1
H
1
1
5
1
:
1
9
9
9
解
説
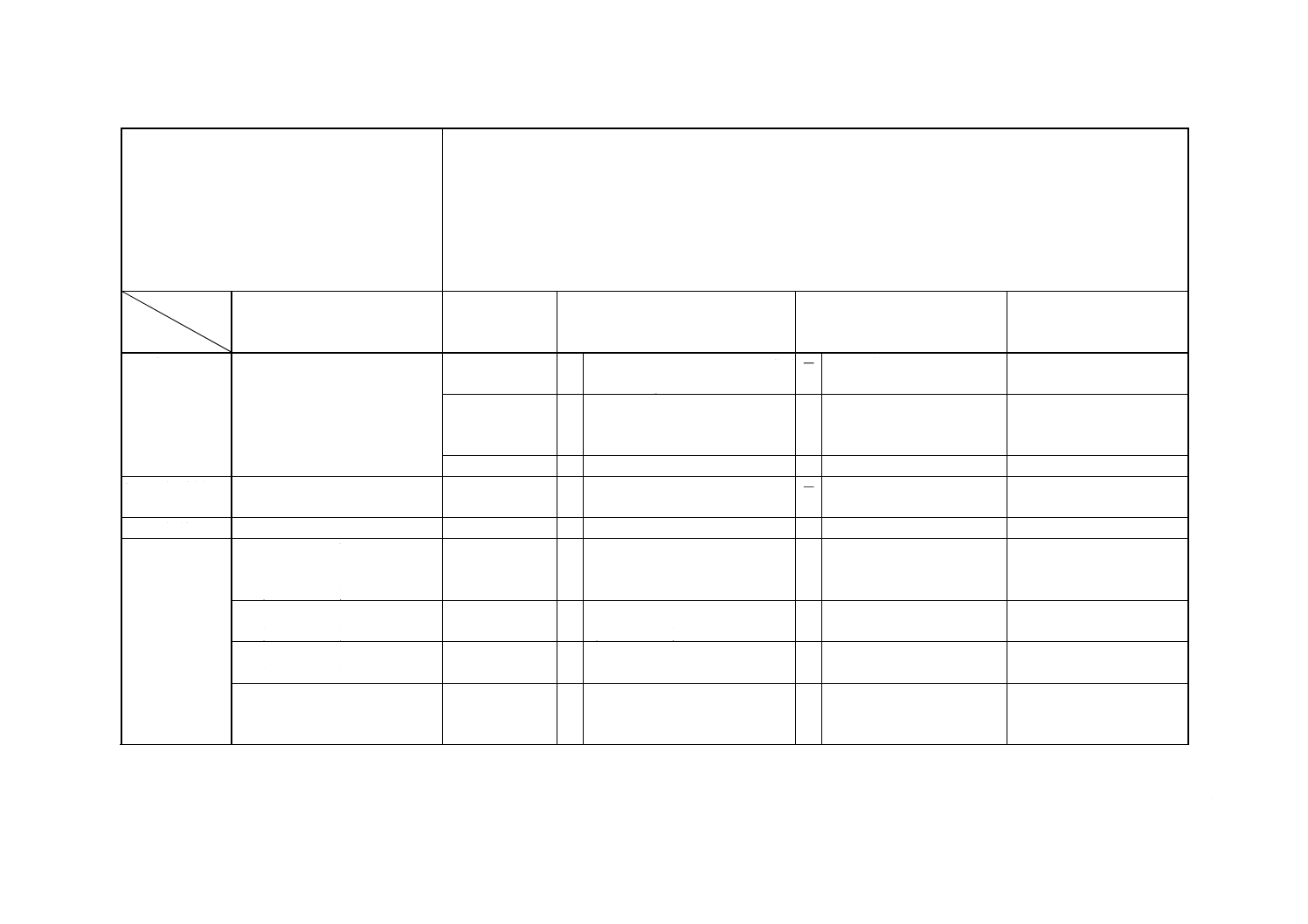
解説表2 JISと対応する国際規格との対比表
JIS H 1151 : 1999 ニッケル地金分析方法
ISO 6351 : 1985 (フレーム原子吸光法による銀,ビスマス,カドミウム,コバルト,銅,鉄,マンガン,鉛及び亜鉛
の定量)
ISO 7523 : 1985 (電気加熱原子吸光法による銀,ひ素,ビスマス,カドミウム,鉛,アンチモン,セレン,すず,テ
ルル及びタリウムの定量)
ISO 7524 : 1985 (燃焼−赤外線吸収法による炭素定量)
ISO 7525 : 1985 (硫化水素気化分離メチレンブルー−吸光光度法による硫黄定量)
ISO 7526 : 1985 (燃焼−赤外線吸収法による硫黄定量)
ISO 7527 : 1985 (燃焼−よう素酸カリウム滴定法による硫黄定量)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
1. 適用範囲
JIS H 2104 : 1997 ニッケル地金 ISO 6351
ISO 7523
○ ISO
6283
Refind
nickel,wrought,cast nickel
= ISOは適用囲が広い。
規格体系の差。現行のままと
する。
ISO 7524
ISO 7526
ISO 7527
○ nickel,ferronickel
nickel alloy
= ISOは適用範囲が広い。
規格体系の差。現行のままと
する。
ISO 7525
○ Refind nickel
=
2. 分析試料
4. 分析試料の取り方及び取扱
い方
6規格
○ Sampling and samples
=
3. 分析値
5. 分析値のまとめ方
6規格
−
−
4. コバルトの
定量
6.2 チオシアン酸・トリオクチル
アミン抽出吸光光度法
(0.001〜0.4%)
−
−
−
6.3 フレーム原子吸光法
(0.001〜1.0%)
ISO 6351
○ フレーム原子吸光法
(0.001〜1.0%)
=
6.4 ICP発光分光分析法
(0.001〜0.5%)
−
−
−
6.5 陰イオン交換分離ICP発光分
光分析法
(0.0001〜0.05%)
−
−
−
2
H
1
1
5
1
:
1
9
9
9
解
説
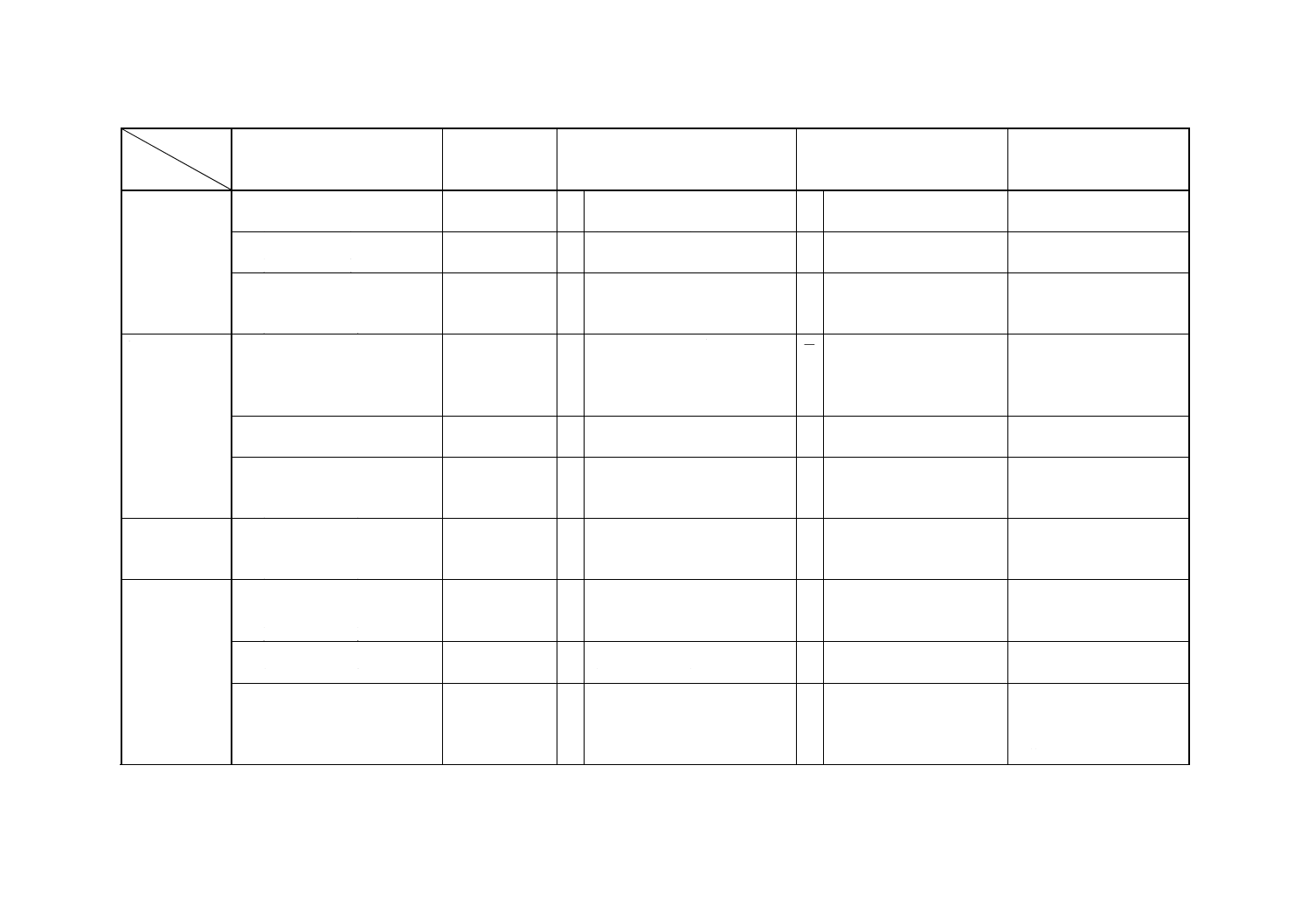
解説表2 JISと対応する国際規格との対比表(続き)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
5. 鉄の定量
7.2 フレーム原子吸光法
(0.000 5〜1.0%)
ISO 6351
○ フレーム原子吸光法
(0.002 5〜0.15%)
=
7.3 ICP発光分光分析法
(0.000 5〜1.0%)
−
−
−
7.4 陰イオン交換分離ICP発光分
光分析法
(0.000 1〜0.05%)
−
−
−
6. 銅の定量
8.2 フレーム原子吸光法
(0.000 5〜1.0%)
ISO 6351
○ フレーム原子吸光法
(0.000 5〜1.0%) (適用範囲では
0.000 2からとなっているが,
0.000 5からの操作しかない。)
=
8.3 ICP発光分光分析法
(0.000 5〜0.5%)
−
−
−
8.4 陰イオン交換分離ICP発光分
光分析法
(0.000 1〜0.05%)
−
−
−
7. 鉛の定量
9.2 水酸化鉄共沈分離フレーム
原子吸光法
(0.000 2〜0.01%)
−
−
−
9.3 水酸化鉄共沈分離ICP発光分
光分析法
(0.000 1〜0.01%)
−
−
−
9.4 フレーム原子吸光法
(0.000 5〜0.01%)
ISO 6351
○ フレーム原子吸光法
(0.000 5〜0.01%)
=
9.5 電気加熱原子吸光法
(0.000 1〜0.001%)
ISO 7523
○ 電気加熱原子吸光法
(0.000 01〜0.001%)
≠ ISOは適用濃度下限が低い。 ISOでは四塩化炭素を使っ
てニッケルを精製している
が,JISではそれを使わない
下限0.000 1とする。
3
H
1
1
5
1
:
1
9
9
9
解
説
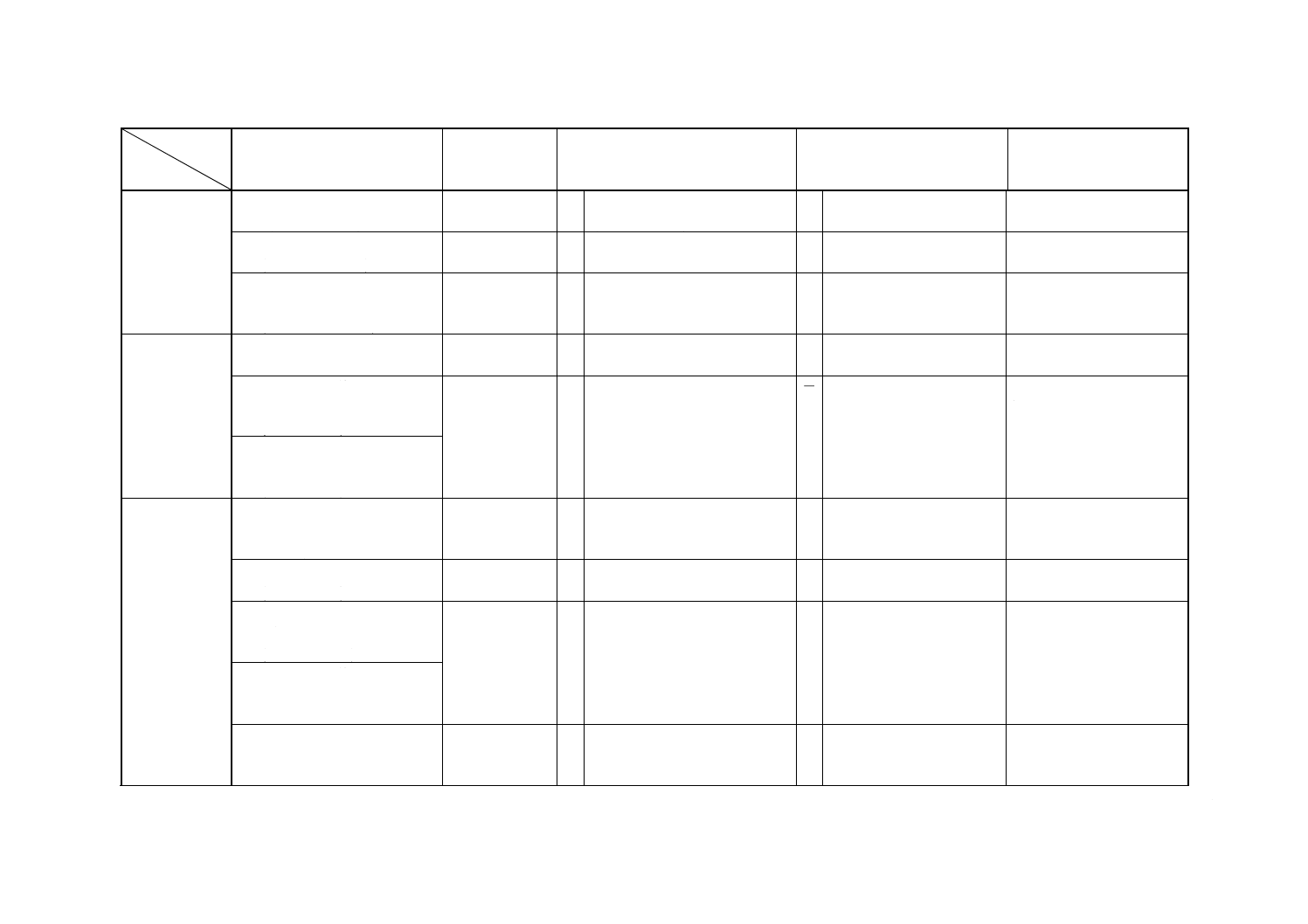
解説表2 JISと対応する国際規格との対比表(続き)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合
が困難な理由及び今後の
対策
8. マンガンの
定量
10.2 フレーム原子吸光法
(0.000 1〜0.2%)
ISO 6351
フレーム原子吸光法
(0.000 5〜0.2%)
=
10.3 ICP発光分光分析法
(0.000 1〜0.004%)
−
−
−
10.4 水酸化鉄共沈分離ICP発光分
光分析法
(0.000 05〜0.004%)
−
−
−
9. 炭素の定量 11.2 燃焼−電量法
(0.001〜0.1%)
−
−
11.3 燃焼−赤外線吸収法(積分
法)
(0.001〜0.1%)
ISO 7524
○ 燃焼−赤外線吸収法
(0.001〜2.0%)
= ISOは適用濃度上限が高い。 ISOはフェロニッケルも対
象としているため。
11.4 燃焼−赤外線吸収法(循環
法)
(0.001〜0.1%)
10. 硫黄の定量 12.2 硫化水素気化分離メチレン
ブルー吸光光度法 (0.0001〜
0.002%)
ISO 7525
○ 硫化水素気化分離メチレンブル
ー吸光光度法 (0.000 1〜0.002%)
=
12.3 燃焼−電量法
(0.001〜0.1%)
−
−
−
12.4 燃焼−赤外線吸収法(秘分
法)
(0.000 1〜0.1%)
ISO 7526
○ 燃焼−赤外線吸収法
(0.001〜0.3%)
= ISOは適用濃度上限が高い。 ISOはフェロニッケルも対
象としているため。
12.5 燃焼−赤外線吸収法(循環
法)
(0.001〜0.1%)
12.6 燃焼−よう素酸カリウム滴
定法
(0.001〜0.3%)
ISO 7527
○ 燃焼−よう素酸カリウム滴定法
(0.001〜0.3%)
=

4
H
1
1
5
1
:
1
9
9
9
解
説
解説表2 JISと対応する国際規格との対比表(続き)
対比項目
規定項目
(I) JISの規定内容
(II) 国際規格番
号
(III) 国際規格の規定内容
(IV) JISと国際規格との相違点
(V) JISと国際規格との整合が
困難な理由及び今後の対
策
11. けい素の定
量
13.2 モリブドけい酸抽出吸光光
度法
(0.000 2〜0.01%)
−
−
−
13.3 モリブドけい酸抽出分離モ
リブドけい酸吸光光度法
(0.000 8〜0.01%)
−
−
−
13.4 ICP発光分光分析法
(0.000 2〜0.01%)
−
−
12. 精度
解説に記載
6規格
Precision
− ISOは技術的規定内容でな
いことからJISに規定してい
ない。
13. 報告書
−
6規格
Test report
− ISOは技術的規定内容でな
いことからJISに規定してい
ない。
備考1. 対比項目(III)の小欄で“○”は該当する項目を規定している場合,“−”は規定していない場合を示す。
2. 対比項目(IV)の小欄の記号の意味は,次のとおり。
“=”:JISと国際規格との技術的内容は同等である。ただし,軽微な技術上の差異がある。
“−”:該当項目がない場合。
“≠”:JISは,国際規格と技術的内容が同等でない。