A 1967:2015
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 2
3 用語及び定義 ··················································································································· 2
4 原理······························································································································· 2
5 試薬・材料 ······················································································································ 3
5.1 揮発性有機化合物(VOC) ······························································································ 3
5.2 希釈溶媒 ······················································································································ 3
5.3 吸着剤 ························································································································· 3
5.4 検量線用標準 ················································································································ 3
5.5 標準空気 ······················································································································ 3
5.6 標準空気添加サンプラ ···································································································· 3
5.7 液体添加用溶液の調製 ···································································································· 4
5.8 標準液体添加サンプラ ···································································································· 4
6 装置······························································································································· 5
6.1 サンプラ ······················································································································ 5
6.2 サンプラエンドキャップ ································································································· 5
6.3 サンプラサンプリングキャップ ························································································ 5
6.4 シリンジ ······················································································································ 5
6.5 ガスクロマトグラフ ······································································································· 5
6.6 加熱脱離装置 ················································································································ 5
6.7 標準サンプラ調製のための注入装置··················································································· 6
7 サンプラの前処理 ············································································································· 6
8 サンプリング ··················································································································· 6
9 手順······························································································································· 7
9.1 安全上の注意 ················································································································ 7
9.2 脱離及び分析 ················································································································ 7
9.3 検量線 ························································································································· 8
9.4 試料濃度の測定 ············································································································· 9
9.5 脱離効率の測定 ············································································································· 9
9.6 拡散取込み速度の算出 ···································································································· 9
10 計算 ····························································································································· 9
10.1 分析対象成分の質量濃度 ································································································ 9
10.2 分析対象成分の体積比 ··································································································· 9
10.3 拡散取込み速度u及びu'の関係 ······················································································ 10
A 1967:2015 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
11 妨害 ···························································································································· 10
12 分析特性 ······················································································································ 10
13 試験報告書 ··················································································································· 10
14 品質管理 ······················································································································ 11
附属書A(参考)パッシブサンプリングの作用原理 ··································································· 20
附属書B(参考)吸着剤の種類 ······························································································ 25
附属書C(参考)吸着剤選択の手引き ····················································································· 26
附属書D(参考)吸着剤使用の手引き ····················································································· 27
附属書E(参考)総合不確かさ,精度,偏り及び保管要領 ··························································· 28
附属書JA(参考)日本におけるパッシブサンプリングの実測データ ············································· 30
参考文献 ···························································································································· 31
附属書JB(参考)JISと対応国際規格との対比表 ······································································ 35
附属書JC(参考)技術上重要な改正に関する新旧対照表 ···························································· 38
A 1967:2015
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づき,日本工業標準調査会の審議を経て,国土交通大臣が改正した日本
工業規格である。
これによって,JIS A 1967:2005は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。国土交通大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
A 1967:2015
室内空気中の揮発性有機化合物(VOC)の
吸着捕集・加熱脱離・キャピラリー
ガスクロマトグラフィーによるサンプリング及び
分析−パッシブサンプリング
Indoor air-Sampling and analysis of volatile organic compounds by sorbent
tube/thermal desorption/capillary gas chromatography-Diffusive sampling
序文
この規格は,2003年に第1版として発行されたISO 16017-2を基とし,国内の実情を反映させるため,
技術的内容を変更して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,対応国際規格を変更している事項である。
変更の一覧表にその説明を付けて,附属書JBに示す。また,技術上重要な改正に関する新旧対照表を,
附属書JCに示す。
1
適用範囲
この規格は,空気中の揮発性有機化合物(VOC)のサンプリング及び分析について一般的な指針を示す。
この規格は,室内,大気及び労働環境の空気に適用する。
この規格は,炭化水素類,ハロゲン化炭化水素類,エステル類,グリコールエーテル類,ケトン類及び
アルコール類を含む広範囲なVOCに適用する。これらのVOCの測定にはそれぞれの適用範囲が異なるサ
ンプラ1) を用いるとよい。ただし,高極性物質は誘導体化が必要で,低沸点化合物はその一部分しか吸着
剤に捕集されないため,定性的な評価だけ可能である。また,準揮発性化合物は吸着剤に全て吸着される
が,一部分しか回収することができない。
この規格は,空気を汚染しているVOCの測定に適用し,暴露時間8時間の場合2〜1×105 μg/m3,暴露
時間4週間の場合0.3〜300 μg/m3の濃度範囲で適用可能である。
使用可能な範囲の上限は,使用する吸着剤の吸着容量及びガスクロマトグラフのカラム・検出器の直線
領域又は使用する分析機器の能力によって設定する。使用可能な範囲の下限は,検出器のノイズレベル及
び分析系又はサンプラからの妨害物質による。妨害物質が1 ng以下の吸着剤の代表例としては,適切に調
製されたTenax GR,Carbopack/Carbotrapなどの炭素系吸着剤,カーボンモレキュラーシーブ及びSpherocarb
などの純活性炭の例があり,数ngレベルの例としてはTenax TA,また,5〜50 ngレベルについては
Chormosorb,Porapackなどの多孔質ポリマーがある。
注記 この規格の対応国際規格及びその対応の程度を示す記号を,次に示す。
ISO 16017-2:2003,Indoor, ambient and workplace air−Sampling and analysis of volatile organic
2
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
compounds by sorbent tube/thermal desorption/capillary gas chromatography−Part 2: Diffusive
sampling(MOD)
なお,対応の程度を表す記号“MOD”は,ISO/IEC Guide 21-1に基づき,“修正している”
ことを示す。
注1)
附属書Bに示した吸着剤及びこの規格にある吸着剤(商品名)は,この規格に規定する性能を
もつ吸着剤として知られているものであり,使用者の便宜のために,一般に入手できるものと
して挙げたが,これらを推奨するわけではない。同じ結果が得られることを証明することがで
きれば,これらと同等の他のものを用いてもよい。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS A 1966 室内空気中の揮発性有機化合物(VOC)の吸着捕集・加熱脱離・キャピラリーガスクロ
マトグラフィーによるサンプリング及び分析−ポンプサンプリング
ISO 6141:2000,Gas analysis−Requirements for certificates for calibration gases and gas mixtures
ISO 6145-1,Gas analysis−Preparation of calibration gas mixtures using dynamic volumetric methods−Part 1:
Methods of calibration
ISO 6145-4,Gas analysis−Preparation of calibration gas mixtures using dynamic volumetric methods−Part 4:
Continuous syringe injection method
ISO 6145-5,Gas analysis−Preparation of calibration gas mixtures using dynamic volumetric methods−Part 5:
Capillary calibration devices
ISO 6145-6,Gas analysis−Preparation of calibration gas mixtures using dynamic volumetric methods−Part 6:
Critical orifices
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS A 1966による。
4
原理
パッシブサンプラを測定期間中空気に暴露する。サンプリング速度は事前の標準空気による校正によっ
て決定する(9.6)。
VOCのガスは拡散によってサンプラ内を移動し,吸着剤に捕集される。サンプラに捕集したガスを加熱
脱離し,不活性キャリヤーガスによってキャピラリーカラム及び水素炎イオン化検出器又は他の適切な検
出器を装備したガスクロマトグラフで分析する。校正は,サンプラに添加した液体又は気体を用いて行う。
吸着剤層の想定される飽和状態,変動及び面風速に関する情報を附属書Aに記載する。また,拡散取込
み速度は汚染物質の濃度レベル及びサンプリング時間に影響され,表1及び表2に示す値と異なることが
ある。パッシブサンプラ性能に関する理論の詳細情報は,prEN 13528-3[1]に規定されている。
参考として,附属書JAに日本におけるパッシブサンプリングの実測データを示す。
3
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5
試薬・材料
分析には,高純度の分析用試薬だけを用いる。検量線用混合溶液は,1週間ごとに調製することが望ま
しい。また,アルコールとカルボン酸間との縮合反応などの劣化が顕著に示される場合は,更に間隔を短
くして調製する。
5.1
揮発性有機化合物(VOC)
VOCは,サンプラへの標準液体添加(5.7〜5.8)又は標準空気添加(5.4〜5.6)に使用する検量線用試薬
として必要である。
5.2
希釈溶媒
標準液体添加(5.7)に使用する検量線用混合溶液の調製のために希釈溶媒が必要である。クロマトグラ
フ用品質のものとする。分析対象成分のピークと重なる成分を含まないものとする。
注記 通常,メタノールが使用される。化学反応又はクロマトグラフ的に重なる可能性が特になけれ
ば,代わりに他の溶媒が使用できる。
5.3
吸着剤
吸着剤は,粒径0.18〜0.25 mm(60〜80メッシュ)のもので,サンプラに充塡する前に不活性ガス流の
もとで,吸着剤の最高使用温度より少なくとも25 ℃程度低い温度で一晩加熱して,前処理する。これら
を清浄な空気のもとで室温まで冷却した後サンプラに充塡し,保管するのがよい。可能であれば,分析時
の脱離温度は前処理条件よりも低く保つことが望ましい。あらかじめ充塡された市販のサンプラの入手が
可能である場合は,前処理だけが必要である。
注記 吸着剤選択の種類を附属書Cに記載する。吸着剤は,附属書に記載する種類以外の同等の吸着
剤を使用してもよい。吸着剤の前処理及び分析時の脱離条件を,附属書Dに記載する。
5.4
検量線用標準
検量線用標準は,実際のサンプリング状態に合わせるため,必要量の対象成分が含まれた標準空気を添
加することによって調製することが望ましい(5.5及び5.6参照)。
この調製方法が使えない場合,次のいずれかの方法によって精度を確認した液体添加法(5.7〜5.8参照)
を用いて,検量線用標準を調製してもよい。
a) 質量及び/又は体積の一次標準に完全にトレーサブルな添加量を与える手順が確立されている方法
b) 標準物質によって確認されている方法
c) 標準空気を使用して作成された標準によって確認されている方法
d) 標準測定方法での結果によって確認されている方法
5.5
標準空気
標準空気は,既知濃度の対象成分から独自の方法によって調製する。ISO 6141及びISO 6145規格群に
よる方法が適切である。この方法で,生成濃度が一次標準(質量及び/又は体積)へ十分なトレーサビリ
ティを確立できない場合,又は発生システムの化学的不活性が保証できない場合,独自の方法で濃度を確
認しなければならない。
注記 標準空気については,参考文献[59]〜[64]を参照。
5.6
標準空気添加サンプラ
正確に測定した体積の標準空気を,例えば,ポンプを用いてサンプラ内を通過させることによって,標
準添加サンプラを調製する。サンプリングする空気の体積は,吸着剤の破過容量を超えてはならない。添
加後,サンプラを外して密栓する。試料ロットごとに新しい標準を調製する。
例えば,対象成分が10 mg/m3及び100 μg/m3となるよう,標準空気を調製する。作業場の空気の測定で
4
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
は,サンプラに10 mg/m3の空気を100 mL,200 mL,400 mL,1 L,2 L又は4 L添加する。大気又は室内
空気の測定では,サンプラに100 μg/m3の空気を100 mL,200 mL,400 mL,1 L,2 L,4 L又は10 L添加
する。
5.7
液体添加用溶液の調製
5.7.1
各液体成分約10 mg/mLを含む溶液
100 mLの全量フラスコに分析対象成分約1 gを正確にはかりとる。最も揮発性の少ない物質から計量を
始める。希釈溶媒(5.2)で100 mLにし,栓をして振り混ぜる。
5.7.2
各液体成分約1 mg/mLを含む溶液
100 mLの全量フラスコに希釈溶媒50 mLを入れる。5.7.1の溶液10 mLを加える。希釈溶媒で100 mL
にし,栓をして振り混ぜる。
5.7.3
各液体成分約100 μg/mLを含む溶液
100 mLの全量フラスコに分析対象成分約10 mgを正確にはかりとる。最も揮発性の少ない物質から計量
を始める。希釈溶媒(5.2)で100 mLにし,栓をして振り混ぜる。
5.7.4
各液体成分約10 μg/mLを含む溶液
100 mLの全量フラスコに希釈溶媒50 mLを入れる。5.7.3の溶液10 mLを加える。希釈溶媒(5.2)で100
mLにし,栓をして振り混ぜる。
5.7.5
気体成分約1 mg/mLを含む溶液
例えば,酸化エチレンのようなガスについては,高濃度の検量線用溶液を,次の方法で調製してもよい。
純ガスの高圧ガス容器から小形の樹脂製ガス袋にガスを充塡し,大気圧下のガスを得る。1 mLのガスタイ
トシリンジに純ガス1 mLを満たし,シリンジの弁を閉じる。2 mLのセプタムバイアルに2 mLの希釈溶
媒を加え,セプタムキャップで栓をする。セプタムキャップを通してシリンジの針の先端を希釈溶媒に挿
入する。弁を開きプランジャーを少し引き,希釈溶媒をシリンジに導入する。ガスが希釈溶媒に溶けるこ
とで,中が負圧となり,シリンジは溶媒で満たされる。溶液をバイアルに戻す。シリンジを溶液で2回洗
浄し,洗液をバイアルに戻す。気体の法則,すなわち,気体の標準状態(温度:273.15 ℃,圧力:1 013.25
hPa)における1 molの気体は22.4 Lであることを利用して,加えたガスの質量を計算する。
5.7.6
気体成分約10 μg/mLを含む溶液
例えば,酸化エチレンのようなガスについては,低濃度の検量線用溶液を,次の方法で調製してもよい。
高圧ガス容器から小形の樹脂製ガス袋に充塡し,大気圧下のガスを得る。10 μLのガスタイトシリンジに
純ガス10 μLを満たし,シリンジの弁を閉じる。2 mLのセプタムバイアルに2 mLの希釈溶媒を加え,セ
プタムキャップで栓をする。セプタムキャップを通してシリンジの針の先端を希釈溶媒に挿入する。弁を
開きプランジャーを少し引き,希釈溶媒をシリンジに導入する。ガスが希釈溶媒に溶けることで,中が負
圧となり,シリンジは溶媒で満たされる。溶液をバイアルに戻す。シリンジを溶液で2回洗浄し,洗液を
バイアルに戻す。気体の法則,すなわち,気体の標準状態において1 molの気体は22.4 Lであることを利
用し,加えたガスの質量を計算する。
5.8
標準液体添加サンプラ
標準液体添加サンプラは,清浄なサンプラに標準溶液を分取,注入して調製する。サンプラを注入装置
(6.7)に取り付け,そこに不活性パージガスを流し,適切な標準溶液1〜4 μLを分取し,セプタムを通し
て注入する。適切な時間が経過後,サンプラを取り外し密栓する。試料の各ロットごとに新しい標準サン
プラを調製する。作業環境については,5.7.3,5.7.4又は5.7.6の溶液の1〜5 μLをサンプラに添加する。
注記 希釈溶媒がメタノールの場合,サンプラから溶媒を除去するパージガスを100 mL/minで5分間
5
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
流す方法が適切であった。他の希釈溶媒を用いる場合,パージ条件は実験で決定するほうがよ
い。
6
装置
6.1
サンプラ
サンプラは,使用する加熱脱離装置(6.6)と互換性がなければならない。例えば,ステンレス鋼製サン
プラで,外径6.3 mm,内径5 mm,及び長さ90 mmのものが一般的であるが,これに限るものではない。
これ以外の寸法のサンプラを使用してもよいが,表1及び表2に示す拡散取込み速度は,この寸法のサン
プラに基づいている。ただし,硫黄化合物などの不安定物質については,ガラスコーティング及びガラス
製のサンプラ(通常は,内径4 mm)を用いて行う。
なお,この場合,取込み速度はステンレス製の場合と異なるので注意する。ステンレス製のときは,取
込み速度を別途求める。
このサンプラの一方の端には,例えば,サンプリング端末から約10 mmにリング状の刻みなどのマーク
がある。サンプラには前処理した吸着剤が充塡されており,極低流量時の拡散侵入による誤差を最小とす
るために,吸着剤充塡部は加熱ゾーン内にあり,かつ,約14 mmの空隙が各端に設けられている。
表1の拡散取込み速度は,少なくとも14 mmの空隙(吸着剤層とサンプリング端末キャップとの間)の
サンプラによるものである。実際は充塡済みのサンプラ寸法は異なる(参考文献[2])。
サンプラは,吸着剤の密度にもよるが,200〜1 000 mgの吸着剤が充塡される。通常,多孔性ポリマー,
カーボンモレキュラーシーブ又はグラファイトカーボンなどの吸着剤が充塡される。吸着剤は,サンプラ
の一種でステンレス鋼製金網,非シラン化グラスウールプラグなどを用いて固定されている。
6.2
サンプラエンドキャップ
サンプラは,例えば,ポリテトラフルオロエチレン(PTFE)シール付金属スクリューキャップなどを用
いて密閉する。
6.3
サンプラサンプリングキャップ
サンプリングキャップは6.2と同様であるが,ガスの通過が可能な金網をもち,開口部のサイズはサン
プラの断面と同じである。
水分の多いときは,金網の内側にシリコーン膜が組み込まれているサンプリングキャップを用いる。そ
の場合は,拡散取込み速度を別途求める。
6.4
シリンジ
0.1 μLまで読取りが可能な10 μLの精密液体シリンジ,0.1 μLまで読取りが可能な10 μLの精密ガスタ
イトシリンジ,及び0.01 mLまで読取りが可能な1 mLの精密ガスタイトシリンジを用いる。
6.5
ガスクロマトグラフ
水素炎イオン化検出器,光イオン化検出器,質量分析計又は他の適切な検出器付きのガスクロマトグラ
フで,最低5:1のS/N比でトルエン1 ngの注入を検出できるものが適している。
キャピラリーカラムは,分析対象成分が他の成分から分離できるものを使う。
6.6
加熱脱離装置
サンプラを2段階で加熱脱離し,脱離した気体を不活性ガスによってガスクロマトグラフに送り込む装
置である。典型的なものは,加熱脱離されるサンプラを保持すると同時に,不活性キャリヤーガスでパー
ジする機能をもつ。脱離温度及び時間は,キャリヤーガス流量と同様,調整可能である。また,自動サン
プルチューブ装塡,漏れ試験,脱離成分を濃縮する移送管(トランスファーライン)の冷却トラップなど
6
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の機能を付加できるものがよい。パージガスに含まれた脱離成分は,加熱したトランスファーラインを通
ってガスクロマトグラフのキャピラリーカラムに送られる。
6.7
標準サンプラ調製のための注入装置
ガスクロマトグラフの注入口を,標準サンプラの調製に使用してもよい。この場合,そのまま使用する
ことも,また,分離・据え付けての使用も可能である。注入口へのキャリヤーガスラインは維持されてい
ることが望ましい。サンプラを取り付けるため必要であれば,注入装置の後部を改造するのがよい。この
場合,Oリングシールで圧着する方法が便利である。
7
サンプラの前処理
前処理が必要なサンプラは使用に先立って,分析脱離温度又はこれより僅かに高い温度(附属書D参照)
で処理する。例えば,キャリヤーガスを流速100 mL/minで10分間流す。キャリヤーガスは,サンプリン
グ時と反対方向に流す。
その後,通常の分析パラメータを用いてサンプラを分析して,加熱脱離ブランク値が十分小さいことを
確認し,このブランク値が容認できない場合,この手順を繰り返してサンプラを再処理することが望まし
い。
一旦,試料の分析が済めば,サンプラは他の試料の捕集に直ちに再使用してもよい。ただし,サンプラ
が再使用前に長期間放置されていた場合又は異なる分析対象成分をサンプリングする場合には,加熱脱離
ブランク値の確認をする必要がある。サンプリングを行わない場合又は前処理済みの場合,サンプラは,
適切なPTFEフェラル付きの金属スクリューキャップで密閉し,気密容器に保管するのが望ましい。
注記 妨害ピークが,対象成分を分析した際の面積の10 %以内であれば,サンプラのブランク値は許
容される。
8
サンプリング
分析対象成分,又は混合物に適したサンプラを選択する。適切な吸着剤についての指針を,表1及び表
2に規定する。また,吸着剤の種類は,附属書Bに記載する。
サンプリング直前に,サンプラの仕様に基づき密閉キャップを外し,サンプラキャップと取り替える。
サンプラキャップが正しく取り付けられ,サンプラ密閉キャップが所定の位置にあることを確認する。
個人暴露量測定サンプリングに使用する場合は,サンプラは呼吸域に取り付けることが望ましい。固定
位置でサンプリングに使用する場合は,適切なサンプリング位置を選択する(室内空気については,JIS A
1960[59]参照)。推奨する空気のサンプリング場所の選定及び悪環境条件からの試料の保護については,附
属書Aに記載する。また,参考文献としてprEN 13528-3[1]がある。
次の三つの主要な要件に注意しなければならない。風速,降雨からの保護及び安全のための保護手段で
ある。主要要件については,次項以降で詳細に説明する。パッシブサンプリングの作用原理は,附属書A
に記載し,参考文献として[1]がある。
サンプラは,面速に関する要件を満たしている条件の下で暴露する。サンプリングキャップ(6.3)付き
のサンプラについては,風速の影響はない。他の装置には,最低風速も含め異なる条件をもつものもある。
注記 この項目の内容は,JIS A 1960[59]と同等である。
風速が非常に大きい場合(12 m/s以上),性能特性が十分把握されていないため,使用者はこれらの影響
の可能性についても注意する必要がある。
サンプリング開始時,終了時に時間,必要に応じ気温及び気圧を記録する。
7
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
サンプラサンプリングキャップをサンプラ密閉キャップと取り替え,しっかりと確実に密閉する。サン
プラは識別のためラベルを付ける。溶剤を含む塗料及びマーカ又は粘着ラベルを,サンプラのラベルに用
いてはならない。
試料を8時間以内に分析しない場合は,清潔でコーティングされていない密閉した金属又はガラス製の
容器内に保管する。規定条件に換算した濃度を表示したいときは,サンプリング中定期的に気温及び気圧
を記録する(10.1)。
現場ブランク(トラベルブランクと同じである。)は,サンプリングに使用したものと同じサンプラを用
いて,また,実際にサンプリングした期間以外のサンプラと同様の取扱い手順で処理する。これらのサン
プラには,ブランクであることのラベルを付け識別する。
9
手順
9.1
安全上の注意
この規格は,使用に関する全ての安全性に関して規定しているわけではない。この規格の使用者は,事
前に,適切な健康及び安全性のための手順を確立し,規制条件を決めなければならない。
9.2
脱離及び分析
9.2.1
脱離
サンプラを適合する加熱脱離装置内に設置する。空気をサンプラから不活性ガスによってパージし,吸
着剤及びガスクロマトグラフの固定相の加熱酸化から生じる生成物による妨害を防ぐ。次にサンプラを加
熱し,気化したガスを,キャリヤーガス流によってガスクロマトグラフに導入する。この段階でガスの流
れの方向は,サンプリング時とは逆とする。サンプラのサンプリング側の端,すなわち,マークを付けた
ほうをガスクロマトグラフカラム入口に接続する。最適脱離効率を示すサンプラ内のガス流量は,30〜50
mL/minである。
初回のエアパージでは,サンプラ内の空気量(2〜3 mL)を完全に置き換えるため,不活性ガスはサン
プラ容積の10倍(20〜30 mL)が通常必要とされる。しかし,親水性の強い吸着剤を使用する場合は,冷
却トラップに氷が形成されないよう,吸着した空気と水とを除くより多くのパージが必要となる。パージ
中は,サンプラの加熱が最小となるよう注意する。
脱離試料は数mLのガス量となるので,キャピラリーガスクロマトグラフ分析前に濃縮が必要である。
濃縮は小形の二次吸着剤冷却トラップを用いれば可能で,このトラップは低流量(5 mL/min未満)で十分
急速に脱離され,成分幅を最小にし,かつ,キャピラリーに適したピークをつくる。若しくは,空の二次
トラップ又はガラスベッドなどの不活性材料を含むものを,試料の予備濃縮に用いてもよいが,これらの
トラップは−100 ℃以下での冷却を必要とする。また,脱離試料を直接ガスクロマトグラフ(一段階脱離)
へ直接通過させ,そこで再フォーカスしてもよい。一般的には,高相比のカラム(例えば,膜厚5 μm,内
径0.2〜0.32 mm)と,初期温度を室温に設定することが要求される。
二次吸着剤冷却トラップが使用できない場合であって氷点下のキャピラリークライオフォーカスを分析
対象成分の予備濃縮に用いる場合,キャピラリーチューブを詰まらせたり,加熱脱離プロセスを停止させ
る氷の生成を防ぐため,脱離前に,試料サンプラの水を完全に除去するのが望ましい。
注記1 二次冷却トラップが使用できず,試料サンプラの最適脱離流量を30〜50 mL/minとする場合,
高分解能キャピラリーカラムでの分析のためには,最低30:1から50:1のスプリット比が
必要となる。このため,一段加熱脱離では検出感度が制約される。
脱離条件は,試料サンプラからの脱離が完全で,かつ,二次冷却トラップ中で試料成分の損失がないよ
8
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
うに条件を選択することが望ましい。一般的なパラメータを,次に示す。
− 冷却トラップ吸着剤 使用する場合は,一般的にサンプラと同じもの,40〜100 mg
− 冷却トラップ最高温度 −180〜+20 ℃,冷却トラップのタイプによる。
− 脱離温度 250〜325 ℃
− 脱離時間 5〜15 min
− 脱離流量 30〜50 mL/min
− キャリヤーガス ヘリウム又は窒素(冷却の程度によって選択する。)
− スプリット比 試料サンプラ,二次冷却トラップ間及び二次冷却トラップと分析カラム(あれば)間
のスプリット比は,想定される空気中の濃度に応じて選択する(詳細は,加熱脱離装置製造業者の手
引きを参照)。
注記2 脱離温度は,分析対象成分と使用する吸着剤によって決まる。推奨値を表1〜表6に示すが,
特定の吸着剤に対しては附属書C及び附属書Dとで与えられる最高脱離温度を守ることが望
ましい。熱的に不安定な,2級及び3級の揮発性アミン,及び炭素原子数が1個又は2個の
ある種のポリハロゲン化合物,特に臭素化合物は,多少熱分解する可能性がある。
9.2.2
分析
分析対象成分の凝縮を防ぐために試料流路の温度(移送ライン温度)は十分高く設定する。ただし,熱
分解を起こすほど高くしてはならない。室温で十分揮発する分析対象成分は,通常,流路の温度を150 ℃
以上にする必要はない。ある種の装置及び成分によっては,より高温を必要とすることもある。
VOCのガスクロマトグラフ分析条件を設定する。各種のガスクロマトグラフ用カラムが使用できる。カ
ラムは多くの場合,どのような妨害成分が存在するかによって選択する。
注記1 一般的な例として,カラムは,ポリジメチルシロキサン結合相の厚い膜厚(1〜5 μm),長さ
50 m,内径0.22 mmの溶融シリカカラム又は7 %シアノプロピル,7 %フェニル,86 %メチ
ルシロキサン結合相の長さ50 mのカラムである。これらのカラムにあっては,初期ホールド
時間を50 ℃で10分間とし,5 ℃/minで50〜250 ℃まで昇温させる温度プログラムが,一
般的な設定条件となる。
キャピラリーカラム又はなるべく全長がコーティングされていない不活性溶融シリカ製カラムを,加熱
脱離装置からガスクロマトグラフへトランスファーラインを通じて一体化させる。また,冷却トラップ内
の吸着剤にできるだけ密着させるか又は一段階脱離器のチューブにできるだけ接近させるのがよい。
内部チューブは不活性とし,デッドボリュームを最小限とする。スプリットバルブを二次トラップの入
口及び/又は出口の適切な位置に設置する。二次トラップ出口のスプリットバルブは,トランスファーラ
インの入口又は出口のいずれに設置してもよい。スプリット比は対象成分への適応性によって設定する。
注記2 低スプリット比は大気(普通は1:1〜10:1),室内空気及びある種の作業場空気(普通は1:
1〜20:1)測定に適しており,高スプリット比(普通は100:1〜1 000:1)は,ほとんどの
作業環境空気測定に適している。
注記3 単一のカラムにおいて保持時間が一致しただけでは,同定の根拠とならない。
9.3
検量線
各標準サンプラ(5.6又は5.8)を加熱脱離し,ガスクロマトグラフによって分析する。
5.7の標準溶液又は5.5の標準空気に対応した標準サンプラを分析し,μgで表示した分析対象成分の質
量の常用対数を横軸に,分析対象成分のピーク面積の対数を縦軸にプロットして,検量線用グラフを作成
する。
9
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 検量線範囲が1桁以内であるならば,そのデータの対数をとる必要はない。
9.4
試料濃度の測定
9.3で規定したように,試料及びトラベルブランクを分析する。ピーク面積を求め,脱離した試料中の分
析対象成分の質量を検量線から読み取る(参考文献[3])。
9.5
脱離効率の測定
脱離効率は,標準サンプラ(9.3)測定のクロマトグラフのピーク面積又は高さと,標準溶液又は標準空
気を直接ガスクロマトグラフに注入して測定したピーク面積又は高さを比較して求める。脱離効率は,標
準サンプラによるピーク面積又は高さを,標準液を直接注入したときのピーク面積又は高さで除した値で
ある。脱離効率が95 %以下の場合は,脱離条件を変更する。
ある種の加熱脱離装置には,直接液体注入部がないものもある。この場合,標準サンプラを混合空気か
ら調製するとき,脱離効率は,分析対象成分の検量線をn-へキサン(6.1)の検量線と比較して確認するこ
とが望ましい。分析対象成分の検量線の傾きに対するn-ヘキサンの傾きの比は,対象成分の相対感度と同
じでなければならない。他の化合物の検出感度は,有効炭素数からおおよそ計算できる(参考文献[1])。
検量線の傾きの比が相対感度と10 %以内で合致しない場合は,脱離条件を変更する。
9.6
拡散取込み速度の算出
表1及び表2の拡散取込み速度(Diffusive uptake rate)は,例えば,6.1の寸法のサンプラで6.3の膜の
ないサンプラサンプリングキャップを使用した場合の拡散取込み速度である。他の異なる仕様の拡散取込
み速度測定については,EN 838[4]又はEN 13528-2[5]がある。
注記 拡散取込み速度は,吸着剤の選択に依存する(prEN 13528-3[1]参照)。
10
計算
10.1
分析対象成分の質量濃度
式(1)によって,試料空気中の分析対象成分の質量濃度を計算する。
······································································ (1)
ここに,
ρm: 試料空気中の分析対象成分の質量濃度(μg/m3)
ma: 9.4で求めた試料中の分析対象成分の質量(μg)
mb: ブランクサンプラ中の分析対象成分の質量(μg)
u: 拡散取込み速度(cm3/min)(表1又は9.6)
t: 暴露時間(min)
注記1 ma及びmbがmg単位で表される場合,結果として濃度ρmの単位はmg/m3で表される。
注記2 例えば,25 ℃,101.3 kPaのような,特定条件における濃度に換算する必要がある場合は,
式(2)を用いる。
····························································· (2)
ここに,
ρc: 特定条件に換算した試料空気中の分析対象成分の濃度
P: サンプリング時の気圧(kPa)
T: サンプリング時の気温(℃)
10.2
分析対象成分の体積比
分析対象成分の体積比を求める場合は,式(3)による。
···································································· (3)
6
b
a
m
10
×
×
−
=
t
u
m
m
ρ
298
273
3.
101
m
c
+
×
×
=
T
P
ρ
ρ
6
b
a
10
×
×′
−
=
t
u
m
m
ϕ
ρ
10
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
ρφ: 空気中の分析対象成分の体積分率(μL/m3)
u': 拡散取込み速度[ng/(μL/L) /min(表1又は9.6)]
t: 暴露時間(min)
注記 ma及びmbがmg単位で表される場合,結果として濃度ρφの単位はmL/m3で表される。
10.3
拡散取込み速度u及びu'の関係
取込み速度u(cm3/min)及びu'[ng/(μL/L)/min]は,式(4)の関係にある。
···································································· (4)
ここに,
M: 分析対象成分の分子量
24.0: 20 ℃,101.3 kPaにおけるモル体積
11
妨害
ガスクロマトグラフ分析において,分析対象成分と保持時間とが同一,又は近接する有機成分によって
妨害が生じることがある。ガスクロマトグラフカラム及び条件の適切な選択並びにサンプラ又は分析シス
テムの使用前の適切な調整によって,妨害を最小限に抑えることができる。
この方法は,多孔質ポリマー及びCarbopack/Carbotrapのような全ての疎水性吸着剤について,相対湿度
95 %までの空気に適用できる。
注記 純活性炭又はカーボモレキュラーシーブのような,疎水性の低い強力な吸着剤を,相対湿度
65 %以上の空気に用いる場合,分析過程で,水による妨害を避けるための手段を講じなければ
ならない。水分を除去又は減少させる適切な方法としては,試料のスプリット,二次冷却トラ
ップの“水分のパージ”,シリコーン膜付きサンプラサンプリングキャップ(6.3)の使用及び
サンプリング時間の短縮がある。
サンプラが,当初良好なレベルのブランク値を示していても,後に妨害物質を形成することがある。水
(参考文献[6])の存在下において,オゾン(参考文献[7])及び窒素酸化物は,Tenax TAを損傷することが
ある。これらの反応によってベンズアルデヒド及びアセトフェノンが発生する可能性がある。
オゾン及び窒素酸化物は,分析対象成分と反応する可能性があるので,試料空気中にこれらの気体を大
量に含んでいることが予想される場合は,サンプリング体積をできるだけ小さくしなければならない。反
応性のガスの存在によってTenax TAを用いて必要な安定性が得られない場合,吸着剤としてCarbopack(参
考文献[8]〜[10])などを用いてもよい。
環境適用性について,主に安定な化合物に対し試験が行われた(表2)。特に,気温及びオゾン濃度が高
い場合,副次的な物質の生成又は捕集した不安定なVOCの分解が生じる可能性がある(参考文献[4])。こ
のような場合は,パッシブサンプリングが適切であることを明らかにするために,例えば,オキシダント
トラップ付きサンプラを使用するなど,影響の少ない手段を代わりに用いて,ポンプサンプリングを実施
する必要がある。
12
分析特性
この規格の手順で試験をした場合の,分析特性(総合的な不確かさ,精度,偏り,保管安定性及びブラ
ンクレベル)に関する記述を含む性能特性の例を,附属書E及び表3〜表8に示す。
13
試験報告書
試験報告書は,少なくとも次の情報を含まなければならない。
3.
101
0.
24
293
×
×
×
×
×
=
′
T
P
M
u
u
11
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 試料内容の明確な識別
b) この規格番号及び参照した他の補足規格
c) サンプリングの場所及び時間並びに暴露時間
d) 必要な場合は,気圧及び気温
e) 試験結果
f)
測定中に観察された特記事項
g) この規格若しくは参照した規格に含まれない操作,又は任意とみなされる操作
14
品質管理
適切なレベルの品質管理を行うことが望ましい(参考文献[18]又は同等の方法)。
サンプラのブランクは,妨害成分のピーク面積が分析対象成分の通常値の10 %以下であることが望まし
い。
表1及び表2に示す拡散取込み速度は,サンプラの耐用期間中は,一定に維持されると想定される。
ただし,過剰な振動によって吸着剤が漏れ出すことがあり,このため吸着剤が劣化することがある。し
たがって,サンプラを頻繁に目視で点検し,100回使用後,又は使用頻度が少ない場合は2年に一度,新
しい吸着剤に詰め替える。
表1−サンプラ(6.1,膜なし)の拡散取込み速度(4〜8時間サンプリングに適用)
化合物
吸着剤
レベルa)
拡散取込み速度b)
cm3/min
ng/(μL/L)/min
炭化水素類
1,3-ブタジエン
Molecular Sieve 13X
A
0.59
1.30
n-ペンタン
Chromosorb 106
A
0.50
1.46
Carbopack B c)
B
0.60
1.77
n-ヘキサン
Chromosorb 106
A
0.50
1.77
ベンゼン
Tenax TA d)
A
0.41
1.3
Porapak Q
A
0.42
1.37
Tenax GR
B
0.57
1.81
Chromosorb 106 e)
B
0.54
1.72
n-ヘプタン
Chromosorb 106
A
0.48
1.95
Tenax TA e)
A
0.43
1.77
Carbotrap B
B
0.47
1.94
トルエン
Tenax TA e)
B
0.44
1.67
Tenax GR
B
0.56
2.12
Chromosorb 106
B
0.52
1.94
Carbopack B
B
0.55
2.06
n-オクタン
Chromosorb 106
A
0.46
2.13
Tenax TA e)
A
0.43
2.00
キシレン
Tenax TA e)
B
0.42
1.82
Chromosorb 106
B
0.48
2.10
Tenax GR
B
0.57
2.48
エチルベンゼン
Tenax TA e)
B
0.46
2.0
Tenax GR
B
0.56
2.43
Chromosorb 106
B
0.44
1.9
Porapak Q
D
0.55
2.38

12
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−サンプラ(6.1,膜なし)の拡散取込み速度(4〜8時間サンプリングに適用)(続き)
化合物
吸着剤
レベルa)
拡散取込み速度b)
cm3/min
ng/(μL/L)/min
スチレン
Tenax TA e)
A
0.47
2.0
Chromosorb 106
B
0.51
2.15
n-ノナン
Chromosorb 106
A
0.46
2.40
Tenax TA e)
A
0.40
2.12
イソプロピルベンゼン
Chromosorb 106
C
0.46
2.26
Tenax TA e)
C
0.46
2.26
Porapak Q
D
0.51
2.5
トリメチルベンゼン
Chromosorb 106
C
0.48
2.37
Tenax TA e)
C
0.48
2.37
n-デカン
Tenax TA
A
0.40
2.3
ハロゲン化炭化水素類
塩化メチレン
Spherocarb
B
0.63
1.3
塩化ビニル
Spherocarb
B
0.78
2.0
1,1-ジクロロエタン
Spherocarb
B
0.63
2.5
トリクロロトリフルオロエタン
Chromosorb 102
B
0.46
3.5
クロロトリフルオロメタン
Chromosorb 102
B
0.42
1.8
ジクロロメタン
Chromosorb 106
B
0.43
1.56
Chromosorb 102
B
0.45
1.56
1,2-ジクロロエタン
Chromosorb 102
B
0.47
1.9
ハロサン(=2-ブロモ-2-クロロ
-1,1,1-トリフルオロエタン)
Tenax TA
B
0.32
2.59
Chromosorb 102
B
0.45
3.6
エンフルラン[=2-クロロ-1-(ジ
フルオロメトキシ)-1,1,2-トリフ
ルオロエタン]
Tenax TA
B
0.33
2.29
イソフルラン[=2-クロロ-2-(ジ
フルオロメトキシ)-1,1,1-トリフ
ルオロエタン]
Tenax TA
B
0.32
2.20
ブロモエタン
Chromosorb 106
E
0.55
2.45
クロロホルム
Tenax GR
B
0.45
2.18
Chromosorb 102
B
0.48
2.35
四塩化炭素
Tenax GR
B
0.59
3.72
Chromosorb 102
B
0.48
2.87
トリクロロエチレン
Chromosorb 106
B
0.47
2.66
Chromosorb 102
B
0.43
2.3
1,1,1-トリクロロエタン
Chromosorb 106
B
0.42
2.3
Chromosorb 102
B
0.42
2.3
Tenax GR
B
0.54
2.92
テトラクロロエチレン
Chromosorb 106
B
0.46
3.1
Tenax TA
B
0.41
2.8
Chromosorb 102
B
0.38
2.6
エピクロルヒドリン
Chromosorb 106
E
0.65
2.45
ペルフルオロジメチルシクロブ
タンd)
Carbotrap
B
0.25
−
ペルフルオロメチルシクロペン
タンd)
Carbotrap
B
0.25
−
13
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−サンプラ(6.1,膜なし)の拡散取込み速度(4〜8時間サンプリングに適用)(続き)
化合物
吸着剤
レベルa)
拡散取込み速度b)
cm3/min
ng/(μL/L)/min
ペルフルオロメチルシクロヘキ
サンd)
Carbotrap
B
0.25
−
エステル類及びグリコールエーテル類
酢酸エチル
Chromosorb 106 e)
B
0.49
2.00
Tenax TA
B
0.40
1.60
酢酸ブチル
Tenax TA
B
0.61
2.26
メタクリル酸メチル
Porapak Q
B
0.49
2.0
アクリル酸ブチル
Tenax TA
B
0.51
2.6
2-メトキシエタノール
Porapak Q
A
0.48
1.5
Chromosorb 106
B
0.51
2.1
2-エトキシエタノール
Tenax
A
0.44
1.8
酢酸2-メトキシエチル
Porapak Q
A
0.58
2.8
酢酸2-エトキシエチル
Chromosorb 106
B
0.39
2.3
Tenax TA
B
0.36
2.10
2-ブトキシエタノール
Chromosorb 106
B
0.35
2.1
Tenax TA
B
0.31
1.9
2-メトキシプロパノール
Chromosorb 106 e)
B
0.45
1.85
Tenax TA
B
0.37
1.52
酢酸2-ブトキシエチル
Tenax
A
0.38
2.8
アルデヒド類及びケトン類
メチルイソブチルケトン
Tenax TA
B
0.42
1.71
Chromosorb 106 e)
B
0.49
2.01
シクロヘキサノン
Tenax TA
D
0.57
2.3
フルフラール
Tenax TA
A
0.63
2.5
アルコール類
2-プロパノール
Spherocarb
C
0.81
2.0
その他
アクリロニトリル
Porapak N
A
0.62
1.35
アセトニトリル
Porapak N
A
0.60
1.0 (2 h)
Porapak N
A
0.48
0.8 (8 h)
プロピオニトリル
Porapak N
A
0.53
1.4 (2 h)
Porapak N
A
0.49
1.3 (8 h)
二硫化炭素
Spherocarb
A
0.83
2.6
一酸化二窒素
Molecular Sieve 5A
B
0.70
1.25
エチレンオキサイド
Spherocarb
B
0.88
1.6
1,4-ジオキサン
Spherocarb
C
0.84
3.0
注a) レベルA=EN 838[4]のレベル1 Aと同等性の確認。
レベルB=部分的にEN 482[13]と同等性の確認。
レベルC=計算−理想数値。
レベルD=動的破過容量から算出。
レベルE=吸着等温式から算出。
b) 特記事項がない限り,数値は作業場における4〜8時間の暴露に適用。
c) 拡散取込み速度が変化するためにサンプラの例は特に挙げていない。
d) ステンレス鋼製金網の代わりにニッケル製金網を使用。
e) 望ましい吸着剤のタイプ。

14
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表2−サンプラ(6.1,膜なし)の拡散取込み速度(1〜4週間サンプリングに適用)
化合物
吸着剤
暴露
期間
週
取込み速度
(標準偏差)
cm3/min
取込み速度
(標準偏差)
ng/(μL/L)/min
評価回数
参考文献
ベンゼン
Tenax TA
1
0.46
1.45
実験室比較
[19]
ベンゼン
Tenax TA
2
0.32
(0.01)
1.03
(0.04)
1実験室/
1現地比較
[19], [20]
ベンゼン
Tenax TA
4
0.22
(0.03)
0.70
(0.09)
2実験室/
1現地比較
[19]〜[21]
ベンゼン
Carbograph TD-1又は
Carbopack B
1
0.67
(0.06)
2.14
(0.21)
1実験室/
1現地比較
[11], [22]
ベンゼン
Carbograph TD-1又は
Carbopack B
2
0.63
(0.07)
2.02
(0.22)
1実験室/
3現地比較
[11], [20],
[22]
ベンゼン
Carbograph TD-1又は
Carbopack B
4
0.58
(0.05)
1.85
(0.15)
1実験室/
3現地比較
[11], [20],
[22]
ベンゼン
Chromosorb 106
1
0.48
(0.03)
1.52
(0.08)
1実験室/
1現地比較
[11], [22]
ベンゼン
Chromosorb 106
2
0.47
(0.06)
1.47
(0.22)
1実験室/
4現地比較
[11], [20],
[22]〜[24]
ベンゼン
Chromosorb 106
4
0.40
(0.08)
1.28
(0.25)
1実験室/
3現地比較
[11], [20],
[22], [24]
ベンゼン
Ambersorb XAD-4
1
0.38
1.21
1実験室比較
[26]
トルエン
Tenax TA
2
0.32
1.22
1現地比較
[20]
トルエン
Tenax TA
4
0.27
(0.07)
1.03
(0.26)
1実験室/
1現地比較
[20], [21]
トルエン
Carbograph TD-1又は
Carbopack B
1
0.57
(0.15)
2.16
(0.58)
1実験室/
1現地比較
[11], [22]
トルエン
Carbograph TD-1又は
Carbopack B
2
0.56
(0.06)
2.13
(0.24)
1実験室/
3現地比較
[11], [20],
[22]
トルエン
Carbograph TD-1又は
Carbopack B
4
0.55
(0.07)
2.07
(0.26)
1実験室/
3現地比較
[11], [20],
[22]
トルエン
Chromosorb 106
1
0.54
(0.15)
2.05
(0.57)
1実験室/
1現地比較
[11], [22]
トルエン
Chromosorb 106
2
0.51
(0.05)
1.91
(0.18)
1実験室/
4現地比較
[11], [20],
[22]〜[24]
トルエン
Chromosorb 106
4
0.48
(0.06)
1.82
(0.24)
1実験室/
3現地比較
[11], [20],
[22], [24]
トルエン
Ambersorb XAD-4
1
0.43
1.62
1実験室比較
[26]
キシレン
Tenax TA
2
0.34
1.49
1実験室/
1現地比較
[19], [20]
キシレン
Tenax TA
4
0.33
(0.15)
1.46
(0.67)
2実験室/
1現地比較
[19]〜[21]
キシレン
Carbograph TD-1又は
Carbopack B
1
0.54
(0.06)
2.37
(0.29)
1実験室/
1現地比較
[11], [22]
キシレン
Carbograph TD-1又は
Carbopack B
2
0.47
(0.04)
2.07
(0.21)
1実験室/
3現地比較
[11], [20],
[22]
キシレン
Carbograph TD-1又は
Carbopack B
4
0.44
(0.07)
1.94
(0.29)
1実験室/
3現地比較
[11], [20],
[22]
キシレン
Chromosorb 106
1
0.55
(0.07)
2.42
(0.30)
1実験室/
1現地比較
[11], [22]
15
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表2−サンプラ(6.1,膜なし)の拡散取込み速度(1〜4週間サンプリングに適用)(続き)
化合物
吸着剤
暴露
期間
週
取込み速度
(標準偏差)
cm3/min
取込み速度
(標準偏差)
ng/(μL/L)/min
評価回数
参考文献
キシレン
Chromosorb 106
2
0.47
(0.07)
2.09
(0.29)
1実験室/
4現地比較
[11], [20],
[22]〜[24]
キシレン
Chromosorb 106
4
0.43
(0.08)
1.91
(0.35)
1実験室/
3現地比較
[11], [20],
[22], [24]
エチルベンゼン
Chromosorb 106
2
0.53
(0.02)
2.31
(0.07)
2現地比較
[23], [24]
エチルベンゼン
Chromosorb 106
4
0.52
2.24
1現地比較
[24]
エチルベンゼン
Carbopack B
2
0.50
2.19
1現地比較
[26]
エチルベンゼン
Carbopack B
4
0.52
2.30
1現地比較
[26]
トリメチルベンゼ
ン
Tenax TA
4
0.54
2.67
1実験室比較
[21]
トリメチルベンゼ
ン
Carbopack B
2
0.44
2.20
1現地比較
[26]
トリメチルベンゼ
ン
Carbopack B
4
0.47
2.30
1現地比較
[26]
デカン
Tenax TA
4
0.51
2.93
1実験室比較
[21]
ウンデカン
Tenax TA
4
0.53
3.34
1実験室比較
[21]
注記1 多くの場合,大気の拡散取込み速度は,都市のバックグラウンドレベルを代表する濃度(例えば,数μg/m3
のベンゼン,トルエン又はキシレン)で決まる。拡散取込み速度は,独立した方法(通常,ポンプサンプ
リング)で確認した実験室の試験用空気で決定する。一方,現場においては,独立した方法(ポンプサン
プリング)で一つ又は幾つかのサンプラで直接サンプリングした空気によって測定されている。参考文献
[19]は,約1 mg/m3のベンゼン及び約2 mg/m3のTVOCを試験用空気として調製し,その濃度レベルにおけ
る拡散取込み速度を記載している。
注記2 附属書Aのように,いろいろな種類の空気の拡散取込み速度は暴露時間と濃度とに依存するため,同じと
は限らない。この影響は,Tenax TAなどの非理想的な吸着剤ではより顕著になる。
表3−添加したサンプラからのベンゼン,トルエン及びキシレンの回収率
調査名称
Chromosorb 106
Carbograph TD-1
ベンゼン
トルエン
キシレン
ベンゼン
トルエン
キシレン
1. 英国の調査
平均回収率%
82.7
87.5
95.9
95.1
100.1
100.6
±標準偏差
8.3
6.7
10.4
12.1
4.4
10.0
n数
20
19
19
19
20
20
2. VOC空気ネットワーク 平均回収率%
93.1
99.1
100.5
98.7
100.3
98.5
±標準偏差
11.9
7.9
5.0
3.0
2.7
2.0
n数
13
13
13
13
13
13
3. 世界的な調査
平均回収率%
104.8
105.9
98.7
103.7
100.7
100.1
±標準偏差
11.3
10.1
7.8
4.6
3.2
2.3
n数
16
16
16
16
16
16
1.〜3.の単純平均
%
93.5
97.5
98.3
99.2
100.4
99.7
注記1 調査No.1では,添加された炭化水素の量はいずれも約80 ng,調査No.2及びNo.3では約200 ng。
注記2 サンプラを現場(1回ではあるが世界規模の調査)に送付し,1か月間並行して暴露(密閉)した後,実験
室に返送して分析した。
16
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表4−パッシブサンプラ測定値と“英国VOC空気ネットワーク”測定値との比較
単位 μg/m3
場所
(都市)
ベンゼン
トルエン
キシレン
パッシブ
VOC空気
ネットワーク
パッシブ
VOC空気
ネットワーク
パッシブ
VOC空気
ネットワーク
Leeds
2.27±0.07
3.33
5.30±0.01
6.75
3.44±0.07
3.99
Belfast
2.10±0.36
2.94
4.44±0.15
6.82
3.34±0.14
3.55
Cardiff
3.70±0.46
9.5
7.90±0.12
8.38
5.67±0.10
5.15
Bristol
2.90±0.33
6.11 a)
6.4 ±0.86
2.25 a)
4.7 ±0.26
2.70
Eltham
2.55±0.35
4.65
5.92±0.69
10.36
3.55±0.30
3.10
Liverpool
2.12±0.16
1.23
4.55±0.19
0.93
3.86±0.19
0.84
Middlesborough
2.35±0.27
2.93
4.23±0.22
4.59
2.66±0.14
−
Southampton
3.87±0.73
1.90
8.12±0.68
11.14
5.50±0.20
5.49
UCL (London)
4.06±0.14
6.05
9.67±0.16
10.87
6.79±0.27
7.79
Edinburgh
1.29±0.20
1.81
3.29±0.42
3.78
2.00±0.27
1.89
Harwell
0.66±0.03
0.90
1.46±0.31
0.98
0.60±0.04
−
Birmingham
1.87±0.27
2.54
4.76±0.31
6.75
4.46±0.27
5.59
注記 パッシブサンプリング期間中のポンプサンプリングのデータが少ないため,VOC空気ネットワークのデータ
は単に傾向を示すものである。
注a) インターネットアーカイブによるベンゼン/トルエン値の置換の可能性。
表5−先導的世界規模の調査/2種類のサンプラによる平均大気濃度
場所
(国名)
平均大気濃度
μg/m3±標準偏差
n
ベンゼン
トルエン
キシレン
スウェーデン
1.95±0.08
5.63±0.77
3.49±0.29
4
デンマーク
0.99±0.04
1.44±0.03
0.80±0.07
4 a)
アメリカ
0.43±0.07
0.76±0.09
0.47±0.09
4
オーストラリア
1.94±0.2
5.23±0.69
3.87±0.28
4
ハンガリー
2.65±0.09
4.7 ±0.19
3.27±0.18
4
ドイツ
1.75±0.15
5.85±0.56
3.74±0.33
4
中国
12.3 ±1.08
23.04±2.32
11.64±0.51
4
フィンランド
0.84±0.16
2.16±0.6
1.58±0.03
4
オランダ
1.55±0.14
3.51±0.27
2.28±0.14
4
イスラエル
1.42±0.17
3.24±0.1
2.79±0.08
4
メキシコ
3.05±0.51
23.43±1.28
8.79±0.25
4
イタリア
1.59±0.15
5.39±0.11
3.36±0.41
4
ブラジル
0.42±0.33
2.00±0.59
1.81±0.15
4 b)
フランス
1.81±0.25
7.32±0.31
3.87±0.13
4
北アイルランド
2.10±0.36
4.44±0.15
3.34±0.14
4
イングランド
1.37±0.25
4.24±0.09
3.36±0.11
4
注a) トルエン及びキシレンは,n=3
b) トルエンは,n=3
17
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表6−Chromosorb 106及びCarboxen 569における試験化合物の分析及び保管の精度(参考文献[15])
(サンプラへの添加レベルは1 μg)
有機化合物
分析精度(変動係数)
%
保管後の回収率
%
Chromosorb
Carboxen
Chromosorb
Carboxen
プロパン
−
1.8
−
115
ペンタン
1.7
−
112
−
ヘキサン
2.1;3.6
−
104
−
ベンゼン
2.9
−
100
−
ジクロロメタン
1.9
−
114
−
1,1,1-トリクロロエタン
2.4
−
101
−
メタノール
−
1.7
−
64
エタノール
5.9
−
96
−
ブタノール
1.3
−
101
−
酢酸メチル
1.8
−
113
−
メトキシエタノール
5.7
−
121
−
メチルエチルケトン
2.2
−
103
−
アセトニトリル
4.1
−
112
−
酢酸ブチル
3.4
−
104
−
α-ピネン
4.2;2.5
−
104
−
デカン
4.2
−
104
−
エチレンオキサイド
−
−
−
−
プロピレンオキサイド
3.6
−
103
−
ヘキサナール
3.5
−
98
−
注記 サンプラへの添加レベルは1 μg
18
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7−Tenaxサンプラにおける溶剤の分析及び保管の精度
有機化合物
添加量
μg
添加直後(保管
無)の変動係数a)
% CV
5か月保管
11か月保管
平均回収率b)
%
変動係数
% CV
平均回収率b)
%
変動係数
% CV
炭化水素類
ヘキサン
7.8
10.7
93.6
17.9
100.8
26.1
ヘプタン
8.4
2.4
99.5
2.1
100.0
1.3
オクタン
8.6
2.4
100.1
1.8
100.0
0.5
ノナン
12.0
0.8
nd (12)
nd (12)
101.0
0.4
デカン
9.2
2.2
100.4
1.5
100.2
0.5
ウンデカン
9.1
2.3
100.7
1.5
100.2
0.2
ドデカン
9.9
2.8
101.8
1.5
101.5
0.4
ベンゼン
11.0
2.5
98.7
2.0
98.6
0.8
トルエン
10.9
2.6
(100.0)
1.8
(100.0)
0.6
p-キシレン
5.3
2.5
99.9
1.7
99.8
0.7
o-キシレン
11.0
2.4
100.0
1.7
98.8
0.7
エチルベンゼン
10.0
0.5
99.6
0.4
97.9
1.3
プロピルベンゼン
10.5
2.3
99.7
1.5
98.5
0.7
クメン
10.9
2.3
98.9
1.8
97.2
1.3
m-+p-エチルトルエン
10.5
2.3
98.8
1.7
96.9
1.2
o-エチルトルエン
5.4
2.2
100.1
1.6
98.9
0.7
1,2,4-トリメチルベンゼン
10.8
2.2
100.1
1.3
99.1
0.5
1,3,5-トリメチルベンゼン
10.7
2.2
100.0
1.5
99.1
0.5
トリメチルベンゼン
10.2
1.7
101.6
0.5
101.3
0.8
エステル類及びグリコールエーテル類
酢酸エチル
10.3
0.6
97.6
1.0
100.0
2.5
酢酸プロピル
10.9
2.4
100.5
1.7
99.1
0.8
酢酸イソプロピル
9.4
1.0
97.0
0.4
100.0
1.4
酢酸ブチル
10.8
2.4
100.3
1.6
99.9
0.6
酢酸イソブチル
10.7
2.3
100.2
1.4
99.8
0.7
メトキシエタノール
8.9
5.4
87.3
5.7
93.1
1.6
エトキシエタノール
10.4
4.2
97.6
2.5
97.2
3.3
ブトキシエタノール
10.0
2.6
100.6
4.1
100.1
3.0
メトキシプロパノール
10.4
2.4
95.3
3.6
99.0
1.2
酢酸メトキシエチル
12.5
2.1
100.6
1.4
98.9
1.4
酢酸エトキシエチル
11.4
0.9
99.8
2.2
98.7
2.6
酢酸ブトキシエチル
11.5
2.3
101.3
1.3
99.9
1.1
ケトン類
メチルエチルケトン
9.2
0.9
97.4
0.8
99.1
0.6
メチルイソブチルケトン
9.3
0.6
100.7
0.6
100.7
0.5
シクロヘキサノン
10.9
0.8
102.4
1.2
100.7
0.6
2-メチルシクロヘキサノン
10.7
0.7
101.1
0.5
101.1
1.3
3-メチルシクロヘキサノン
10.5
0.8
103.6
1.0
103.0
0.7
4-メチルシクロヘキサノン
10.6
0.9
103.6
1.4
102.7
0.6
3,5,5-トリメチルシクロヘキ
サ-2-エンオン
10.6
2.3
101.4
0.9
97.7
1.2

19
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7−Tenaxサンプラにおける溶剤の分析及び保管の精度(続き)
有機化合物
添加量
μg
添加直後(保管
無)の変動係数a)
% CV
5か月保管
11か月保管
平均回収率b)
%
変動係数
% CV
平均回収率b)
%
変動係数
% CV
アルコール類
ブタノール
9.0
1.1
94.8
3.0
96.9
1.2
イソブタノール
8.9
1.0
93.6
3.5
96.4
1.0
注a) 繰返し数6回。
b) トルエンを100とした場合の相対値。トルエンの安定性については,BCR(Bureau Communautaire de Reference)
相互比較で確立されている(参考文献[17])。
nd=測定できず。
表8−Chromosorb 106及びCarbograph TD-1におけるベンゼン,トルエン及び
キシレンのブランクレベル
調査名称
Chromosorb 106
Carbograph TD-1
ベンゼン
トルエン
キシレン
ベンゼン
トルエン
キシレン
1. 英国の調査
平均 ng
7.69
1.39
3.23
7.22
2.04
5.59
±標準偏差
1.96
0.55
1.64
2.75
0.78
2.28
n
20
20
20
18
19
19
2. VOC空気ネット
ワーク
平均 ng
10.38
3.26
1.46
6.88
3.34
1.39
±標準偏差
2.28
2.55
1.44
2.7
1.3
1.39
n
14
14
14
14
14
14
3. 世界的な調査
平均 ng
5.63
2.09
0.96
2.61
4.39
1.63
±標準偏差
3.04
2.36
0.51
1.13
6.19
1.17
n
16
16
16
16
16
16
20
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
パッシブサンプリングの作用原理
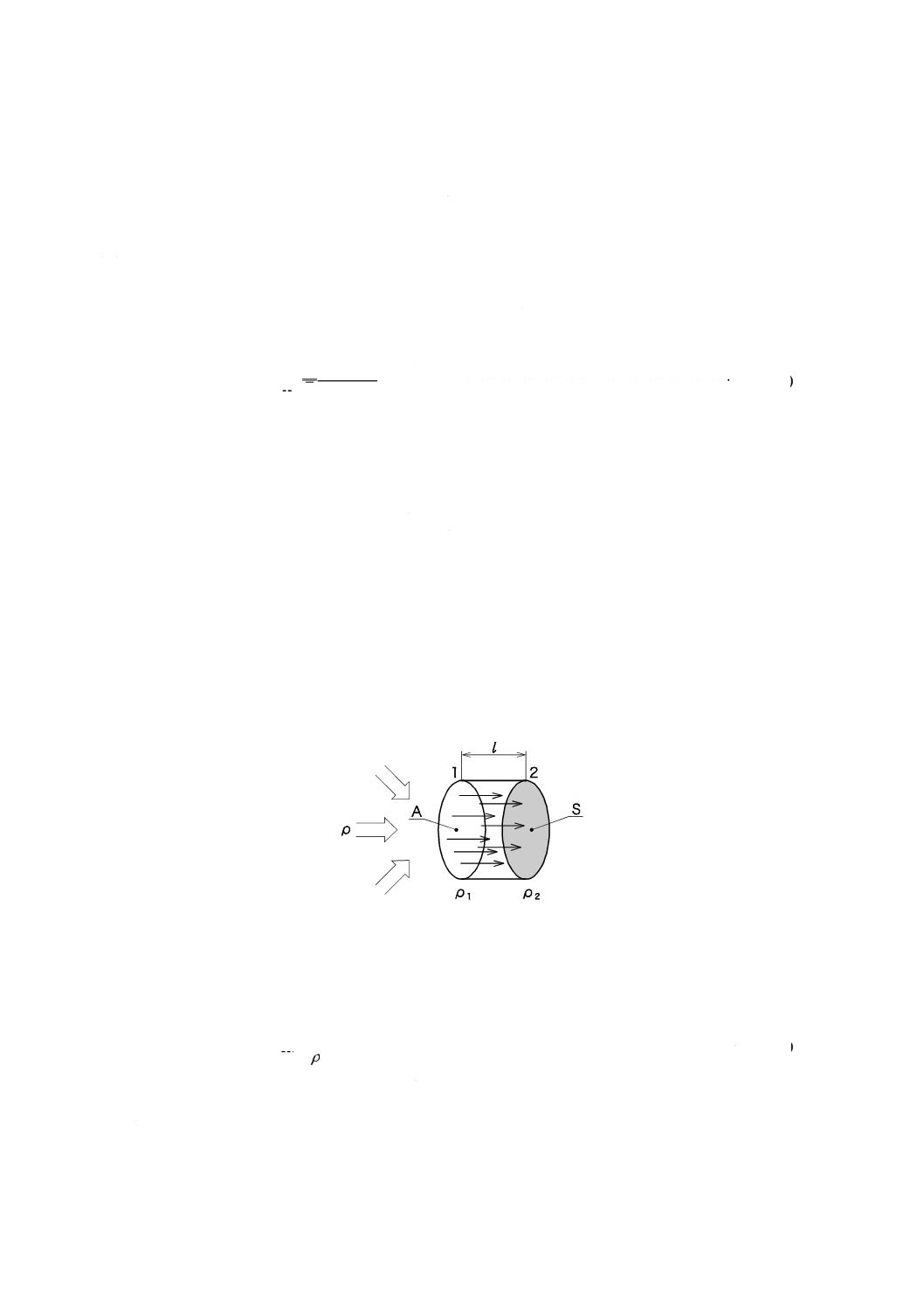
A.1 原理
概要は,参考文献[27]にある。
一定時間内に適切な吸着剤に向かって拡散移動する分析対象成分の質量は,フィックの第一法則に基づ
き式(A.1)から計算される。
··························································· (A.1)
ここに,
A: 断面積
D: 拡散係数
ρ1: 拡散キャップにおける分析対象成分の濃度
ρ2: 吸着剤ベッドにおける分析対象成分の濃度
mf: 分析対象成分の質量
l: 吸着剤表面から拡散キャップまでの距離
t: 時間
この式はρ2がゼロでない場合も考慮している。理想的な状態は,ρ1がパッシブサンプラ外の空気中の分
析対象成分の濃度と等しく,ρ2がゼロである(ゼロシンク状態)。その場合,拡散取込み速度A×D/[式(A.1)
参照]は分析対象成分の拡散係数とパッシブサンプラとの形だけに左右される(図A.1参照)。
位置1における断面Aをもつサンプラの入口が,ρ1の濃度をもつ分析対象成分の拡散経路の始まりを示
す。吸着剤は位置2において吸着若しくは化学反応によって分析対象成分の濃度ρ2を理想的にはゼロまで
減少させ,距離lの間における拡散の駆動力として働く。
図A.1−パッシブサンプリングの原理図
A.2 拡散取込み速度の単位
一定濃度ρ(μg/m3)のガス又は蒸気の拡散取込み速度u(cm3/min)は,式(A.2)で求める。
··············································································· (A.2)
ここに,
mf: 分析対象成分の質量(pg)
t: 暴露時間(min)
注記1 取込み速度uの単位はcm3/minであるが,これはpg/μg/m3/minから求めたものである。また,
これは,分析対象成分を含む空気の実際の流量を表すわけではない。
l
t
D
A
m
×
−
×
×
=
)
(
2
1
f
ρ
ρ
t
m
u
×
=ρf
21
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記2 拡散取込み速度は,pg/ppb/minの単位で表されることが多い。これらは実用的な単位である。
ほとんどの環境分析化学者は,ガス及び蒸気の濃度にppb 1) を用いているからである。取込
み速度の温度及び圧力への依存性については,次に述べる(A.4.1)。このように,あるppb
オーダの(ガス又は蒸気の)濃度(ppb)に対する取込み速度は,式(A.3)で求める。
t
m
u
×
=
′
1
f
φ
·············································································· (A.3)
ここに,
φ1: 109分の1
注1) ppbの使用はISOで推奨されていない。正しい単位は,体積分率で109分の1。
注記3 理想的かつ実用的な拡散取込み速度u'(ng/ppb/min)は,次の式(A.4)で求める。
3.
101
0.
24
293
×
×
×
×
×
=
′
T
P
M
u
u
·································································· (A.4)
ここに,
M: モル質量(g)
T: 絶対温度(K)
P: 大気圧(kPa)
A.3 非理想的な吸着剤の選択による偏り
パッシブサンプラの性能は,吸着剤又は高吸着効率をもつ捕集媒体の選択と使い方に大きく依存してい
る。分析対象成分の,吸着剤表面における残留蒸気圧(ρ2)は,雰囲気濃度と比べて非常に小さくなり,
そこで観察される取込み速度は理想的な定常状態における値に近くなる。この理想的な定常状態における
値は通常,サンプラの形と空気中の分析対象成分との拡散係数から計算できる。
弱い吸着剤を用いると,式(A.1)のρ2とmfとがサンプリング時間の経過とともに減少する。したがって,
式(A.2)のuも,また,サンプリング時間の経過とともに減少する。この影響の大きさは,分析対象成分,
吸着剤の吸着など温線によって決定され,コンピュータモデルは,参考文献[28],[29]を用いて計算するこ
とができる。
弱い吸着剤の影響がもたらすもう一つの現象に,逆拡散がある。これは,サンプリングの開始からある
程度の時間が経過したときに,分析対象成分の吸着剤表面における蒸気圧ρ2が外部の濃度ρ1より大きいと
きに起こる可能性がある。例えば,サンプラが最初に高濃度にさらされ,その後ずっと低い又はゼロの濃
度にさらされたときなどである。
この種の暴露はある種のアプリケーションにおいてよくみられ,誤差の程度は,高濃度暴露が,サンプ
リング期間の始め,中間及び最後の,どの時期に起こったかによって決まる。この現象は,参考文献[30]
〜[32]で解説しており,パルス的な暴露と一定濃度での暴露との間の最大偏りを予測するための簡単な試
験(参考文献[33])が提案され,一定濃度での暴露における評価はサンプラの校正のもととなる。
この試験はEN 838でも採用されており,高濃度に30分間暴露した後,清浄な空気に7.5時間の暴露を
行うものである。大気での使用では,実際には高濃度と低濃度との交互の暴露が想定される。例えば,7
日間のLV(限界値)の2倍値とゼロ濃度とに12時間ずつ交互にさらされるものが,日中で濃度変動が生
じることが一般的である典型的な応用例となる。また,濃度は,昼間に変動することが多い。逆拡散の程
度もまた,モデルを用いて理論的に算定することが可能である(参考文献[29], [34])。
A.4 サンプラの性能に影響を及ぼす環境要因
A.4.1 温度及び圧力
22
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
マックスウェルの式から,拡散係数Dは,絶対温度及び圧力の関数である。
(
)
1
2/3,
−
=
p
T
f
D
······································································ (A.5)
しかし,一般的な気体の法則から,
nRT
p=
V
············································································· (A.6)
RT
p
V
n
c
=
=
······································································ (A.7)
ここに,
n: モル数
R: 気体定数
式(A.1)に,式(A.5)及び式(A.6)を代入すると,
(
)
()
2/1
2/3
f
,
T
f
p
T
T
p
f
m
=
=
································································· (A.8)
したがって,mfは圧力に依存しないが絶対温度の平方根に依存する。1 ℃当たり0.2 %上昇する。
弱い吸着剤を用いる場合は,温度もまた吸着能力に影響を与えることがあり,捕集容量を僅かに減少さ
せる。例えば,Tenaxが捕集するベンゼンの取込み速度は,20〜60 ℃の範囲において1.33 ng/ppm/minか
ら1.23 ng/ppm/minへ減少する(参考文献[35])。
A.4.2 湿度
高湿度も,活性炭,モレキュラーシーブなどの疎水性吸着剤の吸着能力に影響を与える。これによって
通常は,式(A.1)のρ2が増大するため,取込み速度が非線形になることから,(ある濃度において)吸着剤
が飽和状態になるまでのサンプリング期間が減少する。また,チューブ式サンプラの露出された内壁の吸
着性にも変化を与える。凝縮が起こったときには,特に影響が大きい。
A.4.3 変動
フィックの第一法則の単純な適用は,定常状態を想定している。しかし,パッシブサンプラを実際使用
するときには汚染物質の大気濃度レベルが大きく変動することがあり得る。サンプラが真に積分された捕
集量を与えることができるか(吸着剤の影響は無視。A.4.1参照),それとも吸着剤に捕集される前に生じ
る短時間の変動を逃してしまうのかが問題となる。
このことは,合計サンプリング時間が,パッシブサンプラの時定数を大幅に上回る,例えば,10倍,と
すれば,理論的(参考文献[30], [35]〜[37]),かつ,実際的(参考文献[35], [38], [39])に検証されており,
問題とならないことが示されている。ただし,定数τは,定常状態において分子がサンプラに拡散するの
に要する時間をいう。一般的なサンプラのほとんどの時定数は,およそ1〜10秒で,τは式(A.9)で求める。
D
lR
=
τ
··············································································· (A.9)
A.4.4 気流速度の影響
A.4.4.1 低気流速度及び高気流速度の影響
周辺空気の面風速及び方向は,有効拡散距離に影響を及ぼすので,パッシブサンプラの特性に影響を及
ぼす(参考文献[40]〜[43])。
サンプラの拡散質量取込み式(A.1)は,距離lと,サンプラ内の拡散間隙の断面積Aの関数である。通常
の拡散距離はサンプラの形によって決まり,吸着剤表面とサンプラ内の外表面との間の距離である。また,
断面積も,サンプラの形によって決まり,拡散間隙の断面が拡散距離全体にわたって一定でない場合は,
最も狭い部分で決まる。有効距離lは,必ずしも見掛けの距離と一致するわけではなく,状況によっても
っと長い場合も短い場合もある。
23
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
外部風速が低いときには,有効拡散距離が増加することがある(参考文献[42], [43])。これは,サンプラ
内の停滞空気と外部の乱流空気との間に“境界層”(参考文献[40], [41])が生まれ,有効拡散距離lにきい
てくるためである。サンプラの外で停滞空気及び乱流空気の遷移する部分は,実際存在する。しかし,こ
れは停滞空気が余分にもつ距離δlに相当し,トータルのlの値に含まれている。δlの値はサンプラの外形
に依存するが,サンプラの捕集表面に直行する断面積とほぼ比例しており,短くて太いサンプラが大きな
影響を受けるのに対し,内部に長い空気間隙があっても比較的気流の影響を受けない。このことは,実際
の例からもいえるし,また,異なる長さのサンプラを使った実験(参考文献[42], [43])でも証明されてい
る。気流速度が低いときには拡散取込み速度は低くなるが,境界層の影響が問題にならなくなると,一定
値まで上昇する。
外部風速が高い状態では,有効拡散距離が減少することがある(参考文献[42], [44]〜[48])。これは乱流
がサンプラ内の停滞空気層を乱すためで,それによって有効空気間隙が因子δlによって減少する。δlの値
は小さいが,サンプラ空気間隙の距離の直径に対する割合が2.5〜3以上になる(参考文献[42])。これは,
ステンレススチールスクリーン又はプラスチック膜などの通気性シールドを入れることによって,完全に
ではなくてもかなり防止することができる(参考文献[47], [48])。
A.4.4.2 異なる形のサンプラの影響
チューブ形のサンプラは低い気流速度の影響を受けにくいが(参考文献[35], [49], [50]),通気性シールド
のないサンプラは大きい気流速度に影響されることがある。
A.4.5 運搬
ほとんどのサンプラは,サンプリング場所から実験室まで運搬しなければならない。したがって,この
間,試料を元のままの状態に維持することが重要である。次の手順を推奨する。
a) 移動中に汚染物質の侵入及び試料の損失が起こらないよう,シールが完全に閉じていることを確認す
る。温度変動が大きい場合,金属とプラスチックとのシールが緩みやすい。
b) 外部からの汚染物質の侵入を最小限にするため,サンプラを不活性で密閉された容器に入れておく。
c) 試料を空輸する場合は,荷物庫などで,負圧にさらされることのないようにする。
d) 移動中は,車のトランク内など,高温になるところを避ける。
e) 可能であればサンプラを凝縮しない程度の低温で保存し,石油,航空燃料などの汚染源から遠ざける。
上記の幾つかの問題があるかを確認するため,試料とともに運ぶ適切な数の現場ブランク(トラベルブ
ランク)も確保する。
A.5 不利な環境条件からの保護
大気環境で実際使用するときには,気流,降雨及びセキュリティの三つに細心の注意を払う。
A.5.1 全般的な注意
調査地点の壁,ドア及び窓からの距離,並びに最小と最大の高さについて推奨する値が,prEN 13528-3[1]
に示されている。
A.5.2 気流の影響
気流速度の起こし得る影響については,既にA.4.4で述べた。気流速度が低くなるのは,ほとんどが室
内で,強制換気のないところでは約10 cm/s(参考文献[49], [51])になる。ヨーロッパでの月平均大気風速
は,およそ1〜10 m/sの範囲にある(参考文献[52])が,安定した気象条件下でも一時的に0.5 m/s以下に
急減することがある。山間部では局地的に急減することがある(参考文献[53], [54])。
そのうえ,少なくとも局地的発生源では汚染物質濃度は,気流速度と反比例するので(参考文献[52]),
24
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
低い気流速度におけるサンプリング誤差は,時間加重平均で拡大される。
空気移動の少ない室内では(5〜10 cm/s),サンプラは設計どおりの性能を発揮することが確認されてい
る。屋外では,降水,直射日光,高い気流速度が,サンプラの性能に悪影響を及ぼすことがあり,シェル
タによる保護が必要となる(参考文献[57], [58])。
A.5.3 降雨からの保護
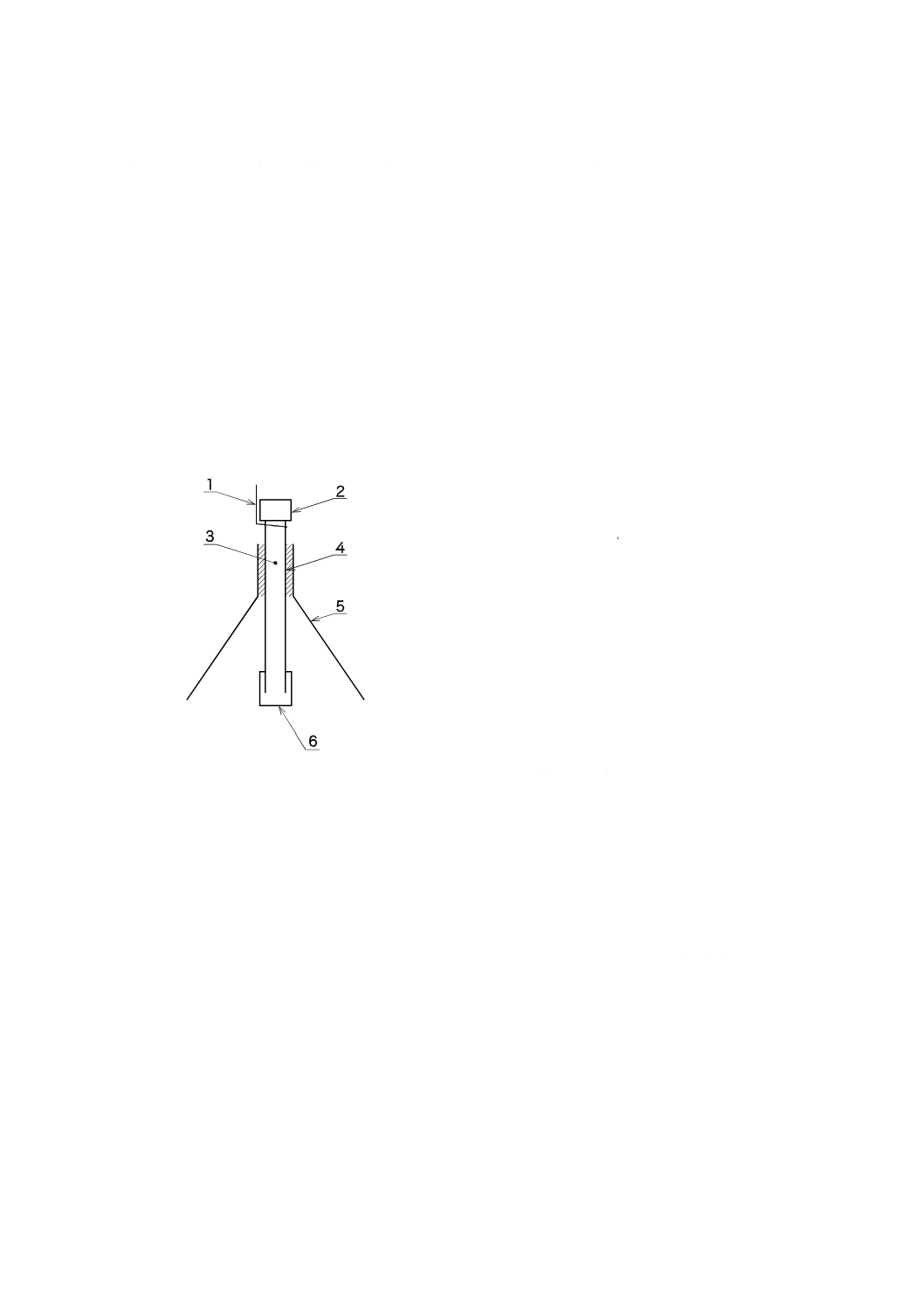
降雨からの保護は,全ての種類のサンプラにとって重要である。雨又は溶けた雪がサンプラ表面を塞ぐ
ことがあり(参考文献[55]),特に垂直(微粒子の侵入を防ぐための通常の位置)に下を向いたチューブ形
サンプラは塞がれやすい。チューブ形サンプラのための簡単なシェルタとしては,逆にしたプラスチック
漏斗がある。漏斗は,サンプラが漏斗の噴射口にぴったりと収容され(必要ならば短く切断してもよい。),
サンプラの開口部が漏斗の開口部の下に目で見えるように装着する(参考文献[56])。図A.2に例を示す。
もう一つの方法は,化学変化のない“巣箱”で,底に穴をあける。サンプラは箱の内部に入れ,やはり
サンプラの開口部が箱の開口部の下に目で見えるように装着する。
記号
1 ひも(糸)
2 スクリューキャップ
3 吸着剤
4 サンプラ
5 漏斗
6 金網付き拡散キャップ
図A.2−野外用カバー付きチューブ形サンプラ
シェルタの代わりにパッシブサンプラを改造する方法もある。例えば,サンプラの端末キャップにアル
ミ製のつばを付けて,雨水が拡散面を塞ぐのを防止する(参考文献[25])。しかし,このような改造によっ
て,必要とされる最小気流速度などのサンプラの特性が変わる。
A.5.4 セキュリティ
公共の場所にサンプラを長期間置く場合,盗難及び破壊から守るセキュリティについても考慮する。サ
ンプラはできるだけ手の届かない,目立たない位置に置くべきである。また,例えば,巣箱に似せるなど
の対策もある。
25
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
吸着剤の種類
吸着剤名
吸着剤の種類
Ambersorb XAD-4
スチレン/ジビニルベンゼン共重合体
Carbotrap B/C
グラファイトカーボン
Carbopack B/C
グラファイトカーボン
Carbosieve S-III
カーボンモレキュラーシーブ
Carboxen 569
カーボンモレキュラーシーブ
Carboxen 1000
カーボンモレキュラーシーブ
Chromosorb 102
スチレン/ジビニルベンゼン
Chromosorb 106
ポリスチレン
Carbograph TD-1
グラファイトカーボン
Porapak N
ビニルピロリドン
Porapak Q
エチルビニルベンゼン/ジビニルベンゼン
Spherocarb
カーボンモレキュラーシーブ
Tenax TA
ポリ(2.6-ジフェニル-p-フェニレンオキシド)
Tenax GR
グラファイト化ポリ(フェニレンオキシド)
注記 CarbotrapTM,CarbopackTM,Carbosieve S-IIITM及びCarboxenTMは,Supelco, Inc., USAの登録商標
である。TenaxTMは,Enka Research Institute, NV, NLの登録商標である。ChromosorbTMは,Manville
Corp, USAの登録商標である。PorapakTMは,Waters Associates Inc., USAの登録商標である。
SpherocarbTMは,Analabs Inc., USAの登録商標である。AmbersorbTMは,Rohm&Haas Co., USA
の登録商標である。CarbographTMは,Alltech Associated, USAの登録商標である。
この情報はこの規格の利用者の便宜を図るものであり,この名称の製品を推奨するものではない。その
他のものが同じ性能を示すものであれば用いてもよい。
26
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(参考)
吸着剤選択の手引き
サンプラ用吸着剤a)
分析成分
揮発性の範囲
最高使用温度
℃
表面積
m2/g
対象成分例
CarbotrapTM C
CarbopackTM C
n-C8からn-C20
400を超え
12
n-C8からn-C16の揮発性範囲のアルキ
ルベンゼン及び脂肪族
TenaxTM TA
沸点100〜400 ℃
n-C6からn-C26
350
35
芳香族,無極性化合物(沸点>100 ℃)
及び揮発性の低い極性化合物(沸点>
150 ℃)
Tenax GR
沸点100〜450 ℃
n-C7からn-C30
350
35
アルキルベンゼン,ガス状多環芳香族
炭化水素,ポリ塩化ビフェニル,上記
Tenax TAと同じ成分
CarbotrapTM B
CarbographTM TD-1
CarbopackTM
(n-C4) n-C5から
n-C14
400を超え
100
ケトン,アルコール及びアルデヒド(沸
点>75 ℃),揮発性のある全ての無極
性化合物,パーフルオロカーボントレ
ーサーガス
ChromosorbTM 102
沸点50〜200 ℃
250
350
含酸素化合物,塩化メチレンより揮発
性の低いハロホルムを含む広範の
VOC
ChromosorbTM 106
沸点50〜200 ℃
250
750
n-C5からn-C12の炭化水素を含む広範
のVOC,揮発性酸化化合物
PorapakTM Q
沸点50〜200 ℃
n-C5からn-C12
250
550
含酸化化合物を含む広範なVOC
PorapakTM N
沸点50〜150 ℃
n-C5からn-C8
180
300
揮発性含窒素化合物
SpherocarbTM b)
沸点−30〜150 ℃
C3からn-C8
400を超え
1 200
塩化ビニル,酸化エチレン,二硫化炭
素,高揮発性化合物(例:CH2CL2),
揮発性化合物(例:メタノール,エタ
ノール及びアセトン)
CarbosieveTM S-III b) 又は
CarboxenTM 1000 b)
沸点−60〜80 ℃
400
800
C3,C4炭化水素,揮発性ハロホルム及
びフレオンのような高揮発性化合物
Molecular Sieve c)
沸点−60〜80 ℃
350
1,3-ブタジエン及び亜酸化窒素
注a) 登録商標の表示は,附属書Bに示す。
b) これらの吸着剤は,幾らかの水分を保持する。相対湿度が高い(90 %を超える。)場合は,安全試料採取量は
1/10に減らすことが望ましい。
c) かなり親水性である。特別な対策をしないで高湿度雰囲気下で使用しない。
27
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D
(参考)
吸着剤使用の手引き
サンプラ用吸着剤
最高使
用温度
℃
疎水性
前処理時の
温度a) 及び
ガス流量
脱離時の温度
及びガス流量
推奨冷却トラップ充塡剤
Carbotrap C
Carbopack C
400を
超え
あり
350 ℃
100 mL/min
325 ℃
30 mL/min
Tenax又はCarbopack C
Tenax TA
350
あり
330 ℃
100 mL/min
300 ℃
30 mL/min
Tenax
Tenax GR
350
あり
330 ℃
100 mL/min
300 ℃
30 mL/min
Tenax
Carbotrap B
Carbopack
400を
超え
あり
350 ℃
100 mL/min
325 ℃
30 mL/min
Tenax又はCarbopack B
Chromosorb 102
250
あり
250 ℃
100 mL/min
250 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ又はChromosorb 102の二層
充塡
Chromosorb 106
250
あり
250 ℃
100 mL/min
250 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ又はChromosorb 106の二層
充塡
Porapak Q
250
あり
250 ℃
100 mL/min
225 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ又はPorapak Qの二層充塡
Porapak N
180
あり
180 ℃
100 mL/min
180 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ又はPorapak Nの二層充塡
Spherocarb b)
400を
超え
なし
400 ℃
100 mL/min
390 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ又はSpherocarb二層充塡
Carbosieve S-III b)
Carboxen 1000 b) などの
カーボンモレキュラー
シーブ
400
なし
350 ℃
100 mL/min
325 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ二層充塡又はカーボンモレ
キュラーシーブ単層
モレキュラーシーブc)
350
なし
330 ℃
100 mL/min
300 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ二層充塡又はカーボンモレ
キュラーシーブ単層
Tenax/Carbopack B複合
サンプラ
350
あり
330 ℃
100 mL/min
300 ℃
30 mL/min
Tenax
Carbopack B b)/カーボ
ンモレキュラーシーブ
複合サンプラ
400
なし
350 ℃
100 mL/min
325 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ二層充塡
Carboxen 1000シリーズ
複合サンプラ
400
なし
350 ℃
100 mL/min
325 ℃
30 mL/min
Carbopack B+カーボンモレキュラ
ーシーブ二層充塡
注記 登録商標の表示は,附属書Bに示す。
注a) 前処理時の温度は,最初の処理温度と同一ではない(5.3参照)。
b) これらの吸着剤は,幾らかの水分を保持する。相対湿度が高い(90 %を超える。)ときは,安全試料採取量を
1/10に減らすことが望ましい。
c) 親水性である。特別な対策をしないで高湿度雰囲気下で使用しない。
28
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書E
(参考)
総合不確かさ,精度,偏り及び保管要領
E.1
総合不確かさ
適切な吸着剤を充塡したサンプラ(6.1)を用いた,EN 838[4]による実験室試験結果から得られた,個々
の有機化合物の拡散取込み速度を次に示す。また,適切な吸着剤を充塡したサンプラ(6.1)を用いて,EN
13528-2[5]による実験室試験及び現地試験結果から得た,個々の有機化合物の拡散取込み速度を表2に示
す。ただし,サンプラサンプリングキャップ内にシリコーン膜がないものを用いた。シリコーン膜がある
場合は,約10 %ほど低い結果となる。多くの場合,それぞれ異なった数値は,長期間サンプリングの大気
及び室内測定の場合よりも,作業場における短期間モニタリングの場合に適用する。これらの評価の結果
は,様々な出典については参考文献[10]に要約されている。表1には結果のまとめが,表2には出典が示
されている。拡散取込み速度は,パッシブサンプラの製造方法の違い,膜の有無又は使用される吸着剤の
種類によって異なるが,他のシステムの一般的な性能は,ここに示す性能と同様であると推測される(9.6
参照)。
調製したサンプラを,現場(1回であるが世界規模の調査)に送付し,1か月間同時に暴露(密閉)した
後,実験室に返送して,ベンゼン,トルエン及びキシレンについてブランクレベルを測定した(参考文献
[11])。サンプラChromosorb 106及びCarbograph TD-1の結果を,表8に示す。いずれの吸着剤も,回収率
は低ngの範囲であり,新たに調製した(参考文献[1])Carbographより,若干高い値となった。
E.2
データの精度及び偏り
6.1に規定するサンプラを使用して実験室での保管実験をしたデータの概要を示す。
実験室試験(参考文献[2])によって,Tenax又はChromosorb 106のいずれかを用いてサンプリングした
芳香族炭化水素,塩素化炭化水素,ケトン,エステル及びグリコールエーテルを含む14の代表的なVOC
について,同一ロット内の繰返し再現性(繰返し6回の変動係数σn−1)は,0.5〜2 %であることが示され
た。試料は,総合不確かさが3 %以内に調製した基準大気からサンプリングした。ロット間の変動率(す
なわち,暴露時間2〜4時間,濃度LVの0.5〜2.0倍の異なる条件での評価)は2〜12 %であった。変動率
は化合物によって異なり,暴露条件の違いは取込み速度の僅かな変化に反映する。吸着剤及び分析対象成
分の組合せが理想的でない場合は,比較的高い数値を示す。ロット間の変動率は,6〜24 %の総合不確か
さに等しい。
ベンゼン,トルエン,キシレンを80 ng又は200 ngのレベルで吸着した液体添加サンプラに対する実験
室試験(参考文献[11])の結果の概要を示す。サンプラを,現場(1回であるが世界規模の調査)に送付し,
1か月間同時に暴露(密閉)した後,実験室に返送して分析した。Chromosorb 106及びCarbograph TD-1の
サンプラの回収率は,82.7〜105.9 %であった。精度は,吸着剤及び分析対象成分によって異なるが,変動
係数で3.2〜12.1 %の範囲であった。英国における多数の現地試験(参考文献[11])において,サンプラ及
び固定式炭化水素モニタリング装置を用いて計測したところ,ベンゼン,トルエン,キシレンは,大気濃
度1〜10 μg/m3であった。パッシブサンプリングの平均精度は,変動係数で表すと,ベンゼン11 %,トル
エン7 %,キシレン5 %であった[サンプラの数n=4(Chromosorb 106及びCarbograph各2本),4週間暴
露]。結果の詳細を表4に示す。“英国VOC空気ネットワーク”のデータは,パッシブサンプリング期間
29
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
中のポンプサンプリングのデータが少ないため,参考にとどめることが望ましい。ブラジルを除く世界各
国の多数の同様な現地試験(参考文献[11])から,パッシブサンプリングの平均精度は,変動係数で表す
と,ベンゼン11 %,トルエン8 %,キシレン6 %であった(表5)。
英国における現地試験(参考文献[12])から,同一地点についてパッシブサンプラ及び固定式炭化水素
モニタリング装置を用いて計測したところ,ベンゼンの大気濃度は1〜2.5 μg/m3であった。年平均値の比
較では,パッシブサンプラを用いた場合が1.76 μg/m3であったのに対して,モニタリング装置を用いた場
合は,1.95 μg/m3であった。
表1にはレベルA又はレベルBの結果が示されている。EN 482[13]に定義する総合不確かさは30 %よ
りも良好である。表2のデータについては,EN 13528-1[14]に定義する総合不確かさは,30 %より良好と
期待される。
注記 作業場の計測の場合(表1),レベルAはEN 838[4]に従って十分な妥当性が確認されている。
レベル1Aは,作業方法の実験室評価である(レベル1Bは,標準に基づき妥当性が確認された
化合物の同族体について考慮している。)。表1のレベルBは,EN 482に基づき一時的に許容
されている部分的な妥当性確認である(EN 838の試験項目中,保証されていない項目がある。)。
大気の測定の場合(表2),EN 13528-2[5]による十分な妥当性確認には,実験室試験及び現地試
験の両方が要求され,表2のデータはいずれも十分な評価を行ったものではない。EN 13528-1
が認めるレベルBに相当するものはない。しかしながら,上記のデータから,環境サンプリン
グの現地での精度は,5〜11 %の範囲であると想定され,取込み速度計測結果の再現性(表4)
は約10 %である。したがって,それらを合わせた精度は約15 %,総合不確かさは30 %である。
E.3
保管に関するデータ
6.1に規定するサンプラを使用した実験室保存性試験をしたデータの概要を,表6及び表7に示す。
Chromosorb 106及びCarboxen 569を充塡したサンプラに,約1.0 μgの添加レベルで,特定の化合物を液
体添加し,室温で2週間保管した実験室試験(参考文献[15])結果の概要を次に示す。Chromosorb 106に
よる平均回収率は105.6 %であった。サンプラTenax TAに約10 μgの添加レベルで,より多種の化合物を
液体添加し,室温で5か月保管した実験室試験(参考文献[16])の結果の概要を示す。ヘキサン及びメト
キシエタノール以外の平均回収率は99.7 %であった。平均変動係数(σn−1)は2 %であった。同様の結果
が,11か月間保管した後にも得られており,ヘキサン及びメトキシエタノール以外の平均回収率は99.4 %
であり,平均変動係数は0.9 %であった。
“CRM112”(参考文献[17])による検定中,ベンゼン,トルエン,m-キシレンを添加したサンプラの安
定性について,温度0〜40 ℃で25か月まで調査した。保管温度0〜4 ℃で,14か月後における3種類の
化合物の回収率は101〜103 %であった。同様の条件での常温及び40 ℃での回収率は,それぞれ102〜
104 %,100〜104 %であった。25か月後も不安定ではなかったが,回収率は報告されていない。
密栓は,冷蔵中に熱収縮の差によって緩みを生じることがある。試料損失又は外部汚染の侵入を防ぐた
め定期的に密栓をチェックする必要がある。冷蔵は吸着されたVOC同士の相互反応を減らすのに役立つ。
30
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JA
(参考)
日本におけるパッシブサンプリングの実測データ
JA.1
日本におけるパッシブサンプリング実測データ
日本では,パッシブサンプリング法としては,国土交通省通達による測定が主流である。実際の居室に
おいて,24時間のパッシブサンプリングを行った実測データを次に示す。
化合物
吸着剤
暴露時間
取込みa)
(標準偏差)
cm3/min
取込みa)
(標準偏差)
ng/(μL/L)/min
データ数
トルエン
Tenax TA
24
0.42
(0.04)
1.6
(0.14)
6
キシレン
Tenax TA
24
0.43
(0.03)
1.8
(0.11)
6
エチルベンゼン
Tenax TA
24
0.41
(0.04)
1.8
(0.17)
6
スチレン
Tenax TA
24
0.46
(0.08)
1.9
(0.18)
6
p-ジクロロベンゼン
Tenax TA
24
0.45
(0.04)
2.7
(0.21)
6
注a) サンプラ(6.1,膜なし)の取込み速度(暴露時間24時間の場合に適用)
31
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] prEN 13528-3:2001,Ambient air quality−Diffusive samplers for the determination of gases vapours−Part 3:
Guide to selection, use and maintenance
[2] WRIGHT, M. D. (1993). Diffusive uptake rates for the Perkin Elmer tube−BCR air sampling intercomparison
at Vito. Mol, Belgium Feb 1991−April 1992. HSE internal report, IR/L/IA/93/3, Health and Safety Laboratory,
Broad Lande, Sheffield S3 7HQ, UK.
[3] STERNBERG, J. C. et al. The mechanism of response of flame ionization detectors. 3rd Intern. Symp. Gas
Chromat., (1960), pp.231-267.
[4] EN 838,Workplace exposure−Procedures for measuring gases and vapours using diffusive samplers−
Requirements and test methods.
[5] EN 13528-2,Ambient air quality. Diffusive samplers for the determination of concentrations of gases and
vapours−Requirements and test methods. Part 2: Specific requirements and test methods
[6] KNOEPPEL, H., VERSINO, B., SCHLITT, H., PEIL, A., SCHAUENBURG, H., VISSERS, H. Organics in air.
Sampling and identification. Proceedings of the first European Symposium on physico-chemical behaviour of
atomospheric pollutants. ISPRA, 16-18 October 1979, 25-40. Commission of the European Communities,
Brussels-Luxemburg 1980
[7] DULSON, W. Organisch-chemische Fremdstoffe in atomosphärischer Luft. Schriftenrihe des Vereins für
Wasser-, Boden-und Lufthygiene Nr. 47. Stuttgart: Gustav-Fischer-Verlag (1978)
[8] BERTONI, G., BRUNER, F., LIBERTI, A., PERRINO, C. Some critical parameters in collection, recovery and
chromatogaraphic analysis of orgainc pollutants in ambient air using light absorbents. J. Chromatogr., 203
(1981), pp.263-270.
[9] VIDAL-MADJAR, C., GONNORD, M.-F., BENCHAH, F., GUIOCHON, G.: Performances of vaious
adsorbents for the trapping and analysis of organohalogenated air pollutants by gas chromatography. J.
Chromatogr. Sci. 16 (1978), pp.190-196
[10] Health and Safety Excutive (1995). Methods for the Determination of Hazardous Substances. Volatile organic
compounds in air−Laboratory method using diffusive solid sorbent tubes, thermal desorption and gas
chromatography. MDHS 80, HSE:London.
[11] PLANT, N. T., WRIGHT, M. D European Diffusive Sampling Initiative: Project report with status at March
1998. IACS 98/01. Health and Safety Laboratory, Sheffield S3 7HQ, UK
[12] BOYLE, W. A. A comparison of benzene data from an automatic monitoring instrument and diffusive samplers.
The Diffusive Monitor, 10 (1998), p.12
[13] EN 482,Workplace atomospheres−General requirements for perfomance of procedures for the measurement of
chemical agents
[14] EN 13528-1,Ambient air quality. Diffusive samplers for the determination of concentrations of gases and
vapours. Requirements and test methods. Part 1:General requirements
[15] Study of sorbing agents for the sampling of volatile compounds from air. EC Contract MAT1CT92-0038. Final
Report (1995).
[16] Health and Safety Excutive (1993). Methods for the Determination of Hazardous Substances. Volatile organic
32
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
compounds in air−Laboratory method using pumped solid sorbent tubes, thermal desorption and gas
chromatography. MDHS 72, HSE: London.
[17] VANDENDRIESSCHE, S., GRIEPINK, B., HOLLANDER, J. C. Th., GIELEN. J. W. J., LANGELAAN, F. G.
G. M., SAUNDERS, K. J. and BROWN, R. H. Certification of a Reference Material for Aromatic
Hydrocarbons in Tenax Samplers. Analyst, 116 (1991), pp.437-441.
[18] Health and Safety Excutive (1991). Methods for the Determination of Hazardous Substances. Analytical quality
in workplace air monitoring. MDHS 71. HSE: Health and Safety Laboratory, Broad Lane Sheffield, S3 7HQ,
UK
[19] PETERS, R., HAFKENSCHEID, T. Testing of tube-type diffusive samplers for monitoring VOC in ambient air.
The Diffusive Monitor, 7 (1995), pp.8-9
[20] ERLAP data in reference [11]
[21] BROWN, V. M., CRUMP, D. R., GARDINER, D., YU, C. W. F. Long-term diffusive sampling of volatile
organic compounds in indoor air. Environ. Technol., 14 (1993), pp.771-777
[22] NMI field data in reference[11]
[23] BP data in DOWNING, G. C. E. H., CAMPBELL, G. W., BAILEY, J.C. A survey of sulphur dioxide, ammonia
and hydrocarbon concentrations in the United Kingdom, using diffusion tubes: July to December 1992. UK
Environmental Technology Executive Agency, Report LR 964 Contract no PECD/7/12/76 (1994)
[24] PFEFFER H.-U., BREUER L., ELLERMANN, K. Materialien No.46. Validation of Passive Samplers for
Measurements of Hydrocarbons in Ambient Air. Editor: Landesumweltamt Nordrhein-Westfalen (North
Rhine-Westphalia State Environment Agency), Wallneyer Str. 6, Essen, Germany, p.92 (1998) (ISSN
0947-5206)
[25] MOWRER, J., POTTER, A., LINDBERG, A. Diffusive monitoring of C6-C9 hydrocarbons in urban air in
Sweden. Analyst, 121 (1996), pp.1295-1300
[26] NMI field data in reference[11]
[27] BROWN, R. H. Diffusive sampling. Clean Air at Work. Eds. Brown, R. H. Curtis, M., Saunders, K. J. and
Vandendriessche, S. EC Publ. no. EUR 14214, Brussels-Luxembourg (1992), pp141-148.
[28] VAN DEN HOED, N., VAN ASSELEN, O. L. J. A computer model for calculating effective uptake rates of
tube-type diffusive air samplers. Ann. occup. Hyg., 35 (1991), pp.273-285
[29] NORDSTRAND, E., KRISTENSSON, J. A computer model for simulating the performance of thick-bed
diffusive samplers. Am. Ind. Hyg. Assoc. J., 55 (1994), pp.935-941
[30] BARTLEY, D. L., DOEMENY, L. J. and TAYLOR, D. G. Diffusive monitoring of fluctuating concentrations.
Am. Ind. Hyg. Assoc. J., 44 (1983), pp.214-217
[31] BARTLEY, D. L. Diffusive monitoring of fluctuating concentrations using weak sorbents. Am. Ind. Hyg. Assoc.
J., 44 (1983), pp.879-885
[32] BARTLEY, D. L., WOEBKENBERG, M. L. and POSNER, J. C. Performance of thick-sorbent diffusive
samplers. Ann. occup. Hyg., 32 (1988), pp.333-343
[33] BARTLEY, D. L., DEYE, G. J. and WOEBKENBERG, M. L. Diffusive monitor test: performance under
transient conditions. Appl. Ind. Hyg. 2 (1987), pp.119-122
[34] POSNER, J. S., MOORE, G. A thermodynamic treatment of passive monitors. Am. Ind. Hyg. Assoc. J., 46
(1985), pp.227-285
33
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[35] BROWN, R. H., CHARLTON, J., SAUNEERS, K. J. The developement of an improved diffusive sampler. Am.
Ind. Hyg. Assoc. J., 42 (1981), pp.865-869
[36] HEARL, F. J., MANNING, M. P. Transient response of diffusive dosimeters. Am. Ind. Hyg. Assoc. J., 41 (1980),
pp.778-783
[37] UNDERHILL, D. W.: Unbiased passive sampling. Am. Ind. Hyg. Assoc. J., 44 (1983), pp.237-239
[38] HORI, H., TANAKA, I.: Response characteristics of the diffusive sampler at fluctuating vapor concentrations.
Am. Ind. Hyg. Assoc. J., 54 (1993), pp.95-101
[39] COMPTON, J. R., DWIGGINS, G. A., FELGLEY, C. E., LUDWIG, D. A.: The effect of square-wave exposure
profiles upon the performance of passive organic vapor monitoring badges. Am. Ind. Hyg. Assoc. J., 45 (1984),
pp.446-450
[40] TOMPKINS F. C. and GOLDSMITH, R. L.: A new personal dosimeter for the monitoring of industrial
pollutants. Am, Ind. Hyg. Assoc. J., 38 (1977), 371-377
[41] UNDERHILL, D. W., FEIGLEY, C. E.: Boundary layer effect in diffusive monitoring. Anal. Chem. 63 (1991),
1011-1013
[42] POZZOLI, L., COTTICA, D. An overview of the effects of temperature, pressure, humidity, storage and face
velocity. Diffusive Sampling, an alternative approach to workplace air monitoring. Eds Berlin, A., Brown, R. H.
and Saunders, K. J., EC Publ. No. 10555 EN, Brussels-Luxembourg (1987), pp.119-130
[43] ZURLO, N., ANDREOLETTI, F.: Effect of air turbulence on diffusive sampling. Diffusive Sampling, an
alternative approach to workplace air monitoring. Eds Berlin, A., Brown, R. H. and Saunders, K. J., EC Publ.
No. 10555 EN, Brussels-Luxembourg (1987), pp.174-176
[44] COLEMAN, S. R. A tube-type diffusive monitor for sulphur dioxide. Am. Ind. Hyg. Assoc. J., 44 (1983),
pp.929-936
[45] PALMES, E. D., GUNNISON, A. F., DIMATTIO, J., TOMCZYK, C. Personal sampler for nitrogen dioxide.
Am. Ind. Hyg. Assoc. J., 37 (1976), pp.570-577
[46] GAIR, A. J., PENKETT, S. A. The effects of wind speed and turbulence on the performance of diffusion tube
samplers. Atmos. Env. 29 (1995), pp.2529-2533
[47] DOWNING, C. E. C., CAMPBELL, G. W., BAILEY, J. C. A survey of sulphur dioxide, ammonia and
hydrocarbon concentrations in the United Kingdom using diffusion tubes: July to December 1992. Warren
Spring Laboratory, Stevenage, UK, Report LR 964 (1994)
[48] FERM, M. A sensitive diffusional sampler. Institutet for Vatten och Luftvardsforskning, Sweden, Report B 1020
(1991)
[49] PANNWITZ, K. H. Influence of air currents on the sampling of organic solvent vapours with diffusive samplers.
In Diffusive Sampling, an alternative approach to workplace air monitoring. Eds Berlin, A., Brown, R. H. and
Saunders, K. J., EC Publ. No. 10555 EN, Brussels-Luxembourg (1987), pp.157-160
[50] MARK, D., ROBERTSON, A., GIBSON, H., BORZUCKI, G., CHERRIE, B., MACLAREN, W. M. Evaluation
of diffusive samplers for monitoring toxic gases and vapours in coalmines. Institute of Occupational Medicine,
Edinburgh, Report TM/90/11 (1990)
[51] MATTHEWS, T. W., THOMPSON, C. V., WILSON, D. L., HAWTHORNE, A. R., MAGE, D. T. Air velocities
inside domestic environments: an important parameter for passive monitoring. Indoor air '87−Proc. 4th Intern.
Conf. on Indoor Air Quality and Cimate. Eds. B Seifert et. al. Inst. for Water, Soil and Air Hygiene, Berlin, 1
34
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1987), pp.174-178
[52] PLANT, N. T., WRIGHT, M. D. European Diffusive Sampling Initiative: Pilot surveys of Sheffield. Health and
Safety Laboratory, Sheffield, Reprot IACS/96/1 (1996)
[53] TROEN, I., PETERSEN, E. L. European-windatlas, RISO National Laboratory, Roskilde, Denmark (1990)
ISBN 87-550-1636-7
[54] CHRISTOFFER, J., ULBRICHT-EISSING, M. Die bodennahen Windverhältnisse in der BR Deutschland,
Berichte des Deutschen Wetterdienstes 147, Offenbach-a.-M. (1989)
[55] HAFKENSCHEID, T. L., MOWRER, J. Intercomparison of tube-type diffusive sampling for the detemination of
volatile hydrocarbons in ambient air. Analyst, 121 (1996), pp.1249-1252
[56] PLANT, N. T., WRIGHT, M. D. European Diffusive Sampling Initiative:UK national survey of BTX. Health and
Safety Laboratory, Sheffield, Report IACS/97/6 (1997), Health and Safety Laboratory, Broad Lane, Sheffield S3
7HQ, UK
[57] VDI 2119, Part 4. Measurement of particulate percipitations: microscopic differentiation and size fraction
determination of particle deposition on adhesive collection plates: Sigma-2 sampler (1997)
[58] REMPLER, P. und Kosmus, W. Integrale Langzeitmethode zur Bestimmung von Ozon in der Atmosphäre.
Fresenius Z. Anal. Chem., 329 (1998), pp.871-874
[59] JIS A 1960 室内空気のサンプリング方法通則
注記 ISO 16000-1:2004,Indoor air−Part 1: General aspects of sampling strategy(MOD)
35
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JB
(参考)
JISと対応国際規格との対比表
JIS A 1967:2015 室内空気中の揮発性有機化合物(VOC)の吸着捕集・加熱脱離・
キャピラリーガスクロマトグラフィーによるサンプリング及び分析−パッシブサン
プリング
ISO 16017-2:2003,Indoor, ambient and workplace air−Sampling and analysis of
volatile organic compounds by sorbent tube/thermal desorption/capillary gas
chromatography−Part 2: Diffusive sampling
(I)JISの規定
(II)
国際規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇条
ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
2 引用規
格
3 用語及
び定義
この規格で用いる
主な用語及び定義
は,JIS A 1966によ
る。
−
−
追加
関連するJIS(ISO規格)との
記載の統一化
技術的な差異はない。
次回のISO規格の改正時に修正
を提案する。
5 試薬・材
料
試薬,標準溶液の調
製方法などを規定。
4
JISとほぼ同じ
変更
ケトンをカルボン酸に修正し
た。
アルコールと縮合するのはケト
ンではなくカルボン酸であるた
め修正した。次回のISO規格の改
正時に提案を検討する。
5.2 希釈溶媒
4.2 希釈溶媒
削除
酢酸エチル,シクロヘキサンな
どはVOCの成分になる場合が
あるので削除した。
次回のISO規格の改正時に修正
を提案する。
6 装置
6.1 サンプラ
5
5.1 サンプラ
追加
実質的な差異はない。
次回のISO規格の改正時に修正
を提案する。
6.3 サンプラサンプ
リングキャップ
5.3 サンプラサンプリン
グキャップ
追加
実質的な差異はない。
6.5 ガスクロマトグ
ラフ
トルエン1 ng
5.5 ガスクロマトグラフ
トルエン0.5 ng
変更
条件の緩和
8 サンプ
リング
現場ブランク
7
トラベルブランク
変更
実質的な差異はない。
技術的な差異はない。
3
5
A
1
9
6
7
:
2
0
1
5
36
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇条
ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
9.2.1 脱離 キャリヤーガスに
ヘリウム又は窒素
を用いる。
8.2.1
キャリヤーガスにヘリウ
ムを用いる。
追加
JISでは,冷却の程度によって
窒素を選択できることとした。
次回のISO規格の改正時に修正
を提案する。
9.6 拡散
取込み速
度の算出
拡散取込み速度
(Diffusive uptake
rate)
8.6
−
追加
拡散取込み速度の意味を明確
化するため,原文を引用した。
実質的な差異はない。
10.1 分析
対象成分
の質量濃
度
記号の変更
9.1
JISとほぼ同じ
変更
関連するJIS(ISO規格)の記
号の統一化
技術的な差異はない。
次回のISO規格の改正時に修正
を提案する。
10.2 分析
対象成分
の体積比
記号の変更
9.2
JISとほぼ同じ
変更
関連するJIS(ISO規格)の記
号の統一化
技術的な差異はない。
次回のISO規格の改正時に修正
を提案する。
10.3 拡散
取込み速
度u及び
u'の関係
記号の変更
9.3
JISとほぼ同じ
変更
関連するJIS(ISO規格)の記
号の統一化
技術的な差異はない。
次回のISO規格の改正時に修正
を提案する。
14 品質管
理
表7 単位μg
13
JISとほぼ同じ
mg
変更
mgをμgに変更した。
ISO規格の誤り。
次回のISO規格の改正時に修正
を提案する。
附属書A
A.1 原理
附属書JA
(参考)
3
6
A
1
9
6
7
:
2
0
1
5

37
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JISと国際規格との対応の程度の全体評価:ISO 16017-2:2003,MOD
注記1 箇条ごとの評価欄の用語の意味は,次による。
− 削除……………… 国際規格の規定項目又は規定内容を削除している。
− 追加……………… 国際規格にない規定項目又は規定内容を追加している。
− 変更……………… 国際規格の規定内容を変更している。
注記2 JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD…………… 国際規格を修正している。
3
7
A
1
9
6
7
:
2
0
1
5
38
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JC
(参考)
技術上重要な改正に関する新旧対照表
現行規格(JIS A 1967:2015)
旧規格(JIS A 1967:2005)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
1 適用範
囲
この規格は,室内,大気及び労働環境の空気に適用す
る。
1. 適用範
囲
この規格は,室内空気に適用する。
ISO 16017-2:2003への整合
ただし,高極性物質は誘導体化が必要で,低沸点化合
物は…
ただし,低沸点化合物は…
ISO 16017-2:2003への整合
暴露時間8時間の場合2〜1×105 μg/m3,…
暴露時間8時間の場合20〜1×105 μg/m3,…
ISO 16017-2:2003への整合
2 引用規
格
JIS A 1966
2.
−
関連JIS間での記載の統一
化
3 用語及
び定義
この規格で用いる主な用語及び定義は,JIS A 1966に
よる。
−
−
関連JIS間での記載の統一
化
5.7.6 気体
成分約10
μg/mLを
含む溶液
10 μLのガスタイトシリンジに純ガス10 μLを満たし
…
4.7.6 気体
成分約10
μg/mLを
含む溶液
10 μLのガスタイトシリンジに純ガス1 μLを満たし,
…
ISO 16017-2:2003への整合
5.8 標準
液体添加
サンプラ
作業環境については,5.7.3,5.7.4又は5.7.6の溶液の
1〜5 μLをサンプラに添加する。
4.8 標準
液体添加
サンプラ
−
ISO 16017-2:2003への整合
6.1 サン
プラ
表1の拡散取込み速度は,少なくとも14 mmの空隙
…
5.1 サン
プラ
表1の拡散取込み速度は,公称15 mmの空げき(隙)
…
ISO 16017-2:2003への整合
6.5 ガス
クロマト
グラフ
…最低5:1のS/N比でトルエン1 ngの注入を…
5.5 ガス
クロマト
グラフ
…最低5:1のSN比でトルエン0.5 ngの注入を…
ISO 16017-2:2003への整合
9.2.1 脱離 キャリヤーガス ヘリウム又は窒素(冷却の程度によ
って選択する。)
8.2.1 脱離 キャリヤーガス:ヘリウム
JIS A 1966への整合
3
8
A
1
9
6
7
:
2
0
1
5
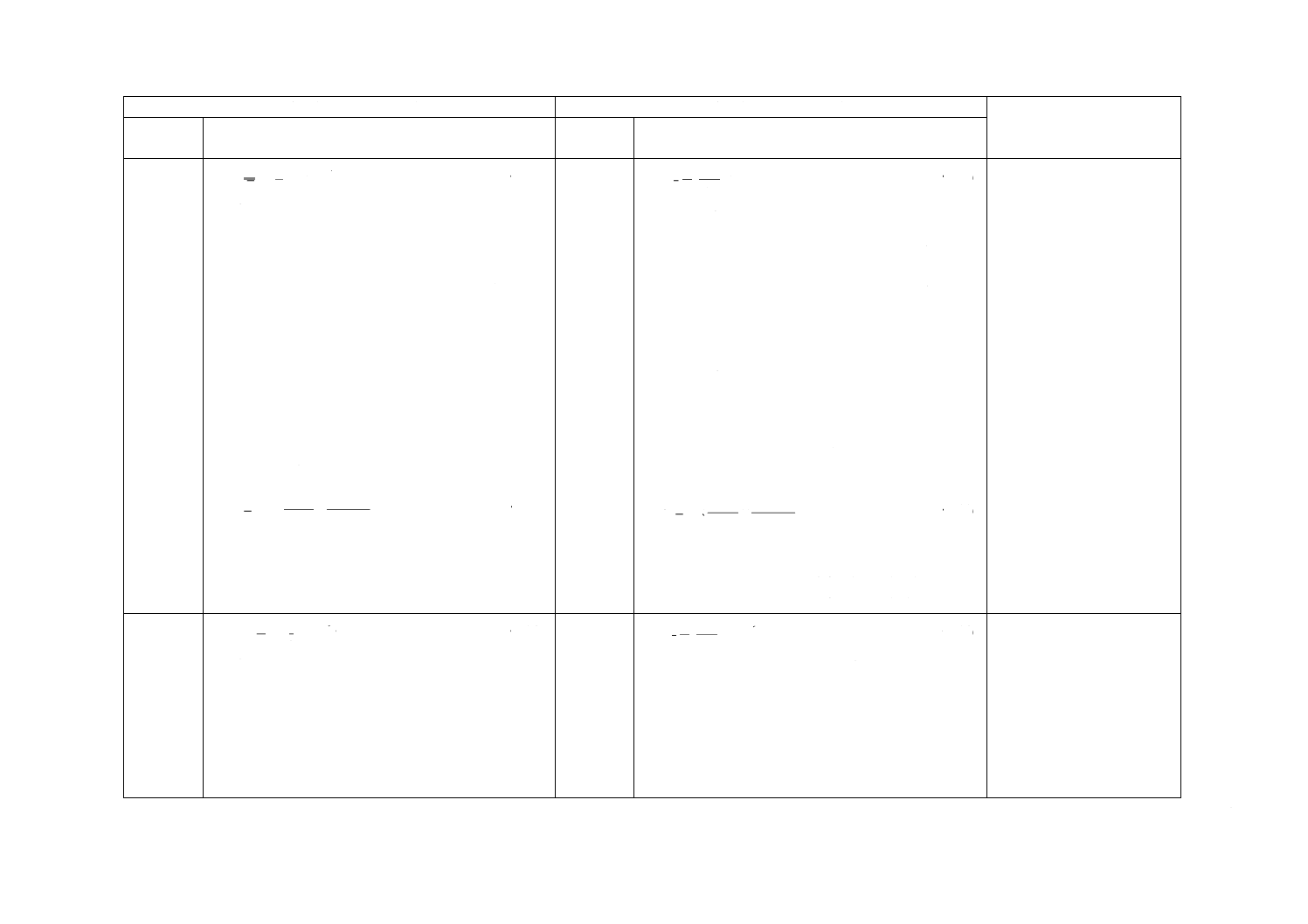
39
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格(JIS A 1967:2015)
旧規格(JIS A 1967:2005)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
10.1 分析
対象成分
の質量濃
度
6
b
a
m
10
×
×
−
=
t
u
m
m
ρ
······································ (1)
ここに, ρm: 試料空気中の分析対象成分の質量濃度
(μg/m3)
ma: 9.4で求めた試料中の分析対象成分の
質量(μg)
mb: ブランクサンプラ中の分析対象成分の
質量(μg)
u: 拡散取込み速度(cm3/min)(表1又は
9.6)
t: 暴露時間(min)
注記1 ma及びmbがmg単位で表される場合,結果と
して濃度ρmの単位はmg/m3で表される。
注記2 例えば,25 ℃,101.3 kPaのような,特定条
件における濃度に換算する必要がある場合
は,式(2)を用いる。
298
273
3.
101
m
c
+
×
×
=
T
P
ρ
ρ
······························ (2)
ここに, ρc: 特定条件に換算した試料空気中の分析
対象成分の濃度
P: サンプリング時の気圧(kPa)
T: サンプリング時の気温(℃)
9.1 分析
対象成分
の質量濃
度
6
v
b
a
10
×
×
−
=
t
q
m
m
ρ
········································ (1)
ここに, ρ: 試料空気中の分析対象成分の質量濃度
(μg/m3)
ma: 8.4で求めた試料中の分析対象成分の
質量(μg)
mb: ブランクサンプラ中の分析対象成分の
質量(μg)
qv: 拡散取込み速度(cm3/min)(表1又は
8.6)
t: 暴露時間(min)
備考1. ma及びmbがmg単位で表される場合,結果と
して濃度ρの単位はmg/m3で表される。
2. 例えば,25 ℃,101.3 kPaのような,特定条
件における濃度に換算する必要がある場合
は,式(2)による。
298
273
3.
101
c
+
×
×
=
T
P
ρ
ρ
································ (2)
ここに, ρc: 特定条件に換算した試料空気中の分析
対象成分濃度
P: サンプリング時の気圧(kPa)
T: サンプリング時の気温(℃)
関連JIS間での記号の統一
10.2 分析
対象成分
の体積比
6
b
a
10
×
×′
−
=
t
u
m
m
ϕ
ρ
······································· (3)
ここに, ρφ: 空気中の分析対象成分の体積分率
(μL/m3)
u': 拡散取込み速度[ng/(μL/L)min(表1又
は9.6)]
t: 暴露時間(min)
注記 ma及びmbがmg単位で表される場合,結果とし
て濃度ρφの単位はmL/m3で表される。
9.2 分析
対象成分
の体積濃
度
6
v
b
a
10
×
×
−
=
t
q
m
m
φ
········································ (3)
ここに, φ: 空気中の分析対象成分の体積分率
(μL/m3)
qv: 拡散取込み速度[ng/(μL/L)min(表1又
は8.6)]
t: 暴露時間(min)
備考 ma及びmbがmg単位で表される場合,結果とし
て濃度φの単位はmL/m3で表される。
関連JIS間での記号の統一
3
9
A
1
9
6
7
:
2
0
1
5
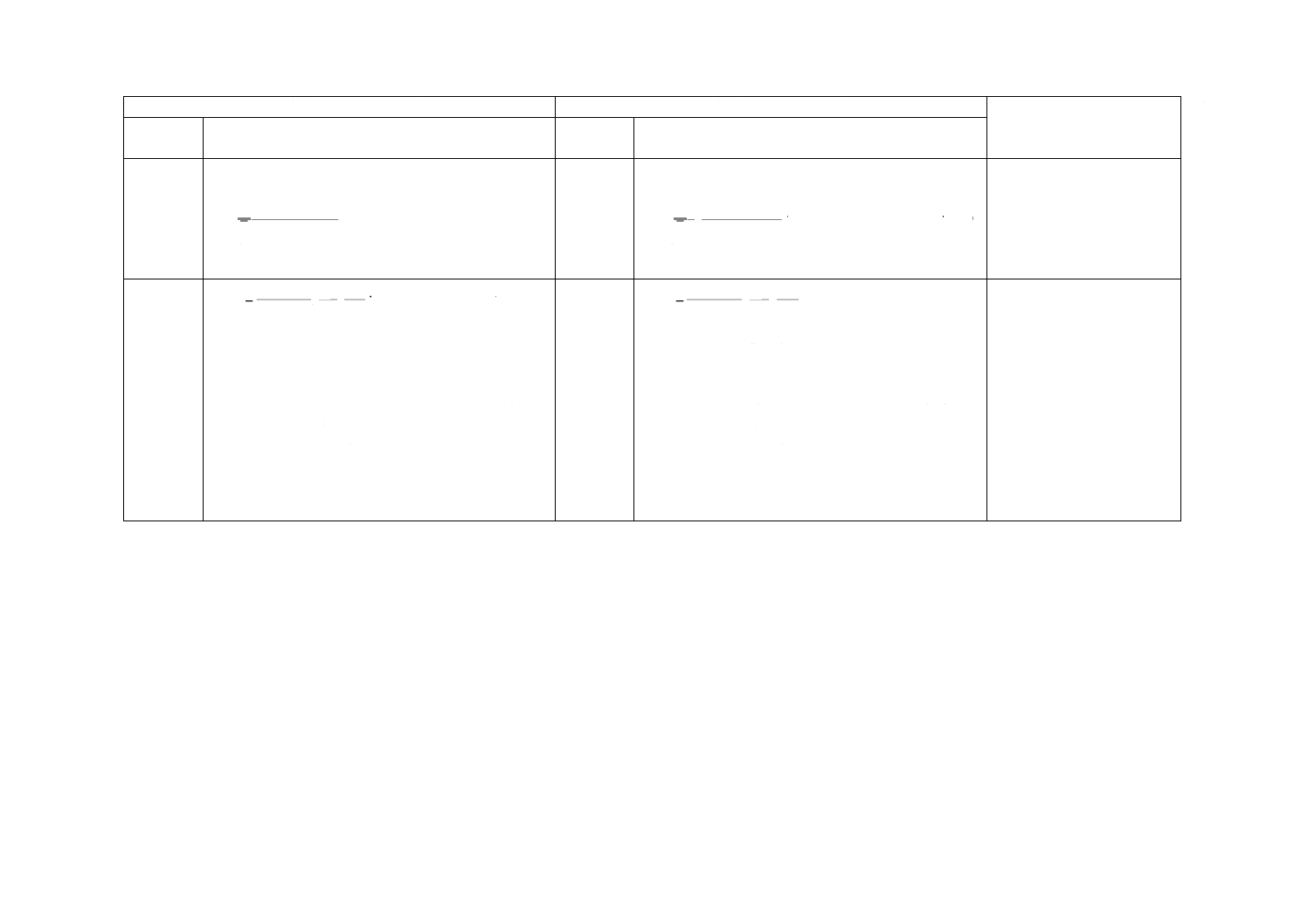
40
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格(JIS A 1967:2015)
旧規格(JIS A 1967:2005)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
10.3 拡散
取込み速
度u及び
u'の関係
取込み速度u(cm3/min)及びu'[ng/(μL/L)/min]は,
式(4)の関係にある。
3.
101
0.
24
293
×
×
×
×
×
=
′
T
P
M
u
u
····································· (4)
ここに, M: 分析対象成分の分子量
24.0: 20 ℃,101.3 kPaにおけるモル体積
9.3 拡散
取込み速
度qv及び
qv' の関係
取込み速度qv(cm3/min)及びqv'[ng/(μL/L)/min]は,
式(4)の関係にある。
3.
101
5.
24
293
v
v
×
×
×
×
×
=
′
T
P
M
q
q
··································· (4)
ここに, M: 分析対象成分の分子量
24.5: 25 ℃,101.3 kPaにおけるモル体積
関連JIS間での記号の統一
及びISO規格への整合
A.1 原理
l
t
D
A
m
×
−
×
×
=
)
(
2
1
f
ρ
ρ
····························· (A.1)
ここに, A: 断面積
D: 拡散係数
ρ1: 拡散キャップにおける分析対象成分の
濃度
ρ2: 吸着剤ベッドにおける分析対象成分の
濃度
mf: 分析対象成分の質量
l: 吸着剤表面から拡散キャップまでの距
離
t: 時間
A.1 原理
l
t
D
A
m
×
−
×
×
=
)
(
2
1
s
ρ
ρ
ここに, A: 断面積
D: 拡散係数
ρ1: 拡散キャップにおける分析対象成分の
濃度
ρ2: 吸着剤ベッドにおける分析対象成分の
濃度
ms: 分析対象成分の質量
l: 吸着剤表面から拡散キャップまでの距
離
t: 時間
関連JIS間での記号の統一
4
0
A
1
9
6
7
:
2
0
1
5
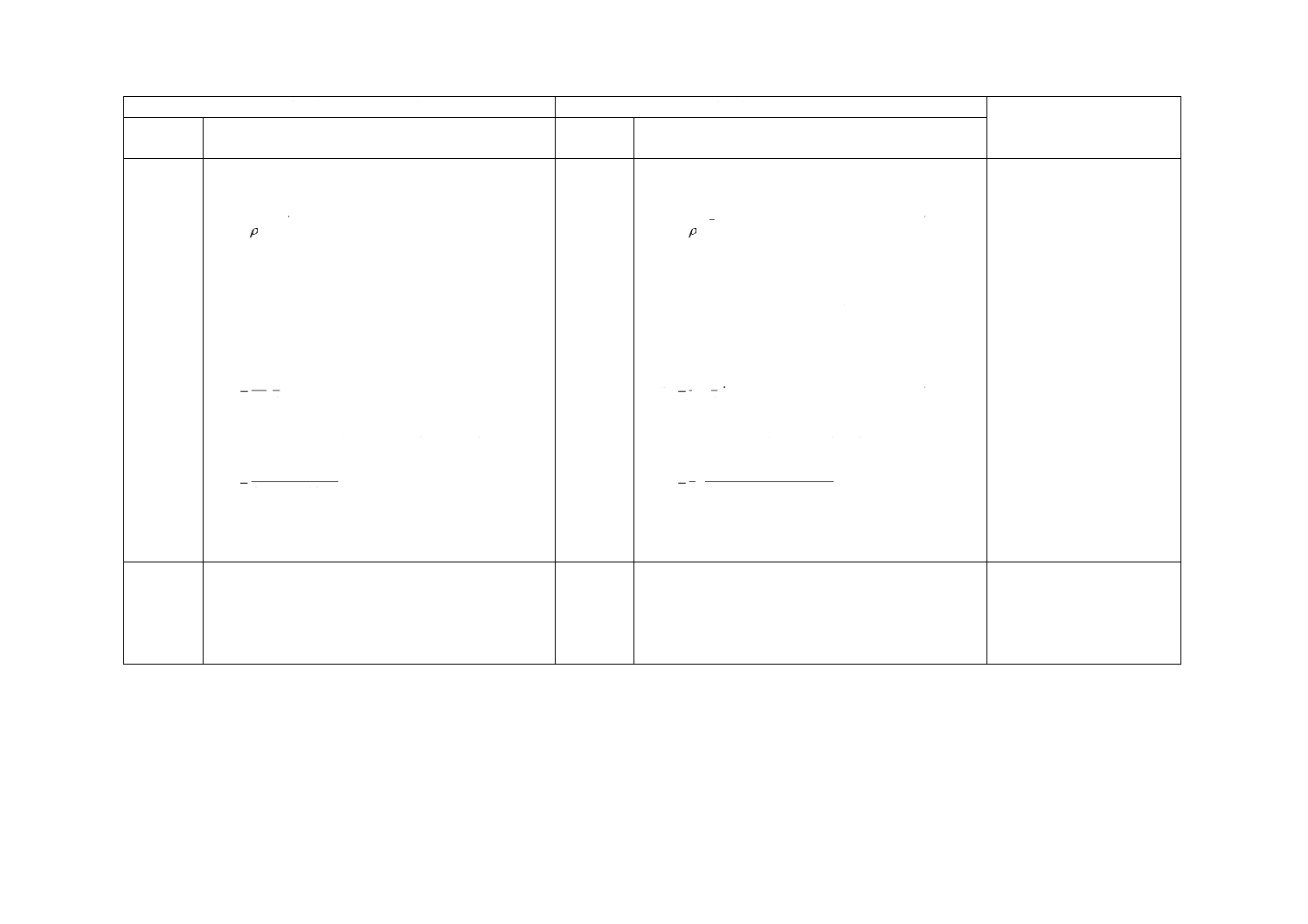
41
A 1967:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格(JIS A 1967:2015)
旧規格(JIS A 1967:2005)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
A.2 拡散
取込み速
度の単位
一定濃度ρ(μg/m3)のガス又は蒸気の拡散取込み速度
u(cm3/min)は,式(A.2)で求める。
t
m
u
×
=ρf ··············································· (A.2)
ここに, mf: 分析対象成分の質量(pg)
t: 暴露時間(min)
注記1 取込み速度uの単位はcm3/minであるが,こ
れはpg/μg/m3/minから求めたものである。ま
た,これは,分析対象成分を含む空気の実際
の流量を表すわけではない。
t
m
u
×
=
′
1
f
φ
·············································· (A.3)
注記3 理想的かつ実用的な拡散取込み速度u'
(ng/ppb/min)は,次の式(A.4)で求める。
3.
101
0.
24
293
×
×
×
×
×
=
′
T
P
M
u
u
·································· (A.4)
ここに, M: モル質量(g)
T: 絶対温度(K)
P: 大気圧(kPa)
A.2 拡散
取込み速
度の単位
一定濃度ρ(μg/m3)のガス又は蒸気の拡散取込み速度
qV(cm3/min)は式(A.2)で求める。
t
m
q
×
=ρs
V
············································· (A.2)
ここに, ms: 分析対象成分の質量(pg)
t: 暴露時間(min)
備考1. 取込み速度qVの単位はcm3/minであるが,こ
れはpg/μg/m3/minから求めたものである。ま
た,これは,分析対象成分を含む空気の実際
の流量を表すわけではない。
t
m
q
×
=
′
1
s
V
φ
············································· (A.3)
3. 理想的,かつ,実用的な拡散取込み速度qV'
(ng/ppb/min)は次の式で求める。
101
0.
24
293
V
V
×
×
×
×
×
=
′
T
P
molarmass
q
q
····················· (A.4)
ここに, molarmass: モル質量(g)
T: 絶対温度(K)
P: 大気圧(kPa)
関連JIS間での記号の統一
A.3 非理
想的な吸
着剤の選
択による
偏り
弱い吸着剤を用いると,式(A.1)のρ2とmfとがサンプ
リング時間の経過とともに減少する。したがって,式
(A.2)のuも,また,サンプリング時間の経過とともに
減少する。
A.3 非理
想的な吸
着剤の選
択による
偏り
弱い吸着剤を用いると,式(A.1)のρ2とmsとがサンプ
リング時間の経過とともに減少する。したがって,式
(A.2)のqVもまたサンプリング時間の経過とともに減
少する。
関連JIS間での記号の統一
4
1
A
1
9
6
7
:
2
0
1
5