G 1327-1:2010
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 一般事項························································································································· 1
4 定量方法の区分 ················································································································ 1
5 イオン交換分離水酸化ナトリウム滴定法 ··············································································· 1
5.1 要旨 ···························································································································· 1
5.2 試薬 ···························································································································· 2
5.3 器具 ···························································································································· 2
5.4 試料はかりとり量 ·········································································································· 4
5.5 操作 ···························································································································· 4
5.6 空試験 ························································································································· 4
5.7 計算 ···························································································································· 5
5.8 許容差 ························································································································· 5
6 ICP発光分光法 ················································································································ 5
6.1 要旨 ···························································································································· 5
6.2 試薬 ···························································································································· 5
6.3 試料はかりとり量 ·········································································································· 6
6.4 操作 ···························································································································· 6
6.5 空試験 ························································································································· 6
6.6 検量線の作成 ················································································································ 6
6.7 計算 ···························································································································· 6
6.8 許容差 ························································································································· 7
G 1327-1:2010
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,日本フェロアロイ協会(JFA)及び財団法人
日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出があり,日本工業
標準調査会の審議を経て,経済産業大臣が制定した日本工業規格である。
これによって,JIS G 1327:1992は廃止され,その一部を分割して制定したこの規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に
抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許
権,出願公開後の特許出願,実用新案権及び出願公開後の実用新案登録出願にかかわる確認について,責
任はもたない。
JIS G 1327の規格群には,次に示す部編成がある。
JIS G 1327-1 第1部:ほう素定量方法
JIS G 1327-2 第2部:炭素定量方法
JIS G 1327-3 第3部:けい素定量方法
JIS G 1327-4 第4部:アルミニウム定量方法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1327-1:2010
フェロボロン分析方法−第1部:ほう素定量方法
Method for chemical analysis of ferroboron-
Part 1: Methods for determination of boron content
序文
JIS G 1327は,1968年に制定され,その後1992年に2回目の改正が行われた。今回,分析技術の進展
に対応するために,JIS G 1327:1992を廃止し,その規格の一部を分割して,ほう素定量方法として制定し
た。
なお,対応国際規格は現時点で制定されていない。
1
適用範囲
この規格は,フェロボロン中のほう素の定量方法について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS G 1301 フェロアロイ−分析方法通則
JIS K 8001 試薬試験方法通則
3
一般事項
分析方法に共通な一般事項は,JIS G 1301による。
4
定量方法の区分
ほう素の定量方法は,次のいずれかによる。
a) イオン交換分離水酸化ナトリウム滴定法 この方法は,ほう素含有率14.0 %(質量分率)以上23.0 %
(質量分率)以下の試料に適用する。
b) ICP発光分光法 この方法は,ほう素含有率14.0 %(質量分率)以上23.0 %(質量分率)以下の試料
に適用する。
5
イオン交換分離水酸化ナトリウム滴定法
5.1
要旨
試料を硝酸と塩酸との混酸で分解し,未分解残さをこし分けて過酸化ナトリウム及び炭酸ナトリウムで
融解した後,水に溶解し,ろ液に合わせる。イオン交換樹脂で共存する鉄,アルミニウムなどを分離した
後,マンニトールを加えて水酸化ナトリウム溶液で滴定する。
2
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2
試薬
試薬は,次による。
5.2.1
塩酸(1+1,1+2,1+100)
5.2.2
混酸(塩酸1+硝酸3) 使用の都度,調製する。
5.2.3
水酸化ナトリウム溶液(40 g/L,4 g/L)
5.2.4
融解合剤(過酸化ナトリウム7+炭酸ナトリウム3)
5.2.5 マンニトール 溶存している炭酸ガスを加熱して煮沸した後,冷却して除去した水に溶解したとき,
溶解前後で水のpHが変化しないものを用いる。
5.2.6
0.1 mol/L水酸化ナトリウム溶液 調製方法は,JIS K 8001のJA.5.2(滴定用溶液の調製,標定及
び計算)r) 3)による。この溶液の標定は,次による。
水300 mLを入れたビーカー(500 mL)に,ほう素標準液(B:1 081 μg/mL)を25.0 mLとり,水を加
えて液量を約200 mLとした後,pH計を用いて水酸化ナトリウム溶液(40 g/L)でpHを3.0〜3.5に調節す
る。以下,5.5.3のb)〜c)の手順に従って操作し,次の式によって,0.1 mol/L水酸化ナトリウム溶液1 mL
に相当するほう素量を求める。
V
f
25
081
001
.0
×
=
ここに,
f: 0.1 mol/L 水酸化ナトリウム溶液1 mLに相当する
ほう素量(g/mL)
V: 0.1 mol/L 水酸化ナトリウム溶液の使用量(mL)
なお,標定操作で使用するほう素標準液(B:1 081 μg/mL)の調製は,次による。
過塩素酸マグネシウムを入れた真空デシケータ中で保存してある恒量としたほう酸6.184 gを熱水約500
mLに溶解し,常温まで冷却した後,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める。
5.2.7
p-ニトロフェノール溶液 p-ニトロフェノール0.2 gをエタノール(95)50 mLに溶解し,水で液量
を100 mLに薄める。
5.3
器具
器具は,通常,次による。
5.3.1
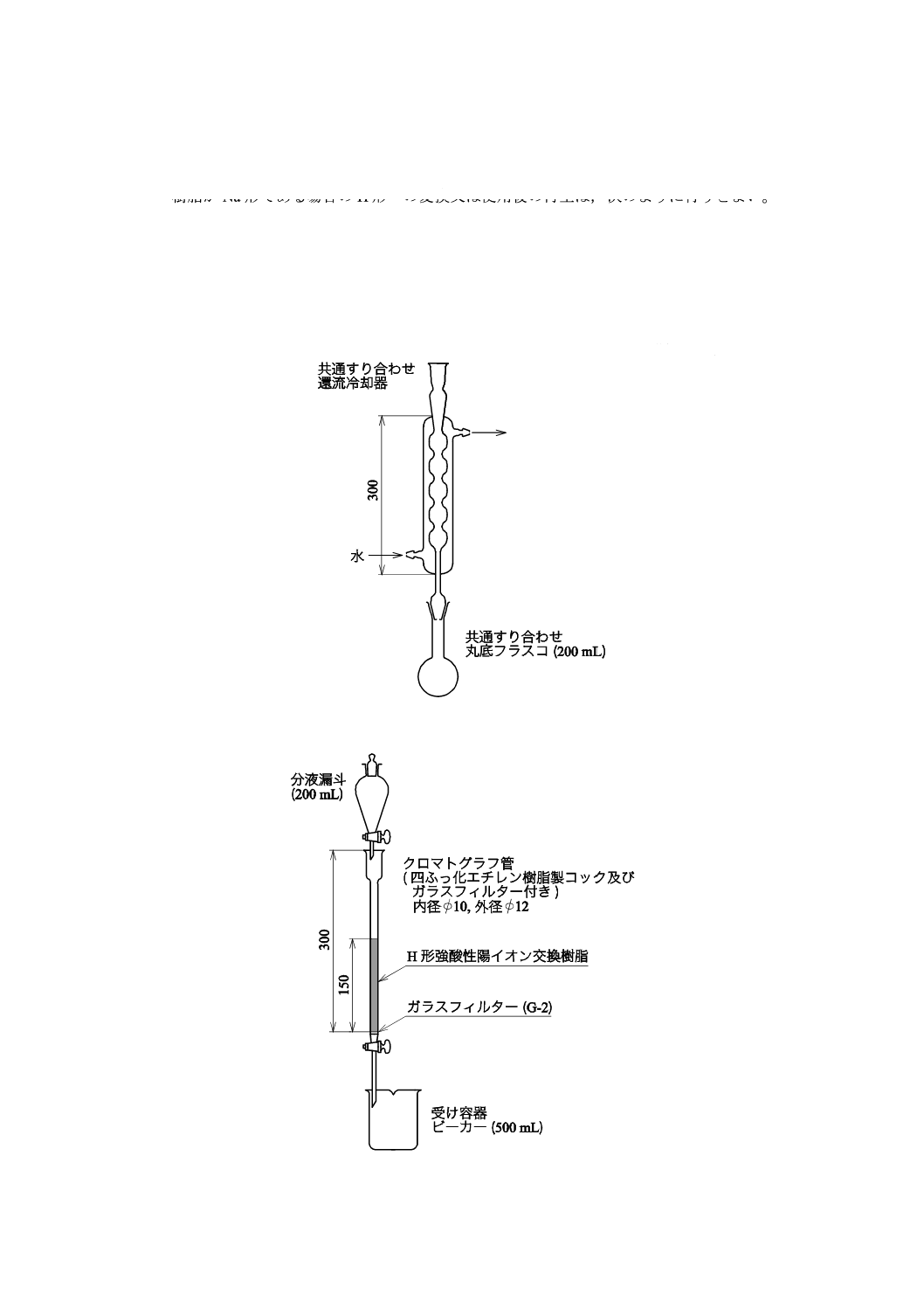
試料分解用容器 共通すり合わせ丸底フラスコ(200 mL)及び共通すり合わせ還流冷却器からな
り,共通すり合わせ丸底フラスコの上部に還流冷却器を接続して内部に冷却水を流す(図1参照)。
5.3.2
イオン交換カラム 粒径250 μm以下のH形強酸性陽イオン交換樹脂1) 2) を,適量の水でスラリー
状態とした後,先端部を太くしたポリエチレン製のこま込めピペットを用いて吸い上げ,四ふっ化エチレ
ン樹脂製コック及びガラスフィルターが付いたクロマトグラフ管(長さ約300 mm,内径約10 mm)の上
部から,クロマトグラフ管を傾斜させて穏やかに注ぎ込み,管内に気泡が生じないように,適量の水を流
し入れて沈降させながらイオン交換樹脂層の高さを約150 mmとする。クロマトグラフ管の上部に試料溶
液及び洗浄水の滴下速度を調節するための分液漏斗(200 mL)を取り付け,下部には受け容器のビーカー
(500 mL)を置く(図2参照)。
なお,粒径250 μm以下のイオン交換樹脂がない場合は,磁製乳鉢で樹脂を少量ずつ粉砕し,目開き250
μmのふるいを用いてふるい分け,ふるい分けした樹脂約150 mLをビーカー(500 mL)に移し入れ,水約
300 mLを加え,十分膨潤させる。かき混ぜてしばらく静置した後,上層の細かい樹脂をデカンテーション
で分離して除去する。水約300 mLを加え,かき混ぜてしばらく静置した後,上層の細かい樹脂をデカン
3
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
テーションで分離して除去する操作を,洗浄も兼ねて更に数回繰り返して整粒した樹脂を用いる。
注1) 強酸性陽イオン交換樹脂として,例えば,ポリスチレンスルホン酸形交換樹脂がある。
2) 樹脂がNa形である場合のH形への変換又は使用後の再生は,次のように行うとよい。
ビーカー(500 mL)に膨潤させた樹脂又は使用後の樹脂約200 mLを入れ,塩酸(1+2)約
300 mLを加え,約5分間よくかき混ぜた後,樹脂と塩酸(1+2)とをデカンテーションで分離
する。さらに,水を加え,かき混ぜて洗浄し,デカンテーションで樹脂と洗液とを分離する操
作を,青色リトマス試験紙に洗液を滴下したとき,リトマス試験紙が赤に変色しなくなるまで
繰り返す。
単位 mm
図1−試料分解用容器の例
単位 mm
図2−イオン交換カラムの例
4
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.4
試料はかりとり量
試料はかりとり量は,1.0 gとする。
5.5
操作
5.5.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって共通すり合わせ丸底フラスコ(200 mL)に移し入れ,図1に示すように共通すり
合わせ還流冷却器を接続してその上部から水20 mLを加え,次に混酸(5.2.2)10 mLを少量ずつ加え
る。始めの激しい反応が終わった後,加熱して約1時間穏やかに煮沸しながら試料を分解する。
b) 放冷した後,共通すり合わせ還流冷却器の上部から水約20 mLを加え,共通すり合わせ還流冷却器の
内壁を洗って取り外す。共通すり合わせ丸底フラスコ中の試料溶液をろ紙(5種B)を用いてビーカー
(300 mL)にろ過し,塩酸(1+100)で4,5回洗浄した後,ろ液及び洗液を合わせて,主液として
保存する。
c) 残さは,ろ紙とともにニッケルるつぼ(30 mL)に入れ,ろ紙を乾燥した後,徐々に加熱してろ紙を
灰化する。るつぼを放冷した後,残さをよくほぐしてから,融解合剤(5.2.4)約2 gを加えて残さと
よく混合する。始めは,低温で加熱した後,徐々に温度を上げて赤熱状態で残さを完全に融解する。
d) 放冷した後,るつぼに温水約20 mLを2回に分けて加え,融成物を溶解した後,溶液をろ紙(5種B)
を用いてビーカー(200 mL)にろ過し,るつぼ,ろ紙及び残さを水で十分に洗浄し,ろ液及び洗液を
合わせる。
e) ろ液及び洗液に,p-ニトロフェノール溶液(5.2.7)を指示薬として3,4滴加え,溶液が黄から無色に
変色するまで塩酸(1+1)を加えた後,b)で保存した主液に合わせて,水を用いて500 mLの全量フ
ラスコに移し入れ,水で標線まで薄める。
5.5.2
イオン交換分離
5.5.1 e)で得た試料溶液100 mLを分液漏斗(200 mL)(図2参照)に分取し,イオン交換カラム(5.3.2)
に1分間当たり5 mLの流量で通し,次に水200 mLを用いて数回に分けて分液漏斗(200 mL)を洗浄し,
その都度,洗液をイオン交換カラムに1分間当たり5 mLの流量で通す。流出液は,ビーカー(500 mL)
に受ける。
5.5.3
滴定
滴定は,次の手順によって行う。
a) 5.5.2で得た溶液のpHを,pH計を用いて,水酸化ナトリウム溶液(40 g/L)でpHを3.0〜3.5に調節
する。
b) 溶液を共通すり合わせ三角フラスコ(500 mL)に移し入れ,共通すり合わせ還流冷却器(図1)を接
続し,加熱して約2分間煮沸した後,室温まで冷却する。溶液をビーカー(500 mL)に水を用いて移
し入れ,pH計を用いて,水酸化ナトリウム溶液(4 g/L)と塩酸(1+100)とでpHを6.9に調節する。
c) 溶液にマンニトール(5.2.5)20 gを加え,よくかき混ぜて溶解した後,pH計を用いて,溶液のpHが
再び6.9になるまで0.1 mol/L水酸化ナトリウム溶液(5.2.6)で滴定し,0.1 mol/L水酸化ナトリウム溶
液(5.2.6)の使用量を求める。
5.6
空試験
試薬だけを用いて,5.5.1 a)〜5.5.3の手順に従って試料と並行して操作する。

5
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.7
計算
試料中のほう素含有率を,次の式によって算出する。
(
)
100
5
1
2
1
×
×
×
−
=
m
f
V
V
B
ここに,
B: 試料中のほう素含有率[%(質量分率)]
V1: 5.5.3 c)で得た0.1 mol/L水酸化ナトリウム溶液の使用
量(mL)
V2: 5.6で得た0.1 mol/L水酸化ナトリウム溶液の使用量
(mL)
f: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当するほう
素量(g/mL)
m: 試料はかりとり量(g)
5.8
許容差
許容差は,表1による。
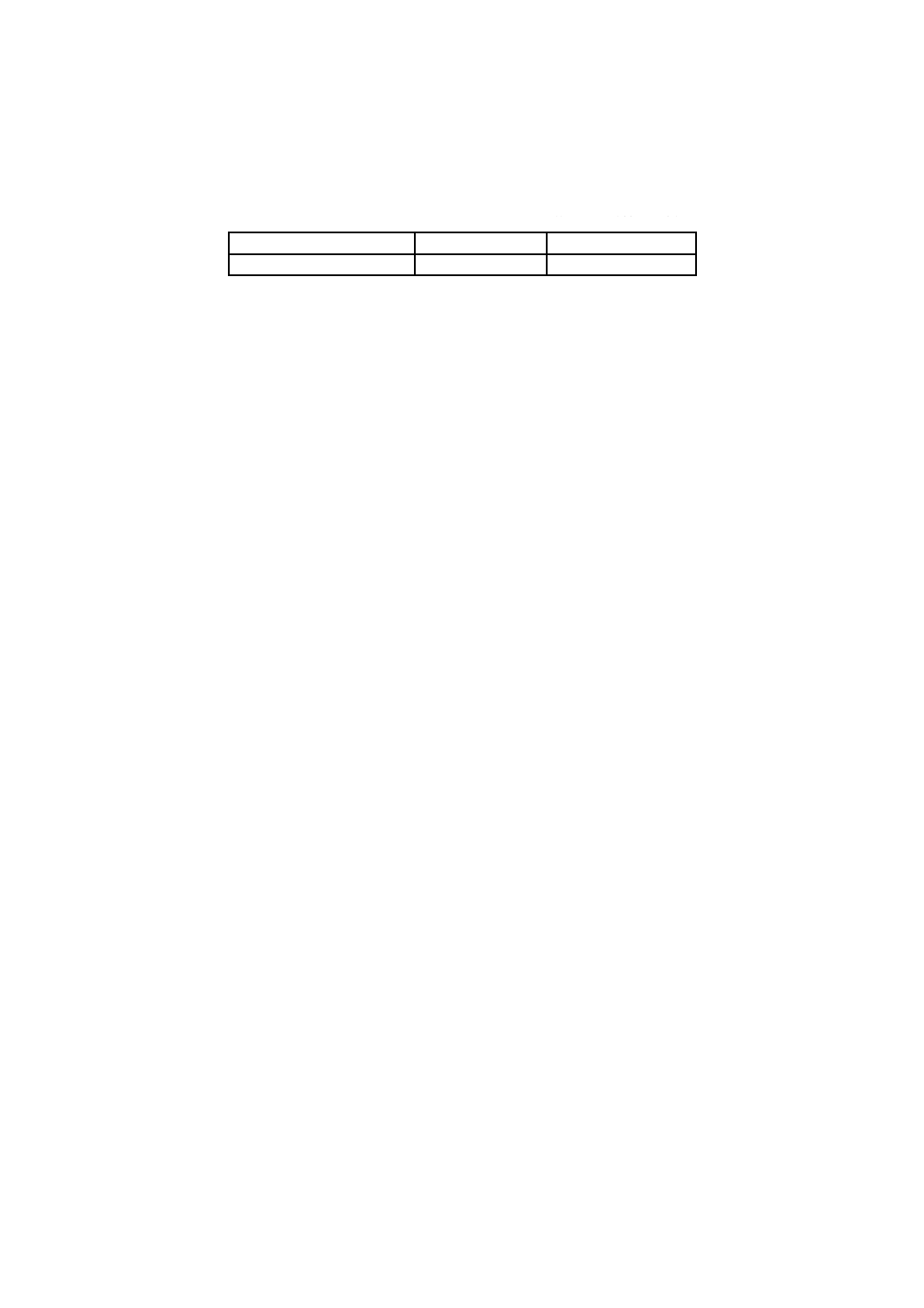
表1−許容差
単位 %(質量分率)
試料中のほう素含有率範囲
室内再現許容差
室間再現許容差
17〜20
0.2
0.5
6
ICP発光分光法
6.1
要旨
試料を過酸化ナトリウム及び炭酸ナトリウムで融解し,融成物を硫酸で溶解する。イットリウムを添加
した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,ほう素及びイットリウムの発光強
度を測定する。
6.2
試薬
試薬は,次による。
6.2.1
硫酸(1+4)
6.2.2
過酸化ナトリウム
6.2.3
炭酸ナトリウム
6.2.4
鉄溶液(Fe:1.6 mg/mL) 鉄[純度99.9 %(質量分率)以上]0.40 gを塩酸(1+1)5 mL及び硝
酸(1+1)10 mLで加熱分解し,250 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
6.2.5
イットリウム溶液(Y:100 μg/mL) 酸化イットリウム(III)[99.9 %(質量分率)以上]1.27 g
を塩酸(1+1)50 mLで加えて加熱して分解し,常温まで冷却した後,溶液を1 000 mLの全量フラスコに
水を用いて移し入れ,水で標線まで薄めて原液(Y:1 000 μg/mL)とする。この原液10 mLを,使用の都
度,100 mLの全量フラスコにとり,水で標線まで薄めてイットリウム溶液とする。
6.2.6
ほう素標準液(B:200 μg/mL) 過塩素酸マグネシウムを入れた真空デシケータ中で保存してあ
る恒量としたほう酸[純度99.9 %(質量分率)以上]5.72 gをはかりとってビーカー(300 mL)に移し入
れ,水を加えて溶解し,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で溶解して標線まで薄
めて原液(1 000 μg/mL)とする。この原液20 mLを,使用の都度,100 mLの全量フラスコにとり,水で
標線まで薄めてほう素標準液とする。
6
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3
試料はかりとり量
試料はかりとり量は,0.20 gとする。
6.4
操作
6.4.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとり,炭酸ナトリウム2.0 gを入れたニッケルるつぼに移し入れ,過酸化ナトリウム3.0 g
を加え,よく混合する。
b) ニッケルるつぼを最初は暗赤熱状態(400〜500 ℃)まで徐々に加熱し,融剤が融け始めたら急激な反
応が生じないように,ニッケルるつぼを回してよく振り混ぜ,反応が穏やかになったら,温度を700
〜800 ℃に上げて,るつぼの底に試料が付着しないように,試料が完全に融解するまで更に揺り動か
しながら融解した後,放冷する。
c) ニッケルるつぼをビーカー(300 mL)中に入れ,時計皿で覆い,硫酸(1+4)100 mLを少量ずつ加
えて可溶性塩類を溶解する。溶解が不十分な場合は,加熱して溶解する。時計皿の下面及びニッケル
るつぼを水で洗浄し,ニッケルるつぼを取り出す。
d) 常温まで冷却した後,溶液を200 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
この溶液10 mLを100 mLの全量フラスコに分取し,イットリウム溶液(6.2.5)を正確に5 mL加え,
水で標線まで薄める。
6.4.2
発光強度比の測定
6.4.1 d)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長208.96 nmに
おけるほう素及び371.03 nmにおけるイットリウムの発光強度を測定し,ほう素含有率とイットリウムと
の発光強度の比を求める。
6.5
空試験
試薬だけを用いて,6.4.1 a)〜6.4.2の手順に従って試料と同じ操作を試料と並行して操作する。ただし,
6.4.1 d)の操作においては,イットリウム溶液(6.2.5)5.0 mLを加えた後,更に鉄溶液(6.2.4)5.0 mLを加
え,水で標線まで薄める。
6.6
検量線の作成
ほう素標準液(6.2.6)0〜15.0 mL(ほう素として0〜3 000 μg)を段階的に数個の100 mLの全量フラス
コにとり,イットリウム溶液(6.2.5)を正確に5.0 mL及び鉄溶液(6.2.4)5.0 mLを加え,水で標線まで
薄める。溶液の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,6.4.2と同一測定条件でほう
素とイットリウムとの発光強度を試料溶液と並行して測定してほう素含有率とイットリウムとの発光強度
の比を求め,得たほう素とイットリウムとの強度の比とほう素含有率との関係線を求め,検量線とする。
6.7
計算
試料中のほう素含有率を,次の式によって算出する。
100
200
102
1
×
×
−
m
A
A
B=
ここに,
B: 試料中のほう素含有率[%(質量分率)]
A1: 分取した試料溶液中のほう素検出量(g)
A2: 分取した空試験液中のほう素検出量(g)
m: 試料はかりとり量(g)
7
G 1327-1:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.8
許容差
許容差は,表2による。
表2−許容差
単位 %(質量分率)
試料中のほう素含有率範囲
室内再現許容差
室間再現許容差
18〜20
0.3
0.6