2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1413-1996
銅ニッケル抵抗材分析方法
Methods of chemical analysis for copper nickel resistance material
1. 適用範囲 この規格は,JIS C 2521に規定された化学成分(銅,ニッケル,コバルト,マンガン)の
分析方法について規定する。
備考 この規格の引用規格を,次に示す。
JIS C 2521 電気抵抗用銅ニッケル線,帯,条及び板
JIS H 0321 非鉄金属材料の検査通則
JIS H 2107 亜鉛地金
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 8005 容量分析用標準物質
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050及びJIS K 0115による。
3. 分析試料の採り方及び取扱い方
3.1
試料の採り方は,JIS H 0321の2.3による。
3.2
鋳込試料を採るときは,その平均品質を代表する試料を得るため,1融解ごとに二つ以上(1融解量
が特に少ないときは一つ)の試料を採る。鋳込試料は,できるだけ完全に製品と同一な品質を得るよう,
特に偏析のないように注意しなければならない。
3.3
試料の削り方は,次による。
(1) 試料の表面に付着物などがある場合は,紙やすりなどを用いて取り除き清浄にする。
(2) きりそのほかの工具類は,アルコールなどを用いて清浄にする。
(3) 鋳込試料から試料を削り取るときは,中央部及び両端に近い部分などの片面から直角にきりもみして
貫通させるか,両面から少なくとも中心部に達するまできりもみするか,又はそのほか適当な方法に
よる。
(4) 線・帯及び板などの製品試料から試料を削り取るには,きり又は適当な工具を用い,分析操作に適当
な大きさに削り取る。
(5) きりもみするときは,発熱のため削り片の表面が酸化することがあるから,酸化させない程度の圧力
と回転数をきりに与えて行う。この際油類そのほかの減摩剤を用いたり,冷却のための水などを注加
したりしてはならない。
また削り片に,きりの摩耗粉が混入しないように注意する。
(6) 削り片の大きさは,あまり厚くならない程度とし,長さを約5mm以下とする。
2
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.4
試料の取扱い方
(1) 削り取った試料は,その全部(通常50g以上)を集め,強力な磁石を用いて混入した鉄粉などを注意
深く取り除き,最後に良く混ぜ合わせて分析用試料とする。
(2) 分析試料の採取方法が上記規定によりがたい場合は,注文者と製造業者との協定によって別途に定め
ることができる。
(3) 分析用試料はデシケーター中に入れ,1時間以上放置した後はかり取る。
3.5
試料のはかり方 試料のはかり方は,次による。
(1) 分析試料のはかり取りに際しては,試料を良くかき混ぜて平均組成を表すように注意しなければなら
ない。
(2) 分析試料のはかり取りには,原則として化学はかりを用い,規定された量に近い量を分析値の表示け
た数を参考として,必要な位まではかり取る。
4. 分析値の表し方と操作上の注意
4.1
分析値の表し方 分析値は百分率で表し,JIS C 2521に規定された位までにJIS Z 8401によって丸
める。
4.2
分析操作上の注意 分析操作上の注意は,次による。
(1) 分析は同一試料について2回以上行って結果を確かめる。
(2) 分析に当たっては全操作を通じて空試験を行い,測定値を補正しなければならない。
5. 銅定量方法
5.1
方法の区分 銅の定量方法は電解重量法による。
5.2
電解重量法
5.2.1
要旨 試料を硝酸と硫酸の混液で分解した後,白金電極を用いて電解を行い,陰極に銅を析出させ
てその質量をはかる(電解残液は,ニッケルの定量に用いることができる。)。
5.2.2
試薬 試薬は,次による。
(1) 混酸 水20mlにかき混ぜながら硫酸5mlを加え,冷却後硝酸5mlを加えて良く混合する。
(2) エチルアルコール (95v/v%)
5.2.3
装置及び器具 装置及び器具は,原則として次のものを用いる。
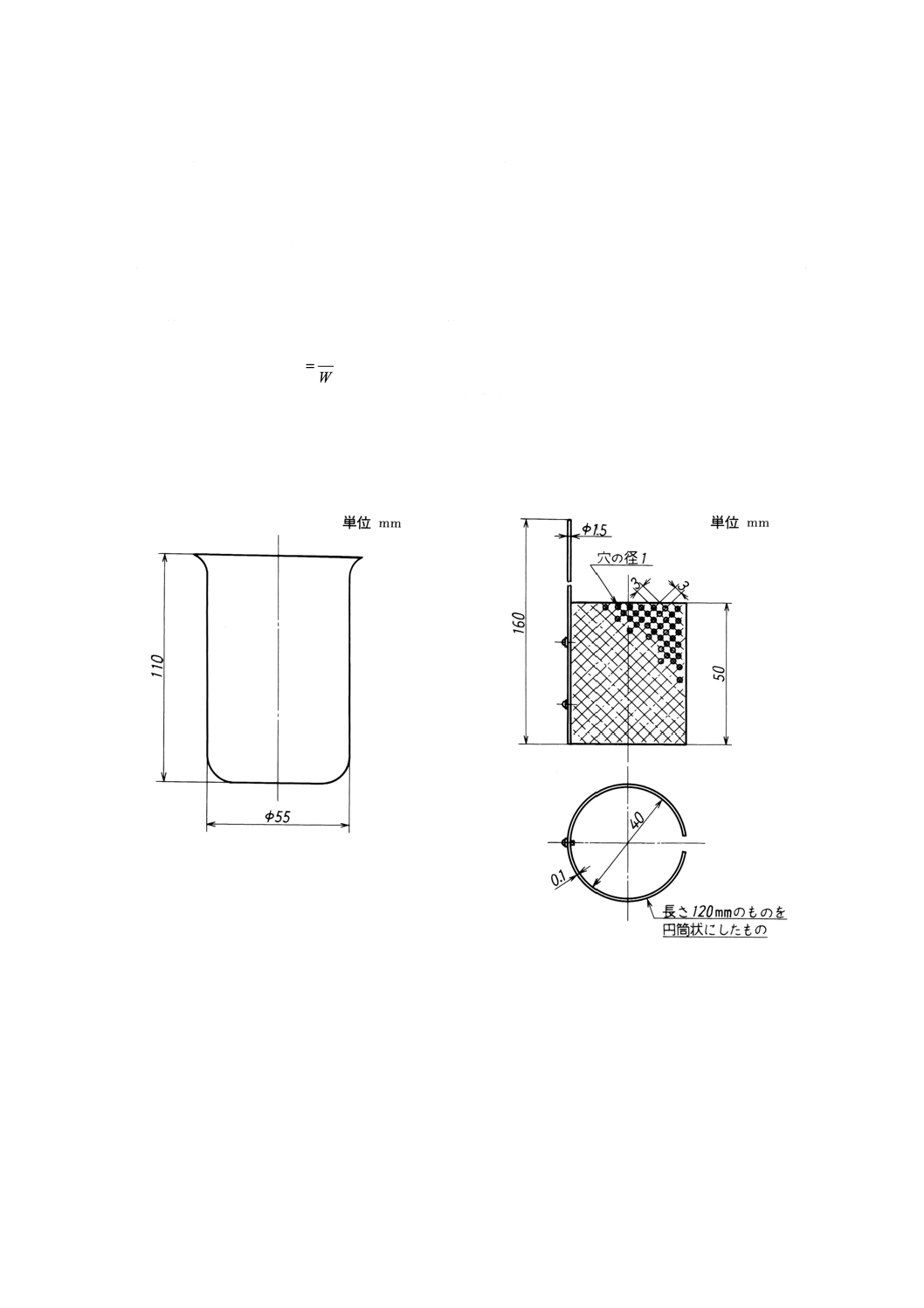
(1) 電解用ビーカー(図1参照)
(2) 白金電極A(図2参照)
(3) 白金電極B(図3参照)
(4) 半円形時計皿(図4参照)
5.2.4
試料はかり取り量 試料は1gをはかり取る。
5.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,電解用ビーカーに移し入れ時計皿で覆い,混酸30mlを加えてなるべく低温で静
かに分解させる。分解が終われば注意して加熱し完全に溶液とし,酸化窒素を追い出す。時計皿の下
面及びビーカーの内壁を洗った後,水を加えて約150mlとする。
(2) あらかじめ質量をはかった白金電極Aを陰極とし,白金電極Bを陽極に用い,2個の半円形時計皿で
覆い,20〜30℃の液温(液温が20℃を下がるときは適当な加熱装置でビーカーを熱する)で0.3〜0.4A
の電流を通じて1夜間電解する。
3
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 少量の水で時計皿の下面,ビーカーの内壁及び電極の柄の液面に露出した部分を洗い,その洗浄水に
よって電解液面を約5mm上昇させ,更に約30分間電解を続ける。
(4) 新しく電解液中に浸った陰極の柄に,もはや銅が析出しなくなれば,電流を通じたまま水洗しながら
両極を徐々に引き上げ,最後は手早く新たな水中に浸して陰極を外す(電解残液は,ニッケルの定量
に用いることができる。)。
(5) 陰極は数回上下して水洗後,エチルアルコールを用いて十分に洗い,直ちに約80℃の空気浴内で速や
かに乾燥し,デシケーター中で約30分間放冷後その質量をはかる。
5.2.6
計算 試料中の銅含有率を次の式によって算出する。
100
×
=Ww
Cu
ここに,
Cu: 試料中の銅含有率 [% (m/m)]
w: 陰極に析出した銅の質量 (g)
W: 試料はかり取り量 (g)
図1 電解用ビーカー
図2 白金電極A
4
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図3 白金電極B
図4 半円形時計皿
6. ニッケル定量方法
6.1
方法の区分 ニッケルの定量方法は,次のいずれかによる。
(1) 滴定法
(2) 重量法
6.2
滴定法
6.2.1
要旨 試料を混酸で分解し,4.に準じて電解を行い,銅などを分離する。電解残液に酒石酸及び塩
化アンモニウムを加えてアンモニア水でアルカリ性とし,ジメチルグリオキシムを加えてニッケルを沈殿
させた後,塩酸で洗い加熱して沈殿を溶解させる。これにEDTA, EBTを加え亜鉛標準溶液で滴定する。
6.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+1, 1+50)
(2) 混酸 5.2.2(1)による。
(3) アンモニア水
(4) アンモニア水 (1+1)
(5) 塩化アンモニウム溶液 (250g/l)
(6) 酒石酸溶液 (250g/l)
(7) ジメチルグリオキシム溶液 ジメチルグリオキシム1.0gを水酸化ナトリウム溶液 (10g/l) 100mlに溶
解する。
(8) 0.02mol/l亜鉛標準溶液 金属亜鉛[99.99%以上,JIS H 2107の特種相当品]1.308gを正しくはかり取
り,なるべく少量の塩酸 (1+1) で加熱分解し,冷却後1 000mlの全量フラスコに移し入れ,水で標線
まで薄める。
5
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(9) 0.02mol/lエチレンジアミン四酢酸二ナトリウム (EDTA) 標準溶液 エチレンジアミン四酢酸二ナト
リウム(2水塩)7.45gを水に溶解して正しく1lとする。力価の標定は,次のように行う。
0.02mol/lEDTA標準溶液を25ml分取し,塩化アンモニウム溶液 (250g/l) 10ml及びEBT指示薬0.1ml
を加え,水で液量を約100mlに薄めた後,溶液が青色になるまでアンモニア水 (1+1) を滴加し,こ
れを0.02mol/l亜鉛標準溶液で滴定し,溶液の青色が赤紫色になった点を終点とし,0.02mol/lEDTA標
準溶液の力価を次の式によって算出する。
25
V
F=
ここに,
F: 0.02mol/lEDTA標準溶液の力価
V: 0.02mol/l亜鉛標準溶液の使用量 (ml)
(10) エリオクロムブラックT (EBT) 溶液 エリオクロムブラックT0.5g及び塩酸ヒドロキシルアミン4.5g
をエチルアルコールに溶解して100mlとする。この溶液は,約6か月間使用することができる。
6.2.3
試料はかり取り量 試料は,1gをはかり取る。
6.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,電解用ビーカーに移し入れ時計皿で覆い,混酸30mlを加え,加熱分解した後,
5.2銅電解重量法に準じて電解を行い,銅を除去(1)する。
(2) 電解残液は250mlの全量フラスコに移し入れ,水で標線まで薄めた後一定量(2)(3)をビーカー (500ml)
に分取する。
(3) これに酒石酸溶液10ml(4)及び塩化アンモニウム溶液20mlを加え,次に溶液があざやかな青色を呈す
るまでアンモニア水を滴加し,更にその過剰に5mlを加え,水で約250mlとする。
(4) 溶液を約90℃に加熱し,かき混ぜながらジメチルグリオキシム溶液をニッケル予想含有量10mgに付
き7mlの割合で加え,更に5mlを過剰に加えて十分にかき混ぜた後,約20分間放置してニッケルを
沈殿させる。
(5) 沈殿をろ紙(5種A)を用いてこし分け,温水で十分に洗浄する。ろ紙上の沈殿は,温水及び熱塩酸 (1
+1) 10mlを注いで元のビーカーに洗い落とし,ろ紙は温水及び温塩酸 (1+50) で数回洗浄し,静か
に加熱して沈殿を溶解させる。
(6) この溶液にニッケル予想含有量10mgに付き0.02mol/lEDTA標準溶液10mlを正しく加えた後,更にそ
の5mlを過剰に加えて2〜3回振り混ぜ,EBT溶液を指示薬として0.1mlを加え,溶液が青色となるま
でアンモニア水 (1+1) を滴加し(5),直ちに0.02mol/l亜鉛標準溶液で滴定(6)し,溶液が赤紫色を呈す
るに至った点を終点とする。
6.2.5
計算 試料中のニッケル含有率を次の式によって算出する。
(
)
100
174
001
.0
2
1
×
×
×
−
×
B
W
V
F
V
Ni=
ここに,
Ni: 試料中のニッケル含有率 [%(m/m)]
V1: 0.02mol/lEDTA標準溶液の使用量 (ml)
F: 0.02mol/lEDTA標準溶液の力価
V2: 0.02mol/l亜鉛標準溶液の使用量 (ml)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
注(1) 銅の除去は,次のアルミニウム還元法によることができる。
試料0.1gをはかり取りビーカー (300ml) に移し入れ時計皿で覆い,硝酸 (1+1) 10mlを加え
6
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
て加熱分解し,分解後,過塩素酸10mlを加えて,白煙が十分に発生するまで加熱する。放冷後,
水約100mlを加えて可溶性塩類を溶解した後,これにアルミニウム板(99.5%以上でニッケルを
含まないもの)の小片を投入し,約8分間静かに煮沸を続けて銅を還元析出させる。
ろ過速度の早いろ紙を用いて速やかにこし分け,温水で十分に洗う。
(2) 滴定法の場合は15ml,重量法の場合は25mlを分取する。
(3) けい素定量方法のろ液を用いてもよい。この場合滴定法では0.05g,重量法では0.1g相当を分
取する。
(4) 注(1)のアルミニウム還元法によるときは,酒石酸溶液の使用量を25mlとする。
(5) このときのpHは,約8.0に調節することが必要である。
(6) このときの液温は,30〜40℃を保持することが必要である。
6.3
重量法
6.3.1
要旨 試料を混酸で分解し,4.に準じて電解を行い,銅などを分離する。電解残液に酒石酸及び塩
化アンモニウムを加えてアンモニア水でアルカリ性とし,ジメチルグリオキシムを加えてニッケルを沈殿
させた後,沈殿を洗浄,乾燥した後,その質量をはかる。
6.3.2
試薬 試薬は次による。
(1) 6.2.2(2)〜(6)による。
6.3.3
試料はかり取り量 試料は,1gをはかり取る。
6.3.4
操作 次の手順によって行う。
(1) 6.2.4(1)〜(4)による。
(2) (4)沈殿をあらかじめ恒量としたガラスろ過器(G3形)を用いてこし分け,温水で十分に洗浄した後,
120〜130℃の空気浴内で恒量となるまで乾燥し,デシケーター中で放冷後,ニッケルジメチルグリオ
キシム [Ni (C4H7N2O2)2] としてその質量をはかる。
6.3.5
試料中のニッケル含有率を次の式によって算出する。
100
2
203
.0
×
×
×
B
W
w
Ni=
ここに,
Ni: 試料中のニッケル含有率 [%(m/m)]
w: ニッケルジメチルグリオキシムの質量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
7. コバルト定量方法
7.1
方法の区分 コバルトの定量方法は,次のいずれかによる。
(1) 重量法 この方法は,コバルト含有率0.1%以上の試料に適用する。
(2) 吸光光度法 この方法は,コバルト含有率0.5%以下の試料に適用する。
7.2
重量法
7.2.1
要旨 試料を硝酸で分解し,塩酸ヒドロキシルアミンを加えて電解を行い,銅を除去する。
電解残液に硝酸を加えて酸化し,水酸化ナトリウムと酸化亜鉛乳を加えて鉄などを沈殿させ,こし分け
る。ろ液を塩酸酸性とし,α‐ニトロソ‐β‐ナフトールを加えてコバルトを沈殿させ,こし分けた後,沈
殿を強熱し,放冷後その質量をはかる。
7.2.2
試薬 試薬は,次による。
7
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸 (1+1)
(4) 水酸化ナトリウム溶液 (100g/l)
(5) 酸化亜鉛乳 乳鉢で良くすりつぶした酸化亜鉛約50gをビーカー (500ml) に入れ,水300mlを加えて
十分にかき混ぜる。
(6) 塩酸ヒドロキシルアミン溶液 (100g/l)
(7) α‐ニトロソ‐β‐ナフトール溶液 α‐ニトロソ‐β‐ナフトールの粉末1gを酢酸15mlに溶解し,こ
し分けて使用する。この試薬は,使用の都度調製する。
7.2.3
装置及び器具 装置及び器具は,原則として5.の5.2.3のものを用いる。
(1) 電解用ビーカー
(2) 白金電極A
(3) 白金電極B
(4) 半円形時計皿
7.2.4
試料はかり取り量 試料は,5gをはかり取る。
7.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料(7)をはかり取り,ビーカー (300ml) に移し入れ時計皿で覆い,硝酸 (1+1) 40mlを加えて静かに
加熱分解させる。引き続き加熱して溶液がシロップ状となるまで蒸発した後,室温まで冷却する。
(2) これに水約150mlを加えて塩類を溶解し,塩酸ヒドロキシルアミン溶液5mlを加えて電解用ビーカー
に移し入れ,白金電極Aを陰極,白金電極Bを陽極として半円形時計皿で覆い,室温において約1A
の電流で約1時間電解する。
(3) 電流を通じたまま水洗し,両極を電解液から分離し,電解液はビーカー (500ml) に移し入れる。
(4) 硝酸 (1+1) 2mlを加えて約2分間煮沸し鉄などを酸化した後,室温近くまで冷却する。
水酸化ナトリウム溶液でニッケルなどの沈殿がわずかに出現する手前まで中和し,この溶液に酸化
亜鉛乳を少量ずつ加えて溶液が少し白濁する程度とし,鉄などを完全に沈殿させる。
約15分間温所に静置し,冷却後ろ紙(5種A)を用いてこし分け,水で数回洗浄する。
(5) ろ液及び洗液は,塩酸5mlを加え,水で約400mlに薄め,煮沸近くまで加熱し,かき混ぜながらα‐
ニトロソ‐β‐ナフトール溶液をコバルト予想含有量10mgに付き6ml(8)の割合で加え,ときどきかき
混ぜながら約30分間放置する。
沈殿はろ紙(5種A)でこし分け,はじめは水で,次に塩酸 (1+1) 及び水で交互に数回洗い,最後
に温水で十分に洗浄する。
(6) 沈殿はろ紙と共にあらかじめ恒量とした磁器るつぼに移し入れ,乾燥後始めは弱く加熱して炭化物を
焼失させ,次に750〜850℃に強熱して,デシケーター中で放冷後,四三酸化コバルト (Co3O4)(9)とし
てその質量をはかり,恒量となるまでこの操作を繰り返す。
7.2.6
計算 試料中のコバルト含有率を,次の式によって算出する。
100
2
734
.0
×
×
W
w
CO=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
w: 四三酸化コバルトの質量 (g)
W: 試料はかり取り量 (g)
8
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(7) 試料のはかり取り量は,なるべく0.01〜0.03g程度のコバルトを含有するように適当に加減する。
(8) 試料のコバルト予想含有量が不明の場合は,溶液がα‐ニトロソ‐β‐ナフトール溶液の過剰に
より茶褐色を示すまで加える。
(9) 沈殿が不純と思われる場合は,塩酸5mlに溶解し,水で約400mlとし,α‐ニトロソ‐β‐ナフ
トール溶液を沈殿生成に使用したときと同量を加え,7.2.5(5)以降の操作に準じて再沈殿を行う。
7.3
吸光光度法
7.3.1
要旨 試料を硝酸で分解し,過塩素酸を加えて白煙を発生させ,アルミニウムを加えて銅を還元し
分離する。溶液の一定量を2個のビーカーに分取し,1個に酢酸ナトリウム及びニトロソR塩を加えた後,
硝酸を加えて煮沸し,試料液とする。他の1個には酢酸ナトリウム,硝酸及びニトロソR塩を加えた後,
これを対照液として試料液の吸光度を測定する。
7.3.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) 過塩素酸
(4) 水酸化ナトリウム溶液 (200g/l)
(5) アルミニウム板 厚さ1mm,幅10mm,長さ50〜60mmのもの。
(6) 酢酸ナトリウム溶液 (500g/l)
(7) ニトロソR塩溶液 (20g/l)
(8) 標準コバルト溶液 (0.1mgCo/ml) 金属コバルト(99%以上)0.100gを硝酸 (1+1) 10mlに分解し,硫
酸 (1+5) 5mlを加え,白煙の発生するまで静かに加熱する。冷却後,水50mlを加えて溶解し,1 000ml
の全量フラスコに移し入れ,水で標線まで薄める。
7.3.3
試料はかり取り量 試料は,2gをはかり取る。
7.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,ビーカー (200ml) に移し入れ時計皿で覆い硝酸 (1+1) 30mlを加えて加熱分解し,
過塩素酸10mlを加えて引き続き白煙が発生するまで加熱する。
(2) 冷却後,水約80mlを加えて塩類を溶解し,この溶液にアルミニウム板を入れ,突沸に注意しながら
静かに加熱する。約3分間加熱し,直ちにろ紙(5種A)を用いて,析出した銅をアルミニウム板と
共にろ紙上に移し,温水で約4回洗浄する。
(3) ろ液は約70mlになるまで濃縮し,冷却後100mlの全量フラスコに移し入れ,水で標線まで薄める。
これから2個のビーカー (200ml) にコバルト含有量に応じ,一定量(10)を分取し,各々の溶液に酢酸
ナトリウム溶液15mlを加え,pH6±0.1になるように水酸化ナトリウム溶液で調節する。
(4) この溶液の1個のビーカーにニトロソR塩溶液10mlを加え,加熱して1〜2分間煮沸する。次に硝酸
10mlを加え,更に約1分間煮沸し,冷却後100mlの全量フラスコに移し入れ,水で標線まで薄めて試
料液とする。
(5) 他の1個のビーカーに硝酸10mlを加え,加熱して1〜2分間煮沸した後,ニトロソR塩溶液10mlを
加え,更に約1分間煮沸して冷却する。この溶液を100mlの全量フラスコに移し入れ,水で標線まで
薄めて,対照液とする。
(6) 対照液を対照として,波長530nm付近の吸光度を測定する。
7.3.5
計算 7.3.6で作成した検量線からコバルト量を求め,試料中のコバルト含有率を次の式によって
算出する。
9
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
×
×B
W
A
CO=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A: 分取した試料溶液中のコバルト検出量 (g)
B: 試料溶液の分取比
W: 試料はかり取り量 (g)
注(10) コバルトとして30〜300μgとなるように分取する。
7.3.6
検量線の作成 標準コバルト溶液の各種液量 (0〜5ml) をビーカー (100ml) に取り,7.3.4(4)以降
に従って操作し,それぞれの吸光度を測定し,得た吸光度とコバルト量との関係線を作成して検量線とす
る。
8. マンガン定量方法
8.1
方法の区分 マンガンの定量方法は,次のいずれかによる。
(1) 滴定法
(2) 吸光光度法
8.2
滴定法
8.2.1
要旨 試料を混酸で分解し,過硫酸アンモニウムを加えてマンガンを酸化し,冷却後亜ひ酸ナトリ
ウム標準溶液で滴定する。
8.2.2
試薬 試薬は,次による。
(1) 混酸 水435ml中に硫酸150mlを徐々に加え,冷却後硝酸250mlとりん酸150ml及び硝酸銀溶液
(200g/l) 15mlを混合する。
(2) 過硫酸アンモニウム溶液 (200g/l) この溶液は,使用の都度調製する。
(3) 亜ひ酸ナトリウム標準溶液 三酸化ひ素(JIS K 8005の標準試薬)0.5gをビーカー (200ml) に正しく
はかり取り,水酸化ナトリウム溶液 (40g/l) 20mlと水約100mlを加えて加熱溶解し,冷却した後1 000ml
の全量フラスコに移す。フェノールフタレインを指示薬として硫酸 (1+35) を加えて微酸性とし,こ
れに炭酸水素ナトリウム溶液 (50g/l) 20mlを加え,水で標線まで薄める。この標準溶液のマンガン相
当量の決定方法は,次のとおりとする。
電気銅と電解ニッケルとを試料中の含有量に近い比率にはかり取り,本文に準じて混酸に溶解し,
これに標準マンガン溶液の一定量を正しく加え,以下8.2.4(2)以降に準じて操作し滴定を行い,亜ひ酸
ナトリウム標準溶液1ml当たりのマンガン相当量を次の式から求める。
2
1
2
000
.0
V
V
f
×
=
ここに,
f: 亜ひ酸ナトリウム標準溶液1mlのマンガン相当量 (g)
V1: 標準マンガン溶液の使用量 (ml)
V2: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
(4) 標準マンガン溶液 (0.2mgMn/ml) 金属マンガン(99.9%以上)0.100gをビーカー (200ml) にはかり
取り,硫酸 (1+4) 50mlを加えて加熱分解し,冷却後500mlの全量フラスコに移し入れ,水で標線ま
で薄める。
8.2.3
試料はかり取り量 試料はマンガン含有率に応じ,原則として表1に従ってはかり取る。
表1 試料はかり取り量
マンガン含有率%
試料はかり取り量g

10
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
マンガン含有率%
試料はかり取り量g
0.5未満
1.0
0.5以上 1.0未満
0.5
1.0以上
0.2
8.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,三角フラスコ (500ml) に移し入れ混酸30mlを加えて加熱分解する。反応が終わ
ればフラスコの内壁を洗い,再び煮沸して酸化窒素を追い出す。
(2) これに温水約200mlを加えて加熱を続け,煮沸し始めたときに過硫酸アンモニウム溶液 (200g/l) 10ml
を加えて小気泡が大気泡となるまで2〜3分間煮沸し,過硫酸アンモニウムを完全に分解するとともに,
マンガンを十分に酸化して過マンガン酸とした後,流水中で25℃以下に冷却する。
(3) 冷却後速やかに亜ひ酸ナトリウム標準溶液で滴定する。
8.2.5
計算 試料中のマンガンの含有率を次の式によって算出する。
100
×
×
W
f
V
Mn=
ここに,
Mn: 試料中のマグネシウム含有率 [% (m/m)]
V: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
f: 亜ひ酸ナトリウム標準溶液1mlのマンガン相当量 (g)
W: 試料はかり取り量 (g)
8.3
吸光光度法
8.3.1
要旨 試料を混酸で分解し,過硫酸アンモニウムを加えてマンガンを呈色させ,流水中で冷却した
後,吸光度を測定する。
8.3.2
試薬 試薬は,次による。
(1) 混酸8.2.2(1)による。
(2) 過硫酸アンモニウム溶液 (200g/l) 8.2.2(2)による。
(3) 標準マンガン溶液 (0.2mgMn/ml) 8.2.2(4)による。
8.3.3
試料はかり取り量 試料はマンガン含有率に応じ,原則として表2に従ってはかり取る。
表2 試料はかり取り量
マンガン含有率%
試料はかり取り量g
0.10未満
1
0.10以上 1.00未満
0.25
1.0以上
0.1
8.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,ビーカー (300ml) に移し入れ時計皿で覆い,混酸20mlを加えて静かに加熱分解
し,煮沸して酸化窒素などを除去し,温水100mlを加える。
(2) これを加熱し,煮沸し始めたときに過硫酸アンモニウム溶液10mlを加え,引き続き2〜3分間煮沸し
てマンガンを過マンガン酸に酸化した後,直ちに流水中で冷却する。次に250mlの全量フラスコに移
し入れ,水で正しく標線まで薄める。
(3) この溶液の一部を光度計の吸収セルに取り,波長530nm付近における吸光度を測定する。
8.3.5
計算 8.3.6で作成した検量線からマンガン量を求め,試料中のマンガン含有率を次の式によって
算出する。
100
×
WA
Mn=
11
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
Mn: 試料中のマンガン含有率 [% (m/m)]
A: 試料中のマンガン検出量 (g)
W: 試料はかり取り量 (g)
8.3.6
検量線の作成 電気銅と電解ニッケルを試料中の含有量に近い比率に数個はかり取り,本文同様に
分解する。これに8.3.2(3)の標準マンガン溶液の各種液量0〜15mlを加え,8.3.4(2)の操作に準じて呈色し
得た吸光度とマンガン量との関係線を作成して検量線とする。
12
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考
次に記載するけい素及び鉄の定量方法は参考のために示すものであって,規格の一部ではない。
1. けい素定量方法
1.1
方法の区分 けい素の定量方法は,重量法による。
1.2
要旨 試料を硝酸で分解し,過塩素酸を加えて加熱蒸発して白煙を発生させ,放冷後水で可溶性塩
類を溶解し,こし分ける。沈殿は強熱後はかる。
1.3
試薬 試薬は,次による。
(1) 塩酸 (1+3)
(2) 硝酸 (1+1)
(3) 過塩素酸
(4) ふっ化水素酸
(5) 硫酸 (1+3)
(6) 過酸化水素水 (1+9)
1.4
試料はかり取り量 試料は,2gをはかり取る。
1.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,ビーカー (300ml) に移し入れ時計皿で覆い,硝酸 (1+1) 30mlを加えて静かに加
熱して完全に分解する。これに過塩素酸30mlを加え加熱して蒸発し,過塩素酸の白煙が発生し始め
てから,更に約10分間加熱を続ける。
(2) これを放冷後温水150mlを加え,振り混ぜて可溶性塩類を溶解し(1),ろ紙(5種B)を用いてこし分
け,温水と温塩酸 (1+3) で交互に洗浄し,過塩素酸イオンのなくなるまで十分に洗浄する。
(3) 沈殿は,ろ紙と共に白金るつぼに入れ,乾燥後灰化し,1 100℃以上で約30分間強熱して恒量とし,
デシケーター中で室温まで放冷して第1回のひょう量をする。
(4) 次に残さを硫酸 (1+3) で湿し,ふっ化水素酸(2)約2mlを加え,注意して加熱し,けい酸及び硫酸を
揮散させた後,1 100℃以上で強熱して恒量とし,デシケーター中で室温まで放冷して第2回のひょう
量をする。
1.6
計算 試料中のけい素含有率を次の式によって算出する。
(
)
100
4
467
.0
2
1
×
×
−
W
w
w
Si=
ここに,
Si: 試料中のけい素含有率 [% (m/m)]
w1: 第1回のひょう量 (g)
w2: 第2回のひょう量 (g)
W: 試料はかり取り量 (g)
注(1) ここで二酸化マンガンの沈殿を認めた場合には,少量の過酸化水素水(約3%)を加えて沈殿を
溶解し,煮沸して過酸化水素を分解してからこし分ける。
(2) 使用量と同量のふっ化水素酸及び硫酸の強熱残さを求め,操作中の揮散減量に加算しなければ
ならない。ただし,ふっ化水素酸は,その1mlに付き強熱残さ量が0.04mgを超えてはならない。
13
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. 鉄定量方法
2.1
方法の区分 鉄の定量方法は,吸光光度法による。
2.2
要旨 試料を硝酸で分解し,アンモニア水と硫酸とを用いて酸濃度を調節し,スルホサリチル酸を
加えて呈色させ,吸光度を測定する。
2.3
試薬 試薬は,次による。
(1) 硝酸(1+1)
(2) 硫酸 (1+5)
(3) アンモニア水 (1+3)
(4) スルホサリチル酸溶液 スルホサリチル酸(2水塩)10gを水50mlに溶解して70〜80mlとした後,
アンモニア水 (1+3) を用いてpH2.2±0.1に調節し,水で100mlに薄める。
(5) 標準鉄溶液 (0.1mgFe/ml) 純鉄(99.5%以上)0.100gをビーカーにはかり取り,硝酸 (1+3) 15mlで
分解後,酸化窒素を追い出し,室温に冷却した後水で正しく1 000mlとする。
2.4
試料はかり取り量 試料は1gをはかり取る。
2.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り,ビーカー (200ml) に移し入れ硝酸 (1+1) 15mlを加え,静かに加熱して完全に分
解し,反応が終わればビーカーの内壁を洗い,再び煮沸して酸化窒素を追い出す。
(2) これに水を加えて約60mlに薄め,この溶液に水酸化物の沈殿がわずかに生成し始めるまでアンモニ
ア水 (1+3) を加え,次に硫酸 (1+5) とアンモニア水 (1+3) を用いてpH2.2±0.1に調節する。
(3) この溶液を100mlの全量フラスコに移し,スルホサリチル酸溶液3mlを加えて水で標線まで薄め,良
く振り混ぜて鉄を呈色させる。
(4) 溶液の一部を光度計の吸収セルにとり,波長520nm付近の吸光度を測定する。
2.6
計算 2.7で作成した検量線を用いて鉄量を求め,試料中の鉄含有率を次の式によって算出する。
100
×
WA
Fe=
ここに,
Fe: 試料中の鉄含有率 [% (m/m)]
A: 試料溶液中の鉄検出量 (g)
W: 試料はかり取り量 (g)
2.7
検量線の作成 電解銅,電解ニッケルを試料中の含有量に近い比率に数個はかり取り,本文同様に
分解する。これに2.3(5)の標準鉄溶液の各種液量0〜20mlを加え,2.5(2)以降に従って操作し,それぞれの
吸光度を測定し,得た吸光度と鉄量との関係線を作成して検量線とする。
14
H 1413-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
非鉄金属部会 電熱材及び抵抗材分析方法専門委員会 構成表(昭和42年2月1日改正のとき)
氏名
所属
(委員会長)
後 藤 秀 弘
東北大学
俣 野 宣 久
金属材料技術研究所
菅 谷 宏
鉄道技術研究所
服 部 只 雄
古河電気工業株式会社
森 田 義 男
三菱電機株式会社
山 田 栄 一
東京芝浦電気株式会社
従 野 睦 秀
赤羽冶金株式会社
菅 井 三 郎
王子合金株式会社
渡 部 武 利
細川製線株式会社
大 森 茂 生
株式会社東京ワイヤー製作所
角 健 蔵
東海高熱工業株式会社
杉 本 正 勝
日本金属工業株式会社
中 田 重 徳
古河特殊金属工業株式会社
野 崎 松 郎
日立熱器具株式会社
望 月 平 一
日本冶金工業株式会社
(事務局)
石 井 清 次
工業技術院標準部材料規格課
種 橋 誠 治
工業技術院標準部材料規格課
(事務局)
廣 瀬 浩 二
工業技術院標準部材料規格課(平成8年3月1日改正のとき)
斎 藤 充
工業技術院標準部材料規格課(平成8年3月1日改正のとき)