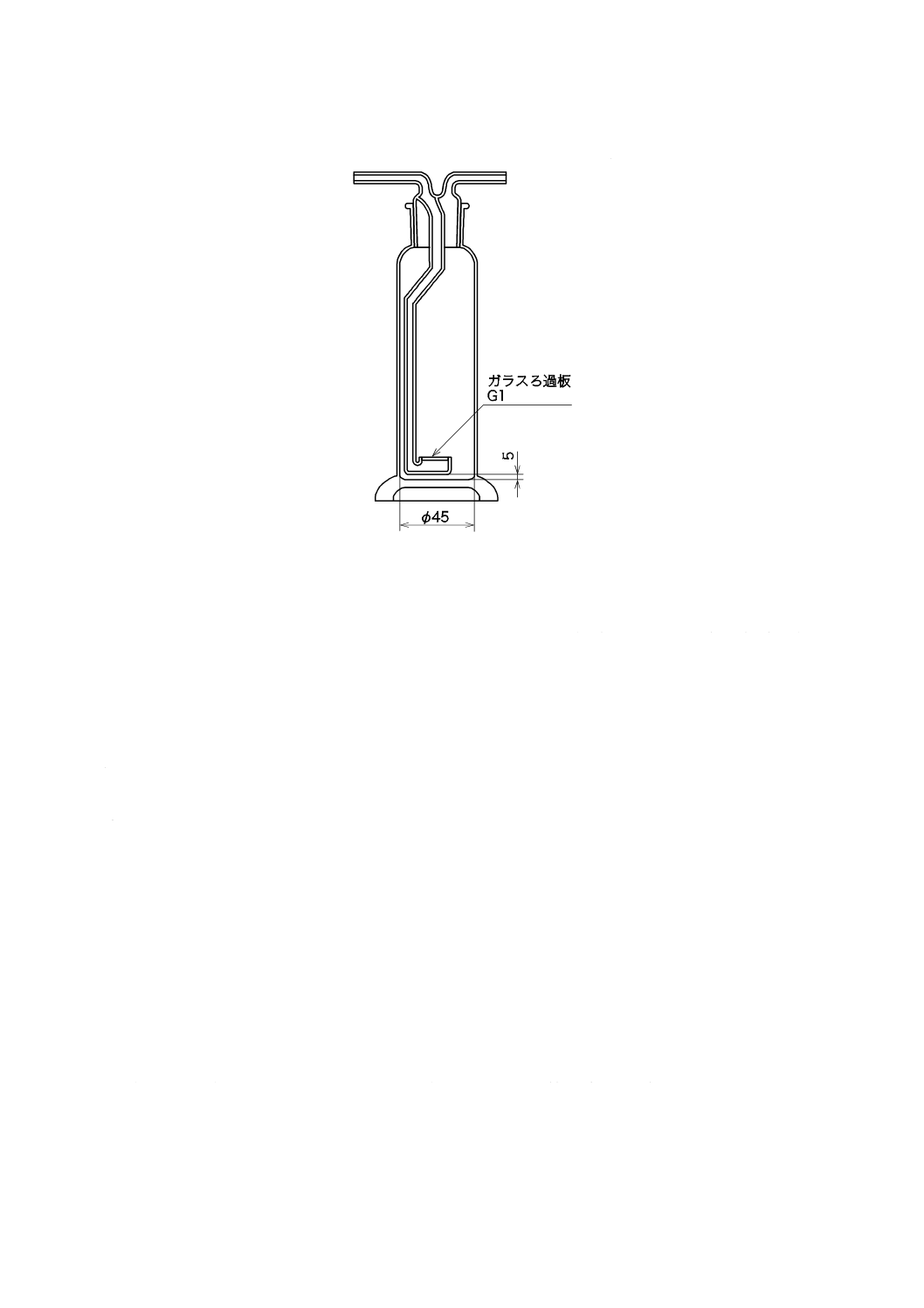
e) 吸収瓶 吸収瓶は,250〜300 mLとし,例を図8に示す。この場合,材質はほうけい酸ガラスとする。
吸収瓶はあらかじめ硝酸(1+13)又は塩酸(1+11),及び水で洗浄して乾燥しておく。吸収瓶は2
本以上を用意する。第3の吸収瓶を使う場合には,ガス状の化合物が漏れ出していないことを第1及
び第2吸収瓶と同様の定量操作を行うことによって確認する。第3の吸収瓶の定量値が空試験値より
も大きくなった場合には,その結果を第1,第2吸収瓶の値に加える。次の吸収液を用いる。
なお,第3吸収瓶の値が全体の10 %以上となった場合には,測定をやり直す。
1) 水素化ひ素などのガス状ひ素化合物の吸収液 それぞれの吸収瓶にa) 1) の過マンガン酸カリウム
溶液(5 g/L)を50 mLずつ入れる。
2) セレン化水素などのガス状セレン化合物の吸収液 それぞれの吸収瓶にa) 2) の臭素飽和臭化水素
酸を50 mLずつ入れる。
f)
冷却槽 冷却槽は,吸収瓶を収納し,吸収液の温度を0〜10 ℃に保つものとする。一般に氷を入れて
冷却する。
12
K 0083:2017
単位 mm
図8−吸収瓶(一例)
5.2.4
試料の採取
試料の採取は,図7に示す試料採取装置を用い,JIS K 0095の7.(化学分析による場合の試料採取)に
よって行う。
なお,粒子状物質中のセレンと,ガス状セレン化合物とを同時に測定する際に,各々の採取ガス流量が
異なる場合には,近接する二点の一方で粒子状物質中のセレンを採取し,もう一方でガス状セレン化合物
を採取するか,又は附属書Bのサイドストリームサンプリングによって試料を採取する。
a) 吸引ガス量の測定 試料採取のため吸引したガス量は,JIS K 0095の7.1.7(試料ガス採取量)によっ
て測定する。採取流量は,1 L/min程度にする。
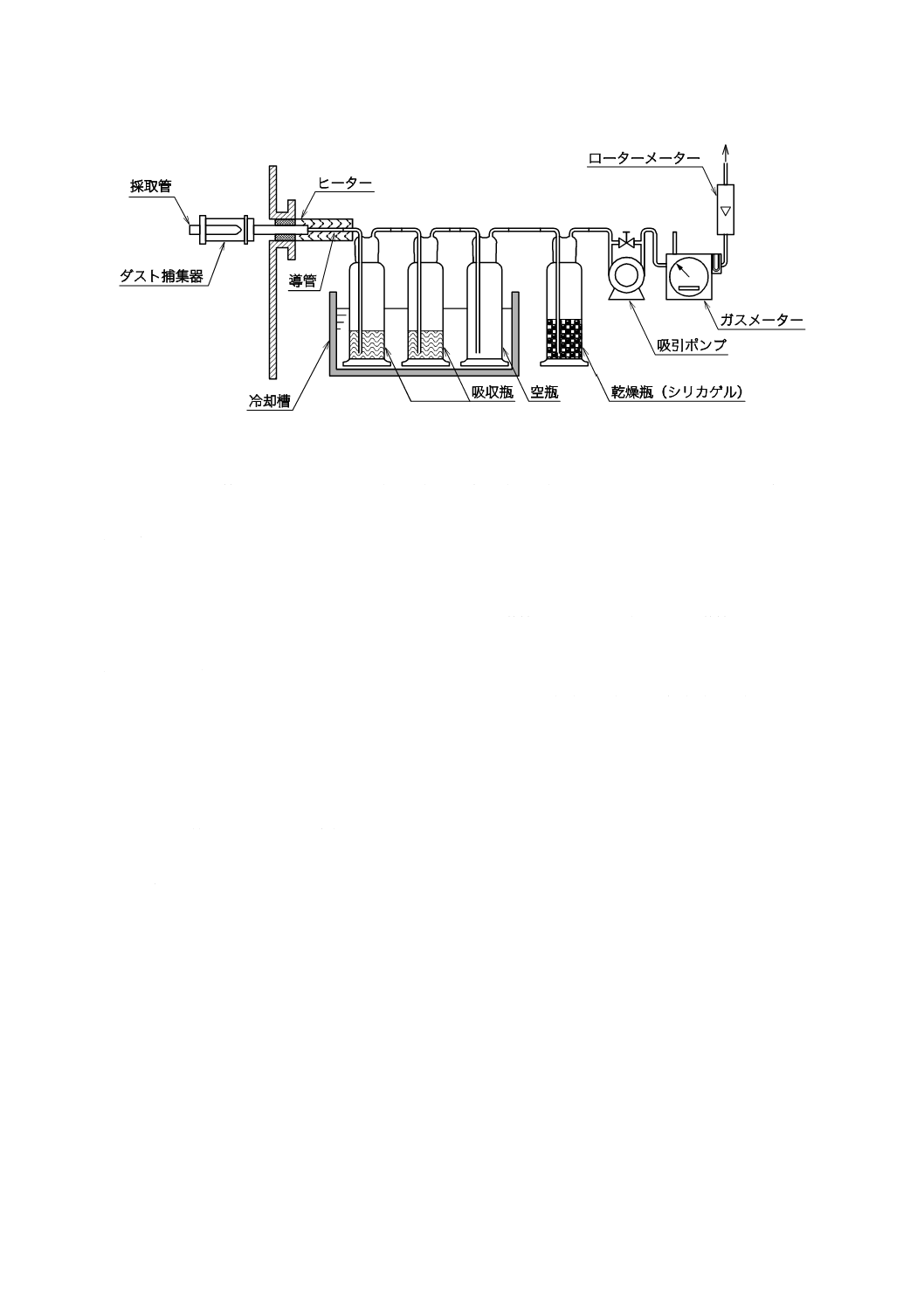
5.3
二酸化セレンなどのガス状セレン化合物の試料採取方法
5.3.1
測定位置の選定
測定位置は,JIS K 0095の5.1(採取位置)による。
5.3.2
測定点の選定
測定点は,JIS K 0095の5.2(採取点)による。
5.3.3
試料採取装置
試料採取装置は,次による。
a) 試料採取装置の構成 試料採取装置の構成は,JIS K 0095の6.1(装置の構成)による。試料採取装置
の構成例を図9に示す。ダスト捕集器の位置は,ダスト内に置く1形とし,吸収瓶のバイパスライン
は設置しない。
注記 測定前にバイパスラインにてガス置換を行うと,採取管,ダスト捕集器などにガス状二酸化セ
レンが付着し,測定誤差となるため,バイパスラインは設置しない。
13
K 0083:2017
図9−試料採取装置の構成例(一例)
b) 採取管 採取管は,あらかじめ硝酸(1+13)又は塩酸(1+11),及び水で洗浄し,乾燥して保存する。
通常,ほうけい酸ガラス,シリカガラス又は四ふっ化エチレン樹脂製とする。
c) ダスト捕集器 ダスト捕集器は,JIS Z 8808の9.3.1.2 b)(ダスト捕集器)に規定したダスト捕集器を
使用する。ろ紙には,シリカ繊維のろ紙を用いる。
d) 導管 導管は,ダスト捕集器から吸収瓶までの間を接続するもので,材質は,通常,ほうけい酸ガラ
ス,シリカガラス又は四ふっ化エチレン樹脂を用いる。導管はできるだけ短くする。導管は,約130 ℃,
又は露点温度より約20 ℃高い温度のいずれか高い方に保温又は加熱する。
e) 吸収瓶 吸収瓶は,250〜300 mLとし,例を図10に示す。材質は,ほうけい酸ガラス,シリカガラス
又は四ふっ化エチレン樹脂とする。吸収瓶は,あらかじめ硝酸(1+13)又は塩酸(1+11),及び水で
洗浄し,乾燥しておく。吸収瓶は2本以上を用意する。第3の吸収瓶を使う場合には,ガス状の化合
物が漏れ出していないことを第1及び第2吸収瓶と同様の定量操作を行うことによって確認する。第
3の吸収瓶の定量値が空試験値よりも大きくなった場合には,その結果を第1,第2吸収瓶の値に加え
る。各吸収瓶に次の吸収液を100 mL入れる。
なお,第3吸収瓶の値が全体の10 %以上となった場合には,測定をやり直す。
1) 吸収液 ビーカーに,水約500 mLをとり,JIS K 8541に規定する硝酸又はそれ以上の純度の硝酸
50 mL及びJIS K 8230に規定する過酸化水素330 mLを加え,かくはんしながら,水を加えて1 000
mLとする。
14
K 0083:2017
単位 mm
図10−吸収瓶の例
f)
空瓶 容量は250〜300 mLとし,材質は,ほうけい酸ガラス,シリカガラス,四ふっ化エチレン樹脂
又はポリプロピレンとする。空瓶は,あらかじめ硝酸(1+13)又は塩酸(1+11),及び水で洗浄し,
乾燥しておく。
注記 空瓶は,吸収液が飛散した場合の吸引ポンプの保護のため設置している。
g) 乾燥瓶 容量は250〜300 mLとし,材質は,ほうけい酸ガラス,シリカガラス,四ふっ化エチレン樹
脂又はポリプロピレンとする。乾燥瓶は,あらかじめ硝酸(1+13)又は塩酸(1+11),及び水で洗浄
し,乾燥しておく。乾燥瓶にシリカゲルを約150 g充塡し,吸収瓶後の空瓶と連結し,吸引ポンプの
上流に設置する。
h) 冷却槽 冷却槽は,吸収瓶及び空瓶を収納し,吸収液を0〜10 ℃に保つ能力をもつものとする。一般
に氷を入れて冷却する。
5.3.4
試料の採取
試料の採取は,図9に示す試料採取装置を用い,JIS K 0095の7.(化学分析による場合の試料採取)に
よって行う。
なお,粒子状物質中のセレンと,ガス状セレン化合物とを同時に測定する際に,各々の採取ガス流量が
異なる場合には,近接する二点の一方で粒子状物質中のセレンを採取し,もう一方でガス状セレン化合物
を採取するか,又は附属書Bのサイドストリームサンプリングによって試料を採取する。
a) 吸引ガス量の測定 試料採取のため吸引したガス量は,JIS K 0095の7.1.7(試料ガス採取量)によっ
て測定する。採取流量は,1 L/min程度にする。
b) 洗浄試料の保管 試料ガス採取後のダスト捕集器,導管及び吸収瓶を密閉し保管する。
6
試料溶液の調製
6.1
カドミウム,鉛,ニッケル,マンガン及びバナジウムの試料溶液の調製
15
K 0083:2017
6.1.1
試薬
試薬は,次による。
a) 硝酸(1+1) JIS K 8541に規定する硝酸又はそれ以上の純度のものを用いて調製する。
b) 過酸化水素 JIS K 8230に規定するもの。
c) 硝酸(2+98) JIS K 8541に規定する硝酸又はそれ以上の純度のものを用いて調製する。
6.1.2
操作
a) 吸収瓶を設けないでろ紙だけに試料採取した場合 操作は,次の1)〜7) 又はA.2.2 a) の1)〜4) の方
法を用いる。
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものを集める。管の内面
に付着した粉末状のものは振り出し,次に少量の硝酸(2+98)で洗い,全てをビーカー200 mLに
集める。ろ紙は適正な大きさに切り,全てを上記のビーカーに入れ,硝酸(1+1)30 mL及び過酸
化水素5 mLを加え,時計皿で覆い,加熱板又は沸騰水浴上で1時間加熱する。
2) 室温まで放冷後,時計皿を温水10 mLで洗い,洗液をろ紙5種Bでろ過する。次に,固形物がなる
べくビーカー内に残るように注意しながら,上澄み液をろ紙5種Bを用いてろ過する。
3) ビーカー内の残留物に硝酸(1+1)20 mLを加え,加熱板又は沸騰水浴上で10分間加熱する。
4) 放冷後,ろ紙5種Bでろ過し,さらに,ビーカー内の残留物を温水で洗い,洗液をろ紙5種Bでろ
過する。全てのろ液をビーカー100 mLに移す。
5) 水浴上で蒸発乾固する。
6) 水10 mL,硝酸(1+1)2 mLを加え,加熱板又は沸騰水浴上で加熱して溶かし,冷却後,全量フラ
スコ100 mLに移し入れ,水を標線まで加え,これを試料溶液とする。
7) 別に,ろ紙について1)〜6) と同様に操作して空試験溶液を調製する。
b) 吸収瓶を設けてろ紙とともに溶液にも吸収させて試料採取した場合 操作は,次の1)〜5) 又はA.2.2
b) の1)〜5) の方法を用いる。
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものについては,a) 1)〜
4) による。
2) 一方,吸収瓶中の溶液については,これをビーカー300 mLに移し入れ,導管及び吸収瓶を水10〜
20 mLで洗い,洗液をビーカー300 mLに合わせた後,硝酸(1+1)10 mL及び過酸化水素5 mLを
加え,加熱板又は沸騰水浴上で乾固近くまで加熱する。
3) 有機物の分解が困難なときには,液が淡黄色又は無色となるまで2) の操作を繰り返した後,内容
物をろ過し,ろ液をa) 4) のビーカー100 mLに入れ,ビーカー300 mL内の残留物を少量の水で洗い,
洗液もろ過してビーカー100 mLに合わせる。
4) 次に,a) 5)〜6) の操作を行う。
5) 別に,ろ紙及び吸収液について1)〜4) と同様に操作して空試験溶液を調製する。
6.2
クロム試料溶液の調製
6.2.1
試薬
試薬は,次による。
a) 硫酸(1+2) 水2容をビーカーにとり,冷却しながらJIS K 8951に規定する硫酸又はそれ以上の純
度の硫酸1容をかき混ぜながら徐々に加える。
b) ふっ化水素酸 JIS K 8819に規定するもの。
c) 炭酸ナトリウム JIS K 8625に規定するもの。
16
K 0083:2017
d) 硝酸ナトリウム JIS K 8562に規定するもの。
e) 硝酸(1+1) 6.1.1 a) による。
f)
過酸化ナトリウム JIS K 8231に規定するもの。
6.2.2
器具及び装置 器具及び装置は,次による。
a) 磁器るつぼ
b) 白金るつぼ 内容量20〜30 mLのものとする。
c) 電気炉
6.2.3
操作
操作は,次による。
a) 吸収瓶を設けないでろ紙だけに試料採取した場合
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものを集める。管の内面
に付着した粉末状のものは振り出し,ろ紙は適正な大きさに切り,磁器るつぼに入れ,電気炉を用
いて徐々に温度を上げて550 ℃で2時間加熱し,試料中の有機物を灰化分解する。
2) るつぼの内容物を白金るつぼに移す。これにふっ化水素酸20 mL及び硫酸数滴を徐々に加え,ドラ
フト内において加熱板上で硫酸の白煙が発生し始めるまで加熱する。放冷した後,ふっ化水素酸10
mLを加え,硫酸の白煙がほとんどなくなるまで加熱し,試料中のけい酸を四ふっ化けい素として
揮散除去する。さらに,白金るつぼを直火で徐々に温度を上げ,硫酸の白煙が発生しなくなるまで
加熱し,放冷する。
3) 白金るつぼに炭酸ナトリウム5 g及び硝酸ナトリウム0.3 gを加えよく混合し,蓋をした後,直火で
徐々に温度を上げ,約900 ℃で時々るつぼを揺り動かし,内容物をよく混ぜ合わせ,約20分間加
熱する。
4) 放冷後,白金るつぼに温水を加え,融成物をビーカー200 mLに移す。融成物を白金るつぼから取り
出すのが困難なときは,白金るつぼ及び蓋を温水約50 mLを加えたビーカー200 mLに入れ,5) の
操作を行う。ろ過する前に,るつぼ及び蓋は水洗して取り出しておく。洗液は,ビーカーに加える。
5) ビーカーを水浴上で加熱してクロム酸塩を浸出する。これをろ紙5種Bを用いてろ過し,ろ紙上の
沈殿物を温水で洗浄する。
なお,不溶解物中にクロム分が残存するおそれのあるときは,不溶解物は,ろ紙ごと乾燥した後,
再び灰化処理を行い,この灰分について融解操作を繰り返す。
6) ビーカー200 mLにろ液と洗液とを合わせ,硫酸(1+2)を加えて中和する。これを煮沸して二酸化
炭素を追い出すとともに,完全に溶解し,液量を80 mL程度以下とする。冷却後,全量フラスコ100
mLに移し,ビーカー洗液,水を標線まで加え,試料溶液とする。
なお,吸光光度法以外によって定量するときは次の操作を行う。
ろ液と洗液とをビーカー200 mLにとり,硝酸(1+1)20 mLを加え,これを煮沸して二酸化炭素
を追い出すとともに,完全に溶解し,液量を80 mL程度以下とする。冷却後,全量フラスコ100 mL
に移し,水を標線まで加え,試料溶液とする。
7) 別に,ろ紙について1)〜6) と同様に操作して空試験溶液を調製する。
注記1 灰化操作によってアルカリ融解時の激しい反応による融成物の損失を防ぐとともに白金るつ
ぼの損傷を避けることができる。
注記2 ふっ化水素酸処理の操作によってアルカリ融解反応を促進させることができる。
注記3 クロム酸塩の浸出液のろ過操作に長時間をかけると,負の誤差を生じやすいので,70〜80 ℃
17
K 0083:2017
でろ過するとよい。
注記4 空試験溶液を調製する際に,3) の加熱操作は,るつぼの内容物が融解状態となるところまで
でよい。
b) 吸収瓶を設けてろ紙とともに吸収液にも吸収させて試料採取した場合
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものについては,a) 1) の
操作を行った後,るつぼの内容物を白金るつぼに移す。
2) 一方,吸収瓶中の溶液については,ビーカー300 mLに入れ,導管及び吸収瓶を水10〜20 mLで洗い,
洗液をビーカー300 mLに合わせた後,硝酸(1+1)10 mL及び硫酸(1+2)1 mLを加え,加熱板
で乾固近くまで加熱する。このとき,有機物の分解が困難な場合には,液が淡黄色又は無色となる
まで硝酸(1+1)10 mLを加えて,加熱板上で乾固近くまで加熱を繰り返す。冷却後,内容物を1) の
白金るつぼに合わせる。
3) 白金るつぼにふっ化水素酸20 mLを徐々に加え,ドラフト内において加熱板上で硫酸の白煙が発生
し始めるまで加熱する。放冷した後,ふっ化水素酸10 mLを加え,硫酸の白煙がほとんどなくなる
まで加熱し,試料中のけい酸を四ふっ化けい素として揮散除去する。さらに,白金るつぼを直火で
徐々に温度を上げ,硫酸の白煙が発生しなくなるまで加熱し,放冷する。
4) 以下,a) の3)〜6) の操作を行う。
5) 別に,ろ紙及び吸収液について1)〜4) と同様に操作して空試験溶液を調製する。
6) 採取した試料は,次の操作によって処理し,試料溶液を調製してもよい。
吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものを集める。管の内
面に付着した粉末状のものは振り出し,ろ紙は適正な大きさに切り,ニッケルるつぼに入れ,電気
炉を用いて徐々に温度を上げて550 ℃で2時間加熱する。
放冷後,過酸化ナトリウム約5 gを加えよく混合し,更に少量の過酸化ナトリウムで表面を覆い,
バーナーの直火で徐々に加熱し,内容物が溶融状となってからは温度を上げ,約3分間赤熱状(余
り高温にしない)として融解する。
放冷後,るつぼを,50 mLの水を加えたビーカー300 mLに入れた後,温水50 mLを注意しながら
少しずつ加え,加熱してるつぼの内容物を浸出する。
次に,るつぼを水で洗って取り出し,浸出液をかき混ぜながら過酸化ナトリウムを少量ずつ加え
て加熱・煮沸して,クロムを完全にクロム(VI)に酸化するとともに過剰の過酸化ナトリウムを分
解する。
全量フラスコ250 mLに沈殿物ごと移し,水を標線まで加えてよく振り混ぜた後,放置する。上
澄み液をろ紙5種Bでろ過し,初めのろ液10 mLは捨て,次のろ液を試料溶液とする。
別に,ろ紙について同様に操作し,空試験溶液を調製する。
また,ニッケルるつぼのほか,鉄,アルミナ又はジルコニア製のるつぼを用いることができる。
ステンレス鋼製のるつぼはさみは使用しない。
6.3
ベリリウム試料溶液の調製
6.3.1
試薬
試薬は,次による。
a) 硝酸 JIS K 8541に規定する硝酸又はそれ以上の純度のもの。
b) 硝酸(2+98) 6.1.1 c) による。
c) ふっ化水素酸 JIS K 8819に規定するもの。
18
K 0083:2017
d) 硫酸 JIS K 8951に規定する硫酸又はそれ以上のもの。
e) 過塩素酸(60 %) JIS K 8223に規定する過塩素酸又はそれ以上の純度のもの。
f)
塩酸(2+1) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
6.3.2
操作
操作は,次による。
a) 吸収瓶を設けないでろ紙だけに試料採取した場合
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものを集める。管の内面
に付着した粉末状のものは振り出し,次に少量の硝酸(2+98)で洗い,全てを四ふっ化エチレン樹
脂ビーカー200 mLに集める。ろ紙は適正な大きさに切り,全てを上記のビーカーに入れ,硝酸30 mL,
ふっ化水素酸10 mL及び硫酸2 mLを加えて加熱板上で硫酸の白煙が発生するまで穏やかに加熱す
る。
2) 放冷後,硝酸10 mL及び過塩素酸(60 %)5 mLを加え,徐々に温度を上げて濃厚な硫酸の白煙が
発生するまで加熱する。
3) 試料中の有機物の分解が困難なときには,液が淡黄色又は無色となるまで2) の操作を繰り返した
後,蒸発乾固する。
4) 放冷後,塩酸(2+1)10 mLを加え,時計皿で覆い,水浴上で約1時間加熱溶解し,ろ紙5種Aを
用いてろ過し,ろ液を全量フラスコ50 mLに移す。ろ紙上の残留物を水洗し,洗液をろ液に合わせ,
水を標線まで加え,これを試料溶液とする。
なお,試験方法としてICP質量分析法を用いる場合は,塩酸(2+1)の替わりに硝酸(2+1)を
用いる。
5) 別に,ろ紙について1)〜4) と同様に操作して空試験溶液を調製する。
b) 吸収瓶を設けてろ紙とともに吸収液にも吸収させて試料採取した場合
1) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものについては,a) 1) の
操作を行った後,放冷する。
2) 一方,吸収瓶中の溶液については,これをビーカー300 mLに入れ,導管及び吸収瓶を水10〜20 mL
で洗い,洗液をビーカー300 mLに合わせた後,加熱板上で加熱し,約50 mLまで濃縮する。内容
物を1) の四ふっ化エチレン樹脂ビーカー100 mLに入れ,少量の水で洗い,洗液も四ふっ化エチレ
ン樹脂ビーカー100 mLに合わせる。
3) 以下,a) 2)〜4) の操作を行う。
4) 別に,ろ紙及び吸収液について1)〜3) と同様に操作して空試験溶液を調製する。
6.4
ひ素及びセレンの試料溶液の調製
6.4.1
試薬
試薬は,次による。
a) 硝酸 6.3.1 a) による。
b) 硝酸(2+98) 6.1.1 c) による。
c) 硫酸(1+1) 水1容をビーカーにとり,冷却しながらJIS K 8951に規定する硫酸又はそれ以上の純
度の硫酸1容をかき混ぜながら徐々に加える。
d) 過塩素酸(60 %) 6.3.1 e) による。
6.4.2
操作
操作は,次による。
19
K 0083:2017
a) 吸引ノズルからろ紙までの管の内面に付着したもの,及びろ紙に捕集したものを集める。管の内面に
付着した粉末状のものは振り出し,次に少量の硝酸(2+98)で洗い,全てをビーカー250 mLに集め
る。ろ紙は適正な大きさに切り,全てを上記のビーカーに入れ,硝酸50 mL及び硫酸(1+1)5 mL
を加えて静かに10分間加熱分解し,温水25 mLを加えて静置した後,その上澄み液を別のビーカー
300 mLに移す。
b) ビーカー250 mL内の残留物に硝酸50 mLを加え,徐々に温度を上げて10分間加熱した後,温水25 mL
を加えてろ紙5種Aを用いてろ過し,ろ液をa) のビーカー300 mLに合わせる。
c) 一方,吸収瓶中の溶液については,これを先のビーカー300 mLに入れ,導管及び吸収瓶を水10〜20 mL
で洗い,洗液をビーカー300 mLに合わせる。
d) 硝酸5 mL及び過塩素酸(60 %)5 mLを加えて,再び徐々に温度を上げ,過塩素酸の白煙が発生し始
めたら時計皿で覆い,過塩素酸がビーカーの器壁を流下する状態を保って有機物を分解する。
e) 試料中の有機物の分解が困難な場合には,液が淡黄色又は無色となるまでd) の操作を繰り返す。ビ
ーカーの器壁を少量の水で洗い,再び白煙が発生するまで有機物を分解する。
f)
放冷後,水100 mLを加えて可溶性塩を溶かし,ろ紙5種Aを用いてろ過し,全量フラスコ250 mL
にろ液を移す。ろ紙上の残留物を水洗し,洗液をろ液に合わせ,水を標線まで加え,試料溶液とする。
g) 別に,ろ紙及び吸収液についてa)〜f) と同様に操作して空試験溶液を調製する。
6.5
ガス状ひ素化合物及びガス状セレン化合物(水素化ひ素,セレン化水素など)の試料溶液の調製
6.5.1
試薬
試薬は,次による。
a) 塩化ヒドロキシルアンモニウム溶液(100 g/L) JIS K 8201に規定する塩化ヒドロキシルアンモニウ
ム10 gを水に溶かして100 mLとする。
b) 塩酸(1+120) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
c) 硝酸(1+120) JIS K 8541に規定する硝酸又はそれ以上の純度のものを用いて調製する。
6.5.2
操作
操作は,次による。
a) 試料を採取した吸収瓶の内容液を全量フラスコ250 mLに移し,吸収瓶,接続管などを水で洗浄して,
洗液を全量フラスコに合わせ,塩化ヒドロキシルアンモニウム溶液(100 g/L)を過マンガン酸カリウ
ム又は臭素の色が消えるまで滴加した後,塩酸(1+120)を標線まで加える。これを試料溶液とする。
なお,試験方法としてICP質量分析法を用いる場合は,塩酸(1+120)の替わりに硝酸(1+120)
を用いる。また,添加した塩化ヒドロキシルアンモニウムに起因する分子イオン(40Ar35Cl)の75As
信号強度に及ぼす影響が十分小さいことを確認して分析する。
b) 別に,吸収液についてa) と同様に操作して空試験溶液を調製する。
6.6
燃焼排ガス中ガス状セレン化合物(二酸化セレンなど)の試料溶液の調製
6.6.1
試薬
試薬は,次による。
a) 硫酸 6.3.1 d) による。
b) 過マンガン酸カリウム JIS K 8247に規定するもの。
c) 硫酸(1.8 mol/L) 全量フラスコ1 000 mLに,水を500 mL入れ,振り混ぜながら硫酸100 mLを除々
に加え,さらに水を標線まで加える。
d) ガス採取装置の洗浄液 全量フラスコ1 000 mLに硫酸(1.8 mol/L)を入れ,振り混ぜながら過マンガ
20
K 0083:2017
ン酸カリウム10 gを加え,さらに硫酸(1.8 mol/L)を標線まで加える。調製した溶液は,着色ガラス
瓶に保存する。これをガス採取装置の洗浄液とする。
6.6.2
操作
吸収液の試料溶液の調製は,次による。
a) ビーカー300 mLに試料ガス採取後の吸収液を移し,5.3.3 e) 1) の吸収液で導管,空瓶及び吸収瓶を洗
浄し,洗液をビーカーに入れる。
b) 5.3.4 b) にて保管した,ガス採取後のダスト捕集器,導管及び吸収瓶を6.6.1 d) のガス採取装置の洗
浄液40 mLで洗浄し,洗液をa) のビーカーに入れる。
c) 全量フラスコ500 mLに,ビーカーの内容液を5.3.3 e) 1) の吸収液で洗い移し,吸収液を標線まで加え
る。これを試料溶液とする。
d) 別に,5.3.3 e) 1) の吸収液460 mLに6.6.1 d) のガス採取装置の洗浄液を40 mL加え,これを空試験溶
液とする。
7
カドミウムの分析方法
7.1
一般
カドミウムの分析には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量
分析法を適用する。
7.2
フレーム原子吸光法
試料溶液をアセチレン−空気フレーム中に噴霧し,カドミウムによる原子吸光を波長228.8 nmで測定し
てカドミウムを定量する。
− 定量範囲 Cd 0.05〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
2) カドミウム標準液(Cd 0.1 mg/mL) カドミウム(99.9 %以上)0.100 gをとり,硝酸(1+1)[6.1.1
a) による。]20 mLに溶かす。煮沸して窒素酸化物を追い出し,放冷した後,全量フラスコ1 000 mL
に移し,水を標線まで加える。又は,国家計量標準へのトレーサビリティが確保されたもの又はそ
れを一定濃度にうすめたものを用いる。
3) カドミウム標準液(Cd 10 μg/mL) 2) のカドミウム標準液(Cd 0.1 mg/mL)50 mLを全量フラスコ
500 mLにとり,硝酸(1+1)[6.1.1 a) による。]10 mLを加えた後,水を標線まで加える。
4) メタクレゾールパープル溶液(1 g/L) JIS K 8889に規定するメタクレゾールパープル0.1 gをJIS
K 8102に規定するエタノール(95)50 mLに加温して溶かし,水を加えて100 mLとする。
5) くえん酸水素二アンモニウム溶液(100 g/L) JIS K 8284に規定するくえん酸水素二アンモニウム
10 gを水約80 mLに溶かし,アンモニア水(1+1)を滴加してpHを約7に調節した後,水を加え
て100 mLとする。
6) アンモニア水(1+1) JIS K 8085に規定するアンモニア水又はそれ以上の純度のものを用いて調製
する。
7) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L) JIS K 8454に規定するN,N-ジエチルジチオ
カルバミド酸ナトリウム三水和物1.3 gを水に溶かして100 mLとする。着色瓶に保存し,2週間以
21
K 0083:2017
上経過したものは使用しない。
8) 酢酸ブチル JIS K 8377に規定するもの。
9) 4-メチル-2-ペンタノン(メチルイソブチルケトン) JIS K 8903に規定するもの。
10) 硫酸アンモニウム溶液(飽和) JIS K 8960に規定する硫酸アンモニウムを用いて調製する。
11) 1-ピロリジンカルボジチオ酸アンモニウム(ピロリジン-N-ジチオカルバミド酸アンモニウム)
(APDC)溶液(10 g/L) 1-ピロリジンカルボジチオ酸アンモニウム1.0 gを水に溶かして100 mL
とする。着色瓶に保存し,2週間以上経過したものは使用しない。
12) 2,6-ジメチル-4-ヘプタノン(DIBK)
13) 臭化水素酸 JIS K 8509に規定するもの。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) カドミウム中空陰極ランプ
c) 操作(溶媒抽出操作を伴わない場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,分
析波長228.8 nmで指示値(吸光度又はその比例値)を読み取る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た指示値
を補正する。
3) 検量線からカドミウムの量を求め,試料溶液中のカドミウムの濃度(Cd mg/L)を算出する。検量
線の作成は,次による。
検量線 カドミウム標準液(Cd 10 μg/mL)0.5〜20 mLを全量フラスコ100 mLに段階的にとり,試
料溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作
を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を
加えた後,1) の操作を行って標準液について得た指示値を補正し,カドミウム(Cd)の量と指示値
との関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(溶媒抽出を伴う場合) 操作は,次による。
1) 6.1で調製した試料溶液の適量をとり,くえん酸水素二アンモニウム溶液(100 g/L)10 mL及び指示
薬としてメタクレゾールパープル溶液(1 g/L)2,3滴を加えた後,アンモニア水(1+1)を溶液の
色が僅かに紫になるまで加える。ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)5 mLを加
えて振り混ぜた後,酢酸ブチル20 mLを加え,約1分間激しく振り混ぜて静置する。酢酸ブチル層
を分離し,ビーカー100 mLに入れる。水層に酢酸ブチル5 mLを加え抽出操作を繰り返す。抽出し
た酢酸ブチル層は先のビーカーに合わせる。抽出した酢酸ブチル層に酢酸ブチルを加えて液量を一
定量にしたもの,又は抽出条件を一定にして,1回抽出を行った酢酸ブチル層をそのまま噴霧し,
分析波長228.8 nmで指示値(吸光度又はその比例値)を読み取る。
注記 酢酸ブチルに代え,4-メチル-2-ペンタノンを用いてもよい。
2) 1) の操作の代わりに,6.1で調製した試料溶液の適量をとり,pHを3.5〜4.0に調節する。硫酸アン
モニウム溶液(飽和)20 mLを加える。1-ピロリジンカルボジチオ酸アンモニウム溶液(10 g/L)5 mL
を加え,静かに振り混ぜた後,3分間放置する。次に,4-メチル-2-ペンタノン10 mLを加え,3分
間激しく振り混ぜた後,有機層と水層とを完全に分離する。有機層をそのまま噴霧し,分析波長228.8
nmで指示値(吸光度又はその比例値)を読み取る。
注記 4-メチル-2-ペンタノンの代わりに2,6-ジメチル-4-ヘプタノンを用いてもよい。この場合は,
22
K 0083:2017
2,6-ジメチル-4-ヘプタノンは,水との相互溶解がほとんどないので,その添加量を少なく
してもよい。
3) 空試験として,空試験溶液を試料溶液と同様に1) 又は2) の操作を行って,試料溶液について得た
指示値を補正する。
4) 検量線からカドミウムの量を求め,試料溶液中のカドミウムの濃度(Cd mg/L)を算出する。検量
線の作成は,次による。
検量線 カドミウム標準液(Cd 10 μg/mL)を適切な濃度(Cd 0.1〜1 μg/mL)にうすめ,その0.5〜
20 mLを段階的にとり,約100 mLとした後,試料溶液と同じ条件になるように酸を加えた後,1) 又
は2) の操作を行う。別に,空試験として同量の水を用いて検量線の作成に用いた標準液と同じ条
件になるように酸を加えた後,1) 又は2) の操作を行って標準液について得た指示値を補正し,カ
ドミウム(Cd)の量と指示値との関係線を作成する。検量線は,試料測定時に作成する。
e) 操作(試料溶液中に多量の亜鉛,銅などが含まれている場合) 操作は,次による。
試料溶液の適量に臭化水素酸を加えて約0.5 mol/Lの臭化水素酸溶液とし,その50 mLに対してト
リオクチルアミン(N,N-ジオクチルオクタンアミン)の4-メチル-2-ペンタノン溶液(1 vol %)10 mL
を加えて振り混ぜ,カドミウムを抽出する。抽出した4-メチル-2-ペンタノン層をそのまま噴霧して原
子吸光分析する。また,アルカリ金属のハロゲン化物が多量に存在すると,その分子吸収,光散乱な
どによって正の誤差を生じる。このような場合には,あらかじめカドミウムを分離するか,又はバッ
クグラウンド補正装置を用いる。
7.3
電気加熱原子吸光法
試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム(II)を加え,電気加熱炉
で原子化し,カドミウムによる原子吸光を波長228.8 nmで測定してカドミウムを標準添加法によって定量
する。
− 定量範囲 Cd 0.5〜10 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試
料に適用し,測定時の酸濃度は一定となるように調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸パラジウム(II)溶液(Pd 1 mg/mL) 原子吸光分析用マトリックスモディファイヤーの硝酸
パラジウム(II)の溶液を希釈して用いる。
4) カドミウム標準液(Cd 0.1 μg/mL) 7.2 a) 3) のカドミウム標準液(Cd 10 μg/mL)10 mLを全量フ
ラスコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) カドミウム中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.1によって処理する。
23
K 0083:2017
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,カドミウム標準液(Cd 0.1
μg/mL)を加えないものと,0.1〜2 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)及びそれと同体積の硝酸パラジウム(II)溶液(Pd 1 mg/mL)
を,マイクロピペットを用いて電気加熱炉に注入する。JIS K 0121の8.(操作方法)の操作に従っ
て,乾燥(100〜120 ℃,30〜40秒間),灰化(500〜800 ℃,30〜40秒間),原子化1)(1 600〜2 200 ℃,
3〜6秒間)し,波長228.8 nmの指示値(吸光度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) カドミウムの添加量と指示値との関係線を作成し,カドミウムの量を求め,試料溶液中のカドミウ
ムの濃度(Cd μg/L)を算出する。
注1) 乾燥,灰化,原子化の条件は装置によって異なる。また,試料の注入量及び共存する塩類の濃
度によっても異なることがある。
7.4
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,カドミウムによる発光を波長214.438 nmで測定して,カドミ
ウムを定量する。
− 定量範囲 Cd 0.010〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+10) JIS K 8541に規定する硝酸又はそれ以上の純度のものを用いて調製する。
3) カドミウム標準液(Cd 10 μg/mL) 7.2 a) 2) のカドミウム標準液(Cd 0.1 mg/mL)50 mLを全量フ
ラスコ500 mLにとり,硝酸(1+1)[6.1.1 a) による。]10 mLを加えた後,水を標線まで加える。
4) 混合標準液[(Cd 10 μg,Pb 10 μg,Ni 10 μg,Mn 10 μg,V 10 μg)/mL] 7.2 a) 2) のカドミウム標
準液(Cd 0.1 mg/mL)50 mL,8.2 a) 3) の鉛標準液(Pb 0.1 mg/mL)50 mL,9.2 a) 10) のニッケル標
準液(Ni 0.1 mg/mL)50 mL,10.2 a) 5) のマンガン標準液(Mn 0.1 mg/mL)50 mL及び11.2 a) 6) の
バナジウム標準液(V 0.1 mg/mL)50 mLのそれぞれを全量フラスコ500 mLにとり,硝酸(1+1)
[6.1.1 a) による。]10 mLを加えて,水を標線まで加える。この溶液は使用の都度調製する。
5) イットリウム溶液(Y 50 μg/mL) 酸化イットリウム(III)0.318 gをとり,JIS K 8180に規定する
塩酸又はそれ以上の純度の塩酸5 mLを加え加熱して溶かし,冷却後,全量フラスコ250 mLに移し
入れ,水を標線まで加える。この溶液10 mLを全量フラスコ200 mLにとり,水を標線まで加える。
6) アンモニア水(1+1) 7.2 a) 6) による。
7) 酢酸−酢酸ナトリウム緩衝液(pH5) JIS K 8371に規定する酢酸ナトリウム三水和物19.2 gとJIS
K 8355に規定する酢酸3.4 mLとを水に溶かして1 Lとする。
8) 1-ピロリジンカルボジチオ酸アンモニウム(ピロリジン-N-ジチオカルバミド酸アンモニウム)
(APDC)溶液(20 g/L) 1-ピロリジンカルボジチオ酸アンモニウム2.0 gを水に溶かして100 mL
とする。着色瓶に保存し,2週間以上経過したものは使用しない。
9) ヘキサメチレンアンモニウムヘキサメチレンカルバモジチオ酸(ヘキサメチレンアンモニウムヘキ
24
K 0083:2017
サメチレンジチオカルバミド酸)のメタノール溶液(20 g/L) ヘキサメチレンアンモニウムヘキサ
メチレンカルバモジチオ酸2.0 gをJIS K 8891に規定するメタノールに溶かして100 mLとする。着
色瓶に保存し,2週間以上経過したものは使用しない。
10) キシレン JIS K 8271に規定するもの。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
波長214.438 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線からカドミウムの量を求め,試料溶液中のカドミウムの濃度(Cd mg/L)を算出する。検量
線の作成は,次による。
検量線 カドミウム標準液(Cd 10 μg/mL)0.1〜20 mL 3) を全量フラスコ100 mLに段階的にとり,
試料溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操
作を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸
を加えた後,1) の操作を行って,標準液について得た発光強度を補正し,カドミウム(Cd)の量と
発光強度との関係線を作成する。検量線は,試料測定時に作成する。
なお,塩の濃度が高い試料溶液で,検量線法が適用できない場合には,JIS K 0116の4.7.3 b)(標
準添加法)に規定する標準添加法を用いるとよい。ただし,この場合は,試料溶液の種類によらず
バックグラウンド補正を行う必要がある。
注2) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。ま
た,精度,正確さを確認してあれば,他の波長を用いてもよい。
注3) カドミウム,鉛,ニッケル,マンガン及びバナジウムを同時に試験する場合には7.4 a) 4) の混
合標準液を用いて,それぞれの金属元素の試験条件で検量線を作成するとよい。
d) 操作(内標準法の場合) 操作は,次による。
1) 6.1で調製した試料溶液の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50 μg/mL)10
mLを加え,c) 1) の試料溶液と同じ条件になるように,酸を加えた後,水を標線まで加える。この
溶液についてc) 1) の噴霧操作を行って,波長214.438 nm及び371.029 nm(イットリウム)の発光
強度を測定し,カドミウムとイットリウムとの発光強度の比を求める。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度の比を補正する。
3) 検量線からカドミウムの量を求め,試料溶液中のカドミウムの濃度(Cd mg/L)を算出する。検量
線の作成は,次による。
検量線 カドミウム標準液(Cd 10 μg/mL)0.1〜20 mL 3) を全量フラスコ100 mLに段階的にとり,
イットリウム溶液(Y 50 μg/mL)10 mLをそれぞれ加え,1) の試料溶液と同じ条件になるように酸
を加えた後,水を標線まで加える。この溶液について1) の噴霧操作を行って,波長214.438 nm及
び371.029 nmの発光強度を測定し,カドミウムの濃度に対するカドミウムとイットリウムとの発光
強度の比を求める。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になる
ように酸を加えた後,1) の操作を行って,標準液について得た発光強度の比を補正し,カドミウム
(Cd)の量と発光強度比との関係線を作成する。検量線は,試料測定時に作成する。
25
K 0083:2017
注記 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
e) 操作(溶媒抽出を伴う場合) 操作は,次による。
1) 6.1で調製した試料溶液の適量をビーカーにとり,酢酸−酢酸ナトリウム緩衝液(pH5)10 mLを加
え,アンモニア水(1+1)又は硝酸(1+10)でpHを5.2に調節する。この溶液を分液漏斗1 L(又
は200〜500 mL)に移し,1-ピロリジンカルボジチオ酸アンモニウム溶液(20 g/L)2 mL,ヘキサメ
チレンアンモニウムヘキサメチレンカルバモジチオ酸のメタノール溶液(20 g/L)2 mLを加えて混
合した後,キシレンの一定量(5〜20 mL)を加えて約5分間激しく振り混ぜ静置する。水層を捨て
キシレン層を共栓試験管に入れる。キシレン層をプラズマ中に噴霧し,波長214.438 nmの発光強度
を測定する。
なお,この操作に用いる酢酸−酢酸ナトリウム緩衝液(pH5)は,使用前に1-ピロリジンカルボ
ジチオ酸アンモニウム溶液(20 g/L)2 mL,ヘキサメチレンアンモニウムヘキサメチレンカルバモ
ジチオ酸のメタノール溶液(20 g/L)2 mL及びキシレン5〜20 mLを加えて振り混ぜ,精製したも
のを用いる。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た指示値
を補正する。
3) 検量線からカドミウムの量を求め,試料溶液中のカドミウムの濃度(Cd mg/L)を算出する。検量
線の作成は,次による。
検量線 カドミウム標準液(Cd 10 μg/mL)3) を適切な濃度(Cd 0.1〜1 μg/mL)にうすめ,その0.1
〜20 mLを段階的にとり,約100 mLとした後,試料溶液と同じ条件になるように酸を加えた後,
1) の操作を行う。別に,空試験として同量の水を用いて検量線の作成に用いた標準液と同じ条件に
なるように酸を加えた後,1) の操作を行って標準液について得た指示値を補正し,カドミウム(Cd)
の量と指示値との関係線を作成する。検量線は,試料測定時に作成する。
注記 この溶媒抽出法は,試料溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃度
が高く,カドミウムの濃度が低い場合に適用するとよい。カドミウム,鉛,ニッケル,マンガ
ン及びバナジウムの定量に用いることができる。
7.5
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,カドミウム及び内標
準物質のそれぞれの質量/荷電数におけるイオンカウントを測定し,カドミウムのイオンカウントと内標準
物質のイオンカウントとの比を求めてカドミウムを定量する。
− 定量範囲 Cd 0.3〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.4 a) 5) のイットリウム溶液(Y 50 μg/mL)20 mLを全量フラス
コ1 000 mLにとり,硝酸(1+1)1.5 mLを加え,水を標線まで加える。使用時に調製する。
4) カドミウム標準液(Cd 1 μg/mL) 7.2 a) 2) のカドミウム標準液(Cd 0.1 mg/mL)10 mLを全量フラ
スコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。
5) カドミウム標準液(Cd 50 ng/mL) 4) のカドミウム標準液(Cd 1 μg/mL)50 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
26
K 0083:2017
6) 混合標準液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL] 7.4 a) 4) の混合標準液100 mL
を全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。使用時に
調製する。
7) 混合標準液[(Cd 50 ng,Pb 50 ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL] 6) の混合標準液50 mL
を全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。使用時に
調製する。
b) 装置 ICP質量分析装置
注記1 イオン源として,誘導結合プラズマと同等の性能をもつものを用いてもよい。
注記2 サンプリングコーン及びスキマーコーンの材質からの汚染が認められないことを確認す
るのがよい。
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Cdとして0.03〜50 μgを含む。)を全量フラスコ100 mLにとり,
イットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるよう
に硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
カドミウムとイットリウムとの質量/荷電数4) における指示値(イオンカウント又はその比例値)
を読み取り,カドミウムの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,カドミウムの指示値とイ
ットリウムの指示値との比を求め,試料について得たカドミウムの指示値とイットリウムの指示値
との比を補正する。
4) 検量線からカドミウムの量を求め,試料中のカドミウムの濃度(Cd μg/L)を算出する。検量線の作
成は,次による。
検量線 カドミウム標準液(Cd 50 ng/mL又はCd 1 μg/mL)0.6〜50 mLを全量フラスコ100 mLに
段階的にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるよう
に硝酸(1+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。
別に,空試験として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の
試料と同じ酸の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って
標準液について得た指示値の比を補正した後,カドミウム(Cd)の量に対する指示値とイットリウ
ムの指示値との比の関係線を作成する。検量線は,試料測定時に作成する。
注記1 分析者からの汚染がないように注意する。JIS T 9107に規定するゴム手袋(打粉のないもの)
などを用いるとよい。
注記2 妨害物質の存在が不明の場合には,定量の前にICP質量分析計による定性分析を行うことに
よって目的とする元素及び内標準物質の測定質量数に対する妨害を推定することができる。
妨害が認められる場合には,測定質量数若しくは内標準物質の変更,又は試料の希釈若しく
は前処理を行って妨害の軽減を図る。内標準物質としてはイットリウム以外に,試料溶液に
含まれず,同重体イオン,分子イオンなどによるスペクトル干渉が無視できる元素であって,
測定元素と質量数とが近い元素を用いるとよい。妨害物質の影響が無視できる試料の場合は,
内標準物質の添加を省略して検量線法によって定量してもよい。
注記3 カドミウム,鉛,ニッケル,マンガン及びバナジウムを同時に定量する場合には,混合標準
液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL]又は混合標準液[(Cd 50 ng,Pb 50
27
K 0083:2017
ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL]を用いて,それぞれの金属元素の試験条件で検量線
を作成するとよい。
注4) 質量数を設定するには,表2を参考にするとよい。安定同位体がある場合,複数の同位体の質
量/荷電数を用いて測定を行うことによってスペクトル干渉による妨害を推定することができ
る。分子イオンが妨害する場合には,コリジョン・リアクションセルなどを用いて分子イオン
の生成を抑えてもよい。それでも影響を受ける場合は,適切な分離方法を用いて妨害となるマ
トリックスを除去した後,測定を行う。
表2−測定質量の一例
元素名
質量数
妨害イオンの例
カドミウム
111
114
95Mo16O+,94Mo16OH+,94Zr16OH+
114Sn+,98Mo16O,97Mo16OH
鉛
208
206
207
−
ニッケル
58
60
62
116Sn++,58Fe+,42Ca16O+,44Ca14N+,23Na35Cl+,24Mg34S+
120Sn++,44Ca16O+,43Ca16OH+,25Mg35Cl+,23Na37Cl+
124Sn++,46Ca16O+,48Ca14N+
マンガン
55
39K16O+,40Ar14NH+,38Ar16OH+,23Na32S+
バナジウム
51
35Cl16O+,37Cl14N+,34S16OH+,36Ar14NH+
クロム
53
52
50
37Cl16O+,36Ar16OH+
40Ar12C+,36Ar16O+,36S16O+,35Cl16OH+
50Ti+,50V+,36Ar14N+,34S16O+
ベリリウム
9
−
ひ素
75
40Ar35Cl+,40Ca35Cl+
セレン
78
82
77
78Kr+,38Ar40Ar+,43Ca35Cl+,62Ni16O+
82Kr+,40Ar40ArH2+,34S16O16O16O+,H81Br+,66Zn16O+
40Ar37Cl+,36Ar40ArH+,40Ca37Cl+,61Ni16O+
8
鉛の分析方法
8.1
一般
鉛の分析には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を
適用する。
8.2
フレーム原子吸光法
試料溶液をアセチレン−空気のフレーム中に噴霧し,鉛による原子吸光を波長283.3 nmで測定して,鉛
を定量する。
− 定量範囲 Pb 1〜20 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 6.1.1 a) による。
3) 鉛標準液(Pb 0.1 mg/mL) JIS K 8701に規定する鉛(特級)0.100 gをとり,硝酸(1+3)[JIS K 8541
に規定する硝酸又はそれ以上の純度の硝酸を用いて調製する。]40 mLを加えて溶かし,加熱して窒
28
K 0083:2017
素化合物を追い出し,放冷した後,全量フラスコ1 000 mLに移し入れ,水を標線まで加えるか,又
はJIS K 8563に規定する硝酸鉛(II)0.160 gをとり,硝酸(1+1)20 mL及び適量の水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又は,国家計量標準へのトレーサビリテ
ィが確保されたもの又はそれを一定濃度にうすめたものを用いる。
4) 鉛標準液(Pb 10 μg/mL) 3) の鉛標準液(Pb 0.1 mg/mL)50 mLを全量フラスコ500 mLにとり,
硝酸(1+1)10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) 鉛中空陰極ランプ
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,波
長283.3 nmの指示値(吸光度又はその比例値)を読み取る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た指示値
を補正する。
3) 検量線から鉛の量を求め,試料溶液中の鉛の濃度(Pb mg/L)を算出する。検量線の作成は,次によ
る。
検量線 鉛標準液(Pb 0.1 mg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料溶液と
同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を行う。
別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を加えた後,
1) の操作を行って標準液について得た指示値を補正し,鉛(Pb)の量と指示値との関係線を作成す
る。検量線は,試料測定時に作成する。
なお,溶媒抽出法を適用するときは,7.2 d) の操作を行う。ただし,カドミウムを鉛と読み替え,
分析波長は283.3 nmを用いる。また,検量線の作成には,鉛標準液(Pb 0.1 mg/mL)を適切な濃度
(Pb 1〜10 μg/mL)にうすめ,その1〜20 mLを段階的にとり,約100 mLとしたものを用いる。
8.3
電気加熱原子吸光法
試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム(II)を加え,電気加熱炉
で原子化し,鉛による原子吸光を波長283.3 nmで測定して鉛を標準添加法によって定量する。
− 定量範囲 Pb 5〜100 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試
料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸パラジウム(II)溶液(Pd 1 mg/mL) 7.3 a) 3) による。
4) 鉛標準液(Pb 1 μg/mL) 8.2 a) 3) の鉛標準液(Pb 0.1 mg/mL)10 mLを全量フラスコ1 000 mLにと
り,硝酸(1+1)20 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
29
K 0083:2017
3) 鉛中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.1によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,鉛標準液(Pb 1 μg/mL)
を加えないものと,0.1〜2 mLの範囲で段階的に3段階以上添加したものとを調製し,それぞれの
溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)及びそれと同体積の硝酸パラジウム(II)溶液(Pd 1 mg/mL)
とを,マイクロピペットを用いて電気加熱炉に注入する。JIS K 0121の8.(操作方法)の操作に従
って,乾燥(100〜120 ℃,30〜40秒間),灰化(500〜800 ℃,30〜40秒間),原子化1)(1 800〜2 500 ℃,
3〜6秒間)し,波長283.3 nmの指示値(吸光度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) 鉛の添加量と指示値との関係線を作成し,鉛の量を求め,試料溶液中の鉛の濃度(Pb μg/L)を算出
する。
8.4
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,鉛による発光を波長220.351 nmで測定して,鉛を定量する。
− 定量範囲 Pb 0.1〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 鉛標準液(Pb 10 μg/mL) 8.2 a) 4) による。
3) 混合標準液[(Cd 10 μg,Pb 10 μg,Ni 10 μg,Mn 10 μg,V 10 μg)/mL] 7.4 a) 4) による。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し
波長220.351 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線から鉛の量を求め,試料溶液中の鉛の濃度(Pb mg/L)を算出する。検量線の作成は,次によ
る。
検量線 鉛標準液(Pb 10 μg/mL)1〜20 mL 3) を全量フラスコ100 mLに段階的にとり,試料溶液と
同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を行う。
別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を加えた後,
1) の操作を行って,標準液について得た発光強度を補正し,鉛(Pb)の量と発光強度との関係線を
作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムを鉛と読み替え,鉛の分析
30
K 0083:2017
波長は220.351 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10 μg/mL)0.1〜20 mL
の代わりに鉛標準液(Pb 10 μg/mL)1〜20 mLを用いる。
注記 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
e) 操作(溶媒抽出を伴う場合) 操作は,次による。
溶媒抽出を適用するときは,7.4 e) の操作を行う。ただし,カドミウムを鉛と読み替え,鉛の分析
波長は220.351 nmを用いる。また,検量線の作成には,鉛標準液(Pb 0.1 mg/mL)を適切な濃度(Pb
1〜10 μg/mL)にうすめ,その1〜20 mLを段階的にとり,約100 mLとしたものを用いる。
注記 この溶媒抽出法は,試料溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃
度が高く,鉛の濃度が低い場合に適用するとよい。カドミウム,鉛,ニッケル,マンガン及
びバナジウムの定量に用いることができる。
8.5
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,鉛及び内標準物質の
それぞれの質量/荷電数におけるイオンカウントを測定し,鉛のイオンカウントと内標準物質のイオンカウ
ントとの比を求めて鉛を定量する。
− 定量範囲 Pb 0.3〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) 鉛標準液(Pb 1 μg/mL) 8.3 a) 4) による。
5) 鉛標準液(Pb 50 ng/mL) 4) の鉛標準液(Pb 1 μg/mL)50 mLを全量フラスコ1 000 mLにとり,硝
酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL] 7.5 a) 6) による。
7) 混合標準液[(Cd 50 ng,Pb 50 ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL] 7.5 a) 7) による。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Pbとして0.03〜50 μgを含む。)を全量フラスコ100 mLにとり,イ
ットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるように
硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
鉛及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を読み取り,
鉛の指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,鉛の指示値とイットリウ
ムの指示値との比を求め,試料について得た鉛の指示値とイットリウムの指示値との比を補正する。
4) 検量線から鉛の量を求め,試料中の鉛の濃度(Pb μg/L)を算出する。検量線の作成は,次による。
検量線 鉛標準液(Pb 50 ng/mL又はPb 1 μg/mL)0.6〜50 mLを全量フラスコ100 mLに段階的にと
り,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝酸(1
31
K 0083:2017
+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。
別に,空試験として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の
試料と同じ酸の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って
標準液について得た指示値の比を補正した後,鉛(Pb)の量に対する指示値とイットリウムの指示
値との比の関係線を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 7.5 c) の注記3参照。
9
ニッケルの分析方法
9.1
一般
ニッケルの分析には,ジメチルグリオキシム吸光光度法,フレーム原子吸光法,電気加熱原子吸光法,
ICP発光分光分析法又はICP質量分析法を適用する。
9.2
ジメチルグリオキシム吸光光度法
試料溶液にくえん酸塩を加え,アンモニア水で微アルカリ性とした後,ジメチルグリオキシム(2,3-ブ
タンジオンジオキシム)を加えて生成したニッケル錯体をクロロホルムで抽出し,これを希塩酸で逆抽出
する。抽出液に臭素及びアンモニア水を加えてニッケルを酸化し,再びジメチルグリオキシムを加えて生
じる赤褐色のニッケル錯体の吸光度を測定して,ニッケルを定量する。
− 定量範囲 Ni 2〜50 μg
− 繰返し分析精度 変動係数で2〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸(1+20) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
3) アンモニア水(1+1),(1+5),(1+50) JIS K 8085に規定するアンモニア水又はそれ以上の純度
のものを用いて調製する。
4) 臭素水(飽和) JIS K 8529に規定する臭素3〜4 mLを水100 mLに加えて激しく振り混ぜ,放置後
その上澄み液を用いる。
5) くえん酸水素二アンモニウム溶液(100 g/L) 7.2 a) 5) による。
6) フェノールフタレイン溶液(5 g/L) JIS K 8799に規定するフェノールフタレイン0.5 gをとり,JIS
K 8102に規定するエタノール(95)50 mLに溶かし,水を加えて100 mLとする。
7) ジメチルグリオキシムエタノール溶液(10 g/L) JIS K 8498に規定するジメチルグリオキシム(2,3-
ブタンジオンジオキシム)1 gをJIS K 8102に規定するエタノール(95)に溶かして100 mLとする。
8) ジメチルグリオキシム水酸化ナトリウム溶液(10 g/L) JIS K 8498に規定するジメチルグリオキシ
ム(2,3-ブタンジオンジオキシム)1 gを水酸化ナトリウム溶液(10 g/L)[JIS K 8576に規定する水
酸化ナトリウムを用いて調製する。]に溶かし,水酸化ナトリウム溶液(10 g/L)を加えて100 mL
とする。不溶解物があるときはろ過する。
9) クロロホルム JIS K 8322に規定するもの。
10) ニッケル標準液(Ni 0.1 mg/mL) JIS K 9062に規定するニッケル(99.9 %以上)0.100 gをとり,
硝酸(1+1)[6.1.1 a) による。]20 mLに溶かし,煮沸して窒素酸化物を追い出し,放冷した後,
全量フラスコ1 000 mLに移し,水を標線まで加える。又は,国家計量標準へのトレーサビリティ
32
K 0083:2017
が確保されたもの又はそれを一定濃度にうすめたものを用いる。
11) ニッケル標準液(Ni 5 μg/mL) 10) のニッケル標準液(Ni 0.1 mg/mL)50 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)[6.1.1 a) による。]20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗
2) 吸光光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Niとして2〜50 μgを含む。)を分液漏斗にとり,くえん酸水素二
アンモニウム溶液(100 g/L)5 mLと指示薬としてフェノールフタレイン溶液(5 g/L)2,3滴とを
加え,次にアンモニア水(1+5)を溶液の色が僅かに赤になるまで滴加する。さらに,アンモニア
水(1+5)2,3滴及び水を加えて約100 mLとする。
2) ジメチルグリオキシムエタノール溶液(10 g/L)2 mLとクロロホルム10 mLとを加え,1分間激し
く振り混ぜて放置した後,クロロホルム層を別の分液漏斗に入れる。水層にクロロホルム5 mLを
加え,1分間激しく振り混ぜて抽出し,放置後,クロロホルム層をとり,先の分液漏斗に合わせる。
この操作を更に1回繰り返す。
3) クロロホルム層を入れた分液漏斗にアンモニア水(1+50)10〜20 mLを加えて30秒間振り混ぜ,
放置後,クロロホルム層を別の分液漏斗に移す。
4) クロロホルム層を入れた分液漏斗に塩酸(1+20)10 mLを加え,1分間激しく振り混ぜ,ニッケル
を逆抽出する。放置後,クロロホルム層を別の分液漏斗に入れる。クロロホルム層に,再び塩酸(1
+20)5 mLを加え,逆抽出を繰り返す。クロロホルム層は捨て,塩酸層は先の塩酸層に合わせて全
量フラスコ25 mLに入れる。
5) 臭素水(飽和)2 mLを加えて振り混ぜ,約1分間放置する。次に,アンモニア水(1+1)を加えて
中和し,さらに,アンモニア水(1+1)2 mLを過剰に加え,流水で室温以下に冷却する。
6) ジメチルグリオキシム水酸化ナトリウム溶液(10 g/L)2 mLを加えて振り混ぜ,ニッケルを発色さ
せた後,水を標線まで加える。
7) この溶液の一部を吸収セルに移し,波長450 nm付近の吸光度を測定する。
8) 別に,空試験溶液について1)〜7) の操作を行って吸光度を求め,試料溶液について得た吸光度を補
正する。
9) 検量線からニッケルの量を求め,試料溶液中のニッケルの濃度(Ni mg/L)を算出する。検量線の作
成は,次による。
検量線 ニッケル標準液(Ni 5 μg/mL)0.4〜10 mLを全量フラスコ25 mLに段階的にとり,5)〜8) の
操作を行ってニッケル(Ni)の量と吸光度との関係線を作成する。
9.3
フレーム原子吸光法
試料溶液をアセチレン−空気のフレーム中に噴霧し,ニッケルによる原子吸光を波長232.0 nmで測定し
て,ニッケルを定量する。
− 定量範囲 Ni 0.3〜6 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) ニッケル標準液(Ni 0.1 mg/mL) 9.2 a) 10) による。
33
K 0083:2017
3) ニッケル標準液(Ni 10 μg/mL) 2) のニッケル標準液(Ni 0.1 mg/mL)50 mLを全量フラスコ500 mL
にとり,硝酸(1+1)[6.1.1 a) による。]10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) ニッケル中空陰極ランプ
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,波
長232.0 nmの指示値(吸光度又はその比例値)を読み取る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た指示値
を補正する。
3) 検量線からニッケルの量を求め,試料溶液中のニッケルの濃度(Ni mg/L)を算出する。検量線の作
成は,次による。
検量線 ニッケル標準液(Ni 10 μg/mL)3〜60 mLを全量フラスコ100 mLに段階的にとり,試料溶
液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を行
う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように,酸を加
えた後,1) の操作を行って標準液について得た指示値を補正し,ニッケル(Ni)の量と指示値との
関係線を作成する。検量線は,試料測定時に作成する。
注記1 溶媒抽出法を適用するときは,次の操作を行うとよい。
試料溶液の適量を分液漏斗にとり,9.2 c) 1) 及び2) に準じて操作し,ニッケルをジメ
チルグリオキシム錯体として,クロロホルム層に抽出する。クロロホルム層を合わせ,塩
酸(1+20)10 mLを加え,振り混ぜてニッケルを逆抽出する。水層を分離した後,クロロ
ホルム層に塩酸(1+20)5 mLを加えて逆抽出を行う。逆抽出液を合わせ,全量フラスコ
25 mLに移し入れ,水を標線まで加え,これをニッケルの定量に用いる。
注記2 注記1とは別の溶媒抽出法として,次の操作を行ってもよい。
7.2 d) の操作を行う。ただし,カドミウムをニッケルと読み替え,分析波長は232.0 nm
を用いる。また,検量線の作成には,ニッケル標準液(Ni 0.1 mg/mL)を適切な濃度(Ni 1
〜10 μg/mL)にうすめ,その1〜20 mLを段階的にとり,約100 mLとしたものを用いる。
9.4
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,ニッケルによる原子吸光を波長232.0 nmで測定してニッ
ケルを標準添加法によって定量する。
− 定量範囲 Ni 5〜50 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試
料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) ニッケル標準液(Ni 1 μg/mL) 9.2 a) 10) のニッケル標準液(Ni 0.1 mg/mL)10 mLを全量フラスコ
1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
34
K 0083:2017
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) ニッケル中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.1によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,ニッケル標準液(Ni 1
μg/mL)を加えないものと,0.1〜1 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) 1) の操作を行った試料の一定量(例えば,10〜50 μL)をマイクロピペットを用いて電気加熱炉に
注入し,JIS K 0121の8.(操作方法)の操作に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(600
〜1 000 ℃,30〜40秒間)し,原子化1)(2 200〜2 700 ℃,3〜6秒間)し,波長232.0 nmの指示値
(吸光度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) ニッケルの添加量と指示値との関係線を作成し,ニッケルの量を求め,試料溶液中のニッケルの濃
度(Ni μg/L)を算出する。
9.5
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,ニッケルによる発光を波長221.647 nmで測定して,ニッケル
を定量する。
− 定量範囲 Ni 0.04〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) ニッケル標準液(Ni 10 μg/mL) 9.3 a) 3) による。
3) 混合標準液[(Cd 10 μg,Pb 10 μg,Ni 10 μg,Mn 10 μg,V 10 μg)/mL] 7.4 a) 4) による。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
波長221.647 nmの発光強度を測定する2)。
2) 空試験として空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強度
を補正する。
3) 検量線からニッケルの量を求め,試料溶液中のニッケルの濃度(Ni mg/L)を算出する。検量線の作
成は,次による。
検量線 ニッケル標準液(Ni 10 μg/mL)0.4〜20 mL 3) を全量フラスコ100 mLに段階的にとり,試
料溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作
を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を
加えた後,1) の操作を行って,標準液について得た発光強度を補正し,ニッケル(Ni)の量と発光
35
K 0083:2017
強度との関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムをニッケルと読み替え,ニ
ッケルの分析波長は221.647 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10 μg/mL)
0.1〜20 mLの代わりにニッケル標準液(Ni 10 μg/mL)0.4〜20 mLを用いる。
注記 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
e) 操作(溶媒抽出を伴う場合) 操作は,次による。
溶媒抽出を適用するときは,7.4 e) の操作を行う。ただし,カドミウムをニッケルと読み替え,ニ
ッケルの分析波長は221.647 nmを用いる。また,検量線の作成には,ニッケル標準液(Ni 10 μg/mL)
を適切な濃度(Ni 0.2〜1 μg/mL)にうすめ,その0.4〜20 mLを段階的にとり,約100 mLとしたもの
を用いる。
注記 この溶媒抽出法は,試料溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃
度が高く,ニッケルの濃度が低い場合に適用するとよい。カドミウム,鉛,ニッケル,マン
ガン及びバナジウムの定量に用いることができる。
9.6
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,ニッケル及び内標準
物質のそれぞれの質量/荷電数におけるイオンカウントを測定し,ニッケルのイオンカウントと内標準物質
のイオンカウントとの比を求めてニッケルを定量する。
− 定量範囲 Ni 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) ニッケル標準液(Ni 1 μg/mL) 9.4 a) 3) による。
5) ニッケル標準液(Ni 50 ng/mL) 4) のニッケル標準液(Ni 1 μg/mL)50 mLを全量フラスコ1 000 mL
にとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL] 7.5 a) 6) による。
7) 混合標準液[(Cd 50 ng,Pb 50 ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL] 7.5 a) 7) による。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Niとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,イ
ットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるように
硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
ニッケル及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を
読み取り,ニッケルの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,ニッケルの指示値とイッ
36
K 0083:2017
トリウムの指示値との比を求め,試料について得たニッケルの指示値とイットリウムの指示値との
比を補正する。
4) 検量線からニッケルの量を求め,試料中のニッケルの濃度(Ni μg/L)を算出する。検量線の作成は,
次による。
検量線 ニッケル標準液(Ni 50 ng/mL又はNi 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階的
にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝酸
(1+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。別に,空試験と
して全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃
度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って標準液について得
た指示値の比を補正した後,ニッケル(Ni)の量に対する指示値とイットリウムの指示値との比の
関係線を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 7.5 c) の注記3参照。
10
マンガンの分析方法
10.1
一般
マンガンの分析には,過よう素酸吸光光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分
光分析法又はICP質量分析法を用いる。
10.2
過よう素酸吸光光度法
試料溶液を硫酸酸性とした後,過よう素酸カリウムを加え,加熱して赤紫の過マンガン酸イオンを生成
させ,その吸光度を測定してマンガンを定量する。
− 定量範囲 Mn 40〜500 μg
− 繰返し分析精度 変動係数で3〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硫酸(1+1) 6.4.1 c) による。
3) りん酸 JIS K 9005に規定するもの。
4) 過よう素酸カリウム JIS K 8249に規定するもの。
5) マンガン標準液(Mn 0.1 mg/mL) JIS K 8247に規定する過マンガン酸カリウム0.288 gをとり,水
150 mLに硫酸(1+1)10 mLを加えた溶液に溶かす。亜硫酸水素ナトリウム溶液(JIS K 8059に規
定する亜硫酸水素ナトリウム10 gを水に溶かして100 mLとする。)を滴加し,かき混ぜて脱色した
後,煮沸して過剰の二酸化硫黄を追い出す。放冷後,全量フラスコ1 000 mLに移し,水を標線まで
加える。又は,マンガン(99.9 %以上)0.100 gをとり,硫酸(1+3)[JIS K 8951に規定する硫酸
又はそれ以上の純度の硫酸を用いて調製する。]20 mLに加熱して溶かし,放冷後,全量フラスコ
1 000 mLに移し入れ,水を標線まで加える。又は,国家計量標準へのトレーサビリティが確保され
たもの又はそれを一定濃度にうすめたものを用いる。
6) マンガン標準液(Mn 20 μg/mL) 5) のマンガン標準液(Mn 0.1 mg/mL)50 mLを全量フラスコ250
mLにとり,水を標線まで加える。
b) 装置 装置は,分光光度計又は光電光度計を用いる。
37
K 0083:2017
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Mnとして40〜500 μgを含む。)をとり,硫酸(1+1)10 mLを加
え,加熱して硫酸の白煙を発生させ,ハロゲン化物を除去する。
2) 放冷後,水約20 mLとりん酸1 mLとを加え,加熱して内容物を溶かす。不溶解物があるときには,
ろ別し,ろ紙及び沈殿を温水で洗った後,ろ液及び洗液を合わせ,水を加えて約45 mLにする。
3) 過よう素酸カリウム0.5 gを加え,沸騰水浴中で30分間加熱して5) 発色させる。
4) 流水で冷却した後,全量フラスコ50 mLに移し,水を標線まで加える。
5) この溶液の一部を吸収セルに移し,波長525 nm付近又は545 nm付近の吸光度を測定する。
6) 別に,空試験溶液について硫酸(1+1)10 mLとりん酸1 mLとを加えた後,3)〜5) の操作を行っ
て吸光度を求め,試料溶液について得た吸光度を補正する。
7) 検量線からマンガンの量を求め,試料溶液中のマンガンの濃度(Mn μg/L)を算出する。検量線の
作成は,次による。
検量線 マンガン標準液(Mn 20 μg/mL)2〜25 mLをビーカー100 mLに段階的にとり,水を加えて
液量を約30 mLとし,硫酸(1+1)10 mLとりん酸1 mLとを加えた後,3)〜6) の操作を行ってマ
ンガン(Mn)の量と吸光度との関係線を作成する。
注5) 加熱時間が長すぎると,生成した過マンガン酸イオンが分解するおそれがあるから,加熱時間
は正しく守る。
10.3
フレーム原子吸光法
試料溶液をアセチレン−空気のフレーム中に噴霧し,マンガンによる原子吸光を波長279.5 nmで測定し
て,マンガンを定量する。
− 定量範囲 Mn 0.1〜4 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) マンガン標準液(Mn 10 μg/mL) 10.2 a) 5) のマンガン標準液(Mn 0.1 mg/mL)50 mLを全量フラ
スコ500 mLにとり,硝酸(1+1)[6.1.1 a) による。]10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) マンガン中空陰極ランプ
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,波
長279.5 nmの指示値(吸光度又はその比例値)を読み取る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た指示値
を補正する。
3) 検量線からマンガンの量を求め,試料溶液中のマンガンの濃度(Mn mg/L)を算出する。検量線の
作成は,次による。
検量線 マンガン標準液(Mn 10 μg/mL)1〜40 mLを全量フラスコ100 mLに段階的にとり,試料
溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を
行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を加
えた後,1) の操作を行って標準液について得た指示値を補正し,マンガン(Mn)の量と指示値と
38
K 0083:2017
の関係線を作成する。検量線は,試料測定時に作成する。
注記1 シリカを多量に含む場合には,干渉抑制剤としてカルシウム(又はマグネシウム)を試料
溶液1 L当たり200 mg程度加えておくとよい。
注記2 マンガンの濃度が低い場合には,次の鉄共沈法で濃縮した後,c) の操作を行ってもよい。
試料溶液の適量をとり,約90 ℃に加熱し,硫酸アンモニウム鉄(III)溶液(Fe 2 mg/mL)
[JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水1.8 gをとり,硝酸(1+6)[JIS
K 8541に規定する硝酸又はそれ以上のものを用いて調製する。]10 mLと水に溶かして100
mLとする。]5 mLとJIS K 8230に規定する過酸化水素5〜10 mLとを加え,この溶液をか
き混ぜながらアンモニア水(1+1)[7.2 a) 6) による。]又は水酸化ナトリウム溶液(100 g/L)
[JIS K 8576に規定する水酸化ナトリウムを用いて調製する。]を加えて,水酸化鉄(III)
の沈殿を生成させる。沈殿が沈降した後,ろ紙5種Aを用いてろ過し,温水で洗う。沈殿
をできるだけ元のビーカーに移し,ろ紙に付着した沈殿は,過酸化水素水を少量加えた塩
酸(1+2)の少量に溶かし,ろ紙は温水で洗浄する。ろ液及び洗液を合わせ,0.1〜1 mol/L
の塩酸酸性溶液の一定量とする。
10.4
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,マンガンによる原子吸光を波長279.5 nmで測定してマン
ガンを標準添加法によって定量する。
− 定量範囲 Mn 1〜30 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) マンガン標準液(Mn 1 μg/mL) 10.3 a) 2) のマンガン標準液(Mn 10 μg/mL)10 mLを全量フラス
コ100 mLにとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
4) マンガン標準液(Mn 0.1 μg/mL) 3) のマンガン標準液(Mn 1 μg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) マンガン中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.1によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った適量を,それぞれ全量フラスコ20 mLにとり,マンガン標準液(Mn 0.1 μg/mL)
を加えないものと,0.2〜6 mLの範囲で段階的に3段階以上添加したものとを調製し,それぞれの
溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)をマイクロピペットを用いて発熱体に注入し,JIS K 0121
39
K 0083:2017
の8.(操作方法)に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(600〜1 000 ℃,30〜40秒
間),原子化1)(2 200〜2 700 ℃,3〜6秒間)し,波長279.5 nmの指示値(吸光度又はその比例値)
を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) マンガンの添加量と指示値との関係線を作成し,マンガンの量を求め,試料溶液中のマンガンの濃
度(Mn μg/L)を算出する。
10.5
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,マンガンによる発光を波長257.610 nmで測定して,マンガン
を定量する。
− 定量範囲 Mn 0.01〜5 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) マンガン標準液(Mn 10 μg/mL) 10.3 a) 2) による。
3) 混合標準液[(Cd 10 μg,Pb 10 μg,Ni 10 μg,Mn 10 μg,V 10 μg)/mL] 7.4 a) 4) による。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
波長257.610 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に,1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線からマンガンの量を求め,試料溶液中のマンガンの濃度(Mn mg/L)を算出する。検量線の
作成は,次による。
検量線 マンガン標準液(Mn 10 μg/mL)0.1〜2 mL(又は2〜50 mL)3) を全量フラスコ100 mLに
段階的にとり,試料溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液に
ついて1) の操作を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件
になるように酸を加えた後,1) の操作を行って,標準液について得た発光強度を補正し,マンガン
(Mn)の量と発光強度との関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムをマンガンと読み替え,マ
ンガンの分析波長は257.610 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10 μg/mL)
0.1〜20 mLの代わりにマンガン標準液(Mn 10 μg/mL)0.1〜2 mL(又は2〜50 mL)を用いる。
注記 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
e) 操作(溶媒抽出を伴う場合) 操作は,次による。
溶媒抽出を適用するときは,7.4 e) の操作を行う。ただし,カドミウムをマンガンと読み替え,マ
ンガンの分析波長は257.610 nmを用いる。また,検量線の作成には,マンガン標準液(Mn 10 μg/mL)
を適切な濃度(Mn 0.1〜1 μg/mL)にうすめ,その0.1〜2 mL(又は2〜50 mL)を段階的にとり,約
100 mLとしたものを用いる。
40
K 0083:2017
注記 この溶媒抽出法は,試料溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃
度が高く,マンガンの濃度が低い場合に適用するとよい。カドミウム,鉛,ニッケル,マン
ガン及びバナジウムの定量に用いることができる。
10.6
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,マンガン及び内標準
物質のそれぞれの質量/荷電数におけるイオンカウントを測定し,マンガンのイオンカウントと内標準物質
のイオンカウントとの比を求めてマンガンを定量する。
− 定量範囲 Mn 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) マンガン標準液(Mn 1 μg/mL) 10.4 a) 3) による。
5) マンガン標準液(Mn 50 ng/mL) 4) のマンガン標準液(Mn 1 μg/mL)50 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL] 7.5 a) 6) による。
7) 混合標準液[(Cd 50 ng,Pb 50 ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL] 7.5 a) 7) による。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Mnとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,
イットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるよう
に硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
マンガン及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を
読み取り,マンガンの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,マンガンの指示値とイッ
トリウムの指示値との比を求め,試料について得たマンガンの指示値とイットリウムの指示値との
比を補正する。
4) 検量線からマンガンの量を求め,試料中のマンガンの濃度(Mn μg/L)を算出する。検量線の作成
は,次による。
検量線 マンガン標準液(Mn 50 ng/mL又はMn 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階
的にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝
酸(1+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。別に,空試験
として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の
濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って標準液について
得た指示値の比を補正した後,マンガン(Mn)の量に対する指示値とイットリウムの指示値との比
の関係線を作成する。検量線は,試料測定時に作成する。
41
K 0083:2017
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 7.5 c) の注記3参照。
11
バナジウムの分析方法
11.1
一般
バナジウムの分析には,N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法,フレーム原子吸光法,
電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を用いる。
11.2
N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法
試料溶液を過マンガン酸カリウムで酸化してバナジウム(V)とし,これにN-ベンゾイル-N-フェニルヒ
ドロキシルアミン(BPHA)を加えて生成した赤紫のバナジウム錯体を塩酸酸性溶液からクロロホルムで
抽出し,その吸光度を測定してバナジウムを定量する。
− 定量範囲 V 2〜50 μg
− 繰返し分析精度 変動係数で3〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸 JIS K 8180に規定する塩酸又はそれ以上のもの。
3) 銅溶液(10 g/L) JIS K 8660に規定する銅(99.9 %以上)1 gを硝酸(1+1)[6.1.1 a) による。]10
mLに溶解し,過塩素酸(60 %)(JIS K 8223に規定する過塩素酸又はそれ以上の純度のもの)20 mL
を加えて加熱蒸発して白煙を十分に発生させる。放冷後,水でうすめて100 mLとする。
4) BPHA−クロロホルム溶液(2 g/L) JIS K 9569に規定するBPHA 0.2 gをJIS K 8322に規定するク
ロロホルム100 mLに溶かす。
5) 過マンガン酸カリウム溶液(3 g/L) JIS K 8247に規定する過マンガン酸カリウム0.3 gを水に溶か
して100 mLとする。
6) バナジウム標準液(V 0.1 mg/mL) JIS K 8747に規定するバナジン(V)酸アンモニウム(メタバ
ナジン酸アンモニウム)(純度が99.0 %以上)0.230 gをとり,硫酸(1+1)[6.4.1 c) による。]10 mL
と熱水200 mLとに溶かす。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
7) バナジウム標準液(V 2 μg/mL) 6) のバナジウム標準液(V 0.1 mg/mL)20 mLを全量フラスコ1 000
mLにとり,硫酸(1+1)[6.4.1 c) による。]10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗
2) 吸光光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Vとして2〜50 μgを含む。)をビーカー100 mLにとり,銅溶液(10
g/L)1 mLを加える6)。
2) 過マンガン酸カリウム溶液(3 g/L)を溶液の色がうすい赤となるまで滴加し,更に1滴を過剰に加
えて約5分間放置し,バナジウムを酸化する7)。
3) 分液漏斗に移し入れ,水で約50 mLとする。
4) BPHA−クロロホルム溶液(2 g/L)10 mLを加える。次に,塩酸20 mL 8) を加えて,過剰の過マン
ガン酸を還元し,直ちに9) 約1分間振り混ぜてバナジウム錯体を抽出する。
42
K 0083:2017
5) 放置した後,クロロホルム層を乾いたろ紙でろ過する。
6) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長530 nm付近の吸光度を
測定する。
7) 別に,空試験溶液について銅溶液(10 g/L)1 mLを加えた後,2)〜6) の操作を行って吸光度を求め,
試料溶液について得た吸光度を補正する。
8) 検量線からバナジウムの量を求め,試料溶液中のバナジウムの濃度(V μg/L)を算出する。検量線
の作成は,次による。
検量線 バナジウム標準液(V 2 μg/mL)1〜25 mLを分液漏斗に段階的にとり,銅溶液(10 g/L)1 mL
を加えた後,2)〜7) の操作を行って,バナジウム(V)の量と吸光度との関係線を作成する。
注6) マンガン(II)が正の誤差の原因となるから,あらかじめ銅イオンを共存させておき,影響を除
くのがよい。
7) バナジウムを5価に酸化するために過マンガン酸カリウムを多く用いると,マンガン量が多く
なるので,必要以上には用いないのがよい。
8) バナジウム抽出のための塩酸最適濃度は,3.5〜4 mol/Lである。
9) 塩酸酸性では,バナジウムが徐々に還元されて低値を示しやすいので,手早く抽出を行う。特
に高濃度の塩酸酸性では還元されやすい。
11.3
フレーム原子吸光法
試料溶液をアセチレン−一酸化二窒素フレーム中に噴霧し,バナジウムによる原子吸光を波長318.4 nm
で測定し,バナジウムを定量する。
− 定量範囲 V 1〜20 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸 6.3.1 a) による。
3) 硝酸アルミニウム溶液(400 g/L) JIS K 8544に規定する硝酸アルミニウム九水和物70 gをとり,
少量の水を加え加熱して溶かす。放冷後,水を加えて100 mLにする。
4) バナジウム標準液(V 0.1 mg/mL) 11.2 a) 6) による。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) バナジウム中空陰極ランプ
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Vとして0.1〜2 mgを含む。)を全量フラスコ100 mLにとり,硝酸
1 mLを加えた後,水を標線まで加える。
2) この溶液50 mLを乾いたビーカーにとり,硝酸アルミニウム溶液(400 g/L)1 mLを加える。
3) 2) の溶液をJIS K 0121の8.(操作方法)の操作に従って,アセチレン−一酸化二窒素フレーム中
に噴霧し10),波長318.4 nmの指示値(吸光度又はその比例値)を読み取る。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って指示値を読み取り,試料溶液
について得た指示値を補正する。
5) 検量線からバナジウムの量を求め,試料溶液中のバナジウムの濃度(V mg/L)を算出する。検量線
の作成は,次による。
43
K 0083:2017
検量線 バナジウム標準液(V 0.1 mg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料
溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について,1)〜3) の操
作を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸
を加えた後,1)〜3) の操作を行って標準液について得た指示値を補正し,バナジウム(V)の量と
指示値との関係線を作成する。検量線は,試料測定時に作成する。
注10) 多燃料フレームの方が高感度が得られる。感度の最も高い部分は,フレームのごく限られた位
置であるから,この位置を確かめておくとよい。
11.4
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,バナジウムによる原子吸光を波長318.4 nmで測定してバ
ナジウムを標準添加法によって定量する。
− 定量範囲 V 10〜200 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) バナジウム標準液(V 1 μg/mL) 11.2 a) 6) のバナジウム標準液(V 0.1 mg/mL)10 mLを全量フラ
スコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) バナジウム中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.1によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,バナジウム標準液(V 1
μg/mL)を加えないものと,0.2〜4 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) 1) の操作を行った試料の一定量(例えば,10〜50 μL)をマイクロピペットを用いて発熱体に注入
し,JIS K 0121の8.(操作方法)の操作に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(500
〜600 ℃,30〜40秒間),原子化1)(2 700〜3 000 ℃,5〜10秒間),波長318.4 nmの指示値(吸光
度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) バナジウムの添加量と指示値との関係線を作成し,バナジウムの量を求め,試料溶液中のバナジウ
ムの濃度(V μg/L)を算出する。
44
K 0083:2017
11.5
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,バナジウムによる発光を波長309.311 nmで測定して,バナジ
ウムを定量する。
− 定量範囲 V 0.02〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) バナジウム標準液(V 10 μg/mL) 11.2 a) 6) のバナジウム標準液(V 0.1 mg/mL)10 mLを全量フラ
スコ100 mLにとり,硝酸(1+1)[6.1.1 a) による。]2 mLを加えて,水を標線まで加える。
3) 混合標準液[(Cd 10 μg,Pb 10 μg,Ni 10 μg,Mn 10 μg,V 10 μg)/mL] 7.4 a) 4) による。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.1で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
波長309.311 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線からバナジウムの量を求め,試料溶液中のバナジウムの濃度(V mg/L)を算出する。検量線
の作成は,次による。
検量線 バナジウム標準液(V 10 μg/mL)0.2〜20 mL 3) を全量フラスコ100 mLに段階的にとり,
試料溶液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操
作を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸
を加えた後,1) の操作を行って標準液について得た発光強度を補正し,バナジウム(V)の量と発
光強度との関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムをバナジウムと読み替え,
バナジウムの分析波長は309.311 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10
μg/mL)0.1〜20 mLの代わりにバナジウム標準液(V 10 μg/mL)0.2〜20 mLを用いる。
注記 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
e) 操作(溶媒抽出を伴う場合) 操作は,次による。
溶媒抽出を適用するときは,7.4 e) の操作を行う。ただし,カドミウムをバナジウムと読み替え,
バナジウムの分析波長は309.311 nmを用いる。また,検量線の作成には,バナジウム標準液(V 10
μg/mL)を適切な濃度(V 0.1〜1 μg/mL)にうすめ,その0.2〜20 mLを段階的にとり,約100 mLとし
たものを用いる。
注記 この溶媒抽出法は,試料溶液のナトリウム,カリウム,マグネシウム,カルシウムなどの濃
度が高く,バナジウムの濃度が低い場合に適用するとよい。カドミウム,鉛,ニッケル,マ
ンガン及びバナジウムの定量に用いることができる。
11.6
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,バナジウム及び内標
準物質のそれぞれの質量/荷電数におけるイオンカウントを測定し,バナジウムのイオンカウントと内標準
物質のイオンカウントとの比を求めてバナジウムを定量する。
45
K 0083:2017
− 定量範囲 V 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) バナジウム標準液(V 1 μg/mL) 11.4 a) 3) による。
5) バナジウム標準液(V 50 ng/mL) 4) のバナジウム標準液(V 1 μg/mL)50 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(Cd 1 μg,Pb 1 μg,Ni 1 μg,Mn 1 μg,V 1 μg)/mL] 7.5 a) 6) による。
7) 混合標準液[(Cd 50 ng,Pb 50 ng,Ni 50 ng,Mn 50 ng,V 50 ng)/mL] 7.5 a) 7) による。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.1で調製した試料溶液の適量(Vとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,イ
ットリウム溶液(Y 1 μg/mL)1 mLを加え,硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるよう
に硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
バナジウム及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)
を読み取り,バナジウムの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,バナジウムの指示値とイ
ットリウムの指示値との比を求め,試料について得たバナジウムの指示値とイットリウムの指示値
との比を補正する。
4) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度(V μg/L)を算出する。検量線の作
成は,次による。
検量線 バナジウム標準液(V 50 ng/mL又はV 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階
的にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝
酸(1+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。別に,空試験
として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の
濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って標準液について
得た指示値の比を補正した後,バナジウム(V)の量に対する指示値とイットリウムの指示値との
比の関係線を作成する。検量線の作成は,試料測定時に行う。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 7.5 c) の注記3参照。
12
クロムの分析方法
12.1
一般
クロムの分析には,ジフェニルカルバジド吸光光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP
46
K 0083:2017
発光分光分析法又はICP質量分析法を適用する。
12.2
ジフェニルカルバジド吸光光度法
クロム(III)を過マンガン酸カリウムで酸化してクロム(VI)とする。過剰の過マンガン酸カリウムを
亜硝酸ナトリウムで分解した後,1,5-ジフェニルカルボノヒドラジド(ジフェニルカルバジド)を加え,
生成する赤紫の錯体の吸光度を測定して定量する。
− 定量範囲 Cr 2〜50 μg
− 繰返し分析精度 変動係数で3〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硫酸(1+9) 水9容をビーカーにとり,冷却しながらJIS K 8951に規定する硫酸又はそれ以上の
純度の硫酸1容をかき混ぜながら徐々に加える。
3) 過マンガン酸カリウム溶液(3 g/L) 11.2 a) 5) による。
4) 亜硝酸ナトリウム溶液(20 g/L) JIS K 8019に規定する亜硝酸ナトリウム2 gを水に溶かして100 mL
とする。使用時に調製する。
5) 尿素溶液(200 g/L) JIS K 8731に規定する尿素20 gを水に溶かして100 mLとする。
6) ジフェニルカルバジド溶液(10 g/L) JIS K 8488に規定する1,5-ジフェニルカルボノヒドラジド(ジ
フェニルカルバジド)1 gをJIS K 8034に規定するアセトン100 mLに溶かし,JIS K 8355に規定す
る酢酸1滴を加えて酸性とする。褐色ガラス瓶に入れ,0〜10 ℃の暗所に保存する。
注記 2週間は安定である。
7) クロム標準液(Cr 0.1 mg/mL) JIS K 8005に規定する容量分析用標準物質の二クロム酸カリウムを
150 ℃で約1時間乾燥し,デシケータ中で放冷する。K2Cr2O7 100 %に対してその0.283 gをとり,
少量の水に溶かして,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又は,国家計量標
準へのトレーサビリティが確保されたもの又はそれを一定濃度にうすめたものを用いる。
8) クロム標準液(Cr 2 μg/mL) 7) のクロム標準液(Cr 0.1 mg/mL)20 mLを全量フラスコ1 000 mL
にとり,水を標線まで加える。又は,国家計量標準へのトレーサビリティが確保されたもの又はそ
れを一定濃度にうすめたものを用いる。
9) アジ化ナトリウム溶液(50 g/L) JIS K 9501に規定するアジ化ナトリウム5 gを水に溶かして100 mL
とする。
b) 装置 装置は,分光光度計又は光電光度計を用いる。
c) 操作 操作は,次による。
1) 6.2で調製した試料溶液の適量(Crとして2〜50 μgを含む。)をビーカー100 mLにとり,硫酸(1
+9)3 mLを加え11),数分間煮沸する。
2) 溶液を静かに加熱し,過マンガン酸カリウム溶液(3 g/L)を1滴ずつ加え着色させる。引き続き加
熱し,溶液の赤い色が消えそうになったら,さらに,過マンガン酸カリウム溶液(3 g/L)を滴加し,
常に赤い色を保つようにして数分間煮沸を続ける。
3) 流水で冷却し,尿素溶液(200 g/L)10 mLを加え,激しくかき混ぜながら亜硝酸ナトリウム溶液(20
g/L)を1滴ずつ加えて溶液の赤い色を消し,過剰の過マンガン酸及び酸化マンガン(IV)を分解す
る。
亜硝酸ナトリウム溶液(20 g/L)の代わりにアジ化ナトリウム溶液(50 g/L)を用いることができ
る。この場合,アジ化ナトリウム溶液(50 g/L)を注意して滴加しよく振り混ぜて過マンガン酸を
47
K 0083:2017
分解し,続いて2〜3分間煮沸して過剰のアジ化ナトリウムを分解する。
4) 全量フラスコ50 mLに移し入れ,液温を約15 ℃ 12) に保ち,ジフェニルカルバジド溶液(10 g/L)
1 mLを加え,直ちに振り混ぜる。水を標線まで加え振り混ぜ,約5分間放置する13)。
5) 溶液の一部を吸収セルに移し,波長540 nm付近の吸光度を測定する。
6) 別に空試験溶液について1)〜5) の操作を行って吸光度を測定し,試料溶液について得た吸光度を補
正する。
7) 検量線からクロムの量を求め,試料溶液中のクロムの濃度(Cr μg/L)を算出する。検量線の作成は,
次による。
検量線 クロム標準液(Cr 2 μg/mL)1〜25 mLを段階的にビーカーにとり,それぞれに硫酸(1+9)
3 mLを加え,水を加えて液量を約30 mLとした後,2)〜6) の操作を行ってクロム(Cr)の量と吸
光度との関係線を作成する。
注11) 1,5-ジフェニルカルボノヒドラジドによるクロム(VI)の発色には,硫酸の濃度は0.1 mol/L程
度が適当である。
12) 液温は,発色に影響するので,約15 ℃に保つことが必要である。
13) 発色は2〜3分程で最高となり,その後徐々に退色するが,5〜15分間はほとんど変化しない。
注記1 試料溶液が鉄を含むとき,鉄が多量になるに従って吸光度が低くなるが,発色液50 mL中約
1 mgで一定となる(約20 %低値を示す。)。ただし,ジフェニルカルバジド溶液を加える前
に,二りん酸ナトリウム溶液(JIS K 8785に規定する二りん酸ナトリウム十水和物5 gを水
に溶かし100 mLとする。)2 mLを加えると,鉄2.5 mgまでは影響されなくなる。鉄がクロ
ムより少ない場合には無視してもよい。
注記2 この方法では,モリブデン,水銀,バナジウムなどが影響する。ただし,モリブデンは0.1 mg
までは影響しない。水銀は塩化物イオンの添加によって妨害しなくなる。また,バナジウム
は発色後,10〜15分間経過してから吸光度を測定すれば,その影響は無視できる。
注記3 他の妨害が多い場合には,次の操作を行うとよい。試料溶液の適量(Crとして2〜50 μgを
含む。)を分液漏斗にとり,試料20 mLにつき硫酸(1+1)[6.4.1 c) による。]5 mLを加え,
硫酸の濃度を約1.8 mol/Lとし,これに過マンガン酸カリウム溶液(3 g/L)を滴加し,僅か
に着色させる。これに,クペロン溶液(50 g/L)[JIS K 8289に規定するクペロン5 gを水に
溶かして100 mLとする。]5 mLとJIS K 8322に規定するクロロホルム10 mLを加えて約30
秒間激しく振り混ぜ,鉄その他を抽出し静置する。クロロホルム層を分離し,水層に再びク
ペロン溶液(50 g/L)1 mLとクロロホルム10 mLとを加えて再び抽出し,クロロホルム層を
分離する。水層をビーカー100 mLに移し,蒸発して軽く乾燥する。これに少量の硫酸と硝酸
とを加え,再び蒸発乾固して有機物を分解する。硫酸(1+1)[6.4.1 c) による。]0.3 mL及
び水約30 mLに溶かす。過マンガン酸カリウム溶液(3 g/L)でクロムを酸化した後,c) に
よって操作する。
12.3
フレーム原子吸光法
試料溶液をアセチレン−空気などのフレーム中に噴霧し,クロムによる原子吸光を波長357.9 nmで測定
して,クロムを定量する。
− 定量範囲 Cr 0.2〜5 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
48
K 0083:2017
1) 水 7.2 a) 1) による。
2) クロム標準液(Cr 10 μg/mL) 12.2 a) 7) のクロム標準液(Cr 0.1 mg/mL)50 mLを全量フラスコ500
mLにとり,硝酸(1+1)[6.1.1 a) による。]10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) クロム中空陰極ランプ
c) 操作 操作は,次による。
1) 6.2で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,波
長357.9 nmの指示値(吸光度又はその比例値)を読み取る。
なお,フレームには,少燃料のアセチレン−空気又はアセチレン−一酸化二窒素フレームを用い
る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,指示値を読み取り,試料溶液
について得た指示値を補正する。
3) 検量線からクロムの量を求め,試料溶液中のクロムの濃度(Cr mg/L)を算出する。検量線の作成は,
次による。
検量線 クロム標準液(Cr 10 μg/mL)2〜50 mLを全量フラスコ100 mLに段階的にとり,試料溶液
と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を行う。
別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を加えた後,
1) の操作を行って標準液について得た指示値を補正し,クロム(Cr)の量と指示値との関係線を作
成する。検量線は,試料測定時に作成する。
注記1 クロムの濃度が低い試料溶液で,抽出を妨害する物質を含まない場合には,次の操作で定
量してもよい。
試料溶液の適量(Crとして5〜100 μgを含む。)をビーカー100 mLにとり,硫酸(1+2)
[6.2.1 a) による。]2 mLを加え過マンガン酸カリウム溶液(3 g/L)数滴を加えて加熱す
る。過マンガン酸の色が消えそうになったら,更に過マンガン酸カリウム溶液(3 g/L)を
滴加し,常に溶液の色が微赤を保つように数分間煮沸を続ける。流水で冷却し,これを分
液漏斗に移し,水を加えて約100 mLとする。トリオクチルアミン(N,N-ジオクチルオク
タンアミン)の酢酸ブチル溶液(30 g/L)20 mLを加え,約10分間激しく振り混ぜた後放
置する。酢酸ブチル層をそのままフレーム中に噴霧してクロムを定量する。検量線の作成
にはクロム標準液(Cr 10 μg/mL)を適宜にうすめて用いる。JIS K 8377に規定する酢酸ブ
チルの代わりにJIS K 8903に規定する4-メチル-2-ペンタノンを用いてもよい。
注記2 アセチレン−空気フレームでは,多燃料フレームにすると感度は高くなるが,鉄,ニッケ
ルなど共存物による妨害も大きくなる。この場合,硫酸ナトリウム,二硫酸カリウム又は
二ふっ化水素アンモニウムを1 %程度共存させる。アセチレン−一酸化二窒素フレームで
は妨害の大部分は消滅する。
12.4
電気加熱原子吸光法
試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム(II)を加え,電気加熱炉
で原子化し,クロムによる原子吸光を波長357.9 nm又は359.3 nmで測定して全クロムを標準添加法によ
って定量する。
− 定量範囲 Cr 5〜100 μg/L
49
K 0083:2017
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸パラジウム(II)溶液(Pd 1 mg/mL) 7.3 a) 3) による。
4) クロム標準液(Cr 1 μg/mL) 12.3 a) 2) のクロム標準液(Cr 10 μg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) クロム中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.2によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,クロム標準液(Cr 1
μg/mL)を加えないものと,0.1〜2 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)及びそれと同体積の硝酸パラジウム(II)溶液(Pd 1mg/mL)
を,マイクロピペットを用いて電気加熱炉に注入する。JIS K 0121の8.(操作方法)の操作に従っ
て,乾燥(100〜120 ℃,30〜40秒間),灰化(500〜800 ℃,30〜40秒間),原子化1)(1 800〜2 500 ℃,
3〜6秒間)し,波長357.9 nm又は359.3 nmの指示値(吸光度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) クロムの添加量と指示値との関係線を作成し,クロムの量を求め,試料溶液中のクロムの濃度(Cr
μg/L)を算出する。
12.5
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,クロムによる発光を波長206.149 nmで測定して,クロムを定
量する。
− 定量範囲 Cr 0.02〜4 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) クロム標準液(Cr 10 μg/mL) 12.3 a) 2) による。
b) 装置 ICP発光分光分析装置
c) 操作(検量線法の場合) 操作は,次による。
1) 6.2で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
50
K 0083:2017
波長206.149 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線からクロムの量を求め,試料溶液中のクロムの濃度(Cr mg/L)を算出する。検量線の作成は,
次による。
検量線 クロム標準液(Cr 10 μg/mL)0.2〜40 mLを全量フラスコ100 mLに段階的にとり,試料溶
液と同じ条件になるように酸を加えた後,水を標線まで加える。この溶液について1) の操作を行
う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件になるように酸を加え
た後,1) の操作を行って試料溶液について得た発光強度を補正し,クロム(Cr)の量と発光強度と
の関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムをクロムと読み替え,クロ
ムの分析波長は206.149 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10 μg/mL)
0.1〜20 mLの代わりにクロム標準液(Cr 10 μg/mL)0.2〜40 mLを用いる。
注記1 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
注記2 クロムの濃度が低い試料溶液で,抽出を妨害する物質を含まない場合には,12.3 c) の注記1
の操作で定量してもよい。ただし,フレームをプラズマと読み替える。プラズマトーチとし
ては有機溶媒用のトーチを用いるとよい。
12.6
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,クロム及び内標準物
質のそれぞれの質量/荷電数におけるイオンカウントを測定し,クロムのイオンカウントと内標準物質のイ
オンカウントとの比を求めてクロムを定量する。
− 定量範囲 Cr 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) クロム標準液(Cr 1 μg/mL) 12.4 a) 4) による。
5) クロム標準液(Cr 50 ng/mL) 4) のクロム標準液(Cr 1 μg/mL)50 mLを全量フラスコ1 000 mLに
とり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.2で調製した試料溶液の適量(Crとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,イ
ットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度となるように
硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
クロム及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を読み
51
K 0083:2017
取り,クロムの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,クロムの指示値とイット
リウムの指示値との比を求め,試料について得たクロムの指示値とイットリウムの指示値との比を
補正する。
4) 検量線からクロムの量を求め,試料中のクロムの濃度(Cr μg/L)を算出する。検量線の作成は,次
による。
検量線 クロム標準液(Cr 50 ng/mL又はCr 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階的に
とり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝酸(1
+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。別に,空試験として
全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度に
なるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って標準液について得た指
示値の比を補正した後,クロム(Cr)の量に対する指示値とイットリウムの指示値との比の関係線
を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
13
ベリリウムの分析方法
13.1
一般
ベリリウムの分析には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量
分析法を適用する。
13.2
フレーム原子吸光法
試料溶液をアセチレン−一酸化二窒素のフレーム中に噴霧し,ベリリウムによる原子吸光を波長234.9
nmで測定し,ベリリウムを定量する。
− 定量範囲 Be 0.05〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸(1+1) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
3) ベリリウム標準液(Be 1 mg/mL) ベリリウム(99 %以上)0.100 gをビーカーにとり,塩酸(1+1)
10 mLを少量ずつ加えて溶かし,時計皿で覆い静かに加熱溶解して,冷却後全量フラスコ100 mL
に移す。ビーカー及び時計皿を水で洗い,洗液を全量フラスコ中の溶液に合わせて,水を標線まで
加える。
4) ベリリウム標準液(Be 10 μg/mL) 3) のベリリウム標準液(Be 1 mg/mL)10 mLを全量フラスコ100
mLにとり,塩酸(1+1)10 mLを加えた後,水を標線まで加える。この溶液25 mLを全量フラス
コ250 mLにとり,塩酸(1+1)25 mLを加えた後,水を標線まで加える。
5) ベリリウム標準液(Be 1 μg/mL) 4) のベリリウム標準液(Be 10 μg/mL)10 mLを全量フラスコ100
mLにとり,塩酸(1+1)10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) フレーム原子吸光分析装置
2) ベリリウム中空陰極ランプ
52
K 0083:2017
c) 操作 操作は,次による。
1) 6.3で調製した試料溶液をJIS K 0121の8.(操作方法)の操作に従って,フレーム中に噴霧し,波
長234.9 nmの指示値(吸光度又はその比例値)を読み取る。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,指示値を読み取り,試料溶液
について得た指示値を補正する。
3) 検量線からベリリウムの量を求め,試料溶液中のベリリウムの濃度(Be mg/L)を算出する。検量線
の作成は,次による。
検量線 ベリリウム標準液(Be 10 μg/mL)0.5〜20 mLを全量フラスコ100 mLに段階的にとり,試
料溶液と同じ条件になるように塩酸(1+1)10 mLを加えた後,水を標線まで加える。この溶液に
ついて,1) の操作を行う。別に,空試験として水について検量線の作成に用いた標準液と同じ条件
になるように酸を加えた後,1) の操作を行って標準液について得た指示値を補正し,ベリリウム
(Be)の量と指示値との関係線を作成する。検量線は,試料測定時に作成する。
注記 ベリリウムの濃度が低い試料溶液で,抽出操作を妨害する物質を含まない場合には,次のよ
うに行ってもよい。
試料溶液の適量(Beとして0.2〜5 μgを含む。)を分液漏斗100 mLにとり,くえん酸水素
二アンモニウム溶液(100 g/L)5 mL及び指示薬としてフェノーフタレイン溶液(1 g/L)数
滴を加え,溶液の色が赤になるまでアンモニア水[JIS K 8085で規定するアンモニア水又は
それ以上の純度のもの]を加えた後,水で50 mLとする。エチレンジアミン四酢酸二水素二
ナトリウム二水和物(JIS K 8107に規定するもの)溶液(20 g/L)5 mL及び炭酸ナトリウム
(JIS K 8625に規定するもの)溶液(5 g/L)2 mLを加える。これにJIS K 8027に規定する
アセチルアセトン溶液(50 g/L)を加えてよく混合した後,JIS K 8903に規定する4-メチル-2-
ペンタノン10 mLを加え,3分間激しく振り混ぜる。30分間放置後,4-メチル-2-ペンタノン
層を分離し,c) 1) 及び2) の操作を行ってベリリウムを定量する。検量線の作成は,ベリリ
ウム標準液(Be 1 μg/mL)0.2〜0.5 mLを分液漏斗にとり,以下,試料溶液を同様に操作して,
ベリリウム(Be)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
13.3
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,ベリリウムによる原子吸光を波長234.9 nmで測定してベ
リリウムを標準添加法によって定量する。
− 定量範囲 Be 5〜50 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) ベリリウム標準液(Be 1 μg/mL) 13.2 a) 4) のベリリウム標準液(Be 10 μg/mL)10 mLを全量フラ
スコ100 mLにとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
53
K 0083:2017
3) ベリリウム中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット10〜50 μL又は自動注入装置。
c) 準備操作 試料を6.3によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,ベリリウム標準液(Be 1
μg/mL)を加えないものと,0.1〜1 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) 1) の操作を行った試料の一定量(例えば,10〜50 μL)をマイクロピペットを用いて発熱体に注入
し,JIS K 0121の8.(操作方法)の操作に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(500
〜800 ℃,30〜40秒間),原子化1)(2 000〜2 700 ℃,4〜6秒間)し,波長234.9 nmの指示値(吸
光度又はその比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) ベリリウムの添加量と指示値との関係線を作成し,ベリリウムの量を求め,試料溶液中のベリリウ
ムの濃度(Be μg/L)を算出する。
13.4
ICP発光分光分析法
試料溶液を誘導結合プラズマ中に噴霧し,ベリリウムによる発光を波長313.042 nmで測定して,ベリリ
ウムを定量する。
− 定量範囲 Be 0.01〜2 mg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) ベリリウム標準液(Be 10 μg/mL) 13.2 a) 3) のベリリウム標準液(Be 1 mg/mL)1 mLを全量フラ
スコ100 mLにとり,硝酸(1+1)[6.1.1 a) による。]2 mLを加えた後,水を標線まで加える。
b) 装置 装置は,ICP発光分光分析装置を用いる。
c) 操作(検量線法の場合) 操作は,次による。
1) 6.3で調製した試料溶液をJIS K 0116の箇条4(ICP発光分光分析)に従って,プラズマ中に噴霧し,
波長313.042 nmの発光強度を測定する2)。
2) 空試験として,空試験溶液を試料溶液と同様に1) の操作を行って,試料溶液について得た発光強
度を補正する。
3) 検量線からベリリウムの量を求め,試料溶液中のベリリウムの濃度(Be mg/L)を算出する。検量線
の作成は,次による。
検量線 ベリリウム標準液(Be 10 μg/mL)0.1〜20 mLを全量フラスコ100 mLに段階的にとり,試
料溶液と同じ条件になるように酸を加えた後,水を標準線まで加える。この溶液について1) の操
作を行う。別に,空試験として,水について検量線の作成に用いた標準液と同じ条件になるように
酸を加えた後,1) の操作を行って標準液について得た発光強度を補正し,ベリリウム(Be)の量と
発光強度との関係線を作成する。検量線は,試料測定時に作成する。
d) 操作(内標準法の場合) 操作は,次による。
54
K 0083:2017
内標準法を適用するときは,7.4 d) の操作を行う。ただし,カドミウムをベリリウムと読み替え,
ベリリウムの分析波長は313.042 nmを用いる。また,検量線の作成には,カドミウム標準液(Cd 10
μg/mL)0.1〜20 mLの代わりにベリリウム標準液(Be 10 μg/mL)01〜20 mLを用いる。
注記1 塩の濃度が比較的高い試料溶液には検量線法よりも内標準法が適している。
注記2 ベリリウムの濃度が低い試料溶液で,抽出を妨害する物質を含まない場合には,13.2 c) の
注記の操作で定量してもよい。
13.5
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,ベリリウム及び内標
準物質のそれぞれの質量/荷電数におけるイオンカウントを測定し,ベリリウムのイオンカウントと内標準
物質のイオンカウントとの比を求めてベリリウムを定量する。
− 定量範囲 Be 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる)。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) ベリリウム標準液(Be 1 μg/mL) 13.3 a) 3) による。
5) ベリリウム標準液(Be 50 ng/mL) 4) のベリリウム標準液(Be 1 μg/mL)50 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.3で調製した試料溶液の適量(Beとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,
イットリウム溶液(Y 1 μg/mL)1 mLを加え,硝酸の最終濃度が0.1〜0.5 mol/Lとなるように硝酸(1
+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
ベリリウム及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を
読み取り,ベリリウムの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,ベリリウムの指示値とイ
ットリウムの指示値との比を求め,試料について得たベリリウムの指示値とイットリウムの指示値
との比を補正する。
4) 検量線からベリリウムの量を求め,試料中のベリリウムの濃度(Be μg/L)を算出する。検量線の作
成は,次による。
検量線 ベリリウム標準液(Be 50 ng/mL又はBe 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段
階的にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように
硝酸(1+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。
別に,空試験として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の
試料と同じ酸の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って
標準液について得た指示値の比を補正した後,ベリリウム(Be)の量に対する指示値とイットリウ
55
K 0083:2017
ムの指示値との比の関係線を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
14
ひ素の分析方法
14.1
一般
ひ素及びガス状ひ素化合物(水素化ひ素など)の分析には,ジエチルジチオカルバミド酸銀吸光光度法,
水素化物発生原子吸光法,電気加熱原子吸光法,水素化物発生ICP発光分光分析法又はICP質量分析法を
用いる。
14.2
ジエチルジチオカルバミド酸銀吸光光度法
ひ素を還元して水素化ひ素として発生させ,これをジエチルジチオカルバミド酸銀のクロロホルム溶液
に吸収させ,生成する赤紫の溶液の吸光度を測定し,ひ素を定量する。
− 定量範囲 As 2〜10 μg
− 繰返し分析精度 変動係数で2〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸 JIS K 8180に規定するひ素分析用のもの又はそれ以上の純度のもの。
3) 塩酸(1+1) 2) の塩酸を用いて調製する。
4) 硫酸 6.3.1 d) による。
5) よう化カリウム溶液(200 g/L) JIS K 8913に規定するよう化カリウム20 gを水に溶かして100 mL
とする。
6) 塩化すず(II)溶液 JIS K 8136に規定する塩化すず(II)二水和物40 gを2) の塩酸に溶かし,塩
酸で100 mLとし,JIS K 8580に規定するすずの粒状のもの2〜3粒を加えて着色ガラス瓶に保存す
る。使用時に適量をとり,水で10倍にうすめる。
7) 酢酸鉛(II)溶液(100 g/L) JIS K 8374に規定する酢酸鉛(II)三水和物12 gをJIS K 8355に規
定する酢酸1,2滴及び水に溶かし100 mLとする。
8) 亜鉛 JIS K 8012に規定する亜鉛のひ素分析用を,JIS Z 8801の規格群に規定する試験用ふるいで
ふるい分け,目開き1 400 μmのふるいを通り1 000 μmのふるいにとど(留)まるもの。
9) ジエチルジチオカルバミド酸銀溶液 JIS K 9512に規定するN,N-ジエチルジチオカルバミド酸銀
(N,N-ジエチルカルバモジチオ酸銀)0.25 gとJIS K 8832に規定するブルシンn水和物0.1 gとを
JIS K 8322に規定するクロロホルムに溶かし,クロロホルムで100 mLとする。
10) クロロホルム JIS K 8322に規定するもの。
11) ひ素標準液(As 0.1 mg/mL) JIS K 8044に規定する三酸化二ひ素を105 ℃で約2時間加熱し,デ
シケータ中で放冷する。As2O3 100 %に対してその0.132 gをとり,水酸化ナトリウム溶液(40 g/L)
[JIS K 8576に規定する水酸化ナトリウムを用いて調製する。]2 mLに溶かした後,水を加えて
500 mLとし,次に硫酸(1+10)[JIS K 8951に規定する硫酸又はそれ以上の純度のものを用いて
調製する。]を加えて微酸性とし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又は,
国家計量標準へのトレーサビリティが確保されたもの又はそれを一定濃度にうすめたものを用い
る。
12) ひ素標準液(As 1 μg/mL) 11) のひ素標準液(As 0.1 mg/mL)10 mLを全量フラスコ1 000 mLに
56
K 0083:2017
とり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 水素化ひ素発生装置 図11に一例を示す。
2) 吸光光度計 分光光度計又は光電光度計
単位 mm
A: 水素化ひ素発生瓶100 mL
B: 導管
b: 酢酸鉛(II)溶液(100 g/L)で湿した
ガラスウール
C: 水素化ひ素吸収管
D: ゴム栓
a)
図11−水素化ひ素発生装置の例
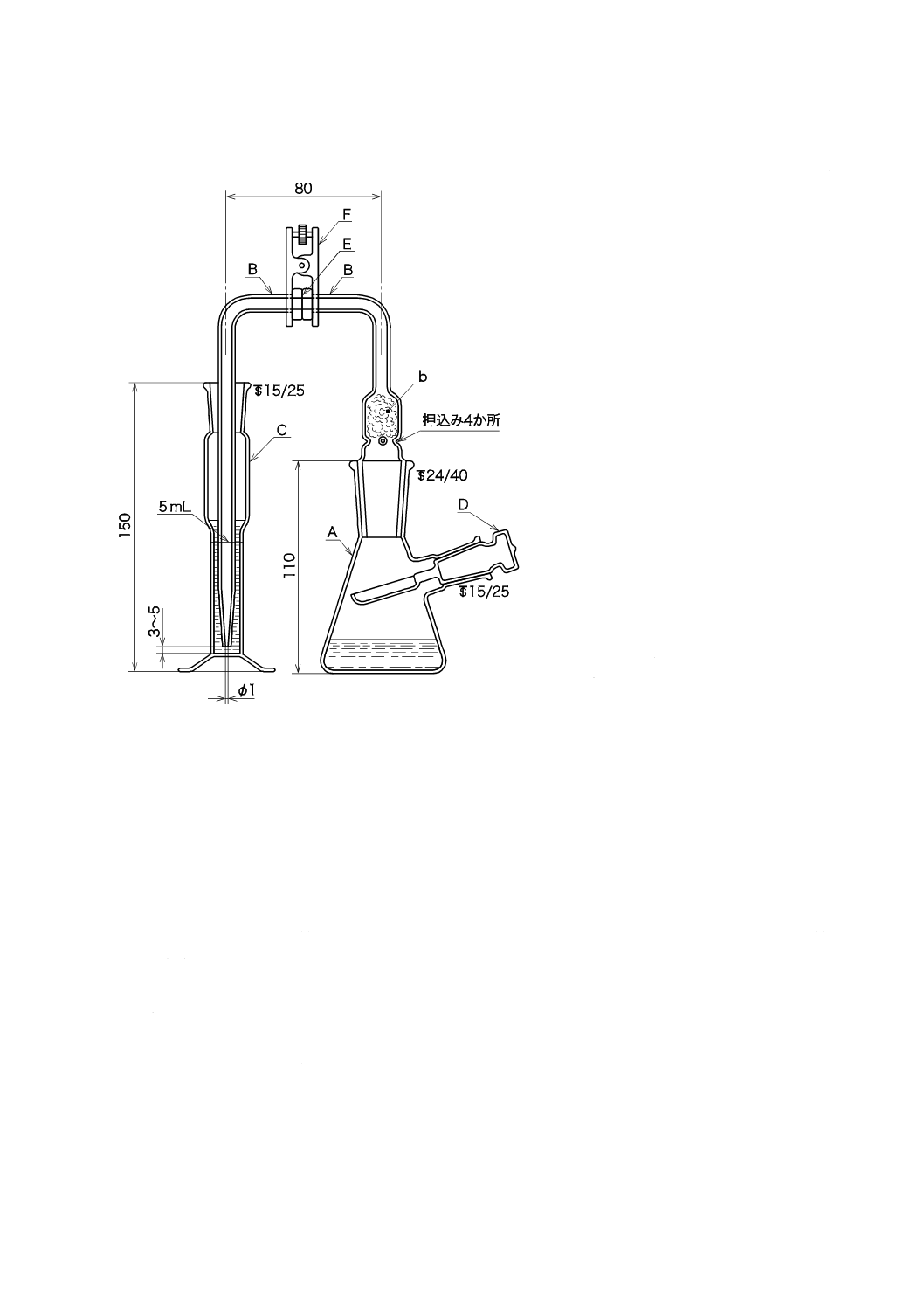
57
K 0083:2017
単位 mm
A: 水素化ひ素発生瓶100 mL
B: 導管
b: 酢酸鉛(II)溶液(100 g/L)で湿した
ガラスウール
C: 水素化ひ素吸収管
D: 亜鉛投入管
E: 平面すり合わせ
F: 押さえばね
b)
図11−水素化ひ素発生装置の例(続き)
c) 操作 操作は,次による。
1) 6.4又は6.5で調製した試料溶液の適量(Asとして2〜10 μgを含む。)を水素化ひ素発生瓶にとり,
硫酸3 mLを加えた後,水で約40 mLとする。
2) 塩酸(1+1)2 mL,よう化カリウム溶液(200 g/L)15 mL及び塩化すず(II)溶液5 mLとを加えて
振り混ぜ,約10分間放置する。
3) 水素化ひ素発生瓶,導管及びジエチルジチオカルバミド酸銀溶液5 mLを入れた水素化ひ素吸収管
を連結した後,水素化ひ素発生瓶に亜鉛約3 gを手早く投入する。水素化ひ素発生瓶を約25 ℃の水
浴中に入れ,約1時間放置してジエチルジチオカルバミド酸銀溶液に水素化ひ素を吸収,発色させ
る。
なお,図11 b) の水素化ひ素発生装置を用いるときは,亜鉛投入管に亜鉛約3 gを入れ吸収管を
連結した後,亜鉛投入管を回転させ,亜鉛を試料中に添加する。
4) この溶液にクロロホルムを加えて5 mLとする。
5) 溶液の一部を吸収セルに移し,クロロホルムを対照液として波長510 nm付近の吸光度を測定する。
6) 別に,空試験溶液について1)〜5) の操作を行って吸光度を求め,試料溶液について得られた吸光度
58
K 0083:2017
を補正する。
7) 検量線からひ素の量を求め,試料溶液中のひ素の濃度(As μg/L)を算出する。検量線の作成は,次
による。
検量線 ひ素標準液(As 1 μg/mL)2〜10 mLを水素化ひ素発生瓶に段階的にとり,硫酸3 mLを加
えた後水で約40 mLとし,2)〜5) の操作を試料溶液と同時に行って吸光度を測定し,ひ素(As)の
量と吸光度との関係線を作成する。検量線は,試料測定時に作成する。
注記 試料溶液中のひ素の濃度が低い場合,又は,水素化ひ素の発生を妨害する物質が含まれてい
る場合には,次の操作によって,ひ素を濃縮し分離して定量してもよい。
試料溶液250 mL当たり,鉄(III)溶液[JIS K 8142に規定する塩化鉄(III)六水和物5 g
又はJIS K 8982に規定する硫酸アンモニウム鉄(III)・12水9 gを塩酸5 mLと水に溶かして
100 mLとする。]2 mL,7.2 a) 4) のメタクレゾールパープル溶液(1 g/L)2,3滴を加え,液
温約80 ℃でかき混ぜながら,アンモニア水(1+2)[JIS K 8085に規定するアンモニア水又
はそれ以上の純度のものを用いて調製する。]を溶液の色が紫(pH約9)になるまで滴加す
る。放置して沈殿が沈降した後,小形のろ紙5種Bでろ別し,温水で2,3回洗浄する。沈
殿は,温硫酸(1+5)18 mL及び塩酸(1+1)2 mLをろ紙上から滴加して溶解し,ろ紙は温
水で洗浄する。ろ液及び洗液を水素化ひ素発生瓶に入れ,水で40 mLとする。よう化カリウ
ム溶液(200 g/L)15 mL及び塩化すず(II)溶液5 mLを加えて振り混ぜ,10分間放置した
後3)〜5) の操作を行って,ひ素を定量する。この方法を用いた場合には,空試験溶液につい
ても同様の操作を行って吸光度を求め,試料溶液について得た吸光度を補正する。
14.3
水素化物発生原子吸光法
試料溶液中のひ素を水素化ひ素とし水素−アルゴンフレーム中に導き,ひ素による原子吸光を波長193.7
nmで測定して,ひ素を定量する。
− 定量範囲 As 5〜50 μg/L
− 繰返し分析精度 変動係数で3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸 JIS K 8180に規定するひ素分析用又はそれ以上の純度のもの。
3) 塩酸(1+1) 2) の塩酸を用いて調製する。
4) 塩酸(1 mol/L) 2) の塩酸を用いて調製する。
5) 硝酸 6.3.1 a) による。
6) 硫酸(1+1) 6.4.1 c) による。
7) 過マンガン酸カリウム溶液(3 g/L) JIS K 8247に規定する過マンガン酸カリウム0.3 gを水に溶か
して100 mLとする。
8) よう化カリウム溶液(200 g/L) 14.2 a) 5) による。
9) テトラヒドロほう酸ナトリウム溶液(10 g/L) テトラヒドロほう酸ナトリウム5 gを水酸化ナトリ
ウム溶液(0.1 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)に溶かして500
mLとする。使用時に調製する。
10) アルゴン JIS K 1105に規定するアルゴン2級。
11) ひ素標準液(As 0.1 μg/mL) 14.2 a) 12) のひ素標準液(As 1 μg/mL)10 mLを全量フラスコ100 mL
にとり,3) の塩酸(1+1)2 mLを加えた後,水を標線まで加える。
59
K 0083:2017
12) アスコルビン酸溶液(100 g/L) JIS K 9502に規定するL(+)-アスコルビン酸10 gを水に溶かして
100 mLとする。
b) 器具及び装置 器具及び装置は,次による。
1) 連続式水素化物発生装置 図12に一例を示す。
2) バッチ式水素化物発生装置 図13に一例を示す。
3) 原子吸光分析装置
4) ひ素中空陰極ランプ
図12−連続式水素化物発生装置の一例
A:コック
B:反応容器
C:貯留器
D:流量計
E:シリンジ
F:セプタム
図13−バッチ式水素化物発生装置の一例
c) 操作(連続式水素化物発生装置を用いる場合) 操作は,次による。
1) 6.4又は6.5で調製した試料溶液の適量(Asとして0.1〜1 μgを含む。)をビーカー100 mLにとり,
硫酸(1+1)1 mL及び硝酸2 mLを加え,さらに過マンガン酸カリウム溶液(3 g/L)を溶液が着色
するまで添加する。
2) 加熱板上で加熱して硫酸の白煙を発生させる。
なお,加熱中に過マンガン酸の色が消えたときは,過マンガン酸カリウム溶液(3 g/L)を追加す
る。
60
K 0083:2017
3) 2) の溶液を室温まで放冷した後,水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及び
アスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全量フラスコ20 mLに洗い
移し,水を標線まで加える。
4) 連続式水素化物発生装置にアルゴンを流しながら,3) の溶液,テトラヒドロほう酸ナトリウム溶液
(10 g/L)及び塩酸(1 mol/L)を,定量ポンプで連続的に装置内に導入し,水素化ひ素を発生させ
る。
5) 発生した水素化ひ素と廃液とを分離した後,水素化ひ素を含む気体を水素−アルゴンフレーム中に
導入し,波長193.7 nmの指示値(吸光度又はその比例値)を読み取る。
6) 空試験として,空試験溶液を試料溶液と同様に,1)〜5) の操作を行った後,指示値を読み取り,試
料について得た指示値を補正する。
7) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。検量線の作成は,次によ
る。
検量線 ひ素標準液(As 0.1 μg/mL)1〜10 mLを段階的に全量フラスコ20 mLにとり,塩酸3 mL,
よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間
静置した後,水を標線まで加え,c) 4) 及び5) の操作を行う。別に,空試験として全量フラスコ20
mLに水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)
0.4 mLを加え,約60分間静置した後,水を標線まで加えたものを用い,c) 4) 及び5) の操作を行
って,ひ素標準液についての指示値を補正し,ひ素(As)の量と指示値との関係線を作成する。検
量線の作成は,試料測定時に行う。
注記1 有機物及び亜硝酸イオンを含まない試料は1)〜3) の代わりに,次のように操作してもよ
い。試料の適量(Asとして0.1〜1 μgを含む。)をビーカー100 mLにとり,塩酸3 mLを加
え,沸騰しない程度に5〜7分間加熱した後,冷却する。次に,よう化カリウム溶液(200
g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全
量フラスコ20 mLに洗い移し,水を標線まで加える。
注記2 多量の有機物を含む場合は,1) 及び2) の代わりに,次のように操作してもよい。試料の
適量に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223に規定する過塩素酸又はそれ以上の
純度の過塩素酸3 mLを加え,加熱して硫酸白煙を発生させ,有機物を分解する。
注記3 ほうけい酸ガラスには,ひ素を含むものがあるので注意する。四ふっ化エチレン樹脂製ビ
ーカーを用いるとよい。
注記4 硝酸が残存すると,分解生成物である窒素酸化物によって水素化ひ素の発生が阻害される
ので,十分に硫酸の白煙を発生させて硝酸を除去する。
注記5 装置によって,最適な塩酸及びテトラヒドロほう酸ナトリウム溶液の流量及び濃度は異な
る。
注記6 水素−アルゴンフレームのアルゴンの代わりにJIS K 1107に規定する窒素2級を用いても
よい。
注記7 水素−アルゴンフレームの代わりに加熱吸収セル方式のものを用いてもよい。ただし,水
素−アルゴンフレームに比べて10〜50倍程度(装置及び操作条件によって異なる。)感度
が良いので,ひ素標準液(As 0.1 μg/mL)の量を,適宜,減らす。
注記8 水素化物発生法は,鉄,ニッケル,コバルト,白金,パラジウムなどの遷移金属によって
発生効率が影響される。また,アンチモン,セレンなどの水素化合物を形成する元素によ
61
K 0083:2017
っても発生効率が低下する。よう化カリウムは,ひ素を5価から3価に還元するために用
いられるが,遷移金属による干渉の低減にも効果がある。
なお,未知試料のように共存物質の影響が不明な場合は,未知試料に一定量の測定対象
元素(ここではひ素)を添加したときに得られる指示値の増加分と,同量の測定元素を含
む検量線用標準液の指示値とを比較することによって,干渉の大きさを知ることができる。
共存物質による干渉がある場合は,標準添加法を適用すると真度が向上する。
d) 操作(バッチ式水素化物発生装置を用いる場合) 操作は,次による。
1) c) 1)〜3) の操作を行った溶液の全量を水素化物発生装置の反応容器に移し入れる。
2) 水素化物発生装置と原子吸光分析装置とを連結し,系内の空気をアルゴンで置換する。
3) コックを回転して,アルゴンで溶液をバブリングする状態にする。
4) セプタムを通してシリンジなどによってテトラヒドロほう酸ナトリウム溶液(10 g/L)2 mLを手早
く加え,発生する水素化物を水素−アルゴンフレーム中に導入し,波長193.7 nmの指示値を読み取
る。
5) 空試験値として,空試験溶液を試料溶液と同様に,1)〜4) の操作を行った後,指示値を読み取り,
試料について得た指示値を補正する。
6) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。検量線の作成は,次によ
る。
検量線 ひ素標準液(As 0.1 μg/mL)1〜10 mLを段階的に全量フラスコ20 mLにとり,塩酸3 mL,
よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間
静置した後,水を標線まで加え,2)〜4) の操作を行う。別に,空試験として全量フラスコ20 mLに
水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4
mLを加え,約60分間静置した後,水を標線まで加えたものを用い,2)〜4) の操作を行って,ひ素
標準液についての指示値を補正し,ひ素(As)の量と指示値との関係線を作成する。検量線の作成
は,試料測定時に行う。
注記9 ごく低濃度の水素化物を分析する目的で,水素化物を濃縮する場合には,ガラスビーズな
どを充塡したU字管を液体窒素中に浸したコールドトラップを用いて水素化物を捕集する。
捕集後,U字管を引き上げて室温に戻し,気化した水素化物をアルゴンでフレーム中に送
り込む。
14.4
電気加熱原子吸光法
試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム(II)及び硝酸マグネシウ
ムを加え,電気加熱炉で原子化し,ひ素による原子吸光を波長193.7 nmで測定してひ素を標準添加法によ
って定量する。
− 定量範囲 As 5〜100 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるよう調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸パラジウム(II)溶液(Pd 1 mg/mL) 7.3 a) 3) による。
62
K 0083:2017
4) 硝酸マグネシウム溶液(Mg 1 mg/mL) 原子吸光分析用マトリックスモディファイヤーの硝酸マグ
ネシウム溶液を希釈して用いる。
5) ひ素標準液(As 10 μg/mL) 14.2 a) 11) のひ素標準液(As 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
6) ひ素標準液(As 1 μg/mL) 5) のひ素標準液(As 10 μg/mL)10 mLを全量フラスコ100 mLにとり,
硝酸(1+1)2 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) ひ素中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット5〜50 μL又は自動注入装置。
c) 準備操作 試料を6.4又は6.5によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,ひ素標準液(As 1 μg/mL)
を加えないものと,0.1〜2 mLの範囲で段階的に3段階以上添加したものとを調製し,それぞれの
溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)及びそれと同体積の硝酸パラジウム(II)溶液(Pd 1 mg/mL)
及び硝酸マグネシウム溶液(Mg 1 mg/mL)14) を,マイクロピペットを用いて電気加熱炉に注入する。
JIS K 0121の8.(操作方法)に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(600〜1 200 ℃,
30〜40秒間),原子化1)(2 000〜2 400 ℃,3〜6秒間)し,波長193.7 nmの指示値(吸光度又はそ
の比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) ひ素の添加量と指示値との関係線を作成し,ひ素の量を求め,試料溶液中のひ素の濃度(As μg/L)
を算出する。
注14) 混合した溶液を用いてもよい。
14.5
水素化物発生ICP発光分光分析法
試料溶液中のひ素を水素化ひ素とし,試料導入部を通して誘導結合プラズマ中に噴霧し,ひ素による発
光を波長193.696 nmで測定してひ素を定量する。
− 定量範囲 As 1〜50 μg/L
− 繰返し分析精度 変動係数で3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,14.3 a) による。
b) 装置 装置は,次による。
1) 連続式水素化物発生装置 14.3 b) 1) による。
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 6.4又は6.5で調製した試料溶液の適量(Asとして0.02〜0.2 μgを含む。)をビーカー100 mLにとり,
硫酸(1+1)1 mL及び硝酸2 mLを加え,さらに過マンガン酸カリウム溶液(3 g/L)を溶液が着色
63
K 0083:2017
するまで滴加する。
2) 加熱板上で加熱して硫酸の白煙を発生させる。
なお,加熱中に過マンガン酸の色が消えたときは,過マンガン酸カリウム溶液(3 g/L)を追加す
る。
3) 2) の溶液を室温まで放冷した後,水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及び
アスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全量フラスコ20 mLに洗い
移し,水を標線まで加える。
4) 連続式水素化物発生装置にアルゴンを流しながら,3) の溶液,テトラヒドロほう酸ナトリウム溶液
(10 g/L)及び塩酸(1 mol/L)を,定量ポンプで連続的に装置内に導入し,水素化ひ素を発生させ
る。
5) 発生した水素化ひ素と廃液とを分離した後,水素化ひ素を含む気体をプラズマ中に導入し,波長
193.696 nmの発光強度を測定する。
6) 空試験として,空試験溶液を試料溶液と同様に,1)〜5) の操作を行い,試料について得た発光強度
を補正する。
7) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。検量線の作成は,次によ
る。
検量線 ひ素標準液(As 0.1 μg/mL)0.2〜10 mLを段階的に全量フラスコ20 mLにとり,塩酸3 mL,
よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間
静置した後,水を標線まで加える。連続式水素化物発生装置にアルゴンを流しながら,この溶液,
テトラヒドロほう酸ナトリウム溶液(10 g/L)及び塩酸(1 mol/L)を,定量ポンプで連続的に装置
内に導入し,水素化ひ素を発生させ,引き続き5) の操作を行う。別に,空試験として全量フラス
コ20 mLに水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100
g/L)0.4 mLを加え,約60分間静置した後,水を標線まで加えたものを用い,4) 及び5) の操作を
行って,標準液について得た発光強度を補正し,ひ素(As)の量と発光強度との関係線を作成する。
検量線の作成は,試料測定時に行う。
注記1 14.3 c) の注記1参照。
注記2 14.3 c) の注記2参照。
注記3 14.3 c) の注記3参照。
注記4 14.3 c) の注記4参照。
注記5 14.3 c) の注記5参照。
注記6 14.3 c) の注記8参照。
注記7 水素化物を発生させるときに副生する水素が,プラズマに導入されることによってプラズ
マが不安定になる場合があるので,特に導入初期には,水素の量が多くなり過ぎないよう
に注意する。
14.6
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,ひ素及び内標準物質
のそれぞれの質量/荷電数におけるイオンカウントを測定し,ひ素のイオンカウントと内標準物質のイオン
カウントとの比を求めてひ素を定量する。
− 定量範囲 As 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
64
K 0083:2017
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) ひ素標準液(As 1 μg/mL) 14.2 a) 11) のひ素標準液(As 0.1 mg/mL)10 mLを全量フラスコ1 000 mL
にとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
5) ひ素標準液(As 50 ng/mL) 4) のひ素標準液(As 1 μg/mL)50 mLを全量フラスコ1 000 mLにとり,
硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(As 1 μg,Se 1 μg)/mL] 14.2 a) 11) のひ素標準液(As 0.1 mg/mL)10 mL,15.2 a) 9)
のセレン標準液(Se 0.2 mg/mL)5 mLをそれぞれ全量フラスコ1 000 mLにとり,硝酸(1+1)20 mL
を加えた後,水を標線まで加える。使用時に調製する。
7) 混合標準液[(As 50 ng,Se 50 ng)/mL] 6) の混合標準液[(As 1 μg,Se 1 μg)/mL]50 mLを全
量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加えた後,水を標線まで加える。使用時に調製
する。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.4又は6.5で調製した試料溶液の適量(Asとして0.05〜50 μgを含む。)を全量フラスコ100 mLに
とり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,硝酸の最終濃度が0.1〜0.5 mol/Lの一定濃度と
なるように硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧し,ひ
素とイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を読み取り,
ひ素の指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,ひ素の指示値とイットリ
ウムの指示値との比を求め,試料について得たひ素の指示値とイットリウムの指示値との比を補正
する。
4) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。検量線の作成は,次によ
る。
検量線 ひ素標準液(As 50 ng/mL又はAs 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階的に
とり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝酸(1
+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。
別に,空試験として全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の
試料と同じ酸の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って
標準液について得た指示値の比を補正した後,ひ素(As)の量に対する指示値とイットリウムの指
示値との比の関係線を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 ひ素,セレンを同時に定量する場合には,混合標準液[(As 1 μg,Se 1 μg)/mL]又は混合
標準液[(As 50 ng,Se 50 ng)/mL]を用いて,それぞれの金属元素の試験条件で検量線を
65
K 0083:2017
作成してもよい。
15
セレンの分析方法
15.1
一般
セレン及び,ガス状セレン化合物の定量には,3,3'-ジアミノベンジジン吸光光度法,水素化合物発生原
子吸光法,電気加熱原子吸光法,ジアミノナフタレン蛍光光度法,水素化合物発生ICP発光分光分析法,
ICP質量分析法又は,水素化合物発生ICP質量分析法を用いる。
15.2
3,3'-ジアミノベンジジン吸光光度法
セレンを水酸化鉄(III)と共沈させ,妨害物質から分離して濃縮する。EDTAを加え,pH1.5〜2.0とし
た後,3,3'-ジアミノベンジジンを加えてセレン錯体を生成させる。溶液のpHを6に調節し,トルエンで
錯体を抽出し,その黄色の吸光度を測定してセレンを定量する。
− 定量範囲 Se 2〜50 μg
− 繰返し分析精度 変動係数で2〜10 %
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸(1+1),(1+3) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
3) アンモニア水(1+1),(1+2) JIS K 8085に規定するアンモニア水又はそれ以上の純度のものを用
いて調製する。
4) 硫酸アンモニウム鉄(III)溶液 JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水9 g及び
硫酸(1+1)[6.4.1 c) による。]4 mLを水に溶かして100 mLとする。
5) EDTA溶液(40 g/L) JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物
4.4 gを水に溶かして100 mLとする。
6) トルエン JIS K 8680に規定するもの。
7) ブロモチモールブルー溶液(1 g/L) JIS K 8842に規定するブロモチモールブルー0.1 gをとり,JIS
K 8102に規定するエタノール(95)50 mLに溶かし,水で100 mLとする。
8) 3,3'-ジアミノベンジジン溶液(5 g/L) 塩化3,3'-ジアミノベンジジニウム0.5 gを水に溶かして100 mL
とする。この溶液は,使用時に調製する。
9) セレン標準液(Se 0.2 mg/mL) JIS K 8598に規定するセレン(99.5 %以上)0.200 gをとり,硝酸(1
+1)[6.1.1 a) による。]20 mLを加え加熱して溶かし,煮沸して窒素酸化物を追い出す。放冷後,
全量フラスコ1 000 mLに移し,水を標線まで加える。
10) セレン標準液(Se 2 μg/mL) 9) のセレン標準液(Se 0.2 mg/mL)10 mLを全量フラスコ1 000 mL
にとり,塩酸(1+100)[JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。]
を標線まで加える。この溶液は,使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 吸光光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして2〜50 μgを含む。)をビーカー200 mLにと
る。
2) 塩酸(1+3)10 mLを加え,沸騰水浴上で10分間加熱する。
66
K 0083:2017
3) 水を加えて液量を約100 mLとし,硫酸アンモニウム鉄(III)溶液2 mL及びブロモチモールブルー
溶液(1 g/L)1 mLとを加え,次に,溶液をかき混ぜながらアンモニア水(1+1)を溶液が青にな
るまで加えた後,静かに加熱して煮沸し,かき混ぜながら溶液が黄色になるまで約30分間煮沸を続
ける。ただし,溶液中に鉄を20 mg以上含む場合は硫酸アンモニウム鉄(III)は加えない。
4) 生成した沈殿が沈降するまで静置し,沈殿をろ紙5種Aでろ別し,温水で洗浄する。
5) 沈殿は元のビーカーに洗い落とし,ろ紙に付着した沈殿は温塩酸(1+1)約2 mLを滴加して溶か
し,沈殿の入っているビーカーに受け,ろ紙は水洗する。加熱して沈殿を溶かし,水で液量を約30
mLとする。
6) 放冷後,EDTA溶液(40 g/L)10 mLを加え,アンモニア水(1+2)を加えて溶液のpHを1.5〜2.0
に調節した後3,3'-ジアミノベンジジン溶液(5 g/L)2 mLを加えて振り混ぜ,沸騰水浴上で約10分
間加熱する。
7) 流水で冷却後,アンモニア水(1+2)を用いて溶液のpHを約6に調節し,分液漏斗に移し,トル
エン10 mLを加えて約30秒間振り混ぜて静置する。
8) トルエン層の一部を吸収セルに移し,トルエンを対照として波長420 nm付近の吸光度を測定する。
9) 別に,空試験溶液について1)〜8) の操作を行って吸光度を求め,試料溶液について得られた吸光度
を補正する。
10) 検量線からセレンの量を求め,試料溶液中のセレンの濃度(Se μg/L)を算出する。検量線の作成
は,次による。
検量線 セレン標準液(Se 2 μg/mL)0〜25 mLを段階的にビーカーにとり,塩酸(1+3)10 mLを
加え,沸騰水浴上で10分間加熱する。以後,3)〜9) の操作を行ってセレン(Se)の量と吸光度と
の関係線を作成する。
注記1 妨害イオンを含まない場合は,操作3)〜5) を省略してもよい。
注記2 7) 以降の操作は,直射日光を避けて行うとよい。
注記3 約30分間煮沸後の溶液のpHは5.4〜5.7となる。
15.3
水素化合物発生原子吸光法
試料溶液中のセレンをセレン化水素とし,水素−アルゴンフレーム中に導き,セレンによる原子吸光を
波長196.0 nmで測定して,セレンを定量する。
− 定量範囲 Se 2〜12 μg/L
− 繰返し分析精度 変動係数で3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸 11.2 a) 2) による。
3) 塩酸(1+1) 2) の塩酸を用いて調製する。
4) 硝酸 6.3.1 a) による。
5) 硫酸(1+1) 6.4.1 c) による。
6) テトラヒドロほう酸ナトリウム溶液(10 g/L) 14.3 a) 9) による。
7) アルゴン JIS K 1105に規定するアルゴン2級のもの。
8) セレン標準液(Se 0.1 μg/mL) 15.2 a) 10) のセレン標準液(Se 2 μg/mL)5 mLを全量フラスコ100 mL
にとり,硫酸(1+100)[JIS K 8951に規定する硫酸又はそれ以上の純度のものを用いて調製する。]
を標線まで加える。この溶液は,使用時に調製する。
67
K 0083:2017
b) 器具及び装置 器具及び装置は,次による。
1) 連続式水素化合物発生装置 図12に一例を示す。
2) バッチ式水素化合物発生装置 図13に一例を示す。
3) 原子吸光分析装置
4) セレン中空陰極ランプ
c) 操作(連続式水素化合物発生装置を用いる場合) 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして0.05〜0.3 μgを含む。)をビーカー100 mL
にとり,硫酸(1+1)1 mL及び硝酸2 mLを加える。
2) 加熱板上で乾固する直前まで加熱する。
3) 放冷した後,塩酸(1+1)20 mLを加え,約90〜100 ℃で約10分間加熱する。
4) 放冷した後,全量フラスコ25 mLに移し入れ,水を標線まで加える。
5) 連続式水素化合物発生装置にアルゴンを流しながら,4) の溶液,テトラヒドロほう酸ナトリウム溶
液(10 g/L)及び塩酸(1 mol/L)[14.3 a) 4) による。]を,定量ポンプで連続的に装置内に導入し,
セレン化水素を発生させる。
6) 発生したセレン化水素と廃液とを分離した後,セレン化水素を含む気体を水素−アルゴンフレーム
中に導入し,波長196.0 nmにおける指示値(吸光度又はその比例値)を読み取る。
7) 空試験として,空試験溶液を試料溶液と同様に,1)〜6) の操作を行った後,指示値を読み取り,試
料について得た指示値を補正する。
8) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。検量線の作成は,次
による。
検量線 セレン標準液(Se 0.1 μg/mL)0.5〜3 mLを段階的にビーカー100 mLにとり,塩酸(1+1)
20 mLを加え,約90〜100 ℃で約10分間加熱する。4)〜6) の操作を行う。別に,空試験としてビ
ーカー100 mLに塩酸(1+1)20 mLをとり,約90〜100 ℃で約10分間加熱する。4)〜6) の操作を
行って標準液について得た指示値を補正し,セレン(Se)の量と指示値との関係線を作成する。検
量線は,試料測定時に作成する。
注記1 有機物を含まない試料は1)〜3) の代わりに,次のように操作してもよい。
試料溶液の適量(Seとして0.05〜0.3 μgを含む。)をビーカー100 mLにとり,試料溶液
と同量の塩酸を加えて,約90〜100 ℃で約10分間加熱する。
注記2 多量の有機物を含む場合は,1) 及び2) の代わりに,次のように操作してもよい。試料の
適量に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223に規定する過塩素酸又はそれ以上の
純度の過塩素酸3 mLを加え,加熱して硫酸白煙を発生させ,有機物を分解する。
注記3 装置によって,最適な塩酸及びテトラヒドロほう酸ナトリウム溶液の流量及び濃度は異な
る。
注記4 水素−アルゴンフレームのアルゴンの代わりにJIS K 1107に規定する窒素2級を用いても
よい。
注記5 水素−アルゴンフレームの代わりに加熱吸収セル方式のものを用いてもよい。ただし,水
素アルゴンフレームに比べて10〜50倍程度(装置及び操作条件によって異なる。)感度が
良いので,セレン標準液(Se 0.1 μg/mL)の量を,適宜,減らす。
注記6 水素化合物発生法は,鉄,ニッケル,コバルト,白金,パラジウムなどの遷移金属によっ
て発生効率が影響される。また,アンチモン,ひ素などの水素化合物を形成する元素によ
68
K 0083:2017
っても発生効率が低下する。塩酸は,セレンを6価から4価に還元するために用いられる。
一般に,塩酸濃度を高くすると遷移金属による干渉が低減することが多い。
なお,未知試料のように共存物質の影響が不明な場合は,未知試料に一定量の測定対象元素(こ
こでは,セレン)を添加したときに得られる指示値の増加分と,同量の測定元素を含む検量線用標
準液の指示値とを比較することによって,干渉の大きさを知ることができる。共存物質による干渉
がある場合は,標準添加法を適用すると真度が向上する。
d) 操作(バッチ式水素化合物発生装置を用いる場合) 操作は,次による。
1) c) 1)〜3) の操作を行った溶液の全量を水素化合物発生装置の反応容器に移し入れる。
2) 水素化合物発生装置と原子吸光分析装置とを連結し,系内の空気をアルゴンで置換する。
3) コックを回転して,アルゴンで溶液をバブリングする状態にする。
4) セプタムを通してシリンジなどによってテトラヒドロほう酸ナトリウム溶液(10 g/L)2 mLを手早
く加え,発生する水素化物を水素−アルゴンフレーム中に導入し,波長196.0 nmの指示値を読み取
る。
5) 空試験値として,空試験溶液を試料溶液と同様に,1)〜4) の操作を行った後,指示値を読み取り,
試料について得た指示値を補正する。
6) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。検量線の作成は,次
による。
検量線 セレン標準液(Se 0.1 μg/mL)0.5〜3 mLを段階的にビーカー100 mLにとり,塩酸(1+1)
20 mLを加え,約90〜100 ℃で約10分間加熱する。放冷した後,全量フラスコ25 mLに移し入れ,
水を標線まで加える。2)〜4) の操作を行う。別に,空試験としてビーカー100 mLに塩酸(1+1)
20 mLをとり,約90〜100 ℃で約10分間加熱する。放冷した後,全量フラスコ25 mLに移し入れ,
水を標線まで加えたものを用い,2)〜4) の操作を行って,セレン標準液についての指示値を補正し,
セレン(Se)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注記7 ごく低濃度の水素化合物を分析する目的で,水素化合物を濃縮する場合には,ガラスビー
ズなどを充塡したU字管を液体窒素中に浸したコールドトラップを用いて水素化合物を捕
集する。捕集後,U字管を引き上げて室温に戻し,気化した水素化合物をアルゴンでフレ
ーム中に送り込む。
15.4
電気加熱原子吸光法
試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム(II)及び硝酸マグネシウ
ムを加え,電気加熱炉で原子化し,セレンによる原子吸光を波長196.0 nmで測定してセレンを標準添加法
によって定量する。
− 定量範囲 Se 20〜200 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
なお,この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少な
い試料に適用し,測定時の酸濃度は一定となるように調製する。
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸パラジウム(II)溶液(Pd 1 mg/mL) 7.3 a) 3) による。
4) 硝酸マグネシウム溶液(Mg 1 mg/mL) 14.4 a) 4) による。
69
K 0083:2017
5) セレン標準液(Se 10 μg/mL) 15.2 a) 9) のセレン標準液(Se 0.2 mg/mL)10 mLを全量フラスコ200
mLにとり,硝酸(1+1)4 mLを加えた後,水を標線まで加える。
6) セレン標準液(Se 1 μg/mL) 5) のセレン標準液(Se 10 μg/mL)10 mLを全量フラスコ100 mLにと
り,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 電気加熱原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
2) 発熱体 黒鉛製又は耐熱金属製のもの。
3) セレン中空陰極ランプ
4) フローガス JIS K 1105に規定するアルゴン2級のもの。
5) マイクロピペット JIS K 0970に規定するピストン式ピペット5〜50 μL又は自動注入装置。
c) 準備操作 試料を6.4,6.5又は6.6によって処理する。
d) 操作 操作は,次による。
1) c) の準備操作を行った試料の適量をそれぞれ全量フラスコ20 mLにとり,セレン標準液(Se 1
μg/mL)を加えないものと0.4〜4 mLの範囲で段階的に3段階以上添加したものとを調製し,それ
ぞれの溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) この溶液の一定量(例えば,10〜50 μL)及びこれと同体積の硝酸パラジウム(II)溶液(Pd 1 mg/mL)
及び硝酸マグネシウム溶液(Mg 1 mg/mL)15) を,マイクロピペットを用いて電気加熱炉に注入する。
JIS K 0121の8.(操作方法)に従って,乾燥(100〜120 ℃,30〜40秒間),灰化(800〜1 300 ℃,
30〜40秒間),原子化1)(1 900〜2 400 ℃,3〜6秒間)し,波長196.0 nmの指示値(吸光度又はそ
の比例値)を読み取る。
3) 引き続いて少なくとも2) の操作を3回繰り返し,指示値が合うことを確認する。
4) 空試験として,空試験溶液を試料溶液と同様に1)〜3) の操作を行って,試料溶液について得た指示
値を補正する。
5) セレンの添加量と指示値との関係線を作成し,セレンの量を求め,試料溶液中のセレンの濃度(Se
μg/L)を算出する。
15.5
ジアミノナフタレン蛍光光度法
試料溶液に塩酸を加えセレンを4価に還元した後,pHを1.0〜1.5に調節し,EDTA溶液及びふっ化ナト
リウムを加えた後,2,3-ジアミノナフタレンを加えてセレン錯体を生成させ,シクロヘキサンに抽出し,
蛍光強度を測定してセレンを定量する。
− 定量範囲 Se 0.1〜1 μg
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸(1+3),(1+120) JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調製する。
3) アンモニア水(1+1) 7.2 a) 6) による。
4) ふっ化ナトリウム溶液(4 g/L) JIS K 8821に規定するふっ化ナトリウム0.4 gを水に溶かして100
mLとする。
5) EDTA溶液(40 g/L) 15.2 a) 5) による。
6) 2,3-ジアミノナフタレン溶液 2,3-ジアミノナフタレン0.1 gをとり,JIS K 8201に規定する塩化ヒ
ドロキシルアンモニウム0.5 g及び塩酸(1+120)100 mLを加え,50 ℃の水浴上で20分間加熱し
70
K 0083:2017
て,溶かす。冷却後,JIS K 8464に規定するシクロヘキサン10 mLを加え振り混ぜた後,有機層を
捨てる。さらに,シクロヘキサン10 mLを加え振り混ぜた後,有機層を捨て,水層をろ紙を用いて
ろ過する。この溶液は,使用時に調製する。
7) シクロヘキサン JIS K 8464に規定するもの。
8) セレン標準液(Se 0.1 μg/mL) 15.2 a) 10) のセレン標準液(Se 2 μg/mL)5 mL及びJIS K 8180に規
定する塩酸又はそれ以上の純度の塩酸1 mLを全量フラスコ100 mLにとり,水を標線まで加える。
この溶液は,使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 蛍光分光光度計
2) 蛍光分析用10 mmセル
c) 操作 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして0.1〜1 μgを含む。)をビーカー200 mLにと
り,塩酸(1+3)10 mLを加える。
2) 水浴上で10分間加熱する。
3) EDTA溶液(40 g/L)2 mLを加え,アンモニア水(1+1)を加えて溶液のpHを1.0〜1.5に調節し
た後,ふっ化ナトリウム溶液0.5 mLを加え,さらに塩酸(1+120)を加えて液量を約100 mLとす
る。
4) 2,3-ジアミノナフタレン溶液5 mLを加える。
5) 時計皿でビーカーを覆い,50 ℃の水浴中で30分間加熱する。
6) 放冷後,分液漏斗に入れ,シクロヘキサン10 mLを加え,5分間激しく振り混ぜる。有機層に塩酸
(1+120)25 mLを加え30秒間振り混ぜて分離した水層を捨て,さらに,有機層に塩酸(1+120)
25 mLを加え30秒間振り混ぜ,水層を分離する。
7) 有機層を乾燥させたろ紙を用いてろ過し,ろ液の一部をセルに入れる。
8) 励起波長378 nmに設定し,蛍光波長520 nm付近における蛍光強度を測定する。
9) 空試験として,空試験溶液を試料溶液と同様に1)〜8) の操作を行って蛍光強度を求め,試料につい
て得られた蛍光強度を補正する。
10) 検量線からセレンの量を求め,試料溶液中のセレン濃度(Se μg/L)を算出する。検量線の作成は,
次による。
検量線 セレン標準液(Se 0.1 μg/mL)1〜10 mLを段階的にとり,塩酸(1+3)10 mLを加えた後,
2)〜8) の操作を行ってセレンの量と蛍光強度との関係線を作成する。検量線は,試料測定時に作
成する。
注記 6) の第2文以降の操作は,直射日光を避け,できるだけ速やかに行うとよい。
15.6
水素化合物発生ICP発光分光分析法
試料溶液中のセレンをセレン化水素とし,試料導入部を通して誘導結合プラズマ中に噴霧し,セレンに
よる発光を波長196.026 nmで測定してセレンを定量する。
− 定量範囲 Se 1〜20 μg/L
− 繰返し分析精度 変動係数で3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,15.3 a) による。
b) 装置 装置は,次による。
1) 連続式水素化合物発生装置 図12に一例を示す。
71
K 0083:2017
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして0.025〜0.5 μgを含む。)をビーカー100 mL
にとり,硫酸(1+1)1 mL及び硝酸2 mLを加える。
2) 加熱板上で乾固する直前まで加熱する。
3) 放冷した後,塩酸(1+1)20 mLを加え,約90〜100 ℃で約10分間加熱する。
4) 放冷した後,全量フラスコ25 mLに移し入れ,水を標線まで加える。
5) 連続式水素化合物発生装置にアルゴンを流しながら,4) の溶液,テトラヒドロほう酸ナトリウム溶
液(10 g/L)及び塩酸(1 mol/L)[14.3 a) 4) による。]を,定量ポンプで連続的に装置内に導入し,
セレン化水素を発生させる。
6) 発生したセレン化水素と廃液とを分離した後,セレン化水素を含む気体をプラズマ中に導入し,波
長196.026 nmの発光強度を測定する。
7) 空試験として,空試験溶液を試料溶液と同様に,1)〜6) の操作を行った後,指示値を読み取り,試
料について得た指示値を補正する。
8) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。検量線の作成は,次
による。
検量線 セレン標準液(Se 0.1 μg/mL)0.25〜5 mLを段階的にビーカー100 mLにとり,塩酸(1+1)
20 mLを加え,約90〜100 ℃で約10分間加熱する。4)〜6) の操作を行う。別に,空試験としてビ
ーカー100 mLに塩酸(1+1)20 mLをとり,約90〜100 ℃で約10分間加熱する。4)〜6) の操作を
行って標準液について得た指示値を補正し,セレン(Se)の量と指示値との関係線を作成する。検
量線は,試料測定時に作成する。
注記1 15.3 c) の注記1参照。
注記2 15.3 c) の注記2参照。
注記3 15.3 c) の注記3参照。
注記4 15.3 c) の注記6参照。
注記5 水素化合物を発生させるときに副生する水素が,プラズマに導入されることによってプラ
ズマが不安定になる場合があるので,特に導入初期には,水素の量が多くなり過ぎないよ
うに注意する。
15.7
ICP質量分析法
試料溶液に内標準物質を加え,試料導入部を通して誘導結合プラズマ中に噴霧し,セレン及び内標準物
質のそれぞれの質量/荷電数におけるイオンカウントを測定し,セレンのイオンカウントと内標準物質のイ
オンカウントとの比を求めてセレンを定量する。
− 定量範囲 Se 0.5〜500 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 硝酸(1+1) 7.3 a) 2) による。
3) イットリウム溶液(Y 1 μg/mL) 7.5 a) 3) による。
4) セレン標準液(Se 1 μg/mL) 15.2 a) 9) のセレン標準液(Se 0.2 mg/mL)5 mLを全量フラスコ1 000
mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
72
K 0083:2017
5) セレン標準液(Se 50 ng/mL) 4) のセレン標準液(Se 1 μg/mL)50 mLを全量フラスコ1 000 mLに
とり,硝酸(1+1)20 mLを加え,水を標線まで加える。使用時に調製する。
6) 混合標準液[(As 1 μg,Se 1 μg)/mL] 14.6 a) 6) による。
7) 混合標準液[(As 50 ng,Se 50 ng)/mL] 14.6 a) 7) による。
b) 装置 ICP質量分析装置
注記1 7.5 b) の注記1参照。
注記2 7.5 b) の注記2参照。
c) 操作 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして0.05〜50 μgを含む。)を全量フラスコ100 mL
にとり,イットリウム溶液(Y 1 μg/mL)1 mLを加え硝酸の最終濃度が0.1〜0.5 mol/Lとなるように
硝酸(1+1)を加えた後,水を標線まで加える。
2) ICP質量分析装置を作動できる状態にし,1) の溶液を試料導入部を通してプラズマ中に噴霧して,
セレン及びイットリウムの質量/荷電数4) における指示値(イオンカウント又はその比例値)を読み
取り,セレンの指示値とイットリウムの指示値との比を求める。
3) 空試験として,空試験溶液を試料溶液と同様に1) 及び2) の操作を行い,セレンの指示値とイット
リウムの指示値との比を求め,試料について得たセレンの指示値とイットリウムの指示値との比を
補正する。
4) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。検量線の作成は,次
による。
検量線 セレン標準液(Se 50 ng/mL又はSe 1 μg/mL)1〜50 mLを全量フラスコ100 mLに段階的に
とり,イットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度になるように硝酸(1
+1)を加えた後,水を標線まで加える。この溶液について2) の操作を行う。別に,空試験として
全量フラスコ100 mLにイットリウム溶液(Y 1 μg/mL)1 mLを加え,1) の試料と同じ酸の濃度に
なるように硝酸(1+1)を加え,水を標線まで加えた後,2) の操作を行って標準液について得た指
示値の比を補正した後,セレン(Se)の量に対する指示値とイットリウム(Y)の指示値との比の
関係線を作成する。検量線は,試料測定時に作成する。
注記1 7.5 c) の注記1参照。
注記2 7.5 c) の注記2参照。
注記3 ひ素,セレンを同時に定量する場合には,混合標準液[(As 1 μg,Se 1 μg)/mL]又は混合
標準液[(As 50 ng,Se 50 ng)/mL]を用いて,それぞれの金属元素の試験条件で検量線を
作成してもよい。
15.8
水素化合物発生ICP質量分析法
試料溶液中のセレンをセレン化水素とし,試料導入部を通して誘導結合プラズマ中に導き,セレンの質
量/荷電数におけるイオンカウントを測定してセレンを定量する。
− 定量範囲 Se 0.1〜100 μg/L
− 繰返し分析精度 変動係数で2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 7.2 a) 1) による。
2) 塩酸 JIS K 8180に規定する塩酸又はそれ以上の純度のもの。
3) 塩酸(1+1) 2) の塩酸を用いて調製する。
73
K 0083:2017
4) 硝酸 6.3.1 a) による。
5) 硫酸(1+1) 6.4.1 c) による。
6) テトラヒドロほう酸ナトリウム溶液(10 g/L) 14.3 a) 9) による。
7) アルゴン 15.3 a) 7) による。
8) セレン標準液(Se 1 μg/mL) 15.2 a) 9) のセレン標準液(Se 0.2 mg/mL)5 mLを全量フラスコ1 000
mLにとり,硫酸(1+100)[JIS K 8951に規定する硫酸又はそれ以上の純度のものを用いて調製す
る。]を標線まで加える。この溶液は,使用時に調製する。
9) セレン標準液(Se 0.1 μg/mL) 8) のセレン標準液(Se 1 μg/mL)10 mLを全量フラスコ100 mLに
とり,硫酸(1+100)[JIS K 8951に規定する硫酸又はそれ以上の純度のものを用いて調製する。]
を標線まで加える。この溶液は,使用時に調製する。
b) 装置 装置は,次による。
1) 連続式水素化合物発生装置 図12に一例を示す。
2) ICP質量分析装置
c) 操作 操作は,次による。
1) 6.4,6.5又は6.6で調製した試料溶液の適量(Seとして0.002 5〜2.5 μgを含む。)をビーカー100 mL
にとり,硫酸(1+1)1 mL及び硝酸2 mLを加える。
2) 加熱板上で乾固する直前まで加熱する。
3) 放冷した後,塩酸(1+1)20 mLを加え,約90〜100 ℃で約10分間加熱する。
4) 放冷した後,全量フラスコ25 mLに移し入れ,水を標線まで加える。
5) 連続式水素化合物発生装置にアルゴンを流しながら,4) の溶液,テトラヒドロほう酸ナトリウム溶
液(10 g/L)及び塩酸(1 mol/L)(JIS K 8180に規定する塩酸又はそれ以上の純度のものを用いて調
製する。)を,定量ポンプで連続的に装置内に導入し,セレン化水素を発生させる。
6) 発生したセレン化水素と廃液とを分離した後,セレン化水素を含む気体をプラズマ中に導入し,セ
レンの質量/荷電数4) における指示値(イオンカウント又はその比例値)を読み取る。
7) 空試験として,空試験溶液を試料溶液と同様に,1)〜6) の操作を行った後,指示値を読み取り,試
料について得た指示値を補正する。
8) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。検量線の作成は,次
による。
検量線 セレン標準液(Se 0.1 μg/mL又はSe 1 μg/mL)0.025〜2.5 mLを段階的にビーカー100 mL
にとり,塩酸(1+1)20 mLを加え,約90〜100 ℃で約10分間加熱する。4)〜6) の操作を行う。
別に,空試験としてビーカー100 mLに塩酸(1+1)20 mLをとり,約90〜100 ℃で約10分間加熱
する。4)〜6) の操作を行って標準液について得た指示値を補正し,セレン(Se)の量と指示値との
関係線を作成する。検量線は,試料測定時に作成する。
16
計算
試料ガス中の金属濃度は,次の式によって算出する。
s
1
000
1
V
v
A
C
×
×
=
ここに,
C: 標準状態における乾き排ガス中の金属濃度
[mg/m3(273.15 K,101.325 kPa)]
74
K 0083:2017
A: 箇条7〜箇条15で求めた試料溶液中の金属濃度(mg/L)
v: 箇条6で調製した試料溶液の量(mL)
Vs: 箇条5で採取した標準状態における乾き排ガス量
[m3(273.15 K,101.325 kPa)]
75
K 0083:2017
附属書A
(規定)
マイクロ波加熱圧力容器による試料の前処理方法
A.1 適用範囲
この附属書は,箇条6のマイクロ波加熱圧力容器による試料の前処理方法について規定する。
A.2 マイクロ波加熱容器による試料溶液の調製
A.2.1 試薬
試薬は,次による。
1) ふっ化水素酸 JIS K 8819に規定するもの。
2) 硝酸 JIS K 8541に規定する硝酸又はそれ以上の純度のもの。
3) 硝酸(1+1) 2) を用いて調製する。
4) 過酸化水素 JIS K 8230に規定するもの。
A.2.2 操作
操作は,次による。
a) 吸収瓶を設けないでろ紙だけに試料採取した場合
1) 試料ガスからの捕集物の付着したろ紙を適正な大きさに切り,密閉式四ふっ化エチレン樹脂分解容
器に入れ,ふっ化水素酸3 mLを加え,試料ろ紙の大部分を溶解した後,硝酸6 mL及び過酸化水素
1 mLを加え,密閉した分解容器をマイクロ波加熱によって完全に分解する1)。
2) 室温まで冷却後,分解容器を濃縮加熱用ローターで,穏やかに約15分〜25分加熱し,蒸発乾固さ
せる2)。
3) 分解容器内に水10 mL及び硝酸(1+1)2 mLを加え,加熱板又は沸騰水浴上で加熱して溶かし,冷
却後,全量フラスコ100 mLに移し入れ,水を標線まで加え,これを試料溶液とする。
4) 別に,ろ紙について1)〜3) と同様に操作して,空試験溶液を調製する。
注1) 分解条件は,マイクロ波の機種によって異なる。高温・高圧条件下で酸分解を行うため,分解
容器は耐薬品性及び耐圧力に優れた材質のものを用い,容器内部温度のモニタリングを行え
る機種が望ましい。
2) 濃縮条件は,マイクロ波機種によって異なる。
b) 吸収瓶を設けてろ紙とともに溶液にも吸収させて試料採取した場合
1) 試料ガスからの捕集物の付着ろ紙については,a) 1)〜4) による。
2) 一方,吸収瓶中の溶液については,これをビーカーに移し入れ,導管及び吸収瓶を水10〜20 mLで
洗い,洗液も合わせる。この溶液を,複数の密閉式四ふっ化エチレン樹脂分解容器に約20 mLずつ
分け入れ,各分解容器に硝酸5 mLを加え,マイクロ波加熱によって処理する1)。
3) 室温まで放冷後,分解容器を濃縮加熱用ローターで,穏やかに約15分〜25分加熱し,蒸発乾固寸
前まで濃縮する2)。
4) 各分解容器内に水5 mL,硝酸(1+1)1 mLを加え,加熱板又は沸騰水浴上で加熱して溶かし,冷
却後,全量フラスコ100 mLに合わせる。水を標線まで加え,これを試料溶液とする。
5) 別に,ろ紙及び吸収液について,1)〜4) と同様に操作して空試験溶液を調製する。
76
K 0083:2017
附属書B
(規定)
サイドストリームサンプリングによる排ガス中のセレンの試料採取方法
B.1
要旨
排ガス中の粒子状物質中のセレンと二酸化セレンなどのガス状セレン化合物とを同時に採取する際,粒
子状物質中のセレンの測定に必要な吸引ガス量又は採取流量が,ガス状セレン化合物の測定に必要な吸引
ガス量又は採取流量と異なる場合には,サイドストリームサンプリングを適用できる。この附属書は,サ
イドストリームサンプリングによる排ガス中のセレン分析方法を説明する。
B.2
試料採取装置
ダスト捕集器とガス洗浄瓶との間の導管を分岐し,ガス吸引部を接続する。試料採取装置の構成例を図
B.1に示す。吸引ノズルは5.1.3に規定するものとし,採取管,ダスト捕集器,導管,吸収瓶,空瓶,乾燥
瓶及び冷却槽は,5.3.3に規定するものとする。
図B.1−試料採取装置の構成例(一例)
B.3
試料の採取
B.3.1 吸引ガス採取流量
ガス状セレン化合物を採取するための吸引ポンプ②による採取流量は1 L/min程度にする。粒子状物質
中のセレンを採取するための吸引ポンプ①による採取流量は,等速吸引のための吸引流量からガス状セレ
ン化合物を採取するための採取流量を引いた採取流量にする。
B.3.2 試料ガス採取操作
吸引ポンプ①を起動させ,等速吸引のための吸引流量に調整する。吸引ポンプ②を起動させ,ガス状セ
レン化合物の採取流量に調整するとともに,吸引ポンプ①の吸引量を粒子状物質中のセレンを採取するた
77
K 0083:2017
めの採取流量に調整する。
B.4
試料の保管
試料ガス採取後のダスト捕集器,三方コックまでの導管,三方コック,三方コックから吸収瓶までの導
管及び吸収瓶を,それぞれ分けて密閉して保管する。
B.5
試料溶液の調製
B.5.1 粒子状物質中のセレンの試料溶液調製
ダスト捕集器のろ紙の捕集物を,6.4に規定する方法にて試料溶液を調製する。
B.5.2 ガス状セレン化合物の試料溶液調製
a) 6.6.1 d) のガス採取装置の洗浄液でダスト捕集器,三方コックまでの導管及び三方コックを洗浄し,
洗液をビーカーに入れて保管する。
b) 試料採取後の吸収液をa) とは別のビーカーに移し,5.3.3 e) 1) の吸収液で三方コックから吸収瓶まで
の導管,空瓶及び吸収瓶を洗浄し,洗液をビーカーに入れる。
c) 三方コックから吸収瓶までの導管を6.6.1 d) のガス採取装置の洗浄液で洗浄し,洗液をb) のビーカ
ーに入れる。
B.6
試料溶液中のセレンの定量方法
粒子状物質中のセレンの試料溶液1種類及びガス状セレン化合物の試料溶液2種類を,それぞれ箇条15
に規定する方法にて試料溶液中のセレンを定量する。
B.7
排ガス中濃度の計算方法
排ガス中の粒子状物質中のセレン及びガス状セレン化合物の濃度を次の式によって算出する。排ガス中
のセレン濃度は,粒子状物質中のセレン濃度とガス状セレン化合物濃度との和として算出する。
g
s
C
C
C
+
=
··········································································· (B.1)
gSD
sSD
s
s
s
1
000
1
V
V
v
A
C
+
×
×
=
························································ (B.2)
gSD
gA
gA
gSD
sSD
gR
gR
g
1
000
1
1
000
1
V
v
A
V
V
v
A
C
×
×
+
+
×
×
=
······························ (B.3)
ここに,
C: 標準状態における乾き排ガス中のセレン濃度
[mg/m3(273.15 K,101.325 kPa)]
Cs: 標準状態における乾き排ガス中の粒子状物質中のセレ
ン濃度[mg/m3(273.15 K,101.325 kPa)]
Cg: 標準状態における乾き排ガス中のガス状セレン濃度
[mg/m3(273.15 K,101.325 kPa)]
As: B.5.1の試料溶液中のセレン濃度(mg/L)
AgR: B.5.2 a) の試料溶液中のセレン濃度(mg/L)
AgA: B.5.2 c) の試料溶液中のセレン濃度(mg/L)
vs: B.5.1にて調製した試料溶液の量(mL)
vgR: B.5.2 a) にて調製した試料溶液の量(mL)
vgA: B.5.2 c) にて調製した試料溶液の量(mL)
78
K 0083:2017
VsSD: 粒子状物質中のセレンを採取したガスメーター①の吸
引ガス量の標準状態における乾き排ガス量
[m3(273.15 K,101.325 kPa)]
VgSD: ガス状セレンを採取したガスメーター②の標準状態に
おける乾き排ガス量[m3(273.15 K,101.325 kPa)]
参考文献 JIS T 9107 使い捨て手術用ゴム手袋