1
H
1
2
8
3
:
1
9
9
8
解
説
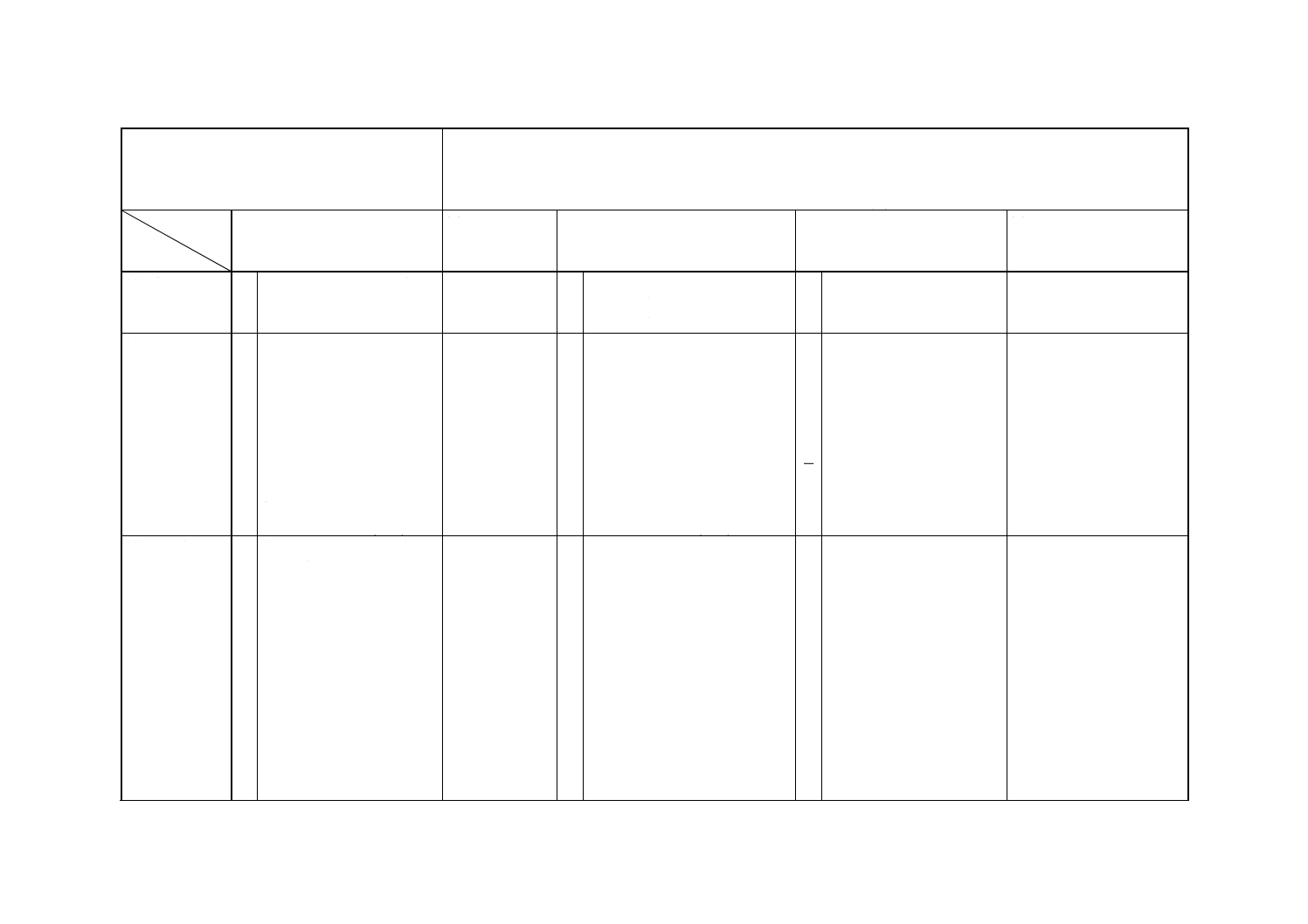
解説表1 JISと対応する国際規格との対比表
JIS H 1283 : 1999 ニッケル及びニッケル合金中の
コバルト定量方法
ISO 6351 : 1985 ニッケル−銀,ビスマス,カドミウム,コバルト,銅,鉄,マンガン,鉛及び亜鉛含有量の定量
ISO 7530-1 : 1990 ニッケル合金−フレーム原子吸光分析方法 Part 1:一般要求事項及び試料溶解
ISO 7530-2:1990 ニッケル合金−フレーム原子吸光分析方法 Part 2:コバルト含有量の定量
ISO 9389 : 1989 ニッケル合金−コバルト含有量の定量−ヘキサシアノ鉄(III)酸カリウム電位差滴定法
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
(1) 適用範囲
○ ニッケル及びニッケル合金 ISO 6351
ISO 7530-2
ISO 9389
○
○
○
ニッケル
ニッケル合金
ニッケル合金
≡
(2) 定量方法の
区分
○ a) 銅分離ニトロソR塩吸光
光度法
0.02〜1.0% (m/m)
○ b) ヘキサシアノ鉄(III)酸カ
リウム電位差滴定法
2〜25% (m/m)
ISO 9389
○ ヘキサシアノ鉄(III)酸カリウム電
位差滴定法
2〜25% (m/m)
≡
○ c) 原子吸光A法
0.01〜2.0% (m/m)
ISO 6351
○ フレーム原子吸光法
0.01〜2.0% (m/m)
≡
○ d) 原子吸光B法
0.01〜4.0% (m/m)
ISO 7530-2
○ フレーム原子吸光法
0.01〜4.0% (m/m)
≡
(3) 分析方法
○ a) 銅分離ニトロソR塩吸光
光度法
(試料を硝酸と硫酸との混
酸で分解し,銅を電解して分
離した後,溶液を蒸発乾固す
る。水と硫酸を加えて可溶性
塩類を溶解し,酢酸ナトリウ
ムとニトロソR塩を加え,煮
沸してコバルトを呈色させ
た後,硝酸を加えて煮沸す
る。冷却した後,光度計を用
いて,その吸光度を測定す
る。)
2
H
1
2
8
3
:
1
9
9
8
解
説
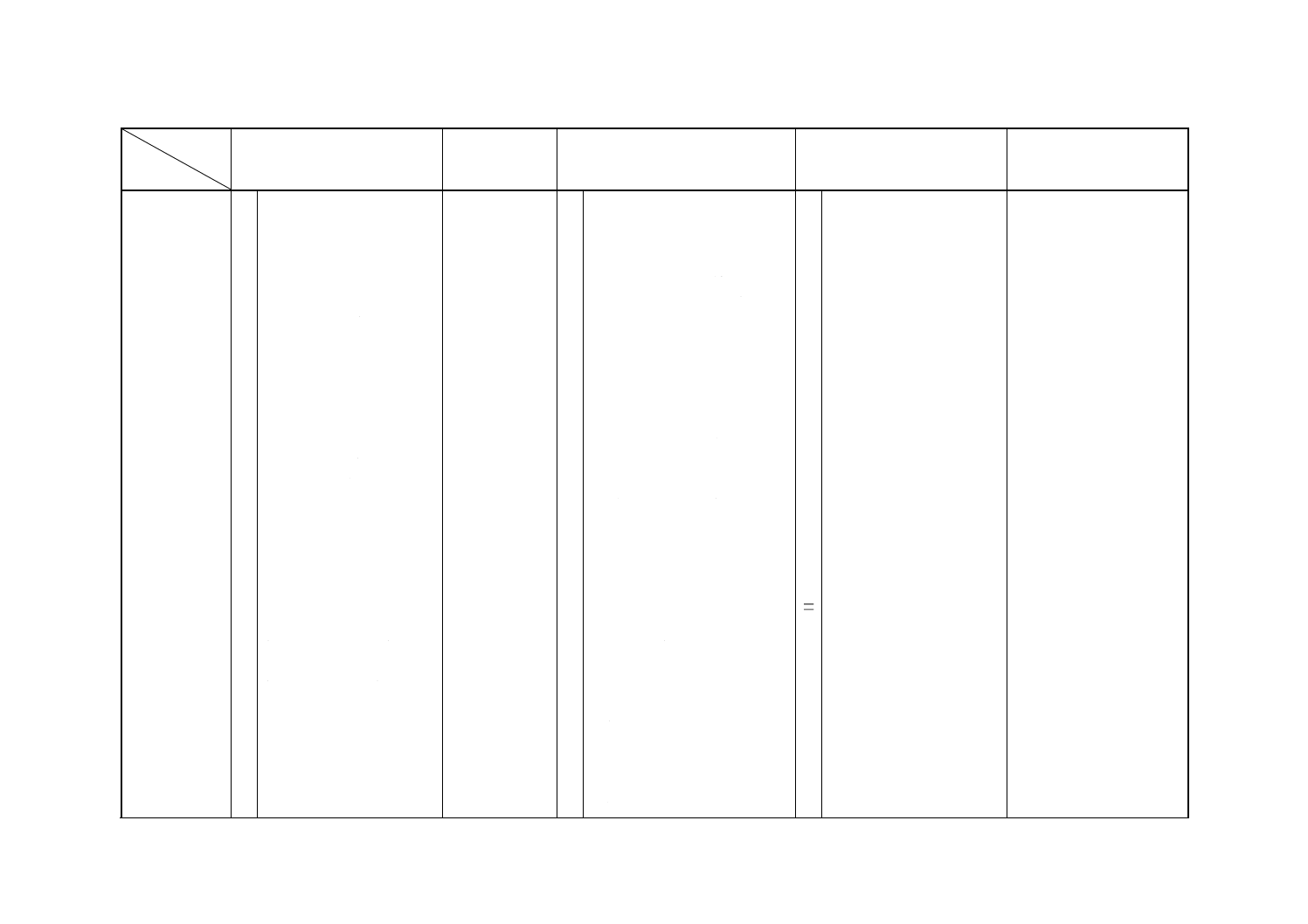
解説表1 JISと対応する国際規格との対比表(続き)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
(3) 分析方法
○ b) ヘキサシアノ鉄(III)酸カ
リウム電位差滴定法
[試料を硝酸と塩酸との混
酸で分解し,過塩素酸を加
え,白煙が発生するまで加熱
濃縮し,クロムを酸化した
後,マンガンの錯体を形成さ
せるために二りん酸ナトリ
ウムを加え,窒素ガスを通じ
て溶け込んでいる塩素や酸
素を除去する。試料溶液にく
えん酸アンモニウム溶液,硫
酸アンモニウム溶液,アンモ
ニア水及び一定量のヘキサ
シアノ鉄(III)酸カリウム標準
溶液を加え電位差滴定装置
を用いて,一定量のヘキサシ
アノ鉄(III)酸カリウム標準溶
液を標準コバルト溶液で滴
定する。]
ISO 9389
○ ヘキサシアノ鉄(III)酸カリウム電
位差滴定法
[試料を硝酸と塩酸との混酸で
分解し,過塩素酸を加え,白煙が
発生するまで加熱濃縮し,クロム
を酸化する。マンガンの錯体を形
成させるために二りん酸ナトリ
ウムを加え,窒素ガスを流して溶
け込んでいる塩素ガスや酸素を
除去する。試料溶液にくえん酸ア
ンモニウム溶液,硫酸アンモニウ
ム溶液,アンモニア水及び過剰の
ヘキサシアノ鉄(III)酸カリウム標
準溶液を加え,標準コバルト溶液
を用いて過剰のヘキサシアノ鉄
(III)酸カリウム標準溶液を電位差
滴定する。]
≡
○ c) 原子吸光A法
(試料を硝酸で分解した後,
溶液を原子吸光光度計の空
気・アセチレンフレーム中に
噴霧し,その吸光度を測定す
る。)
ISO 6351
○ フレーム原子吸光法
(試料を硝酸で分解し,過剰の窒
素酸化物を追い出した後,水を用
いて定容する。溶液を原子吸光計
の空気・アセチレンフレーム中に
噴霧する。元素のスペクトルの共
鳴線エネルギーの吸光度を測定
し,マトリックスのニッケルを試
料と同量共存させた定量元素の
検量用溶液の吸光度と比較す
る。)
≡
3
H
1
2
8
3
:
1
9
9
8
解
説
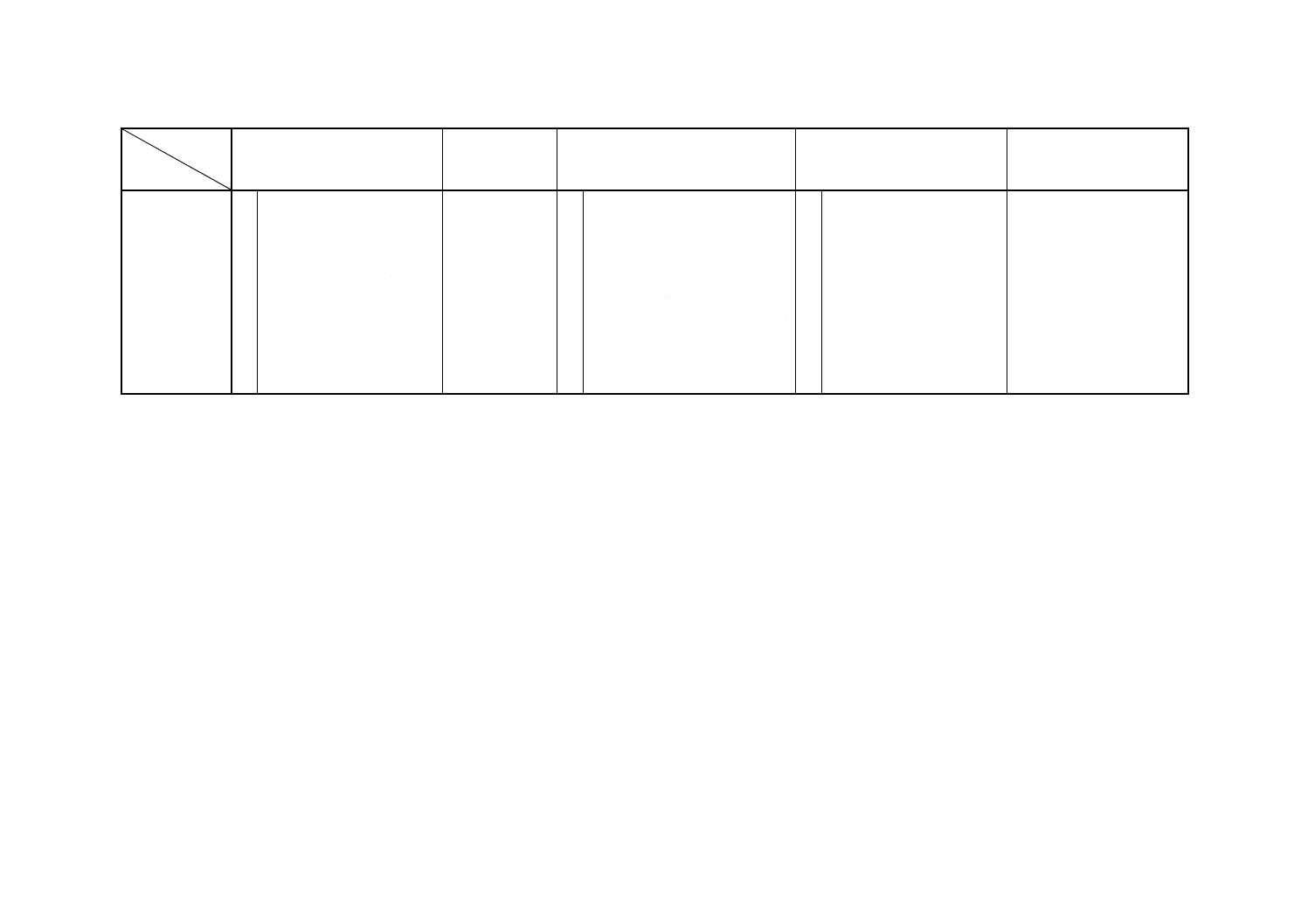
解説表1 JISと対応する国際規格との対比表(続き)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
(3) 分析方法
○ d) 原子吸光B法
(試料を塩酸と硝酸との混
酸で分解し,乾固近くまで加
熱する。塩酸を加え,乾固近
くまで加熱した後,塩酸及び
塩化ストロンチウムを加え
る。この溶液を原子吸光光度
計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を
測定する。)
ISO 7530-2
○ フレーム原子吸光法
(試料を酸で分解し,溶液を原子
吸光光度計の空気・アセチレンフ
レーム中に噴霧する。240.7nmの
波長におけるコバルトのスペク
トルの共鳴線エネルギーの吸光
度を測定し,検量用溶液の吸光度
と比較する。)
≡
備考1. 対比項目(I)及び(III)の小欄で,“○”は該当する項目を規定している場合を示す。
2. 対比項目(IV)の小欄の記号の意味は,次による。
“≡”:JISと国際規格との技術的内容は,同等である。