H 1405:2016
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 一般事項························································································································· 1
4 試料の採り方及び前処理 ···································································································· 2
4.1 試料の採り方 ················································································································ 2
4.2 試料の前処理 ················································································································ 2
5 分析値のまとめ方 ············································································································· 2
5.1 分析回数 ······················································································································ 2
5.2 分析値の表示 ················································································································ 2
6 鉄定量方法 ······················································································································ 2
6.1 定量方法の区分 ············································································································· 2
6.2 1,10-フェナントロリン吸光光度法 ····················································································· 2
6.3 原子吸光分析法 ············································································································· 5
6.4 ICP発光分光分析法········································································································ 6
7 モリブデン定量方法 ·········································································································· 9
7.1 定量方法の区分 ············································································································· 9
7.2 チオシアン酸吸光光度法 ································································································· 9
7.3 原子吸光分析法 ············································································································ 11
7.4 ICP発光分光分析法······································································································· 13
8 酸化トリウム(IV)定量方法 ···························································································· 15
8.1 定量方法の区分 ············································································································ 15
8.2 塩化水素ガス揮散重量法 ································································································ 15
8.3 水酸化物沈殿分離重量法 ································································································ 17
H 1405:2016
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,タングステン・モリ
ブデン工業会(JTMIA)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規
格を改正すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規
格である。
これによって,JIS H 1405:2001は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
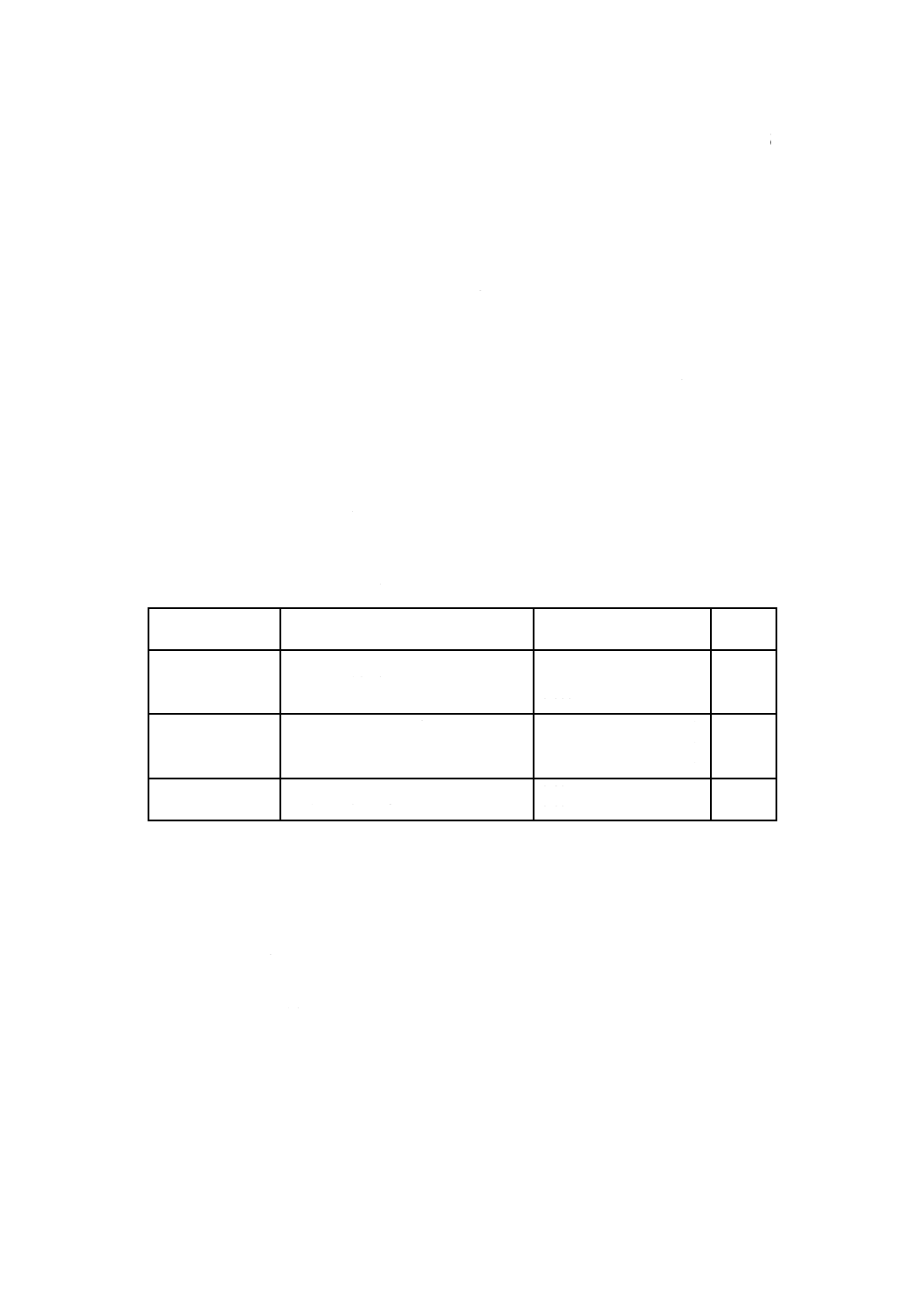
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1405:2016
トリエーテッドタングステン材料の分析方法
Methods for chemical analysis of thoriated tungsten materials
1
適用範囲
この規格は,トリエーテッドタングステン材料(照明,電子機器及び非消耗電極溶接用)中の鉄,モリ
ブデン及び酸化トリウム(IV)の定量方法について規定する。
なお,これらの定量方法は,各成分について表1に規定する含有率範囲の定量に適用する。
警告 この規格に基づいて試験を行う者は,通常の実験室での作業に精通していることを前提とする。
この規格は,その使用に関連して起こる全ての安全上の問題を取り扱おうとするものではない。
この規格の利用者は,各自の責任において安全及び健康に対する適切な措置をとらなければな
らない。
表1−トリエーテッドタングステン中の各種成分の定量方法及び適用含有率範囲
成分
定量方法
適用含有率範囲
%(質量分率)
箇条
番号
鉄
1,10-フェナントロリン吸光光度法
原子吸光分析法
ICP発光分光分析法
0.001 0以上 0.030 以下
0.000 5以上 0.010 以下
0.000 5以上 0.030 以下
6
モリブデン
チオシアン酸吸光光度法
原子吸光分析法
ICP発光分光分析法
0.001 0以上 0.016 以下
0.001 0以上 0.020 以下
0.001 0以上 0.020 以下
7
酸化トリウム(IV) 塩化水素ガス揮散重量法
水酸化物沈殿分離重量法
0.10 以上
0.10 以上
8
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS Z 8402-1 測定方法及び測定結果の精確さ(真度及び精度)−第1部:一般的な原理及び定義
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
3
一般事項
分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116及びJIS K 0121による。
2
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4
試料の採り方及び前処理
4.1
試料の採り方
試料の採り方は,その品質を代表するように,汚染などに注意して,分析に必要な量を採取する。
4.2
試料の前処理
試料の前処理は,次による。
a) 4.1で採取した試料は,水酸化ナトリウム溶液(300 g/L)中に浸して約15分間煮沸した後,水洗する。
b) 清浄なガーゼ,ブラシなどで試料に付着している黒鉛などを拭い去った後,適切な大きさ(JIS Z 8801-1
の付表2に規定する公称目開き710 μm以下)に粉砕し,塩酸(1+1)で約15分間煮沸する。水洗し
た後,更にエタノールで洗浄し,乾燥する。
c) 乾燥した試料は,酸化及び吸湿を防止するために適切な容器に入れ密封し,分析用試料とする。
5
分析値のまとめ方
5.1
分析回数
分析回数は,JIS Z 8402-1の3.14[併行条件(repeatability conditions)]に規定する併行条件で2個を分
析する。
5.2
分析値の表示
分析値は,併行条件で分析した2個の値を平均し,表1の適用含有率範囲の桁まで丸める。
6
鉄定量方法
6.1
定量方法の区分
鉄の定量方法は,次のいずれかによる。
a) 1,10-フェナントロリン吸光光度法 この方法は,鉄含有率0.001 0 %(質量分率)以上0.030 %(質量
分率)以下の試料に適用する。
b) 原子吸光分析法 この方法は,鉄含有率0.000 5 %(質量分率)以上0.010 %(質量分率)以下の試料
に適用する。
c) ICP発光分光分析法 この方法は,鉄含有率0.000 5 %(質量分率)以上0.030 %(質量分率)以下の
試料に適用する。
6.2
1,10-フェナントロリン吸光光度法
6.2.1
要旨
試料を適切な試薬で分解し,酒石酸を加えて,タングステン,鉄などを錯塩とした後,pHを調節する。
L(+)-アスコルビン酸で鉄(III)を鉄(II)に還元し,1,10-フェナントロリンを加えて1,10-フェナント
ロリン・鉄(II)錯体を生成させ,試料溶液をろ過する。分光光度計を用いて波長510 nm付近の吸光度を
測定する。
6.2.2
試薬
試薬は,次による。
6.2.2.1
ほう酸溶液(50 g/L)
6.2.2.2
混酸A(硝酸1,ふっ化水素酸1)
6.2.2.3
アンモニア水
6.2.2.4
水酸化ナトリウム溶液(100 g/L)
6.2.2.5
過酸化水素
3
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.2.6
酢酸
6.2.2.7
酒石酸溶液(500 g/L)
6.2.2.8
L(+)-アスコルビン酸溶液(50 g/L) 使用の都度,調製する。
6.2.2.9
1,10-フェナントロリン溶液 塩化1,10-フェナントロリニウム一水和物(別名:塩酸-o-フェナン
トロリン一水和物)0.24 gを水に溶解し,水で液量を100 mLとするか,又は1,10-フェナントロリン0.2 g
をエタノール(99.5)10 mLに溶解し,水で液量を100 mLとする。
6.2.2.10 鉄標準液(Fe:50 μg/mL) 鉄標準液は,次のいずれかを用いる。
a) 市販の鉄標準液 市販の鉄標準液(Fe:1 000 μg/mL)は,酸濃度,安定剤の有無などが使用目的に合
致することを確認して用い,使用の都度,必要量だけ水で正確に20倍にうすめて使用する。
注記 計量標準供給制度(JCSS:Japan Calibration Service System。以下,JCSSという。)に基づく
鉄標準液がある。
b) 金属を用いて調製した鉄標準液 鉄[純度99.9 %(質量分率)以上]1.000 gをはかりとってビーカー
(200 mL)に移し入れ,時計皿で覆い,塩酸(1+1)20 mLを加え,穏やかに加熱して分解する。過
酸化水素1 mLを加え,煮沸して鉄を酸化するとともに,過剰の過酸化水素を分解する。常温まで冷
却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用
いて移し入れ,水で標線までうすめて原液(Fe:1 000 μg/mL)とする。この原液を使用の都度,必要
量だけ水で正確に20倍にうすめて鉄標準液とする。
6.2.2.11 p-ニトロフェノール溶液(4 g/L)
6.2.3
試料はかりとり量
試料はかりとり量は,1.0 gとし,10 mgの桁まではかる。
6.2.4
操作
6.2.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Aによる分解 混酸Aによる分解は,次による。
1) 試料をはかりとって白金皿(75番又は90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200
mL)又はポリエチレン製ビーカー(100 mL又は200 mL)に移し入れる。
2) 白金製蓋,四ふっ化エチレン樹脂製時計皿又はポリエチレン製時計皿で覆い,混酸A 6 mLを少量
ずつ加え,穏やかに加熱して分解する。ただし,試料の形状が極細線及び細粒の場合は,分解反応
が激しいので,混酸Aに10 mL以下の水を加えて使用してもよい。引き続き加熱して窒素酸化物を
追い出す。
なお,ポリエチレン製ビーカーを用いる場合は,加熱による容器の変形が起こらないように,水
浴上又は水浴中で加熱しながら分解する。
3) 常温まで冷却した後,蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,酒石酸溶液(500
g/L)10 mL及びほう酸溶液(50 g/L)30 mLを白金棒又はプラスチック棒でかき混ぜながら加え,
100 mLの全量フラスコに水を用いて移し入れる。
4) p-ニトロフェノール溶液(4 g/L)数滴を指示薬として加えた後,振り混ぜながら溶液の色が黄にな
るまでアンモニア水を加える。次に,酢酸を溶液が無色となるまで滴加し,更に数滴を過剰に加え
る。
5) L(+)-アスコルビン酸溶液(50 g/L)(6.2.2.8)2 mL及び1,10-フェナントロリン溶液(6.2.2.9)10 mL
を加えて振り混ぜ,水で標線までうすめる。約10分間放置して,呈色を安定させる。
4
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 過酸化水素による分解 過酸化水素による分解は,次による。
1) 試料をはかりとってビーカー(100 mL又は200 mL)に移し入れる。
2) 数mLの水で試料を湿らせ時計皿で覆い,過酸化水素約10 mLを少量ずつ数回に分けて加え,放置
又は加熱して分解する。分解が不十分な場合は,更に過酸化水素を少量ずつ追加する。試料を分解
した後,液量が約10 mLになるまで加熱して酸化タングステン(VI)一水和物を析出させた後,水
でうすめて液量を約20 mLとする。
3) 室温まで冷却した後,水酸化ナトリウム溶液(100 g/L)15 mLを少量ずつ加える。激しい発泡が終
わった後,液量が10〜20 mLになるまで煮沸して未溶解の酸化タングステン(VI)一水和物及び過
剰の過酸化水素を分解し,酒石酸溶液(500 g/L)10 mLを加え,約1分間煮沸する。
4) 常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,100 mLの全量フラスコに水
を用いて移し入れる。
5) 以下,a) 4) 及びa) 5) の手順に従って操作する。
6.2.4.2
吸光度の測定
6.2.4.1のa) 5) 又はb) 5) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mLは捨て,
その後の一部を分光光度計の吸収セル(10 mm)に採り,水を対照液として波長510 nm付近の吸光度を測
定する。
6.2.5
空試験
試料を用いないで試料と同じ操作を試料と併行して行って,空試験液を調製し,6.2.4.2に従って,空試
験液の吸光度を測定する。
6.2.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を6.2.4.1 a) によって行う場合 数個の白金皿(75番又は90番),四ふっ化エチレン
樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100 mL又は200 mL)を用意
し,6.2.4.1のa) 2) 及びa) 3) の手順に従って操作した後,溶液をそれぞれ100 mLの全量フラスコに
水を用いて移し入れる。鉄標準液(Fe:50 μg/mL)(6.2.2.10)0〜6.0 mL(鉄として0〜300 μg)を段
階的に加える。以下,6.2.4.1のa) 4) 及びa) 5) の手順に従って操作した後,得た吸光度と鉄量との関
係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を6.2.4.1 b) によって行う場合 数mLの水及び6.2.4.1 b) 2) で加えた量と同量の過酸
化水素を数個のビーカー(100 mL又は200 mL)にとり,時計皿で覆う。液量が5〜10 mLになるまで
加熱して濃縮した後,水でうすめて液量を約20 mLとし,室温まで冷却する。6.2.4.1のb) 3) 及びb) 4)
の手順に従って操作した後,溶液をそれぞれ100 mLの全量フラスコに水を用いて移し入れ,水で標
線までうすめる。鉄標準液(Fe:50 μg/mL)(6.2.2.10)0〜6.0 mL(鉄として0〜300 μg)を段階的に
加える。以下,6.2.4.1 b) 5) の手順に従って操作した後,得た吸光度と鉄量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
6.2.7
計算
6.2.4.2及び6.2.5で得た吸光度と,6.2.6で作成した検量線の鉄量とから鉄検出量を求め,試料中の鉄含
有率を次の式によって算出する。
100
1
2
1
×
−
=
m
A
A
Fe
ここに,
Fe: 試料中の鉄含有率[%(質量分率)]
5
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 試料溶液中の鉄検出量(g)
A2: 空試験液中の鉄検出量(g)
m1: 試料はかりとり量(g)
6.3
原子吸光分析法
6.3.1
要旨
試料を適切な試薬で分解した後,試料溶液を原子吸光分析装置のアセチレン・空気フレーム中に噴霧し,
その吸光度を測定する。
6.3.2
試薬
試薬は,次による。
6.3.2.1
ほう酸溶液(50 g/L)
6.3.2.2
りん酸(1+1)
6.3.2.3
混酸A(硝酸1,ふっ化水素酸1)
6.3.2.4
過酸化水素
6.3.2.5
タングステン粉 鉄含有率が既知で,かつ,その鉄含有率が適用含有率範囲の下限値[0.000 5 %
(質量分率)]より低いもの。
6.3.2.6
鉄標準液(Fe:50 μg/mL) 6.2.2.10による。
6.3.3
試料はかりとり量
試料はかりとり量は,3.0 gとし,10 mgの桁まではかる。
6.3.4
操作
6.3.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Aによる分解 混酸Aによる分解は,次による。
1) 試料をはかりとって白金皿(75番又は90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200
mL)又はポリエチレン製ビーカー(100 mL又は200 mL)に移し入れる。
2) 白金製蓋,四ふっ化エチレン樹脂製時計皿又はポリエチレン製時計皿で覆い,りん酸(1+1)2 mL
及び混酸A 10 mLを少量ずつ加え,穏やかに加熱して分解する。引き続き加熱して窒素酸化物を追
い出し,常温まで冷却した後,蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,ほう
酸溶液(50 g/L)40 mLを白金棒又はプラスチック棒でかき混ぜながら加え,溶液を100 mLの全量
フラスコに水を用いて移し入れる。
なお,ポリエチレン製ビーカーを用いる場合は,加熱による容器の変形が起こらないように,水
浴上又は水浴中で加熱しながら分解する。
3) 水で標線までうすめる。
b) 過酸化水素による分解 過酸化水素による分解は,次による。
1) 試料をはかりとってビーカー(100 mL又は200 mL)に移し入れる。
2) 時計皿で覆い,数mLの水で試料を湿らせ,りん酸(1+1)2 mL及び過酸化水素約10 mLを少量ず
つ加え,放置又は加熱して分解する。分解が不十分な場合は,更に過酸化水素を少量ずつ追加する。
3) 試料を分解した後,液量が約10 mLになるまで加熱し,常温まで冷却した後,時計皿の下面を水で
洗浄して時計皿を取り除き,溶液を100 mLの全量フラスコに水を用いて移し入れる。
4) 水で標線までうすめる。
6
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3.4.2
吸光度の測定
6.3.4.1のa) 3) 又はb) 4) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mLは捨て,
その後の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・空気フレーム中に噴霧し,
波長248.3 nmにおける吸光度を測定する。
6.3.5
空試験
6.3.6の検量線の作成操作において得られる鉄標準液(Fe:50 μg/mL)を添加しない溶液を空試験液とし,
試料溶液と併行して,6.3.4.2に従って吸光度を測定する。
6.3.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を6.3.4.1 a) によって行う場合
1) タングステン粉(6.3.2.5)を3.0 gずつ10 mgの桁まで数個はかりとり,それぞれ白金皿(75番又は
90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100
mL又は200 mL)に移し入れる。
2) 6.3.4.1 a) 2) の操作を試料と併行して行って得た溶液に,鉄標準液(Fe:50 μg/mL)(6.3.2.6)0〜6.0
mL(鉄として0〜300 μg)を段階的に加え,水で標線までうすめる。
3) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・空気フレー
ム中に噴霧し,波長248.3 nmにおける吸光度を試料溶液と併行して測定し,得た吸光度と鉄量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を6.3.4.1 b) によって行う場合
1) タングステン粉(6.3.2.5)を3.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 6.3.4.1のb) 2) 及びb) 3) の操作を試料と併行して行って得た溶液に,鉄標準液(Fe:50 μg/mL)
(6.3.2.6)0〜6.0 mL(鉄として0〜300 μg)を段階的に加え,水で標線までうすめる。
3) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・空気フレー
ム中に噴霧し,波長248.3 nmにおける吸光度を試料溶液と併行して測定し,得た吸光度と鉄量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
6.3.7
計算
6.3.4.2及び6.3.5で得た吸光度と,6.3.6で作成した検量線の鉄量とから鉄検出量を求め,試料中の鉄含
有率を次の式によって算出する。
(
)100
2
5
4
3
×
−
−
=
m
A
A
A
Fe
ここに,
Fe: 試料中の鉄含有率[%(質量分率)]
A3: 試料溶液中の鉄検出量(g)
A4: 空試験液中の鉄検出量(g)
A5: タングステン粉(6.3.2.5)3.0 g中に含まれる鉄量(g)
m2: 試料はかりとり量(g)
6.4
ICP発光分光分析法
6.4.1
要旨
試料を適切な試薬で分解した後,試料溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
7
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.4.2
試薬
試薬は,次による。
6.4.2.1
ほう酸溶液(50 g/L)
6.4.2.2
りん酸(1+1)
6.4.2.3
混酸B(硝酸1,ふっ化水素酸1,水3)
6.4.2.4
混酸C(塩酸3,硝酸1,水4)
6.4.2.5
過酸化水素
6.4.2.6
タングステン粉 6.3.2.5による。
6.4.2.7
鉄標準液(Fe:50 μg/mL) 6.2.2.10による。
6.4.3
試料はかりとり量
試料はかりとり量は,1.0 gとし,10 mgの桁まではかる。
6.4.4
操作
6.4.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Bによる分解 混酸Bによる分解は,次による。
1) 試料をはかりとって白金皿(75番又は90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200
mL)又はポリエチレン製ビーカー(100 mL又は200 mL)に移し入れる。
2) 白金製蓋,四ふっ化エチレン樹脂製時計皿又はポリエチレン製時計皿で覆い,りん酸(1+1)1 mL
及び混酸B 10 mLを少量ずつ加え,穏やかに加熱して分解する。引き続き加熱して窒素酸化物を追
い出し,常温まで冷却する。
3) ほう酸溶液(50 g/L)40 mLを白金棒又はプラスチック棒でかき混ぜながら加える。
4) 蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,溶液を100 mLの全量フラスコに水
を用いて移し入れ,水で標線までうすめる。
なお,ポリエチレン製ビーカーを用いる場合は,加熱による容器の変形が起こらないように,水
浴上又は水浴中で加熱しながら分解する。
b) 過酸化水素による分解 過酸化水素による分解は,次による。
1) 試料をはかりとってビーカー(100 mL又は200 mL)に移し入れる。
2) 数mLの水で試料を湿らせ時計皿で覆い,りん酸(1+1)1 mL及び過酸化水素約10 mLを少量ずつ
加え,放置又は加熱して分解する。分解が不十分な場合は,更に過酸化水素を少量ずつ追加する。
3) 試料を分解した後,液量が約10 mLになるまで加熱し,常温まで冷却する。
4) 時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100 mLの全量フラスコに水を用いて移し
入れ,水で標線までうすめる。
c) 混酸Cによる分解 混酸Cによる分解は,次による。
1) 試料をはかりとってビーカー(100 mL又は200 mL)に移し入れる。
2) 数mLの水で試料を湿らせ時計皿で覆い,りん酸(1+1)1 mL及び過酸化水素約10 mLを少量ずつ
加え,放置又は加熱して分解する。分解が不十分な場合は,更に過酸化水素を少量ずつ追加する。
試料を分解した後,液量が5〜10 mLになるまで加熱して濃縮し,室温まで冷却する。
3) 混酸C 10 mLを少量ずつ加え,数分間放置し激しい発泡を終わらせ,約5分間煮沸した後,常温ま
で冷却する。
4) 時計皿の下面を水で洗浄して時計皿を取り除き,溶液を50 mLの全量フラスコに水を用いて移し入
8
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
れ,水で標線までうすめる。
6.4.4.2
発光強度の測定
6.4.4.1のa) 4) ,b) 4) 又はc) 4) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mL
は捨て,その後の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長238.204 nm又は
259.940 nmにおける発光強度を測定する。
なお,精確さを確認してあれば,他の波長を用いて測定してもよい。また,高次のスペクトル線が使用
可能な装置では,高次のスペクトル線を用いてもよい。バックグラウンド補正機構が付いている装置では,
バックグラウンド補正機構を用いてもよい。これらの操作を適用した場合は,検量線の作成においても同
様に行う。
6.4.5
空試験
6.4.6の検量線の作成操作において得られる鉄標準液(Fe:50 μg/mL)を添加しない溶液を空試験液とし,
試料溶液と併行して,6.4.4.2に従って発光強度を測定する。
6.4.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を6.4.4.1 a) によって行う場合
1) タングステン粉(6.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれ白金皿(75番又は
90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100
mL又は200 mL)に移し入れる。
2) 6.4.4.1のa) 2) 及びa) 3) の手順に従って操作した後,鉄標準液(Fe:50 μg/mL)(6.4.2.7)0〜6.0 mL
(鉄として0〜300 μg)を段階的に加え,水で標線までうすめる。
3) 以下,6.4.4.2の手順に従って操作した後,得た発光強度と鉄量との関係線を作成して検量線とする。
b) 試料溶液の調製を6.4.4.1 b) によって行う場合
1) タングステン粉(6.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 6.4.4.1のb) 2) 及びb) 3) の手順に従って操作した後,鉄標準液(Fe:50 μg/mL)(6.4.2.7)0〜6.0 mL
(鉄として0〜300 μg)を段階的に加え,水で標線までうすめる。
3) 以下,6.4.4.2の手順に従って操作した後,得た発光強度と鉄量との関係線を作成して検量線とする。
c) 試料溶液の調製を6.4.4.1 c) によって行う場合
1) タングステン粉(6.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 6.4.4.1のc) 2) 及びc) 3) の手順に従って操作した後,鉄標準液(Fe:50 μg/mL)(6.4.2.7)0〜6.0 mL
(鉄として0〜300 μg)を段階的に加え,水で標線までうすめる。
3) 以下,6.4.4.2の手順に従って操作した後,得た発光強度と鉄量との関係線を作成して検量線とする。
6.4.7
計算
6.4.4.2及び6.4.5で得た発光強度と,6.4.6で作成した検量線の鉄量とから鉄検出量を求め,試料中の鉄
含有率を次の式によって算出する。
(
)100
3
8
7
6
×
−
−
=
m
A
A
A
Fe
ここに,
Fe: 試料中の鉄含有率[%(質量分率)]
A6: 試料溶液中の鉄検出量(g)
9
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A7: 空試験液中の鉄検出量(g)
A8: タングステン粉(6.4.2.6)1.0 g中に含まれる鉄量(g)
m3: 試料はかりとり量(g)
7
モリブデン定量方法
7.1
定量方法の区分
モリブデンの定量方法は,次のいずれかによる。
a) チオシアン酸吸光光度法 この方法は,モリブデン含有率0.001 0 %(質量分率)以上0.016 %(質量
分率)以下の試料に適用する。
b) 原子吸光分析法 この方法は,モリブデン含有率0.001 0 %(質量分率)以上0.020 %(質量分率)以
下の試料に適用する。
c) ICP発光分光分析法 この方法は,モリブデン含有率0.001 0 %(質量分率)以上0.020 %(質量分率)
以下の試料に適用する。
7.2
チオシアン酸吸光光度法
7.2.1
要旨
試料を適切な試薬で分解し,水酸化ナトリウムを添加する。くえん酸を加えて,タングステンなどを錯
塩とし,塩酸を加えて酸性とした後,鉄(III),チオシアン酸カリウム,酢酸エチル及び塩化すず(II)を
加えて,モリブデン(VI)を還元するとともに生成したモリブデンのチオシアン酸錯体を酢酸エチルに抽
出し,分光光度計を用いてその吸光度を測定する。
7.2.2
試薬
試薬は,次による。
7.2.2.1
塩酸
7.2.2.2
硝酸
7.2.2.3
混酸A(硝酸1,ふっ化水素酸1)
7.2.2.4
水酸化ナトリウム溶液(100 g/L)
7.2.2.5
過酸化水素
7.2.2.6
タングステン粉 モリブデン含有率が既知で,かつ,そのモリブデン含有率が適用含有率範囲の
下限値[0.001 0 %(質量分率)]より低いもの。
7.2.2.7
塩化すず(II)溶液 塩化すず(II)二水和物150 gを塩酸100 mLに加熱して溶解し,室温まで
冷却した後,水250 mLを加える。小粒の粒状すず2,3個を加えて褐色ガラス製瓶に保存する。
7.2.2.8
鉄(III)溶液 硫酸アンモニウム鉄(III)・12水0.87 gを硫酸(0.1 mol/L)100 mLに溶解する。
この溶液1 mLは,鉄として約1 mgを含む。
7.2.2.9
チオシアン酸カリウム溶液 チオシアン酸カリウム150 gを水350 mLに溶解する。
7.2.2.10 くえん酸溶液 くえん酸一水和物500 gを水800 mLに溶解する。
7.2.2.11 酢酸エチル
7.2.2.12 モリブデン標準液(Mo:50 μg/mL) モリブデン標準液は,次のいずれかを用いる。
a) 市販のモリブデン標準液 市販のモリブデン標準液(Mo:1 000 μg/mL)は,酸濃度,安定剤の有無
などが使用目的に合致することを確認して用い,使用の都度,必要量だけ水で正確に20倍にうすめて
使用する。
注記 JCSSに基づくモリブデン標準液がある。
10
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 酸化物を用いて調製したモリブデン標準液 酸化モリブデン(VI)[純度99.5 %(質量分率)以上]
1.500 gをはかりとってビーカー(200 mL)に移し入れ,時計皿で覆い,水酸化ナトリウム溶液(100 g/L)
10 mLを加え,加熱して分解する。常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り
除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線までうすめて原液(Mo:1 000
μg/mL)とする。この原液を使用の都度,必要量だけ水で正確に20倍にうすめてモリブデン標準液と
する。
7.2.3
試料はかりとり量
試料はかりとり量は,1.0 gとし,10 mgの桁まではかる。
7.2.4
操作
7.2.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Aによる分解 混酸Aによる分解は,次による。
1) 試料をはかりとって白金皿(75番又は90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200
mL)又はポリエチレン製ビーカー(100 mL又は200 mL)に移し入れる。
2) 白金製蓋,四ふっ化エチレン樹脂製時計皿又はポリエチレン製時計皿で覆い,混酸A 6 mLを少量
ずつ加え,穏やかに加熱して分解する。蓋又は時計皿の下面を水で洗浄して,蓋又は時計皿を取り
除き,引き続き加熱して乾固寸前まで濃縮する。さらに,硝酸約1 mLを加え,再び乾固し室温ま
で冷却する。水酸化ナトリウム溶液(100 g/L)15 mLを加え,蓋又は時計皿で覆い加熱して塩類を
溶解する。
なお,ポリエチレン製ビーカーを用いる場合は,加熱による容器の変形が起こらないように,水
浴上又は水浴中で加熱しながら分解する。
3) 常温まで冷却した後,蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,溶液を100 mL
の全量フラスコに水を用いて移し入れ,水で標線までうすめる。
b) 過酸化水素による分解 過酸化水素による分解は,次による。
1) 試料をはかりとってビーカー(100 mL又は200 mL)に移し入れる。
2) 数mLの水で試料を湿らせ時計皿で覆い,過酸化水素約10 mLを少量ずつ加え,放置又は加熱して
分解する。分解が不十分な場合は,更に過酸化水素を少量ずつ追加し,分解する。
3) 室温まで冷却した後,水酸化ナトリウム溶液(100 g/L)15 mLを加える。激しい発泡が終わった後,
液量が10〜20 mLになるまで煮沸し,過剰の過酸化水素を分解する。
4) 常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100 mLの全量フラ
スコに水を用いて移し入れ,水で標線までうすめる。
7.2.4.2
抽出
抽出は,次の手順によって行う。
a) 7.2.4.1のa) 3) 又はb) 4) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mLは捨て,
その後の50 mLを分液漏斗(200 mL)に分取する。
b) くえん酸溶液(7.2.2.10)14 mL,塩酸6 mL及び鉄(III)溶液(7.2.2.8)1 mLを加え常温まで冷却し
た後,チオシアン酸カリウム溶液(7.2.2.9)4 mL,酢酸エチル15 mL及び塩化すず(II)溶液(7.2.2.7)
4 mLを順次加え,約1分間激しく振り混ぜる。2分間以上放置した後,下層の水相を捨てる。
7.2.4.3
吸光度の測定
7.2.4.2 b) で得た有機相の一部を,乾いたろ紙(5種A)を用いてろ過し,最初の数 mLは捨て,次のろ
11
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
液を分光光度計の吸収セル(10 mm)に採り,酢酸エチルを対照液として波長500 nm付近の吸光度を測定
する。
なお,吸光度の測定は,60分間以内に行う。
7.2.5
空試験
空試験は,行わない。
7.2.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を7.2.4.1 a) によって行う場合
1) タングステン粉(7.2.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれ白金皿(75番又は
90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100
mL又は200 mL)に移し入れ,7.2.4.1 a) 2) の操作を行う。
2) 常温まで冷却した後,蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,溶液を100 mL
の全量フラスコに水を用いて移し入れる。
3) モリブデン標準液(Mo:50 μg/mL)(7.2.2.12)0〜3.2 mL(モリブデンとして0〜160 μg)を段階的
に加え,水で標線までうすめる。これらの溶液を50 mLずつ分取し,それぞれを分液漏斗(200 mL)
に移し入れ,以下,7.2.4.2 b) 及び7.2.4.3の手順に従って操作する。
4) 得た吸光度とモリブデン標準液として加えたモリブデン量の1/2の量との関係線を作成し,この関
係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を7.2.4.1 b) によって行う場合
1) タングステン粉(7.2.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れ,7.2.4.1 b) 2) の操作を行う。
2) 常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100 mLの全量フラ
スコに水を用いて移し入れる。
3) モリブデン標準液(Mo:50 μg/mL)(7.2.2.12)0〜3.2 mL(モリブデンとして0〜160 μg)を段階的
に加え,水で標線までうすめる。これらの溶液を50 mLずつ分取し,それぞれを分液漏斗(200 mL)
に移し入れ,以下,7.2.4.2 b) 及び7.2.4.3の手順に従って操作する。
4) 得た吸光度とモリブデン標準液として加えたモリブデン量の1/2の量との関係線を作成し,この関
係線を原点を通るように平行移動して検量線とする。
7.2.7
計算
7.2.4.3で得た吸光度と7.2.6で作成した検量線のモリブデン量とからモリブデン検出量を求め,試料中の
モリブデン含有率を,次の式によって算出する。
100
2
1
4
9
×
×
=
m
A
Mo
ここに,
Mo: 試料中のモリブデン含有率[%(質量分率)]
A9: 分取した試料溶液中のモリブデン検出量(g)
m4: 試料はかりとり量(g)
7.3
原子吸光分析法
7.3.1
要旨
試料を適切な試薬で分解した後,試料溶液を原子吸光分析装置のアセチレン・一酸化二窒素フレーム中
12
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に噴霧し,その吸光度を測定する。
7.3.2
試薬
試薬は,次による。
7.3.2.1
ほう酸溶液(50 g/L)
7.3.2.2
りん酸(1+1)
7.3.2.3
混酸A(硝酸1,ふっ化水素酸1)
7.3.2.4
過酸化水素
7.3.2.5
タングステン粉 7.2.2.6による。
7.3.2.6
モリブデン標準液(Mo:50 μg/mL)7.2.2.12による。
7.3.3
試料はかりとり量
試料はかりとり量は,3.0 gとし,10 mgの桁まではかる。
7.3.4
操作
7.3.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Aによる分解 混酸Aによる分解は,6.3.4.1 a) による。
b) 過酸化水素による分解 過酸化水素による分解は,6.3.4.1 b) による。
7.3.4.2
吸光度の測定
7.3.4.1のa) 又はb) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mLは捨て,その
後の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・一酸化二窒素フレーム中に噴
霧し,波長313.3 nmにおける吸光度を測定する。
7.3.5
空試験
7.3.6の検量線の作成操作において得られるモリブデン標準液を添加しない溶液を空試験液とし,試料溶
液と併行して,7.3.4.2に従って吸光度を測定する。
7.3.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を7.3.4.1 a) によって行う場合
1) タングステン粉(7.3.2.5)を3.0 gずつ10 mgの桁まで数個はかりとり,それぞれ白金皿(75番又は
90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100
mL又は200 mL)に移し入れる。
2) 6.3.4.1 a) 2) の操作を試料と併行して行った後,溶液を100 mLの全量フラスコに水を用いて移し入
れる。
3) モリブデン標準液(Mo:50 μg/mL)(7.3.2.6)0〜12.0 mL(モリブデンとして0〜600 μg)を段階的
に加え,水で標線までうすめる。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・一酸化二窒
素フレーム中に噴霧し,波長313.3 nmにおける吸光度を試料溶液と併行して測定し,得た吸光度と
モリブデン量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を7.3.4.1 b) によって行う場合
1) タングステン粉(7.3.2.5)を3.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 6.3.4.1 b) 2) の操作を試料と併行して行った後,溶液を100 mLの全量フラスコに水を用いて移し入
13
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
れる。
3) モリブデン標準液(Mo:50 μg/mL)(7.3.2.6)0〜12.0 mL(モリブデンとして0〜600 μg)を段階的
に加え,水で標線までうすめる。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光分析装置のアセチレン・一酸化二窒
素フレーム中に噴霧し,波長313.3 nmにおける吸光度を試料溶液と併行して測定し,得た吸光度と
モリブデン量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7.3.7
計算
7.3.4.2及び7.3.5で得た吸光度と,7.3.6で作成した検量線のモリブデン量とからモリブデン検出量を求
め,試料中のモリブデン含有率を次の式によって算出する。
(
)
100
5
12
11
10
×
−
−
=
m
A
A
A
Mo
ここに,
Mo: 試料中のモリブデン含有率[%(質量分率)]
A10: 試料溶液中のモリブデン検出量(g)
A11: 空試験液中のモリブデン検出量(g)
A12: タングステン粉(7.3.2.5)3.0 g中に含まれるモリブデン
量(g)
m5: 試料はかりとり量(g)
7.4
ICP発光分光分析法
7.4.1
要旨
試料を適切な試薬で分解した後,試料溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
7.4.2
試薬
試薬は,次による。
7.4.2.1
ほう酸溶液(50 g/L)
7.4.2.2
りん酸(1+1)
7.4.2.3
混酸B(硝酸1,ふっ化水素酸1,水3)
7.4.2.4
混酸C(塩酸3,硝酸1,水4)
7.4.2.5
過酸化水素
7.4.2.6
タングステン粉 7.2.2.6による。
7.4.2.7
モリブデン標準液(Mo:50 μg/mL) 7.2.2.12による。
7.4.3
試料はかりとり量
試料はかりとり量は,1.0 gとし,10 mgの桁まではかる。
7.4.4
操作
7.4.4.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸Bによる分解 混酸Bによる分解は,6.4.4.1 a) による。
b) 過酸化水素による分解 過酸化水素による分解は,6.4.4.1 b) による。
c) 混酸Cによる分解 混酸Cによる分解は,6.4.4.1 c) による。
7.4.4.2
発光強度の測定
7.4.4.1のa) ,b) 又はc) で得た溶液を,乾いたろ紙(5種C)を用いてろ過し,最初の10 mLは捨て,
その後の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長202.030 nm又は277.540 nm
14
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
における発光強度を測定する。
なお,精確さを確認してあれば,他の波長を用いて測定してもよい。また,高次のスペクトル線が使用
可能な装置では,高次のスペクトル線を用いてもよい。バックグラウンド補正機構が付いている装置では,
バックグラウンド補正機構を用いてもよい。これらの操作を適用した場合は,検量線の作成においても同
様に行う。
7.4.5
空試験
7.4.6の検量線の作成操作において得られるモリブデン標準液を添加しない溶液を空試験液とし,試料溶
液と併行して,7.4.4.2に従って発光強度を測定する。
7.4.6
検量線の作成
検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を7.4.4.1 a) によって行う場合
1) タングステン粉(7.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれ白金皿(75番又は
90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)又はポリエチレン製ビーカー(100
mL又は200 mL)に移し入れる。
2) 7.4.4.1 a) の手順に従って操作した後,モリブデン標準液(Mo:50 μg/mL)(7.4.2.7)0〜4.0 mL(モ
リブデンとして0〜200 μg)を段階的に加え,水で標線までうすめる。
3) 以下,7.4.4.2の手順に従って操作した後,得た発光強度とモリブデン量との関係線を作成して検量
線とする。
b) 試料溶液の調製を7.4.4.1 b) によって行う場合
1) タングステン粉(7.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 7.4.4.1 b) の手順に従って操作した後,モリブデン標準液(Mo:50 μg/mL)(7.4.2.7)0〜4.0 mL(モ
リブデンとして0〜200 μg)を段階的に加え,水で標線までうすめる。
3) 以下,7.4.4.2の手順に従って操作した後,得た発光強度とモリブデン量との関係線を作成して検量
線とする。
c) 試料溶液の調製を7.4.4.1 c) によって行う場合
1) タングステン粉(7.4.2.6)を1.0 gずつ10 mgの桁まで数個はかりとり,それぞれビーカー(100 mL
又は200 mL)に移し入れる。
2) 7.4.4.1 c) の手順に従って操作した後,モリブデン標準液(Mo:50 μg/mL)(7.4.2.7)0〜4.0 mL(モ
リブデンとして0〜200 μg)を段階的に加え,水で標線までうすめる。
3) 以下,7.4.4.2の手順に従って操作した後,得た発光強度とモリブデン量との関係線を作成して検量
線とする。
7.4.7
計算
7.4.4.2及び7.4.5で得た発光強度と,7.4.6で作成した検量線のモリブデン量とからモリブデン検出量を
求め,試料中のモリブデン含有率を次の式によって算出する。
(
)100
6
15
14
13
×
−
−
=
m
A
A
A
Mo
ここに,
Mo: 試料中のモリブデン含有率[%(質量分率)]
A13: 試料溶液中のモリブデン検出量(g)
A14: 空試験液中のモリブデン検出量(g)
15
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A15: タングステン粉(7.4.2.6)1.0 g中に含まれるモリブデン
量(g)
m6: 試料はかりとり量(g)
8
酸化トリウム(IV)定量方法
8.1
定量方法の区分
酸化トリウム(IV)の定量方法は,次のいずれかによる。
a) 塩化水素ガス揮散重量法 この方法は,酸化トリウム(IV)含有率0.10 %(質量分率)以上の試料に
適用する。
b) 水酸化物沈殿分離重量法 この方法は,酸化トリウム(IV)含有率0.10 %(質量分率)以上の試料に
適用する。
8.2
塩化水素ガス揮散重量法
8.2.1
要旨
試料を加熱して酸化物にし,塩化水素ガス及び酸素(又は圧縮空気)を通じて揮発分を揮散させ,残分
となる酸化トリウム(IV)の質量をはかる。
8.2.2
試薬
試薬は,次による。
8.2.2.1
塩酸
8.2.2.2
硫酸
8.2.2.3
液化塩化水素(必要な場合に用いる。)
8.2.2.4
酸素(又は圧縮空気)
8.2.3
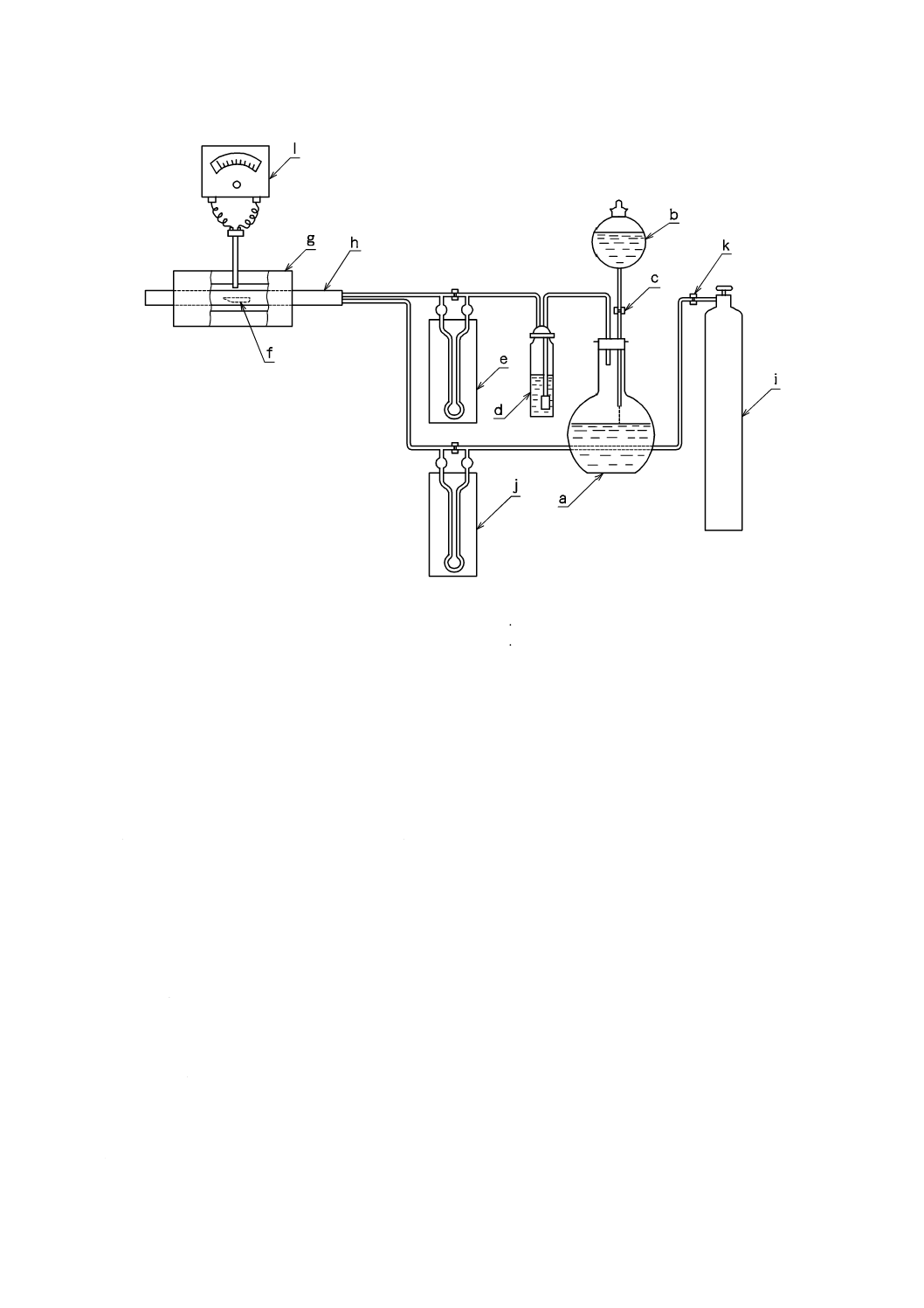
装置 酸化トリウム(IV)定量装置は,通常,図1に示すものを用いる。
酸化トリウム(IV)定量装置は,塩化水素発生部及び反応部からなる。
a) 塩化水素発生部 塩化水素発生部は,塩酸を入れた塩化水素発生用フラスコ(a),塩化水素を発生さ
せるために滴下する硫酸を入れた硫酸滴下漏斗(b),発生した塩化水素を清浄にするための硫酸を入
れた塩化水素洗浄瓶(d)及び塩化水素流量計(e)からなる。または,液化塩化水素ボンベを用いて
もよい。
b) 反応部 反応部は,電気抵抗加熱炉(g),反応管(h)及び熱電温度計(l)からなり,炉の中央部に
おいて約950 ℃の温度を保つことのできるもので,反応管(h)の加熱中央部の温度を熱電温度計(l)
を用いて測定できるもの。
16
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a 塩化水素発生用フラスコ
h 反応管(石英ガラス製)
b 硫酸滴下漏斗
i 酸素(又は圧縮空気)ボンベ
c 硫酸滴下量調整コック
j 酸素(又は圧縮空気)流量計
d 塩化水素洗浄瓶
k 酸素(又は圧縮空気)流量調整コック
e 塩化水素流量計
l 熱電温度計
f 白金ボート又は石英ガラス製ボート
g 電気抵抗加熱炉
図1−酸化トリウム(IV)定量装置の例
8.2.4
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
8.2.5
操作
8.2.5.1
準備操作
酸化トリウム(IV)定量装置(8.2.3)の電気抵抗加熱炉(g)に通電して反応管(h)を加熱し,管内温
度を750〜800 ℃に保持する。
なお,熱電温度計(l)の指示値は,一般的に管内温度と異なるので,あらかじめその差を求めておき,
その差を補正して熱電温度計の指示値を設定する。
8.2.5.2
試料の酸化
試料をはかりとって,白金ボート又は石英ガラス製ボート(f)に移し入れ,8.2.5.1で昇温した反応管(h)
の中央部に挿入し,約1時間加熱して酸化する。
8.2.5.3
揮発分の揮発
揮発分の揮発は,次の手順によって行う。
a) 塩化水素発生部[8.2.3 a)]と反応管(h)とを接続し,塩化水素発生用フラスコ(a)の塩酸中に硫酸
17
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
滴下漏斗(b)の硫酸滴下量調整コック(c)を開いて硫酸を滴下し,塩化水素を発生させ塩化水素流
量計(e)によって反応管(h)の断面積(cm2)当たり20〜40 mL/分になるように硫酸滴下量調整コ
ック(c)を調節する。また,酸素(又は圧縮空気)が,酸素(又は圧縮空気)流量計(j)によって
反応管(h)の断面積(cm2)当たり3〜6 mL/分になるように酸素(又は圧縮空気)流量調整コック
(k)を調節する。
なお,塩化水素発生部に液化塩化水素ボンベを用いる場合には,ボンベと塩化水素流量計(e)とを
接続し,反応管(h)の断面積(cm2)当たり20〜40 mL/分になるように塩化水素流量を調整する。
注記 内径35 mmの反応管を使用した場合,塩化水素の流量は,200〜400 mL/分,酸素(又は圧
縮空気)の流量は,30〜60 mL/分となる。
b) 塩化水素を流してから2〜2.5時間経過して酸化タングステン(VI)による黄色が認められないことを
確認し,揮発が完結したことを確かめた後,管内温度を上昇させ,900〜950 ℃に保持し約2時間加熱
を続けた後,白金ボート又は石英ガラス製ボート(f)を取り出し,デシケーターに移し入れ常温まで
放冷する。
8.2.5.4
ひょう量
ひょう量は,次の手順によって行う。
a) 8.2.5.3 b) で常温まで放冷した白金ボート又は石英ガラス製ボートの質量を0.1 mgの桁まではかる。
b) 白金ボート又は石英ガラス製ボートの内容物をはけなどで払い出した後,白金ボート又は石英ガラス
製ボートの質量を0.1 mgの桁まではかる。
8.2.6
空試験
空試験は,行わない。
8.2.7
計算
試料中の酸化トリウム(IV)含有率を,次の式によって算出する。
100
7
9
8
×
−
=
m
m
m
ThO2
ここに,
ThO2: 試料中の酸化トリウム(IV)含有率[%(質量分率)]
m8: 8.2.5.4 a) で得た質量(g)
m9: 8.2.5.4 b) で得た質量(g)
m7: 試料はかりとり量(g)
8.3
水酸化物沈殿分離重量法
8.3.1
要旨
試料をふっ化水素酸及び硝酸で分解し,水酸化ナトリウムでアルカリ性として水酸化トリウム(IV)を
沈殿させ,こし分けて強熱した後,二硫酸カリウムで融解し,熱水で融成物を溶解する。次に,アンモニ
ア水でアルカリ性として水酸化トリウム(IV)を沈殿させ,こし分けて塩酸で溶解した後,しゅう酸を加
えてしゅう酸トリウム(IV)を沈殿させ,強熱して酸化トリウム(IV)とし,その質量をはかる。
8.3.2
試薬
試薬は,次による。
8.3.2.1
塩酸(1+2)
8.3.2.2
混酸D(硝酸1,ふっ化水素酸1,水1)
8.3.2.3
アンモニア水
8.3.2.4
アンモニア水(1+49)
18
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.3.2.5
水酸化ナトリウム溶液(200 g/L)
8.3.2.6
二硫酸カリウム
8.3.2.7
しゅう酸二水和物
8.3.2.8
しゅう酸溶液 しゅう酸二水和物5 gを水約150 mLに溶解し,塩酸10 mLを加えて,水で250 mL
にうすめる。
8.3.3
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
8.3.4
操作
8.3.4.1
準備操作
白金るつぼ(20番又は30番)を8.3.4.4 b) の手順に従って処理して恒量(強熱前後の質量差が0.3 mg
以下)とした後,その質量を0.1 mgの桁まではかる。
8.3.4.2
試料の分解及び分離
試料の分解及び分離は,次の手順によって行う。
a) 試料をはかりとって白金皿(75番又は90番),四ふっ化エチレン樹脂製ビーカー(100 mL又は200 mL)
又はポリエチレン製ビーカー(100 mL又は200 mL)に移し入れる。
b) 白金製蓋,四ふっ化エチレン樹脂製時計皿又はポリエチレン製時計皿で覆い,混酸D 15 mLを少量ず
つ加え,穏やかに加熱して分解する。引き続き加熱して窒素酸化物を追い出す。
なお,ポリエチレン製ビーカーを用いる場合は,加熱による容器の変形が起こらないように,水浴
上又は水浴中で加熱しながら分解する。
c) 室温まで冷却した後,蓋又は時計皿の下面を水で洗浄して蓋又は時計皿を取り除き,加熱して酸化タ
ングステン(VI)一水和物が僅かに析出する程度(3〜5 mL)に濃縮する。
d) この溶液に水10 mL,次に水酸化ナトリウム溶液(200 g/L)30 mLを加え,白金棒又はプラスチック
棒でかき混ぜ,約10分間加熱する。
e) ろ紙(6種)でこし分け,沈殿を熱水で洗浄する。沈殿はろ紙とともに白金るつぼ(20番又は30番)
に移し入れ,徐々に加熱して乾燥し,ろ紙が炭化してから注意して強熱して灰化する。
f)
二硫酸カリウム3 gを加え蓋で覆い,初めは低温で加熱し,次第に温度を上げて融解し放冷する。白
金るつぼ及びその蓋をビーカー(200 mL)に入れ,熱水約50 mLを加え,白金るつぼ中の融成物を完
全に溶解し,白金るつぼ及びその蓋を水で洗浄して取り除き,水で液量を約100 mLとする。
8.3.4.3
沈殿の生成
沈殿の生成は,次の手順によって行う。
a) 8.3.4.2 f) で得た溶液にアンモニア水を加えて微アルカリ性とし,2〜3分間煮沸した後,沈殿をろ紙(5
種B)でこし分け,40〜60 ℃に温めたアンモニア水(1+49)で洗浄する。元のビーカーに塩酸(1
+2)30 mLを加えて加熱した後,ろ紙上に注いで沈殿を溶解し,温水で洗浄する。溶液は,ビーカー
(200 mL)に受け,水で約80 mLにうすめる。
b) この溶液にしゅう酸二水和物5 gを加え,水浴上で1〜2時間加熱した後,約12時間静置してしゅう
酸トリウム(IV)粒子を熟成させる。沈殿をろ紙(5種C)でこし分け,しゅう酸溶液(8.3.2.8)で数
回洗浄する。
8.3.4.4
灰化及びひょう量
灰化及びひょう量は,次の手順によって行う。
a) 8.3.4.3 b) で得た沈殿を,ろ紙とともに8.3.4.1で恒量とした白金るつぼ(20番又は30番)に移し入れ,
19
H 1405:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
徐々に加熱して乾燥し,ろ紙が炭化してから注意して強熱して灰化する。
b) 約1 000 ℃で約1時間加熱した後,デシケーター中で常温まで放冷し,その質量を0.1 mgの桁までは
かる。この操作を恒量になるまで繰り返す。
8.3.5
空試験
空試験は,行わない。
8.3.6
計算
試料中の酸化トリウム(IV)含有率を,次の式によって算出する。
100
10
12
11
×
−
=
m
m
m
ThO2
ここに,
ThO2: 試料中の酸化トリウム(IV)含有率[%(質量分率)]
m11: 8.3.4.4 b) で得た質量(g)
m12: 8.3.4.1で得た質量(g)
m10: 試料はかりとり量(g)