2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1121-1995
鉛地金分析方法
Methods for chemical analysis of lead metal
1. 適用範囲 この規格は,JIS H 2105に規定する銀,銅,ビスマス,アンチモン,ひ素,すず,鉄及び
亜鉛の定量方法について規定する。
備考 この規格の引用規格を,次に示す。
JIS H 2105 鉛地金
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8012 亜鉛(試薬)
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116及びJIS K 0121によ
る。
3. 分析試料の採り方及び取扱い方
3.1
試料の採り方 試料の採り方は,次による。
(1) 鋳込試料又は製品試料から切粉を採るときは,削り取った試料がその鋳込試料又は製品試料の品質を
代表するように,採取する箇所は,試料の中央部,周辺に近い部分などとする。
(2) ボーリングによって切粉試料を採るときは,あらかじめドリルその他の工具類をエタノールなどを用
いて清浄にする。試料採取箇所を清浄にし,次に,油類その他の減磨剤を用いないで,切粉が酸化し
ない程度の力を与えて,試料面に直角にボーリングして貫通させる。
この際,ドリルの圧力及び回転数などを加減して,極端に発熱しないようにしなければいけないが,
冷却するために水などを注いではならない。
(3) 切粉試料は,その全部を集め,強力な磁石を用いて鉄粉などを除去した後,清浄なはさみなどを用い
て約5mm以下に切断し,よく混ぜ合わせて分析用試料とする。
(4) 分析用試料の採取と調製が,(1)〜(3)の規定によることができない場合には,受渡当事者間の協議によ
って定める。
3.2
試料の取扱い方 試料の取扱い方は,次による。
(1) 分析用試料は,異物などによる汚染を防止するため,適当なふた付きガラス容器などに入れ,密封し
2
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
て保存する。
(2) 分析用試料は,その表面に油などが付着しているおそれのあるときは,あらかじめエタノール,アセ
トンなどで洗浄して乾燥する。
(3) 分析用試料を鉄の定量に用いる場合には,あらかじめ次の操作を行う。
分析用試料の必要量をビーカーに取り,塩酸 (1+10) を試料片が沈む程度に加え,加熱して約5分
間煮沸するか,又は約80℃で約30分間加熱して,表面に付着又は混入した鉄分を溶解する。水で洗
浄した後,エタノール,アセトンで順次洗浄して乾燥する。
3.3
試料のはかり方 試料のはかり方は,次による。
(1) 分析試料のはかり取りに際しては,平均組成を代表するように注意しなければならない。
(2) 分析試料のはかり取りには,原則として化学はかりを用いる。
4. 分析値のまとめ方
4.1
分析回数 原則として,同一分析所において2回の繰返し分析を行う。
4.2
空試験 分析にあたっては,空試験を行い,測定値を補正する
4.3
分析値の表示 分析値は,質量百分率で表し,JIS H 2105に規定された数値の次の二けたまで算出
し,JIS Z 8401によってJIS H 2105に規定された数値の次の位に丸める。
5. 銀定量方法
5.1
定量方法の区分 銀の定量方法は,次のいずれかによる。
(1) 原子吸光法 この方法は,銀含有率0.000 2% (m/m) 以上0.004% (m/m) 以下の試料に適用する。
(2) 誘導結合プラズマ発光分光法 この方法は,銀含有率0.000 1% (m/m) 以上0.004% (m/m) 以下の試料
に適用する。
5.2
原子吸光法
5.2.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
5.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上で銀を含有しないもの,又は銀含有率が既知で,かつ,試料中の銀含有率より
低いもの。
(3) 酒石酸溶液 (500g/l)
(4) 標準銀溶液A (100μgAg/ml) 銀[99.99% (m/m) 以上]0.100gをはかり取り,ビーカー (300ml) に移
し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,加熱して分解する。常温まで冷却した後,時計皿
の下面を水で洗って時計皿を取り除く。溶液を1 000mlの褐色の全量フラスコに水を用いて移し入れ,
水で標線まで薄めて標準銀溶液Aとする。
(5) 標準銀溶液B (10μgAg/ml) 標準銀溶液A [(4)] を使用の都度,水で正しく10倍に薄めて標準銀溶液
Bとする。
5.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
5.2.4
操作
5.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり取って,ビーカー (300ml) に移し入れる。
3
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 時計皿で覆い,酒石酸溶液10ml及び硝酸 (1+4) 35mlを加え,穏やかに加熱して分解した後,煮沸し
て窒素酸化物などを追い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。
(3) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(1)。
注(1) この溶液を用いて,原子吸光法によって銅,ビスマス,アンチモン,鉄及び亜鉛を定量するこ
とができる。
5.2.4.2
吸光度の測定 5.2.4.1(3)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の
空気・アセチレンフレーム中に噴霧し,波長328.1nmにおける吸光度を測定する。
5.2.5
空試験 5.2.6の検量線の作成操作において得られる標準銀溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
5.2.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [5.2.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(2) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(3) 標準銀溶液A [5.2.2(4)] 及び標準銀溶液B [5.2.2(5)] の各種液量(銀として0〜200μg)を段階的に加え,
水で標線まで薄める。
(4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中
に噴霧し,波長328.1nmにおける吸光度を試料と並行して測定し,得た吸光度と銀量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
5.2.7
計算 5.2.4.2及び5.2.5で得た吸光度と5.2.6で作成した検量線とから銀量を求め,試料中の銀含有
率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Ag
ここに, Ag: 試料中の銀含有率% (m/m)
A1: 試料溶液中の銀検出量 (g)
A2: 空試験液中の銀検出量 (g)
A3: 鉛 [5.2.2(2)] 5.0g中に含まれる銀量 (g)
m: 試料はかり取り量 (g)
5.3
誘導結合プラズマ発光分光法
5.3.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を誘導結合プラズマ発光分光装置のアル
ゴンプラズマ中に噴霧し,その発光強度を測定する。
5.3.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 5.2.2(2)による
(3) 酒石酸溶液 (500g/l)
(4) 標準銀溶液A (100μgAg/ml) 5.2.2(4)による。
(5) 標準銀溶液B (10μgAg/ml) 5.2.2(5)による。
5.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
5.3.4
操作
5.3.4.1
試料溶液の調製 試料溶液の調製は,5.2.4.1による。
5.3.4.2
発光強度の測定 5.3.4.1で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズ
マ中に噴霧し,波長328.068nmにおける発光強度を測定する(2)。
注(2) 精度及び正確さを確認してあれば,他の波長を用いて測定してもよい。
4
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
また,高次のスペクトル線が使用可能な装置では高次のスペクトル線を用いてもよい。バッ
クグラウンド補正機能がついている装置では,バックグラウンド補正機能を用いてもよい。
5.3.5
空試験 5.3.6の検量線の作成操作において得られる標準銀溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
5.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [5.3.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(2) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(3) 標準銀溶液A [5.3.2(4)] 及び標準銀溶液B [5.3.2(5)] の各種液量(銀として0〜200μg)を段階的に加え,
水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
328.068nmにおける発光強度を試料と並行して測定し,得た発光強度と銀量との関係線を作成し,そ
の関係線を原点を通るように平行移動して検量線とする。
5.3.7
計算 5.3.4.2及び5.3.5で得た発光強度と5.3.6で作成した検量線とから銀量を求め,試料中の銀含
有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Ag
ここに, Ag: 試料中の銀含有率% (m/m)
A1: 試料溶液中の銀検出量 (g)
A2: 空試験液中の銀検出量 (g)
A3: 鉛 [5.3.2 (2) ] 5.0g中に含まれる銀量 (g)
m: 試料はかり取り量 (g)
6. 銅定量方法
6.1
定量方法の区分 銅の定量方法は,次のいずれかによる
(1) 原子吸光法 この方法は,銅含有率0.000 5% (m/m) 以上0.05% (m/m) 以下の試料に適用する。
(2) 鉛分離誘導結合プラズマ発光分光法 この方法は,銅含有率0.000 1% (m/m) 以上0.05% (m/m) 以下の
試料に適用する。
6.2
原子吸光法
6.2.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
6.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上で銅を含有しないもの,又は銅含有率が既知で,かつ,試料中の銅含有率より
低いもの。
(3) 酒石酸溶液 (500g/l)
(4) 標準銅溶液A (100μgCu/ml) 銅[99.99% (m/m) 以上]0.100gをはかり取り,ビーカー (300ml) に移
し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,加熱して分解する。常温まで冷却した後,時計皿
の下面を水で洗って時計皿を取り除く。溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で
標線まで薄めて標準銅溶液Aとする。
(5) 標準銅溶液B (10μgCu/ml) 標準銅溶液A [(4)] を使用の都度,水で正しく10倍に薄めて標準銅溶液
Bとする。
5
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.2.4
操作
6.2.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中の銅含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(3)。
注(3) この溶液を用いて,原子吸光法によって銀,ビスマス,アンチモン,鉄及び亜鉛を定量するこ
とができる。
(2) 試料中の銅含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。溶液20mlを分取して
50mlの全量フラスコに移し入れ,酒石酸溶液3ml及び硝酸 (1+4) 10mlを加え,水で標線まで薄め
る(4)。
注(4) この溶液を用いて,原子吸光法によってビスマス,アンチモン,鉄及び亜鉛を定量することが
できる。
6.2.4.2
吸光度の測定 6.2.4.1の(1)(b)又は(2)(b)で得た溶液の一部を,水を用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長324.8nmにおける吸光度を測定する。
6.2.5
空試験 6.2.6の検量線の作成操作において得られる標準銅溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
6.2.6
検量線の作成 検量線の作成は,次のいずれかによる。
(1) 試料中の銅含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合
(a) 鉛 [6.2.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(c) 標準銅溶液A [6.2.2(4)] 及び標準銅溶液B [6.2.2(5)] の各種液量(銅として0〜1 000μg)を段階的に
加え,水で標線まで薄める。
(d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長324.8nmにおける吸光度を試料と並行して測定し,得た吸光度と銅量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
(2) 試料中の銅含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合
(a) 鉛 [6.2.2(2)] を2.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 6.2.6(1)の(b)〜(d)の手順に従って操作する。
6.2.7
計算 計算は,次のいずれかによる。
(1) 試料中の銅含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合 6.2.4.2及び6.2.5で得た吸光度と
6.2.6(1)で作成した検量線とから銅量を求め,試料中の銅含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Cu
ここに, Cu: 試料中の銅含有率% (m/m)
A1: 試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
A3: 鉛 [6.2.2(2)] 5.0g中に含まれる銅量 (g)
m: 試料はかり取り量 (g)
6
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 試料中の銅含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合 6.2.4.2及び6.2.5で得た吸光度と
6.2.6(2)で作成した検量線とから銅量を求め,試料中の銅含有率を次の式によって算出する.
100
50
20
)
(
6
5
4
×
×
−
−
=
m
A
A
A
Cu
ここに, Cu: 試料中の銅含有率% (m/m)
A4: 分取した試料溶液中の銅検出量 (g)
A5: 空試験液中の銅検出量 (g)
A6: 鉛 [6.2.2(2)] 2.0g中に含まれる銅量 (g)
m: 試料はかり取り量 (g)
6.3
鉛分離誘導結合プラズマ発光分光法
6.3.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
6.3.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 6.2.2(2)による。
(4) 酒石酸溶液 (500g/l)
(5) 標準銅溶液A (100μgCu/ml) 6.2.2(4)による。
(6) 標準銅溶液B (10μgCu/ml) 6.2.2(5)による。
6.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.3.4
操作
6.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり取って,ビーカー (300ml) に移し入れる。
(2) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(3) 塩酸20mlを加え,水で標線まで薄めた後,約30分間放置し,塩化鉛の沈殿を沈降させる。
(4) 溶液を乾いたろ紙(5種C)を用いてろ過し,最初のろ液25mlを捨て,その後のろ液が25ml以上と
なるまでろ過する。
(5) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(5)。
注(5) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,ビスマス,アンチモン,ひ
素,すず,鉄及び亜鉛を定量することができる。
6.3.4.2
発光強度の測定 6.3.4.1(5)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラ
ズマ中に噴霧し,波長324.754nmにおける発光強度を測定する(2)。
6.3.5
空試験 6.3.6の検量線の作成操作において得られる標準銅溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
6.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [6.3.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 時計皿で覆い,酒石酸溶液8ml及び硝酸 (1+4) 25mlを加え,穏やかに加熱して分解した後,煮沸し
て窒素酸化物などを追い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。
(3) これらの溶液を100mlの全量フラスコに水を用いて移し入れ,塩酸20mlを加える。
7
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 標準銅溶液A [6.3.2(5)] 及び標準銅溶液B [6.3.2(6) ] の各種液量(銅として0〜2 000μg)を段階的に加
え,水で標線まで薄める。
(5) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
324.754nmにおける発光強度を試料と並行して測定し,得た発光強度と銅量との関係線を作成し,そ
の関係線を原点を通るように平行移動して検量線とする。
6.3.7
計算 6.3.4.2及び6.3.5で得られた発光強度と6.3.6で作成した検量線とから銅量を求め,試料中の
銅含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Cu
ここに, Cu: 試料中の銅含有率% (m/m)
A1: 分取した試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
A3: 鉛 [6.3.2(3)] 0.09g中に含まれる銅量 (g)
m: 試料はかり取り量 (g)
7. ビスマス定量方法
7.1
定量方法の区分 ビスマスの定量方法は,次のいずれかによる。
(1) 原子吸光法 この方法は,ビスマス含有率0.001% (m/m) 以上0.15% (m/m) 以下の試料に適用する。
(2) 鉛分離誘導結合プラズマ発光分光法 この方法は,ビスマス含有率0.000 1% (m/m) 以上0.15% (m/m)
以下の試料に適用する。
7.2
原子吸光法
7.2.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
7.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上でビスマスを含有しないもの,又はビスマス含有率が既知で,かつ,試料中の
ビスマス含有率より低いもの。
(3) 酒石酸溶液 (500g/l)
(4) 標準ビスマス溶液A (200μgBi/ml) ビスマス[99.99% (m/m) 以上]0.200gをはかり取り,ビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,加熱して分解する。常温まで冷却した
後,時計皿の下面を水で洗って,時計皿を取り除く。溶液を1 000mlの全量フラスコに水を用いて移
し入れ,水で標線まで薄めて標準ビスマス溶液Aとする。
(5) 標準ビスマス溶液B (10μgBi/ml) 標準ビスマス溶液A [(4)] を使用の都度,水で正しく20倍に薄め
て標準ビスマス溶液Bとする。
7.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
7.2.4
操作
7.2.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中のビスマス含有率が0.000 1% (m/m) 以上0.06% (m/m) 未満の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(6)。
8
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(6) この溶液を用いて,原子吸光法によって銀,銅,アンチモン,鉄及び亜鉛を定量することがで
きる。
(2) 試料中のビスマス含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。溶液20mlを分取して
50mlの全量フラスコに移し入れ,酒石酸溶液3ml及び硝酸 (1+4) 10mlを加え,水で標線まで薄め
る(7)。
注(7) この溶液を用いて,原子吸光法によって銅,アンチモン,鉄及び亜鉛を定量することができる。
7.2.4.2
吸光度の測定 7.2.4.1の(1)(b)又は(2)(b)で得た溶液の一部を,水を用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長223.1nmにおける吸光度を測定する。
7.2.5
空試験 7.2.6の検量線の作成操作において得られる標準ビスマス溶液を添加しない溶液の吸光
度を,空試験の吸光度とする。
7.2.6
検量線の作成 検量線の作成は,次のいずれかによる。
(1) 試料中のビスマス含有率が0.001% (m/m) 以上0.06% (m/m) 未満の場合
(a) 鉛 [7.2.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(c) 標準ビスマス溶液A [7.2.2(4)] 及び標準ビスマス溶液B [7.2.2(5)] の各種液量(ビスマスとして0〜
3 000μg)を段階的に加え,水で標線まで薄める。
(d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長223.1nmにおける吸光度を試料と並行して測定し,得た吸光度とビスマス量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
(2) 試料中のビスマス含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合
(a) 鉛 [7.2.2(2)] を2.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 7.2.6(1)の(b)〜(d)の手順に従って操作する。
7.2.7
計算 計算は,次のいずれかによる。
(1) 試料中のビスマス含有率が0.001% (m/m) 以上0.06% (m/m) 未満の場合 7.2.4.2及び7.2.5で得た吸
光度と7.2.6(1)で作成した検量線とからビスマス量を求め,試料中のビスマス含有率を次の式によって
算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Bi
ここに, Bi: 試料中のビスマス含有率% (m/m)
A1: 試料溶液中のビスマス検出量 (g)
A2: 空試験液中のビスマス検出量 (g)
A3: 鉛 [7.2.2(2)] 5.0g中に含まれるビスマス量 (g)
m: 試料はかり取り量 (g)
(2) 試料中のビスマス含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合 7.2.4.2及び7.2.5で得た吸光
度と7.2.6(2)で作成した検量線とからビスマス量を求め,試料中のビスマス含有率を次の式によって算
出する。
100
50
20
)
(
6
5
4
×
×
−
−
=
m
A
A
A
Bi
9
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, Bi: 試料中のビスマス含有率% (m/m)
A4: 分取した試料溶液中のビスマス検出量 (g)
A5: 空試験液中のビスマス検出量 (g)
A6: 鉛 [7.2.2(2) ] 2.0g中に含まれるビスマス量 (g)
m: 試料はかり取り量 (g)
7.3
鉛分離誘導結合プラズマ発光分光法
7.3.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
7.3.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 7.2.2(2)による。
(4) 酒石酸溶液 (500g/l)
(5) 標準ビスマス溶液A (200μgBi/ml) 7.2.2(4)による。
(6) 標準ビスマス溶液B (10μgBi/ml) 7.2.2(5)による。
7.3.3
試料はかり取り量 試料はかり取り量は,5. 0gとし,10mgのけたまではかる。
7.3.4
操作
7.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する。
(2) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え・水で標線まで薄める(8)。
注(8) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,アンチモン,ひ素,す
ず,鉄及び亜鉛を定量することができる。
7.3.4.2
発光強度の測定 7.3.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラ
ズマ中に噴霧し,波長223.061nmにおける発光強度を測定する(2)。
7.3.5
空試験 7.3.6の検量線の作成操作において得られる標準ビスマス溶液を添加しない溶液の発光強
度を空試験の発光強度とする。
7.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [7.3.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作する。
(3) 標準ビスマス溶液A [7.3.2(5)] 及び標準ビスマス溶液B [7.3.2(6)] の各種液量(ビスマスとして0〜
6 000μg)を段階的に加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
223.061nmにおける発光強度を試料と並行して測定し,得た発光強度とビスマス量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
7.3.7
計算 7.3.4.2及び7.3.5で得た発光強度と7.3.6で作成した検量線とからビスマス量を求め,試料中
のビスマス含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Bi
ここに, Bi: 試料中のビスマス含有率% (m/m)
10
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 分取した試料溶液中のビスマス検出量 (g)
A2: 空試験液中のビスマス検出量 (g)
A3: 鉛 [7.3.2(3)] 0.09g中に含まれるビスマス量 (g)
m: 試料はかり取り量 (g)
8. アンチモン定量方法
8.1
定量方法の区分 アンチモンの定量方法は,次のいずれかによる。
(1) 酸化マンガン (IV) 共沈・塩化物抽出分離ローダミンB吸光光度法 この方法は,アンチモン含有率
0.000 1% (m/m) 以上0.15% (m/m) 以下の試料に適用する。
(2) 原子吸光法 この方法は,アンチモン含有率0.001% (m/m) 以上0.15% (m/m) 以下の試料に適用する。
(3) 鉛分離誘導結合プラズマ発光分光法 この方法は,アンチモン含有率0.000 1% (m/m) 以上0.15%
(m/m) 以下の試料に適用する。
8.2
酸化マンガン (IV) 共沈・塩化物抽出分離ローダミンB吸光光度法
8.2.1
要旨 試料を硝酸で分解した後,硝酸マンガン及び過マンガン酸カリウムを加えて,アンチモンを
酸化マンガン (IV) と共沈させてこし分ける。沈殿を硫酸と過酸化水素とで溶解し,臭化水素酸及び臭素
を加え,ジイソプロピルエーテルでタリウムを抽出して除去する。水相の酸濃度を塩酸及び硫酸で調節し
た後,硫酸セリウム (IV) でアンチモン (III) をアンチモン (V) に酸化する。ジイソプロピルエーテルで
アンチモンを抽出し,ローダミンBを加えてローダミンBアンチモン錯体を生成させ,光度計を用いて有
機層の吸光度を測定する。
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) 硝酸 (1+4)
(4) 臭化水素酸
(5) 硫酸 (1+2)
(6) 硝酸・過酸化水素溶液 硝酸 (1+1) 100mlに過酸化水素3mlを加える。この溶液は,使用の都度調製
する。
(7) 硫酸・過酸化水素溶液 硫酸 (1+6) 100mlに過酸化水素2mlを加える。この溶液は,使用の都度調製
する。
(8) 臭素水(飽和)
(9) 硝酸マンガン溶液 硝酸マンガン六水和物10gを水に溶解して液量を100mlとする。
(10) 過マンガン酸カリウム溶液 (20g/l)
(11) 硫酸セリウム (IV) 溶液 硫酸セリウム (IV) 四水和物3gを硫酸 (1+35) 50mlに溶解し,硫酸 (1+35)
で液量を100mlとする。
(12) ローダミンB溶液 (0.2g/l) ローダミンB0.02gを硫酸 (1+35) 100mlに溶解する。この溶液は,使用
の都度約80℃に加熱し,常温まで冷却した後用いる。
(13) ジイソプロピルエーテル
(14) 標準アンチモン溶液 (5μgSb/ml) アンチモン[99.9% (m/m) 以上]0.100gをはかり取り,ビーカー
(300ml) に移し入れ,時計皿で覆い,硫酸25mlを加え,加熱して分解する。常温まで冷却した後,時
計皿の下面を硫酸 (1+6) で洗って,時計皿を取り除く。溶液を1000mlの全量フラスコに硫酸 (1+
11
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6) を用いて移し入れ,硫酸 (1+6) で標線まで薄めて原液 (100μgSb/ml) とする。この原液を使用の
都度,硫酸 (1+6) で正しく20倍に薄めて標準アンチモン溶液とする。
8.2.3
試料はかり取り量 試料はかり取り量は,10.0gとし,10mgのけたまではかる。
8.2.4
操作
8.2.4.1
試料の分解及びアンチモンの共沈分離 試料の分解及びアンチモンの共沈分離は,次の手順によ
って行う。
(1) 試料をはかり取ってビーカー (500ml) に移し入れ,時計皿で覆い,硝酸 (1+4) 70mlを加えて穏やか
に加熱して分解した後,水を加えて液量を約250mlとする。
(2) 硝酸マンガン溶液 [8.2.2(9)] 10mlを加え,煮沸する程度に加熱した後,溶液をかき混ぜながら過マン
ガン酸カリウム溶液10mlを加え,5〜10分間穏やかに煮沸して,酸化マンガン (IV) を沈殿させる。
時計皿の下面を水で洗って時計皿を取り除く。
(3) 沈殿をろ紙(5種B)を用いてこし分け,温水で数回洗浄した後,元のビーカーに洗い移す。元のビ
ーカーを漏斗の下に置き,ろ紙上から温硝酸・過酸化水素溶液 [8.2.2(6)] 20mlを少量ずつ加えて,ろ
紙上及びビーカーに残留した沈殿を溶解する。ろ紙は温水で洗浄し,洗液は,元のビーカーに合わせ
る。
(4) 時計皿で覆い,溶液を加熱して過酸化水素を十分に追い出した後(9),水を加えて液量を約250mlとす
る。煮沸する程度に加熱した後,この溶液をかき混ぜながら過マンガン酸カリウム溶液を少量ずつ加
えて,溶液が紅色となってから更に10mlを加え,5〜10分間穏やかに煮沸し,酸化マンガン (IV) を
沈殿させる。時計皿の下面を水で洗って時計皿を取り除く。
注(9) 蒸発によって残留硝酸量が少なくなったときは硝酸を追加して,硝酸が4〜5mlとなるように調
節する。
(5) 沈殿をろ紙(5種B)を用いてこし分け,温水で数回洗浄した後,元のビーカーに洗い移す。元のビ
ーカーを漏斗の下に置き,ろ紙上から温硫酸・過酸化水素溶液 [8.2.2(7)] 50mlを少量ずつ加えて,ろ
紙上及びビーカー中の沈殿を溶解し,ろ紙は温水で洗浄する。洗液を元のビーカーに合わせた後,加
熱して硫酸の白煙を発生させ,乾固近くまで濃縮し(10),室温まで放冷する。
注(10) 硫酸白煙の発生がほとんどやむ程度とする。
8.2.4.2
タリウムの分離(11) 8.2.4.1(5)で得た塩類に,臭化水素酸10ml及び臭素水5mlを加えて溶解し,
水を用いて分液漏斗 (100ml) に移し入れ,水で液量を50mlとする(12)。ジイソプロピルエーテル25mlを
加え,約3分間激しく振り混ぜ,静置して2相に分離した後,水相を元のビーカーに移し入れる(13)。有機
相は捨てる。水相に硝酸5ml及び硫酸 (1+2) 10mlを加え,加熱し硫酸の白煙を発生させ,乾固近くまで
濃縮し(10),室温まで放冷する。
注(11) 試料中にタリウムを含まない場合には,この8.2.4.2の操作は行わない。
(12) このときの臭化水素酸の濃度は,1.0〜2.4mol/lの範囲とする。
(13) タリウム含有量が多い場合には,水相を別の分液漏斗に移し入れ,ジイソプロピルエーテル10ml
を加えて抽出を繰り返した後,水相を元のビーカーに移し入れる。
8.2.4.3
試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中のアンチモン含有率が0.000 1% (m/m) 以上0.001 5% (m/m) 未満の場合
8.2.4.1(5)又は8.2.4.2で得た塩類に塩酸10ml及び硫酸 (1+2) 20mlを加え,加熱して溶解する。常温
まで冷却した後,溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(2) 試料中のアンチモン含有率が0.001 5% (m/m) 以上0.015% (m/m) 未満の場合
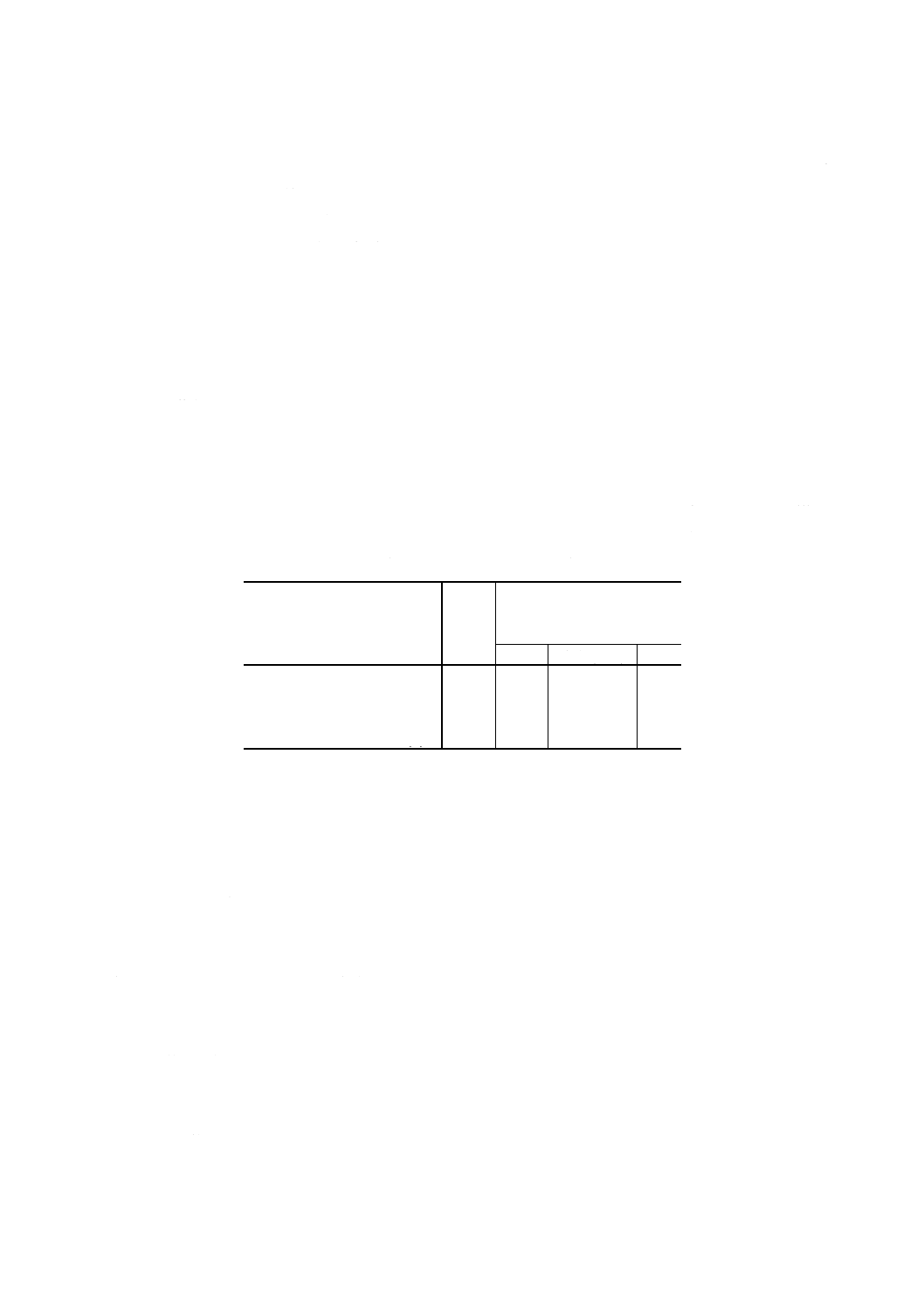
12
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) (1)の操作を行う。
(b) (a)で得た溶液から正確に10mlを100mlの全量フラスコに分取し,塩酸18ml及び硫酸 (1+2) 36ml
を加えた後,水で標線まで薄める。
(3) 試料中のアンチモン含有率が0.015% (m/m) 以上0.15% (m/m) 未満の場合
(a) (2)の(a)及び(b)の手順に従って操作する。
(b) (a)で得た溶液から正確に10mlを100mlの全量フラスコに分取し,塩酸18ml及び硫酸 (1+2) 36ml
を加えた後,水で標線まで薄める。
8.2.4.4
アンチモンの抽出分離及び呈色 アンチモンの抽出分離及び呈色は,次の手順によって行う。
(1) 8.2.4.3の(1),(2)(b)又は(3)(b)で得た溶液から試料中のアンチモン含有率に応じて表1に規定された量
を100mlの分液漏斗に分取し,表1に規定された量の塩酸,硫酸 (1+2) 及び水を加える。
(2) 溶液が黄色となるまで,硫酸セリウム (IV) 溶液 [8.2.2(11)] を少量ずつ加え,更に0.5mlを少量ずつ
加える。約3分間放置した後,塩酸22mlを加え,次に,硫酸セリウム (IV) 溶液 [8.2.2(11)] 1mlを加
える。
(3) 室温まで放冷した後,ジイソプロピルエーテル30mlを加え,約5分間激しく振り混ぜ,静置して2
相に分離した後,水相を取り除く。有機相にローダミンB溶液 [8.2.2(12)] 5mlを加え,約2分間激し
く振り混ぜ,静置して2相に分離した後,水相を取り除き,有機相を目盛付試験管に移し入れる。
表1 分取量並びに塩酸,硫酸 (1+2) 及び水の添加量
アンチモン含有率
% (m/m)
分取量
ml
8.2.4.4(1)での塩酸,硫酸 (1+
2)及び水の添加量
ml
塩酸
硫酸 (1+2)
水
0.000 1 以上
0.000 5 未満(14)
25
0
0
0
0.000 5 以上
0.001 5 未満(14)
10
3
6
6
0.001 5 以上
0.015
未満(15)
10
3
6
6
0.015
以上
0.15
未満(16)
10
3
6
6
注(14) 8.2.4.3(1)で調製した溶液からの分取量
(15) 8.2.4.3(2)で調製した溶液からの分取量
(16) 8.2.4.3(3)で調製した溶液からの分取量
8.2.4.5
吸光度の測定 8.2.4.4(3)で得た有機相の入っている試験管を温水(約60℃)中で気泡が発生する
まで加熱する。流水中で常温まで冷却した後,ジイソプロピルエーテルで液量を正しく30mlとし,ただち
にこの溶液の一部を光度計の吸収セル (10mm) に取り,ジイソプロピルエーテルを対照液として,波長
550nm付近の吸光度を測定する。
8.2.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
8.2.6
検量線の作成 標準アンチモン溶液 [8.2.2(14)] 0〜6.0ml(アンチモンとして0〜30μg)を段階的に
数個の分液漏斗 (100ml) に取り,硫酸 (1+2) 10ml及び塩酸5mlを加え,水で液量を25mlとする。以下
8.2.4.4(2)〜8.2.4.5の手順に従って試料と並行して操作し,得た吸光度とアンチモン量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
8.2.7
計算 計算は,次のいずれかによる。
(1) 試料中のアンチモン含有率が0.000 1% (m/m) 以上0.001 5% (m/m) 未満の場合 8.2.4.5及び8.2.5で
得た吸光度と8.2.6で作成した検量線とからアンチモン量を求め,試料中のアンチモン含有率を次の式
によって算出する。
13
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
50
2
1
×
×
−
=
B
m
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率 % (m/m)
A1: 分取した試料溶液中のアンチモン検出量 (g)
A2: 分取した空試験液中のアンチモン検出量 (g)
m: 試料はかり取り量 (g)
B: 8.2.4.4(1)で分取した試料溶液及び空試験液の量 (ml)
(2) 試料中のアンチモン含有率が0.001 5% (m/m) 以上0.015% (m/m) 未満の場合 8.2.4.5及び8.2.5で得
た吸光度と8.2.6で作成した検量線からアンチモン量を求め,試料中のアンチモン含有率を次の式によ
って算出する。
100
100
10
50
10
2
1
×
×
×
−
=
m
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率% (m/m)
A1: 分取した試料溶液中のアンチモン検出量 (g)
A2: 分取した空試験液中のアンチモン検出量 (g)
m: 試料はかり取り量 (g)
(3) 試料中のアンチモン含有率が0.015% (m/m) 以上0.15% (m/m) 未満の場合 8.2.4.5及び8.2.5で得た
吸光度と8.2.6で作成した検量線からアンチモン量を求め,試料中のアンチモン含有率を次の式によっ
て算出する。
100
100
10
100
10
50
10
2
1
×
×
×
×
−
=
m
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率% (m/m)
A1: 分取した試料溶液中のアンチモン検出量 (g)
A2: 分取した空試験液中のアンチモン検出量 (g)
m: 試料はかり取り量 (g)
8.3
原子吸光法
8.3.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
8.3.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上でアンチモンを含有しないもの,又はアンチモン含有率が既知で,かつ,試料
中のアンチモン含有率より低いもの。
(3) 酒石酸溶液 (500g/l)
(4) 標準アンチモン溶液A (200μgSb/ml) アンチモン [99.9% (m/m)] 0.200gをはかり取り,ビーカー
(300ml) に移し入れ,時計皿で覆い,酒石酸溶液 [(3)] 10ml及び硝酸 (1+1) 25mlを加え,加熱して分
解する。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。溶液を1 000mlの全量
フラスコに水を用いて移し入れ,水で標線まで薄めて標準アンチモン溶液Aとする。
(5) 標準アンチモン溶液B (10μgSb/ml) 標準アンチモン溶液A [(4)] を使用の都度,100mlの全量フラス
コに正確に5ml取り,酒石酸溶液 [(3)] 10mlを加え,水で標線まで薄めて標準アンチモン溶液Bとす
る。
14
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
8.3.4
操作
8.3.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中のアンチモン含有率が0.001% (m/m) 以上0.06% (m/m) 未満の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(17)。
注(17) この溶液を用いて,原子吸光法によって銀,銅,ビスマス,鉄及び亜鉛を定量することができ
る。
(2) 試料中のアンチモン含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。溶液20mlを分取して
50mlの全量フラスコに移し入れ,酒石酸溶液3ml及び硝酸 (1+4) 10mlを加え,水で標線まで薄め
る(18)。
注(18) この溶液を用いて,原子吸光法によって銅,ビスマス,鉄及び亜鉛を定量することができる。
8.3.4.2
吸光度の測定 8.3.4.1の(1)(b)又は(2)(b)で得た溶液の一部を,水を用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長217.6nmにおける吸光度を測定する。
8.3.5
空試験 8.3.6の検量線の作成操作において得られる標準アンチモン溶液を添加しない溶液の吸光
度を,空試験の吸光度とする。
8.3.6
検量線の作成 検量線の作成は,次のいずれかによる。
(1) 試料中のアンチモン含有率が0.001% (m/m) 以上0.06% (m/m) 未満の場合
(a) 鉛 [8.3.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(c) 標準アンチモン溶液A [8.3.2(4)] 及び標準アンチモン溶液B [8.3.2(5)] の各種液量(アンチモンとし
て0〜3 000μg)を段階的に加え,水で標線まで薄める。
(d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長217.6nmにおける吸光度を試料と並行して測定し,得た吸光度とアンチモン量と
の関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
(2) 試料中のアンチモン含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合
(a) 鉛 [8.3.2(2)] を2.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 8.3.6(1)の(b)〜(d)の手順に従って操作する。
8.3.7
計算 計算は,次のいずれかによる
(1) 試料中のアンチモン含有率が0.001% (m/m) 以上0.06% (m/m) 未満の場合 8.3.4.2及び8.3.5で得た
吸光度と8.3.6(1)で作成した検量線とからアンチモン量を求め,試料中のアンチモン含有率を次の式に
よって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率% (m/m)
A1: 試料溶液中のアンチモン検出量 (g)
A2: 空試験液中のアンチモン検出量 (g)
A3: 鉛 [8.3.2(2)] 5.0g中に含まれるアンチモン量 (g)
m: 試料はかり取り量 (g)
15
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 試料中のアンチモン含有率が0.06% (m/m) 以上0.15% (m/m) 以下の場合 8.3.4.2及び8.3.5で得た吸
光度と8.3.6(2)で作成した検量線とからアンチモン量を求め,試料中のアンチモン含有率を次の式によ
って算出する。
100
50
20
)
(
6
5
4
×
×
−
−
=
m
A
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率% (m/m)
A4: 分取した試料溶液中のアンチモン検出量 (g)
A5: 空試験液中のアンチモン検出量 (g)
A6: 鉛 [8.3.2(2)] 2.0g中に含まれるアンチモン量 (g)
m: 試料はかり取り量 (g)
8.4
鉛分離誘導結合プラズマ発光分光法
8.4.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
8.4.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 8.3.2(2)による。
(4) 酒石酸溶液 (500g/l)
(5) 標準アンチモン溶液A (200μgSb/ml) 8.3.2(4)による。
(6) 標準アンチモン溶液B (10μgSb/ml) 8.3.2(5)による。
8.4.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
8.4.4
操作
8.4.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する。
(2) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(19)。
注(19) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,ビスマス,ひ素,すず,
鉄及び亜鉛を定量することができる。
8.4.4.2
発光強度の測定 8.4.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラ
ズマ中に噴霧し,波長217.581nmにおける発光強度を測定する(2)。
8.4.5
空試験 8.4.6の検量線の作成操作において得られる標準アンチモン溶液を添加しない溶液の発光
強度を,空試験の発光強度とする。
8.4.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [8.4.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作する。
(3) 標準アンチモン溶液A [8.4.2(5)] 及び標準アンチモン溶液B [8.4.2(6)] の各種液量(アンチモンとして
0〜6 000μg)を段階的に加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
217.581nmにおける発光強度を試料と並行して測定し,得た発光強度とアンチモン量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
16
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.7
計算 8.4.4.2及び8.4.5で得た発光強度と8.4.6で作成した検量線とからアンチモン量を求め,試料
中のアンチモン含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Sb
ここに, Sb: 試料中のアンチモン含有率% (m/m)
A1: 分取した試料溶液中のアンチモン検出量 (g)
A2: 空試験液中のアンチモン検出量 (g)
A3: 鉛 [8.4.2(3)] 0.09g中に含まれるアンチモン量 (g)
m: 試料はかり取り量 (g)
9. ひ素定量方法
9.1
定量方法の区分 ひ素の定量方法は,次のいずれかによる。
(1) 鉛分離三水素化ひ素気化分離ジエチルジチオカルバミン酸銀吸光光度法 この方法は,ひ素含有率
0.000 1% (m/m) 以上0.01% (m/m) 以下の試料に適用する。
(2) 鉛分離誘導結合プラズマ発光分光法 この方法は,ひ素含有率0.000 1% (m/m) 以上0.01% (m/m) 以下
の試料に適用する。
9.2
鉛分離三水素化ひ素気化分離ジエチルジチオカルバミン酸銀吸光光度法
9.2.1
要旨 試料を硝酸で分解した後,硫酸を加えて硫酸鉛を沈殿させ,ろ過する。ろ液に,硫酸を加え
て酸濃度を調節した後,よう化カリウム,塩化すず (II) 及び亜鉛を加えてひ素を三水素化ひ素として気化
させ,ジエチルジチオカルバミン酸銀(以下,Ag-DDTCという。)・ブルシン−クロロホルムに吸収させ,
光度計を用いて,その吸光度を測定する。
9.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 硫酸 (1+1, 1+3, 1+50)
(3) 亜鉛 JIS K 8012[亜鉛(試薬)]のひ素分析用(砂状)を用いる。
(4) 塩化すず (II) 溶液 塩化すず (II) 二水和物40gを塩酸[ひ素含有率が0.000 01% (m/m) 以下のものを
用いる。]100mlに溶解し,これに金属すずの小粒を加えて保存する。この溶液は,使用の都度,必要
量だけ水で正しく10倍に薄める。
(5) よう化カリウム溶液 (200g/l) この溶液は,使用の都度調製する。
(6) 酢酸鉛溶液 酢酸鉛三水和物10gを酢酸1,2滴を含む水に溶解した後,100mlとする。
(7) Ag-DDTC・ブルシン‐クロロホルム溶液 Ag-DDTC0.25gとブルシン二水和物0.05gとを100mlのク
ロロホルムに溶解する。この溶液は,使用の都度調製する。
(8) Ag-DDTC・ピリジン溶液 Ag-DDTC0.5gを100mlのピリジンに溶解する。
(9) クロロホルム
(10) 標準ひ素溶液 (1μgAs/ml) 三酸化二ひ素 (JIS K 8005) 0.132gを水酸化ナトリウム溶液 (40g/l) 2mlに
溶解し,水で薄め,フェノールフタレイン溶液[調製方法は,JIS K 8001の4.4(指示薬)の表7によ
る]1,2滴を指示薬として加え,硫酸 (1+10) で微酸性とした後,溶液を1 000mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄めて原液 (100μgAs/ml) とする。この原液を使用の都度,水で
正しく100倍に薄めて標準ひ素溶液とする。
9.2.3
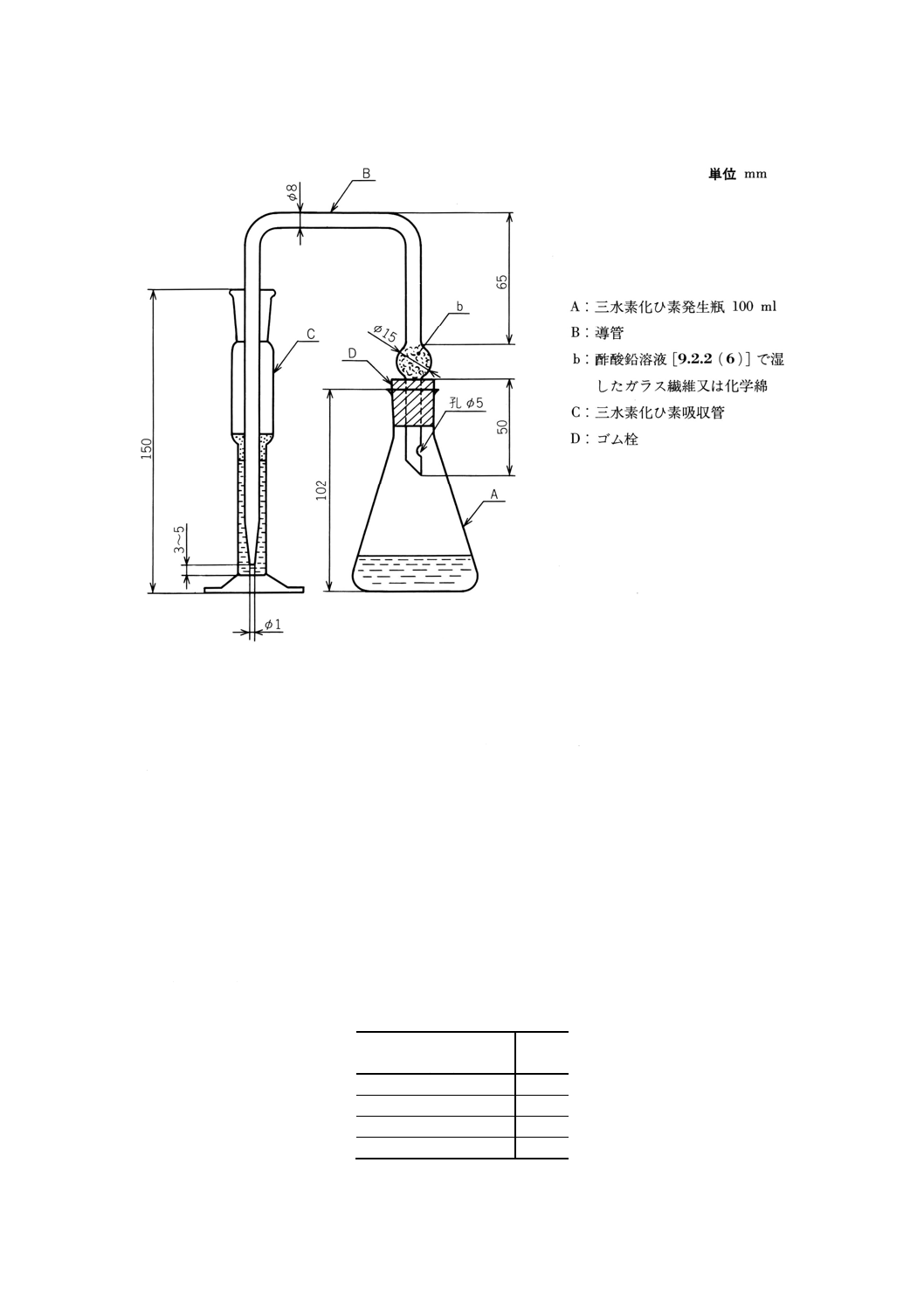
装置及び器具 装置及び器具は,三水素化ひ素発生器及び吸収管を用いる(図1参照)。
17
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 三水素化ひ素発生器及び吸収管の例
9.2.4
試料はかり取り量 試料はかり取り量は,10.0gとし,10mgのけたまではかる。
9.2.5
操作
9.2.5.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をビーカー (300ml) にはかり取り,時計皿で覆い,硝酸 (1+4) 70mlを加え,穏やかに加熱して
分解する。
(2) 溶液をかき混ぜながら,硫酸 (1+3) 30mlを加え,静置し,放冷した後,時計皿の下面を水で洗って
時計皿を取り除く。溶液をろ紙(5種B)を用いてろ過し,硫酸 (1+50) で数回洗浄する。
(3) 常温まで冷却した後,ろ液及び洗液を250mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
(4) この溶液を試料中のひ素含有率に応じて,表2に従ってビーカー (300ml) に分取する。
(5) 硫酸 (1+1) 5mlを加え,加熱濃縮して硫酸の白煙を十分に発生させる。放冷した後,水10ml及び硫
酸 (1+1) 5mlを加え,加熱して塩類を溶解し,常温まで冷却する。少量の水を用いて三水素化ひ素発
生瓶(図1のA)に移し入れる。
表2 分取量
ひ素含有率
% (m/m)
分取量
ml
0.000 1 以上 0.001 未満
100
0.001 以上 0.002 未満
50
0.002 以上 0.004 未満
25
0.004 以上 0.01 以下
10
18
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.5.2 三水素化ひ素の気化分離及び呈色 三水素化ひ素の気化分離及び呈色は,次の手順によって行う。
(1) 9.2.5.1(5)で得た三水素化ひ素発生瓶中の溶液に水を加えて液量を約40mlとする。
(2) よう化カリウム溶液 [9.2.2(5)] 2ml及び塩化すず (II) 溶液 [9.2.2(4)] 5mlを加えて振り混ぜ,約15分
間室温に放置する。
(3) 亜鉛 [9.2.2(3)] 5gを加え,手早く発生瓶(図1のA),導管(図1のB)及びAg-DDTC・ブルシン−
クロロホルム溶液 [9.2.2(7)](20)5mlを入れた吸収管(図1のC)を連結し,発生瓶を約25℃の水中で,
約1時間放置する。
注(20) Ag-DDTCピリジン溶液 [9.2.2(8)] を使用してもよい。この場合は,次の(4)の操作は行わない。
(4) 三水素化ひ素吸収管中の溶液にクロロホルムを加えて液量を正確に5mlとし,振り混ぜる。
9.2.5.3
吸光度の測定 9.2.5.2(4)で得た溶液の一部を光度計の吸収セル (10mm) に取り,Ag-DDTC・ブ
ルシン−クロロホルム溶液 [9.2.2(7)] を対照液として,波長510nm付近の吸光度を測定する(21)。
注(21) 9.2.5.2(3)で注(20)を適用したときには,Ag-DDTCピリジン溶液 [9.2.2(8)] を対照液とし,波長
530nm付近の吸光度を測定する。
9.2.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
9.2.7
検量線の作成 標準ひ素溶液 [9.2.2(10)] 0〜15.0ml(ひ素として0〜15μg)を段階的に数個の三水
素化ひ素発生瓶(図1のA)に取り,硫酸 (1+1) 10mlを加え,水を加えて液量を約40mlとする。以下,
9.2.5.2(2)〜9.2.5.3の手順に従って試料と並行して操作し,得た吸光度とひ素量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
9.2.8
計算 9.2.5.3及び9.2.6で得た吸光度と9.2.7で作成した検量線とからひ素量を求め,試料中のひ素
含有率を次の式によって算出する。
100
250
2
1
×
×
−
=
B
m
A
A
As
ここに, As: 試料中のひ素含有率% (m/m)
A1: 分取した試料溶液中のひ素検出量 (g)
A2: 分取した空試験液中のひ素検出量 (g)
m: 試料はかり取り量 (g)
B: 9.2.5.1(4)で分取した試料溶液及び空試験液の量 (ml)
9.3
鉛分離誘導結合プラズマ発光分光法
9.3.1
要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
9.3.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 99.99% (m/m) 以上でひ素を含有しないもの,又はひ素含有率が既知で,かつ,試料中のひ素含有
率より低いもの。
(4) 酒石酸溶液 (500g/l)
(5) 標準ひ素溶液A (100μgAs/ml) 9.2.2(10)の原液 (100μgAs/ml) を標準ひ素溶液Aとする。
(6) 標準ひ素溶液B (10μgAs/ml) 標準ひ素溶液A [(5)] を使用の都度,水で正しく10倍に薄めて標準ひ
素溶液Bとする。
19
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
9.3.4
操作
9.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する。
(2) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(22)。
注(22) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,ビスマス,アンチモン,
すず,鉄及び亜鉛を定量することができる。
9.3.4.2
発光強度の測定 9.3.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラ
ズマ中に噴霧し,波長228.812nmにおける発光強度を測定する(2)。
9.3.5 空試験 9.3.6の検量線の作成操作において得られる標準ひ素溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
9.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [9.3.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作する。
(3) 標準ひ素溶液A [9.3.2(5)] 及び標準ひ素溶液B [9.3.2(6)] の各種液量(ひ素として0〜400μg)を段階的
に加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
228.812nmにおける発光強度を試料と並行して測定し,得た発光強度とひ素量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
9.3.7
計算 9.3.4.2及び9.3.5で得た発光強度と9.3.6で作成した検量線とからひ素量を求め,試料中のひ
素含有率を次の式によって算出する。
100
50
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
As
ここに, As: 試料中のひ素含有率% (m/m)
A1: 分取した試料溶液中のひ素検出量 (g)
A2: 空試験液中のひ素検出量 (g)
A3: 鉛 [9.3.2(3)] 0.09g中に含まれるひ素量 (g)
m: 試料はかり取り量 (g)
10. すず定量方法
10.1 定量方法の区分 すずの定量方法は,次のいずれかによる。
(1) 酸化マンガン (IV) 共沈・チオシアン酸抽出分離フェニルフルオロン吸光光度法 この方法は,すず
含有率0.000 5% (m/m) 以上0.15% (m/m) 以下の試料に適用する。
(2) 鉛分離誘導結合プラズマ発光分光法 この方法は,すず含有率0.000 1% (m/m) 以上0.15% (m/m) 以下
の試料に適用する。
10.2 酸化マンガン (IV) 共沈・チオシアン酸抽出分離フェニルフルオロン吸光光度法
20
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.1 要旨 試料を硝酸で分解した後,硝酸マンガン及び過マンガン酸カリウムを加えて,すずを酸化マ
ンガン (IV) と共沈させてこし分ける。沈殿を硫酸と過酸化水素とで溶解し,過マンガン酸カリウムを加
えてすずを酸化した後,過剰のマンガン (VII) をL (+) −アスコルビン酸で還元する。塩酸及びくえん酸
を加え,アンモニア水で酸濃度を調整した後,ポリビニルアルコール及びフェニルフルオロンを加えてフ
ェニルフルオロンすず錯体を生成させ,光度計を用いて,その吸光度を測定する。
10.2.2 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+11)
(3) 硝酸
(4) 硝酸 (1+4)
(5) 硫酸 (1+1)
(6) 硝酸・過酸化水素水溶液 硝酸 (1+1) 100mlに過酸化水素3mlを加える。この溶液は,使用の都度調
製する。
(7) 硫酸・過酸化水素水溶液 硫酸 (1+6) 100mlに過酸化水素2mlを加える。この溶液は,使用の都度調
製する。
(8) アンモニア水 (1+1)
(9) 硝酸マンガン溶液 硝酸マンガン六水和物10gを水100mlに溶解する。
(10) 過マンガン酸カリウム溶液 (20g/l)
(11) チオシアン酸アンモニウム溶液 (500g/l)
(12) チオシアン酸アンモニウム洗浄液 チオシアン酸アンモニウム76gを水約300mlに溶解し,硫酸 (1
+35) 500mlを加えた後,水で液量を約1 000mlとする。
(13) L (+) −アスコルビン酸
(14) くえん酸溶液 くえん酸一水和物10gを水100mlに溶解する。
(15) 酒石酸溶液 (100g/l)
(16) エチレンジアミン四酢酸二水素二ナトリウム (EDTA2Na) 溶液 EDTA2Na二水和物20gに水100ml
を加え,アンモニア水を少量ずつ加えて溶解した後,水で液量を200mlとする。
(17) ポリビニルアルコール溶液 (5g/l) ポリビニルアルコール(けん化度80%のK形)0.5gを水に溶解
し,水で液量を100mlとする。
(18) フェニルフルオロン溶液 フェニルフルオロン0.05gをエタノール (99.5) 及び塩酸 (1+2) 10mlに溶
解した後,エタノール (99.5) で液量を500mlとする。
(19) 酢酸エチル
(20) 標準すず溶液 (5μgSn/ml) すず [99.9% (m/m)] 0.100gをはかり取り,ビーカー (300ml) に移し入れ,
時計皿で覆い,塩酸 (1+1) 100mlを加え,加熱して分解する。常温まで冷却した後,時計皿の下面を
塩酸 (1+1) で洗って時計皿を取り除く。溶液を1000mlの全量フラスコに塩酸 (1+1) を用いて移し
入れ,塩酸 (1+1) で標線まで薄めて原液 (100μgSn/ml) とする。この原液を使用の都度,塩酸 (1+
1) で正しく20倍に薄めて標準すず溶液とする。
(21) ブロモクレゾールグリーン溶液 ブロモクレゾールグリーン0.04gをエタノール (99.5) 20mlに溶解し
た後,水で液量を100mlとする。
10.2.3 試料はかり取り量 試料はかり取り量は,10.0gとし,10mgのけたまではかる。
10.2.4 操作

21
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.4.1 試料の分解及びすずの共沈分離 試料の分解及びすずの共沈分離は,8.2.4.1の(1)〜(5)の手順に従
って操作する。
10.2.4.2 すずの抽出分離(23) すずの抽出分離は,次の手順によって行う。
注(23) 試料中のアンチモン又はビスマスの含有率が0.01% (m/m) 以下の場合には,この抽出分離操作
は省略してもよい。
(1) 10.2.4.1で得た乾固物の入っているビーカーに酒石酸溶液10ml及び水20mlを加え,加熱して塩類を
溶解する。硫酸 (1+1) 1.5mlを加え,常温mまで冷却した後,溶液を少量の水を用いて分液漏斗
(100ml) に移し入れる。
(2) チオシアン酸アンモニウム溶液10mlを加え,水で液量を60〜70mlとし,酢酸エチル約20mlを加え,
約2分間激しく振り混ぜ,静置して2相に分離した後,水相を取り除く。
(3) 有機相にチオシアン酸アンモニウム洗浄液 [10.2.2(12)] 10mlを加え,約30秒間振り混ぜ,静置して2
相に分離した後,水相を取り除く。
(4) 有機相をビーカー (100ml) に移し入れ,硫酸 (1+1) 5mlを加え,加熱して酢酸エチルを揮散させた後,
白煙を発生させる。硝酸5mlを加え,加熱して遊離の硫黄,炭素などを分解し,更に加熱を続けて乾
固近くまで濃縮し,放冷する。
10.2.4.3 試料溶液の調製 10.2.4.1又は10.2.4.2(4)で得た乾固物の入っているビーカーに塩酸10ml及び水
を加え,加熱して塩類を溶解し,常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入
れ,水で標線まで薄める(24)。
注(24) 試料中のすず含有率が0.02% (m/m) 以上の場合は,この溶液から正確に10mlを100mlの全量フ
ラスコに分取し,塩酸9mlを加えた後,水で標線まで薄める。
10.2.4.4 呈色 呈色は,次の手順によって行う。
(1) 10.2.4.3で得た溶液から,試料中のすず含有率に応じて,表3に規定された量を50mlの全量フラスコ
及びビーカー (200ml) に分取する。
(2) ビーカー中の溶液に水を加えて液量を約50mlとし,ブロモクレゾールグリーン溶液 [10.2.2(21)] (25)2,
3滴を指示薬として加え,アンモニア水 (1+1) で滴定し,溶液の色が黄から青に変わる点を終点とし,
その滴定量をTmlとする。
注(25) pH3〜5の変色域をもつ他の指示薬を用いてもよい。
(3) 全量フラスコ中の溶液に溶液が微紅色を保つまで過マンガン酸カリウム溶液を少量ずつ加え,約5分
間放置する。少量のL (+) −アスコルビン酸を加えて過マンガン酸イオンの紅色を消した後,塩酸 (1
+11) 1.5ml,くえん酸溶液 [10.2.2(14)] 5ml,アンモニア水 (1+1) Tml,ポリビニルアルコール溶液
[10.2.2(17)] 5mlを順次加え,水で液量を約40mlとする。フェニルフルオロン溶液 [10.2.2(18)] を正確
に5ml加え,約20分間放置した後,EDTA2Na溶液 [10.2.2(16)] 1mlを加え,水で標線まで薄める。
表3 分取量
すず含有率
% (m/m)
分取量
ml
0.000 5 以上 0.004 未満
20
0.004 以上 0.02 未満
5
0.02 以上 0.1 未満(26)
10
0.1
以上 0.15 以下(26)
5
注(26) 注(24)に従って調製した溶液
からの分取量
22
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.4.5 吸光度の測定 10.2.4.4(3)で得た溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液と
して波長510nm付近の吸光度を測定する。
10.2.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
10.2.6 検量線の作成 標準すず溶液 [10.2.2(20)] 0〜10.0ml(すずとして0〜50μg)を段階的に数個の50ml
の全量フラスコ及びビーカー (200ml) に取り,以下,10.2.4.4(2)〜10.2.4.5の手順に従って試料と並行して
操作し,得た吸光度とすず量との関係線を作成し,この関係線を原点を通るように平行移動して検量線と
する。
10.2.7 計算 計算は,次のいずれかによる。
(1) 注(24)を適用しなかった場合 10.2.4.5及び10.2.5で得た吸光度と10.2.6で得た検量線とからすず量を
求め,試料中のすず含有率を次の式によって算出する。
100
100
2
1
×
×
−
=
B
m
A
A
Sn
ここに, Sn: 試料中のすず含有率% (m/m)
A1: 分取した試料溶液中のすず検出量 (g)
A2: 分取した空試験液中のすず検出量 (g)
m: 試料はかり取り量 (g)
B: 10.2.4.4(1)で分取した試料溶液及び空試験液の量 (ml)
(2) 注(24)を適用した場合 10.2.4.5及び10.2.5で得た吸光度と10.2.6で得た検量線とからすず量を求め,
試料中のすず含有率を次の式によって算出する。
100
100
10
100
2
1
×
×
×
−
=
B
m
A
A
Sn
ここに, Sn: 試料中のすず含有率% (m/m)
A1: 分取した試料溶液中のすず検出量 (g)
A2: 分取した空試験液中のすず検出量 (g)
m: 試料はかり取り量 (g)
B: 10.2.4.4(1)で分取した試料溶液及び空試験液の量 (ml)
10.3 鉛分離誘導結合プラズマ発光分光法
10.3.1 要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
10.3.2 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 99.99% (m/m) 以上ですずを含有しないもの,又はすず含有率が既知で,かつ,試料中のすず含有
率より低いもの。
(4) 酒石酸溶液 (500g/l)
(5) 標準すず溶液A (200μgSn/ml) すず[99.9% (m/m) 以上]0.200gをはかり取り,ビーカー (300ml) に
移し入れ,時計皿で覆い,酒石酸溶液20ml及び混酸(塩酸3,硝酸1)10mlを加え,加熱して分解す
る。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。溶液を1 000mlの全量フラ
スコに水を用いて移し入れ,水で標線まで薄めて標準すず溶液Aとする。
23
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(6) 標準すず溶液B (10μgSn/ml) 標準すず溶液A [(5)] を使用の都度,100mlの全量フラスコに正確に5ml
取り,酒石酸溶液10mlを加え,水で標線まで薄めて標準すず溶液Bとする。
10.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
10.3.4 操作
10.3.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する。
(2) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(27)。
注(27) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,ビスマス,アンチモン,
ひ素,鉄及び亜鉛を定量することができる。
10.3.4.2 発光強度の測定 10.3.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプ
ラズマ中に噴霧し,波長189.950nmにおける発光強度を測定する(2)。
10.3.5 空試験 10.3.6の検量線の作成操作において得られる標準すず溶液を添加しない溶液の発光強度
を,空試験の発光強度とする。
10.3.6 検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [10.3.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作する。
(3) 標準すず溶液A [10.3.2(5)] 及び標準すず溶液B [10.3.2(6)] の各種液量(すずとして0〜6 000μg)を段
階的に加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
189.950nmにおける発光強度を試料と並行して測定し,得た発光強度とすず量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
10.3.7 計算 10.3.4.2及び10.3.5で得た発光強度と10.3.6で作成した検量線とからすず量を求め,試料中
のすず含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Sn
ここに, Sn: 試料中のすず含有率% (m/m)
A1: 分取した試料溶液中のすず検出量 (g)
A2: 空試験液中のすず検出量 (g)
A3: 鉛 [10.3.2(3)] 0.09g中に含まれるすず量 (g)
m: 試料はかり取り量 (g)
11. 鉄定量方法
11.1 定量方法の区分 鉄の定量方法は,次のいずれかによる。
(1) 1,10−フェナントロリン・過塩素酸抽出吸光光度法 この方法は,鉄含有率0.000 1% (m/m) 以上0.05%
(m/m) 以下の試料に適用する。
(2) 原子吸光法 この方法は,鉄含有率0.000 5% (m/m) 以上0.05% (m/m) 以下の試料に適用する。
(3) 鉛分離誘導結合プラズマ発光分光法 この方法は,鉄含有率0.000 1% (m/m) 以上,0.05% (m/m) 以下
の試料に適用する。
11.2 1,10−フェナントロリン・過塩素酸抽出吸光光度法
24
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.2.1 要旨 試料を硝酸で分解した後,塩化ヒドロキシルアンモニウム,酢酸アンモニウム,1,10−フ
ェナントロリン及び過塩素酸ナトリウムを加え,生成した1,10−フェナントロリン鉄錯体の過塩素酸塩
を1,2−ジクロロエタンで抽出し,光度計を用いて,有機相の吸光度を測定する。
11.2.2 試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 硝酸 (1+4)
(3) 過塩素酸ナトリウム溶液 (100g/l)
(4) 酢酸
(5) 塩化ヒドロキシルアンモニウム溶液 (100g/l)
(6) 酢酸アンモニウム (500g/l)
(7) 1,10−フェナントロリン溶液 塩化1,10−フェナントロリニウム一水和物1.2gを水500mlに溶解
するか,又は1,10−フェナントロリン一水和物1gをエタノール (95) 50mlに溶解し,水で液量を500ml
とする。
(8) 1,2−ジクロロエタン
(9) 標準鉄溶液 (5μgFe/ml) 鉄[99.9% (m/m) 以上]0.100gをはかり取り,ビーカー (300ml) に移し入
れ,時計皿で覆い,硝酸 (1+1) 20ml及び塩酸 (1+1) 5mlを加え,加熱して分解する。常温まで冷却
した後,時計皿の下面を水で洗って時計皿を取り除く。溶液を1 000mlの全量フラスコに水を用いて
移し入れ,水で標線まで薄めて原液 (100μgFe/ml) とする。この原液を使用の都度,硝酸 (1+100) で
正しく20倍に薄めて標準鉄溶液とする。
11.2.3 試料はかり取り量 試料はかり取り量は,10.0gとし,10mgのけたまではかる。
11.2.4 操作
11.2.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+4) 70mlを加え,穏やか
に加熱して分解する。
(2) 常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を100mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄める(28)。
注(28) 試料中の鉄含有率が0.01% (m/m) 以上の場合は,この溶液から正確に10mlを100mlの全量フラ
スコに分取し,硝酸 (1+4) 63mlを加えた後,水で標線まで薄める。
11.2.4.2 呈色 呈色は,次の手順によって行う。
(1) 11.2.4.1(2)で得た溶液を試料中の鉄の含有率に応じて,表4に従ってビーカー (100ml) に分取する。
(2) 塩化ヒドロキシルアンモニウム溶液5ml及び酢酸アンモニウム溶液10mlを加え,酢酸を用いてpHを
4〜5に調節した後,溶液を分液漏斗 (100ml) に水を用いて移し入れる。
(3) 1,10−フェナントロリン溶液 [11.2.2(7)] 2ml及び過塩素酸ナトリウム溶液2mlを加えて振り混ぜ,約
3分間放置した後,1,2−ジクロロエタンを正確に10ml加え,約2分間激しく振り混ぜる。静置して
2相に分離した後,水相を取り除く。

25
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表4 分取量
鉄含有率
% (m/m)
分取量
ml
0.000 1 以上 0.002 未満
20
0.002 以上 0.01 未満
5
0.01
以上 0.05 以下(29)
10
注(29) 注(28)で調製した溶液からの
分取量
11.2.4.3 吸光度の測定 11.2.4.2(3)で得た有機相の水分を除き(30),その一部を光度計の吸収セル (10mm)
に取り,1,2‐ジクロロエタンを対照液として,波長510nm付近の吸光度を測定する。
注(30) 脱脂綿又は乾いたろ紙を用いて脱水する。
11.2.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
11.2.6 検量線の作成 標準鉄溶液 [11.2.2(9)] 0〜6.0ml(鉄として0〜30μg)を段階的に数個のビーカーに
取り,以下,11.2.4.2(2)〜11.2.4.3の手順に従って試料と並行に操作し,得た吸光度と鉄量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
11.2.7 計算 計算は,次のいずれかによる。
(1) 注(28)を適用しなかった場合 11.2.4.3及び11.2.5で得た吸光度と11.2.6で得た検量線とから鉄量を求
め,試料中の鉄含有率を次の式によって算出する。
100
100
2
1
×
×
−
=
B
m
A
A
Fe
ここに, Fe: 試料中の鉄含有率% (m/m)
A1: 分取した試料溶液中の鉄検出量 (g)
A2: 分取した空試験液中の鉄検出量 (g)
m: 試料はかり取り量 (g)
B: 11.2.4.2(1)で分取した試料溶液及び空試験液の量 (ml)
(2) 注(28)を適用した場合 11.2.4.3及び11.2.5で得た吸光度と11.2.6で得た検量線とから鉄量を求め,試
料中の鉄含有率を次の式によって算出する。
100
100
10
100
10
2
1
×
×
×
−
=
m
A
A
Fe
ここに, Fe: 試料中の鉄含有率% (m/m)
A1: 分取した試料溶液中の鉄検出量 (g)
A2: 分取した空試験液中の鉄検出量 (g)
m: 試料はかり取り量 (g)
11.3 原子吸光法
11.3.1 要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
11.3.2 試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上で鉄を含有しないもの,又は鉄含有率が既知で,かつ,試料中の鉄含有率より
低いもの。
(3) 酒石酸溶液 (500g/l)
26
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 標準鉄溶液A (100μgFe/ml) 11.2.2(9)の原液 (100μgFe/ml) を標準鉄溶液Aとする。
(5) 標準鉄溶液B (5μgFe/ml) 11.2.2(9)による。
11.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
11.3.4 操作
11.3.4.1 試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中の鉄含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(31)。
注(31) この溶液を用いて,原子吸光法によって銀,銅,ビスマス,アンチモン及び亜鉛を定量するこ
とができる。
(2) 試料中の鉄含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。溶液20mlを分取して
50mlの全量フラスコに移し入れ,酒石酸溶液3ml及び硝酸 (1+4) 10mlを加え,水で標線まで薄め
る(32)。
注(32) この溶液を用いて,原子吸光法によって銅,ビスマス,アンチモン及び亜鉛を定量することが
できる。
11.3.4.2 吸光度の測定 11.3.4.1の(1)(b)又は(2)(b)で得た溶液の一部を,水を用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長248.3nmにおける吸光度を測定する。
11.3.5 空試験 11.3.6の検量線の作成操作において得られる標準鉄溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
11.3.6 検量線の作成 検量線の作成は,次のいずれかによる。
(1) 試料中の鉄含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合
(a) 鉛 [11.3.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(c) 標準鉄溶液A [11.3.2(4)] 及び標準鉄溶液B [11.3.2(5)] の各種液量(鉄として0〜1 000μg)を段階的
に加え,水で標線まで薄める。
(d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長248.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉄量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
(2) 試料中の鉄含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合
(a) 鉛 [11.3.2(2)] を2.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) (1)の(b)〜(d)の手順に従って操作する。
11.3.7 計算 計算は,次のいずれかによる。
(1) 試料中の鉄含有率が0.000 5% (m/m) 以上0.02% (m/m) 未満の場合 11.3.4.2及び11.3.5で得た吸光度
と11.3.6(1)で作成した検量線とから鉄量を求め,試料中の鉄含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率% (m/m)
A1: 試料溶液中の鉄検出量 (g)
27
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A2: 空試験液中の鉄検出量 (g)
A3: 鉛 [11.3.2(2)] 5.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
(2) 試料中の鉄含有率が0.02% (m/m) 以上0.05% (m/m) 以下の場合 11.3.4.2及び11.3.5で得た吸光度と
11.3.6(2)で作成した検量線とから鉄量を求め,試料中の鉄含有率を次の式によって算出する。
100
50
20
)
(
6
5
4
×
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率% (m/m)
A4: 分取した試料溶液中の鉄検出量 (g)
A5: 空試験液中の鉄検出量 (g)
A6: 鉛 [11.3.2(2)] 2.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
11.4 鉛分離誘導結合プラズマ発光分光法
11.4.1 要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
11.4.2 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 11.3.2(2)による
(4) 酒石酸溶液 (500g/l)
(5) 標準鉄溶液A (100μgFe/ml) 11.2.2(9)の原液 (100μgFe/ml) を標準鉄溶液Aとする。
(6) 標準鉄溶液B (5μgFe/ml) 11.2.2(9)による。
11.4.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
11.4.4 操作
11.4.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する(33)。
注(33) 6.3.4.1(4)のろ過操作の際に,乾いたろ紙(5種C)から鉄の溶出が認められる場合には,ろ過操
作を行わず,遠心分離操作を行って,塩化鉛の沈殿と溶液を分離する。
(2) ろ液(34)から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(35)。
注(34) 注(33)を適用した場合には,遠心分離後の上澄液を用いる。
(35) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,ビスマス,アンチモン,
ひ素,すず及び亜鉛を定量することができる。
11.4.4.2 発光強度の測定 11.4.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴン中に
噴霧し,波長259.934nmにおける発光強度を測定する(2)。
11.4.5 空試験 11.4.6の検量線の作成操作において得られる標準鉄溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
11.4.6 検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [11.4.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作する。
28
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 標準鉄溶液A [11.4.2(5)] 及び標準鉄溶液B [11.4.2(6)] の各種液量(鉄として0〜2 000μg)を段階的に
加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
259.934nmにおける発光強度を試料と並行して測定し,得た発光強度と鉄量との関係線を作成し,そ
の関係線を原点を通るように平行移動して検量線とする。
11.4.7 計算 11.4.4.2及び11.4.5で得た発光強度と11.4.6で作成した検量線とから鉄量を求め,試料中の
鉄含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率% (m/m)
A1: 分取した試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: 鉛 [11.4.2(3)] 0.09g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
12. 亜鉛定量方法
12.1 定量方法の区分 亜鉛の定量方法は,次のいずれかによる。
(1) 原子吸光法 この方法は,亜鉛含有率0.000 5% (m/m) 以上0.015% (m/m) 以下の試料に適用する。
(2) 鉛分離誘導結合プラズマ発光分光法 この方法は,亜鉛含有率0.000 1% (m/m) 以上0.015% (m/m) 以
下の試料に適用する。
12.2 原子吸光法
12.2.1 要旨 試料を酒石酸の存在下で,硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフ
レーム中に噴霧し,その吸光度を測定する。
12.2.2 試薬 試薬は,次による。
(1) 硝酸 (1+4)
(2) 鉛 99.99% (m/m) 以上で亜鉛を含有しないもの,又は亜鉛含有率が既知で,かつ,試料中の亜鉛含有
率より低いもの。
(3) 酒石酸溶液 (500g/l)
(4) 標準亜鉛溶液A (100μgZn/ml) 亜鉛[99.99% (m/m) 以上]0.100gをはかり取り,ビーカー (300ml) に
移し入れ,時計皿で覆い,硝酸 (1+1) 10mlを加え,加熱して分解する。常温まで冷却した後,時計
皿の下面を水で洗って取り除く。溶液を1 000mlの全量フラスコに水を用いて移し入れ水で標線まで
薄めて標準亜鉛溶液Aとする。
(5) 標準亜鉛溶液B (10μgZn/ml) 標準亜鉛溶液A [(4)] を使用の都度,水で正しく10倍に薄めて標準亜
鉛溶液Bとする。
12.2.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
12.2.4 操作
12.2.4.1 試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 試料中の鉄含有率が0.000 5% (m/m) 以上0.006% (m/m) 未満の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(36)。
29
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(36) この溶液を用いて,原子吸光法によって銀,銅,ビスマス,アンチモン及び鉄を定量すること
ができる。
(2) 試料中の鉄含有率が0.006% (m/m) 以上0.015% (m/m) 以下の場合
(a) 5.2.4.1の(1)及び(2)の手順に従って操作する。
(b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。溶液20mlを分取して
50mlの全量フラスコに移し入れ,酒石酸溶液3ml及び硝酸 (1+4) 10mlを加え,水で標線まで薄め
る(37)。
注(37) この溶液を用いて,原子吸光法によって銅,ビスマス,アンチモン及び鉄を定量することがで
きる。
12.2.4.2 吸光度の測定 12.2.4.1の(1)(b)又は(2)(b)で得た溶液の一部を,水を用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長213.9nmにおける吸光度を測定する。
12.2.5 空試験 12.2.6の検量線の作成操作において得られる標準鉄溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
12.2.6 検量線の作成 検量線の作成は,次のいずれかによる。
(1) 試料中の亜鉛含有率が0.000 5% (m/m) 以上0.006% (m/m) 未満の場合
(a) 鉛 [12.2.2(2)] を5.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) 5.2.4.1(2)に従って操作した後,溶液を100mlの全量フラスコに水を用いて移し入れる。
(c) 標準亜鉛溶液A [12.2.2(4)] 及び標準亜鉛溶液B [12.2.2(5)] の各種液量(亜鉛として0〜300μg)を段
階的に加え,水で標線まで薄める。
(d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長213.9nmにおける吸光度を試料と並行して測定し,得た吸光度と亜鉛量との関係
線を作成し,その関係線を原点を通るように平行移動して検量線とする。
(2) 試料中の亜鉛含有率が0.006% (m/m) 以上0.015% (m/m) 以下の場合
(a) 鉛 [12.2.2(2)] を2.0gずつ数個はかり取り,それぞれビーカー (300ml) に移し入れる。
(b) (1)の(b)〜(d)の手順に従って操作する。
12.2.7 計算 計算は,次のいずれかによる。
(1) 試料中の亜鉛含有率が0.000 5% (m/m) 以上0.006% (m/m) 未満の場合 12.2.4.2及び12.2.5で得た吸
光度と12.2.6(1)で作成した検量線とから亜鉛量を求め,試料中の亜鉛含有率を次の式によって算出す
る。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Zn
ここに, Zn: 試料中の亜鉛含有率% (m/m)
A1: 試料溶液中の亜鉛検出量 (g)
A2: 空試験液中の亜鉛検出量 (g)
A3: 鉛 [12.2.2(2)] 5.0g中に含まれる亜鉛量 (g)
m: 試料はかり取り量 (g)
(2) 試料中の亜鉛含有率が0.006% (m/m) 以上0.015% (m/m) 以下の場合 12.2.4.2及び12.2.5で得た吸光
度と12.2.6(2)で作成した検量線とから亜鉛量を求め,試料中の亜鉛含有率を次の式によって算出する。
100
50
20
)
(
6
5
4
×
×
−
−
=
m
A
A
A
Zn
30
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, Zn: 試料中の亜鉛含有率% (m/m)
A4: 分取した試料溶液中の亜鉛検出量 (g)
A5: 分取した空試験液中の亜鉛検出量 (g)
A6: 鉛 [12.2.2(2)] 2.0g中に含まれる亜鉛量 (g)
m: 試料はかり取り量 (g)
12.3 鉛分離誘導結合プラズマ発光分光法
12.3.1 要旨 試料を酒石酸の存在下で,硝酸で分解した後,塩酸を加えて塩化鉛を沈殿させ,ろ過する。
ろ液に塩酸を加え,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定
する。
12.3.2 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸 (1+4)
(3) 鉛 12.2.2(2)による。
(4) 酒石酸溶液 (500g/l)
(5) 標準亜鉛溶液A (100μgZn/ml) 12.2.2(4)による。
(6) 標準亜鉛溶液B (10μgZn/ml) 12.2.2(5)による。
12.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
12.3.4 操作
12.3.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 6.3.4.1の(1)〜(4)の手順に従って操作する。
(2) ろ液から20mlを25mlの全量フラスコに分取し,塩酸2mlを加え,水で標線まで薄める(38)。
注(38) この溶液を用いて,鉛分離誘導結合プラズマ発光分光法によって,銅,ビスマス,アンチモン,
ひ素,すず及び鉄を定量することができる。
12.3.4.2 発光強度の測定 12.3.4.1(2)で得た溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプ
ラズマ中に噴霧し,波長213.856nmにおける発光強度を測定する(2)。
12.3.5 空試験 12.3.6の検量線の作成操作において得られる標準亜鉛溶液を添加しない溶液の発光強度
を,空試験の発光強度とする。
12.3.6 検量線の作成 検量線の作成は,次の手順によって行う。
(1) 鉛 [12.3.2(3)] を0.09gずつ数個はかり取り,それぞれビーカー (200ml) に移し入れる。
(2) 6.3.6の(2)及び(3)の手順に従って操作を行う。
(3) 標準亜鉛溶液A [12.3.2(5)] 及び標準亜鉛溶液B [12.3.2(6)] の各種液量(亜鉛として0〜600μg)を段階
的に加え,水で標線まで薄める。
(4) これらの溶液の一部を,誘導結合プラズマ発光分光装置のアルゴンプラズマ中に噴霧し,波長
213.856nmにおける発光強度を試料と並行して測定し,得た発光強度と亜鉛量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
12.3.7 計算 12.3.4.2及び12.3.5で得た発光強度と12.3.6で作成した検量線とから亜鉛量を求め,試料中
の亜鉛含有率を次の式によって算出する。
100
25
20
)
(
3
2
1
×
×
−
−
=
m
A
A
A
Zn
ここに, Zn: 試料中の亜鉛含有率% (m/m)
31
H 1121-1995
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 分取した試料溶液中の亜鉛検出量 (g)
A2: 空試験液中の亜鉛検出量 (g)
A3: 鉛 [12.3.2(3)] 0.09g中に含まれる亜鉛量 (g)
m: 試料はかり取り量 (g)
JIS H 1121原案作成委員会 構成表(順不動)
氏名
所属
(委員長)
奥 谷 忠 雄
日本大学理工学部
増 田 聰 博
通商産業省資源エネルギー庁
高 木 譲 一
工業技術院材料規格課
加 藤 金 夫
大蔵省造幣局東京支局
藤 貫 正
社団法人日本分析化学会
束 原 巌
古河電気工業株式会社
森 本 良 幸
社団法人日本蓄電池工業会
森 孝 夫
日本鉛亜鉛需要研究会
小 島 昌 夫
株式会社小島半田製造所
石 橋 達 也
古河電池株式会社
尾 上 喬
同和鉱業株式会社
永 井 巌
住友金属鉱山株式会社
丹 野 一 雄
東邦亜鉛株式会社
中 村 靖
株式会社ジャパンエナジー
佐 山 恭 正
三菱マテリアル株式会社
渡 部 武 雄
三井金属鉱業株式会社
稲 垣 勝 彦
日本鉱業協会
(関係者)
細 矢 一 仁
同和鉱業株式会社
村 井 幸 男
株式会社ジャパンエナジー
松 岡 俊 和
三井金属鉱業株式会社