1
G 1326 : 2000
解説
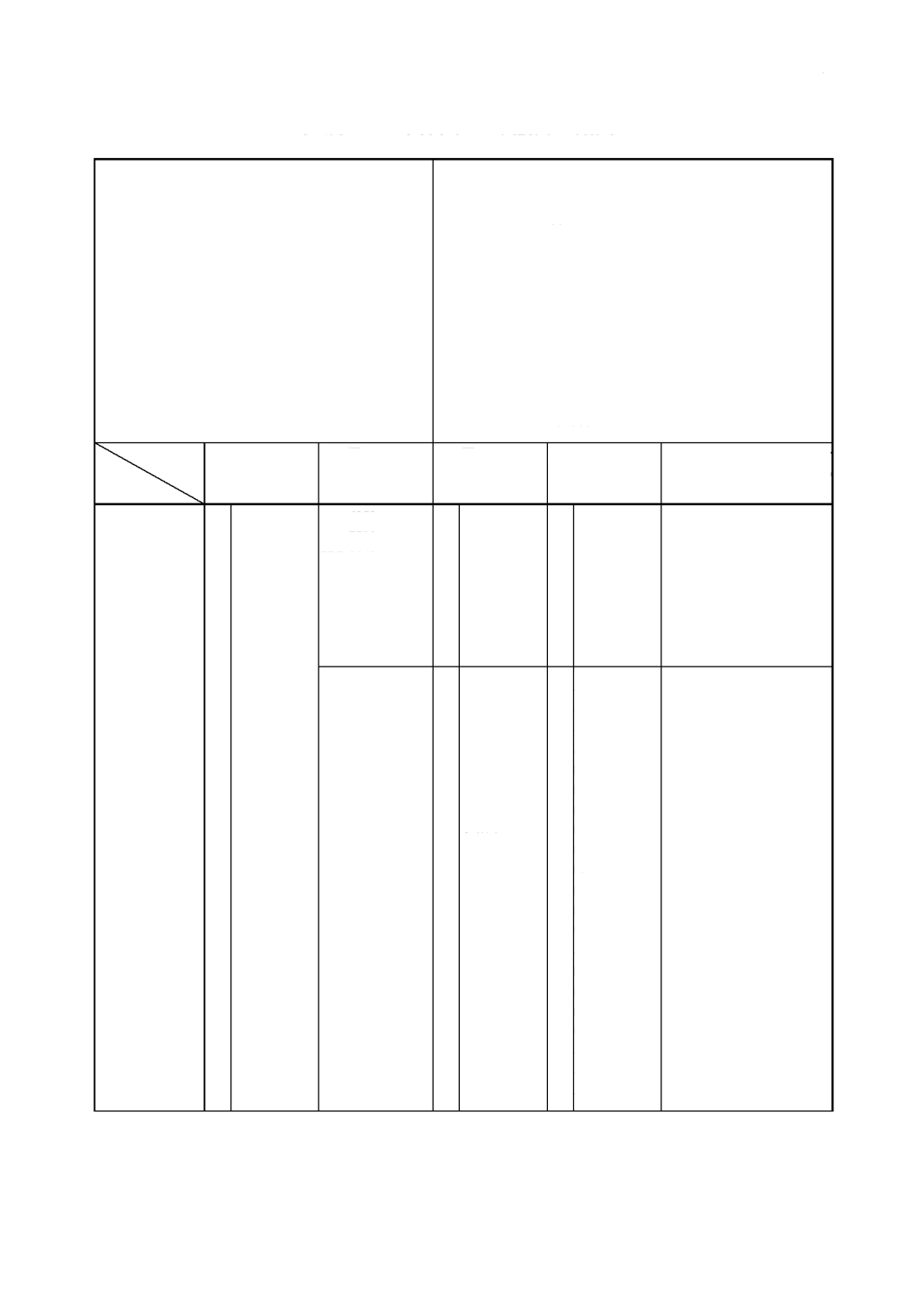
解説表
2
JIS
と対応する国際規格との対比表
JIS G 1326
: 2000
フェロニッケル分析方法
ISO 6352
: 1985
フェロニッケル−ニッケルの定量−ジメチル
グリオキシム重量法
ISO 7520
: 1985
フェロニッケル−コバルトの定量−原子吸光
法
ISO 7524
: 1985
ニッケル,フェロニッケル及びニッケル合金−
炭素の定量−燃焼赤外線吸収法
ISO 7526
: 1985
ニッケル,フェロニッケル及びニッケル合金−
硫黄の定量−燃焼赤外線吸収法
ISO 7527
: 1985
ニッケル,フェロニッケル及びニッケル合金−
硫黄の定量−燃焼よう素酸カリウム滴定法
ISO 8343
: 1985
フェロニッケル−けい素の定量−重量法
ISO 11400
: 1992
ニッケル,フェロニッケル及びニッケル合金
−りんの定量−りんバナドモリブデン酸吸光
光度法
対比項目
規定項目
(I)
JIS
の規定内
容
(II)
国 際 規 格 番
号
(III)
国 際 規 格 の
規定内容
(IV)
JIS
と国際規
格との相違点
(V)
JIS
と国際規格との整合
が困難な理由及び今後の
対策
1.
適用範囲
○
フ ェ ロ ニ ッ
ケ ル 中 の ニ
ッケル,コバ
ルト,炭素,
けい素,マン
ガン,りん,
硫黄,クロム
及 び 銅 の 定
量 方 法 に つ
いて規定。
ISO 6352
ISO 7520
ISO 8343
○ フ ェ ロ ニ ッ
ケ ル 中 の ニ
ッケル,コバ
ル ト 及 び け
い 素 の 含 有
率 を 定 量 す
る 方 法 を 規
定
≡
ISO 7524
ISO 7526
ISO 7527
ISO 11400
○ ニッケル,フ
ェ ロ ニ ッ ケ
ル 及 び ニ ッ
ケ ル 合 金 中
の炭素,硫黄
及 び り ん の
含 有 率 を 定
量 す る 方 法
を規定
= ニ ッ ケ ル 及
び ニ ッ ケ ル
合 金 を 対 象
として,
ISO
7524
に
JIS H
1151
と
H
1275
が,
ISO
7526
と
7527
に
JIS H 1151
と
H 1277
が,
ま た ,
ISO
11400
に
JIS
H 1278
が対
応 す る 。 ま
た ,
JIS H
1151
,
H 1275
,
H 1277
及び
H 1278
はそ
れ ぞ れ 整 合
化 さ れ て い
るので,ニッ
ケ
2
G 1326 : 2000
解説
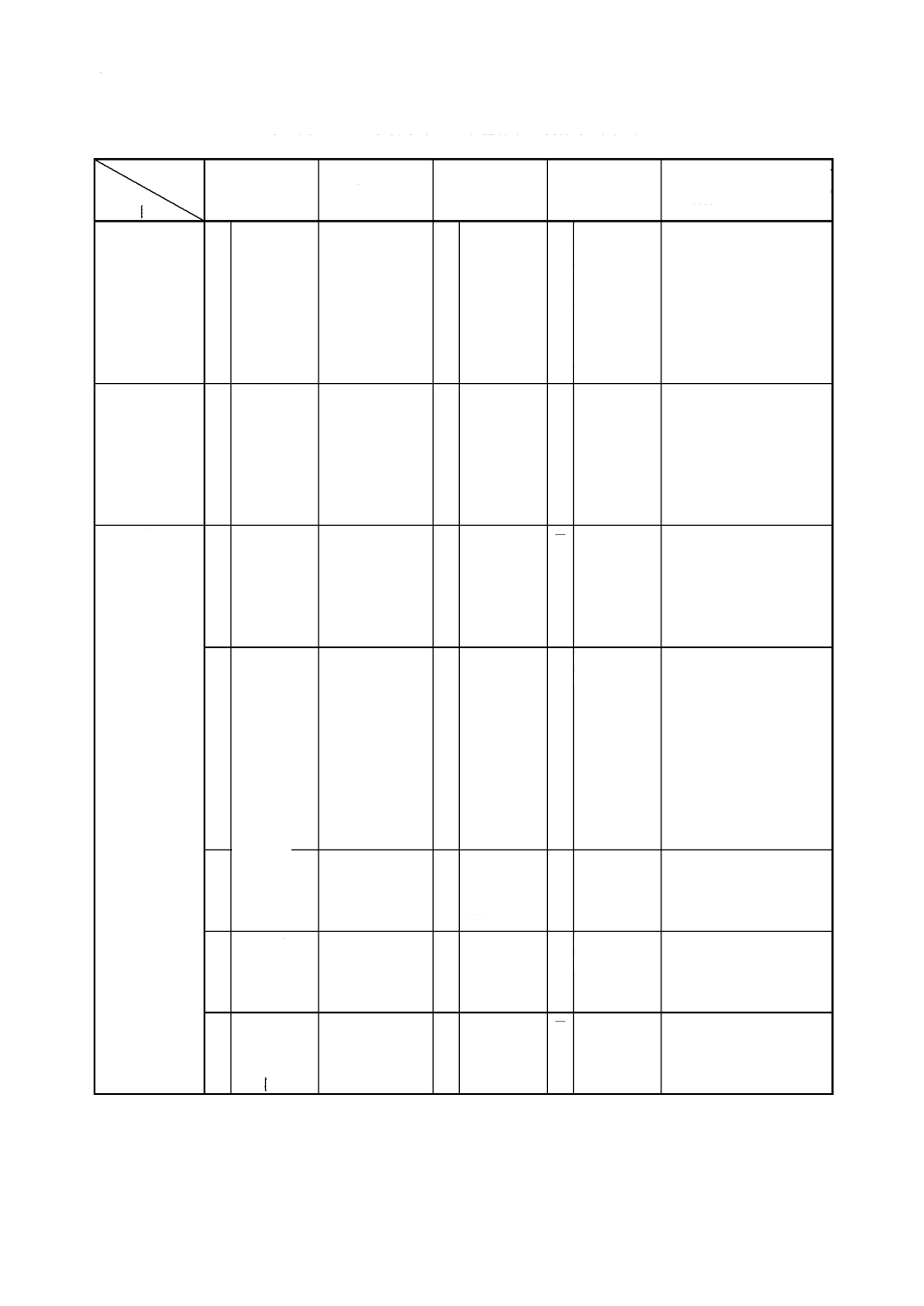
解説表
2
JIS
と対応する国際規格との対比表(続き)
対比項目
規定項目
(I)
JIS
の規定内
容
(II)
国 際 規 格 番
号
(III)
国 際 規 格 の
規定内容
(IV)
JIS
と国際規
格との相違点
(V)
JIS
と国際規格との整合
が困難な理由及び今後の
対策
ル 及 び ニ ッ
ケ ル 合 金 を
削除し,フェ
ロ ニ ッ ケ ル
に つ い て だ
け 附 属 書 方
式 で 採 用 し
た。
2.
一般事項
○
JIS G 1301
に
よる。
ただし,
附属
書
1
,
3
,
5
,
6
,
10
,
11
及び
12
に は 適 用 し
ない
−
−
3.
定 量 方 法 の
区分
○
附属書
1
ニッ
ケ ル 定 量 方
法 − ジ メ チ
ル グ リ オ キ
シム重量法
ISO 6352
○ ジ メ チ ル グ
リ オ キ シ ム
重 量 法 に よ
る
ニ ッ ケ ル の
定量方法
≡
○
附属書
2
ニッ
ケ ル 定 量 方
法 − ジ メ チ
ル グ リ オ キ
シ ム 沈 殿 分
離 エ チ レ ン
ジ ア ミ ン 四
酢 酸 二 水 素
二 ナ ト リ ウ
ム滴定法
−
−
ISO
に規定さ
れていない
○
附属書
3
コバ
ル ト 定 量 方
法 − 原 子 吸
光法
ISO 7520
○ 原 子 吸 光 法
に よ る コ バ
ル ト の 定 量
方法
≡
○
附属書
4
炭素
定 量 方 法 −
燃 焼 − 電 量
法
−
−
ISO
に規定さ
れていない
○
附属書
5
炭素
定 量 方 法 −
燃 焼 − 赤 外
線吸収法
ISO 7524
○ 燃 焼 赤 外 線
吸 収 法 に よ
る 炭 素 の 定
量方法
≡
3
G 1326 : 2000
解説
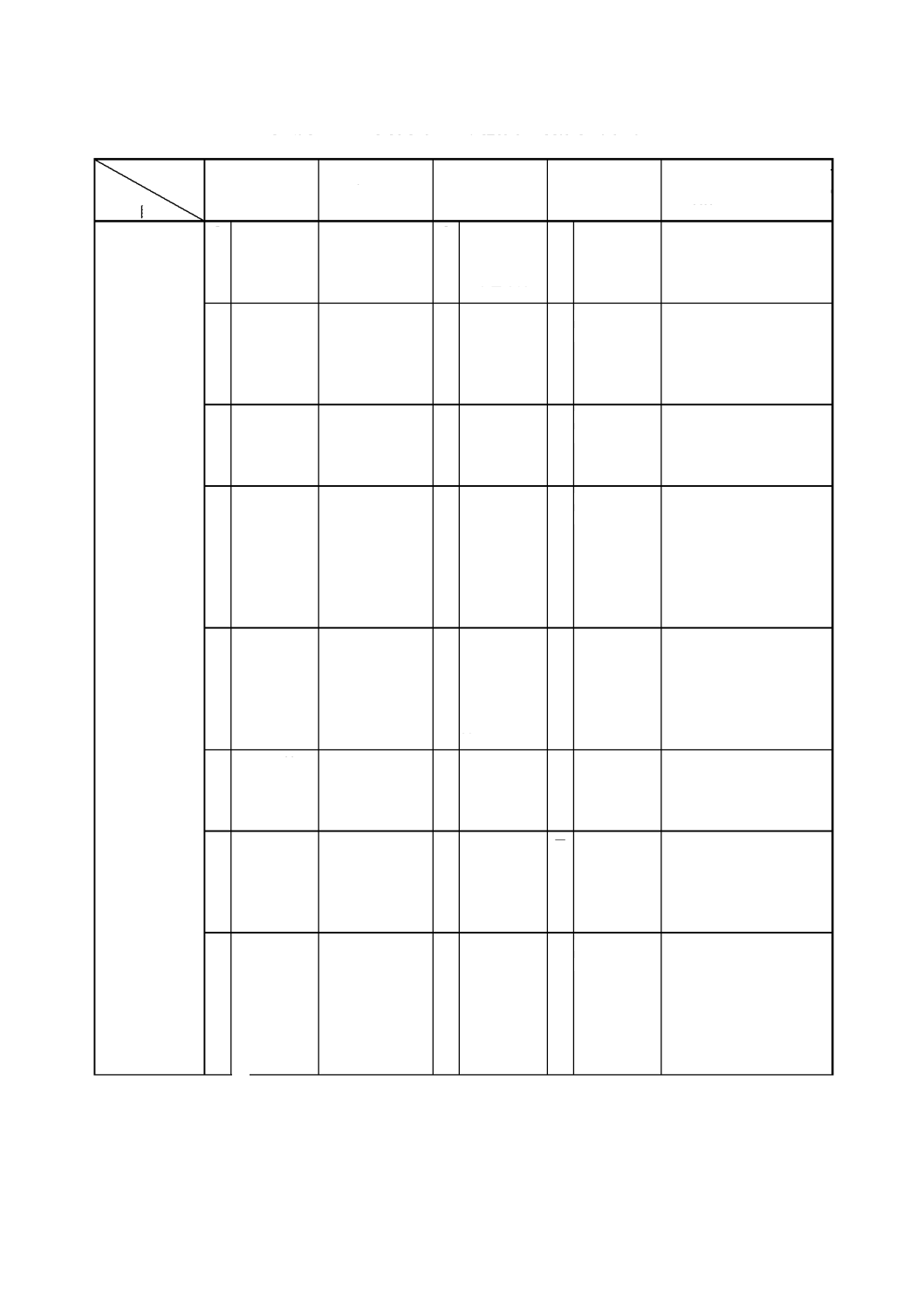
解説表
2
JIS
と対応する国際規格との対比表(続き)
対比項目
規定項目
(I)
JIS
の規定内
容
(II)
国 際 規 格 番
号
(III)
国 際 規 格 の
規定内容
(IV)
JIS
と国際規
格との相違点
(V)
JIS
と国際規格との整合
が困難な理由及び今後の
対策
3.
定 量 方 法 の
区分
○
附属書
6
けい
素 定 量 方 法
− 二 酸 化 け
い素重量法
ISO 8343
○ 二 酸 化 け い
素 重 量 法 に
よ る け い 素
の定量方法
≡
○
附属書
7
けい
素 定 量 方 法
− モ リ ブ ド
け い 酸 青 吸
光光度法
−
−
ISO
に規定さ
れていない
○
附属書
8
マン
ガ ン 定 量 方
法 − 原 子 吸
光法
−
−
ISO
に規定さ
れていない
○
附属書
9
りん
定 量 方 法 −
モ リ ブ ド り
ん 酸 抽 出 分
離 モ リ ブ ト
り ん 酸 青 吸
光光度法
−
−
ISO
に規定さ
れていない
○
附属書
10
り
ん 定 量 方 法
− モ リ ブ ド
バ ナ ド り ん
酸 抽 出 吸 光
光度法
ISO 11400
○ り ん バ ナ ド
モ リ ブ デ ン
酸 吸 光 光 度
法 に よ る り
ん の 定 量 方
法
≡
○
附属書
11
硫
黄 定 量 方 法
− 燃 焼 − 赤
外線吸収法
ISO 7526
○ 燃 焼 赤 外 線
吸 収 法 に よ
る 硫 黄 の 定
量方法
≡
○
附属書
12
硫
黄 定 量 法 −
燃 焼 − よ う
素 酸 カ リ ウ
ム滴定法
ISO 7527
○ 燃 焼 よ う 素
酸 カ リ ウ ム
滴 定 法 に よ
る 硫 黄 の 定
量方法
≡
○
附属書
13
硫
黄 定 量 方 法
− 硫 化 水 素
気 化 分 離 メ
チ レ ン ブ ル
ー 吸 光 光 度
法
−
−
ISO
に規定さ
れていない
4
G 1326 : 2000
解説
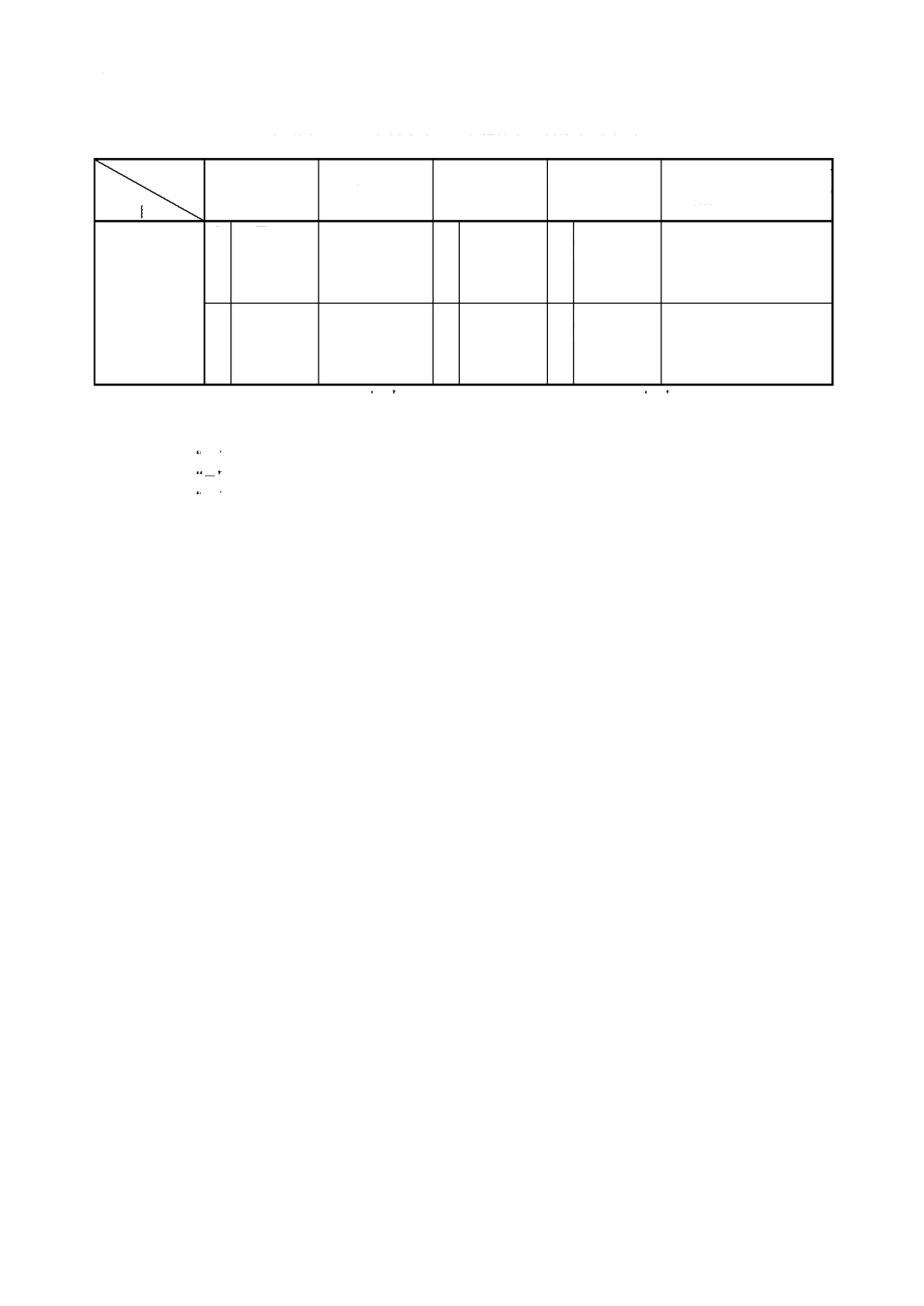
解説表
2
JIS
と対応する国際規格との対比表(続き)
対比項目
規定項目
(I)
JIS
の規定内
容
(II)
国 際 規 格 番
号
(III)
国 際 規 格 の
規定内容
(IV)
JIS
と国際規
格との相違点
(V)
JIS
と国際規格との整合
が困難な理由及び今後の
対策
3.
定 量 方 法 の
区分
○
附 属 書
14
ク ロ ム 定 量
方 法 − 原 子
吸光法
−
−
ISO
に規定さ
れていない
○
附 属 書
15
銅 定 量 方 法
− 原 子 吸 光
法
−
−
ISO
に規定さ
れていない
備考
1.
対比項目
(I)
及び
(III)
の小欄で, ○
は該当する項目を規定している場合, −
は規定していない場合を
示す。
2.
対比項目
(IV)
の小欄の記号の意味は,次による。
≡ :
JIS
と国際規格との技術的内容は同等である。
= :
JIS
と国際規格との技術的内容は同等である。ただし,軽微な技術上の差異がある。
− :該当項目がない場合。